 Maxemiliano V. Vargas
Maxemiliano V. Vargas Retsina Meyer
Retsina Meyer Arabo A. Avanes
Arabo A. Avanes Mark Rus2
Mark Rus2 David E. Olson
David E. Olson
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 04 October 2021
Sec. Psychological Therapies
Volume 12 - 2021 | https://doi.org/10.3389/fpsyt.2021.727117
This article is part of the Research Topic Can Psychedelic Therapies open a New Frontier in Mental Healthcare (Or Will the Bubble Burst?) View all 22 articles
Psychedelics have inspired new hope for treating brain disorders, as they seem to be unlike any treatments currently available. Not only do they produce sustained therapeutic effects following a single administration, they also appear to have broad therapeutic potential, demonstrating efficacy for treating depression, post-traumatic stress disorder (PTSD), anxiety disorders, substance abuse disorder, and alcohol use disorder, among others. Psychedelics belong to a more general class of compounds known as psychoplastogens, which robustly promote structural and functional neural plasticity in key circuits relevant to brain health. Here we discuss the importance of structural plasticity in the treatment of neuropsychiatric diseases, as well as the evidence demonstrating that psychedelics are among the most effective chemical modulators of neural plasticity studied to date. Furthermore, we provide a theoretical framework with the potential to explain why psychedelic compounds produce long-lasting therapeutic effects across a wide range of brain disorders. Despite their promise as broadly efficacious neurotherapeutics, there are several issues associated with psychedelic-based medicines that drastically limit their clinical scalability. We discuss these challenges and how they might be overcome through the development of non-hallucinogenic psychoplastogens. The clinical use of psychedelics and other psychoplastogenic compounds marks a paradigm shift in neuropsychiatry toward therapeutic approaches relying on the selective modulation of neural circuits with small molecule drugs. Psychoplastogen research brings us one step closer to actually curing mental illness by rectifying the underlying pathophysiology of disorders like depression, moving beyond simply treating disease symptoms. However, determining how to most effectively deploy psychoplastogenic medicines at scale will be an important consideration as the field moves forward.
Theories regarding the etiology of depression and related neuropsychiatric diseases have evolved considerably in recent years. One of the oldest and most widely known theories posits that chemical imbalances in the brain are largely responsible for the development of neuropsychiatric diseases. Support for this theory originated with the observation in the 1950's that administration of the natural product reserpine induces depression (1). Reserpine depletes monoamine levels through inhibition of vesicular monoamine transporters (2), leading to subsequent degradation by monoamine oxidase (MAO) (3). Support for the chemical imbalance hypothesis (also known as the monoamine hypothesis) was further bolstered by findings that MAO inhibitors, tricyclics, and selective-serotonin reuptake inhibitors (SSRIs)—compounds that elevate synaptic levels of monoamines—all seemed to alleviate depressive symptoms (4). However, several pieces of evidence have emerged suggesting that the chemical imbalance hypothesis of depression is a drastic oversimplification.
Reduction of monoamines through acute tryptophan or phenylalanine/tyrosine depletion does not induce depression in healthy subjects (5), which questions the causal role of “chemical imbalances” in depression. However, the primary issue with the monoamine hypothesis of depression lies in the temporal discrepancy between the acute effects of traditional antidepressants and their delayed therapeutic responses. Tricyclics, MAO inhibitors, and SSRIs all increase synaptic levels of monoamines in the brain within minutes, while it takes weeks before the antidepressant effects become apparent (Figure 1) (4, 9). In a patient population at heightened risk for suicide, the need for rapid-acting antidepressants is self-evident.
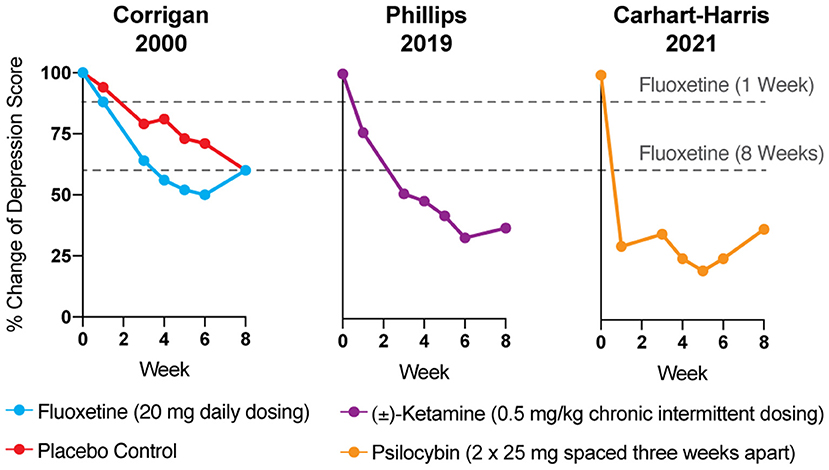
Figure 1. The antidepressant effects of psychoplastogens (e.g., ketamine and psilocybin) are more rapid and sustained than those of traditional antidepressants (e.g., fluoxetine). Data adapted from three clinical trials evaluating the effects of fluoxetine (6), ketamine (7), and psilocybin (8) for treating depression. Dashed lines represent the efficacy of either 1 or 8 weeks of fluoxetine treatment.
While traditional antidepressants acutely increase synaptic levels of monoamines, their chronic administration leads to changes in structural neuroplasticity that are contemporaneous with their clinical therapeutic effects (10–12). In fact, evidence suggests that chronic administration of traditional antidepressants can re-wire the brain, with chronic fluoxetine promoting ocular dominance plasticity in the visual cortex of adult rats (13). Such induced plasticity (iPlasticity) (14) has been hypothesized to play a major role in the actions of essentially all antidepressant treatments including slow-acting traditional antidepressants (15), transcranial magnetic stimulation (16), electroconvulsive therapy (15, 17), exercise (18), and acute sleep deprivation (19). Moreover, people (particularly males) with the brain-derived neurotrophic factor (BDNF) Val66Met single nucleotide polymorphism—a condition that reduces activity-dependent BDNF release (20)—are more likely to experience chronic depression (21, 22). These results lend substantial support to the neuroplasticity hypothesis of depression (sometimes referred to as the neurotrophin hypothesis) (11, 23–36). This hypothesis provides a strong conceptual framework for understanding mental illnesses as disorders of neural circuits induced by a combination of genetic and environmental factors (Figure 2). The corollary being that compounds capable of rectifying these circuit pathologies can potentially serve as a powerful, disease-modifying therapeutics.
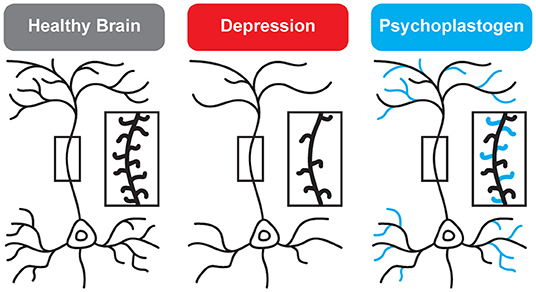
Figure 2. Genetic and environmental factors lead to the cortical atrophy observed in depression, which includes retraction of dendritic branches and loss of dendritic spines. These structural changes are reversed by psychoplastogens (12).
Psychedelics—molecules with “mind-manifesting” properties—include pharmacologically diverse compounds such as dissociatives (e.g., ketamine), classic hallucinogens (e.g., LSD, psilocybin, DMT), and entactogens (e.g., MDMA). Several psychedelics have emerged as some of the most promising treatments for re-wiring pathological neural circuitry. Given their unusually robust abilities to produce rapid and long-lasting changes in neuronal structure and function following a single administration, these compounds have been classified as psychoplastogens—a term we coined to describe this new class of therapeutic compounds (37). Unlike traditional antidepressants, psychoplastogens produce both fast-acting and sustained beneficial behavioral effects after a single administration (Figure 1) (38–42). Here, we present evidence that directly targeting cortical circuits with psychoplastogens has the potential to produce enduring therapeutic responses in depression and co-morbid diseases. First-generation psychoplastogens are all hallucinogenic—they cause people to perceive things that are not real—which has important implications for how these medicines must be administered and how many patients could ultimately benefit from these treatment approaches. In this regard, non-hallucinogenic psychoplastogens offer significant advantages, with the potential to reach much larger patient populations and even replace traditional antidepressants as first-line treatments.
Depression and related neuropsychiatric diseases are often viewed as stress-related disorders given the fact that they can be precipitated or exacerbated by chronic stress (43). In animals, chronic stress results in the prolonged release of glucocorticoids and leads to hypertrophy of the amygdala and nucleus accumbens, atrophy of the hippocampus and prefrontal cortex (PFC), and functional impairment of the PFC (44–55). Given the importance of the PFC in cognition and mediating top-down control over subcortical brain regions (56–66), these changes in neural circuitry are believed to underlie the deficits in learning/memory, mood, motivation, and reward seeking that are characteristic of depression and related disorders (10, 67–74).
Postmortem studies have demonstrated that patients with depression and related mental illnesses have lower BDNF and/or TRKB mRNA levels (75–77), reduced cortical neuron size (75, 78, 79), lower synaptic protein levels (80), decreased mTOR signaling (81), and fewer dendritic spines/synapses (82, 83) in the PFC. Clinical imaging studies have confirmed the results of these studies, demonstrating robust structural and functional deficits in the PFC across a range of disorders including depression, bipolar disorder, anxiety, obsessive compulsive disorder (OCD), schizophrenia, PTSD, alcohol abuse disorder, and substance abuse disorder (84–103). More recently, the advent of [11C]UCB-J (104) and [18F]UCB-J (105) has opened up new opportunities for using positron emission tomography (PET) imaging to measure the density of the synaptic protein SV2A in vivo. Using these new tools, depression severity was found to inversely correlate with SV2A density, and this neuronal atrophy was associated with aberrant network function as measured by magnetic resonance imaging (MRI) functional connectivity (106). Similar results were observed in patients with schizophrenia (107). Thus, substantial evidence points to the PFC as the convergence point underlying the pathophysiology of many neuropsychiatric diseases.
In imaging studies of traditional antidepressant treatment response, increased cortical thickness and cerebral blood flow in the PFC correlate with efficacy (108–112). Next-generation, psychoplastogenic antidepressants also modulate PFC function with both ketamine and psychedelics increasing PFC activation as measured by 18F-FDG PET imaging (113–116). In fact, antidepressant outcomes following ketamine treatment correlate well with PFC activation (117, 118). There is some evidence in humans that psychoplastogens can impact brain structure as well. Chronic use of the psychedelic tisane ayahuasca is associated with thickening of the anterior cingulate cortex (119), and ketamine treatment has been shown to rescue atrophy of the inferior frontal gyrus observed in MDD and PTSD patients (120).
Structural plasticity studies in preclinical animal models support the findings in humans suggesting that cortical neuron structure/function plays a key role in depression and related neuropsychiatric disorders. Cortical neuron atrophy and dysfunction is observed in rodents following chronic corticosterone administration (121), chronic unpredictable mild stress (122, 123), chronic restraint stress (48, 49, 124, 125), and chronic social defeat stress (126–128). These structural changes are accompanied by depressive phenotypes related to motivation (129), anxiety (122), and anhedonia (122, 129). Moreover, antidepressants appear to rectify these structural changes by promoting structural plasticity in the PFC.
Nature uses BDNF to induce structural plasticity in many neuronal populations, and direct administration of BDNF into the rodent brain has been shown to alleviate several depressive phenotypes, curb addiction, and enhance fear extinction (130–133). Conversely, disruption of BDNF signaling in the brain can block the behavioral effects of antidepressants. BDNF heterozygous mice are resistant to the effects of traditional antidepressants (134) and the psychoplastogen ketamine does not produce antidepressant-like effects in BDNF inducible knockout animals (135). The BDNF Val66Met single-nucleotide polymorphism causes major structural atrophy and functional deficits in the PFC and blocks the synaptogenic effects of ketamine (136, 137). Both rodents and humans with BDNF Val66Met polymorphisms exhibit impaired fear extinction learning and reduced mPFC activity during extinction (138), phenotypes that are common in stress-related disorders.
All antidepressant treatments impact BDNF/TrkB signaling in some way. Chronic, but not acute, administration of several traditional antidepressants has been shown to significantly increase mRNA levels of BDNF and/or TrkB as well as increase levels of cAMP response element binding protein (CREB), a transcription factor that regulates the expression of several proteins important for plasticity including BDNF (17, 139). Other studies have found that antidepressants from a variety of chemical families (e.g., SSRIs, SNRIs, tricyclics, etc.) all increase activation of TrkB and its downstream effector CREB (140). Moreover, the effects of the traditional antidepressant fluoxetine on synaptic plasticity and fear extinction have been shown to be dependent on BDNF (141, 142). Recent evidence suggests that several antidepressants, including traditional antidepressants, may directly interact with the TrkB receptor to facilitate TrkB signaling (143, 144). Taken together, the importance of BDNF/TrkB signaling in the therapeutic effects of traditional antidepressants is clear, even if these agents must be administered chronically to achieve robust regulation of this pathway.
Psychoplastogens are also known to impact BDNF/TrkB signaling, but in contrast to traditional antidepressants, they do so rapidly after a single administration. Ketamine increases BDNF protein translation (135, 145) and its antidepressant effects are absent when administered to inducible BDNF knockout mice (135) or homozygous mice harboring the BDNF Val66Met mutation (136). Ketamine and psychedelics modulate cortical neuron function by increasing dendritic spine and synapse density in the PFC (129, 146–149); however, ketamine's effects on structural plasticity appear to last for approximately a week (150) while psilocybin's effects seem to be more durable lasting for at least a month (151, 152). Though the primary molecular targets of ketamine and serotonergic psychedelics are distinct, their downstream pharmacology overlaps, requiring AMPA receptor, TrkB, and mTOR activation to elicit changes in neuronal structure and function (135–137, 148, 153, 154). Moreover, their effects seem to be Cmax driven, as very short stimulation periods (15 min−1 h) are sufficient to induce sustained changes in cortical neuron structure (153). Psilocybin has also been shown to increase the density of SV2A in vivo as measured by PET imaging (155). Importantly, Liston et al. recently used a photoactivatable Rac1 to demonstrate that ketamine-induced spine growth in the PFC was causally related to long-lasting antidepressant effects of the drug in rodents (129). In humans, the subjective effects of ketamine wane after a few hours, but the antidepressant response continues to increase over several days (156). This time course is consistent with what we know about how ketamine and other psychoplastogens alter neuronal structure over time. An exceedingly short stimulation period (<1 h) is sufficient for psychoplastogens to activate cortical neuron growth mechanisms that can last for several days (153).
Several volatile anesthetics, such as isoflurane, nitrous oxide, propofol, and xenon, may produce rapid antidepressant effects (157–165). Xenon is not a small molecule, and by definition, is not a psychoplastogen. Other volatile anesthetics might be considered psychoplastogens if additional studies in larger patient populations confirm that they produce sustained therapeutic effects after a single administration. The data for isoflurane are encouraging as isoflurane produces rapid antidepressant effects after a single administration in both preclinical and in a subset of patients suffering from treatment-resistant depression (157–160). Moreover, these effects appear to be mediated via TrkB signaling and subsequent increases in dendritic spine density in the PFC and hippocampus (164, 165). Thus, preliminary evidence suggests that isoflurane may be considered a psychoplastogen.
While the beneficial effects of psychoplastogens can last for months following a single administration (40, 166, 167), and these medicines have demonstrated efficacy across a range of neuropsychiatric disorders including depression, PTSD, and addiction (156, 168–172), they are not panaceas. Their broad therapeutic utility likely arises from their ability to impact the structure/function of layer V pyramidal neurons in the PFC. As the PFC is a key hub impacted in most neuropsychiatric disorders, it is not surprising that psychoplastogens have proven useful for a variety of indications. Indeed, with the advent of opto- and chemogenetics, systems neurobiology has developed a much deeper understanding of what circuits control behavior. The PFC is known to exert top-down control over a variety of subcortical regions, and recent research has identified a number of circuits originating in the PFC that control behaviors relevant to the treatment of depression, anxiety, and addiction (Figure 3) (56, 173).
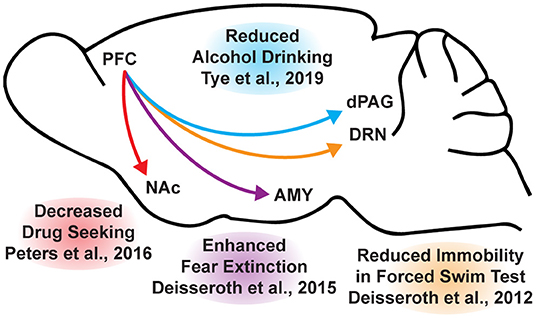
Figure 3. Opto- and chemogenetic experiments have revealed a number of circuits originating in the PFC that are relevant to the treatment of neuropsychiatric disorders. Arrows indicated excitatory projections.
In mice subjected to social defeat stress, optogenetic stimulation of the ventral medial PFC increased social interaction and reduced anhedonia as measured via the sucrose preference test (174). Deisseroth et al. later found that optogenetic stimulation of medial PFC neurons projecting to the dorsal raphe nucleus (DRN) decreased immobility in the forced swim test (175). The forced swim test is a preclinical assay for antidepressant potential with high predictive validity (176, 177), and activation of this PFC → DRN circuit likely mediates the robust effects of psychoplastogens on forced swim test behavior. While the National Institutes of Mental Health (NIMH) has stressed that the FST is not a model of depression, it is a powerful behavioral readout for activation of the PFC → DRN circuit. The DRN is a serotonergic nucleus that has been implicated in major depressive disorder (178), and recent evidence suggests that acute activation of serotoninergic neurons in the DRN increase active coping to inescapable stress (179). Dzirasa et al. demonstrated that optical stimulation of layer V pyramidal neurons in the PFC expressing channelrhodopsin-2 was sufficient to produce an antidepressant-like response in the forced swim test and suppress anxiety-like behavior in the elevated plus maze for over 10 days after the last optical stimulation session (180). This type of long-lasting anxiolytic effect is reminiscent of sustained effects observed following a single administration of a psychoplastogen. Finally, Duman et al. found that optogenetic stimulation of the infralimbic cortex produces rapid and sustained antidepressant-like effects comparable to ketamine in the forced swim, novelty-suppressed feeding, and sucrose preference tests (181). Moreover, microinfusion of ketamine into the infralimbic cortex produces comparable antidepressant-effects as systemic administration of ketamine and inactivation of the infralimbic cortex with muscimol was sufficient to block the antidepressant-like effects of systemic ketamine (181).
In addition to relieving symptoms associated with depression, psychoplastogens have also demonstrated efficacy for treating PTSD—a disorder that can involve dysfunction of the amygdala (AMY). The medial PFC and amygdala are well-connected, and bi-directional communication between these structures is likely involved in modulating responses to emotional stimuli (182). In fact, optogenetic stimulation of ventral medial PFC projections to the basomedial amygdala decreases fear responses and facilitates fear extinction learning (183). Chemogenetic inhibition of PFC neurons projecting to the amygdala is sufficient to impair fear extinction learning (184). As PTSD has often been described as a disorder of impaired fear extinction (73, 185–187), the therapeutic effects of psychoplastogens might result from their ability to strengthen PFC → AMY circuits mediating top-down control of fear responses.
Like fear extinction, drug-cue extinction is believed to involve neurons in the PFC (59). This is perhaps unsurprising given the large body of neuroimaging data suggesting that PFC hypofunction is a hallmark of addiction (68). Chemogenetic activation of ventral medial PFC neurons projecting to the nucleus accumbens (NAc) shell was able to reduce cue-induced reinstatement of drug-seeking behavior (188). Moreover, chronic cocaine self-administration has been shown to decrease the intrinsic excitability of pyramidal neurons in the PFC (189). Optogenetic stimulation of these neurons prevented compulsive drug-seeking while silencing these neurons promoted drug-seeking behavior despite being paired with aversive foot shocks (189). Similarly, pharmacological activation and inactivation of neurons in the infralimbic cortex suppressed and enhanced reinstatement of drug-seeking behavior, respectively (190). Optogenetic experiments have also revealed that ventral medial PFC projections to the NAc shell are involved in suppressing ethanol self-administration in the presence of aversive stimuli (191). A PFC → NAc circuit appears to be involved in compulsive food-seeking behavior as well, given that chemogenetic inhibition of this circuit led to compulsive food seeking even in the presence of aversive foot shocks (192). While PFC → NAc circuits have been well-established in controlling drug-seeking behavior, more recently, Tye et al. demonstrated that a PFC projection to the dorsal periaqueductal gray (dPAG) may also play an important role in addiction (193). Specifically, they showed that optogenetic activation of a PFC → dPAG circuit prevented compulsive alcohol consumption.
Psychoplastogens produce robust, fast-acting, and long-lasting effects on structural plasticity in the PFC. This may explain why they have demonstrated efficacy in many preclinical rodent behavioral tests involving PFC circuitry including the forced swim test and fear extinction learning (151, 194–197). However, achieving circuit-level selectivity is a key challenge in the design of optimized psychoplastogens with minimal to no side effects. The issue with non-selective activation of BDNF/TrkB signaling is apparent from chronic stress studies demonstrating that enhanced BDNF/TrkB signaling in the amygdala leads to maladaptive plasticity resulting in overactivation of this brain region and exacerbated anxiety and fear responses (47, 198–200). Furthermore, compounds that promote plasticity in the mesolimbic pathway could have pro-depressive and/or addictive properties (201, 202).
Psychoplastogens that target the 5-HT2A receptor have advantages over NMDA receptor antagonists like ketamine, as 5-HT2A receptors exhibit a relatively selective expression profile. With the exception of the claustrum, the highest density of 5-HT2A receptors is in layer V pyramidal neurons of the PFC, which are precisely the neurons that are most impacted in stress-related neuropsychiatric diseases. In rodents, this expression pattern has been confirmed using immunohistochemistry (203–206), light and electron microscope immunocytochemistry (203), in situ hybridization (207, 208), receptor autoradiography (209), and transgenic mice expressing EGFP under control of the 5-HT2A receptor promoter (210). A similar pattern of 5-HT2A receptor expression has been shown in human post-mortem tissue using both autoradiography (211) and in situ hybridization (212). Additionally, PET imaging has revealed a high density of 5-HT2 receptors in the frontal and temporal cortices of the human brain (213). The high genetic localization of 5-HT2A receptors to excitatory neurons in layer V of the PFC is perhaps why animals do not typically self-administer classic serotonergic psychedelics (214, 215) and most psychedelics are not considered to be addictive (216, 217).
At high doses, psychedelics reliably induce both hallucinations and mystical-type experiences. Currently, it is unclear if the mystical-type experiences they induce are necessary for their therapeutic effects in humans (218, 219). Moreover, it is unclear if the intoxicating effects of psychedelics can be decoupled from their therapeutic properties. This critical question has profound implications for healthcare, as hallucinogenic treatments will inevitably be more limited in scope given safety and cost considerations. Many patients describe psychedelic-induced “peak” or “mystical” experiences as being among the most meaningful events of their lives, and the intensity of these events correlates with therapeutic responses (40, 220–226). While these events could provide patients with valuable insight relevant to their disease symptoms, it is important to remember that correlation does not imply causation, and mystical-type experiences could simply be an epiphenomenon associated with 5-HT2A receptor activation (227, 228). Activation of 5-HT2A receptors also promotes structural and functional neuroplasticity (148), which could be the primary driver of the sustained behavioral effects following a single administration of ketamine or serotonergic psychedelics.
Given that 5-HT2A receptor activation is associated with both psychoplastogenic effects (148) and mystical-type experiences (227, 228), it is challenging to determine exactly how much each contributes to the therapeutic properties of psychedelics. However, a number of key pieces of evidence suggest that intoxicating subjective effects are not necessary to achieve some level of therapeutic efficacy. In patients treated with ketamine, floating sensations did not correlate with PFC activation as measured by 18F-PET, demonstrating that it is possible to activate cortical circuits to produce antidepressant responses without inducing dissociative effects (117). In fact, several studies have demonstrated that intraoperative ketamine administration can improve postoperative mood even though the patients were unconscious at the time of administration (229–231). This strongly suggests that the dissociative experience itself is not necessary for ketamine to produce antidepressant effects.
Additionally, there is preclinical evidence suggesting that R-ketamine is a potent psychoplastogen that induces longer-lasting antidepressant-like effects than the S-enantiomer (i.e., Spravato®) despite having a lower affinity for the NMDA receptor and producing fewer dissociative effects (232, 233). Similarly, a metabolite of R-ketamine lacking dissociative properties has also demonstrated robust antidepressant-like effects in rodents (234). Future clinical trials using these non-hallucinogenic agents will prove informative when assessing the role of subjective effects in ketamine treatment response.
While R-ketamine is still in the early stages of clinical development, 3,4-methylenedioxymethamphetamine (MDMA) has demonstrated robust results in a recently disclosed Phase III trial (235). As an atypical psychedelic of the entactogen family (236), MDMA produces robust effects on cortical neuron growth (148), and facilitates fear extinction learning (197) through a 5-HT2-dependent mechanism (237) without inducing psilocybin- or ketamine-like perceptual effects or dissociation. In fact, only about 20% of recreational MDMA users report experiencing any visual hallucinations, and these are relatively mild compared to those induced by psilocybin and LSD (236, 238, 239). While MDMA does induce a “blissful state,” likely due to its effects on monoamine efflux, it does not produce mystical-type experiences as measured using a variety of scales related to altered states of consciousness (240, 241). This is in stark contrast to drugs like ketamine and psilocybin.
As an entactogen, MDMA is pharmacologically distinct from classic serotonergic psychedelics like psilocybin and LSD. However, there is clinical evidence suggesting that non-hallucinogenic analogs of classic psychedelics can also produce therapeutic effects. Lisuride, a non-hallucinogenic structural analog of LSD, has been shown to have an antidepressant properties in the clinic (242). Moreover, lisuride has demonstrated some efficacy in preclinical models as well (243). Lisuride has a polypharmacology profile that includes activation of D2 and 5-HT1A receptors in addition to 5-HT2 receptors, and thus, it is currently unclear what receptor(s) mediate its antidepressant effects.
In addition to the clinical evidence supporting the efficacy of non-hallucinogenic psychoplastogens, mounting preclinical data suggest that the beneficial effects of psychoplastogens can be dissociated from their hallucinogenic effects. In 2019, we demonstrated that low doses of DMT produce beneficial effects in rodent behavioral paradigms relevant to treating depression and PTSD comparable to high doses (194, 195). The low dose was predicted to be subhallucinogenic in humans based on allometric scaling while the high dose was predicted to be hallucinogenic. However, the data on human psychedelic microdosing are equivocal, and therapeutic benefit of low doses of psychedelics remains to be demonstrated in a well-controlled clinical study (244–249). Moreover, administration of low doses of hallucinogenic compounds is not an ideal therapeutic strategy, as these compounds still possess the potential for abuse. Fortunately, through careful chemical design, we were able to engineer several non-hallucinogenic analogs of psychedelics with beneficial properties (250–252). These compounds do not induce a head-twitch response—a behavioral proxy for hallucinations in mice that correlates exceptionally well with hallucinogenic potency in humans (253, 254).
Our two most advanced compounds—AAZ and TBG—have demonstrated robust plasticity-promoting properties and produce sustained (>1 week) antidepressant-like effects following a single administration in both environmental (chronic unpredictable stress) and genetic (VMAT2 heterozygous mice) models of depression as measured via behavioral tests relevant to motivation, anhedonia, anxiety, and cognitive flexibility (250, 251, 255). TBG has also been shown to have anti-addictive properties in models of alcohol and opioid use disorders (250). Moreover, a single dose of TBG was able to completely rescue circuit-level dysfunction induced by chronic stress, which included deficits in dendritic spine density, calcium dynamics, and interneuron function (255). The psychoplastogenic effects of TBG and AAZ may involve activation of 5-HT2 receptors (250, 251, 256), though detailed mechanistic studies have not yet been reported.
One of the most important questions to address is the durability of psychoplastogen effects. In both humans and rodents, the antidepressant responses of ketamine appear to last for about 1 week. This correlates well with the effects of the drug on dendritic spine density (129, 150). In contrast, the mood-elevating properties of psilocybin seem to last significantly longer (40–42, 151), as do its effects on neuronal structure (152). Currently, it is unclear exactly how long the effects of non-hallucinogenic psychoplastogens will last following a single administration, and head-to-head comparisons with ketamine and psilocybin are warranted to help establish optimal dosing frequency.
Given that ~20% of American adults suffer from a mental illness in a given year (256), the economic burden of these disorders is estimated to be in the hundreds of billions of dollars annually (257, 258). Moreover, the current standard of care treatments suffer from slow onset (e.g., 7 week average response time with citalopram) (259), low efficacy (only 27% of depressed patients achieved remission after 12 weeks of citalopram treatment as measured by HAM-D) (259), and often intolerable side-effects (260), it is clear that we need to expand psychiatry's arsenal to include a variety of new approaches. Both hallucinogenic and non-hallucinogenic psychoplastogens have important roles to play in the fight against mental illness, but it is critical to identify which patient populations are best served by these types of treatment. First, many patients may not want to participate in psychedelic-assisted therapy (261) given that hallucinogens can induce acute anxiety and may lead to challenging experiences (40). For those who are open to psychedelic-assisted therapy, significant barriers to treatment still exist.
Currently, there are over 300 clinical trials registered on clinicaltrials.gov1 to study the effects of psychedelics in humans with the majority of recent studies focusing on the effects of psilocybin and MDMA (262, 263). However, only a fraction of patients who volunteer for psychedelic-assisted therapy are permitted to participate. Exclusion criteria for these trials typically include cardiovascular and mental health risks that could potentially be exacerbated by psychedelics. For example, psychedelic-assisted therapy is generally contraindicated for people with a family history of psychotic disorders or complex psychiatric comorbidities to avoid the possibility of triggering a first episode of psychosis or precipitating suicidal behaviors (264).
In two recent psilocybin clinical trials, ~95% of all volunteers were eliminated on the basis of exclusion criteria (Figure 4) (8, 42). For comparison, ~25% of participants were excluded from two recent major depressive disorder (MDD) trials of the non-hallucinogenic compound vortioxetine (Figure 4) (265, 266). If these strict exclusion criteria are deemed necessary by the FDA and payers, they will drastically limit the number of patients who could potentially benefit from this treatment paradigm, especially when you consider the high genetic heritability and co-morbidity of neuropsychiatric disorders (Figure 5) (272–278). Between 33 and 66% of patients suffering from MDD have a psychiatric comorbidity that could potentially exclude them from psychedelic-assisted therapy (267–271, 279, 280).
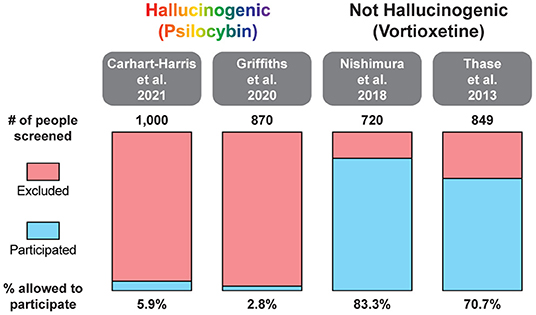
Figure 4. Percentage of patients excluded from recent large MDD trials of the hallucinogenic drug psilocybin and the non-hallucinogenic drug vortioxetine. A comprehensive analysis of all psychedelic trials is beyond the scope of this review. For additional information on the broad range of smaller trials with psychedelics, please see (262, 263).
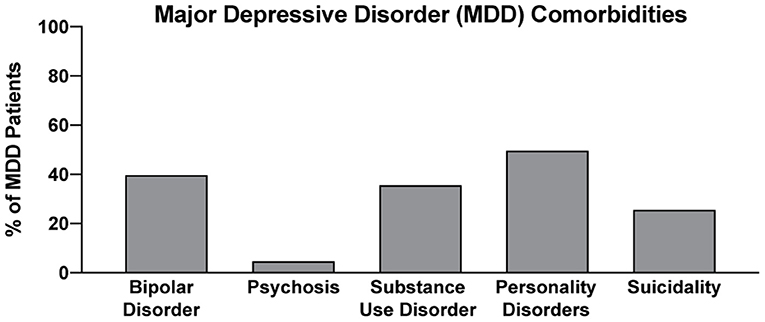
Figure 5. Percentage of MDD with various comorbidities. Data for bipolar disorder, psychosis, substance use disorder, personality disorders, and suicidality were obtained from references (267–271), respectively.
Identifying factors that can predict how patients will respond to psychedelic-assisted therapy will be important for maximizing efficacy and reducing adverse events. For example, patients who exhibit apprehension, preoccupation, or baseline attachment avoidance appear to be more likely to have challenging experiences following administration of a psychedelic drug (281, 282). Using these metrics to pre-screen patients should improve the safety and efficacy of psychedelic-assisted therapy; however, stratification of patients in this manner will inevitably reduce the addressable patient population, again, emphasizing the need for a non-hallucinogenic first-line treatment against depression and related disorders.
Presumably, treatment with non-hallucinogenic psychoplastogens would not be limited by comorbidities or other factors that might exclude someone from participating in psychedelic-assisted therapy. For these reasons, and others described below, it seems reasonable for non-hallucinogenic psychoplastogens to potentically be used as first-line treatments, assuming that they demonstrate greater efficacy in humans than the standard of care (i.e., traditional antidepressants). Psychedelic-assisted therapy could be reserved for patients who have not responded to any other medicine. Indeed, some patients may benefit from the mystical-type experiences occasioned by psychedelics as many people rate these experiences as among the most meaningful in their lives. Such a positive experience could have a variety of effects on patients including, but not limited to, improving the relationship between therapist and patient, helping patients to gain insight about their condition, or producing a powerful placebo effect. It is challenging to design truly double-blind placebo-controlled clinical trials with psychedelics given their profound subjective effects. Despite efforts to employ “active placebos” (e.g., niacin or a low-dose of a psychedelic) (40, 220), many patients and clinicians can still correctly distinguish between a high dose of a psychedelic drug and an active placebo. Thus, the exact role of placebo effects in the overall efficacy of psychedelic-assisted therapy has not been firmly established. Even so, an enhanced placebo effect can potentially be leveraged to treat severely depressed patients who have not been helped by other means (283–285).
In addition to comorbidities and genetic predispositions, the basic mechanics of healthcare systems are likely to be major factors limiting the number of patients who will ultimately be able to receive hallucinogen-based therapeutics. Given their profound effects on perception, these drugs necessitate administration in a clinical setting where the patient can be observed by medical professionals. In 2019, Johnson & Johnson's SpravatoⓇ (esketamine nasal spray) became the first hallucinogenic psychoplastogen treatment approved by the FDA for refractory depression (286), and in 2020 the indication was expanded to include adult major depressive disorder (287). SpravatoⓇ has to be administered in a clinic under supervision due to the known risks of serious adverse outcomes resulting from disassociation and sedation as well as the potential for abuse. After intake and medical screening, the patient is enrolled in the SpravatoⓇ REMS (Risk Evaluation and Mitigation Strategy) program. SpravatoⓇ is self-administered intranasally in the presence of a healthcare professional, and the patient is monitored for the next 2 h. Patients receive two treatments a week for the 1st month, and once a week or once every 2 weeks after that. For racemic ketamine administered intravenously, clinics follow the NIMH trial protocol of six infusions administered over a two-to-3-week period in an outpatient clinic or medical facility. Boosters are given every 3–5 weeks after that. Intravenous infusion takes about 40 min, and guidance is provided not to drive, operate any dangerous machinery, or make any important decisions until the day after a ketamine treatment. The requirement for administration in a clinical setting drastically increases the cost of both racemic ketamine and SpravatoⓇ.
Though psychedelics are expected to be administered less frequently than ketamine given their robust effects, they too must be administered under the care of a healthcare professional. Current research-based psychedelic-assisted therapy has three phases: preparation, treatment, and integration. Given that psychedelics have the potential to cause dangerous behaviors due to potential negative psychological reactions such as anxiety, fear, panic, or psychosis, a team of two professional therapists is required to be in attendance to supervise, but minimally interact with the patients throughout the course of the drug's action (288). Moreover, it is recommended that multiple healthcare workers be involved in all three stages to ensure that professional boundaries are maintained (289). The preparation session establishes the alliance between the therapist and the participant. Treatment sessions, which typically last 6–8 h for psilocybin-assisted therapy (288) allow the participants to have a peak experience within a set and setting thought to be most conducive to optimizing the therapeutic effect. The integration session is meant to help the participant process, rationalize, and gain insight from the hallucinogenic experience. Although psychedelic therapies have demonstrated outstanding benefits in several clinical trials (290), the cost-effectiveness and overall accessibility of such therapies raises major concerns.
The costs associated with treating mental illness with hallucinogenic psychoplastogens is extremely high compared to the standard of care (Figure 6). For example, the initial month of ketamine therapy costs from $4,720 to $6,785, and subsequent monthly therapy can range from $2,360 to $3,540 (291). Additional costs associated with patients taking time off work to receive treatment and to travel to appropriately staffed/equipped clinics must also be considered.
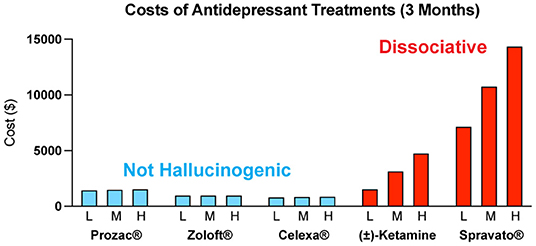
Figure 6. Estimated quarterly (3 months) costs (L = low, M = medium, H = high) for daily administration of non-hallucinogenic take-home traditional antidepressants (blue) and intermittent dosing of clinically administered, dissociative psychoplastogen (ketamine) treatments (red). Note: generic ketamine costs include drug, administration (IV), and required monitoring, while Prozac®, Zoloft®, Celexa®, and Spravato® costs are for the drug only. Retail costs for SSRIs reflect the low and high branded price published on goodrx.com1 with the medium price being calculated as an average of the two. To estimate the total cost, we used medium doses for each drug (Prozac® 20 mg, Zoloft® 50 mg, and Celexa® 20 mg,) and assumed daily dosing for 90 days. Ketamine infusion cost was sourced from ketamineclinicsdirectory.com/ketamine-infusion-cost/.3 This site estimates the low and high cost of a complete set of 4–6 generic ketamine infusions to be $1,600–$4,800, respectively. The medium cost was calculated as an average of the two ($3,200). Our estimate for the 3-month cost of generic ketamine assumes that no additional doses are needed beyond the initial 4–6 doses. Retail Spravato® costs were estimated based on the price of a 56 mg dose ($900) published on goodrx.com3 and dosing was based on the package insert (Revised 7/2020). The label indicates Spravato® should be dosed twice a week during the induction phase (weeks 1–4; $900 × 2 × 4 = $7,200), once a week during the maintenance phase (weeks 5–8; $900 × 1 × 4 = $3,600), and once a week thereafter (weeks 9–12; $900 × 1 × 4 = $3,600). The estimated low cost of Spravato® includes the induction phase only. The estimated medium cost of Spravato® includes the induction phase and the maintenance phase. The estimated high cost of Spravato® includes the induction phase, maintenance phase, and 4 weeks of additional treatment. Note: the cost of administration and monitoring for Spravato are not publicly available, and thus not included, but these costs are anticipated to be significant.
Many people assume that psilocybin-assisted therapy will be cheaper than ketamine treatment as the antidepressant effects following a single administration of psilocybin appear to be more enduring than those of ketamine (292). However, the overall cost of psilocybin treatment is estimated to be close to, if not higher than, the cost of SpravatoⓇ treatment due to the increased session participation of therapists. The low-throughput nature and associated high costs of the psychedelic-assisted therapy model have been acknowledged by the community and have resulted in new studies being launched by non-profit and corporate sponsors to streamline the process and reduce costs through group and virtual therapy sessions (293). However, it is currently unclear if such approaches will be as safe and efficacious as the current model.
The cost associated with rolling out the psychedelic-assisted therapy model poses a great hurdle to its effective implementation in the current mental health ecosystem. In 2019, the Institute for Clinical and Economic Review (ICER) issued a recommendation that deemed SpravatoⓇ to deliver a “low value for money” according to their value-assessment framework (294). Furthermore, the UK price watchdog agency, the National23 Institute for Health and Care Excellence (NICE), refused to endorse SpravatoⓇ therapy for inclusion as a reimbursable drug on the UK's National Health System (NHS) (295). Although NICE acknowledged the drug's efficacy for relieving the symptoms of depression, the agency also commented that the “introduction of esketamine into clinical practice in the NHS will be complex because the structure and delivery of services would need to be changed. Estimates of the costs of providing the clinical service for esketamine were highly uncertain.” As these price watchdog agencies become progressively more influential in the decision-making process of payers, their recommendations will likely lead to payers raising their medical criteria for coverage, potentially jeopardizing patient access to psychedelic-assisted therapy for financial reasons. We are already seeing racemic ketamine being administered off-label for a variety of neuropsychiatric disorders without reimbursement from insurance companies (296). This leads to an inequity issue, where only wealthy individuals who can afford the out-of-pocket costs have access to this type of treatment.
In addition to the financial burdens to the patients, understaffing of qualified psychotherapy practitioners is likely to be one of the biggest issues for nationwide implementation of psychedelic-assisted therapy. The FDA requires all U.S. therapists to have at least a master's degree, and current best practices require a minimum of two therapists to be present during psychedelic sessions (288). Moreover, all the therapists who participate in the psilocybin and MDMA clinical development programs are required to hold a professional license and demonstrate clinical experience in psychotherapy or mental health counseling (288). Although institutes such as the California Institute of Integral Study (CIIS), Multidisciplinary Association for Psychedelic Studies (MAPS), and corporate programs from companies like COMPASS Pathways offer short-term training programs for psychedelic-assisted counseling, there is a huge demand and supply gap for competent therapists considering that the estimated prevalence of treatment-resistant depression (TRD) in the U.S. is around 2.8 million people (258). Moreover, given that set and setting are well-known to influence the subjective effects of psychedelics (288, 297), clinical centers with appropriate facilities will need to be established.
The limitations associated with hallucinogenic medicines could prevent many patients from benefiting from the growing body of psychedelic-inspired research related to pathological circuit remodeling. By eliminating the need to treat patients in the clinic, non-hallucinogenic psychoplastogens—which would presumably be available from retail pharmacies much like traditional antidepressants—would reduce the complexity of treatment administration and have the potential to greatly expand access of patients to psychoplastogenic medicines. These molecules produce the same types of long-lasting structural and functional changes in the brains of preclinical animals that follow administration of ketamine or serotonergic psychedelics. Assuming that they are efficacious in humans, non-hallucinogenic psychoplastogens would not bear the same administrative costs and limitations as first-generation hallucinogenic psychoplastogens. When comparing the known or predicted yearly healthcare costs associated with ketamine, psilocybin, SSRIs, and non-hallucinogenic psychoplastogen treatments, it is clear that the non-hallucinogenic approaches will be more cost-effective (Figure 6).
Non-hallucinogenic psychoplastogens could potentially meet the demands of patients who fail to respond to monoaminergic agents, or even replace them as more effective first-line treatments with fewer side-effects. In preclinical animal models, like in humans, traditional monoaminergic antidepressants require chronic administration to achieve robust efficacy (298). Such chronic dosing paradigms inevitably lead to a host of undesirable side-effects that include weight gain, sexual dysfunction, and gastrointestinal problems (299), with many patients refusing to take traditional antidepressants due to their side-effects (260). In contrast, non-hallucinogenic psychoplastogens, like their hallucinogenic counterparts, produce sustained therapeutic behavioral responses in preclinical animal models after a single administration (250, 251). Thus, the need for chronic dosing in humans will likely be obviated resulting in fewer undesired side-effects. However, a major challenge for the field will be to determine exactly what frequency of dosing will be most effective. Fortunately, new imaging tools have the potential to identify biomarkers of psychoplastogen efficacy. These include relatively new positron emission tomography (PET) tracers (104, 105) that can non-invasively measure the effects of psychoplastogens on synaptic vesicle density in vivo (155).
It is quite possible that insight gained from mystical-type experiences coupled with changes in neurocircuitry is responsible for the large effect size and durability of psychedelic-assisted therapy. However, if even a fraction of the efficacy or durability could be achieved using compounds that do not induce mystical-type experiences or hallucinations, a much larger patient population could benefit. While mystical-type experiences will undoubtedly be beneficial for some patients, they may not be necessary for all patients. Thus, if non-hallucinogenic psychoplastogens can demonstrate efficacy in the clinic as robust as their effects in preclinical models, their advantages over both traditional monoaminergic antidepressants and hallucinogenic psychoplastogens should position them as first-line treatment options (Figure 7).

Figure 7. Comparison of traditional antidepressant treatments with hallucinogenic and non-hallucinogenic psychoplastogen treatments. *Currently, no clinical trials have been conducted with non-hallucinogenic psychoplastogens, and thus, their fast-acting and long-lasting effects refer to preclinical testing only. **The scalability and cost-effectiveness of non-hallucinogenic psychoplastogens are based on the assumption that they will produce clinical efficacy greater than the standard of care (i.e., traditional antidepressants).
The ability to selectively modulate neural circuits using small molecule psychoplastogens opens up new horizons in neuropsychiatry focused on healing pathological neural circuitry rather than masking disease symptoms. This type of circuit-based approach represents a fundamental shift in how we might treat a number of neuropsychiatric diseases and has important implications for the future of CNS drug discovery. Given the history of neuropsychiatry and the intractable nature of brain disorders, we need to take advantage of every available tool in our therapeutic arsenal including both hallucinogenic and non-hallucinogenic psychoplastogens. Ketamine and psilocybin have demonstrated that it is possible to produce long-lasting beneficial changes in neural circuitry using small molecule drugs, and they have forged a path for future, optimized psychoplastogens to take their place. If we ever hope to heal the nearly 20% of the population suffering from a mental illness, we must find innovative ways to reduce healthcare costs and broaden patient access to psychoplastogenic medicines. Non-hallucinogenic psychoplastogens have the potential to be truly scalable solutions to many of the problems facing neuropsychiatry.
DO wrote the manuscript with input from all authors. MV and AA made significant contributions to the introduction and the sections titled Harnessing Structural Plasticity to Treat Mental Illness and Possible Explanations for the Broad Therapeutic Potential of Psychoplastogens. RM and MR made significant contributions to the sections titled Patient Populations Best Suited for Hallucinogenic and Non-Hallucinogenic Approaches, Healthcare System Issues with the Psychedelic-Assisted Psychotherapy Model, and Democratizing Access to Psychoplastogenic Medicines. All authors contributed to the article and approved the submitted version.
This work was supported by funds from the National Institutes of Health (NIH) (R01GM128997 to DO and 5T32GM099608 to MV). The authors declare that this study received funding from Delix Therapeutics Inc. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
DO is a co-founder and chief scientific officer of Delix Therapeutics, Inc. MR and RM are employees of Delix Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. ^Clinical trials. https://clinicaltrials.gov/ [Accessed June 12, 2021]. (2021). Keyword: psychedelic including synonyms hallucinogen and hallucinogenic drugs. We identified 316 studies. Re-querying the database in August 2021 we identified 324 studies of which 151 studies are completed.
2. ^Data were obtained from goodrx.com [accessed June 2021].
3. ^Data for racemic ketamine were obtained from https://ketamineclinicsdirectory.com/ [accessed June 2021].
1. Muller JC, Pryor WW, Gibbons JE, Orgain ES. Depression and anxiety occurring during Rauwolfia therapy. J Am Med Assoc. (1955) 159:836–9. doi: 10.1001/jama.1955.02960260006002
2. Nickell JR, Siripurapu KB, Vartak A, Crooks PA, Dwoskin LP. The vesicular monoamine transporter-2: an important pharmacological target for the discovery of novel therapeutics to treat methamphetamine abuse. Adv Pharmacol. (2014) 69:71–106. doi: 10.1016/B978-0-12-420118-7.00002-0
3. Bortolato M, Chen K, Shih JC. Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv Drug Deliv Rev. (2008) 60:1527–33. doi: 10.1016/j.addr.2008.06.002
4. Racagni G, Popoli M. Cellular and molecular mechanisms in the long-term action of antidepressants. Dialogues Clin Neurosci. (2008) 10:385–400. doi: 10.31887/DCNS.2008.10.4/gracagni
5. Ruhe HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: a meta-analysis of monoamine depletion studies. Mol Psychiatry. (2007) 12:331–59. doi: 10.1038/sj.mp.4001949
6. Corrigan MH, Denahan AQ, Wright CE, Ragual RJ, Evans DL. Comparison of pramipexole, fluoxetine, and placebo in patients with major depression. Depress Anxiety. (2000) 11:58–65. doi: 10.1002/(SICI)1520-6394(2000)11:2<58::AID-DA2>3.0.CO
7. Phillips JL, Norris S, Talbot J, Hatchard T, Ortiz A, Birmingham M, et al. Single and repeated ketamine infusions for reduction of suicidal ideation in treatment-resistant depression. Neuropsychopharmacology. (2020) 45:606–12. doi: 10.1038/s41386-019-0570-x
8. Carhart-Harris R, Giribaldi B, Watts R, Baker-Jones M, Murphy-Beiner A, Murphy R, et al. Trial of psilocybin versus escitalopram for depression. N Engl J Med. (2021) 384:1402–11. doi: 10.1056/NEJMoa2032994
9. Hyman SE, Nestler EJ. Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am J Psychiatry. (1996) 153:151–62. doi: 10.1176/ajp.153.2.151
10. Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. (2008) 33:88–109. doi: 10.1038/sj.npp.1301574
11. Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. (1997) 54:597–606. doi: 10.1001/archpsyc.1997.01830190015002
12. Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. (2012) 338:68–72. doi: 10.1126/science.1222939
13. Maya Vetencourt JF, Sale A, Viegi A, Baroncelli L, De Pasquale R, O'Leary OF. The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science. (2008) 320:385–8. doi: 10.1126/science.1150516
14. Castrén E, Antila H. Neuronal plasticity and neurotrophic factors in drug responses. Mol Psychiatry. (2017) 22:1085–95. doi: 10.1038/mp.2017.61
15. Altar CA, Whitehead RE, Chen R, Wörtwein G, Madsen TM. Effects of electroconvulsive seizures and antidepressant drugs on brain-derived neurotrophic factor protein in rat brain. Biol Psychiatry. (2003) 54:703–9. doi: 10.1016/S0006-3223(03)00073-8
16. Müller MB, Toschi N, Kresse AE, Post A, Keck ME. Long-term repetitive transcranial magnetic stimulation increases the expression of brain-derived neurotrophic factor and cholecystokinin mRNA, but not neuropeptide tyrosine mRNA in specific areas of rat brain. Neuropsychopharmacology. (2000) 23:205–15. doi: 10.1016/S0893-133X(00)00099-3
17. Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. (1995) 15:7539–47. doi: 10.1523/JNEUROSCI.15-11-07539.1995
18. Gómez-Pinilla F, Ying Z, Roy RR, Molteni R, Edgerton VR. Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplasticity. J Neurophysiol. (2002) 88:2187–95. doi: 10.1152/jn.00152.2002
19. Giese M, Beck J, Brand S, Muheim F, Hemmeter U, Hatzinger M, et al. Fast BDNF serum level increase and diurnal BDNF oscillations are associated with therapeutic response after partial sleep deprivation. J Psychiatr Res. (2014) 59:1–7. doi: 10.1016/j.jpsychires.2014.09.005
20. Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. (2003) 112:257–69. doi: 10.1016/S0092-8674(03)00035-7
21. Lee Y, Lim SW, Kim SY, Chung JW, Kim J, Myung W, et al. Association between the BDNF Val66Met polymorphism and chronicity of depression. Psychiatry Investig. (2013) 10:56–61. doi: 10.4306/pi.2013.10.1.56
22. Verhagen M, van der Meij A, van Deurzen PA, Janzing JG, Arias-Vásquez A, Buitelaar JK, et al. Meta-analysis of the BDNF Val66Met polymorphism in major depressive disorder: effects of gender and ethnicity. Mol Psychiatry. (2010) 15:260–71. doi: 10.1038/mp.2008.109
23. Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. (2006) 59:1116–27. doi: 10.1016/j.biopsych.2006.02.013
24. Duman RS. Role of neurotrophic factors in the etiology and treatment of mood disorders. Neuromolecular Med. (2004) 5:11–25. doi: 10.1385/NMM:5:1:011
25. Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. (2002) 34:13–25. doi: 10.1016/S0896-6273(02)00653-0
26. Castrén E, Rantamäki T. Role of brain-derived neurotrophic factor in the aetiology of depression: implications for pharmacological treatment. CNS Drugs. (2010) 24:1–7. doi: 10.2165/11530010-000000000-00000
27. Castrén E, Rantamäki T. The role of BDNF and its receptors in depression and antidepressant drug action: reactivation of developmental plasticity. Dev Neurobiol. (2010) 70:289–97. doi: 10.1002/dneu.20758
28. Castrén E, Võikar V, Rantamäki T. Role of neurotrophic factors in depression. Curr Opin Pharmacol. (2007) 7:18–21. doi: 10.1016/j.coph.2006.08.009
29. Martinowich K, Manji H, Lu B. New insights into BDNF function in depression and anxiety. Nat Neurosci. (2007) 10:1089–93. doi: 10.1038/nn1971
30. Duman RS Li N. A neurotrophic hypothesis of depression: role of synaptogenesis in the actions of NMDA receptor antagonists. Philos Trans R Soc Lond B Biol Sci. (2012) 367:2475–84. doi: 10.1098/rstb.2011.0357
31. Castrén E. Neurotrophic effects of antidepressant drugs. Curr Opin Pharmacol. (2004) 4:58–64. doi: 10.1016/j.coph.2003.10.004
32. Quevedo J, Carvalho AF, Zarate CA. The neurotrophic hypothesis of depression revisited: new insights and therapeutic implications. In: Neurobiology of Depression: Road to Novel Therapeutics. Cambridge, MA: Academic Press (2019). p. 43–62. doi: 10.1016/B978-0-12-813333-0.00006-8
33. Björkholm C, Monteggia LM. BDNF—a key transducer of antidepressant effects. Neuropharmacology. (2016) 102:72–9. doi: 10.1016/j.neuropharm.2015.10.034
34. Castrén E, Kojima M. Brain-derived neurotrophic factor in mood disorders and antidepressant treatments. Neurobiol Dis. (2017) 97:119–26. doi: 10.1016/j.nbd.2016.07.010
35. Duman RS, Deyama S, Fogaça MV. Role of BDNF in the pathophysiology and treatment of depression: activity-dependent effects distinguish rapid-acting antidepressants. Eur J Neurosci. (2021) 53:126–39. doi: 10.1111/ejn.14630
36. Yang T, Nie Z, Shu H, Kuang Y, Chen X, Cheng J, et al. The role of BDNF on neural plasticity in depression. Front Cell Neurosci. (2020) 14:82. doi: 10.3389/fncel.2020.00082
37. Olson DE. Psychoplastogens: a promising class of plasticity-promoting neurotherapeutics. J Exp Neurosci. (2018) 12:1179069518800508. doi: 10.1177/1179069518800508
38. Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. (2006) 63:856–64. doi: 10.1001/archpsyc.63.8.856
39. McGirr A, Berlim MT, Bond DJ, Fleck MP, Yatham LN, Lam RW, et al. systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychol Med. (2015) 45:693–704. doi: 10.1017/S0033291714001603
40. Griffiths RR, Johnson MW, Carducci MA, Umbricht A, Richards WA, Richards BD, et al. Psilocybin produces substantial and sustained decreases in depression and anxiety in patients with life-threatening cancer: a randomized double-blind trial. J Psychopharmacol. (2016) 30:1181–97. doi: 10.1177/0269881116675513
41. Carhart-Harris RL, Bolstridge M, Day CMJ, Rucker J, Watts R, Erritzoe DE, et al. Psilocybin with psychological support for treatment-resistant depression: six-month follow-up. Psychopharmacology. (2018) 235:399–408. doi: 10.1007/s00213-017-4771-x
42. Davis AK, Barrett FS, May DG, Cosimano MP, Sepeda ND, Johnson MW, et al. Effects of psilocybin-assisted therapy on major depressive disorder: a randomized clinical trial. J Am Med Assoc Psychiatry. (2021) 78:481–9. doi: 10.1001/jamapsychiatry.2020.3285
43. Yang L, Zhao Y, Wang Y, Liu L, Zhang X, Li B, et al. The effects of psychological stress on depression. Curr Neuropharmacol. (2015) 13:494–504. doi: 10.2174/1570159X1304150831150507
44. Averill LA, Fouda S, Murrough JW, Abdallah CG. Chronic stress pathology and ketamine-induced alterations in functional connectivity in major depressive disorder: An abridged review of the clinical evidence. Adv Pharmacol. (2020) 89:163–94. doi: 10.1016/bs.apha.2020.04.003
45. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. (2007) 87:873–904. doi: 10.1152/physrev.00041.2006
46. Rodrigues SM, LeDoux JE, Sapolsky RM. The influence of stress hormones on fear circuitry. Annu Rev Neurosci. (2009) 32:289–313. doi: 10.1146/annurev.neuro.051508.135620
47. Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. (2002) 22:6810–8. doi: 10.1523/JNEUROSCI.22-15-06810.2002
48. Liu RJ, Aghajanian GK. Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. Proc Natl Acad Sci USA. (2008) 105:359–64. doi: 10.1073/pnas.0706679105
49. Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron. (2012) 73:962–77. doi: 10.1016/j.neuron.2011.12.033
50. Myers B, McKlveen JM, Herman JP. Glucocorticoid actions on synapses, circuits, and behavior: implications for the energetics of stress. Front Neuroendocrinol. (2014) 35:180–96. doi: 10.1016/j.yfrne.2013.12.003
51. Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. (2011) 13:22–37. doi: 10.1038/nrn3138
52. Licznerski P, Jonas EA. BDNF signaling: harnessing stress to battle mood disorder. Proc Natl Acad Sci USA. (2018) 115:3742–4. doi: 10.1073/pnas.1803645115
53. Mizoguchi K, Ishige A, Aburada M, Tabira T. Chronic stress attenuates glucocorticoid negative feedback: involvement of the prefrontal cortex and hippocampus. Neuroscience. (2003) 119:887–97. doi: 10.1016/S0306-4522(03)00105-2
54. Campioni MR, Xu M, McGehee DS. Stress-induced changes in nucleus accumbens glutamate synaptic plasticity. J Neurophysiol. (2009) 101:3192–8. doi: 10.1152/jn.91111.2008
55. Bessa JM, Morais M, Marques F, Pinto L, Palha JA, Almeida OFX, et al. Stress-induced anhedonia is associated with hypertrophy of medium spiny neurons of the nucleus accumbens. Transl Psychiatry. (2013) 3:e266. doi: 10.1038/tp.2013.39
56. Riga D, Matos MR, Glas A, Smit AB, Spijker S, Van den Oever MC. Optogenetic dissection of medial prefrontal cortex circuitry. Front Syst Neurosci. (2014) 8:230. doi: 10.3389/fnsys.2014.00230
57. Crews FT, Boettiger CA. Impulsivity, frontal lobes and risk for addiction. Pharmacol Biochem Behav. (2009) 93:237–47. doi: 10.1016/j.pbb.2009.04.018
58. Kalivas PW. Addiction as a pathology in prefrontal cortical regulation of corticostriatal habit circuitry. Neurotox Res. (2008) 14:185–9. doi: 10.1007/BF03033809
59. Peters J, Kalivas PW, Quirk GJ. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem. (2009) 16:279–88. doi: 10.1101/lm.1041309
60. Gass JT, Chandler LJ. The plasticity of extinction: contribution of the prefrontal cortex in treating addiction through inhibitory learning. Front Psychiatry. (2013) 4:46. doi: 10.3389/fpsyt.2013.00046
61. Kouneiher F, Charron S, Koechlin E. Motivation and cognitive control in the human prefrontal cortex. Nat Neurosci. (2009) 12:939–45. doi: 10.1038/nn.2321
62. Marek R, Sun Y, Sah P. Neural circuits for a top-down control of fear and extinction. Psychopharmacology. (2019) 236:313–20. doi: 10.1007/s00213-018-5033-2
63. Gilmartin MR, Balderston NL, Helmstetter FJ. Prefrontal cortical regulation of fear learning. Trends Neurosci. (2014) 37:455–64. doi: 10.1016/j.tins.2014.05.004
64. Kennerley SW, Walton ME. Decision making and reward in frontal cortex: complementary evidence from neurophysiological and neuropsychological studies. Behav Neurosci. (2011) 125:297–317. doi: 10.1037/a0023575
65. Duverne S, Koechlin E. Rewards and cognitive control in the human prefrontal cortex. Cereb Cortex. (2017) 27:5024–39. doi: 10.1093/cercor/bhx210
66. Arnsten AF. Stress signaling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. (2009) 10:410–22. doi: 10.1038/nrn2648
67. Duman RS, Aghajanian GK, Sanacora G, Krystal JH. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med. (2016) 22:238–49. doi: 10.1038/nm.4050
68. Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci. (2011) 12:652–69. doi: 10.1038/nrn3119
69. Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry. (2002) 159:1642–52. doi: 10.1176/appi.ajp.159.10.1642
70. Volkow ND, Fowler JS, Wang GJ, Goldstein RZ. Role of dopamine, the frontal cortex and memory circuits in drug addiction: insight from imaging studies. Neurobiol Learn Mem. (2002) 78:610–24. doi: 10.1006/nlme.2002.4099
71. Pahng AR, McGinn MA, Paulsen RI, Edwards S. The prefrontal cortex as a critical gate of negative affect and motivation in alcohol use disorder. Curr Opin Behav Sci. (2017) 13:139–43. doi: 10.1016/j.cobeha.2016.11.004
72. Arnsten AF, Raskind MA, Taylor FB, Connor DF. The effects of stress exposure on prefrontal cortex: translating basic research into successful treatments for post-traumatic stress disorder. Neurobiol Stress. (2015) 1:89–99. doi: 10.1016/j.ynstr.2014.10.002
73. Koenigs M, Grafman J. Posttraumatic stress disorder: the role of medial prefrontal cortex and amygdala. Neuroscientist. (2009) 15:540–8. doi: 10.1177/1073858409333072
74. Hare BD, Duman RS. Prefrontal cortex circuits in depression and anxiety: contribution of discrete neuronal populations and target regions. Mol Psychiatry. (2020) 25:2742–58. doi: 10.1038/s41380-020-0685-9
75. Khundakar A, Morris C, Oakley A, McMeekin W, Thomas AJ. Morphometric analysis of neuronal and glial cell pathology in the dorsolateral prefrontal cortex in late-life depression. Br J Psychiatry. (2009) 195:163–9. doi: 10.1192/bjp.bp.108.052688
76. Ray MT, Shannon Weickert C, Webster MJ. Decreased BDNF and TrkB mRNA expression in multiple cortical areas of patients with schizophrenia and mood disorders. Transl Psychiatry. (2014) 4:e389. doi: 10.1038/tp.2014.26
77. Tripp A, Oh H, Guilloux JP, Martinowich K, Lewis DA, Sibille E. Brain-derived neurotrophic factor signaling and subgenual anterior cingulate cortex dysfunction in major depressive disorder. Am J Psychiatry. (2012) 169:1194–202. doi: 10.1176/appi.ajp.2012.12020248
78. Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. (1999) 45:1085–98. doi: 10.1016/S0006-3223(99)00041-4
79. Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb Cortex. (2002) 12:386–94. doi: 10.1093/cercor/12.4.386
80. Feyissa AM, Chandran A, Stockmeier CA, Karolewicz B. Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog Neuropsychopharmacol Biol Psychiatry. (2009) 33:70–5. doi: 10.1016/j.pnpbp.2008.10.005
81. Jernigan CS, Goswami DB, Austin MC, Iyo AH, Chandran A, Stockmeier CA, et al. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. (2011) 35:1774–9. doi: 10.1016/j.pnpbp.2011.05.010
82. Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med. (2012) 18:1413–7. doi: 10.1038/nm.2886
83. Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. (2000) 57:65–73. doi: 10.1001/archpsyc.57.1.65
84. Dolan RJ, Bench CJ, Brown RG, Scott LC, Frackowiak RS. Neuropsychological dysfunction in depression: the relationship to regional cerebral blood flow. Psychol Med. (1994) 24:849–57. doi: 10.1017/S0033291700028944
85. Breier A, Buchanan RW, Elkashef A, Munson RC, Kirkpatrick B, Gellad F. Brain morphology and schizophrenia. A magnetic resonance imaging study of limbic, prefrontal cortex, and caudate structures. Arch Gen Psychiatry. (1992) 49:921–6. doi: 10.1001/archpsyc.1992.01820120009003
86. Meyer-Lindenberg A, Tost H. Neuroimaging and plasticity in schizophrenia. Restor Neurol Neurosci. (2014) 32:119–27. doi: 10.3233/RNN-139014
87. Soares JC, Mann JJ. The functional neuroanatomy of mood disorders. J Psychiatr Res. (1997) 31:393–432. doi: 10.1016/S0022-3956(97)00016-2
88. Bearden CE, Hoffman KM, Cannon TD. The neuropsychology and neuroanatomy of bipolar affective disorder: a critical review. Bipolar Disord. (2001) 3:106–50. doi: 10.1034/j.1399-5618.2001.030302.x
89. Cannistraro PA, Rauch SL. Neural circuitry of anxiety: evidence from structural and functional neuroimaging studies. Psychopharmacol Bull. (2003) 37:8–25.
90. Drevets WC, Price JL, Simpson JR Jr, Todd RD, Reich T, Vannier M, et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. (1997) 386:824–7. doi: 10.1038/386824a0
91. Kühn S, Gallinat J. Gray matter correlates of posttraumatic stress disorder: a quantitative meta-analysis. Biol Psychiatry. (2013) 73:70–4. doi: 10.1016/j.biopsych.2012.06.029
92. Zhang Y, Yang Y, Zhu L, Zhu Q, Jia Y, Zhang L, et al. Volumetric deficit within the fronto-limbic-striatal circuit in first-episode drug naïve patients with major depression disorder. Front Psychiatry. (2021) 11:600583. doi: 10.3389/fpsyt.2020.600583
93. Wang J, Fan Y, Dong Y, Ma M, Ma Y, Dong Y, et al. Alterations in brain structure and functional connectivity in alcohol dependent patients and possible association with impulsivity. PLoS ONE. (2016) 11:e0161956. doi: 10.1371/journal.pone.0161956
94. Asensio S, Morales JL, Senabre I, Romero MJ, Beltran MA, Flores-Bellver M, et al. Magnetic resonance imaging structural alterations in brain of alcohol abusers and its association with impulsivity. Addict Biol. (2016) 21:962–71. doi: 10.1111/adb.12257
95. Yuan Y, Zhu Z, Shi J, Zou Z, Yuan F, Liu Y, et al. Gray matter density negatively correlates with duration of heroin use in young lifetime heroin-dependent individuals. Brain Cogn. (2009) 71:223–8. doi: 10.1016/j.bandc.2009.08.014
96. Lyoo IK, Pollack MH, Silveri MM, Ahn KH, Diaz CI, Hwang J, et al. Prefrontal and temporal gray matter density decreases in opiate dependence. Psychopharmacology. (2006) 184:139–44. doi: 10.1007/s00213-005-0198-x
97. Liu H, Hao Y, Kaneko Y, Ouyang X, Zhang Y, Xu L, et al. Frontal and cingulate gray matter volume reduction in heroin dependence: optimized voxel-based morphometry. Psychiatry Clin Neurosci. (2009) 63:563–8. doi: 10.1111/j.1440-1819.2009.01989.x
98. Schwartz DL, Mitchell AD, Lahna DL, Luber HS, Huckans MS, Mitchell SH, et al. Global and local morphometric differences in recently abstinent methamphetamine-dependent individuals. Neuroimage. (2010) 50:1392–401. doi: 10.1016/j.neuroimage.2010.01.056
99. Franklin TR, Acton PD, Maldjian JA, Gray JD, Croft JR, Dackis CA, et al. Decreased gray matter concentration in the insular, orbitofrontal, cingulate, and temporal cortices of cocaine patients. Biol Psychiatry. (2002) 51:134–42. doi: 10.1016/S0006-3223(01)01269-0
100. Matochik JA, London ED, Eldreth DA, Cadet JL, Bolla KI. Frontal cortical tissue composition in abstinent cocaine abusers: a magnetic resonance imaging study. Neuroimage. (2003) 19:1095–102. doi: 10.1016/S1053-8119(03)00244-1
101. Sim ME, Lyoo IK, Streeter CC, Covell J, Sarid-Segal O, Ciraulo DA, et al. Cerebellar gray matter volume correlates with duration of cocaine use in cocaine-dependent subjects. Neuropsychopharmacology. (2007) 32:2229–37. doi: 10.1038/sj.npp.1301346
102. Li H, Hu X, Gao Y, Cao L, Zhang L, Bu X, et al. Neural primacy of the dorsolateral prefrontal cortex in patients with obsessive-compulsive disorder. Neuroimage Clin. (2020) 28:102432. doi: 10.1016/j.nicl.2020.102432
103. Shin YW, Yoo SY, Lee JK, Ha TH, Lee KJ, Lee JM, et al. Cortical thinning in obsessive compulsive disorder. Hum Brain Mapp. (2007) 28:1128–35. doi: 10.1002/hbm.20338
104. Nabulsi NB, Mercier J, Holden D, Carré S, Najafzadeh S, Vandergeten MC, et al. Synthesis and preclinical evaluation of 11C-UCB-J as a PET tracer for imaging the synaptic vesicle glycoprotein 2A in the brain. J Nucl Med. (2016) 57:777–84. doi: 10.2967/jnumed.115.168179
105. Li S, Cai Z, Zhang W, Holden D, Lin SF, Finnema SJ, et al. Synthesis and in vivo evaluation of [18F]UCB-J for PET imaging of synaptic vesicle glycoprotein 2A (SV2A). Eur J Nucl Med Mol Imaging. (2019) 46:1952–65. doi: 10.1007/s00259-019-04357-w
106. Holmes SE, Scheinost D, Finnema SJ, Naganawa M, Davis MT, DellaGioia N, et al. Lower synaptic density is associated with depression severity and network alterations. Nat Commun. (2019) 10:1529. doi: 10.1038/s41467-019-09562-7
107. Onwordi EC, Halff EF, Whitehurst T, Mansur A, Cotel MC, Wells L, et al. Synaptic density marker SV2A is reduced in schizophrenia patients and unaffected by antipsychotics in rats. Nat Commun. (2020) 11:246. doi: 10.1038/s41467-019-14122-0
108. Bartlett EA, DeLorenzo C, Sharma P, Yang J, Zhang M, Petkova E, et al. Pretreatment and early-treatment cortical thickness is associated with SSRI treatment response in major depressive disorder. Neuropsychopharmacology. (2018) 43:2221–30. doi: 10.1038/s41386-018-0122-9
109. Bench CJ, Frackowiak RS, Dolan RJ. Changes in regional cerebral blood flow on recovery from depression. Psychol Med. (1995) 25:247–61. doi: 10.1017/S0033291700036151
110. Ishizaki J, Yamamoto H, Takahashi T, Takeda M, Yano M, Mimura M. Changes in regional cerebral blood flow following antidepressant treatment in late-life depression. Int J Geriatr Psychiatry. (2008) 23:805–11. doi: 10.1002/gps.1980
111. Koenig J, Westlund Schreiner M, Klimes-Dougan B, Ubani B, Mueller BA, Lim KO, et al. Increases in orbitofrontal cortex thickness following antidepressant treatment are associated with changes in resting state autonomic function in adolescents with major depression - preliminary findings from a pilot study. Psychiatry Res Neuroimag. (2018) 281:35–42. doi: 10.1016/j.pscychresns.2018.08.013
112. Nemati S, Abdallah CG. Increased cortical thickness in patients with major depressive disorder following antidepressant treatment. Chronic Stress. (2020) 4:2470547019899962. doi: 10.1177/2470547019899962
113. Vollenweider FX, Leenders KL, Scharfetter C, Antonini A, Maguire P, Missimer J, et al. Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG). Eur Neuropsychopharmacol. (1997) 7:9–24. doi: 10.1016/S0924-977X(96)00039-9
114. Långsjö JW, Salmi E, Kaisti KK, Aalto S, Hinkka S, Aantaa R, et al. Effects of subanesthetic ketamine on regional cerebral glucose metabolism in humans. Anesthesiology. (2004) 100:1065–71. doi: 10.1097/00000542-200405000-00006
115. Vollenweider FX, Leenders KL, Scharfetter C, Maguire P, Stadelmann O, Angst J. Positron emission tomography and fluorodeoxyglucose studies of metabolic hyperfrontality and psychopathology in the psilocybin model of psychosis. Neuropsychopharmacology. (1997) 16:357–72. doi: 10.1016/S0893-133X(96)00246-1
116. Gouzoulis-Mayfrank E, Schreckenberger M, Sabri O, Arning C, Thelen B, Spitzer M, et al. 3,4-methylenedioxyethylamphetamine (MDE) and d-methamphetamine in healthy volunteers. A double-blind, placebo-controlled PET study with [18F]FDG. Neuropsychopharmacology. (1999) 20:565–81. doi: 10.1016/S0893-133X(98)00089-X
117. Li CT, Chen MH, Lin WC, Hong CJ, Yang BH, Liu RS, et al. The effects of low-dose ketamine on the prefrontal cortex and amygdala in treatment-resistant depression: a randomized controlled study. Hum Brain Mapp. (2016) 37:1080–90. doi: 10.1002/hbm.23085
118. Abdallah CG, Averill LA, Collins KA, Geha P, Schwartz J, Averill C, et al. Ketamine treatment and global brain connectivity in major depression. Neuropsychopharmacology. (2017) 42:1210–9. doi: 10.1038/npp.2016.186
119. Bouso JC, Palhano-Fontes F, Rodríguez-Fornells A, Ribeiro S, Sanches R, Crippa JA, et al. Long-term use of psychedelic drugs is associated with differences in brain structure and personality in humans. Eur Neuropsychopharmacol. (2015) 25:483–92. doi: 10.1016/j.euroneuro.2015.01.008
120. Dai D, Lacadie CM, Holmes SE, Cool R, Anticevic A, Averill C, et al. Ketamine normalizes the structural alterations of inferior frontal gyrus in depression. Chronic Stress. (2020) 4:2470547020980681. doi: 10.1177/2470547020980681
121. Wellman CL. Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J Neurobiol. (2001) 49:245–53. doi: 10.1002/neu.1079
122. Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. (2011) 69:754–61. doi: 10.1016/j.biopsych.2010.12.015
123. Bachis A, Cruz MI, Nosheny RL, Mocchetti I. Chronic unpredictable stress promotes neuronal apoptosis in the cerebral cortex. Neurosci Lett. (2008) 442:104–8. doi: 10.1016/j.neulet.2008.06.081
124. Brown SM, Henning S, Wellman CL. Mild, short-term stress alters dendritic morphology in rat medial prefrontal cortex. Cereb Cortex. (2005) 15:1714–22. doi: 10.1093/cercor/bhi048
125. Cook SC, Wellman CL. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J Neurobiol. (2004) 60:236–48. doi: 10.1002/neu.20025
126. Colyn L, Venzala E, Marco S, Perez-Otaño I, Tordera RM. Chronic social defeat stress induces sustained synaptic structural changes in the prefrontal cortex and amygdala. Behav Brain Res. (2019) 373:112079. doi: 10.1016/j.bbr.2019.112079
127. Qu Y, Yang C, Ren Q, Ma M, Dong C, Hashimoto K. Regional differences in dendritic spine density confer resilience to chronic social defeat stress. Acta Neuropsychiatr. (2018) 30:117–22. doi: 10.1017/neu.2017.16
128. Zhang J, Qu Y, Chang L, Pu Y, Hashimoto K. (R)-ketamine rapidly ameliorates the decreased spine density in the medial prefrontal cortex and hippocampus of susceptible mice after chronic social defeat stress. Int J Neuropsychopharmacol. (2019) 22:675–9. doi: 10.1093/ijnp/pyz048
129. Moda-Sava RN, Murdock MH, Parekh PK, Fetcho RN, Huang BS, Huynh TN, et al. Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science. (2019) 364:eaat8078. doi: 10.1126/science.aat8078
130. Siuciak JA, Lewis DR, Wiegand SJ, Lindsay RM. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacol Biochem Behav. (1997) 56:131–7. doi: 10.1016/S0091-3057(96)00169-4
131. Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. (2002) 22:3251–61. doi: 10.1523/JNEUROSCI.22-08-03251.2002
132. Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW Jr, Miller SW, et al. infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur J Neurosci. (2007) 26:757–66. doi: 10.1111/j.1460-9568.2007.05692.x
133. Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. (2010) 328:1288–90. doi: 10.1126/science.1186909
134. Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald E, et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci. (2003) 23:349–57. doi: 10.1523/JNEUROSCI.23-01-00349.2003
135. Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, et al. receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. (2011) 475:91–5. doi: 10.1038/nature10130
136. Liu RJ, Lee FS Li XY, Bambico F, Duman RS, Aghajanian GK. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry. (2012) 71:996–1005. doi: 10.1016/j.biopsych.2011.09.030
137. Pattwell SS, Bath KG, Perez-Castro R, Lee FS, Chao MV, Ninan I. The BDNF Val66Met polymorphism impairs synaptic transmission and plasticity in the infralimbic medial prefrontal cortex. J Neurosci. (2012) 32:2410–21. doi: 10.1523/JNEUROSCI.5205-11.2012
138. Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, Pattwell SS, et al. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science. (2010) 327:863–6. doi: 10.1126/science.1181886
139. Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci. (1996) 16:2365–72. doi: 10.1523/JNEUROSCI.16-07-02365.1996
140. Rantamäki T, Hendolin P, Kankaanpää A, Mijatovic J, Piepponen P, Domenici E, et al. Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cgamma signaling pathways in mouse brain. Neuropsychopharmacology. (2007) 32:2152–62. doi: 10.1038/sj.npp.1301345
141. Karpova NN, Pickenhagen A, Lindholm J, Tiraboschi E, Kulesskaya N, Agústsdóttir A, et al. Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science. (2011) 334:1731–4. doi: 10.1126/science.1214592
142. Bath KG, Jing DQ, Dincheva I, Neeb CC, Pattwell SS, Chao MV, et al. Val66Met impairs fluoxetine-induced enhancement of adult hippocampus plasticity. Neuropsychopharmacology. (2012) 37:1297–304. doi: 10.1038/npp.2011.318
143. Jang SW, Liu X, Chan CB, Weinshenker D, Hall RA, Xiao G, et al. Amitriptyline is a TrkA and TrkB receptor agonist that promotes TrkA/TrkB heterodimerization and has potent neurotrophic activity. Chem Biol. (2009) 16:644–56. doi: 10.1016/j.chembiol.2009.05.010
144. Casarotto PC, Girych M, Fred SM, Kovaleva V, Moliner R, Enkavi G, et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell. (2021) 184:1299–313.e19. doi: 10.1016/j.cell.2021.01.034
145. Zhou W, Wang N, Yang C, Li XM, Zhou ZQ, Yang JJ. Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur Psychiatry. (2014) 29:419–23. doi: 10.1016/j.eurpsy.2013.10.005
146. Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. (2010) 329:959–64. doi: 10.1126/science.1190287
147. Browne CA, Lucki I. Antidepressant effects of ketamine: mechanisms underlying fast-acting novel antidepressants. Front Pharmacol. (2013) 4:161. doi: 10.3389/fphar.2013.00161
148. Ly C, Greb AC, Cameron LP, Wong JM, Barragan EV, Wilson PC, et al. Psychedelics promote structural and functional neural plasticity. Cell Rep. (2018) 23:3170–82. doi: 10.1016/j.celrep.2018.05.022
149. de la Fuente Revenga M, Zhu B, Guevara CA, Naler LB, Saunders JM, Zhou Z, et al. Prolonged epigenetic and synaptic plasticity alterations following single exposure to a psychedelic in mice. bioRxiv. (2021). doi: 10.1101/2021.02.24.432725
150. Phoumthipphavong V, Barthas F, Hassett S, Kwan AC. Longitudinal effects of ketamine on dendritic architecture in vivo in the mouse medial frontal cortex. eNeuro. (2016) 3:ENEURO.0133-15.2016. doi: 10.1523/ENEURO.0133-15.2016
151. Hibicke M, Landry AN, Kramer HM, Talman ZK, Nichols CD. Psychedelics, but not ketamine, produce persistent antidepressant-like effects in a rodent experimental system for the study of depression. ACS Chem Neurosci. (2020) 11:864–71. doi: 10.1021/acschemneuro.9b00493
152. Shao LX, Liao C, Gregg I, Davoudian PA, Savalia NK, Delagarza K, et al. Psilocybin induces rapid and persistent growth of dendritic spines in frontal cortex in vivo. Neuron. (2021). doi: 10.1101/2021.02.17.431629
153. Ly C, Greb AC, Vargas MV, Duim WC, Grodzki ACG, Lein PJ, et al. Transient stimulation with psychoplastogens is sufficient to initiate neuronal growth. ACS Pharmacol Transl Sci. (2020) 4:452–60. doi: 10.1021/acsptsci.0c00065
154. Gregorio DD, Popic J, Enns JP, Inserra A, Skalecka A, Markopoulos A, et al. Lysergic acid diethylamide (LSD) promotes social behavior through mTORC1 in the excitatory neurotransmission. Proc Natl Acad Sci USA. (2021) 118:e2020705118. doi: 10.1073/pnas.2020705118
155. Raval NR, Johansen A, Donovan LL, Ros NF, Ozenne B, Hansen HD, et al. Single dose of psilocybin increases synaptic density and decreases 5-HT2A receptor density in the pig brain. Int J Mol Sci. (2021) 22:835. doi: 10.3390/ijms22020835
156. Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. (2000) 47:351–4. doi: 10.1016/S0006-3223(99)00230-9
157. Langer G, Neumark J, Koinig G, Graf M, Schönbeck G. Rapid psychotherapeutic effects of anesthesia with isoflurane (ES narcotherapy) in treatment-refractory depressed patients. Neuropsychobiology. (1985) 14:118–20. doi: 10.1159/000118216
158. Engelhardt W, Carl G, Hartung E. Intra-individual open comparison of burst-suppression-isoflurane-anaesthesia versus electroconvulsive therapy in the treatment of severe depression. Eur J Anaesthesiol. (1993) 59:113–8. doi: 10.1097/00132586-199312000-00059
159. Langer G, Karazman R, Neumark J, Saletu B, SchönbeckG, Grünberger J, et al. Isoflurane narcotherapy in depressive patients refractory to conventional antidepressant drug treatment. Neuropsychobiology. (1995) 31:182–94. doi: 10.1159/000119190
160. Weeks HR, Tadler SC, Smith KW, Iacob E, Saccoman M, White AT, et al. Antidepressant and neurocognitive effects of isoflurane anesthesia versus electroconvulsive therapy in refractory depression. PLoS ONE. (2013) 8:1–8. doi: 10.1371/journal.pone.0069809
161. Nagele P, Duma A, Kopec M, Gebara MA, Parsoei A, Walker M, et al. Nitrous oxide for treatment-resistant major depression: a proof-of-concept trial. Biol Psychiatry. (2015) 78:10–8. doi: 10.1016/j.biopsych.2014.11.016
162. Mickey BJ, White AT, Arp AM, Leonardi K, Torres MM, Larson AL, et al. Propofol for treatment-resistant depression: a pilot study. Int J Neuropsychopharmacol. (2018) 21:1079–89. doi: 10.1093/ijnp/pyy085
163. Shao J, Meng L, Yang Z, Yu P, Song L, Gao Y, et al. Xenon produces rapid antidepressant- and anxiolytic-like effects in lipopolysaccharide-induced depression mice model. Neuroreport. (2020) 31:387–93. doi: 10.1097/WNR.0000000000001415
164. Antila H, Ryazantseva M, Popova D, Sipilä P, Guirado R, Kohtala S, et al. Isoflurane produces antidepressant effects and induces TrkB signaling in rodents. Sci Rep. (2017) 7:7811. doi: 10.1038/s41598-017-08166-9
165. Zhang SS, Tian YH, Jin SJ, Wang WC, Zhao JX Si XM, et al. Isoflurane produces antidepressant effects inducing BDNF-TrkB signaling in CUMS mice. Psychopharmacology. (2019) 236:3301–15. doi: 10.1007/s00213-019-05287-z
166. Kyzar EJ, Nichols CD, Gainetdinov RR, Nichols DE, Kalueff AV. Psychedelic drugs in biomedicine. Trends Pharmacol Sci. (2017) 38:992–1005. doi: 10.1016/j.tips.2017.08.003
167. Barrett FS, Doss MK, Sepeda ND, Pekar JJ, Griffiths RR. Emotions and brain function are altered up to one month after a single high dose of psilocybin. Sci Rep. (2020) 10:2214. doi: 10.1038/s41598-020-59282-y
168. Krupitsky E, Burakov A, Romanova T, Dunaevsky I, Strassman R, Grinenko A. Ketamine psychotherapy for heroin addiction: immediate effects and 2-year follow-up. J Subst Abuse Treat. (2002) 23:273–83. doi: 10.1016/S0740-5472(02)00275-1
169. Feder A, Parides MK, Murrough JW, Perez AM, Morgan JE, Saxena S, et al. Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder: a randomized clinical trial. JAMA Psychiatry. (2014) 71:681–8. doi: 10.1001/jamapsychiatry.2014.62
170. Zarate CA Jr, Brutsche NE, Ibrahim L, Franco-Chaves J, Diazgranados N, Cravchik A, et al. Replication of ketamine's antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol Psychiatry. (2012) 71:939–46. doi: 10.1016/j.biopsych.2011.12.010
171. Dos Santos RG, Osório FL, Crippa JA, Riba J, Zuardi AW, Hallak JE. Antidepressive, anxiolytic, and antiaddictive effects of ayahuasca, psilocybin and lysergic acid diethylamide (LSD): a systematic review of clinical trials published in the last 25 years. Ther Adv Psychopharmacol. (2016) 6:193–213. doi: 10.1177/2045125316638008
172. Bogenschutz MP, Johnson MW. Classic hallucinogens in the treatment of addictions. Prog Neuropsychopharmacol Biol Psychiatry. (2016) 64:250–8. doi: 10.1016/j.pnpbp.2015.03.002
173. Lammel S, Tye KM, Warden MR. Progress in understanding mood disorders: optogenetic dissection of neural circuits. Genes Brain Behav. (2014) 13:38–51. doi: 10.1111/gbb.12049
174. Covington HE 3rd, Lobo MK, Maze I, Vialou V, Hyman JM, Zaman S, et al. Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci. (2010) 30:16082–90. doi: 10.1523/JNEUROSCI.1731-10.2010
175. Warden MR, Selimbeyoglu A, Mirzabekov JJ, Lo M, Thompson KR, Kim SY, et al. A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature. (2012) 492:428–32. doi: 10.1038/nature11617
176. Yankelevitch-Yahav R, Franko M, Huly A, Doron R. The forced swim test as a model of depressive-like behavior. J Vis Exp. (2015) (97):52587. doi: 10.3791/52587
177. Slattery DA, Cryan JF. Using the rat forced swim test to assess antidepressant-like activity in rodents. Nat Protoc. (2012) 7:1009–14. doi: 10.1038/nprot.2012.044
178. Maes M, Meltzer H. The serotonin hypothesis of major depression. Psychopharmacology. (1995) 1995:933–44.
179. Nishitani N, Nagayasu K, Asaoka N, Yamashiro M, Andoh C, Nagai Y, et al. Manipulation of dorsal raphe serotonergic neurons modulates active coping to inescapable stress and anxiety-related behaviors in mice and rats. Neuropsychopharmacology. (2019) 44:721–32. doi: 10.1038/s41386-018-0254-y
180. Kumar S, Black SJ, Hultman R, Szabo ST, DeMaio KD, Du J, et al. Cortical control of affective networks. J Neurosci. (2013) 33:1116–29. doi: 10.1523/JNEUROSCI.0092-12.2013
181. Fuchikami M, Thomas A, Liu R, Wohleb ES, Land BB, DiLeone RJ, et al. Optogenetic stimulation of infralimbic PFC reproduces ketamine's rapid and sustained antidepressant actions. Proc Natl Acad Sci USA. (2015) 112:8106–11. doi: 10.1073/pnas.1414728112
182. Hübner C, Bosch D, Gall A, Lüthi A, Ehrlich I. Ex vivo dissection of optogenetically activated mPFC and hippocampal inputs to neurons in the basolateral amygdala: implications for fear and emotional memory. Front Behav Neurosci. (2014) 8:64. doi: 10.3389/fnbeh.2014.00064
183. Adhikari A, Lerner TN, Finkelstein J, Pak S, Jennings JH, Davidson TJ, et al. Basomedial amygdala mediates top-down control of anxiety and fear. Nature. (2015) 527:179–85. doi: 10.1038/nature15698
184. Bloodgood DW, Sugam JA, Holmes A, Kash TL. Fear extinction requires infralimbic cortex projections to the basolateral amygdala. Transl Psychiatry. (2018) 8:60. doi: 10.1038/s41398-018-0106-x
185. Jovanovic T, Norrholm SD, Blanding NQ, Davis M, Duncan E, Bradley B, et al. Impaired fear inhibition is a biomarker of PTSD but not depression. Depress Anxiety. (2010) 27:244–51. doi: 10.1002/da.20663
186. Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research–past, present, and future. Biol Psychiatry. (2006) 60:376–82. doi: 10.1016/j.biopsych.2006.06.004
187. Milad MR, Rauch SL, Pitman RK, Quirk GJ. Fear extinction in rats: implications for human brain imaging and anxiety disorders. Biol Psychol. (2006) 73:61–71. doi: 10.1016/j.biopsycho.2006.01.008
188. Augur IF, Wyckoff AR, Aston-Jones G, Kalivas PW, Peters J. Chemogenetic activation of an extinction neural circuit reduces cue-induced reinstatement of cocaine seeking. J Neurosci. (2016) 36:10174–80. doi: 10.1523/JNEUROSCI.0773-16.2016
189. Chen BT, Yau HJ, Hatch C, Kusumoto-Yoshida I, Cho SL, Hopf FW, et al. Rescuing cocaine-induced prefrontal cortex hypoactivity prevents compulsive cocaine seeking. Nature. (2013) 496:359–62. doi: 10.1038/nature12024
190. Peters J, LaLumiere RT, Kalivas PW. Infralimbic prefrontal cortex is responsible for inhibiting cocaine seeking in extinguished rats. J Neurosci. (2008) 28:6046–53. doi: 10.1523/JNEUROSCI.1045-08.2008
191. Halladay LR, Kocharian A, Piantadosi PT, Authement ME, Lieberman AG, Spitz NA, et al. Prefrontal regulation of punished ethanol self-administration. Biol Psychiatry. (2020) 87:967–78. doi: 10.1016/j.biopsych.2019.10.030
192. Domingo-Rodriguez L, Ruiz de Azua I, Dominguez E, Senabre E, Serra I, Kummer S, et al. A specific prelimbic-nucleus accumbens pathway controls resilience versus vulnerability to food addiction. Nat Commun. (2020) 11:782. doi: 10.1038/s41467-020-14458-y
193. Siciliano CA, Noamany H, Chang CJ, Brown AR, Chen X, Leible D, et al. A cortical-brainstem circuit predicts and governs compulsive alcohol drinking. Science. (2019) 366:1008–12. doi: 10.1126/science.aay1186
194. Cameron LP, Benson CJ, Dunlap LE, Olson DE. Effects of N, N-dimethyltryptamine on rat behaviors relevant to anxiety and depression. ACS Chem Neurosci. (2018) 9:1582–90. doi: 10.1021/acschemneuro.8b00134
195. Cameron LP, Benson CJ, DeFelice BC, Fiehn O, Olson DE. Chronic, intermittent microdoses of the psychedelic N,N-dimethyltryptamine (DMT) produce positive effects on mood and anxiety in rodents. ACS Chem Neurosci. (2019) 10:3261–70. doi: 10.1021/acschemneuro.8b00692
196. Catlow BJ, Song S, Paredes DA, Kirstein CL, Sanchez-Ramos J. Effects of psilocybin on hippocampal neurogenesis and extinction of trace fear conditioning. Exp Brain Res. (2013) 228:481–91. doi: 10.1007/s00221-013-3579-0
197. Young MB, Andero R, Ressler KJ, Howell LL. 3,4-Methylenedioxymethamphetamine facilitates fear extinction learning. Transl Psychiatry. (2015) 5:e634. doi: 10.1038/tp.2015.138
198. Vyas A, Jadhav S, Chattarji S. Prolonged behavioral stress enhances synaptic connectivity in the basolateral amygdala. Neuroscience. (2006) 143:387–93. doi: 10.1016/j.neuroscience.2006.08.003
199. Vyas A, Pillai AG, Chattarji S. Recovery after chronic stress fails to reverse amygdaloid neuronal hypertrophy and enhanced anxiety-like behavior. Neuroscience. (2004) 128:667–73. doi: 10.1016/j.neuroscience.2004.07.013
200. Lakshminarasimhan H, Chattarji S. Stress leads to contrasting effects on the levels of brain derived neurotrophic factor in the hippocampus and amygdala. PLoS ONE. (2012) 7:e30481. doi: 10.1371/journal.pone.0030481
201. Eisch AJ, Bolaños CA, de Wit J, Simonak RD, Pudiak CM, Barrot M, et al. Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: a role in depression. Biol Psychiatry. (2003) 54:994–1005. doi: 10.1016/j.biopsych.2003.08.003
202. Niehaus JL, Cruz-Bermudez ND, Kauer JA. Plasticity of addiction: a mesolimbic dopamine short-circuit? Am J Addict. (2009) 18:259–71. doi: 10.1080/10550490902925946
203. Cornea-Hébert V, Riad M, Wu C, Singh SK, Descarries L. Cellular and subcellular distribution of the serotonin 5-HT2A receptor in the central nervous system of adult rat. J Comp Neurol. (1999) 409:187–209. doi: 10.1002/(SICI)1096-9861(19990628)409:2<187::AID-CNE2>3.0.CO
204. Xu T, Pandey SC. Cellular localization of serotonin (2A) (5HT(2A)) receptors in the rat brain. Brain Res Bull. (2000) 51:499–505. doi: 10.1016/S0361-9230(99)00278-6
205. Willins DL, Deutch AY, Roth BL. Serotonin 5-HT2A receptors are expressed on pyramidal cells and interneurons in the rat cortex. Synapse. (1997) 27:79–82. doi: 10.1002/(SICI)1098-2396(199709)27:1<79::AID-SYN8>3.0.CO
206. Miner LA, Backstrom JR, Sanders-Bush E, Sesack SR. Ultrastructural localization of serotonin2A receptors in the middle layers of the rat prelimbic prefrontal cortex. Neuroscience. (2003) 116:107–17. doi: 10.1016/S0306-4522(02)00580-8
207. Amargós-Bosch M, Bortolozzi A, Puig MV, Serrats J, Adell A, Celada P, et al. Co-expression and in vivo interaction of serotonin1A and serotonin2A receptors in pyramidal neurons of prefrontal cortex. Cereb Cortex. (2004) 14:281–99. doi: 10.1093/cercor/bhg128
208. Santana N, Bortolozzi A, Serrats J, Mengod G, Artigas F. Expression of serotonin1A and serotonin2A receptors in pyramidal and GABAergic neurons of the rat prefrontal cortex. Cereb Cortex. (2004) 14:1100–9. doi: 10.1093/cercor/bhh070
209. Quirion R, Richard J, Dam TV. Evidence for the existence of serotonin type-2 receptors on cholinergic terminals in rat cortex. Brain Res. (1985) 333:345–9. doi: 10.1016/0006-8993(85)91590-2
210. Weber ET, Andrade R. Htr2a gene and 5-HT(2A) receptor expression in the cerebral cortex studied using genetically modified mice. Front Neurosci. (2010) 4:36. doi: 10.3389/fnins.2010.00036
211. Pazos A, Probst A, Palacios JM. Serotonin receptors in the human brain–IV. Autoradiographic mapping of serotonin-2 receptors. Neuroscience. (1987) 21:123–39. doi: 10.1016/0306-4522(87)90327-7
212. Burnet PW, Eastwood SL, Lacey K, Harrison PJ. The distribution of 5-HT1A and 5-HT2A receptor mRNA in human brain. Brain Res. (1995) 676:157–68. doi: 10.1016/0006-8993(95)00104-X
213. Wong DF, Lever JR, Hartig PR, Dannals RF, Villemagne V, Hoffman BJ, et al. Localization of serotonin 5-HT2 receptors in living human brain by positron emission tomography using N1-([11C]-methyl)-2-Br-LSD. Synapse. (1987) 1:393–8. doi: 10.1002/syn.890010502
214. Goodwin AK. An intravenous self-administration procedure for assessing the reinforcing effects of hallucinogens in nonhuman primates. J Pharmacol Toxicol Methods. (2016) 82:31–6. doi: 10.1016/j.vascn.2016.07.004
215. Fantegrossi WE, Woods JH, Winger G. Transient reinforcing effects of phenylisopropylamine and indolealkylamine hallucinogens in rhesus monkeys. Behav Pharmacol. (2004) 15:149–57. doi: 10.1097/00008877-200403000-00007
216. Martin WR. Assessment of the abuse-potentiality of amphetamines and LSD-like hallucinogens in man and its relationship to basic animal assessment programs. In: Goldberg L, Hoffmeister F, editors, Bayer-Symposium IV, Psychic Dependence. New York, NY: Springer-Verlag (1973). p. 146–55. doi: 10.1007/978-3-642-87987-6_17
217. Nutt D, King LA, Saulsbury W, Blakemore C. Development of a rational scale to assess the harm of drugs of potential misuse. Lancet. (2007) 369:1047–53. doi: 10.1016/S0140-6736(07)60464-4
218. Olson DE. The subjective effects of psychedelics may not be necessary for their enduring therapeutic effects. ACS Pharmacol Transl Sci. (2020) 2020:c00192. doi: 10.1021/acsptsci.0c00192
219. Yaden DB, Griffiths RR. The subjective effects of psychedelics are necessary for their enduring therapeutic effects. ACS Pharmacol Transl Sci. (2020) 2020:c00194. doi: 10.1021/acsptsci.0c00194
220. Ross S, Bossis A, Guss J, Agin-Liebes G, Malone T, Cohen B, et al. Rapid and sustained symptom reduction following psilocybin treatment for anxiety and depression in patients with life-threatening cancer: a randomized controlled trial. J Psychopharmacol. (2016) 30:1165–80. doi: 10.1177/0269881116675512
221. Bogenschutz MP, Forcehimes AA, Pommy JA, Wilcox CE, Barbosa PC, Strassman RJ. Psilocybin-assisted treatment for alcohol dependence: a proof-of-concept study. J Psychopharmacol. (2015) 29:289–99. doi: 10.1177/0269881114565144
222. Garcia-Romeu A, Griffiths RR, Johnson MW. Psilocybin-occasioned mystical experiences in the treatment of tobacco addiction. Curr Drug Abuse Rev. (2014) 7:157–64. doi: 10.2174/1874473708666150107121331
223. Roseman L, Nutt DJ, Carhart-Harris RL. Quality of acute psychedelic experience predicts therapeutic efficacy of psilocybin for treatment-resistant depression. Front Pharmacol. (2018) 8:974. doi: 10.3389/fphar.2017.00974
224. Sos P, Klirova M, Novak T, Kohutova B, Horacek J, Palenicek T. Relationship of ketamine's antidepressant and psychotomimetic effects in unipolar depression. Neuro Endocrinol Lett. (2013) 34:287–93.
225. Luckenbaugh DA, Niciu MJ, Ionescu DF, Nolan NM, Richards EM, Brutsche NE, et al. Do the dissociative side effects of ketamine mediate its antidepressant effects? J Affect Disord. (2014) 159:56–61. doi: 10.1016/j.jad.2014.02.017
226. Dakwar E, Anerella C, Hart CL, Levin FR, Mathew SJ, Nunes EV. Therapeutic infusions of ketamine: do the psychoactive effects matter? Drug Alcohol Depend. (2014) 136:153–7. doi: 10.1016/j.drugalcdep.2013.12.019
227. Vollenweider FX, Vollenweider-Scherpenhuyzen MF, Bäbler A, Vogel H, Hell D. Psilocybin induces schizophrenia-like psychosis in humans via a serotonin-2 agonist action. Neuroreport. (1998) 9:3897–902. doi: 10.1097/00001756-199812010-00024
228. Preller KH, Burt JB Ji JL, Schleifer CH, Adkinson BD, Stämpfli P, et al. Changes in global and thalamic brain connectivity in LSD-induced altered states of consciousness are attributable to the 5-HT2A receptor. Elife. (2018) 7:e35082. doi: 10.7554/eLife.35082
229. Kudoh A, Takahira Y, Katagai H, Takazawa T. Small-dose ketamine improves the postoperative state of depressed patients. Anesth Analg. (2002) 95:114–8. doi: 10.1097/00000539-200207000-00020
230. Jiang M, Wang MH, Wang XB, Liu L, Wu JL, Yang XL, et al. Effect of intraoperative application of ketamine on postoperative depressed mood in patients undergoing elective orthopedic surgery. J Anesth. (2016) 30:232–7. doi: 10.1007/s00540-015-2096-7
231. Xu R, Zhan Y, Chen S. Effect of intraoperative single administration of sub-anesthesia ketamine on breast cancer patients with depression. Biomedical Res. (2017) 2017:S552–6.
232. Yang C, Shirayama Y, Zhang JC, Ren Q, Yao W, Ma M, et al. R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl Psychiatry. (2015) 5:e632. doi: 10.1038/tp.2015.136
233. Vollenweider FX, Leenders KL, Oye I, Hell D, Angst J. Differential psychopathology and patterns of cerebral glucose utilisation produced by (S)- and (R)-ketamine in healthy volunteers using positron emission tomography (PET). Eur Neuropsychopharmacol. (1997) 7:25–38. doi: 10.1016/S0924-977X(96)00042-9
234. Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. (2016) 533:481–6. doi: 10.1038/nature17998
235. Mitchell JM, Bogenschutz M, Lilienstein A, Harrison C, Kleiman S, Parker-Guilbert K, et al. MDMA-assisted therapy for severe PTSD: a randomized, double-blind, placebo-controlled phase 3 study. Nat Med. (2021) 27:1025–33. doi: 10.1038/s41591-021-01336-3
236. Dunlap LE, Andrews AM, Olson DE. Dark classics in chemical neuroscience: 3,4-methylenedioxymethamphetamine. ACS Chem Neurosci. (2018) 9:2408–27. doi: 10.1021/acschemneuro.8b00155
237. Young MB, Norrholm SD, Khoury LM, Jovanovic T, Rauch SAM, Reiff CM, et al. Inhibition of serotonin transporters disrupts the enhancement of fear memory extinction by 3,4-methylenedioxymethamphetamine (MDMA). Psychopharmacology. (2017) 234:2883–95. doi: 10.1007/s00213-017-4684-8
238. Peroutka SJ, Newman H, Harris H. Subjective effects of 3,4-methylenedioxymethamphetamine in recreational users. Neuropsychopharmacology. (1988) 1:273–7.
239. Liechti ME, Saur MR, Gamma A, Hell D, Vollenweider FX. Psychological and physiological effects of MDMA (“Ecstasy”) after pretreatment with the 5HT(2) antagonist ketanserin in healthy humans. Neuropsychopharmacology. (2000) 23:396–404. doi: 10.1016/S0893-133X(00)00126-3
240. Studerus E, Gamma A, Vollenweider FX. Psychometric evaluation of the altered states of consciousness rating scale (OAV). PLoS ONE. (2010) 5:e12412. doi: 10.1371/journal.pone.0012412
241. Holze F, Vizeli P, Müller F, Ley L, Duerig R, Varghese N, et al. Distinct acute effects of LSD, MDMA, and D-amphetamine in healthy subjects. Neuropsychopharmacology. (2020) 45:462–71. doi: 10.1038/s41386-019-0569-3
242. Hougaku H, Matsumoto M, Hata R, Handa N, Imaizumi M, Sugitani Y, et al. Therapeutic effect of lisuride maleate on post-stroke depression. Nihon Ronen Igakkai Zasshi. (1994) 31:52–9. doi: 10.3143/geriatrics.31.52
243. Nakamura K, Ikoma Y, Kimura K, Nakada Y, Kobayashi S, Yamaguchi M, et al. Effects in animal models of depression of lisuride alone and upon coadministration with antidepressants. Nihon Yakurigaku Zasshi. (1989) 94:81–9. doi: 10.1254/fpj.94.81
244. Szigeti B, Kartner L, Blemings A, Rosas F, Feilding A, Nutt DJ, et al. Self-blinding citizen science to explore psychedelic microdosing. Elife. (2021) 10:e62878. doi: 10.7554/eLife.62878
245. Anderson T, Petranker R, Rosenbaum D, Weissman CR, Dinh-Williams LA, Hui K, et al. Microdosing psychedelics: personality, mental health, and creativity differences in microdosers. Psychopharmacology. (2019) 236:731–40. doi: 10.1007/s00213-018-5106-2
246. Johnstad P. Powerful substances in tiny amounts: an interview study of psychedelic microdosing Nordic Stud Alcohol Drugs. (2018) 35:39–51. doi: 10.1177/1455072517753339
247. Cameron LP, Nazarian A, Olson DE. Psychedelic microdosing: prevalence and subjective effects. J Psychoact Drugs. (2020) 52:113–22. doi: 10.1080/02791072.2020.1718250
248. Prochazkova L, Lippelt DP, Colzato LS, Kuchar M, Sjoerds Z, Hommel B. Exploring the effect of microdosing psychedelics on creativity in an open-label natural setting. Psychopharmacology. (2018) 235:3401–13. doi: 10.1007/s00213-018-5049-7
249. Polito V, Stevenson RJ. A systematic study of microdosing psychedelics. PLoS ONE. (2019) 14:e0211023. doi: 10.1371/journal.pone.0211023
250. Cameron LP, Tombari RJ, Lu J, Pell AJ, Hurley ZQ, Ehinger Y, et al. A non-hallucinogenic psychedelic analogue with therapeutic potential. Nature. (2021) 589:474–9. doi: 10.1038/s41586-020-3008-z
251. Dong C, Ly C, Dunlap LE, Vargas MV, Sun J, Hwang IW, et al. Psychedelic-inspired drug discovery using an engineered biosensor. Cell. (2021) 184:2779–92.e18. doi: 10.1016/j.cell.2021.03.043
252. Dunlap LE, Azinfar A, Ly C, Cameron LP, Viswanathan J, Tombari RJ, et al. Identification of psychoplastogenic N,N-dimethylaminoisotryptamine (isoDMT) analogues through structure-activity relationship studies. J Med Chem. (2020) 63:1142–55. doi: 10.1021/acs.jmedchem.9b01404
253. Halberstadt AL, Chatha M, Klein AK, Wallach J, Brandt SD. Correlation between the potency of hallucinogens in the mouse head-twitch response assay and their behavioral and subjective effects in other species. Neuropharmacology. (2020) 167:107933. doi: 10.1016/j.neuropharm.2019.107933
254. Hanks JB, González-Maeso J. Animal models of serotonergic psychedelics. ACS Chem Neurosci. (2013) 4:33–42. doi: 10.1021/cn300138m
255. Lu J, Tjia M, Mullen B, Cao B, Lukasiewicz K, Shah-Morales S, et al. An analog of psychedelics restores functional neural circuits disrupted by unpredictable stress. Mol Psychiatry. (2021). doi: 10.1038/s41380-021-01159-1. [Epub ahead of print].
256. Substance Abuse and Mental Health Services Administration. Key Substance Use and Mental Health Indicators in the United States: Results From the 2019 National Survey on Drug Use and Health (HHS Publication No. PEP20-07-01-001). (2020). Available online at: https://www.samhsa.gov/data/sites/default/files/reports/rpt29393/2019NSDUHFFRPDFWHTML/2019NSDUHFFR1PDFW090120.pdf (accessed June 17, 2021).
257. Greenberg PE, Fournier AA, Sisitsky T, Pike CT, Kessler RC. The economic burden of adults with major depressive disorder in the United States (2005 and 2010). J Clin Psychiatry. (2015) 76:155–62. doi: 10.4088/JCP.14m09298
258. Zhdanava M, Pilon D, Ghelerter I, Chow W, Joshi K, Lefebvre P, et al. The prevalence and national burden of treatment-resistant depression and major depressive disorder in the United States. J Clin Psychiatry. (2021) 82:20m13699. doi: 10.4088/JCP.20m13699
259. Gaynes BN, Warden D, Trivedi MH, Wisniewski SR, Fava M, Rush AJ. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatr Serv. (2009) 60:1439–45. doi: 10.1176/ps.2009.60.11.1439
260. Ferguson JM. SSRI antidepressant medications: adverse effects and tolerability. Prim Care Companion J Clin Psychiatry. (2001) 3:22–7. doi: 10.4088/PCC.v03n0105
261. Corrigan K, Haran M, McCandliss C, McManus R, Cleary S, Trant R, et al. Psychedelic perceptions: mental health service user attitudes to psilocybin therapy. Ir J Med Sci. (2021). doi: 10.1007/s11845-021-02668-2. [Epub ahead of print].
262. Siegel AN, Meshkat S, Benitah K, Lipsitz O, Gill H, Lui LMW, et al. Registered clinical studies investigating psychedelic drugs for psychiatric disorders. J Psychiatr Res. (2021) 139:71–81. doi: 10.1016/j.jpsychires.2021.05.019
263. Galvão-Coelho NL, Marx W, Gonzalez M, Sinclair J, de Manincor M, Perkins D, et al. Classic serotonergic psychedelics for mood and depressive symptoms: a meta-analysis of mood disorder patients and healthy participants. Psychopharmacology. (2021) 238:341–54. doi: 10.1007/s00213-020-05719-1
264. Tupper KW, Wood E, Yensen R, Johnson MW. Psychedelic medicine: a re-emerging therapeutic paradigm. CMAJ. (2015) 187:1054–9. doi: 10.1503/cmaj.141124
265. Nishimura A, Aritomi Y, Sasai K, Kitagawa T, Mahableshwarkar AR. Randomized, double-blind, placebo-controlled 8-week trial of the efficacy, safety, and tolerability of 5, 10, and 20 mg/day vortioxetine in adults with major depressive disorder. Psychiatry Clin Neurosci. (2018) 72:64–72. doi: 10.1111/pcn.12565
266. Jain R, Mahableshwarkar AR, Jacobsen PL, Chen Y, Thase ME. A randomized, double-blind, placebo-controlled 6-wk trial of the efficacy and tolerability of 5 mg vortioxetine in adults with major depressive disorder. Int J Neuropsychopharmacol. (2013) 16:313–21. doi: 10.1017/S1461145712000727
267. Angst J, Cui L, Swendsen J, Rothen S, Cravchik A, Kessler RC, et al. Major depressive disorder with subthreshold bipolarity in the National Comorbidity Survey Replication. Am J Psychiatry. (2010) 167:1194–201. doi: 10.1176/appi.ajp.2010.09071011
268. Gaudiano BA, Dalrymple KL, Zimmerman M. Prevalence and clinical characteristics of psychotic versus nonpsychotic major depression in a general psychiatric outpatient clinic. Depress Anxiety. (2009) 26:54–64. doi: 10.1002/da.20470
269. Hunt GE, Malhi GS, Lai HMX, Cleary M. Prevalence of comorbid substance use in major depressive disorder in community and clinical settings, 1990–2019: systematic review and meta-analysis. J Affect Disord. (2020) 266:288–304. doi: 10.1016/j.jad.2020.01.141
270. Van HL, Kool M. What we do, do not, and need to know about comorbid depression and personality disorders. Lancet Psychiatry. (2018) 5:776–8. doi: 10.1016/S2215-0366(18)30260-8
271. Han B, McKeon R, Gfroerer J. Suicidal ideation among community-dwelling adults in the United States. Am J Public Health. (2014) 104:488–97. doi: 10.2105/AJPH.2013.301600
272. Brainstorm Consortium, Anttila V, Bulik-Sullivan B, Walters RK, Bras J, Duncan L, et al. Analysis of shared heritability in common disorders of the brain. Science. (2018) 360:eaap8757. doi: 10.1126/science.aap8757
273. McGuffin P, Rijsdijk F, Andrew M, Sham P, Katz R, Cardno A. The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch Gen Psychiatry. (2003) 60:497–502. doi: 10.1001/archpsyc.60.5.497
274. Rietveld MJ, Hudziak JJ, Bartels M, van Beijsterveldt CE, Boomsma DI. Heritability of attention problems in children: I. Cross-sectional results from a study of twins, age 3-12 years. Am J Med Genet B Neuropsychiatr Genet. (2003) 117B:102–13. doi: 10.1002/ajmg.b.10024
275. Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. (2000) 157:1552–62. doi: 10.1176/appi.ajp.157.10.1552
276. McElroy SL, Altshuler LL, Suppes T, Keck PE Jr, Frye MA, Denicoff KD, et al. Axis I psychiatric comorbidity and its relationship to historical illness variables in 288 patients with bipolar disorder. Am J Psychiatry. (2001) 158:420–6. doi: 10.1176/appi.ajp.158.3.420
277. Zimmerman M, Chelminski I, McDermut W. Major depressive disorder and axis I diagnostic comorbidity. J Clin Psychiatry. (2002) 63:187–93. doi: 10.4088/JCP.v63n0303
278. Goes FS, McCusker MG, Bienvenu OJ, et al. Co-morbid anxiety disorders in bipolar disorder and major depression: familial aggregation and clinical characteristics of co-morbid panic disorder, social phobia, specific phobia and obsessive-compulsive disorder. Psychol Med. (2012) 42:1449–59. doi: 10.1017/S0033291711002637
279. Thaipisuttikul P, Ittasakul P, Waleeprakhon P, Wisajun P, Jullagate S. Psychiatric comorbidities in patients with major depressive disorder. Neuropsychiatr Dis Treat. (2014) 10:2097–103. doi: 10.2147/NDT.S72026
280. Steffen A, Nübel J, Jacobi F, Bätzing J, Holstiege J. Mental and somatic comorbidity of depression: a comprehensive cross-sectional analysis of 202 diagnosis groups using German nationwide ambulatory claims data. BMC Psychiatry. (2020) 20:142. doi: 10.1186/s12888-020-02546-8
281. Aday JS, Davis AK, Mitzkovitz CM, Bloesch EK, Davoli CC. Predicting reactions to psychedelic drugs: a systematic review of states and traits related to acute drug effects. ACS Pharmacol Transl Sci. (2021) 4:424–35. doi: 10.1021/acsptsci.1c00014
282. Stauffer CS, Anderson BT, Ortigo KM, Woolley J. Psilocybin-assisted group therapy and attachment: observed reduction in attachment anxiety and influences of attachment insecurity on the psilocybin experience. ACS Pharmacol Transl Sci. (2020) 4:526–32. doi: 10.1021/acsptsci.0c00169
283. Hartogsohn I. The meaning-enhancing properties of psychedelics and their mediator role in psychedelic therapy, spirituality, and creativity. Front Neurosci. (2018) 12:129. doi: 10.3389/fnins.2018.00129
284. Hartogsohn I. Set and setting, psychedelics and the placebo response: an extra-pharmacological perspective on psychopharmacology. J Psychopharmacol. (2016) 30:1259–67. doi: 10.1177/0269881116677852
285. Olson JA, Suissa-Rocheleau L, Lifshitz M, Raz A, Veissière SPL. Tripping on nothing: placebo psychedelics and contextual factors. Psychopharmacology. (2020) 237:1371–82. doi: 10.1007/s00213-020-05464-5
286. Food and Drug Administration. FDA Approves New Nasal Spray Medication for Treatment-Resistant Depression; Available Only at a Certified Doctor's Office or Clinic. FDA News Release (2019). Available online at: https://www.fda.gov/news-events/press-announcements/fda-approves-new-nasal-spray-medication-treatment-resistant-depression-available-only-certified (accessed June 17, 2021).
287. Janssenlabels. SPRAVATO® [Prescribing Information]. Titusville, NJ: Janssen Pharmaceuticals, Inc. (2020). Available online at: https://www.janssenlabels.com/package-insert/product-monograph/prescribing-information/SPRAVATO-pi.pdf (accessed June 17, 2021).
288. Tai SJ, Nielson EM, Lennard-Jones M, Johanna Ajantaival RL, Winzer R, Richards WA, et al. Development and evaluation of a therapist training program for psilocybin therapy for treatment-resistant depression in clinical research. Front Psychiatry. (2021) 12:586682. doi: 10.3389/fpsyt.2021.586682
289. Johnson MW. Consciousness, religion, and gurus: pitfalls of psychedelic medicine. ACS Pharmacol Transl Sci. (2020) 4:578–81. doi: 10.1021/acsptsci.0c00198
290. Castro Santos H, Gama Marques J. What is the clinical evidence on psilocybin for the treatment of psychiatric disorders? A systematic review. Porto Biomed J. (2021) 6:e128. doi: 10.1097/j.pbj.0000000000000128
291. Statnews. J&J's New Esketamine Drug for Depression May Solve an Unmet Need, But May Not Be Cost-Effective. (2019). Available online at: https://www.statnews.com/pharmalot/2019/03/22/antidepressants-cost-effective-johnson-and-johnson/ (accessed June 17, 2021).
292. Young SN. Single treatments that have lasting effects: some thoughts on the antidepressant effects of ketamine and botulinum toxin and the anxiolytic effect of psilocybin. J Psychiatry Neurosci. (2013) 38:78–83. doi: 10.1503/jpn.120128
293. Trope A, Anderson BT, Hooker AR, Glick G, Stauffer C, Woolley JD. Psychedelic-assisted group therapy: a systematic review. J Psychoactive Drugs. (2019) 51:174–88. doi: 10.1080/02791072.2019.1593559
294. Agboola F, Atlas SJ, Touchette DR, Fazioli K, Pearson SD. The effectiveness and value of esketamine for the management of treatment-resistant depression. J Manag Care Spec Pharm. (2020) 26:16–20. doi: 10.18553/jmcp.2020.26.1.16
295. National Institute for Health and Care Excellence. Appraisal Consultation Document Esketamine for Treating Treatment-Resistant Depression. (2020). Available online at: https://www.nice.org.uk/guidance/gid-ta10371/documents/129 (accessed June 17, 2021).
296. Statnews. Ketamine Gives Hope to Patients With Severe Depression. But Some Clinics Stray From the Science and Hype Its Benefit. (2018). Available online at: https://www.statnews.com/2018/09/24/ketamine-clinics-severe-depression-treatment/ (accessed June 17, 2021).
297. Strickland JC, Garcia-Romeu A, Johnson MW. Set and setting: a randomized study of different musical genres in supporting psychedelic therapy. ACS Pharmacol Transl Sci. (2020) 4:472–8. doi: 10.1021/acsptsci.0c00187
298. Cartwright C, Gibson K, Read J, Cowan O, Dehar T. Long-term antidepressant use: patient perspectives of benefits and adverse effects. Patient Prefer Adherence. (2016) 10:1401–7. doi: 10.2147/PPA.S110632
Keywords: psychoplastogen, ketamine, psilocybin, depression, neuroplasticity, prefrontal cortex, psychedelic, hallucinogenic
Citation: Vargas MV, Meyer R, Avanes AA, Rus M and Olson DE (2021) Psychedelics and Other Psychoplastogens for Treating Mental Illness. Front. Psychiatry 12:727117. doi: 10.3389/fpsyt.2021.727117
Received: 18 June 2021; Accepted: 06 September 2021;
Published: 04 October 2021.
Edited by:
Graham Campbell, Hammersmith Medicines Research, United KingdomReviewed by:
Petter Johnstad, Vestland fylkeskommune, NorwayCopyright © 2021 Vargas, Meyer, Avanes, Rus and Olson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David E. Olson, ZGVvbHNvbkB1Y2RhdmlzLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.