 Ju-Chun Pei
Ju-Chun Pei Da-Zhong Luo
Da-Zhong Luo Shiang-Shin Gau1
Shiang-Shin Gau1 Wen-Sung Lai
Wen-Sung Lai
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 01 October 2021
Sec. Psychopharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fpsyt.2021.742058
This article is part of the Research Topic Modulation of NMDA Receptors: From Bench Side to Clinical Applications in Psychiatry View all 11 articles
Schizophrenia is a severe mental illness that affects ~1% of the world's population. It is clinically characterized by positive, negative, and cognitive symptoms. Currently available antipsychotic medications are relatively ineffective in improving negative and cognitive deficits, which are related to a patient's functional outcomes and quality of life. Negative symptoms and cognitive deficits are unmet by the antipsychotic medications developed to date. In recent decades, compelling animal and clinical studies have supported the NMDA receptor (NMDAR) hypofunction hypothesis of schizophrenia and have suggested some promising therapeutic agents. Notably, several NMDAR-enhancing agents, especially those that function through the glycine modulatory site (GMS) of NMDAR, cause significant reduction in psychotic and cognitive symptoms in patients with schizophrenia. Given that the NMDAR-mediated signaling pathway has been implicated in cognitive/social functions and that GMS is a potential therapeutic target for enhancing the activation of NMDARs, there is great interest in investigating the effects of direct and indirect GMS modulators and their therapeutic potential. In this review, we focus on describing preclinical and clinical studies of direct and indirect GMS modulators in the treatment of schizophrenia, including glycine, D-cycloserine, D-serine, glycine transporter 1 (GlyT1) inhibitors, and D-amino acid oxidase (DAO or DAAO) inhibitors. We highlight some of the most promising recently developed pharmacological compounds designed to either directly or indirectly target GMS and thus augment NMDAR function to treat the cognitive and negative symptoms of schizophrenia. Overall, the current findings suggest that indirectly targeting of GMS appears to be more beneficial and leads to less adverse effects than direct targeting of GMS to modulate NMDAR functions. Indirect GMS modulators, especially GlyT1 inhibitors and DAO inhibitors, open new avenues for the treatment of unmet medical needs for patients with schizophrenia.
Schizophrenia is a devastating mental illness, and the lifetime prevalence of schizophrenia is ~1%. Globally, there were 1.13 million schizophrenia cases and 12.66 million DALYs (disability-adjusted life years) due to schizophrenia in 2017 (1). The global burden of schizophrenia remains large and continues to increase, increasing the burden on health-care systems worldwide. This debilitating brain disorder typically emerges in late adolescence and early adulthood and is characterized by three main symptoms: positive symptoms, negative symptoms, and cognitive deficits (2, 3). Positive symptoms include delusions, hallucinations, and disorganized thoughts and speech typically regarded as manifestations of psychosis. Negative symptoms include reduced affect display, alogia, anhedonia, asociality, avolition, lack of emotional response, and motivation. Cognitive deficits include dysfunctions in working memory, attention, processing speed, visual and verbal learning with substantial deficits in reasoning, planning, abstract thinking, and problem solving. Cognitive impairments and negative symptoms, as the core features of schizophrenia, are enduring and correlate with the degree of disability (4, 5).
Currently, antipsychotic medications are mainstays in the treatment of schizophrenia and a range of other psychotic disorders. Positive symptoms of schizophrenia often respond well to antipsychotic drugs. In contrast, the available antipsychotic medications, which mainly affect the dopamine and serotonin receptor systems, are relatively ineffective in improving negative and cognitive deficits. Negative symptoms of schizophrenia tend to linger or worsen over time and are accompanied by impaired cognitive function in patients with schizophrenia (6). The improvement of cognitive dysfunction is a better predictor of patient quality of life (7, 8). Since existing pharmacological and biological therapeutic modalities fail to improve cognitive symptoms, various cognitive remediation strategies have been adopted (9). In addition, the cognitive deficits in adolescents at risk for schizophrenia and in patients after their first episode of schizophrenia suggest that schizophrenia-related cognitive dysfunction is not the result of chronic illness (10). The US National Institute of Mental Health (NIMH) thus developed the Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS), which significantly raised awareness of the cognitive dysfunction in schizophrenia (11). In addition to the reliance on the dopamine receptor D2 (DRD2) as a conventional therapeutic target (12), a focus on the different symptom domains of schizophrenia may lead to the identification of different endophenotypic markers that can promote the development of novel therapeutics useful for rational cellular and molecular targets.
Similar to those of many other psychiatric disorders, the etiology and pathophysiology of schizophrenia remain unclear. Accumulating evidence from human genetic studies and association studies has revealed several schizophrenia susceptibility loci and genes. A genome-wide association study (GWAS) revealed notable associations relevant to the major hypotheses of the etiology and treatment of schizophrenia, including DRD2 (the main target of many effective antipsychotics) and multiple genes [e.g., metabotropic glutamate receptor 3 (GRM3), glutamate ionotropic receptor NMDA type subunit 2A (GRIN2A), serine racemase (SR), and glutamate receptor, ionotropic, AMPA receptor 1 (GRIA1)] involved in glutamatergic neurotransmission and synaptic plasticity (13). In contrast to the conventional view of dopamine involvement in schizophrenia (i.e., the dopamine hypothesis of schizophrenia), glutamatergic neurotransmission has been gradually attracting attention in the investigation of the pathophysiology and treatment of schizophrenia in recent decades (14–16).
In the central nervous system (CNS), glutamate is the main excitatory neurotransmitter and activates metabotropic and ionotropic glutamate receptors. NMDARs are ionotropic glutamate-gated cation channels with high calcium permeability that play vital roles in synaptic transmission, neuroplasticity, and cognitive functions. Heterotetrameric NMDARs are widely distributed throughout most of the brain and are composed of two obligatory GluN1 (NR1) subunits with either two GluN2 (NR2) subunits or a combination of GluN2 (NR2) and GluN3 (NR3) subunits. As illustrated in the top left panel of Figure 1, activation of NMDARs requires not only the binding of glutamate on the GluN2 subunit but also the binding of the coagonist glycine or D-serine at the glycine modulatory site (GMS, also referred to as the glycine-B site or the strychnine-insensitive glycine site) on the GluN1 subunit (17). Intriguingly, although the endogenous high-potency coagonists glycine and D-serine are present in the extracellular space (18), the GMSs on NMDARs are not saturated in vivo (19). D-serine appears to be the dominant endogenous coagonist for NMDARs and a modulator for NMDAR-related neurotoxicity, even though the levels of glycine are 10-fold higher than those of D-serine (20–22). The activation of NMDARs produces prolonged increases in intracellular calcium concentration and thus triggers downstream signaling cascades involved in the regulation of many physiological and pathophysiological processes (23).
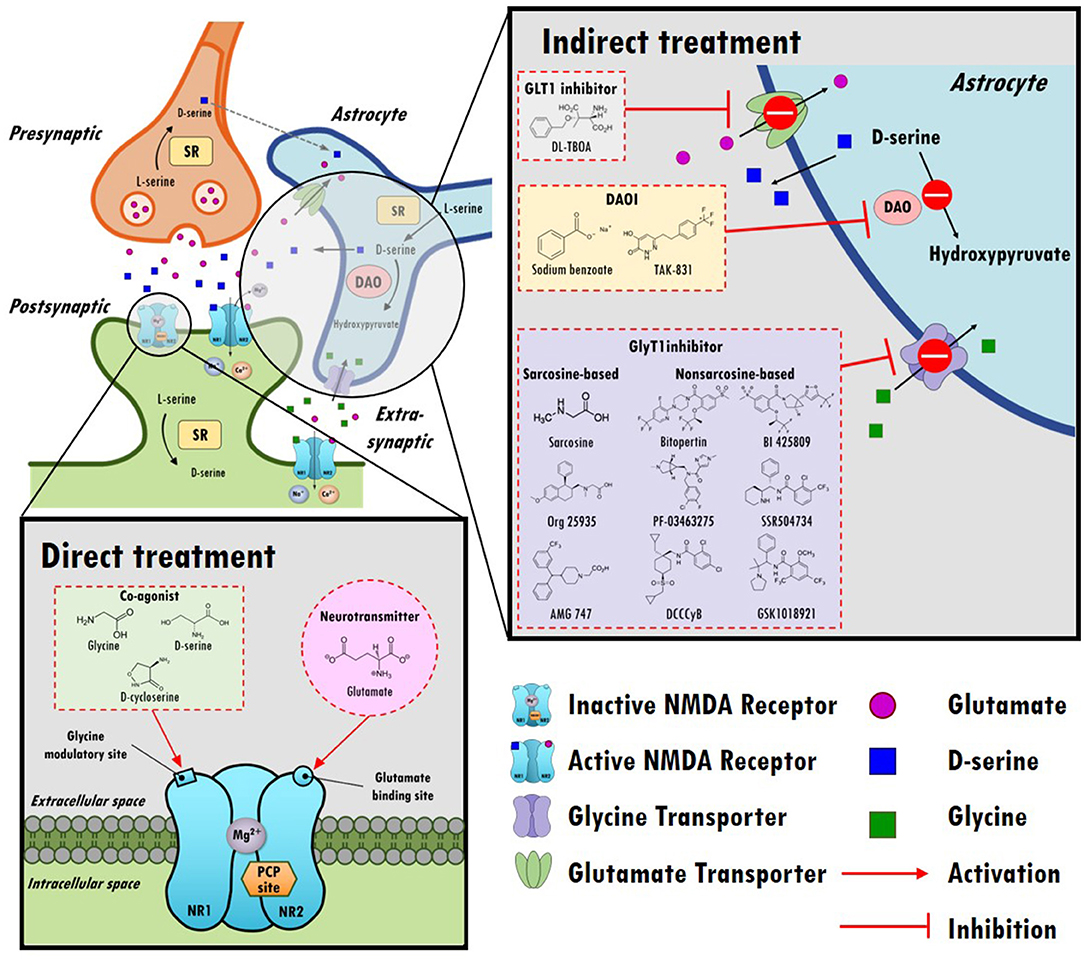
Figure 1. An overview of the hypothesis of N-methyl-D-aspartate receptor (NMDAR) hypofunction in schizophrenia and the direct/indirect treatments in the regulation of NMDAR functioning. Top left panel: A model of glutamatergic trisynapses: pre-synapses, post-synapses, and astrocytes. Activation of NMDAR requires not only the binding of glutamate to the GluN2 (NR2) subunit but also the binding of the coagonist glycine or D-serine at the glycine modulatory site (GMS) of the GluN1 (NR1) subunit. In response to NMDAR activation, the intracellular calcium concentration increases and thereby triggers downstream signaling cascades. After activation, glutamate and glycine are taken up by astrocytes through the glutamate transporter (GLT1) and glycine transporter (GlyT1), respectively. D-serine, another coagonist of GMS, is predominantly produced in neurons, is synthesized from L-serine by serine racemase (SR) and is shuttled to astrocytes, where it is stored and released. NMDARs are critical for synaptic plasticity, cortical maturation, and learning and memory processes. The hypofunction of ionotropic glutamate NMDARs has been proposed to be a model of schizophrenia in humans, and NMDAR hypofunction plays a key role in the pathophysiology of schizophrenia. Bottom left panel: Enhancing NMDAR functions through direct treatments. Glutamate, glycine, D-cycloserine, and D-serine compounds directly target postsynaptic NMDAR and activate NMDAR functioning. Top right panel: Boosting NMDAR functions via indirect treatments (e.g., GLT1 inhibitors, DAO inhibitors, and GlyT1 inhibitors). GLT1 inhibitors block the reuptake of glutamate and increase the synaptic levels of glutamate. D-serine is metabolized into hydroxypyruvate by D-amino acid oxidase (DAO) in astrocytes. DAO inhibitors block the metabolism of D-serine, which prolongs the synaptic concentration of D-serine. GlyT1 inhibitors block the reuptake of glycine and increase synaptic levels of glycine.
NMDAR has been proposed to be an important and potential therapeutic target for many CNS and psychiatric disorders (24). There is increasing evidence acquired through different approaches supports the supposition that NMDAR hypofunction plays a role in schizophrenia. In addition to the abovementioned large-scale GWAS, copy number variant studies have also led to the identification of rare genetic variants in NMDAR-related genes and components related to the postsynaptic density associated with increased risk for schizophrenia (25, 26). Postmortem brain studies have also indicated decreased expression of the NR1 subunit (mRNA and protein) and NR2C subunit (mRNA) in the postmortem dorsolateral prefrontal cortex in schizophrenic patients (27) and reductions in D-serine and serine racemase (SR) levels in patients with schizophrenia (28). A meta-analysis study further indicated significant decreases in the expression of NR1 mRNA and protein in the prefrontal cortex of schizophrenic patients (29). In addition to these genetic and postmortem studies, aberrant NMDAR function has been identified via the use of psychotomimetic agents. Pharmacological studies have revealed that the use of NMDAR antagonists (e.g., phencyclidine (PCP) and ketamine) causes not only positive symptoms of schizophrenia but also negative symptoms and cognitive deficits in healthy humans (30–32). Subanesthetic doses of ketamine not only induce psychotomimetic effects but also increase amphetamine-induced dopamine release in the striatum, which has been observed in schizophrenic patients (33). In addition, positron emission tomography (PET) imaging data have indicated links between glutamatergic system dysfunction and schizophrenia (34). NMDAR hypofunction in parvalbumin (PV) interneurons has also been proposed as a pathological mechanism of schizophrenia (35). Proton magnetic resonance spectroscopy (MRS) studies have revealed increased glutamine levels in the medial prefrontal cortex, anterior cingulate cortex, and thalamus in drug-naïve patients with first-episode psychosis (36, 37), suggesting dysregulation of glutamate neurotransmission (38). Moreover, reduced activation of the prefrontal cortices (i.e., hypofrontality) has been considered to underlie negative symptoms and cognitive deficits in schizophrenia (39–41). Notably, it has been proposed that antipsychotic medications may reduce NMDARs activity and produce dysfunctions in the corticolimbothalamic circuit and hypofrontality in patients with schizophrenia (42). Accordingly, these studies indicate the involvement of NMDARs in the pathophysiology of schizophrenia and provide new potential targets for the treatment of schizophrenia.
Given the importance of glutamate in the NMDAR hypofunction hypothesis for schizophrenia and NMDAR-mediated neurotransmission, one possible strategy to boost NMDAR functions involves either directly or indirectly enhancing glutamate levels in synapses, as illustrated in the bottom left and top right portions of Figure 1. However, excessive glutamate induces high levels of calcium influx, which has been shown to lead to excitotoxicity and neuronal injury in cellular and animal models (43, 44). In addition, indirect enhancement of glutamate via DL-TBOA, a glutamate transporter 1 (GLT1) inhibitor, resulted in attenuated baroreflex control of sympathetic nerve activity and heart rate (45). Apparently, from a safety perspective, neither direct nor indirect enhancement of synaptic glutamate levels is a reasonable therapeutic approach in the regulation of NMDAR functions. Alternatively, agents that act at the GMSs of NMDARs have been proposed to be promising treatments to moderate severe negative symptoms and cognitive impairments.
A unique characteristic of NMDAR is that the GMS must be occupied by glycine and/or D-serine for glutamate to induce channel opening. GMS was first reported by Johnson and Ascher to facilitate the activation of NMDARs in cultured mouse brain neurons (18). It was later demonstrated that glycine is necessary to activate NMDARs (46). Mice carrying targeted point mutations in the GMS of the NMDAR NR1 subunit gene (Grin1) exhibited marked NMDAR hypofunctions and deficits in long-term potentiation and spatial learning (47, 48), as well as impaired social ability and spatial recognition (49). Accumulating evidence has indicated that binding to the GMS can enhance the affinity and efficacy of glutamate neurotransmission (50), and the administration of GMS agonists (e.g., glycine) can benefit schizophrenic patients by regulating NMDAR-mediated neurotransmission (19). The disturbance of GMS modulators found in schizophrenia patients has been identified as a contributor to NMDAR hypofunction. Previous studies have revealed reduced D-serine and SR in schizophrenia (28). In addition, the levels of kynurenic acid, the only known competitively endogenous antagonist of the GMS in NMDAR, are elevated in the postmortem brain tissue (51) and in the cerebrospinal fluid (CSF) of living schizophrenic patients (52), suggesting that GMS occupancy might be shifted toward antagonism in this disorder. Accordingly, modulation of NMDAR through the GMS has been proposed as a possible therapeutic target for the treatment of negative and cognitive symptoms in schizophrenia (53, 54).
Indeed, several agonists have been designed to either directly or indirectly target GMS due to its great potential for the treatment of negative and positive symptoms in schizophrenia. For example, 3-(4,6-dichloro-2-carboxyindol-3-yl) propionic acid, an indole-2-carboxylic acid derivative, has been found to have > 2,100-fold greater affinity for the GMS than glycine (55), and 3-hydroxy-imidazolidin-4-one derivatives are partial agonists of the GMS (56). Additional computational methods that can be used to identify potential agonists have been used (57). In addition to agonists of the GMS, GMS-specific antagonists, such as 7-chlorkynurenic or L-701,324, have been developed for research purposes (58, 59). Although numerous potential agonists and antagonists have been developed or identified, only a few of the candidates are suitable for advancement from preclinical studies to clinical trials. To date, most clinical studies have focused mainly on targeting the GMS using single amino acids as agonists of GMS, including glycine, D-cycloserine, and D-serine, as indicated in the bottom left panel of Figure 1.
Glycine is the simplest amino acid and acts as a neurotransmitter in the CNS. In addition to glycinergic terminals, glycine may be simultaneously released into the synaptic cleft with GABA (60). Extracellular glycine is immediately recycled through glycine transporters, including glycine transporter 1 (GlyT1) in glial cells or glutamatergic neurons, and glycine transporter 2 (GlyT2) in presynaptic neurons (61). Intracellular glycine is then metabolized into L-serine by serine hydroxymethyltransferase in glial cells or catabolized into carbon dioxide and ammonium by the glycine cleavage system in neurons (62). Glycine causes inhibitory and excitatory neural transmission via strychnine-sensitive glycine receptors and NMDA receptors, respectively. Glycine receptors are mainly located in the brainstem and spinal cord. In contrast, NMDARs are present in high density within the cerebral cortices and hippocampus and are thought to be involved in the pathophysiology of schizophrenia (24, 63).
Numerous investigations support decreased glutamatergic signaling and NMDAR hypofunction as pathogenic mechanisms of schizophrenia. Interestingly, it has been reported that glycine is upregulated in patients with schizophrenia. Findings on schizophrenic patients obtained postmortem have revealed increased binding activity of radiolabeled [3H]glycine in the brain, especially in the parietal cortex and occipital cortex (64). Rats treated with a glycine-rich diet for a long period also exhibit schizophrenia-like abnormalities, including altered sensory gating function, enlarged cerebral ventricles, and diminished hippocampal dimensions (65). Similarly, high serum glycine levels have been reported in patients with chronic schizophrenia, and these levels have been associated with impaired sensorimotor gating function in pre-pulse inhibition (66). These findings imply that glycine levels might compensate for alterations in glutamate-NMDAR transmission in patients with chronic schizophrenia. For example, a postmortem study indicated a striking decrease in tyrosine phosphorylation of the GluN2 subunit in the dorsolateral prefrontal cortex of schizophrenic patients, but the postsynaptic density of NMDAR complexes in these patients was, in fact, increased (67). Inconsistently, lower plasma glycine levels have reported in schizophrenic patients compared to healthy controls and have been correlated with negative symptoms of schizophrenia (68). To further elucidate the glycine levels in the brains of schizophrenia patients, it is necessary to measure glycine levels in serum and CSF in a large sample size.
Despite the controversial findings regarding glycine levels in patients with schizophrenia, glycine-induced augmentation of NMDAR-mediated neurotransmission has been considered a potentially safe, and feasible approach for ameliorating negative symptoms of schizophrenia. Glycine appears to be safe, even at dosages of as high as 5 g/kg per day in rats (69) and 0.8 g/kg body weight per day in schizophrenic patients (70). In addition to its high biocompatibility and low toxicity, the effect of glycine on the amelioration of schizophrenia-related symptoms has been demonstrated in animal models of schizophrenia. Subchronic administration of glycine at doses relevant to its clinical effects (71) significantly prevents PCP-induced abnormalities in auditory mismatch negativity (MMN, a neurophysiological characteristic of schizophrenia) (72). Glycine also significantly reduced novelty- and methamphetamine-induced locomotor activity in neonatal ventral hippocampal damaged rats compared with sham rats (73). In addition, microinjection of 1 μmol of glycine into the mouse prefrontal cortex alleviated PCP-induced behavioral deficits in latent learning (74), suggesting the involvement of glycine in the regulation of frontocortical NMDARs and cognitive functions. Glycinamide, a prodrug of glycine, can be converted to glycine in CNS by hydrolysis and it prevented MK-801 (dizocilpine, a non-competitive antagonist of NMDAR)-induced deficits in a novel object recognition task in rabbits (75, 76). Despite contrasting neurochemical profiles, a recent study further proved that partial glycine site agonists and glycine reuptake inhibitors display comparable precognitive effects in rats and therefore have potential relevance as treatments of cognitive impairments in schizophrenia (77).
The effects of glycine on the treatment of schizophrenic symptoms in clinical studies are summarized in Table 1. Briefly, in the late 1980s, a series of open-label clinical studies failed to demonstrate the therapeutic potential of glycine in the amelioration of negative symptoms of schizophrenia (78–80). Milacemide, an acylated prodrug of glycine, did not alleviate schizophrenic symptoms, and psychotic symptoms were worsened (91, 92). Later, glycine was demonstrated to improve negative symptoms at 0.4 g/kg/day (81). Consistently, recent clinical studies have also indicated that a high dose of glycine is associated with improvement in clinical rating scales of schizophrenia, especially scales of negative symptoms (70, 71, 82, 83, 86, 89, 90). However, inconsistent results have been reported and indicate that glycine administered with clozapine had no effect on patients with schizophrenia (84, 85, 87). In a 16-week randomized double-blind, double-dummy, and parallel-group clinical trial conducted at four sites in the United States and one site in Israel, no significant differences were found between the total average scores on the Scale for the Assessment of Negative Symptoms (SANS) of patients treated with glycine or placebo, and no change in the average cognitive scores was apparent (88). The lack of consistency across trials could be due to small sample sizes, different doses of glycine, different trial durations, and different clinical ratings. Notably, glycine is an inhibitory neurotransmitter in glycinergic neurons, and it has been reported to have poor CNS penetration (i.e., rate of permeation across the blood-brain barrier) (93). Therefore, higher doses of glycine might be required for treatment purpose in patients. Unfortunately, systemic administration of high-does glycine is problematic and is not well-tolerated. The administration of high-dose glycine can result in some unwanted adverse effects, such as nausea (71, 83, 87) and sensorimotor gating deficits (94). Thus, these studies suggest that glycine is not a generally effective therapeutic option for treating negative symptoms or cognitive impairments. It seems wise to explore other drug candidates targeting GMS in the glutamatergic system.
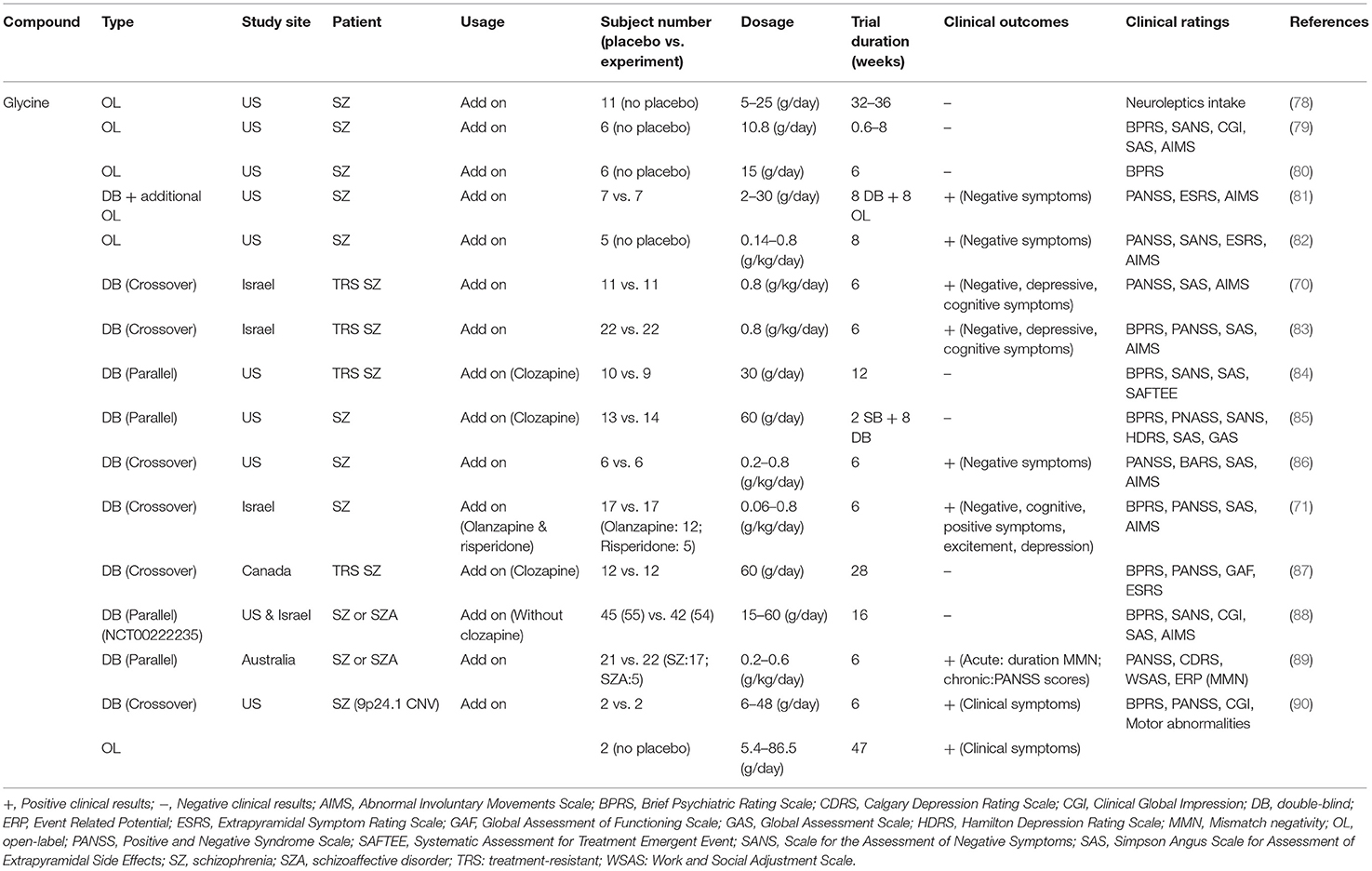
Table 1. Summary of effects of glycine on the treatment of schizophrenic symptoms in clinical studies.
D-cycloserine is a well-known antibiotic metabolite produced by Streptomyces orchidaceus and Streptomyces garyphalus that has therapeutic effects on tuberculosis. D-cycloserine has also been found to act as a partial agonist targeting the GMS of NMDAR (95), and its binding affinity is 100-fold less than that of glycine (96). Similar to glycine, D-cycloserine has been reported to improve cognitive functions through modulation of NMDAR function in animal studies. For example, both systemic administration and intra-amygdala infusions of D-cycloserine facilitated conditioned fear extinction and improved memory consolidation in rats (97, 98). Single administration of D-cycloserine also significantly improved visual recognition memory in rhesus monkeys (99). However, inconsistently, some studies reported that D-cycloserine had no effect on neural activity in a mouse model of schizophrenia (100), MK-801-induced sensorimotor gating dysfunction in mice (101), or acquisition of memory performance in MK-801-treated rats in the radial arm maze and the water maze (102).
Similarly, inconsistent findings have also been reported in clinical studies. Effects of D-cycloserine on the treatment of schizophrenic symptoms in clinical studies are summarized in Table 2. Briefly, some studies indicated that D-cycloserine at a dosage of 50 or 100 mg/day had therapeutic effects in the treatment of negative symptoms and/or cognitive deficits (90, 103, 106, 108, 111, 112, 115–119). In contrast, others reported that D-cycloserine had no effect on patients with schizophrenia (88, 104, 107, 113, 114). There are several possible explanations for the contradictory findings in clinical studies. First, D-cycloserine has a very narrow therapeutic window. The administration of D-cycloserine >100 mg/day has been reported to result in the deterioration of clinical outcomes in patients with schizophrenia (96, 103, 110). It has been shown that D-cycloserine has neurotoxic side effects, including hyperexcitability, depression, anxiety, memory deficits, and even seizures (121). Second, D-cycloserine administered with clozapine can result in drug-drug interactions, which might lead to the exacerbation of symptoms in patients (105, 109). Third, the treatment effect of D-cycloserine might be influenced by heterogeneity caused by differences in onset age and white matter integrity (120). In addition, a study revealed that patients receiving D-cycloserine demonstrated a significant increase in temporal lobe activation, suggesting that the addition of D-cycloserine to conventional neuroleptics may improve negative symptoms through enhanced temporal lobe function (115). Finally, a meta-analysis indicated that full agonists (such as glycine and D-serine) appear to be more effective than partial agonists (such as D-cycloserine) (122, 123). Thus, the therapeutic potential of D-cycloserine appears to be limited and not particularly effective.
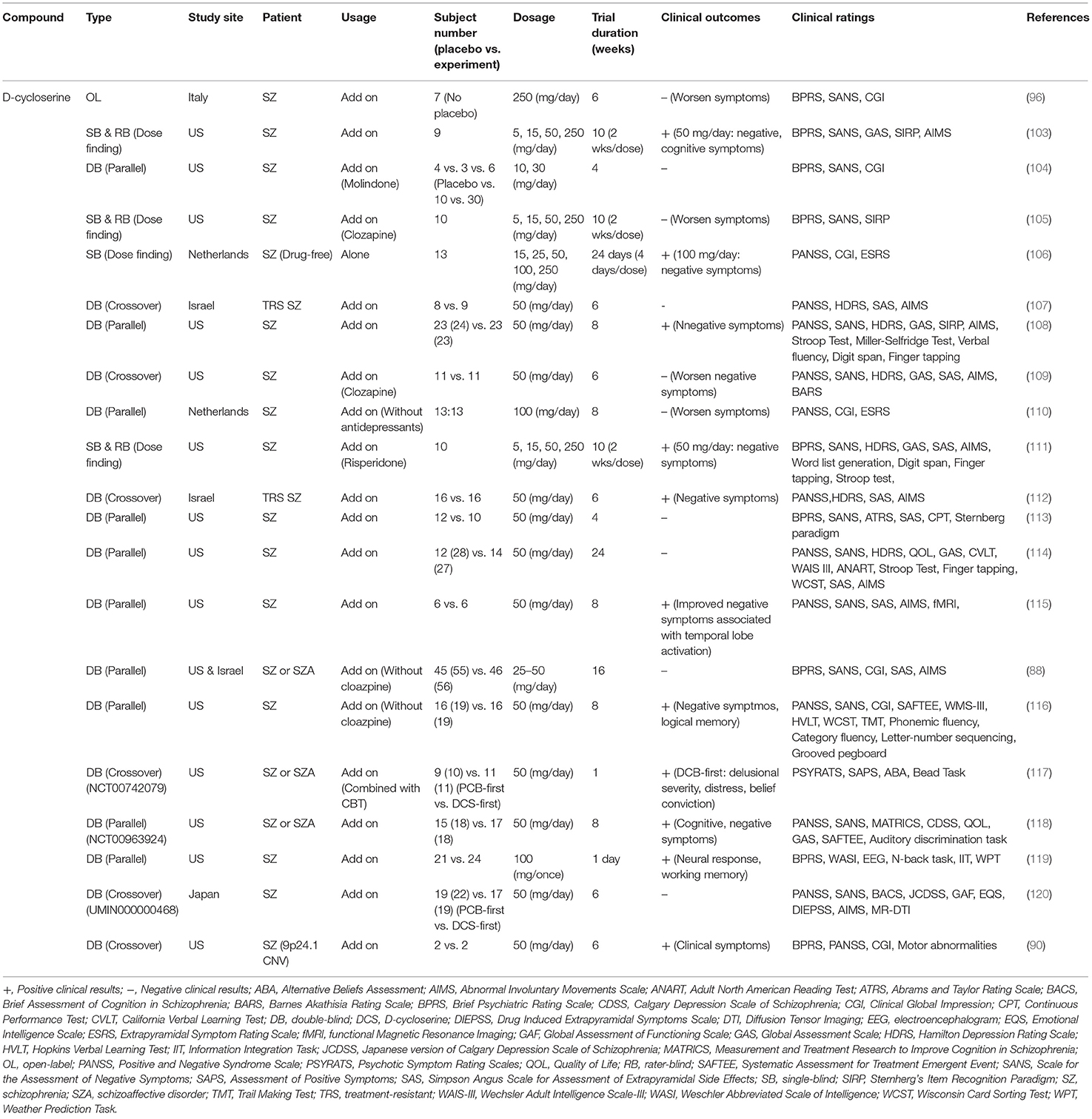
Table 2. Major findings in clinical trials examining effects of D-cycloserine on the treatment of schizophrenic symptoms.
D-serine is enriched in the forebrain and is an endogenous ligand of the GMS on NMDAR (124). Emerging evidence suggests the potential role of D-serine in the regulation of NMDAR functions for the treatment of schizophrenia. For the GluN1/N2 subunits of NMDAR, the binding affinity of D-serine is three-fold more potent than that of glycine (125). D-serine is mainly expressed by glutamatergic neurons, even though there has been considerable controversy regarding the concentration and function of D-serine in glial cells and neurons (126). D-serine is predominantly produced in neurons by the stereoconversion of L-serine (provided by astrocytes) via the PLP-dependent enzyme serine racemase (SR) and is then shuttled to astrocytes, where it is stored and released. Studies using more-selective antibodies have demonstrated that SR and D-serine are prominently expressed in forebrain glutamatergic neurons (127–130). In addition, the distribution of D-serine residues in the brain is similar to that of NMDARs (131). Intriguingly, it has been reported that the deletion of neuronal SR resulted in impaired NMDAR functions and synaptic plasticity, whereas deletion of astrocytic SR had no effect (132). Notably, D-serine is the primary coagonist of synaptic NMDARs, whereas glycine is the primary coagonist of extrasynaptic NMDARs (22). In general, D-serine is an allosteric modulator of brain NMDARs and is predominantly released from glutamatergic neurons.
Emerging evidence suggests that D-serine is involved in the pathophysiology of schizophrenia and is a potential therapeutic agent and/or biomarker for schizophrenia. Indeed, decreased levels of D-serine in serum and CSF have been found in patients with schizophrenia compared to those in healthy controls (133). A CSF and postmortem brain study also revealed a 25% decreases in D-serine levels and the D/L-serine ratio in the CSF of schizophrenia patients, suggesting that reduced brain SR and elevated D-amino acid oxidase (DAO) protein levels may contribute to the lower D-serine levels observed in the CSF of schizophrenic patients (28). A recent study further indicated that poor executive function performance is associated with a lower D-serine/total serine ratio in schizophrenic patients (134). Moreover, accumulating evidence has indicated that alteration of D-serine is associated with neuroplasticity and cognitive deficits in schizophrenia. For example, supplementation with D-serine prevented the onset of cognitive deficits in adult offspring after maternal immune activation in pregnant mice (135), suggesting that early intervention with D-serine may prevent the occurrence of psychosis in high-risk subjects. Decreasing synaptic D-serine by enhancing Na+-independent alanine–serine–cysteine transporter-1 abolished long-term potentiation (LTP) and reduced synaptic NMDAR responses by 60–70% (136). Taking advantage of SR-null mice, a series of studies confirmed that D-serine is required for NMDAR responses, NMDAR-dependent LTP, dendritic spine formation, cognitive functions, and social memory (137–141). However, D-serine is metabolized rapidly by DAO, reducing its bioavailability and requiring the administration of high doses, which may lead to peripheral neuropathies, creating a potential problem for the use of D-serine in treating schizophrenia-related symptoms (142, 143). D-serine levels in blood and urine are sensitive to the presence of kidney dysfunction of different origins. There are also concerns that high concentrations of D-serine augment kidney dysfunction and cause potential nephrotoxicity, which has been reported in rats that have developed acute tubular necrosis associated with higher doses of D-serine (144, 145). Nevertheless, serum D/L-serine levels might provide a measurable biological marker for schizophrenia, and D-serine may be effective for the treatment of negative symptoms and cognitive dysfunction in schizoprhenia. The study of D-serine requires accurate methodologies and specific controls, and a specific guideline for accurate measurement and detection methods has been described previously (146).
Along the same lines, D-serine has been employed alone or as an add-on treatment to standard antipsychotics for improving positive, negative, and cognitive symptoms of schizophrenia in numerous clinical studies (147–159). Effects of D-serine on the treatment of schizophrenic symptoms in clinical studies are summarized in Table 3. Briefly, some clinical studies have demonstrated positive outcomes for D-serine (147, 149–151), and repeated D-serine administrations have been shown to improve MMN and cortical plasticity in patients with schizophrenia (156, 157). However, other studies have revealed negative results (148, 152–155). A meta-analysis indicated that the effect size of D-serine on the treatment of negative symptoms (SMD = −0.319) and positive symptoms (SMD = −0.211) appeared to be small (160). In particular, in the first randomized double-blind placebo-controlled study with 60 mg/kg D-serine in schizophrenia, D-serine led to significant improvement in MMN frequency generation and clinical symptoms (157), which is consistent with another meta-analyses showing significant effects of D-serine on schizophrenia. This study also implied that a minimum daily dose of 3.6 g D-serine is needed to improve negative symptoms. However, high concentrations of D-serine can lead to peripheral neuropathies, such as oxidative damage (161), neurotoxicity (162), and renal toxicity (150, 163). In summary, these studies indicate that the therapeutic benefit of D-serine may be limited due to its adverse effects.
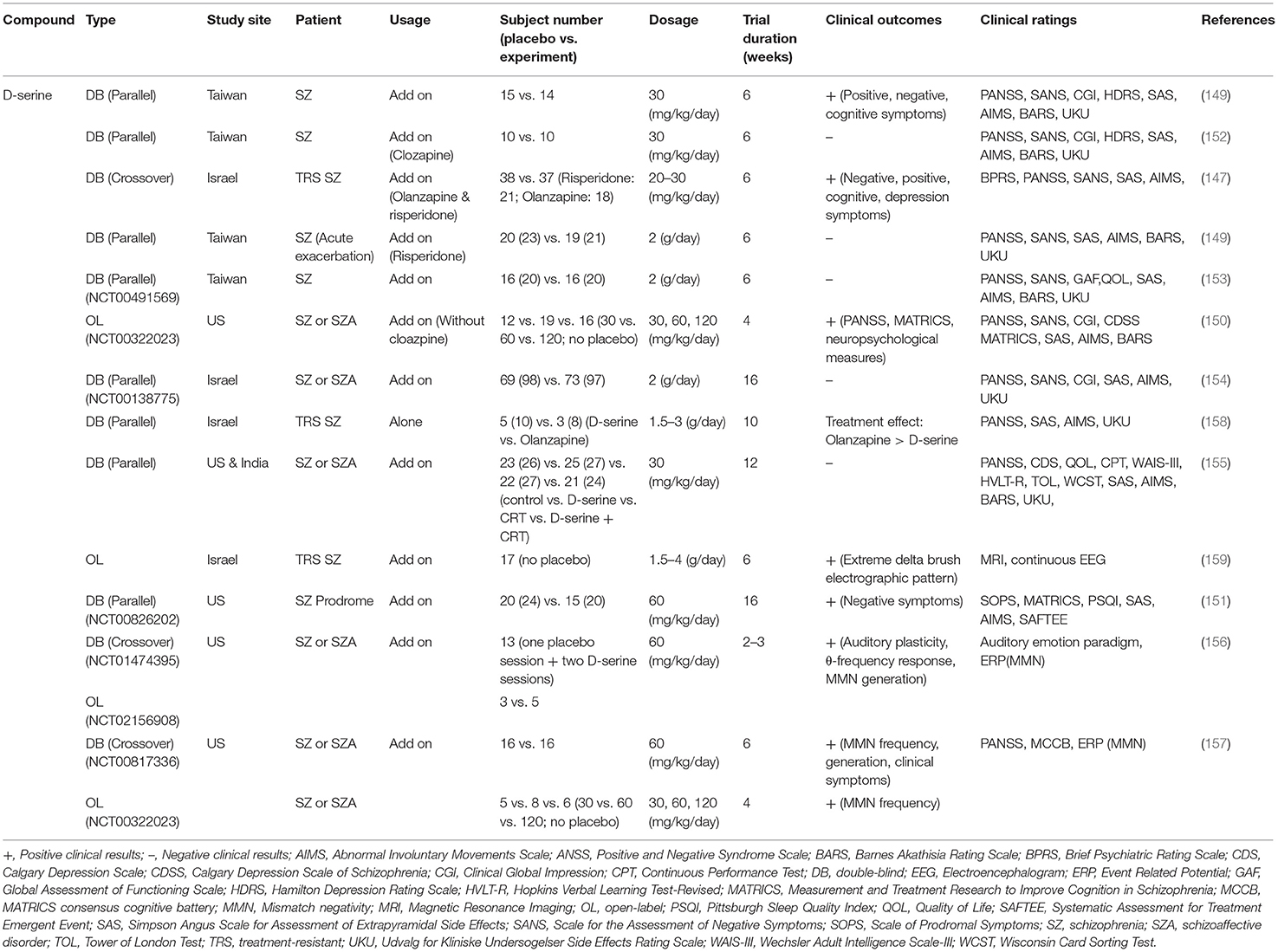
Table 3. Summary of clinical outcomes and benefits related to D-serine in patients with schizophrenia.
As described previously, activation of NMDARs requires the binding of a coagonist, D-serine or glycine, at the GMS of NMDARs. To date, the GMS on NMDAR is one of the most promising therapeutic targets for contributing to the medical needs of patients with schizophrenia. However, the beneficial effect of directly targeting the GMS with D-serine is limited because of the requirements for a high dose, narrow therapeutic window and poor CNS penetration rate, concomitant side effects and potential drug-drug interactions. Alternatively, as illustrated in the right panel of Figure 1, indirectly targeting the GMS of NMDARs via enhancement of synaptic glycine/D-serine levels from in astrocytes provides a new approach to modulate NMDAR functions and to help meet the needs of patients in schizophrenia (164).
A glycine reuptake inhibitor inhibits the reuptake of synaptic glycine by blocking astrocytic glycine transporters and increasing the availability of glycine at the synaptic cleft. Glycine transporter type 1 (GlyT1) is expressed at glutamatergic synapses throughout mammalian brain regions and primarily regulates the synaptic concentrations of glycine (165). GlyT1 is highly colocalized with NMDARs on glial cells and neurons in the cortex, hippocampus, septum and thalamus (166). GlyT1 effectively regulates synaptic glycine reuptake and governs GMS occupancy at NMDARs in excitatory synapses (19). Thus, selective inhibition of astrocytic GlyT1 is a promising new therapeutic target for indirectly enhancing synaptic glycine concentrations and facilitating NMDAR function.
Accumulating evidence from preclinical studies indicates that inhibition of GlyT1 enhances NMDAR functions in animals. Initial studies have revealed that glycyldodecylamide, a non-selective glycine transport antagonist, reverses PCP-induced behavioral deficits (167, 168). Subsequently, a series of studies consistently demonstrated that administration of N[3-(40-fluorophenyl)-3-(40-phenylphenoxy)propyl]-sarcosine (NFPS, also known as Alx5470), a GlyT1 inhibitor, enhanced LTP and behavioral performances in associative learning, spatial and object memory, and social memory (140, 141, 169–171). In agreement with the results obtained with NFPS, a series of studies also indicated that sarcosine, another GlyT1 inhibitor, has promising therapeutic potential in ameliorating behavioral impairments and cognitive deficits in both pharmacological and genetic mouse models of schizophrenia (139, 172, 173). Furthermore, sarcosine has been proven to effectively regulate the surface trafficking of NMDARs, NMDAR-evoked electrophysiological activity, brain glycine levels and MK-801-induced abnormalities in the brain, which might contribute to the therapeutic effect for the treatment of schizophrenia (139). Intriguingly, it has been proven that sarcosine also binds to the GMS of NMDARs and enhances NMDAR functions through more than one mechanism (139, 174). In addition, other GlyT1 inhibitors, such as SSR504734 and ORG 24598, have also displayed similar beneficial effects in sensorimotor gating, learning and memory functions, and schizophrenia-like behaviors (175–178). Furthermore, selective genetic disruption of GlyT1 resulted in enhancement of NMDAR functions, spatial retention memory, selective attention, and procognitive and antipsychotic phenotypic profiles, suggesting that inhibition of GlyT1 might have both cognitive-enhancing and antipsychotic effects (179–181). These studies indicate that GlyT1 is an attractive and promising drug target for the treatment of schizophrenia-related behaviors and cognitive deficits, even though the high binding affinity of the GlyT1 inhibitor can cause unpredictable toxicity leading to a coma-like state, compulsive walking or respiratory distress (15, 182).
With the aim of treating unmet medical needs in schizophrenia, a number of pharmaceutical industries have developed selective GlyT1 inhibitors as novel therapeutic drugs for schizophrenia. Numerous clinical studies have been carried out to evaluate the effects of special GlyT1 inhibitors on the treatment of schizophrenic symptoms. Based on the chemical structures of GlyT1 inhibitors, these clinical studies can be divided into two major structural classes: sarcosine-based and non-sarcosine-based inhibitors, and the summaries of these studies are shown in Tables 4, 5, respectively.
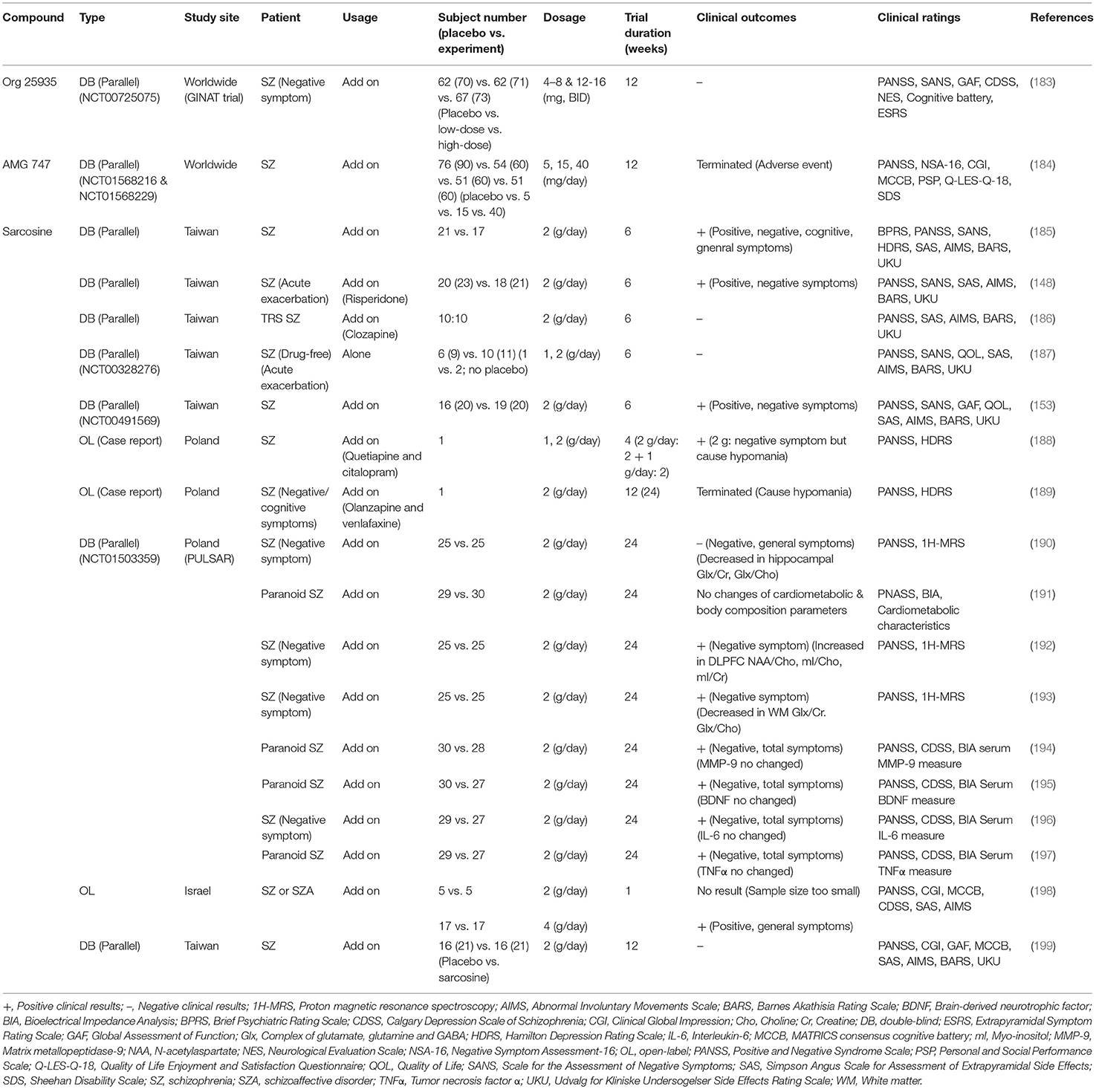
Table 4. Summary of clinical trials evaluating effects of sarcosine-based GlyT1 inhibitors on the treatment of schizophrenic symptoms.
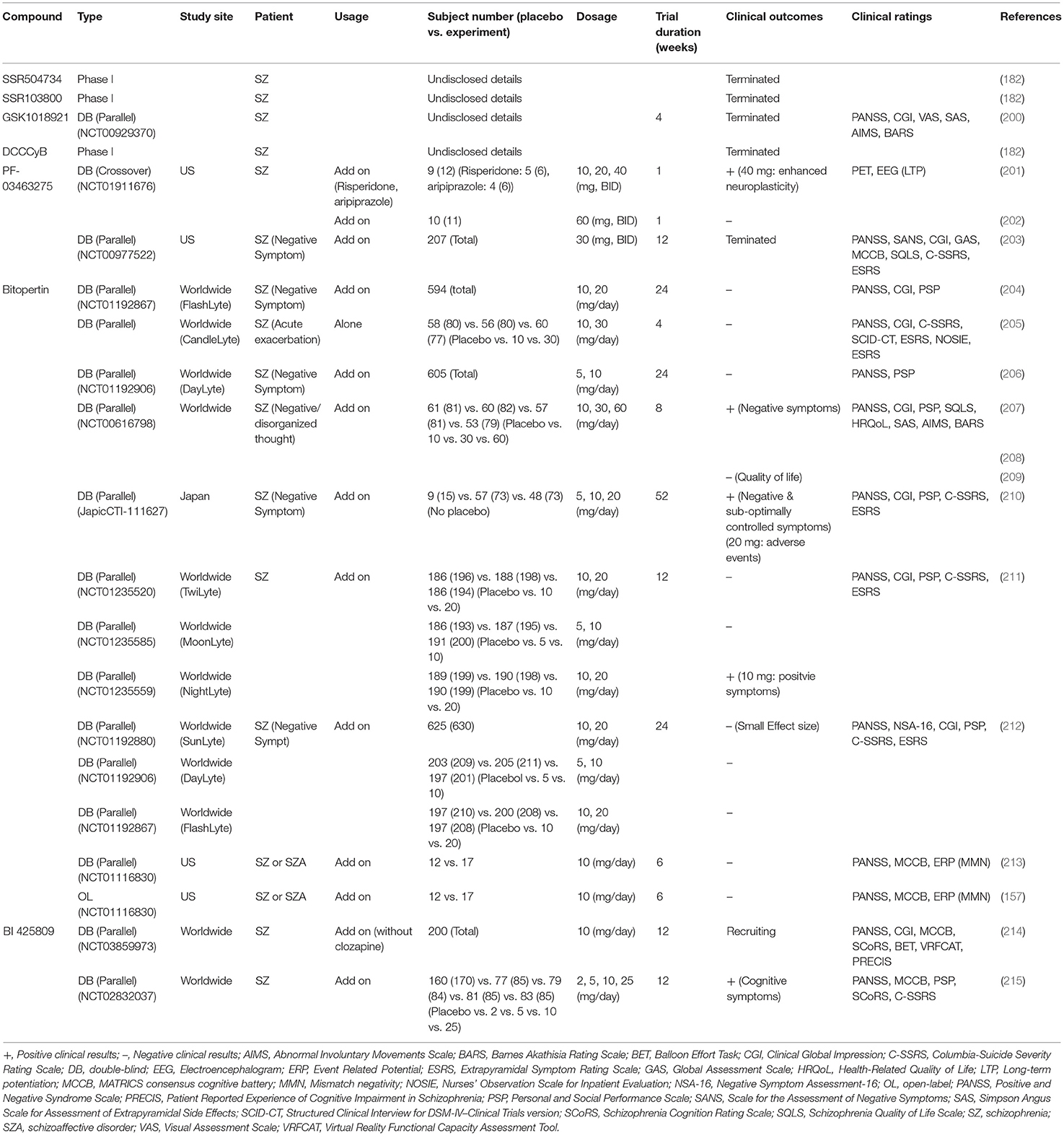
Table 5. Major findings in clinical trials examining effects of non-sarcosine-non-sarcosine-based glycine transporter 1 (GlyT1) inhibitors in patients with schizophrenia.
In the early period of drug discovery, several high-affinity GlyT1 inhibitors derived from sarcosine derivatives [e.g., NFPS (141, 169, 177) and Org 24598 (178)] were produced but caused unexpected toxicity and side effects (15, 178). Only two sarcosine-based GlyT1 inhibitors, AMG 747 (184) and Org 25935 (also known as SCH 900435 or MK-8435) (183), were advanced into clinical trials. Both AMG 747 and Org 25935 trials ended due to unspecified safety events and failure to benefit schizophrenia, respectively (182). Researchers have focused on the low-affinity GlyT1 inhibitor sarcosine as an adjunctive medication to conventional antipsychotics. Off-label use of sarcosine in clinical studies has been demonstrated to improve positive symptoms, negative symptoms, and quality of life with minimal side effects in patients with schizophrenia (148, 153, 185, 198). Moreover, findings from previous clinical trials and moderator analyses further indicated that sarcosine is more efficacious than D-serine in general psychopathology for chronically ill stable schizophrenic patients as well as for schizophrenic patients with acutely exacerbated symptoms of schizophrenia (123, 148, 153). Along the same lines, a series of studies from the Polish Sarcosine Study in Schizophrenia (PULSAR) project illustrated that schizophrenic patients treated with sarcosine for 6 months displayed significant improvements in negative symptoms, general psychopathology and changes in glutamatergic transmission in the brain (190, 192, 193). However, no significant differences in cardiometabolic systems, body composition or neurochemical levels (e.g., BDNF, IL-6 and TNF-α) were found in PULSAR studies (191, 194–197). Double-blind clinical studies revealed no beneficial effect of adjunctive sarcosine in drug-free schizophrenia patients or patients treated with clozapine (186, 187, 199). In terms of the side effects and safety profile of sarcosine, the overall results have been satisfactory in most clinical studies; however, sarcosine administered with glutamatergic and serotoninergic agents may have had a synergistic effect that exacerbated schizophrenic symptoms and hypomania in two case reports (188, 189).
In addition to sarcosine-based inhibitors, non-sarcosine-derived GlyT1 inhibitors are potential alternatives for indirectly modulating the GMS on NMDARs. Compared to sarcosine-based GlyT1 inhibitors, non-sarcosine-based compounds are associated with faster off-rates and less toxic side effects (182). The earliest non-sarcosine-based GlyT1 inhibitors, including SSR504734 (216), SSR103800 (217), GSK1018921 (218), and DCCCyB (219), were developed and have been entered into phase I clinical trials. However, the trials with all these compounds were halted or discontinued for undisclosed reasons (182, 200, 220). In addition, PF-3463275, another non-sarcosine-based GlyT1 inhibitor developed by Pfizer (221), was entered into clinical trials and provided positive results for the enhancement of cognitive remediation in schizophrenia (201). However, the first phase II clinical trial (203) on the use of PF-3463275 as an add-on therapy for the treatment of negative symptoms was terminated because of unspecified scientific reasons and safety concerns. The second phase II clinical trial (202) was initiated in 2013, and although it has remained active, to the best of our knowledge, there has been no recruitment efforts to date.
In addition to the abovementioned non-sarcosine-based GlyT1 inhibitors, bitopertin (also known as RG1678 or RO4917838) is an oral, non-competitive GlyT1 inhibitor that was originally developed by Roche as a potential drug candidate for the treatment of negative symptoms of schizophrenia. Preclinical studies revealed that bitopertin modulated schizophrenia-like behaviors in several naïve and pharmacologically challenged animal models (222, 223). The most promising finding of bitopertin was the result of an 8-week randomized, double-blind, proof-of-concept phase II study, in which bitopertin was proven to be safe, with the results showing an inverted U-shaped dose–response efficacy against the predominant negative symptoms of stable schizophrenia patients (207, 208), but no similar effect was observed in the quality of life of these patients (209). Subsequently, in a phase II/III clinical trial, bitopertin monotherapy improved only the positive subscale score of the PANSS (Positive and Negative Syndrome Scale) with respect to acute exacerbation of schizophrenia (205). In a randomized double-blind phase III study following one-year as an adjunctive treatment, bitopertin was found to be generally safe and well-tolerated for the treatment of Japanese patients with schizophrenia, and all three bitopertin-treated groups showed improvements in all the efficacy endpoints for both “negative symptoms” and “suboptimally controlled symptoms” throughout the duration of the study (210). Except for this study, unfortunately, the superior efficacy over placebo of adjunctive bitopertin at any of the doses tested in patients with persistent predominant negative symptoms of schizophrenia could not be proven in several randomized, double-blind, placebo-controlled phase III trials (204, 206, 211, 212). Furthermore, bitopertin did not significantly affect any symptoms, NMDAR-related biomarkers, or MMN frequency at the doses tested in double-blind clinical trials with patients with schizophrenia (157, 213). Accordingly, the negative results and small improvements associated with bitopertin suggest that adjunctive bitopertin treatment might only offer a modest benefit and that bitopertin might not be a broadly effective or optimal therapeutic candidate for the treatment of schizophrenia. Further study will be needed to elucidate the effect of bitopertin in animal models and clinical trials.
Furthermore, BI 425809 was recently developed by Boehringer Ingelheim as a novel, investigational GlyT1 inhibitor to improve cognitive function and memory in patients with schizophrenia and Alzheimer's disease (224–226). A recent randomized double-blind, placebo-controlled phase II study revealed that BI 425809 improved cognitive functions after 12 weeks in patients with schizophrenia (215), suggesting that BI 425809 can provide an effective treatment for cognitive impairment associated with schizophrenia. Currently, another phase II trial of BI 425809 combined with computerized cognitive training for schizophrenic patients is in progress (214, 227). Further large-scale phase III clinical trials will be necessary to replicate these encouraging findings and to confirm the therapeutic potential of BI 425809 for the treatment of cognitive deficits in schizophrenia.
In summary, both sarcosine-based and non-sarcosine-based GlyT1 inhibitors are generally well-tolerated and exhibit a satisfactory safety profile. GlyT1 inhibitors also exert more-promising therapeutic potential than agonists directly targeting the GMS in the improvement of schizophrenic symptoms. However, in consideration of the etiology and pathophysiology of schizophrenia, no evidence has supported a proposal that GlyT1 is overexpressed in the brains of schizophrenic patients. In contrast, a series of negative findings of association studies have revealed that neither glycine transmission nor GlyT1 is implicated in the pathogenesis of schizophrenia (228–230). As described previously, although concentrations of glycine are 10-fold higher than D-serine, D-serine is considered the dominant endogenous coagonist of NMDARs and a modulator of NMDAR-related neurotoxicity (20, 21). Thus, targeting GlyT1 might not be an optimal strategy for modulation of NMDAR functions. Furthermore, functional distinctions between synaptic and extrasynaptic NMDARs in brain physiology, in which synaptic and extrasynaptic NMDARs are gated by D-serine and glycine, respectively, have been reported (22, 231). D-serine and glycine differentially impact NMDAR membrane diffusion and neuroplasticity (21, 22). Given that glycine, but not D-serine, preferentially gates NMDARs located at extrasynaptic sites and that synaptic, but not extrasynaptic, NMDARs are essential for LTP induction, it is plausible that the efficacy and therapeutic effect of GlyT1 inhibitors might be relatively less effective than those of D-serine. Thus, as an alternative to GlyT1 inhibitors, one of the promising approaches for the development of novel therapeutic compounds to treat schizophrenia is based on increased synaptic D-serine levels realized through the indirect modulation of astrocytic D-serine synthesis.
DAO (or DAAO) encodes D-amino acid oxidase which has a flavin adenine dinucleotide (FAD) as the prosthetic group, and DAO catalyzes the oxidative deamination of a wide range of D-amino acids, including D-serine (232–234). The human DAO gene is located on chromosome 12q24, and DAO is mainly expressed in the liver, kidney and CNS (235). DAO is abundant in both neurons and glial cells in the cerebral cortex, hippocampus and cerebellum and contributes to normal neuronal functioning (236, 237). DAO has been of interest in psychiatry because its major substrate in the brain is D-serine, which modulates NMDAR functions and contributes to NMDAR hypofunction in schizophrenia. D-serine is synthesized from L-serine by SR and is metabolized by DAO and SR through an α, β-elimination reaction. Among DAO substrates in the brain, D-serine is clearly the most abundant. DAO is believed to play a crucial role in the regulation of cellular D-serine concentrations and release (143). In particular, the three-dimensional structure of human DAO is a stable homodimer and it is highly conserved compared to the microorganism sources (238, 239). Human DAO possesses a low FAD binding function and mainly presents in an inactive apoprotein form (238, 240) because of its specific structure. DAO also exhibits a low substrate affinity and catalytic efficiency for D-serine (234, 241). The inactive apoprotein form of human DAO prevents excessive degradation of D-serine in the brain. The active holeenzyme of human DAO is reconstituted by binding of active-site ligands, such as FAD and the substrate stabilizes flavin binding, and thus pushing the acquisition of catalytic competence (238, 242). Intriguingly, it has been reported that DAO inhibitor (e.g., benzoate) increases the holoenzyme reconstitution of human DAO and stabilizes the flavoprotein (243). In addition, human DAO is mainly colocalized with pyramidal neurons in the prefrontal cortex and hippocampus (236). Enhanced DAO activity is considered a potential cause of reduced D-serine and subsequent impairment to NMDAR functioning in schizophrenia (123, 244).
The glutamate hypothesis of schizophrenia suggests that increased DAO activity leads to decreased D-serine levels, which may subsequently lead to NMDAR hypofunction. Supporting evidence from association studies, DAO expression in schizophrenic patients and behavioral outcomes observed in rodent models have suggested potential therapeutic benefits of DAO inhibitors (DAOIs). Accumulating evidence from genetic studies has indicated that DAO and G72 are putative genes related to schizophrenia (235, 245, 246). Schizophrenic patients with genetic variation in DAO and G72 genes also display negative valence and cognitive deficits (247–250). In complementary findings, a recent GWAS revealed that of 108 schizophrenia-associated loci, none were within the DAO or G72 gene regions (13). Although reports on the association of DAO and G72 with schizophrenia are ambiguous, these genes remain candidates in schizophrenia because of their roles in glutamatergic signaling, which has been associated with schizophrenia in multiple lines of research (157, 166, 246). Both G72 mRNA and G72 protein (as known as pLG72) are detected in higher levels in brain and blood of schizophrenia patients (251, 252). Intriguingly, DAO-pLG72 complex was reported to modulate intracellular D-serine concentration in human (233, 238), which suggests a novel avenue to design molecules to regulate human DAO activity and thus NMDAR function for future research. In the same vein, the expression and activity of DAO are significantly increased in patients with schizophrenia (28, 236, 244, 253). Intriguingly, it has been reported that chlorpromazine (i.e., a first-generation antipsychotic) and risperidone (i.e., a second-generation antipsychotic) are potentially active substances that inhibit DAO function (254, 255). In addition, inactivation of DAO in rodents produces behavioral and biochemical effects, suggesting potential therapeutic benefits (143). Indeed, increasing levels of D-serine have been observed in rodents after the administration of DAOIs (256–258). Consistently, PCP- or MK-801-induced pre-pulse inhibition deficits and cognitive deficits relevant to schizophrenia were ameliorated after treatment with DAOIs (256, 259, 260). DAOIs increase the levels of D-alanine, which might also be beneficial for increasing NMDAR function (260). Moreover, ddY/DAO(–) mice, which lack active DAO due to a point mutation, exhibited increased cerebellar NMDAR functions (261), enhanced hippocampal LTP, and improved spatial learning in a water maze (262). Other animal studies have indicated that DAO is involved in the mechanism of D-serine nephrotoxicity (263), which is attenuated by DAOIs (264). D-serine combined with DAOI or DAOI alone might be beneficial for enhancing NMDAR functions in schizophrenia.
In agreement with the abovementioned studies, DAOIs are among the most attractive therapeutic targets for improving cognition and reducing negative symptoms in schizophrenia discovered in recent decades. Basically, DAOIs can be divided into two categories: cofactor-competitive and substrate-competitive inhibitors (238, 240, 241, 265–267). Chlorpromazine, the first antipsychotic medication, is a traditional dopamine D2 receptor antagonist but it has been reported that chlorpromazine is also a FAD-competitive DAO inhibitor (243, 255). Compared to the cofactor-competitive inhibitor of DAO, substrate-competitive DAOIs (such as CBIO and benzoate) are frequently used as scaffolds for developing novel drugs. In the late 2000s, a series of structurally similar molecules (such as ASO57278 (256), Merck compound (257), Pfizer compounds (258), and CBIO (268), displayed a potent inhibition of DAO in vitro but had limited elevation of D-serine in vivo. Especially, it has been reported that acute and chronic administrations of ASO57278 produced inverted U-shaped dose-response curves to reverse PCP-induced PPI deficits (256). And co-administration of CBIO with D-serine also significantly increased D-serine level and attenuated MK-801 induced PPI deficit (259). Thus, these studies imply that DAOIs have beneficial effects in treatment of schizophrenia.
To date, there are at least two potential DAOIs that have been advanced into clinical evaluation, including sodium benzoate and TAK-831. Effects of these two DAOIs on the treatment of schizophrenic symptoms in clinical studies are summarized in Table 6. Sodium benzoate is known as a preservative that is widely used as a food pickling agent. Sodium benzoate is a prototype competitive inhibitor of DAO, and preclinical studies have indicated that it attenuates PCP-induced pre-pulse inhibition deficits as well as D-serine-induced nephrotoxicity (264, 277). The first randomized, double-blind, placebo-controlled trial with chronic schizophrenia patients reported that add-on sodium benzoate relieved positive, negative, and cognitive symptoms as well as improved quality of life (269). Sodium benzoate also showed efficacy and safety for schizophrenic patients who had a poor response to clozapine (270). Moreover, adjunctive sodium benzoate plus sarcosine, but not sarcosine alone, improved the cognitive and global functioning of chronic schizophrenia patients (199). However, a randomized clinical study in Australia indicated that adjunctive use of sodium benzoate had no effect on individuals with early psychosis (271, 272). Two adaptive clinical phase II studies performed to evaluate the safety and efficacy of sodium benzoate in adolescent schizophrenia patients (273) and treatment-resistant schizophrenia patients (274) are currently recruiting. One probable drawback for the development of sodium benzoate as a drug candidate is that it lacks patentability due to its simple chemical structure. More evidence on the therapeutic effect of sodium benzoate, especially in larger-scale clinical trials in schizophernia, is required to prove its effectiveness and applicability. In addition, another highly selective and potent DAOI from Takeda known as TAK-831 is currently being evaluated for schizophrenia in a phase II clinical trial (275, 276, 278). A series of studies of TAK831, including those directed to pharmacokinetics, target occupancy, and D-serine concentrations in the brain, have detected and analyzed a non-linear quantitative multilayer mechanistic model for multilayer biomarker-assisted clinical development with multiple CNS indications (279). Investigations to discover the characteristics and potential development of TAK-831 are needed to determine its efficacy and tolerability in the management of different domains of schizophrenia. In addition to sodium benzoate and TAK-831, there are additional unpublished data on DAOIs for which patent applications have been filed and which have been claimed to have specific therapeutic utility in the treatment of schizophrenia and other neuropsychiatric disorders (280). It is worth further investigating the safety and therapeutic potential of these novel DAOIs for the treatment of unmet medical needs of patients with in schizophrenia in future studies.
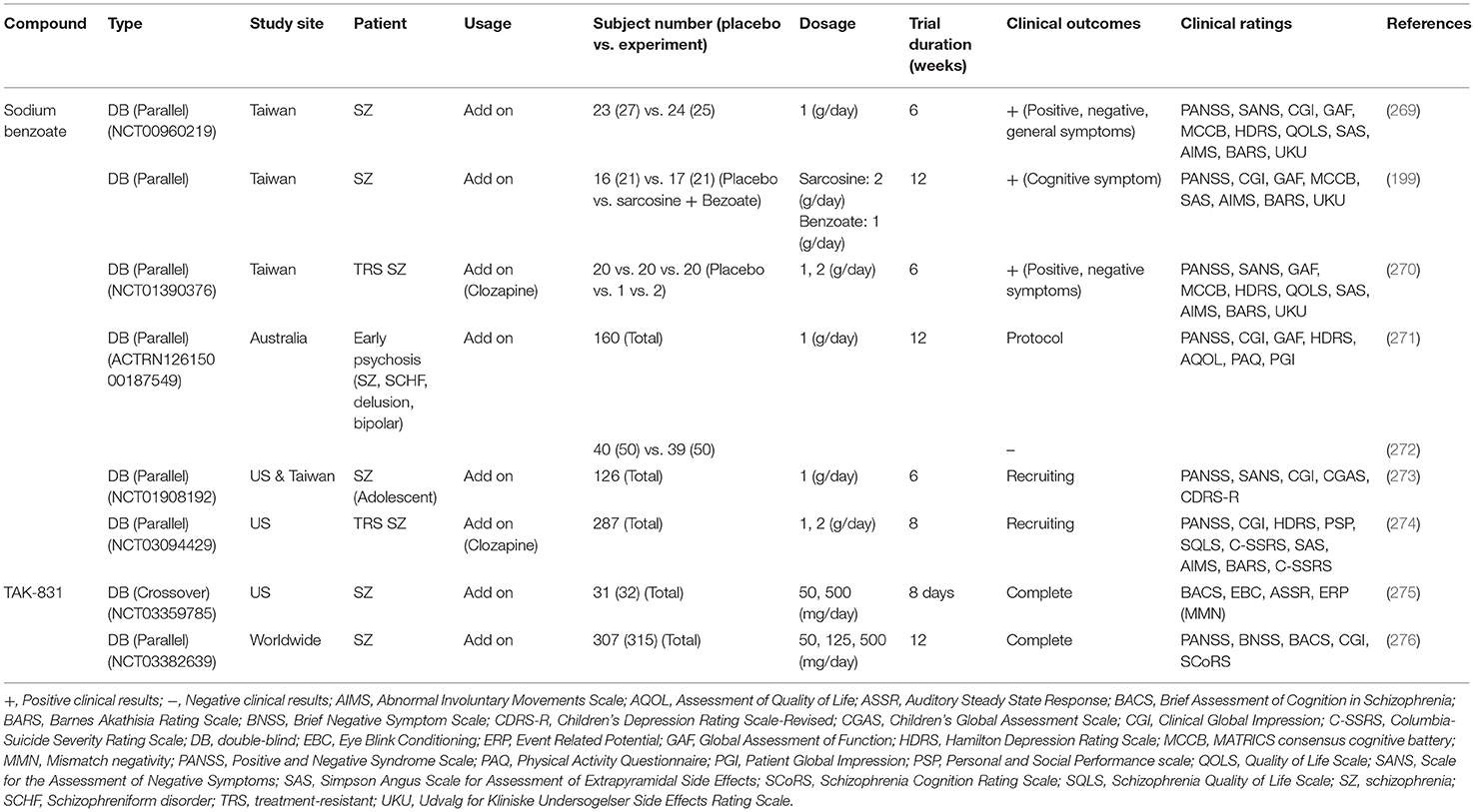
Table 6. Potential clinical efficacy and benefits related to D-amino acid oxidase inhibitors (DAOIs) on the treatment of schizophrenic symptoms.
Data from clinical, genetic, postmortem, and animal studies strongly implicate NMDARs as central hubs for many pathophysiological processes in the brains of schizophrenic patients. Notably, several NMDAR-enhancing agents, particularly those directed to the GMS of NMDARs, result in the significant alleviation of schizophrenia-like behavioral deficits and cognitive dysfunctions in animal models as well as in patients with schizophrenia. There is great interest in identifying potential drug candidates targeting the GMS of NMDARs and to evaluate their therapeutic effectiveness in attenuating the negative and cognitive symptoms of schizophrenia with minimal adverse effects. Modulation of NMDAR functions through the GMS has been proposed as a possible therapeutic approach to drug development, and either direct or indirect activation of GMS results in differential benefits and adverse effects in the treatment of schizophrenia. A summary of the relevant animal study data, as well as those from clinical trials, examining the therapeutic effects and experimental outcomes of direct and indirect GMS modulators is provided in this article. Overall, current findings suggest that indirectly targeting GMS appears to be more beneficial and results in fewer adverse effects than directly targeting GMS to modulate NMDAR functions. In particular, compared with GlyT1 inhibitors, one of the promising approaches to the development of novel therapeutic compounds for treating schizophrenia is to indirectly increase synaptic D-serine levels by targeting DAO. As illustrated in Figure 2, inhibition of DAO via DAOIs not only results in increased synaptic D-serine levels but also the regulation of NMDAR-evoked electrophysiological activity, which contributes to the amelioration of NMDAR hypofunction and restoration of mental functions. There is great interest in further investigating the effects of DAOIs on brain activity, neuromorphology, and cell surface trafficking of NMDARs, which contribute to the amelioration of NMDAR hypofunction and untreated symptoms of schizophrenia. Thus, GMS modulators, especially GlyT1 inhibitors and DAOIs, may open new avenues to the treatment of unmet medical needs in patients with schizophrenia, which is worthy of further investigation. For the development of new antipsychotic drugs, the establishment of safety profiles of these potential compounds will be beneficial and informative, possibly leading to the elucidation of their precise mechanisms of action and the evaluation of their therapeutic effects in both animal models and clinical studies. Notably, however, this review presents an oversimplified summary of the treatment alternatives for an extremely complex psychiatric disorder. Indeed, human diseases are far more complex and only some aspects of human diseases can be partially modeled in animal models. Clinical trials are essential and irreplaceable in drug development. In complementary to human studies, preclinical animal studies are highly valuable and indispensable to the understanding of the underlying mechanism and for the development of new drugs. And we simply focus on discussing the importance of NMDAR functions on excitatory rather than inhibitory neurons in this review article. The role of inhibitory neurons and the impact of NMDAR hypofunction on GABAergic neurons in the pathophysiology of schizophrenia are worth further investigating (281, 282). Because the etiology of schizophrenia remains unclear, disturbances to the GABAergic, cholinergic, and dopaminergic neurotransmitter systems (283, 284), as well as disruptions to astrocyte function (164), are also worthy of further investigation.
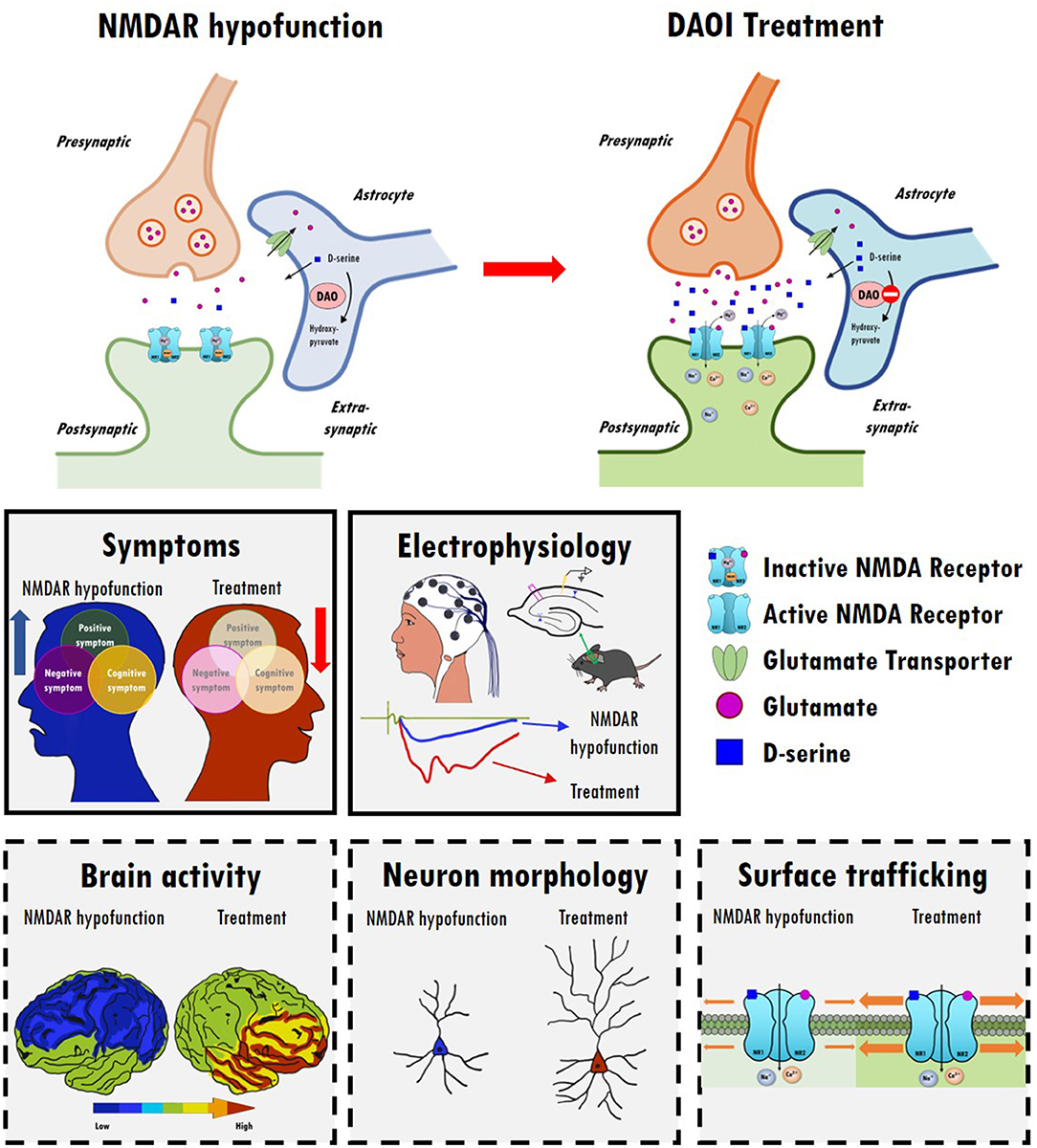
Figure 2. The therapeutic effects and possible underlying mechanisms of D-amino acid oxidase inhibitors (DAOIs) in the treatment of schizophrenia. Top panel: Indirect modulation of NMDAR functions by DAOIs restores NMDAR hypofunction. Inhibition of DAO results in increased synaptic levels of D-serine. Middle panels: DAOIs significantly alleviate positive, negative, and cognitive symptoms in patients with schizophrenia and moderate schizophrenia-like behavioral deficits in animal models. DAOIs enhance NMDAR functions and hippocampal LTP in animal studies. Bottom panels: Possible mechanism of action of DAOIs. The effects of DAOIs on brain activity, neuromorphology, and cell surface trafficking of NMDARs, which contribute to the amelioration of NMDAR hypofunction and restoration of mental functions, are worthy of further investigation.
J-CP and D-ZL are the key persons to wrote and prepare this review article. S-SG collected data and prepared tables. C-YC wrote some paragraphs of this article. W-SL organized, wrote, and modified the whole article. All authors contributed to the article and approved the submitted version.
This research was supported by Grant Nos. 109-2918-I-002-008, 109-2926-I-002-509, 109-2410-H-002-087-MY3, and 110-2410-H-002-235-MY3 from the Ministry of Science and Technology in Taiwan to W-SL and by grant support from National Taiwan University and National Taiwan University Hospital.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank all members of the Laboratory of Integrated Neuroscience and Ethology (LINE) in the Department of Psychology, National Taiwan University, for their assistance and comments.
1. He H, Liu Q, Li N, Guo L, Gao F, Bai L, et al. Trends in the incidence and DALYs of schizophrenia at the global, regional and national levels: results from the global burden of disease study 2017. Epidemiol Psychiatr Sci. (2020) 29:e91. doi: 10.1017/S2045796019000891
2. Lewis DA, Sweet RA. Schizophrenia from a neural circuitry perspective: advancing toward rational pharmacological therapies. J Clin Invest. (2009) 119:706–16. doi: 10.1172/JCI37335
3. Powell CM, Miyakawa T. Schizophrenia-relevant behavioral testing in rodent models: a uniquely human disorder? Biol Psychiatry. (2006) 59:1198–207. doi: 10.1016/j.biopsych.2006.05.008
4. McCleery A, Green MF, Hellemann GS, Baade LE, Gold JM, Keefe RS, et al. Latent structure of cognition in schizophrenia: a confirmatory factor analysis of the MATRICS consensus cognitive battery (MCCB). Psychol Med. (2015) 45:2657–66. doi: 10.1017/S0033291715000641
5. Strauss GP, Horan WP, Kirkpatrick B, Fischer BA, Keller WR, Miski P, et al. Deconstructing negative symptoms of schizophrenia: avolition-apathy and diminished expression clusters predict clinical presentation and functional outcome. J Psychiatr Res. (2013) 47:783–90. doi: 10.1016/j.jpsychires.2013.01.015
6. Elert E. Aetiology: searching for schizophrenia's roots. Nature. (2014) 508:S2–3. doi: 10.1038/508S2a
7. Bowie CR, Harvey PD. Cognitive deficits and functional outcome in schizophrenia. Neuropsychiatr Dis Treat. (2006) 2:531–6. doi: 10.2147/nedt.2006.2.4.531
8. Correll CU, Schooler NR. Negative symptoms in schizophrenia: a review and clinical guide for recognition, assessment, and treatment. Neuropsychiatr Dis Treat. (2020) 16:519–34. doi: 10.2147/NDT.S225643
9. Tripathi A, Kar SK, Shukla R. Cognitive deficits in schizophrenia: understanding the biological correlates and remediation strategies. Clin Psychopharmacol Neurosci. (2018) 16:7–17. doi: 10.9758/cpn.2018.16.1.7
10. Mohamed S, Paulsen JS, O'Leary D, Arndt S, Andreasen N. Generalized cognitive deficits in schizophrenia: a study of first-episode patients. Arch Gen Psychiatry. (1999) 56:749–54. doi: 10.1001/archpsyc.56.8.749
11. Marder SR. The NIMH-MATRICS project for developing cognition-enhancing agents for schizophrenia. Dialogues Clin Neurosci. (2006) 8:109–13. doi: 10.31887/DCNS.2006.8.1/smarder
12. Lally J, MacCabe JH. Antipsychotic medication in schizophrenia: a review. Br Med Bull. (2015) 114:169–79. doi: 10.1093/bmb/ldv017
13. Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. (2014) 511:421–7. doi: 10.1038/nature13595
14. Egerton A, Stone JM. The glutamate hypothesis of schizophrenia: neuroimaging and drug development. Curr Pharm Biotechnol. (2012) 13:1500–12. doi: 10.2174/138920112800784961
15. Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. (2012) 37:4–15. doi: 10.1038/npp.2011.181
16. Uno Y, Coyle JT. Glutamate hypothesis in schizophrenia. Psychiatry Clin Neurosci. (2019) 73:204–15. doi: 10.1111/pcn.12823
17. Parsons CG, Danysz W, Quack G. Glutamate in CNS disorders as a target for drug development: an update. Drug News Perspect. (1998) 11:523–69. doi: 10.1358/dnp.1998.11.9.863689
18. Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. (1987) 325:529–31. doi: 10.1038/325529a0
19. Bergeron R, Meyer TM, Coyle JT, Greene RW. Modulation of N-methyl-D-aspartate receptor function by glycine transport. Proc Natl Acad Sci USA. (1998) 95:15730–4. doi: 10.1073/pnas.95.26.15730
20. Shleper M, Kartvelishvily E, Wolosker H. D-serine is the dominant endogenous coagonist for NMDA receptor neurotoxicity in organotypic hippocampal slices. J Neurosci. (2005) 25:9413–7. doi: 10.1523/JNEUROSCI.3190-05.2005
21. Wolosker H, Balu DT. D-Serine as the gatekeeper of NMDA receptor activity: implications for the pharmacologic management of anxiety disorders. Transl Psychiatry. (2020) 10:184. doi: 10.1038/s41398-020-00870-x
22. Papouin T, Ladepeche L, Ruel J, Sacchi S, Labasque M, Hanini M, et al. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. (2012) 150:633–46. doi: 10.1016/j.cell.2012.06.029
23. Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. (2001) 11:327–35. doi: 10.1016/S0959-4388(00)00215-4
24. Balu DT. The NMDA receptor and schizophrenia: from pathophysiology to treatment. Adv Pharmacol. (2016) 76:351–82. doi: 10.1016/bs.apha.2016.01.006
25. Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. (2014) 506:179–84. doi: 10.1038/nature12929
26. Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. (2012) 17:142–53. doi: 10.1038/mp.2011.154
27. Weickert CS, Fung SJ, Catts VS, Schofield PR, Allen KM, Moore LT, et al. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol Psychiatry. (2013) 18:1185–92. doi: 10.1038/mp.2012.137
28. Bendikov I, Nadri C, Amar S, Panizzutti R, De Miranda J, Wolosker H, et al. A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophr Res. (2007) 90:41–51. doi: 10.1016/j.schres.2006.10.010
29. Catts VS, Lai YL, Weickert CS, Weickert TW, Catts SV. A quantitative review of the postmortem evidence for decreased cortical N-methyl-D-aspartate receptor expression levels in schizophrenia: how can we link molecular abnormalities to mismatch negativity deficits? Biol Psychol. (2016) 116:57–67. doi: 10.1016/j.biopsycho.2015.10.013
30. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. (1991) 148:1301–8. doi: 10.1176/ajp.148.10.1301
31. Krystal JH, Abi-Saab W, Perry E, D'Souza DC, Liu N, Gueorguieva R, et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology. (2005) 179:303–9. doi: 10.1007/s00213-004-1982-8
32. Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. (1994) 51:199–214. doi: 10.1001/archpsyc.1994.03950030035004
33. Kegeles LS, Abi-Dargham A, Zea-Ponce Y, Rodenhiser-Hill J, Mann JJ, Van Heertum RL, et al. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: implications for schizophrenia. Biol Psychiatry. (2000) 48:627–40. doi: 10.1016/S0006-3223(00)00976-8
34. Bressan RA, Pilowsky LS. Imaging the glutamatergic system in vivo–relevance to schizophrenia. Eur J Nucl Med. (2000) 27:1723–31. doi: 10.1007/s002590000372
35. Gonzalez-Burgos G, Lewis DA. NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr Bull. (2012) 38:950–7. doi: 10.1093/schbul/sbs010
36. Bartha R, Williamson PC, Drost DJ, Malla A, Carr TJ, Cortese L, et al. Measurement of glutamate and glutamine in the medial prefrontal cortex of never-treated schizophrenic patients and healthy controls by proton magnetic resonance spectroscopy. Arch Gen Psychiatry. (1997) 54:959–65. doi: 10.1001/archpsyc.1997.01830220085012
37. Theberge J, Bartha R, Drost DJ, Menon RS, Malla A, Takhar J, et al. Glutamate and glutamine measured with 4.0 T proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatry. (2002) 159:1944–6. doi: 10.1176/appi.ajp.159.11.1944
38. Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. (1999) 33:523–33. doi: 10.1016/S0022-3956(99)00029-1
39. Gao WJ, Yang SS, Mack NR, Chamberlin LA. Aberrant maturation and connectivity of prefrontal cortex in schizophrenia-contribution of NMDA receptor development and hypofunction. Mol Psychiatry. (2021). doi: 10.1038/s41380-021-01196-w
40. Wolkin A, Sanfilipo M, Wolf AP, Angrist B, Brodie JD, Rotrosen J. Negative symptoms and hypofrontality in chronic schizophrenia. Arch Gen Psychiatry. (1992) 49:959–65. doi: 10.1001/archpsyc.1992.01820120047007
41. Carter CS, Perlstein W, Ganguli R, Brar J, Mintun M, Cohen JD. Functional hypofrontality and working memory dysfunction in schizophrenia. Am J Psychiatry. (1998) 155:1285–7. doi: 10.1176/ajp.155.9.1285
42. Krzystanek M, Palasz A. NMDA receptor model of antipsychotic drug-induced hypofrontality. Int J Mol Sci. (2019) 20:1442. doi: 10.3390/ijms20061442
43. Muir KW. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr Opin Pharmacol. (2006) 6:53–60. doi: 10.1016/j.coph.2005.12.002
44. Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic–ischemic brain damage. Ann Neurol. (1986) 19:105–11. doi: 10.1002/ana.410190202
45. Yamamoto K, Mifflin S. Inhibition of glial glutamate transporter GLT1 in the nucleus of the solitary tract attenuates baroreflex control of sympathetic nerve activity and heart rate. Physiol Rep. (2018) 6:e13877. doi: 10.14814/phy2.13877
46. Kleckner NW, Dingledine R. Requirement for glycine in activation of NMDA-receptors expressed in xenopus oocytes. Science. (1988) 241:835–7. doi: 10.1126/science.2841759
47. Ballard TM, Pauly-Evers M, Higgins GA, Ouagazzal AM, Mutel V, Borroni E, et al. Severe impairment of NMDA receptor function in mice carrying targeted point mutations in the glycine binding site results in drug-resistant nonhabituating hyperactivity. J Neurosci. (2002) 22:6713–23. doi: 10.1523/JNEUROSCI.22-15-06713.2002
48. Kew JN, Koester A, Moreau JL, Jenck F, Ouagazzal AM, Mutel V, et al. Functional consequences of reduction in NMDA receptor glycine affinity in mice carrying targeted point mutations in the glycine binding site. J Neurosci. (2000) 20:4037–49. doi: 10.1523/JNEUROSCI.20-11-04037.2000
49. Labrie V, Lipina T, Roder JC. Mice with reduced NMDA receptor glycine affinity model some of the negative and cognitive symptoms of schizophrenia. Psychopharmacology. (2008) 200:217–30. doi: 10.1007/s00213-008-1196-6
50. Fadda E, Danysz W, Wroblewski JT, Costa E. Glycine and D-serine increase the affinity of N-methyl-D-aspartate sensitive glutamate binding sites in rat brain synaptic membranes. Neuropharmacology. (1988) 27:1183–5. doi: 10.1016/0028-3908(88)90015-9
51. Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci. (2012) 13:465–77. doi: 10.1038/nrn3257
52. Erhardt S, Blennow K, Nordin C, Skogh E, Lindstrom LH, Engberg G. Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci Lett. (2001) 313:96–8. doi: 10.1016/S0304-3940(01)02242-X
53. Javitt DC, Spencer KM, Thaker GK, Winterer G, Hajos M. Neurophysiological biomarkers for drug development in schizophrenia. Nat Rev Drug Discov. (2008) 7:68–83. doi: 10.1038/nrd2463
54. Lin CH, Huang CL, Chang YC, Chen PW, Lin CY, Tsai GE, et al. Clinical symptoms, mainly negative symptoms, mediate the influence of neurocognition and social cognition on functional outcome of schizophrenia. Schizophr Res. (2013) 146:231–7. doi: 10.1016/j.schres.2013.02.009
55. Salituro FG, Harrison BL, Baron BM, Nyce PL, Stewart KT, McDonald IA. 3-(2-carboxyindol-3-yl)propionic acid derivatives: antagonists of the strychnine-insensitive glycine receptor associated with the N-methyl-D-aspartate receptor complex. J Med Chem. (1990) 33:2944–6. doi: 10.1021/jm00173a003
56. Cordi A, Lacoste JM, Audinot V, Millan M. Design, synthesis and structure-activity relationships of novel strychnine-insensitive glycine receptor ligands. Bioorg Med Chem Lett. (1999) 9:1409–14. doi: 10.1016/S0960-894X(99)00194-8
57. Krueger BA, Weil T, Schneider G. Comparative virtual screening and novelty detection for NMDA-GlycineB antagonists. J Comput Aided Mol Des. (2009) 23:869–81. doi: 10.1007/s10822-009-9304-1
58. Bristow LJ, Flatman KL, Hutson PH, Kulagowski JJ, Leeson PD, Young L, et al. The atypical neuroleptic profile of the glycine/N-methyl-D-aspartate receptor antagonist, L-701,324, in rodents. J Pharmacol Exp Ther. (1996) 277:578–85
59. Witkin JM, Brave S, French D, Geter-Douglass B. Discriminative stimulus effects of R-(+)-3-amino-1-hydroxypyrrolid-2-one, [(+)-HA-966], a partial agonist of the strychnine-insensitive modulatory site of the N-methyl-D-aspartate receptor. J Pharmacol Exp Ther. (1995) 275:1267–73.
60. Jonas P, Bischofberger J, Sandkühler J. Corelease of two fast neurotransmitters at a central synapse. Science. (1998) 281:419–24. doi: 10.1126/science.281.5375.419
61. Zafra F, Giménez C. Glycine transporters and synaptic function. IUBMB Life. (2008) 60:810–7. doi: 10.1002/iub.128
62. Murtas G, Marcone GL, Sacchi S, Pollegioni L. L-serine synthesis via the phosphorylated pathway in humans. Cell Mol Life Sci. (2020) 77:5131–48. doi: 10.1007/s00018-020-03574-z
63. Béchade C, Sur C, Triller A. The inhibitory neuronal glycine receptor. Bioessays. (1994) 16:735–44. doi: 10.1002/bies.950161008
64. Ishimaru M, Kurumaji A, Toru M. Increases in strychnine-insensitive glycine binding sites in cerebral cortex of chronic schizophrenics: evidence for glutamate hypothesis. Biol Psychiatry. (1994) 35:84–95. doi: 10.1016/0006-3223(94)91197-5
65. Waziri R, Baruah S. A hyperglycinergic rat model for the pathogenesis of schizophrenia: preliminary findings. Schizophr Res. (1999) 37:205–15. doi: 10.1016/S0920-9964(98)00169-8
66. Heresco-Levy U, Bar G, Levin R, Ermilov M, Ebstein RP, Javitt DC. High glycine levels are associated with prepulse inhibition deficits in chronic schizophrenia patients. Schizophr Res. (2007) 91:14–21. doi: 10.1016/j.schres.2006.12.003
67. Banerjee A, Wang HY, Borgmann-Winter KE, MacDonald ML, Kaprielian H, Stucky A, et al. Src kinase as a mediator of convergent molecular abnormalities leading to NMDAR hypoactivity in schizophrenia. Mol Psychiatry. (2015) 20:1091–100. doi: 10.1038/mp.2014.115
68. Neeman G, Blanaru M, Bloch B, Kremer I, Ermilov M, Javitt DC, et al. Relation of plasma glycine, serine, and homocysteine levels to schizophrenia symptoms and medication type. Am J Psychiatry. (2005) 162:1738–40. doi: 10.1176/appi.ajp.162.9.1738
69. Shoham S, Javitt DC, Heresco-Levy U. Chronic high-dose glycine nutrition: effects on rat brain cell morphology. Biol Psychiatry. (2001) 49:876–85. doi: 10.1016/S0006-3223(00)01046-5
70. Heresco-Levy U, Javitt DC, Ermilov M, Mordel C, Horowitz A, Kelly D. Double-blind, placebo-controlled, crossover trial of glycine adjuvant therapy for treatment-resistant schizophrenia. Br J Psychiatry. (1996) 169:610–7. doi: 10.1192/bjp.169.5.610
71. Heresco-Levy U, Ermilov M, Lichtenberg P, Bar G, Javitt DC. High-dose glycine added to olanzapine and risperidone for the treatment of schizophrenia. Biol Psychiatry. (2004) 55:165–71. doi: 10.1016/S0006-3223(03)00707-8
72. Um SM, Ha S, Lee H, Kim J, Kim K, Shin W, et al. NGL-2 deletion leads to autistic-like behaviors responsive to NMDAR modulation. Cell Rep. (2018) 23:3839–51. doi: 10.1016/j.celrep.2018.05.087
73. Kato K, Shishido T, Ono M, Shishido K, Kobayashi M, Niwa S. Glycine reduces novelty- and methamphetamine-induced locomotor activity in neonatal ventral hippocampal damaged rats. Neuropsychopharmacology. (2001) 24:330–2. doi: 10.1016/S0893-133X(00)00213-X
74. Mouri A, Noda Y, Noda A, Nakamura T, Tokura T, Yura Y, et al. Involvement of a dysfunctional dopamine-D1/N-methyl-d-aspartate-NR1 and Ca2+/calmodulin-dependent protein kinase II pathway in the impairment of latent learning in a model of schizophrenia induced by phencyclidine. Mol Pharmacol. (2007) 71:1598–609. doi: 10.1124/mol.106.032961
75. Hoffman KL, Basurto E. Clozapine and glycinamide prevent MK-801-induced deficits in the novel object recognition (NOR) test in the domestic rabbit (Oryctolagus cuniculus). Behav Brain Res. (2014) 271:203–11. doi: 10.1016/j.bbr.2014.06.012
76. Alonso ER, Kolesniková L, Białkowska-Jaworska E, Kisiel Z, León I, Guillemin JC, et al. Glycinamide, a glycine precursor, caught in the gas phase: a laser-ablation jet-cooled rotational study. Astrophys J. (2018) 861:70. doi: 10.3847/1538-4357/aac6e9
77. Fone KCF, Watson DJG, Billiras RI, Sicard DI, Dekeyne A, Rivet JM, et al. Comparative pro-cognitive and neurochemical profiles of glycine modulatory site agonists and glycine reuptake inhibitors in the rat: potential relevance to cognitive dysfunction and its management. Mol Neurobiol. (2020) 57:2144–66. doi: 10.1007/s12035-020-01875-9
78. Waziri R. Glycine therapy of schizophrenia. Biol Psychiatry. (1988) 23:210–1. doi: 10.1016/0006-3223(88)90093-5
79. Rosse RB, Theut SK, Banay-Schwartz M, Leighton M, Scarcella E, Cohen CG, et al. Glycine adjuvant therapy to conventional neuroleptic treatment in schizophrenia: an open-label, pilot study. Clin Neuropharmacol. (1989) 12:416–24. doi: 10.1097/00002826-198910000-00006
80. Costa J, Khaled E, Sramek J, Bunney W Jr, Potkin SG. An open trial of glycine as an adjunct to neuroleptics in chronic treatment-refractory schizophrenics. J Clin Psychopharmacol. (1990) 10:71–2. doi: 10.1097/00004714-199002000-00027
81. Javitt DC, Zylberman I, Zukin SR, Heresco-Levy U, Lindenmayer JP. Amelioration of negative symptoms in schizophrenia by glycine. Am J Psychiatry. (1994) 151:1234–6. doi: 10.1176/ajp.151.8.1234
82. Leiderman E, Zylberman I, Zukin SR, Cooper TB, Javitt DC. Preliminary investigation of high-dose oral glycine on serum levels and negative symptoms in schizophrenia: an open-label trial. Biol Psychiatry. (1996) 39:213–5. doi: 10.1016/0006-3223(95)00585-4
83. Heresco-Levy U, Javitt DC, Ermilov M, Mordel C, Silipo G, Lichtenstein M. Efficacy of high-dose glycine in the treatment of enduring negative symptoms of schizophrenia. Arch Gen Psychiatry. (1999) 56:29–36. doi: 10.1001/archpsyc.56.1.29
84. Potkin SG, Jin Y, Bunney BG, Costa J, Gulasekaram B. Effect of clozapine and adjunctive high-dose glycine in treatment-resistant schizophrenia. Am J Psychiatry. (1999) 156:145–7. doi: 10.1176/ajp.156.1.145
85. Evins AE, Fitzgerald SM, Wine L, Rosselli R, Goff DC. Placebo-controlled trial of glycine added to clozapine in schizophrenia. Am J Psychiatry. (2000) 157:826–8. doi: 10.1176/appi.ajp.157.5.826
86. Javitt DC, Silipo G, Cienfuegos A, Shelley AM, Bark N, Park M, et al. Adjunctive high-dose glycine in the treatment of schizophrenia. Int J Neuropsychopharmacol. (2001) 4:385–91. doi: 10.1017/S1461145701002590
87. Diaz P, Bhaskara S, Dursun SM, Deakin B. Double-blind, placebo-controlled, crossover trial of clozapine plus glycine in refractory schizophrenia negative results. J Clin Psychopharmacol. (2005) 25:277–8. doi: 10.1097/01.jcp.0000165740.22377.6d
88. Buchanan RW, Javitt DC, Marder SR, Schooler NR, Gold JM, McMahon RP, et al. The cognitive and negative symptoms in schizophrenia trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am J Psychiatry. (2007) 164:1593–602. doi: 10.1176/appi.ajp.2007.06081358
89. Greenwood LM, Leung S, Michie PT, Green A, Nathan PJ, Fitzgerald P, et al. The effects of glycine on auditory mismatch negativity in schizophrenia. Schizophr Res. (2018) 191:61–9. doi: 10.1016/j.schres.2017.05.031
90. Bodkin JA, Coleman MJ, Godfrey LJ, Carvalho CMB, Morgan CJ, Suckow RF, et al. Targeted treatment of individuals with psychosis carrying a copy number variant containing a genomic triplication of the glycine decarboxylase gene. Biol Psychiatry. (2019) 86:523–35. doi: 10.1016/j.biopsych.2019.04.031
91. Rosse RB, Schwartz BL, Leighton MP, Davis RE, Deutsch SI. An open-label trial of milacemide in schizophrenia: an NMDA intervention strategy. Clin Neuropharmacol. (1990) 13:348–54. doi: 10.1097/00002826-199008000-00010
92. Rosse RB, Schwartz BL, Davis RE, Deutsch SI. An NMDA intervention strategy in schizophrenia with “low-dose” milacemide. Clin Neuropharmacol. (1991) 14:268–72. doi: 10.1097/00002826-199106000-00012
93. Toth E, Lajtha A. Elevation of cerebral levels of nonessential amino acids in vivo by administration of large doses. Neurochem Res. (1981) 6:1309–17. doi: 10.1007/BF00964352
94. O'Neill BV, Croft RJ, Mann C, Dang O, Leung S, Galloway MP, et al. High-dose glycine impairs the prepulse inhibition measure of sensorimotor gating in humans. J Psychopharmacol. (2011) 25:1632–8. doi: 10.1177/0269881110372546
95. Thomas JW, Hood WF, Monahan JB, Contreras PC, O'Donohue TL. Glycine modulation of the phencyclidine binding site in mammalian brain. Brain Res. (1988) 442:396–8. doi: 10.1016/0006-8993(88)91533-8
96. Cascella NG, Macciardi F, Cavallini C, Smeraldi E. d-cycloserine adjuvant therapy to conventional neuroleptic treatment in schizophrenia: an open-label study. J Neural Transm Gen Sect. (1994) 95:105–11. doi: 10.1007/BF01276429
97. Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. (2002) 22:2343–51. doi: 10.1523/JNEUROSCI.22-06-02343.2002
98. Ledgerwood L, Richardson R, Cranney J. Effects of D-cycloserine on extinction of conditioned freezing. Behav Neurosci. (2003) 117:341–9. doi: 10.1037/0735-7044.117.2.341
99. Matsuoka N, Aigner TG. D-cycloserine, a partial agonist at the glycine site coupled to N-methyl-D-aspartate receptors, improves visual recognition memory in rhesus monkeys. J Pharmacol Exp Ther. (1996) 278:891–7. doi: 10.1016/S0021-5198(19)36628-4
100. Yao L, Wang Z, Deng D, Yan R, Ju J, Zhou Q. The impact of D-cycloserine and sarcosine on in vivo frontal neural activity in a schizophrenia-like model. BMC Psychiatry. (2019) 19:314. doi: 10.1186/s12888-019-2306-1
101. Kanahara N, Shimizu E, Ohgake S, Fujita Y, Kohno M, Hashimoto T, et al. Glycine and D-serine, but not D-cycloserine, attenuate prepulse inhibition deficits induced by NMDA receptor antagonist MK-801. Psychopharmacology. (2008) 198:363–74. doi: 10.1007/s00213-008-1151-6
102. Pitkänen M, Sirviö J, MacDonald E, Niemi S, Ekonsalo T, Riekkinen P. The effects of D-cycloserine and MK-801 on the performance of rats in two spatial learning and memory tasks. Eur Neuropsychopharmacol. (1995) 5:457–63. doi: 10.1016/0924-977X(95)80004-L
103. Goff DC, Tsai G, Manoach DS, Coyle JT. Dose-finding trial of D-cycloserine added to neuroleptics for negative symptoms in schizophrenia. Am J Psychiatry. (1995) 152:1213–5. doi: 10.1176/ajp.152.8.1213
104. Rosse RB, Fay-McCarthy M, Kendrick K, Davis RE, Deutsch SI. D-cycloserine adjuvant therapy to molindone in the treatment of schizophrenia. Clin Neuropharmacol. (1996) 19:444–50. doi: 10.1097/00002826-199619050-00008
105. Goff DC, Tsai G, Manoach DS, Flood J, Darby DG, Coyle JT. D-cycloserine added to clozapine for patients with schizophrenia. Am J Psychiatry. (1996) 153:1628–30. doi: 10.1176/ajp.153.12.1628
106. van Berckel BN, Hijman R, van der Linden JA, Westenberg HG, van Ree JM, Kahn RS. Efficacy and tolerance of D-cycloserine in drug-free schizophrenic patients. Biol Psychiatry. (1996) 40:1298–300. doi: 10.1016/S0006-3223(96)00311-3
107. Heresco-Levy U, Javitt DC, Ermilov M, Silipo G, Shimoni J. Double-blind, placebo-controlled, crossover trial of D-cycloserine adjuvant therapy for treatment-resistant schizophrenia. Int J Neuropsychopharmacol. (1998) 1:131–5. doi: 10.1017/S1461145798001242
108. Goff DC, Tsai G, Levitt J, Amico E, Manoach D, Schoenfeld DA, et al. A placebo-controlled trial of D-cycloserine added to conventional neuroleptics in patients with schizophrenia. Arch Gen Psychiatry. (1999) 56:21–7. doi: 10.1001/archpsyc.56.1.21
109. Goff DC, Henderson DC, Evins AE, Amico E. A placebo-controlled crossover trial of D-cycloserine added to clozapine in patients with schizophrenia. Biol Psychiatry. (1999) 45:512–4. doi: 10.1016/S0006-3223(98)00367-9
110. van Berckel BN, Evenblij CN, van Loon BJ, Maas MF, van der Geld MA, Wynne HJ, et al. D-cycloserine increases positive symptoms in chronic schizophrenic patients when administered in addition to antipsychotics: a double-blind, parallel, placebo-controlled study. Neuropsychopharmacology. (1999) 21:203–10. doi: 10.1016/S0893-133X(99)00014-7
111. Evins AE, Amico E, Posever TA, Toker R, Goff DC. D-Cycloserine added to risperidone in patients with primary negative symptoms of schizophrenia. Schizophr Res. (2002) 56:19–23. doi: 10.1016/S0920-9964(01)00220-1
112. Heresco-Levy U, Ermilov M, Shimoni J, Shapira B, Silipo G, Javitt DC. Placebo-controlled trial of D-cycloserine added to conventional neuroleptics, olanzapine, or risperidone in schizophrenia. Am J Psychiatry. (2002) 159:480–2. doi: 10.1176/appi.ajp.159.3.480
113. Duncan EJ, Szilagyi S, Schwartz MP, Bugarski-Kirola D, Kunzova A, Negi S, et al. Effects of D-cycloserine on negative symptoms in schizophrenia. Schizophr Res. (2004) 71:239–48. doi: 10.1016/j.schres.2004.03.013
114. Goff DC, Herz L, Posever T, Shih V, Tsai G, Henderson DC, et al. A six-month, placebo-controlled trial of D-cycloserine co-administered with conventional antipsychotics in schizophrenia patients. Psychopharmacology. (2005) 179:144–50. doi: 10.1007/s00213-004-2032-2
115. Yurgelun-Todd DA, Coyle JT, Gruber SA, Renshaw PF, Silveri MM, Amico E, et al. Functional magnetic resonance imaging studies of schizophrenic patients during word production: effects of D-cycloserine. Psychiatry Res. (2005) 138:23–31. doi: 10.1016/j.pscychresns.2004.11.006
116. Goff DC, Cather C, Gottlieb JD, Evins AE, Walsh J, Raeke L, et al. Once-weekly D-cycloserine effects on negative symptoms and cognition in schizophrenia: an exploratory study. Schizophr Res. (2008) 106:320–7. doi: 10.1016/j.schres.2008.08.012
117. Gottlieb JD, Cather C, Shanahan M, Creedon T, Macklin EA, Goff DC. D-cycloserine facilitation of cognitive behavioral therapy for delusions in schizophrenia. Schizophr Res. (2011) 131:69–74. doi: 10.1016/j.schres.2011.05.029
118. Cain CK, McCue M, Bello I, Creedon T, Tang DI, Laska E, et al. d-Cycloserine augmentation of cognitive remediation in schizophrenia. Schizophr Res. (2014) 153:177–83. doi: 10.1016/j.schres.2014.01.016
119. Forsyth JK, Bachman P, Mathalon DH, Roach BJ, Ye E, Asarnow RF. Effects of augmenting N-Methyl-D-Aspartate receptor signaling on working memory and experience-dependent plasticity in schizophrenia: an exploratory study using acute d-cycloserine. Schizophr Bull. (2017) 43:1123–33. doi: 10.1093/schbul/sbw193
120. Takiguchi K, Uezato A, Itasaka M, Atsuta H, Narushima K, Yamamoto N, et al. Association of schizophrenia onset age and white matter integrity with treatment effect of D-cycloserine: a randomized placebo-controlled double-blind crossover study. BMC Psychiatry. (2017) 17:249. doi: 10.1186/s12888-017-1410-3
121. Zareifopoulos N, Panayiotakopoulos G. Neuropsychiatric effects of antimicrobial agents. Clin Drug Investig. (2017) 37:423–37. doi: 10.1007/s40261-017-0498-z
122. Heresco-Levy U, Javitt DC. Comparative effects of glycine and d-cycloserine on persistent negative symptoms in schizophrenia: a retrospective analysis. Schizophr Res. (2004) 66:89–96. doi: 10.1016/S0920-9964(03)00129-4
123. Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr Pharm Des. (2010) 16:522–37. doi: 10.2174/138161210790361452
124. Schell MJ, Molliver ME, Snyder SH. D-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc Natl Acad Sci USA. (1995) 92:3948–52. doi: 10.1073/pnas.92.9.3948
125. Mothet JP, Le Bail M, Billard JM. Time and space profiling of NMDA receptor co-agonist functions. J Neurochem. (2015) 135:210–25. doi: 10.1111/jnc.13204
126. Papouin T, Henneberger C, Rusakov DA, Oliet SHR. Astroglial versus neuronal d-serine: fact checking. Trends Neurosci. (2017) 40:517–20. doi: 10.1016/j.tins.2017.05.007
127. Kartvelishvily E, Shleper M, Balan L, Dumin E, Wolosker H. Neuron-derived D-serine release provides a novel means to activate N-methyl-D-aspartate receptors. J Biol Chem. (2006) 281:14151–62. doi: 10.1074/jbc.M512927200
128. Miya K, Inoue R, Takata Y, Abe M, Natsume R, Sakimura K, et al. Serine racemase is predominantly localized in neurons in mouse brain. J Comp Neurol. (2008) 510:641–54. doi: 10.1002/cne.21822
129. Wolosker H. Serine racemase and the serine shuttle between neurons and astrocytes. Biochim Biophys Acta. (2011) 1814:1558–66. doi: 10.1016/j.bbapap.2011.01.001
130. Wolosker H, Balu DT, Coyle JT. The rise and fall of the d-serine-mediated gliotransmission hypothesis. Trends Neurosci. (2016) 39:712–21. doi: 10.1016/j.tins.2016.09.007
131. Schell MJ, Brady RO Jr, Molliver ME, Snyder SH D-serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptors. J Neurosci. (1997) 17:1604–15. doi: 10.1523/JNEUROSCI.17-05-01604.1997
132. Benneyworth MA, Li Y, Basu AC, Bolshakov VY, Coyle JT. Cell selective conditional null mutations of serine racemase demonstrate a predominate localization in cortical glutamatergic neurons. Cell Mol Neurobiol. (2012) 32:613–24. doi: 10.1007/s10571-012-9808-4
133. Hashimoto K, Fukushima T, Shimizu E, Komatsu N, Watanabe H, Shinoda N, et al. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry. (2003) 60:572–6. doi: 10.1001/archpsyc.60.6.572
134. Hons J, Zirko R, Vasatova M, Doubek P, Klimova B, Masopust J, et al. Impairment of executive functions associated with lower d-serine serum levels in patients with schizophrenia. Front Psychiatry. (2021) 12:514579. doi: 10.3389/fpsyt.2021.514579
135. Fujita Y, Ishima T, Hashimoto K. Supplementation with D-serine prevents the onset of cognitive deficits in adult offspring after maternal immune activation. Sci Rep. (2016) 6:37261. doi: 10.1038/srep37261
136. Rosenberg D, Artoul S, Segal AC, Kolodney G, Radzishevsky I, Dikopoltsev E, et al. Neuronal D-serine and glycine release via the Asc-1 transporter regulates NMDA receptor-dependent synaptic activity. J Neurosci. (2013) 33:3533–44. doi: 10.1523/JNEUROSCI.3836-12.2013
137. Basu AC, Tsai GE, Ma CL, Ehmsen JT, Mustafa AK, Han L, et al. Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol Psychiatry. (2009) 14:719–27. doi: 10.1038/mp.2008.130
138. Balu DT, Li Y, Puhl MD, Benneyworth MA, Basu AC, Takagi S, et al. Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc Natl Acad Sci USA. (2013) 110:E2400–9. doi: 10.1073/pnas.1304308110
139. Pei JC, Hung WL, Lin BX, Shih MH, Lu LY, Luo DZ, et al. Therapeutic potential and underlying mechanism of sarcosine (N-methylglycine) in N-methyl-D-aspartate (NMDA) receptor hypofunction models of schizophrenia. J Psychopharmacol. (2019) 33:1288–302. doi: 10.1177/0269881119856558
140. Shimazaki T, Kaku A, Chaki S. D-Serine and a glycine transporter-1 inhibitor enhance social memory in rats. Psychopharmacology. (2010) 209:263–70. doi: 10.1007/s00213-010-1794-y
141. Karasawa J, Hashimoto K, Chaki S. D-Serine and a glycine transporter inhibitor improve MK-801-induced cognitive deficits in a novel object recognition test in rats. Behav Brain Res. (2008) 186:78–83. doi: 10.1016/j.bbr.2007.07.033
142. MacKay MB, Kravtsenyuk M, Thomas R, Mitchell ND, Dursun SM, Baker GB. D-Serine: potential therapeutic agent and/or biomarker in schizophrenia and depression? Front Psychiatry. (2019) 10:25. doi: 10.3389/fpsyt.2019.00025
143. Verrall L, Burnet PW, Betts JF, Harrison PJ. The neurobiology of D-amino acid oxidase and its involvement in schizophrenia. Mol Psychiatry. (2010) 15:122–37. doi: 10.1038/mp.2009.99
144. Carone FA, Ganote CE. D-serine nephrotoxicity. The nature of proteinuria, glucosuria, and aminoaciduria in acute tubular necrosis. Arch Pathol. (1975) 99:658–62
145. Suzuki M, Gonda Y, Yamada M, Vandebroek AA, Mita M, Hamase K, et al. Serum D-serine accumulation after proximal renal tubular damage involves neutral amino acid transporter Asc-1. Sci Rep. (2019) 9:16705. doi: 10.1038/s41598-019-53302-2
146. Mothet JP, Billard JM, Pollegioni L, Coyle JT, Sweedler JV. Investigating brain d-serine: advocacy for good practices. Acta Physiol. (2019) 226:e13257. doi: 10.1111/apha.13257
147. Heresco-Levy U, Javitt DC, Ebstein R, Vass A, Lichtenberg P, Bar G, et al. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol Psychiatry. (2005) 57:577–85. doi: 10.1016/j.biopsych.2004.12.037
148. Lane HY, Chang YC, Liu YC, Chiu CC, Tsai GE. Sarcosine or D-serine add-on treatment for acute exacerbation of schizophrenia: a randomized, double-blind, placebo-controlled study. Arch Gen Psychiatry. (2005) 62:1196–204. doi: 10.1001/archpsyc.62.11.1196
149. Tsai G, Yang P, Chung LC, Lange N, Coyle JT. D-serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry. (1998) 44:1081–9. doi: 10.1016/S0006-3223(98)00279-0
150. Kantrowitz JT, Malhotra AK, Cornblatt B, Silipo G, Balla A, Suckow RF, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res. (2010) 121:125–30. doi: 10.1016/j.schres.2010.05.012
151. Kantrowitz JT, Woods SW, Petkova E, Cornblatt B, Corcoran CM, Chen H, et al. D-serine for the treatment of negative symptoms in individuals at clinical high risk of schizophrenia: a pilot, double-blind, placebo-controlled, randomised parallel group mechanistic proof-of-concept trial. The Lancet Psychiatry. (2015) 2:403–12. doi: 10.1016/S2215-0366(15)00098-X
152. Tsai GE, Yang P, Chung LC, Tsai IC, Tsai CW, Coyle JT. D-serine added to clozapine for the treatment of schizophrenia. Am J Psychiatry. (1999) 156:1822–5
153. Lane HY, Lin CH, Huang YJ, Liao CH, Chang YC, Tsai GE. A randomized, double-blind, placebo-controlled comparison study of sarcosine (N-methylglycine) and D-serine add-on treatment for schizophrenia. Int J Neuropsychopharmacol. (2010) 13:451–60. doi: 10.1017/S1461145709990939
154. Weiser M, Heresco-Levy U, Davidson M, Javitt DC, Werbeloff N, Gershon AA, et al. A multicenter, add-on randomized controlled trial of low-dose d-serine for negative and cognitive symptoms of schizophrenia. J Clin Psychiatry. (2012) 73:e728–34. doi: 10.4088/JCP.11m07031
155. D'Souza DC, Radhakrishnan R, Perry E, Bhakta S, Singh NM, Yadav R, et al. Feasibility, safety, and efficacy of the combination of D-serine and computerized cognitive retraining in schizophrenia: an international collaborative pilot study. Neuropsychopharmacology. (2013) 38:492–503. doi: 10.1038/npp.2012.208
156. Kantrowitz JT, Epstein ML, Beggel O, Rohrig S, Lehrfeld JM, Revheim N, et al. Neurophysiological mechanisms of cortical plasticity impairments in schizophrenia and modulation by the NMDA receptor agonist D-serine. Brain. (2016) 139 (Pt. 12):3281–95. doi: 10.1093/brain/aww262
157. Kantrowitz JT, Epstein ML, Lee M, Lehrfeld N, Nolan KA, Shope C, et al. Improvement in mismatch negativity generation during d-serine treatment in schizophrenia: correlation with symptoms. Schizophr Res. (2018) 191:70–9. doi: 10.1016/j.schres.2017.02.027
158. Ermilov M, Gelfin E, Levin R, Lichtenberg P, Hashimoto K, Javitt DC, et al. A pilot double-blind comparison of d-serine and high-dose olanzapine in treatment-resistant patients with schizophrenia. Schizophr Res. (2013) 150:604–5. doi: 10.1016/j.schres.2013.09.018
159. Heresco-Levy U, Durrant AR, Ermilov M, Javitt DC, Miya K, Mori H. Clinical and electrophysiological effects of D-serine in a schizophrenia patient positive for anti-N-methyl-D-aspartate receptor antibodies. Biol Psychiatry. (2015) 77:e27–9. doi: 10.1016/j.biopsych.2014.08.023
160. Cho SE, Na KS, Cho SJ, Kang SG. Low d-serine levels in schizophrenia: a systematic review and meta-analysis. Neurosci Lett. (2016) 634:42–51. doi: 10.1016/j.neulet.2016.10.006
161. da Silva Lde B, Leipnitz G, Seminotti B, Fernandes CG, Beskow AP, Amaral AU, et al. D-serine induces lipid and protein oxidative damage and decreases glutathione levels in brain cortex of rats. Brain Res. (2009) 1256:34–42. doi: 10.1016/j.brainres.2008.12.036
162. Katsuki H, Nonaka M, Shirakawa H, Kume T, Akaike A. Endogenous D-serine is involved in induction of neuronal death by N-methyl-D-aspartate and simulated ischemia in rat cerebrocortical slices. J Pharmacol Exp Ther. (2004) 311:836–44. doi: 10.1124/jpet.104.070912
163. Ganote CE, Peterson DR, Carone FA. The nature of D-serine–induced nephrotoxicity. Am J Pathol. (1974) 77:269–82.
164. Chang CY, Luo DZ, Pei JC, Kuo MC, Hsieh YC, Lai WS. Not just a bystander: the emerging role of astrocytes and research tools in studying cognitive dysfunctions in schizophrenia. Int J Mol Sci. (2021) 22:5343. doi: 10.3390/ijms22105343
165. Zafra F, Gomeza J, Olivares L, Aragon C, Gimenez C. Regional distribution and developmental variation of the glycine transporters GLYT1 and GLYT2 in the rat CNS. Eur J Neurosci. (1995) 7:1342–52. doi: 10.1111/j.1460-9568.1995.tb01125.x
166. Balu DT, Coyle JT. The NMDA receptor 'glycine modulatory site' in schizophrenia: D-serine, glycine, and beyond. Curr Opin Pharmacol. (2015) 20:109–15. doi: 10.1016/j.coph.2014.12.004
167. Javitt DC, Frusciante M. Glycyldodecylamide, a phencyclidine behavioral antagonist, blocks cortical glycine uptake: implications for schizophrenia and substance abuse. Psychopharmacology. (1997) 129:96–8. doi: 10.1007/s002130050168
168. Javitt DC, Sershen H, Hashim A, Lajtha A. Reversal of phencyclidine-induced hyperactivity by glycine and the glycine uptake inhibitor glycyldodecylamide. Neuropsychopharmacology. (1997) 17:202–4. doi: 10.1016/S0893-133X(97)00047-X
169. Hashimoto K, Fujita Y, Ishima T, Chaki S, Iyo M. Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of the glycine transporter-1 inhibitor NFPS and D-serine. Eur Neuropsychopharmacol. (2008) 18:414–21. doi: 10.1016/j.euroneuro.2007.07.009
170. Mao SC, Lin HC, Gean PW. Augmentation of fear extinction by infusion of glycine transporter blockers into the amygdala. Mol Pharmacol. (2009) 76:369–78. doi: 10.1124/mol.108.053728
171. Manahan-Vaughan D, Wildforster V, Thomsen C. Rescue of hippocampal LTP and learning deficits in a rat model of psychosis by inhibition of glycine transporter-1 (GlyT1). Eur J Neurosci. (2008) 28:1342–50. doi: 10.1111/j.1460-9568.2008.06433.x
172. Chen HH, Stoker A, Markou A. The glutamatergic compounds sarcosine and N-acetylcysteine ameliorate prepulse inhibition deficits in metabotropic glutamate 5 receptor knockout mice. Psychopharmacology. (2010) 209:343–50. doi: 10.1007/s00213-010-1802-2
173. Kopec K, Flood DG, Gasior M, McKenna BA, Zuvich E, Schreiber J, et al. Glycine transporter (GlyT1) inhibitors with reduced residence time increase prepulse inhibition without inducing hyperlocomotion in DBA/2 mice. Biochem Pharmacol. (2010) 80:1407–17. doi: 10.1016/j.bcp.2010.07.004
174. Zhang HX, Hyrc K, Thio LL. The glycine transport inhibitor sarcosine is an NMDA receptor co-agonist that differs from glycine. J Physiol. (2009) 587 (Pt. 13):3207–20. doi: 10.1113/jphysiol.2009.168757
175. Nishikawa H, Inoue T, Izumi T, Nakagawa S, Koyama T. SSR504734, a glycine transporter-1 inhibitor, attenuates acquisition and expression of contextual conditioned fear in rats. Behav Pharmacol. (2010) 21:576–9. doi: 10.1097/FBP.0b013e32833d419d
176. Singer P, Feldon J, Yee BK. The glycine transporter 1 inhibitor SSR504734 enhances working memory performance in a continuous delayed alternation task in C57BL/6 mice. Psychopharmacology. (2009) 202:371–84. doi: 10.1007/s00213-008-1286-5
177. Lipina T, Labrie V, Weiner I, Roder J. Modulators of the glycine site on NMDA receptors, D-serine and ALX 5407, display similar beneficial effects to clozapine in mouse models of schizophrenia. Psychopharmacology. (2005) 179:54–67. doi: 10.1007/s00213-005-2210-x
178. Le Pen G, Kew J, Alberati D, Borroni E, Heitz MP, Moreau JL. Prepulse inhibition deficits of the startle reflex in neonatal ventral hippocampal-lesioned rats: reversal by glycine and a glycine transporter inhibitor. Biol Psychiatry. (2003) 54:1162–70. doi: 10.1016/S0006-3223(03)00374-3
179. Tsai G, Ralph-Williams RJ, Martina M, Bergeron R, Berger-Sweeney J, Dunham KS, et al. Gene knockout of glycine transporter 1: characterization of the behavioral phenotype. Proc Natl Acad Sci USA. (2004) 101:8485–90. doi: 10.1073/pnas.0402662101
180. Gabernet L, Pauly-Evers M, Schwerdel C, Lentz M, Bluethmann H, Vogt K, et al. Enhancement of the NMDA receptor function by reduction of glycine transporter-1 expression. Neurosci Lett. (2005) 373:79–84. doi: 10.1016/j.neulet.2004.09.064
181. Yee BK, Balic E, Singer P, Schwerdel C, Grampp T, Gabernet L, et al. Disruption of glycine transporter 1 restricted to forebrain neurons is associated with a procognitive and antipsychotic phenotypic profile. J Neurosci. (2006) 26:3169–81. doi: 10.1523/JNEUROSCI.5120-05.2006
182. Cioffi CL. Glycine transporter-1 inhibitors: a patent review (2011-2016). Expert Opin Ther Pat. (2018) 28:197–210. doi: 10.1080/13543776.2018.1429408
183. Schoemaker JH, Jansen WT, Schipper J, Szegedi A. The selective glycine uptake inhibitor org 25935 as an adjunctive treatment to atypical antipsychotics in predominant persistent negative symptoms of schizophrenia: results from the GIANT trial. J Clin Psychopharmacol. (2014) 34:190–8. doi: 10.1097/JCP.0000000000000073
184. Dunayevich E, Buchanan RW, Chen CY, Yang J, Nilsen J, Dietrich JM, et al. Efficacy and safety of the glycine transporter type-1 inhibitor AMG 747 for the treatment of negative symptoms associated with schizophrenia. Schizophr Res. (2017) 182:90–7. doi: 10.1016/j.schres.2016.10.027
185. Tsai G, Lane HY, Yang P, Chong MY, Lange N. Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry. (2004) 55:452–6. doi: 10.1016/j.biopsych.2003.09.012
186. Lane HY, Huang CL, Wu PL, Liu YC, Chang YC, Lin PY, et al. Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to clozapine for the treatment of schizophrenia. Biol Psychiatry. (2006) 60:645–9. doi: 10.1016/j.biopsych.2006.04.005
187. Lane HY, Liu YC, Huang CL, Chang YC, Liau CH, Perng CH, et al. Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biol Psychiatry. (2008) 63:9–12. doi: 10.1016/j.biopsych.2007.04.038
188. Strzelecki D, Szyburska J, Rabe-Jablonska J. Two grams of sarcosine in schizophrenia - is it too much? A potential role of glutamate-serotonin interaction. Neuropsychiatr Dis Treat. (2014) 10:263–6. doi: 10.2147/NDT.S54024
189. Strzelecki D, Szyburska J, Kotlicka-Antczak M, Kaluzynska O. Hypomania after augmenting venlafaxine and olanzapine with sarcosine in a patient with schizophrenia: a case study. Neuropsychiatr Dis Treat. (2015) 11:533–6. doi: 10.2147/NDT.S75734
190. Strzelecki D, Podgorski M, Kaluzynska O, Gawlik-Kotelnicka O, Stefanczyk L, Kotlicka-Antczak M, et al. Supplmentation of antipsychotic treatment with sarcosine - GlyT1 inhibitor - causes changes of glutamatergic (1)NMR spectroscopy parameters in the left hippocampus in patients with stable schizophrenia. Neurosci Lett. (2015) 606:7–12. doi: 10.1016/j.neulet.2015.08.039
191. Strzelecki D, Kaluzynska O, Szyburska J, Wlazlo A, Wysokinski A. No changes of cardiometabolic and body composition parameters after 6-month add-on treatment with sarcosine in patients with schizophrenia. Psychiatry Res. (2015) 230:200–4. doi: 10.1016/j.psychres.2015.08.040
192. Strzelecki D, Podgorski M, Kaluzynska O, Stefanczyk L, Kotlicka-Antczak M, Gmitrowicz A, et al. Adding sarcosine to antipsychotic treatment in patients with stable schizophrenia changes the concentrations of neuronal and glial metabolites in the left dorsolateral prefrontal cortex. Int J Mol Sci. (2015) 16:24475–89. doi: 10.3390/ijms161024475
193. Strzelecki D, Podgorski M, Kaluzynska O, Gawlik-Kotelnicka O, Stefanczyk L, Kotlicka-Antczak M, et al. Supplementation of antipsychotic treatment with the amino acid sarcosine influences proton magnetic resonance spectroscopy parameters in left frontal white matter in patients with schizophrenia. Nutrients. (2015) 7:8767–82. doi: 10.3390/nu7105427
194. Strzelecki D, Kaluzynska O, Szyburska J, Wysokinski A. MMP-9 serum levels in schizophrenic patients during treatment augmentation with sarcosine (results of the PULSAR study). Int J Mol Sci. (2016) 17:1075. doi: 10.3390/ijms17071075
195. Strzelecki D, Kaluzynska O, Wysokinski A. BDNF serum levels in schizophrenic patients during treatment augmentation with sarcosine (results of the PULSAR study). Psychiatry Res. (2016) 242:54–60. doi: 10.1016/j.psychres.2016.05.019
196. Strzelecki D, Urban-Kowalczyk M, Wysokinski A. Serum levels of interleukin 6 in schizophrenic patients during treatment augmentation with sarcosine (results of the PULSAR study). Hum Psychopharmacol. (2018) 33:e2652. doi: 10.1002/hup.2652
197. Strzelecki D, Urban-Kowalczyk M, Wysokinski A. Serum levels of TNF-alpha in patients with chronic schizophrenia during treatment augmentation with sarcosine (results of the PULSAR study). Psychiatry Res. (2018) 268:447–53. doi: 10.1016/j.psychres.2018.08.002
198. Amiaz R, Kent I, Rubinstein K, Sela BA, Javitt D, Weiser M. Safety, tolerability and pharmacokinetics of open label sarcosine added on to anti-psychotic treatment in schizophrenia - preliminary study. Isr J Psychiatry Relat Sci. (2015) 52:12–5
199. Lin CY, Liang SY, Chang YC, Ting SY, Kao CL, Wu YH, et al. Adjunctive sarcosine plus benzoate improved cognitive function in chronic schizophrenia patients with constant clinical symptoms: a randomised, double-blind, placebo-controlled trial. World J Biol Psychiatry. (2017) 18:357–68. doi: 10.3109/15622975.2015.1117654
200. GlaxoSmithKline. A Repeat Dose Study With GSK1018921 to Assess Safety, Tolerability, Pharmacokinetics, Pharmacodynamics in Healthy Volunteers and Patients With Schizophrenia and to Evaluate Its Effect on PK of Midazolam (GT1110791) U.S.: clinicaltrials.gov. (2009). Available online at: https://clinicaltrials.gov/ct2/show/NCT00929370
201. D'Souza DC, Carson RE, Driesen N, Johannesen J, Ranganathan M, Krystal JH, et al. Dose-Related target occupancy and effects on circuitry, behavior, and neuroplasticity of the glycine transporter-1 inhibitor pf-03463275 in healthy and schizophrenia subjects. Biol Psychiatry. (2018) 84:413–21. doi: 10.1016/j.biopsych.2017.12.019
202. Yale University. Translational Neuroscience Optimization of GlyT1 Inhibitor (NCATS) U.S.: clinicaltrials.gov. (2013). Available online at: https://clinicaltrials.gov/ct2/show/NCT01911676 (accessed January 7, 2021).
203. Pfizer. A Study of PF-03463275 as Add-On Therapy in Outpatients With Persistent Negative Symptoms of Schizophrenia U.S.: clinicaltrials.gov. (2009). Available online at: https://clinicaltrials.gov/ct2/show/NCT00977522 (accessed February 7, 2012).
204. Blaettler T, Bugarski-Kirola D, Fleischhacker WW, Bressan R, Arango C, Abi-Saab D, et al. Efficacy and safety of adjunctive bitopertin (10 and 20mg) versus placebo in subjects with persistent predominant negative symptoms of schizophrenia treated with antipsychotics — results from the phase III FlashLyte study. Schizophr Res. (2014) 158:e2–3. doi: 10.1016/j.schres.2014.07.036
205. Bugarski-Kirola D, Wang A, Abi-Saab D, Blattler T. A phase II/III trial of bitopertin monotherapy compared with placebo in patients with an acute exacerbation of schizophrenia - results from the CandleLyte study. Eur Neuropsychopharmacol. (2014) 24:1024–36. doi: 10.1016/j.euroneuro.2014.03.007
206. Arango C, Nasrallah H, Lawrie S, Lohmann TO, Zhu JL, Garibaldi G, et al. Efficacy and safety of adjunctive bitopertin (5 and 10mg) versus placebo in subjects with persistent predominant negative symptoms of schizophrenia treated with antipsychotics — results from the phase III DayLyte study. Schizophr Res. (2014) 158:e1. doi: 10.1016/j.schres.2014.07.038
207. Umbricht D, Alberati D, Martin-Facklam M, Borroni E, Youssef EA, Ostland M, et al. Effect of bitopertin, a glycine reuptake inhibitor, on negative symptoms of schizophrenia: a randomized, double-blind, proof-of-concept study. JAMA Psychiatry. (2014) 71:637–46. doi: 10.1001/jamapsychiatry.2014.163
208. Edgar CJ, Blaettler T, Bugarski-Kirola D, Le Scouiller S, Garibaldi GM, Marder SR. Reliability, validity and ability to detect change of the PANSS negative symptom factor score in outpatients with schizophrenia on select antipsychotics and with prominent negative or disorganized thought symptoms. Psychiatry Res. (2014) 218:219–24. doi: 10.1016/j.psychres.2014.04.009
209. Rofail D, Regnault A, le Scouiller S, Berardo CG, Umbricht D, Fitzpatrick R. Health-related quality of life in patients with prominent negative symptoms: results from a multicenter randomized phase II trial on bitopertin. Qual Life Res. (2016) 25:201–11. doi: 10.1007/s11136-015-1057-9
210. Hirayasu Y, Sato S, Takahashi H, Iida S, Shuto N, Yoshida S, et al. A double-blind randomized study assessing safety and efficacy following one-year adjunctive treatment with bitopertin, a glycine reuptake inhibitor, in Japanese patients with schizophrenia. BMC Psychiatry. (2016) 16:66. doi: 10.1186/s12888-016-0778-9
211. Bugarski-Kirola D, Iwata N, Sameljak S, Reid C, Blaettler T, Millar L, et al. Efficacy and safety of adjunctive bitopertin versus placebo in patients with suboptimally controlled symptoms of schizophrenia treated with antipsychotics: results from three phase 3, randomised, double-blind, parallel-group, placebo-controlled, multicentre studies in the SearchLyte clinical trial programme. Lancet Psychiatry. (2016) 3:1115–28. doi: 10.1016/S2215-0366(16)30344-3
212. Bugarski-Kirola D, Blaettler T, Arango C, Fleischhacker WW, Garibaldi G, Wang A, et al. Bitopertin in negative symptoms of schizophrenia-results from the phase III FlashLyte and DayLyte studies. Biol Psychiatry. (2017) 82:8–16. doi: 10.1016/j.biopsych.2016.11.014
213. Kantrowitz JT, Nolan KA, Epstein ML, Lehrfeld N, Shope C, Petkova E, et al. Neurophysiological effects of bitopertin in schizophrenia. J Clin Psychopharmacol. (2017) 37:447–51. doi: 10.1097/JCP.0000000000000722
214. Harvey PD, Bowie CR, McDonald S, Podhorna J. Evaluation of the efficacy of BI 425809 pharmacotherapy in patients with schizophrenia receiving computerized cognitive training: methodology for a double-blind, randomized, parallel-group trial. Clin Drug Investig. (2020) 40:377–85. doi: 10.1007/s40261-020-00893-8
215. Fleischhacker WW, Podhorna J, Gröschl M, Hake S, Zhao Y, Huang S, et al. Efficacy and safety of the novel glycine transporter inhibitor BI 425809 once daily in patients with schizophrenia: a double-blind, randomised, placebo-controlled phase 2 study. The Lancet Psychiatry. (2021) 8:191–201. doi: 10.1016/S2215-0366(20)30513-7
216. Depoortere R, Dargazanli G, Estenne-Bouhtou G, Coste A, Lanneau C, Desvignes C, et al. Neurochemical, electrophysiological and pharmacological profiles of the selective inhibitor of the glycine transporter-1 SSR504734, a potential new type of antipsychotic. Neuropsychopharmacology. (2005) 30:1963–85. doi: 10.1038/sj.npp.1300772
217. Boulay D, Pichat P, Dargazanli G, Estenne-Bouhtou G, Terranova JP, Rogacki N, et al. Characterization of SSR103800, a selective inhibitor of the glycine transporter-1 in models predictive of therapeutic activity in schizophrenia. Pharmacol Biochem Behav. (2008) 91:47–58. doi: 10.1016/j.pbb.2008.06.009
218. Ouellet D, Sutherland S, Wang T, Griffini P, Murthy V. First-time-in-human study with GSK1018921, a selective GlyT1 inhibitor: relationship between exposure and dizziness. Clin Pharmacol Ther. (2011) 90:597–604. doi: 10.1038/clpt.2011.154
219. Blackaby WP, Lewis RT, Thomson JL, Jennings AS, Goodacre SC, Street LJ, et al. Identification of an orally bioavailable, potent, and selective inhibitor of GlyT1. ACS Med Chem Lett. (2010) 1:350–4. doi: 10.1021/ml1001085
220. Singer P, Dubroqua S, Yee BK. Inhibition of glycine transporter 1: the yellow brick road to new schizophrenia therapy? Curr Pharm Des. (2015) 21:3771–87. doi: 10.2174/1381612821666150724100952
221. Lowe JA 3rd, Hou X, Schmidt C, David Tingley F 3rd, McHardy S, Kalman M, et al. The discovery of a structurally novel class of inhibitors of the type 1 glycine transporter. Bioorg Med Chem Lett. (2009) 19:2974–6. doi: 10.1016/j.bmcl.2009.04.035
222. Alberati D, Moreau JL, Lengyel J, Hauser N, Mory R, Borroni E, et al. Glycine reuptake inhibitor RG1678: a pharmacologic characterization of an investigational agent for the treatment of schizophrenia. Neuropharmacology. (2012) 62:1152–61. doi: 10.1016/j.neuropharm.2011.11.008
223. Eddins D, Hamill TG, Puri V, Cannon CE, Vivian JA, Sanabria-Bohorquez SM, et al. The relationship between glycine transporter 1 occupancy and the effects of the glycine transporter 1 inhibitor RG1678 or ORG25935 on object retrieval performance in scopolamine impaired rhesus monkey. Psychopharmacology. (2014) 231:511–9. doi: 10.1007/s00213-013-3260-0
224. Rosenbrock H, Desch M, Kleiner O, Dorner-Ciossek C, Schmid B, Keller S, et al. Evaluation of pharmacokinetics and pharmacodynamics of BI 425809, a novel GlyT1 inhibitor: translational studies. Clin Transl Sci. (2018) 11:616–23. doi: 10.1111/cts.12578
225. Moschetti V, Schlecker C, Wind S, Goetz S, Schmitt H, Schultz A, et al. Multiple rising doses of oral BI 425809, a GlyT1 inhibitor, in young and elderly healthy volunteers: a randomised, double-blind, phase i study investigating safety and pharmacokinetics. Clin Drug Investig. (2018) 38:737–50. doi: 10.1007/s40261-018-0660-2
226. Moschetti V, Desch M, Goetz S, Liesenfeld KH, Rosenbrock H, Kammerer KP, et al. Safety, tolerability and pharmacokinetics of oral BI 425809, a glycine transporter 1 inhibitor, in healthy male volunteers: a partially randomised, single-blind, placebo-controlled, first-in-human study. Eur J Drug Metab Pharmacokinet. (2018) 43:239–49. doi: 10.1007/s13318-017-0440-z
227. Boehringer Ingelheim. This Study Tests Whether BI 425809 Together With Brain Training Using a Computer Improves Mental Functioning in Patients With Schizophrenia U.S.: Clinicaltrials.gov. (2019). Available online at: https://clinicaltrials.gov/ct2/show/NCT03859973 (accessed June 29, 2021).
228. Feng J, Craddock N, Jones IR, Cook EH Jr, Goldman D, et al. Systematic screening for mutations in the glycine receptor alpha2 subunit gene (GLRA2) in patients with schizophrenia and other psychiatric diseases. Psychiatr Genet. (2001) 11:45–8. doi: 10.1097/00041444-200103000-00009
229. Tsai SJ, Cheng CY, Hong CJ, Liao DL, Hou SJ, et al. Association study of polymorphisms in glycine transporter with schizophrenia. J Neural Transm (Vienna). (2006) 113:1545–9. doi: 10.1007/s00702-006-0438-1
230. Merk W, Kucia K, Medrala T, Kowalczyk M, Owczarek A, Kowalski J. Association study of the excitatory amino acid transporter 2 (EAAT2) and glycine transporter 1 (GlyT1) gene polymorphism with schizophrenia in a polish population. Neuropsychiatr Dis Treat. (2019) 15:989–1000. doi: 10.2147/NDT.S194924
231. Li Y, Sacchi S, Pollegioni L, Basu AC, Coyle JT, Bolshakov VY. Identity of endogenous NMDAR glycine site agonist in amygdala is determined by synaptic activity level. Nat Commun. (2013) 4:1760. doi: 10.1038/ncomms2779
232. Sacchi S, Cappelletti P, Murtas G. Biochemical properties of human D-amino acid oxidase variants and their potential significance in pathologies. Front Mol Biosci. (2018) 5:55. doi: 10.3389/fmolb.2018.00055
233. Sacchi S, Bernasconi M, Martineau M, Mothet JP, Ruzzene M, Pilone MS, et al. pLG72 modulates intracellular D-serine levels through its interaction with D-amino acid oxidase: effect on schizophrenia susceptibility. J Biol Chem. (2008) 283:22244–56. doi: 10.1074/jbc.M709153200
234. Murtas G, Sacchi S, Valentino M, Pollegioni L. Biochemical properties of human d-amino acid oxidase. Front Mol Biosci. (2017) 4:88. doi: 10.3389/fmolb.2017.00088
235. Chumakov I, Blumenfeld M, Guerassimenko O, Cavarec L, Palicio M, Abderrahim H, et al. Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc Natl Acad Sci USA. (2002) 99:13675–80. doi: 10.1073/pnas.182412499
236. Verrall L, Walker M, Rawlings N, Benzel I, Kew JN, Harrison PJ, et al. d-Amino acid oxidase and serine racemase in human brain: normal distribution and altered expression in schizophrenia. Eur J Neurosci. (2007) 26:1657–69. doi: 10.1111/j.1460-9568.2007.05769.x
237. Moreno S, Nardacci R, Cimini A, Ceru MP. Immunocytochemical localization of D-amino acid oxidase in rat brain. J Neurocytol. (1999) 28:169–85. doi: 10.1023/A:1007064504007
238. Pollegioni L, Piubelli L, Molla G, Rosini E. D-Amino acid oxidase-pLG72 interaction and D-serine modulation. Front Mol Biosci. (2018) 5:3. doi: 10.3389/fmolb.2018.00003
239. Kawazoe T, Tsuge H, Pilone MS, Fukui K. Crystal structure of human D-amino acid oxidase: context-dependent variability of the backbone conformation of the VAAGL hydrophobic stretch located at the si-face of the flavin ring. Protein Sci. (2006) 15:2708–17. doi: 10.1110/ps.062421606
240. Molla G. Competitive inhibitors unveil structure/function relationships in human D-amino acid oxidase. Front Mol Biosci. (2017) 4:80. doi: 10.3389/fmolb.2017.00080
241. Molla G, Sacchi S, Bernasconi M, Pilone MS, Fukui K, Polegioni L. Characterization of human D-amino acid oxidase. FEBS Lett. (2006) 580:2358–64. doi: 10.1016/j.febslet.2006.03.045
242. Caldinelli L, Molla G, Sacchi S, Pilone MS, Pollegioni L. Relevance of weak flavin binding in human D-amino acid oxidase. Protein Sci. (2009) 18:801–10. doi: 10.1002/pro.86
243. Caldinelli L, Molla G, Bracci L, Lelli B, Pileri S, Cappelletti P, et al. Effect of ligand binding on human D-amino acid oxidase: implications for the development of new drugs for schizophrenia treatment. Protein Sci. (2010) 19:1500–12. doi: 10.1002/pro.429
244. Burnet PW, Eastwood SL, Bristow GC, Godlewska BR, Sikka P, Walker M, et al. D-amino acid oxidase activity and expression are increased in schizophrenia. Mol Psychiatry. (2008) 13:658–60. doi: 10.1038/mp.2008.47
245. Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. (2008) 40:827–34. doi: 10.1038/ng.171
246. Jagannath V, Gerstenberg M, Correll CU, Walitza S, Grunblatt E. A systematic meta-analysis of the association of Neuregulin 1 (NRG1), D-amino acid oxidase (DAO), and DAO activator (DAOA)/G72 polymorphisms with schizophrenia. J Neural Transm. (2018) 125:89–102. doi: 10.1007/s00702-017-1782-z
247. Jagannath V, Theodoridou A, Gerstenberg M, Franscini M, Heekeren K, Correll CU, et al. Prediction analysis for transition to schizophrenia in individuals at clinical high risk for psychosis: the relationship of DAO, DAOA, and NRG1 variants with negative symptoms and cognitive deficits. Front Psychiatry. (2017) 8:292. doi: 10.3389/fpsyt.2017.00292
248. Hall J, Whalley HC, Moorhead TW, Baig BJ, McIntosh AM, Job DE, et al. Genetic variation in the DAOA (G72) gene modulates hippocampal function in subjects at high risk of schizophrenia. Biol Psychiatry. (2008) 64:428–33. doi: 10.1016/j.biopsych.2008.03.009
249. Jansen A, Krach S, Krug A, Markov V, Eggermann T, Zerres K, et al. A putative high risk diplotype of the G72 gene is in healthy individuals associated with better performance in working memory functions and altered brain activity in the medial temporal lobe. Neuroimage. (2009) 45:1002–8. doi: 10.1016/j.neuroimage.2008.12.054
250. Opgen-Rhein C, Lencz T, Burdick KE, Neuhaus AH, DeRosse P, Goldberg TE, et al. Genetic variation in the DAOA gene complex: impact on susceptibility for schizophrenia and on cognitive performance. Schizophr Res. (2008) 103:169–77. doi: 10.1016/j.schres.2008.04.020
251. Korostishevsky M, Kaganovich M, Cholostoy A, Ashkenazi M, Ratner Y, Dahary D, et al. Is the G72/G30 locus associated with schizophrenia? Single nucleotide polymorphisms, haplotypes, and gene expression analysis. Biol Psychiatry. (2004) 56:169–76. doi: 10.1016/j.biopsych.2004.04.006
252. Lin CH, Chang HT, Chen YJ, Lin CH, Huang CH, Tun R, et al. Distinctively higher plasma G72 protein levels in patients with schizophrenia than in healthy individuals. Mol Psychiatry. (2014) 19:636–7. doi: 10.1038/mp.2013.80
253. Madeira C, Freitas ME, Vargas-Lopes C, Wolosker H, Panizzutti R. Increased brain D-amino acid oxidase (DAAO) activity in schizophrenia. Schizophr Res. (2008) 101:76–83. doi: 10.1016/j.schres.2008.02.002
254. Abou El-Magd RM, Park HK, Kawazoe T, Iwana S, Ono K, Chung SP, et al. The effect of risperidone on D-amino acid oxidase activity as a hypothesis for a novel mechanism of action in the treatment of schizophrenia. J Psychopharmacol. (2010) 24:1055–67. doi: 10.1177/0269881109102644
255. Iwana S, Kawazoe T, Park HK, Tsuchiya K, Ono K, Yorita K, et al. Chlorpromazine oligomer is a potentially active substance that inhibits human D-amino acid oxidase, product of a susceptibility gene for schizophrenia. J Enzyme Inhib Med Chem. (2008) 23:901–11. doi: 10.1080/14756360701745478
256. Adage T, Trillat AC, Quattropani A, Perrin D, Cavarec L, Shaw J, et al. In vitro and in vivo pharmacological profile of AS057278, a selective d-amino acid oxidase inhibitor with potential anti-psychotic properties. Eur Neuropsychopharmacol. (2008) 18:200–14. doi: 10.1016/j.euroneuro.2007.06.006
257. Duplantier AJ, Becker SL, Bohanon MJ, Borzilleri KA, Chrunyk BA, Downs JT, et al. Discovery, SAR, and pharmacokinetics of a novel 3-hydroxyquinolin-2(1H)-one series of potent D-amino acid oxidase (DAAO) inhibitors. J Med Chem. (2009) 52:3576–85. doi: 10.1021/jm900128w
258. Smith SM, Uslaner JM, Yao L, Mullins CM, Surles NO, Huszar SL, et al. The behavioral and neurochemical effects of a novel D-amino acid oxidase inhibitor compound 8 [4H-thieno [3,2-b]pyrrole-5-carboxylic acid] and D-serine. J Pharmacol Exp Ther. (2009) 328:921–30. doi: 10.1124/jpet.108.147884
259. Hashimoto K, Fujita Y, Horio M, Kunitachi S, Iyo M, Ferraris D, et al. Co-administration of a D-amino acid oxidase inhibitor potentiates the efficacy of D-serine in attenuating prepulse inhibition deficits after administration of dizocilpine. Biol Psychiatry. (2009) 65:1103–6. doi: 10.1016/j.biopsych.2009.01.002
260. Horio M, Fujita Y, Ishima T, Iyo M, Ferraris D, Tsukamoto T, et al. Effects of D-Amino acid oxidase inhibitor on the extracellular d-alanine levels and the efficacy of D-alanine on dizocilpine-induced prepulse inhibition deficits in mice. Open Clin Chem J. (2009) 2:16–21. doi: 10.2174/1874241600902010016
261. Almond SL, Fradley RL, Armstrong EJ, Heavens RB, Rutter AR, Newman RJ, et al. Behavioral and biochemical characterization of a mutant mouse strain lacking D-amino acid oxidase activity and its implications for schizophrenia. Mol Cell Neurosci. (2006) 32:324–34. doi: 10.1016/j.mcn.2006.05.003
262. Maekawa M, Watanabe M, Yamaguchi S, Konno R, Hori Y. Spatial learning and long-term potentiation of mutant mice lacking D-amino-acid oxidase. Neurosci Res. (2005) 53:34–8. doi: 10.1016/j.neures.2005.05.008
263. Maekawa M, Okamura T, Kasai N, Hori Y, Summer KH, Konno R. D-amino-acid oxidase is involved in D-serine-induced nephrotoxicity. Chem Res Toxicol. (2005) 18:1678–82. doi: 10.1021/tx0500326
264. Williams RE, Lock EA. Sodium benzoate attenuates D-serine induced nephrotoxicity in the rat. Toxicology. (2005) 207:35–48. doi: 10.1016/j.tox.2004.08.008
265. Sacchi S, Caldinelli L, Cappelletti P, Pollegioni L, Molla G. Structure-function relationships in human D-amino acid oxidase. Amino Acids. (2012) 43:1833–50. doi: 10.1007/s00726-012-1345-4
266. Hopkins SC, Heffernan ML, Saraswat LD, Bowen CA, Melnick L, Hardy LW, et al. Structural, kinetic, and pharmacodynamic mechanisms of D-amino acid oxidase inhibition by small molecules. J Med Chem. (2013) 56:3710–24. doi: 10.1021/jm4002583
267. Terry-Lorenzo RT, Chun LE, Brown SP, Heffernan ML, Fang QK, Orsini MA, et al. Novel human D-amino acid oxidase inhibitors stabilize an active-site lid-open conformation. Biosci Rep. (2014) 34:e00133. doi: 10.1042/BSR20140071
268. Ferraris D, Duvall B, Ko YS, Thomas AG, Rojas C, Majer P, et al. Synthesis and biological evaluation of D-amino acid oxidase inhibitors. J Med Chem. (2008) 51:3357–9. doi: 10.1021/jm800200u
269. Lane HY, Lin CH, Green MF, Hellemann G, Huang CC, Chen PW, et al. Add-on treatment of benzoate for schizophrenia: a randomized, double-blind, placebo-controlled trial of D-amino acid oxidase inhibitor. JAMA Psychiatry. (2013) 70:1267–75. doi: 10.1001/jamapsychiatry.2013.2159
270. Lin CH, Lin CH, Chang YC, Huang YJ, Chen PW, Yang HT, et al. Sodium benzoate, a D-amino acid oxidase inhibitor, added to clozapine for the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Biol Psychiatry. (2018) 84:422–32. doi: 10.1016/j.biopsych.2017.12.006
271. Ryan A, Baker A, Dark F, Foley S, Gordon A, Hatherill S, et al. The efficacy of sodium benzoate as an adjunctive treatment in early psychosis - CADENCE-BZ: study protocol for a randomized controlled trial. Trials. (2017) 18:165. doi: 10.1186/s13063-017-1908-5
272. Scott JG, Baker A, Lim CCW, Foley S, Dark F, Gordon A, et al. Effect of sodium benzoate vs placebo among individuals with early psychosis: a randomized clinical trial. JAMA Netw Open. (2020) 3:e2024335. doi: 10.1001/jamanetworkopen.2020.24335
273. SyneuRx International (Taiwan) Corp. Adaptive Phase II Study to Evaluate the Safety & Efficacy of NaBen®. U.S: ClinicalTrials.gov (2013). Available online at: https://www.clinicaltrials.gov/ct2/show/NCT01908192 (accessed April 7, 2020).
274. SyneuRx International (Taiwan) Corp. An Adaptive Phase II/III, Two-Part, Double-Blind, Randomized, Placebo-controlled, Dose-Finding, Multi-center Study of the Safety and Efficacy of NaBen®, as an Add-on Therapy With Clozapine, for Residual Symptoms of Refractory Schizophrenia in Adults U.S.: Clinicaltrials.gov. (2017). Available online at: https://www.clinicaltrials.gov/ct2/show/NCT03094429 (accessed April 7, 2020).
275. Neurocrine Biosciences. A Study to Evaluate Efficacy, Tolerability, Pharmacodynamic and Pharmacokinetics of Multiple Oral Doses of TAK-831 in Adults With Schizophrenia U.S.: ClinicalTrials.gov. (2017). Available online at: https://www.clinicaltrials.gov/ct2/show/NCT03359785
276. Neurocrine Biosciences. A Study to Evaluate Efficacy, Safety, Tolerability, and Pharmacokinetics of 3 Dose Levels of TAK-831 in Adjunctive Treatment of Adult Participants With Negative Symptoms of Schizophrenia U.S.: clinicaltrials.gov. (2017). Available online at: https://www.clinicaltrials.gov/ct2/show/NCT03382639
277. Matsuura A, Fujita Y, Iyo M, Hashimoto K. Effects of sodium benzoate on pre-pulse inhibition deficits and hyperlocomotion in mice after administration of phencyclidine. Acta Neuropsychiatr. (2015) 27:159–67. doi: 10.1017/neu.2015.1
278. Krogmann A, Peters L, von Hardenberg L, Bodeker K, Nohles VB, Correll CU. Keeping up with the therapeutic advances in schizophrenia: a review of novel and emerging pharmacological entities. CNS Spectr. (2019) 24:38–69. doi: 10.1017/S109285291900124X
279. Yoneyama T, Sato S, Sykes A, Fradley R, Stafford S, Bechar S, et al. Mechanistic multilayer quantitative model for nonlinear pharmacokinetics, target occupancy and pharmacodynamics (PK/TO/PD) relationship of d-amino acid oxidase inhibitor, TAK-831 in mice. Pharm Res. (2020) 37:164. doi: 10.1007/s11095-020-02893-x
280. Tseng YFJ, Liu YL, Sun CM, Hwu HG, Liu CM, Lai WS. Use of Known Compounds as D-Amino Acid Oxidase Inhibitors U.S. (2014). Available online at: https://patents.google.com/patent/US9868975B2/en
281. Cohen SM, Tsien RW, Goff DC, Halassa MM. The impact of NMDA receptor hypofunction on GABAergic neurons in the pathophysiology of schizophrenia. Schizophr Res. (2015) 167:98–107. doi: 10.1016/j.schres.2014.12.026
282. Nakazawa K, Zsiros V, Jiang Z, Nakao K, Kolata S, Zhang S, et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology. (2012) 62:1574–83. doi: 10.1016/j.neuropharm.2011.01.022
283. Coyle JT, Balu D, Benneyworth M, Basu A, Roseman A. Beyond the dopamine receptor: novel therapeutic targets for treating schizophrenia. Dialogues Clin Neurosci. (2010) 12:359–82. doi: 10.31887/DCNS.2010.12.3/jcoyle
Keywords: schizophrenia, unmet medical need, negative symptoms, cognitive impairments, glycine modulatory site (GMS), d-serine, glycine transporter 1 (GlyT1) inhibitor, D-amino acid oxidase (DAO) inhibitor
Citation: Pei J-C, Luo D-Z, Gau S-S, Chang C-Y and Lai W-S (2021) Directly and Indirectly Targeting the Glycine Modulatory Site to Modulate NMDA Receptor Function to Address Unmet Medical Needs of Patients With Schizophrenia. Front. Psychiatry 12:742058. doi: 10.3389/fpsyt.2021.742058
Received: 15 July 2021; Accepted: 02 September 2021;
Published: 01 October 2021.
Edited by:
Hsien-Yuan Lane, China Medical University, TaiwanReviewed by:
Qiang Zhou, Peking University, ChinaCopyright © 2021 Pei, Luo, Gau, Chang and Lai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen-Sung Lai, d3NsYWlAbnR1LmVkdS50dw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.