 Maja Milovanovic
Maja Milovanovic Roberto Grujicic
Roberto Grujicic
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Psychiatry , 29 September 2021
Sec. Child and Adolescent Psychiatry
Volume 12 - 2021 | https://doi.org/10.3389/fpsyt.2021.686021
This article is part of the Research Topic Psychiatric Comorbidities in Children and Adolescents with ASD and in Typically Developing Children View all 9 articles
Electroencephalography (EEG) can further out our understanding of autistic spectrum disorders (ASD) neurophysiology. Epilepsy and ASD comorbidity range between 5 and 46%, but its temporal relationship, causal mechanisms and interplay with intellectual disability are still unknown. Epileptiform discharges with or without seizures go as high as 60%, and associate with epileptic encephalopathies, conceptual term suggesting that epileptic activity can lead to cognitive and behavioral impairment beyond the underlying pathology. Seizures and ASD may be the result of similar mechanisms, such as abnormalities in GABAergic fibers or GABA receptor function. Epilepsy and ASD are caused by a number of genetic disorders and variations that induce such dysregulation. Similarly, initial epilepsy may influence synaptic plasticity and cortical connection, predisposing a growing brain to cognitive delays and behavioral abnormalities. The quantitative EEG techniques could be a useful tool in detecting and possibly measuring dysfunctions in specific brain regions and neuronal regulation in ASD. Power spectra analysis reveals a U-shaped pattern of power abnormalities, with excess power in the low and high frequency bands. These might be the consequence of a complicated network of neurochemical changes affecting the inhibitory GABAergic interneurons and their regulation of excitatory activity in pyramidal cells. EEG coherence studies of functional connectivity found general local over-connectivity and long-range under-connectivity between different brain areas. GABAergic interneuron growth and connections are presumably impaired in the prefrontal and temporal cortices in ASD, which is important for excitatory/inhibitory balance. Recent advances in quantitative EEG data analysis and well-known epilepsy ASD co-morbidity consistently indicate a role of aberrant GABAergic transmission that has consequences on neuronal organization and connectivity especially in the frontal cortex.
Autism spectrum disorders (ASD) are manifested by persistent impairments in social communication and interaction, in addition with restricted, repetitive patterns of behavior, interests, and activities (1). The co-morbidity of ASD and epilepsy is now well-documented (2). The prevalence of epilepsy in patients with ASD is substantially greater than in the general population (1.8-60%; 0.5-0.7%, respectively) (3, 4). Furthermore, in patients with autism, the incidence of epileptiform activity (EA) on electroencephalography (EEG) ranges from 23.6 to 60.8% (5–8).
EEG has been the primary method used to record and describe epileptiform paroxysmal activity, which occur more often in ASD. EEG recordings may also be used to examine functional connectivity across various brain areas over time through EEG coherence, which is a quantitative assessment of the connection between two EEG signals' frequency spectra (9). This useful feature can help us better understand impaired interconnections across brain areas that have been revealed by functional MRI studies in patients with ASD (10–16).
The aim of this review was to provide the summary of recent literature on the EEG findings in ASD that contribute to the understanding the neurophysiology of ASD. First, we present available data on the neurobiological mechanisms underlying ASD based on epilepsy and EA comorbidity, and then based on quantitative EEG assessments.
According to a meta-analysis of 23 studies, the prevalence of epilepsy in patients with ASD and intellectual impairment is 21.5% (2,150/10,000) and 8% (800/10,000) in patients with ASD without intellectual disability (17). This is especially important due to the fact that 31% of children with ASD have an intellectual impairment [intelligence quotient (IQ) < 70], and 25% have IQ scores in the borderline range (IQ 71–85) (18). In a sample of 5,185 children with ASD, Viscidi et al. (19) demonstrated a significant connection between seizures and cognitive impairment. They demonstrated an inverse connection between IQ and epilepsy, i.e., for every 1 standard deviation increase in IQ, the probability of developing epilepsy reduced by 47% in children over the age of 10.
ASD patients have high rate of interictal epileptic discharges (IEDs) even in the absence of definite clinical seizures—subclinical epileptic discharges (SEDs). In our previous study of patients with severe ASD of unexplained cause (n = 112), prevalence of epilepsy was 15.2%. IEDs in awake EEG recordings were found in 20.4%, and in sleep EEG recordings in 41.3% of cases (20). SEDs and the diagnosis of epilepsy occurred more frequently in the group of non-verbal ASD patients (15.8%; 21.0%, respectively) compared to verbal ones (7.5%; 11.3%, respectively), but the difference was not statistically significant. Patients with ASD without epilepsy and/or SEDs showed slight tendency to have better motor skills scores on Vineland adaptive behavior scale II, compared to the group with epilepsy and/or SEDs. Other symptoms of ASD didn't differ significantly between the groups. Similarly, according to Turk et al., children with ASD and epilepsy were more likely to have intellectual impairment, motor problems, developmental delays, and demanding behaviors than children with only ASD (21).
Specifically, this comorbidity is reciprocal; according to review of 19 studies, the median overall period prevalence of ASD in individuals with epilepsy was 9.0% (4).
ASD associated with seizures, language regression, or motor impairments might represent clinical subtypes, and could aid genetic research into the etiology of ASD (22). A number of the genes linked to ASD have also been linked to epilepsy. Synaptic transmission and DNA methylation/chromatin remodeling are the major functions of these genes (23).
Both early-onset epilepsy and ASD symptomatology characterize genetic disorders such as Fragile X (FXS), tuberous sclerosis, Rett syndrome, maternal duplications on chromosome 15q11.2-q13.1 (Dup15q), and Phelan-McDermid syndromes (24). Comorbidity with intellectual impairment is also a feature of these hereditary disorders (25). Dup15q syndrome is caused by overexpression of several genes, including ubiquitin ligase E3A (UBE3A) and a cluster of GABAA receptor subunits (26). Single-nucleotide polymorphisms in GABA receptor subunit genes have been linked to ASD and epilepsy in association studies (27, 28).
Additionally, number of existing evidence point to the complex association between ASD and epileptic encephalopathies (EE). EE are a group of neurological disorders that occur in early life and present with characteristic symptoms: different types of seizures and/or EA in EEG, as well as severe cognitive, behavioral, and neurological deficits (Table 1). According to ILAE definition: “EE is a conceptual term suggesting that EA, seizures, or IEDs can lead to cognitive and behavioral impairment, including ASD, above and beyond what might be expected from the underlying pathology” (29). This definition is based on the finding of various mechanisms through which EA affects brain development, such as disruption of anatomical and functional characteristics of the brain at various stages of development (30). Very important question that comes from EE is whether the seizures and IEDs independently worsen the development of ASD, especially if they emerged at the first 2 years, during the period of rapid brain growth and maturation (31, 32).

Table 1. Types of epileptic encephalopathies.
Core ASD features in monogenic EE are repetitive behaviors with a lower order cognitive component (e.g., motor stereotypies) and behaviors with higher cognitive implications (e.g., perseverations and obsessions) (33).
The genetic overlap between ASD and EE might potentially be seen as a connection between the two disorders. According to McTague et al., 62 genes could be connected with the development of EE (34). Out of these, 34 genes are proven to be the significant risk factors for development of ASD. Also, certain types of EE occur more frequently with ASD symptoms compared to others. Especially, EEs associated with mutations in CDKL5 (encoding cyclin-dependent kinase-like 5), SCN1A (encoding sodium voltage-gated channel alpha subunit 1), and SLC6A1 (encoding GABA transporter 1) have a high co-occurrence of ASD features (35–37).
Tuberous sclerosis complex (TSC) is a disorder of mTOR signaling caused by mutations in TSC1 and TSC2, associated with epilepsy (up to 80%), especially infantile spasms (IS) (38–40) and high prevalence rates of ASD (up to 50%) (41, 42). Not all patients with IS develop ASD, and not all individuals with ASD had prior IS, implying that ASD and IS may be two distinct end-results of a shared CNS abnormality in the TSC population (43).
Dravet syndrome (DS) is another example of a condition in which cognitive outcomes do not always correspond to seizure intensity. Syndrome is characterized by myoclonic seizures in infancy, often associated with fever, which progresses to other seizure types (44). DS is frequently caused by mutations in the SCN1A (45), that have been linked to autism, also (46).
In epilepsy-aphasia spectrum, amount of EA are responsible for developmental regression (47). Landau-Kleffner syndrome (LKS) is characterized by language regression and temporal lobe epileptiform discharges (47). It has been linked to mutations in the GRIN2A gene (48), which codes for the GluN2A protein present in speech and language cortical areas. Autistic symptoms may be present in a clinical picture of LKS, in addition to cognitive delay and EE (49).
But there are important differences between developmental regression in LKS and ASD. In LKS, after relatively typical early development, regression primarily affects language between 3 and 9 years of age, but behavioral problems are considerably less common and may be caused by speech impairment or cognitive decline (49). In ASD, language and social skills loss typically begins before the age of three, and the regression might be clinically mild (e.g., loss of single words, reduced gesturing) (50). The epileptic discharges associated with developmental regression in ASD includes focal spikes that can be infrequent (51) (Figure 1). Language loss in LKS is severe, with loss of completely developed language, and EEG is characterized by frequent temporoparietal spikes, which are notably activated by slow wave sleep (SWS), or with the EEG pattern of continuous spike-wave in sleep (CSWS) (Figure 2). CSWS is defined as almost continuous 1.5-2 Hz spike-waves, that takes up >85% of SWS (29). However, even a spike-wave index in sleep (SWI) of more than 50% affect child's development, including the development of autistic symptoms as well as cognitive, behavioral, and/or motor regression (52).
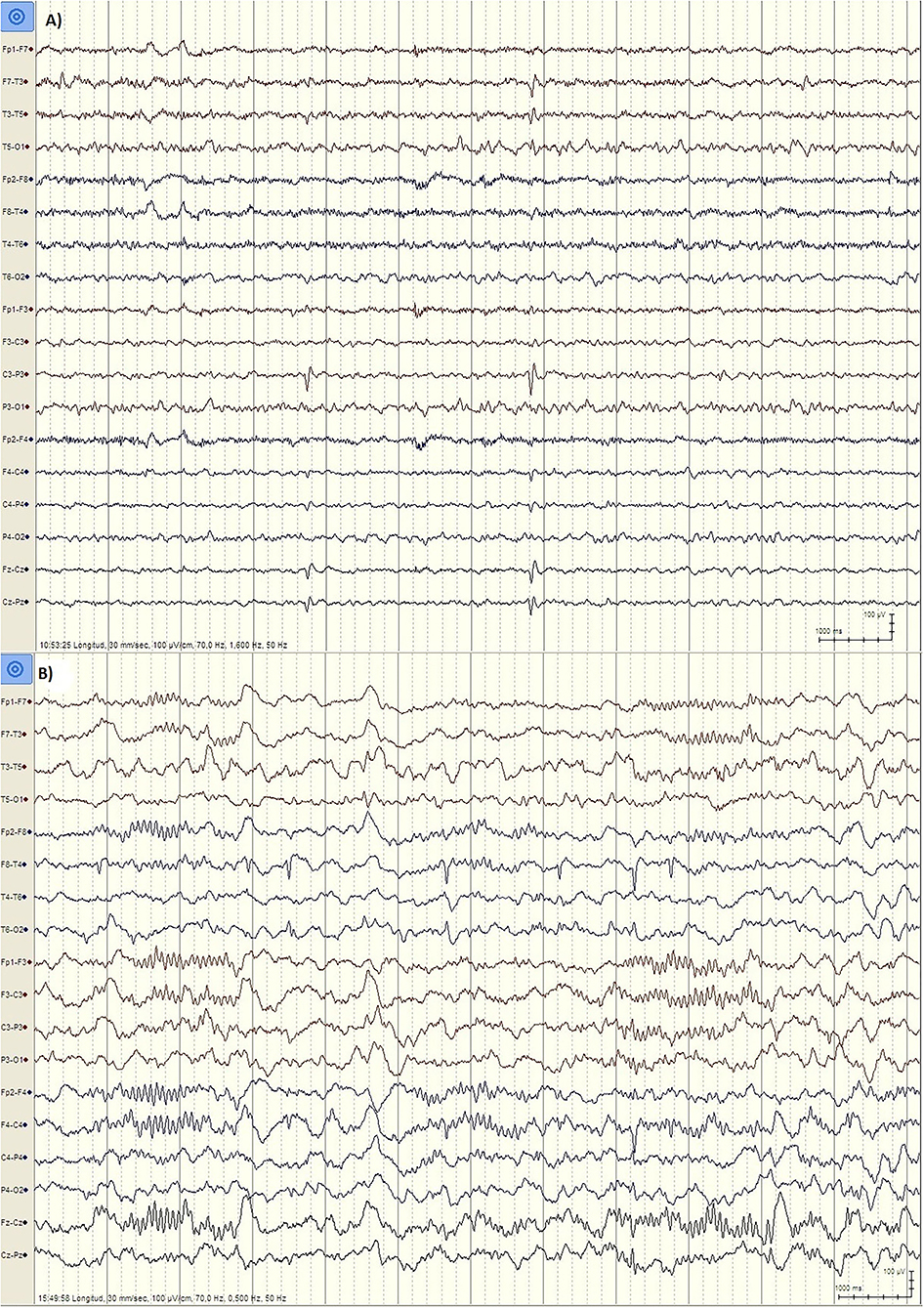
Figure 1. EEG findings in ASD patients. (A) Patient PR, male, 6 years. EEG during wakefulness (Bipolar longitudinal montage, Sensitivity 100 μV/cm, High pass filter 1.6 Hz, Low pass filter 70 Hz): Left centroparietal spikes (P3, C3). (B) Patient JL, male, 5 years old. EEG during N2 stage Sleep (Bipolar longitudinal montage, Sensitivity 100 μV/cm, High pass filter 0.5 Hz, Low pass filter 70 Hz): Right temporal spikes (T4, T6).
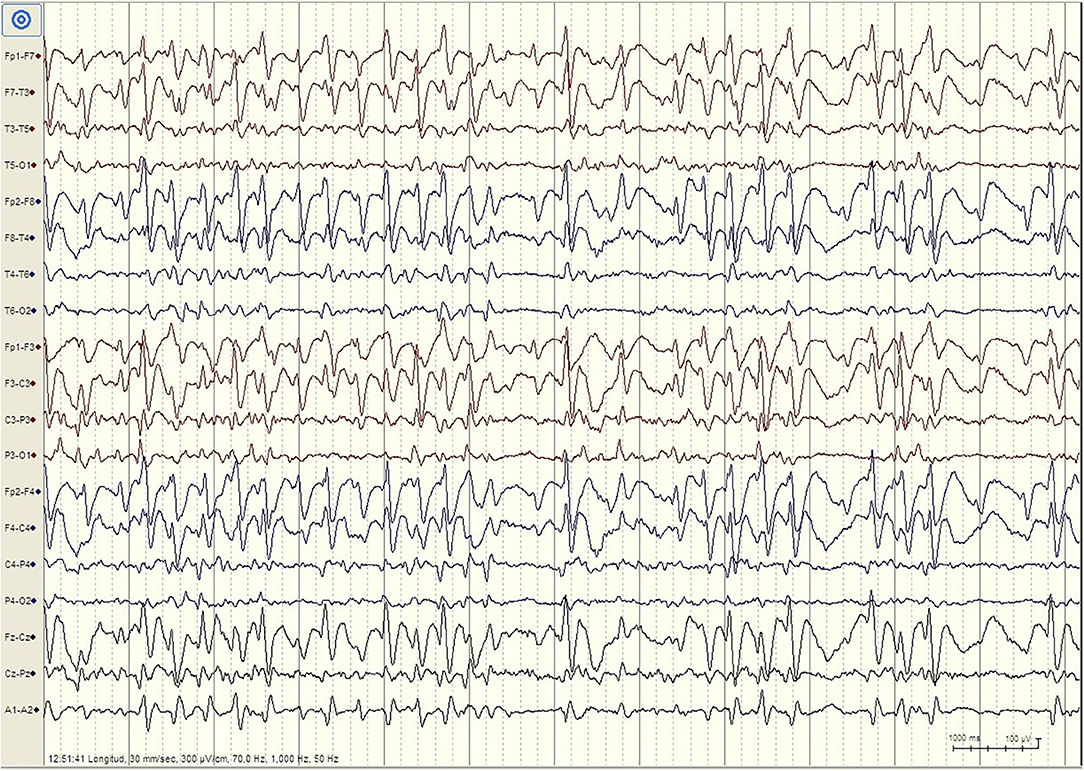
Figure 2. EEG finding in patient with Landau-Kleffner Syndrome. Patient LM, female 6 years. EEG during N2 Stage Sleep (Bipolar Longitudinal montage, Sensitivity 300 μV/cm, High pass filter 1 Hz, Low pass filter 70 Hz): High amplitude generalized spike–waves with variable frequency, mostly <3Hz. There is repeated fragmentation of these discharges, which still occupy more than 80% of the epoch. For the whole sleep period, the spike–waves occupied more than 85% of slow waves sleep, constituting continuous spike-waves during slow sleep (CSWS).
The clinical assessment of patients with developmental regression, epilepsy and ASD has to be comprehensive and needs to include multiple variables. Some of the important variables include the type of regression (language vs. autistic), the age of onset of seizures or IEDs, the detailed location and description, as well as assessing the amount and persistence of EA. These variables can significantly influence further clinical management and treatment plan (25). There is still no existing evidence that strongly supports treatment with antiepileptic drugs in ASD, if the EEG demonstrates infrequent spikes, in absence of seizures. However, there's still an ongoing debate among experts concerning the use of pharmacotherapy in these patients. It is important to note that there are several clinical studies which demonstrated improvement in ASD symptomatology when treated with anticonvulsants (53, 54), but further studies are needed to confirm these findings.
Children with EE are more prone to develop ASD; hence, in addition to urgent pharmacological treatment with protocols for EE, early behavioral, communication, and educational interventions should be addressed as part of their comprehensive management (25).
The quantitative EEG techniques could be a useful tool in detecting and possibly measuring dysfunctions in specific brain regions and neuronal regulation in ASD. Scientific interest in identifying EEG biomarkers of ASD, with a focus on spectral power, coherence, and hemisphere asymmetry, recently raised.
Wang and colleagues, in their review of resting-state EEG studies in ASD, reported a potential “U-shaped" profile of EEG power spectra in ASD as compared to typically developing controls, with excess power in theta and gamma frequency bands and decreased power in alpha frequency band (55). Other research has found alpha band asymmetry (56) between hemispheres in infants at risk for ASD.
EEG coherence, a key approach of EEG functional connectivity research, has been used to investigate how brain regions communicate in real time. When two signals of the same frequency are active and have a constant phase relationship across time, they are termed coherent, meaning there is a high degree of coordinated activity between the brain areas creating them (57–59).
Two EEG coherence investigations in ASD patients found under and over-connectivity in distinct frequency bands (60, 61). Increased local coherence has been found across the frontal area in delta band (62), as well as over the left frontal and temporal regions in the theta band (61). In contrast, reduced intrahemispheric and interhemispheric local coherence in all brain regions has been reported in delta and theta bands (63), and, also, reduced local coherence over the frontal region in delta (62) and alpha bands (63). Machado et al. discovered that ASD children had considerably higher intrahemispheric long-range coherence in the left hemisphere, confirming the notion of over functional connectivity in ASD (64). Carson et al., on the other hand, found that children with ASD had reduced long-distance coherence at the alpha frequency in resting state (65). Children with ASD have decreased interhemispherical and intrahemispherical coherence in the delta and theta frequency bands, according to Coben et al. (63). Wang et al. showed higher coherence of short and long-distance connections in children with ASD compared to controls, which was related to clinical severity scale scores (66).
According to the theory of nerve pruning, throughout normal brain development, neurons matures by the myelination, and is further pruned and modified (67, 68). Pruning and synaptogenesis allow continuous changes in both short- and long-range neuronal circuitry in the normal brain growth, leading to a weakening of the functional connections between the neighboring areas of the brain, and simultaneously strengthening connections between distant brain regions (69). This mechanism may be disrupted in developmental diseases such as autism, resulting in aberrant brain connections. There is evidence that synaptic disruption occurs in ASD at both the local level of single axons and the broader level of brain networks (70, 71). Using EEG coherence to examine electrical connection patterns, researchers may be able to analyze the resulting differences in brain function between persons with and without ASD (72, 73).
The co-morbidity of ASD and epilepsy could be currently explained by the most widely accepted theory of brain hyperexcitability. Various structural and functional defects of genetic, metabolic, immune or environmental etiologies, could permanently compromise balance in excitation (E) and inhibition (I) circuits (74).
The interaction of ASD, epilepsy, and intellectual disability led to the hypothesis that ASD and epilepsy are outcomes of similar processes, such as dysregulation of E/I balance, caused by defects in GABAergic fibers, particularly GABAergic interneurons maturation, or GABA receptor function (75). Several genes involved in the function of ion channels that play key functions in the brain, such as SCN1A or GABAA receptors, have been implicated in ASD and EEs (25). The increased activity of glutamate receptor signaling can also lead to hyperexcitability (76). Primary epilepsy, on the other hand, may affect synaptic plasticity and cortical connection, predisposing a developing brain to cognitive delays and behavioral abnormalities (25, 77, 78).
Based on quantitative EEG findings, U-shaped power spectra profile may be attributed to abnormal functioning of GABAergic tone in inhibitory circuitry, which influences the functional and developmental plasticity of the brain and decrease power of high-frequency and low-frequency bands while increasing the power of middle-range frequencies (79). This profile could be caused by affected GABAergic interneurons that has modulating role on excitatory pyramidal cells (55). The gamma band activity has been related to dendritic GABAergic inhibitory dysfunction (80). GABAergic interneurons synapsis with Nmethyl-D-aspartate receptors (NMDAR) on glutamatergic neurons causes thalamocortical delta oscillations, which are regulated by dopaminergic neurons in the thalamus (81).
Functional connectivity studies using EEG coherence revealed overall local over-connectivity and long-range under-connectivity, as well as increased power of delta frequency in the frontal brain region in individuals with ASD (66, 82, 83). Those findings consistently point to a role of aberrant GABAergic transmission on neuronal organization and connectivity especially in the frontal cortex.
There is evidence that GABAergic interneuron growth and connections in the prefrontal and temporal cortices are altered in ASD (84), which could lead to E/I imbalance (85). In post-mortem brain samples of ASD cases, it was found that neocortical minicolumns, elemental modular microcircuits made up of excitatory pyramidal neurons surrounded by GABAergic inhibitory neurons, were reduced, which could results in inhibitory circuits disruption (84, 86).
GABAergic disorders can affect early development, because prenatally GABA has the role of excitatory trophic factor, leading to the growth and binding of dendrites (87). In the mature brain, GABA acts as an inhibitory transmitter. Defects in GABAergic signaling, especially shifting the E/I balance toward excitatory transmission, may thereby explain some of the characteristics of ASD (88).
Although there is a number of etiological hypotheses, one of the most researched etiological mechanisms in the development of ASD in the last decade is the E/I imbalance in key cortical and subcortical neuronal circuits (75, 89, 90). This hypothesis was first proposed in the seminal work of Rubenstein and Merzenich in 2003 (74). To date, there is an abundance of evidence that support this model both from preclinical (90) and in clinical (89) perspectives.
When processed and analyzed with the most advanced techniques, the EEG might be a valuable approach in clinical and scientific studies of ASD neurophysiological substrates. Nevertheless, the quantitative EEG techniques could be a useful tool in detecting and possibly measuring dysfunctions in specific brain regions and neuronal regulation in ASD. Advances in quantitative EEG analysis in recent years and well-known epilepsy ASD co-morbidity consistently indicate a role of aberrant GABAergic transmission that has consequences on neuronal organization and connectivity especially in the frontal cortex.
Bosl and colleagues in 2011 provided initial data that highlighted the role of brain connectivity in early development (91). The authors investigated EEG complexity in newborns at risk for ASD compared to normal controls, finding that the infants-at-risk has less brain complexity. This study shows that decreased connectivity during early development is linked to the likelihood of ASD, although no definitive diagnostic outcome for infants was obtained in this investigation. As a result, it is uncertain if connectivity has any predictive value for autism risk.
Although, at this point, EEG research in ASD shows promising results in early detection and prediction of atypical brain development (92), EEG is still not a reliable clinical diagnostic tool for ASD due to its low sensitivity or specificity. However, the data presented in this review strongly suggest that EEG should be a complementary technique to the existing methods in diagnostic process.
Also, the available data on this matter indicate that further research is needed to provide better understanding of different electrophysiological features of high importance which could fill in major gaps in understanding pathophysiology and assessment of ASD. Due to the fact that ASD is a neurodevelopmental disorder, primary research focus should be on longitudinal studies which could potentially strengthen the available findings and also define the developmental stages of ASD. We strongly believe that combining the new approaches in EEG methodology with already established ones, could potentially open a new perspective on ASD assessment and eventually lead to new early diagnosis, early intervention and prevention strategies.
MM contributed with literature research, analysis, expertise, and interpretation, as well as the writing and reviewing process. RG contributed with the research, interpretation, and writing of the paper, as well as reviewing and technical work. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Levy SE, Mandell DS, Schultz RT. Autism. Lancet. (2009) 374:1627–38. doi: 10.1016/S0140-6736(09)61376-3
2. Buckley AW, Holmes GL. Epilepsy and autism. Cold Spring Harb Perspect Med. (2016) 6:a022749. doi: 10.1101/cshperspect.a022749
3. Forsgren L, Beghi E, Oun A, Sillanpää M. The epidemiology of epilepsy in Europe—a systematic review. Eur J Neurol. (2005) 12:245–53. doi: 10.1111/j.1468-1331.2004.00992.x
4. Lukmanji S, Manji SA, Kadhim S, Sauro KM, Wirrell EC, Kwon, et al. The co-occurrence of epilepsy and autism: a systematic review. Epilepsy Behav. (2019) 98:238–48. doi: 10.1016/j.yebeh.2019.07.037
5. Rossi PG, Parmeggiani A, Bach V, Santucci M, Visconti P. EEG features and epilepsy in patients with autism. Brain Dev. (1995) 17:169–74. doi: 10.1016/0387-7604(95)00019-8
6. Kawasaki Y, Yokota K, Shinomiya M, Shimizu Y, Niwa S. Brief report: electroencephalographic paroxysmal activities in the frontal area emerged in middle childhood and during adolescence in a follow-up study of autism. J Autism Dev. Disord. (1997) 27:605–20. doi: 10.1023/A:1025886228387
7. Giovanardi Rossi P, Posar A, Parmeggiani A. Epilepsy in adolescents and young adults with autistic disorder. Brain Dev. (2000) 22:102–6. doi: 10.1016/S0387-7604(99)00124-2
8. Hashimoto T, Sasaki M, Sugai K, Hanaoka S, Fukumizu M, Kato T. Paroxysmal discharges on EEG in young autistic patients are frequent in frontal regions. J Med Invest. (2001) 48:175–80.
9. Olejniczak P. Neurophysiologic basis of EEG. J Clin Neurophysiol. (2006) 23:186–9. doi: 10.1097/01.wnp.0000220079.61973.6c
10. Barnea-Goraly N, Kwon H, Menon V, Eliez S, Lotspeich L, Reiss AL. White matter structure in autism: preliminary evidence from diffusion tensor imaging. Biol Psychiatry. (2004) 55:323–6. doi: 10.1016/j.biopsych.2003.10.022
11. Just MA, Cherkassky VL, Keller TA, Minshew NJ. Cortical activation and synchronization during sentence comprehension in high-functioning autism: evidence of underconnectivity. Brain. (2004) 127:1811–21. doi: 10.1093/brain/awh199
12. Herbert MR, Ziegler DA, Deutsch CK, O'Brien LM, Kennedy DN, Filipek PA, et al. Brain asymmetries in autism and developmental language disorder: a nested whole-brain analysis. Brain. (2005) 128:213–26. doi: 10.1093/brain/awh330
13. Alexander AL, Lee JE, Lazar M, Boudos R, DuBray MB, Oakes TR, et al. Diffusion tensor imaging of the corpus callosum in Autism. Neuroimage. (2007) 34:61–73. doi: 10.1016/j.neuroimage.2006.08.032
14. Sundaram SK, Kumar A, Makki MI, Behen ME, Chugani HT, Chugani DC. Diffusion tensor imaging of frontal lobe in autism spectrum disorder. Cereb Cortex. (2008) 18:2659–65. doi: 10.1093/cercor/bhn031
15. Monk CS, Peltier SJ, Wiggins JL, Weng S-J, Carrasco M, Carrasco M, et al. Abnormalities of intrinsic functional connectivity in autism spectrum disorders. Neuroimage. (2009) 47:764–72. doi: 10.1016/j.neuroimage.2009.04.069
16. Wass S. Distortions and disconnections: disrupted brain connectivity in autism. Brain Cogn. (2011) 75:18–28. doi: 10.1016/j.bandc.2010.10.005
17. Amiet C, Gourfinkel-An I, Bouzamondo A, Tordjman S, Baulac M, Lechat P, et al. Epilepsy in autism is associated with intellectual disability and gender: evidence from a meta-analysis. Biol Psychiatry. (2008) 64:577–82. doi: 10.1016/j.biopsych.2008.04.030
18. Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z, et al. Prevalence of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, united states, 2014. MMWR Surveill Summ. (2018) 67:1–23. doi: 10.15585/mmwr.ss6706a1
19. Viscidi EW, Triche EW, Pescosolido MF, McLean RL, Joseph RM, Spence SJ, et al. Clinical characteristics of children with autism spectrum disorder and co-occurring epilepsy. PLoS ONE. (2013) 8:e67797. doi: 10.1371/journal.pone.0067797
20. Milovanovic M, Radivojevic V, Radosavljev-Kircanski J, Grujicic R, Toskovic O, Aleksić-Hil O, et al. Epilepsy and interictal epileptiform activity in patients with autism spectrum disorders. Epilepsy Behav. (2019) 92:45–52. doi: 10.1016/j.yebeh.2018.12.011
21. Turk J, Bax M, Williams C, Amin P, Eriksson M, Gillberg C. Autism spectrum disorder in children with and without epilepsy: impact on social functioning and communication. Acta Paediatr. (2009) 98:675–81. doi: 10.1111/j.1651-2227.2008.01184.x
22. Deonna T, Roulet E. Autistic spectrum disorder: evaluating a possible contributing or causal role of epilepsy. Epilepsia. (2006) 47 (Suppl 2):79–82. doi: 10.1111/j.1528-1167.2006.00697.x
23. Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. (2014) 515:216–21. doi: 10.1038/nature13908
24. Jacob J. Cortical interneuron dysfunction in epilepsy associated with autism spectrum disorders. Epilepsia. (2016) 57:182–93. doi: 10.1111/epi.13272
25. Jeste SS, Tuchman R. Autism spectrum disorder and epilepsy: two sides of the same coin? J Child Neurol. (2015) 30:1963–71. doi: 10.1177/0883073815601501
26. Schroer RJ, Phelan MC, Michaelis RC, Crawford EC, Skinner SA, Cuccaro M, et al. Autism and maternally derived aberrations of chromosome 15q. Am J Med Genet. (1998) 76:327–36.
27. Buxbaum JD, Silverman JM, Smith CJ, Greenberg DA, Kilifarski M, Reichert J, et al. Association between a GABRB3 polymorphism and autism. Mol Psychiatry. (2002) 7:311–6. doi: 10.1038/sj.mp.4001011
28. Collins AL, Ma D, Whitehead PL, Martin ER, Wright HH, Abramson RK, et al. Investigation of autism and GABA receptor subunit genes in multiple ethnic groups. Neurogenetics. (2006) 7:167–74. doi: 10.1007/s10048-006-0045-1
29. Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. (2010) 51:676–85. doi: 10.1111/j.1528-1167.2010.02522.x
30. Marsh ED, Brooks-Kayal AR, Porter BE. Seizures and antiepileptic drugs: does exposure alter normal brain development? Epilepsia. (2006) 47:1999–2010. doi: 10.1111/j.1528-1167.2006.00894.x
31. Tuchman R. Autism and epilepsy: what has regression got to do with it? Epilepsy Curr. (2006) 6:107–11. doi: 10.1111/j.1535-7511.2006.00113.x
32. Knickmeyer RC, Gouttard S, Kang C, Evans D, Wilber K, Smith JK, et al. A structural MRI study of human brain development from birth to 2 years. J Neurosci. (2008) 28:12176–82. doi: 10.1523/JNEUROSCI.3479-08.2008
33. Srivastava S, Sahin M. Autism spectrum disorder and epileptic encephalopathy: common causes, many questions. J Neurodev Disord. (2017) 9:23. doi: 10.1186/s11689-017-9202-0
34. McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. (2016) 15:304–16. doi: 10.1016/S1474-4422(15)00250-1
35. Kwong AK-Y, Fung C-W, Chan S-Y, Wong VC-N. Identification of SCN1A and PCDH19 mutations in Chinese children with Dravet syndrome. PLoS ONE. (2012) 7:e41802. doi: 10.1371/journal.pone.0041802
36. Zhao Y, Zhang X, Bao X, Zhang Q, Zhang J, Cao G, et al. Clinical features and gene mutational spectrum of CDKL5-related diseases in a cohort of Chinese patients. BMC Med. Genet. (2014) 15:24. doi: 10.1186/1471-2350-15-24
37. Carvill GL, McMahon JM, Schneider A, Zemel M, Myers CT, Saykally J, et al. Mutations in the GABA Transporter SLC6A1 Cause Epilepsy with Myoclonic-Atonic Seizures. Am J Hum Genet. (2015) 96:808–15. doi: 10.1016/j.ajhg.2015.02.016
38. Curatolo P, Verdecchia M, Bombardieri R. Tuberous sclerosis complex: a review of neurological aspects. Eur J Paediatr Neurol. (2002) 6:15–23. doi: 10.1053/ejpn.2001.0538
39. Curatolo P, Bombardieri R, Verdecchia M, Seri S. Intractable seizures in tuberous sclerosis complex: from molecular pathogenesis to the rationale for treatment. J Child Neurol. (2005) 20:318–25. doi: 10.1177/08830738050200040901
40. Shields WD. Infantile spasms: little seizures, BIG consequences. Epilepsy Curr. (2006) 6:63–9. doi: 10.1111/j.1535-7511.2006.00100.x
41. Jeste SS, Sahin M, Bolton P, Ploubidis GB, Humphrey A. Characterization of autism in young children with tuberous sclerosis complex. J Child Neurol. (2008) 23:520–5. doi: 10.1177/0883073807309788
42. Richards C, Jones C, Groves L, Moss J, Oliver C. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry. (2015) 2:909–16. doi: 10.1016/S2215-0366(15)00376-4
43. Bolton PF, Park RJ, Higgins JNP, Griffiths PD, Pickles A. Neuro-epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain. (2002) 125:1247–55. doi: 10.1093/brain/awf124
44. Millichap JJ, Koh S, Laux LC, Nordli DRJ. Child neurology: dravet syndrome: when to suspect the diagnosis. Neurology. (2009) 73:e59–62. doi: 10.1212/WNL.0b013e3181b9c880
45. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. (2001) 68:1327–32. doi: 10.1086/320609
46. O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. (2011) 43:585–9. doi: 10.1038/ng.835
47. Tassinari CA, Rubboli G, Volpi L, Meletti S, d'Orsi G, Franca M, et al. Encephalopathy with electrical status epilepticus during slow sleep or ESES syndrome including the acquired aphasia. Clin Neurophysiol. (2000) 111 (Suppl 2):S94–S102. doi: 10.1016/S1388-2457(00)00408-9
48. Lesca G, Rudolf G, Bruneau N, Lozovaya N, Labalme A, Boutry-Kryza N, et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet. (2013) 45:1061–6. doi: 10.1038/ng.2726
49. Pearl PL, Carrazana EJ, Holmes GL. The landau-kleffner syndrome. Epilepsy Curr. (2001) 1:39–45. doi: 10.1111/j.1469-5812.2005.00134.x-i1
50. Rogers SJ. Developmental regression in autism spectrum disorders. Ment Retard Dev Disabil Res Rev. (2004) 10:139–43. doi: 10.1002/mrdd.20027
51. McVicar KA, Ballaban-Gil K, Rapin I, Moshé SL, Shinnar S. Epileptiform EEG abnormalities in children with language regression. Neurology. (2005) 65:129–31. doi: 10.1212/01.wnl.0000167193.53817.0f
52. Fernández IS, Chapman KE, Peters JM, Kothare SV, Nordli DR, Jensen FE, et al. The tower of Babel: survey on concepts and terminology in electrical status epilepticus in sleep and continuous spikes and waves during sleep in North America. Epilepsia. (2013) 54:741–50. doi: 10.1111/epi.12039
53. Rugino TA, Samsock TC. Levetiracetam in autistic children: an open-label study. J Dev Behav Pediatr. (2002) 23:225–30. doi: 10.1097/00004703-200208000-00006
54. Luz-Escamilla L, Morales-González JA. Association between interictal epileptiform discharges and autistic spectrum disorder. Brain Sci. (2019) 9:185. doi: 10.3390/brainsci9080185
55. Wang J, Barstein J, Ethridge LE, Mosconi MW, Takarae Y, Sweeney JA. Resting state EEG abnormalities in autism spectrum disorders. J Neurodev Disord. (2013) 5:24. doi: 10.1186/1866-1955-5-24
56. Gabard-Durnam L, Tierney AL, Vogel-Farley V, Tager-Flusberg H, Nelson CA. Alpha asymmetry in infants at risk for autism spectrum disorders. J Autism Dev. Disord. (2015) 45:473–80. doi: 10.1007/s10803-013-1926-4
57. Duffy FH, Als H. A stable pattern of EEG spectral coherence distinguishes children with autism from neuro-typical controls—a large case control study. BMC Med. (2012) 10:64. doi: 10.1186/1741-7015-10-64
58. Srinivasan R, Winter WR, Ding J, Nunes PL. EEG and MEG coherence: measures of functional connectivity at distinct spatial scales of neocortical dynamics. J Neurosci Methods. (2007) 166:41–52. doi: 10.1016/j.jneumeth.2007.06.026
59. Fries P. A mechanism for cognitive dynamics: neuronal communication through neuronal coherence. Trends Cogn Sci. (2005) 9:474–80. doi: 10.1016/j.tics.2005.08.011
60. Courchesne E, Campbell K, Solso S. Brain growth across the life span in autism: age-specific changes in anatomical pathology. Brain Res. (2011) 1380:138–45. doi: 10.1016/j.brainres.2010.09.101
61. Murias M, Webb SJ, Greenson J, Dawson G. Resting state cortical connectivity reflected in EEG coherence in individuals with autism. Biol Psychiatry. (2007) 62:270–3. doi: 10.1016/j.biopsych.2006.11.012
62. Barttfeld P, Wicker B, Cukier S, Navarta S, Lew S, Sigman M. A big-world network in ASD: dynamical connectivity analysis reflects a deficit in long-range connections and an excess of short-range connections. Neuropsychologia. (2011) 49:254–63. doi: 10.1016/j.neuropsychologia.2010.11.024
63. Coben R, Clarke AR, Hudspeth W, Barry RJ. EEG power and coherence in autistic spectrum disorder. Clin Neurophysiol. (2008) 119:1002–9. doi: 10.1016/j.clinph.2008.01.013
64. Machado C, Estévez M, Leisman G, Melillo R, Rodríguez R, DeFina P, et al. QEEG spectral and coherence assessment of autistic children in three different experimental conditions. J Autism Dev Disord. (2015) 45:406–24. doi: 10.1007/s10803-013-1909-5
65. Carson AM, Salowitz NMG, Scheidt RA, Dolan BK, Van Hecke AV. Electroencephalogram coherence in children with and without autism spectrum disorders: Decreased interhemispheric connectivity in autism. Autism Res. (2014) 7:334–43. doi: 10.1002/aur.1367
66. Wang J, Wang X, Wang X, Zhang H, Zhou Y, Chen L, et al. Increased EEG coherence in long-distance and short-distance connectivity in children with autism spectrum disorders. Brain Behav. (2020) 10:e01796. doi: 10.1002/brb3.1796
67. Chugani HT, Phelps ME, Mazziotta JC. Positron emission tomography study of human brain functional development. Ann Neurol. (1987) 22:487–97. doi: 10.1002/ana.410220408
68. Huttenlocher PR, De Courten C, Garey LJ, Hendrik VDL. Synaptogenesis in human visual cortex—evidence for synapse elimination during normal development. Neurosci Lett. (1982) 33:247–52. doi: 10.1016/0304-3940(82)90379-2
69. Fair DA, Dosenbach NUF, Church JA, Cohen AL, Brahmbhatt S, Miezin FM, et al. Development of distinct control networks through segregation and integration. Proc Natl Acad Sci. (2007) 104:13507–12. doi: 10.1073/pnas.0705843104
70. Ameis SH, Catani M. Altered white matter connectivity as a neural substrate for social impairment in autism spectrum disorder. Cortex. (2015) 62:158–81. doi: 10.1016/j.cortex.2014.10.014
71. De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. (2014) 515:209–15. doi: 10.1038/nature13772
72. Lord V, Opacka-Juffry J. Electroencephalography (EEG) measures of neural connectivity in the assessment of brain responses to salient auditory stimuli in patients with disorders of consciousness. Front Psychol. (2016) 7:397. doi: 10.3389/fpsyg.2016.00397
73. Schwartz S, Kessler R, Gaughan T, Buckley AW. EEG coherence patterns in autism: an updated review. Pediatr Neurol. (2017) 67:7–22. doi: 10.1016/j.pediatrneurol.2016.10.018
74. Rubenstein JLR, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. (2003) 2:255–67. doi: 10.1034/j.1601-183X.2003.00037.x
75. Bozzi Y, Provenzano G, Casarosa S. Neurobiological bases of autism-epilepsy comorbidity: a focus on excitation/inhibition imbalance. Eur J Neurosci. (2018) 47:534–48. doi: 10.1111/ejn.13595
76. Oberman LM. mGluR antagonists and GABA agonists as novel pharmacological agents for the treatment of autism spectrum disorders. Expert Opin Investig Drugs. (2012) 21:1819–25. doi: 10.1517/13543784.2012.729819
77. Brooks-Kayal A. Epilepsy and autism spectrum disorders: are there common developmental mechanisms? Brain Dev. (2010) 32:731–8. doi: 10.1016/j.braindev.2010.04.010
78. Brooks-Kayal AR, Bath KG, Berg AT, Galanopoulou AS, Holmes GL, Jensen FE, et al. Issues related to symptomatic and disease-modifying treatments affecting cognitive and neuropsychiatric comorbidities of epilepsy. Epilepsia. (2013) 54 (Suppl 4):44–60. doi: 10.1111/epi.12298
79. Tierney AL, Gabard-Durnam L, Vogel-Farley V, Tager-Flusberg H, Nelson CA. Developmental trajectories of resting EEG power: an endophenotype of autism spectrum disorder. PLoS ONE. (2012) 7:e39127. doi: 10.1371/journal.pone.0039127
80. Wendling F, Bartolomei F, Bellanger JJ, Chauvel P. Epileptic fast activity can be explained by a model of impaired GABAergic dendritic inhibition. Eur J Neurosci. (2002) 15:1499–508. doi: 10.1046/j.1460-9568.2002.01985.x
81. Zhang Y, Llinas RR, Lisman JE. Inhibition of NMDARs in the nucleus reticularis of the thalamus produces delta frequency bursting. Front Neural Circuits. (2009) 3:20. doi: 10.3389/neuro.04.020.2009
82. Srinivasan R, Nunez PL, Silberstein RB. Spatial filtering and neocortical dynamics: estimates of EEG coherence. IEEE Trans Biomed Eng. (1998) 45:814–26. doi: 10.1109/10.686789
83. Whitford TJ, Rennie CJ, Grieve SM, Clark CR, Gordon E, Williams LM. Brain maturation in adolescence: concurrent changes in neuroanatomy and neurophysiology. Hum Brain Mapp. (2007) 28:228–37. doi: 10.1002/hbm.20273
84. Casanova MF, Buxhoeveden DP, Switala AE, Roy E. Minicolumnar pathology in autism. Neurology. (2002) 58:428–32. doi: 10.1212/WNL.58.3.428
85. Levitt P. Disruption of interneuron development. Epilepsia. (2005) 46 (Suppl 7):22–8. doi: 10.1111/j.1528-1167.2005.00305.x
86. Casanova MF, Buxhoeveden D, Gomez J. Disruption in the inhibitory architecture of the cell minicolumn: implications for autism. Neuroscientist. (2003) 9:496–507. doi: 10.1177/1073858403253552
87. Owens DF, Kriegstein AR. Developmental neurotransmitters? Neuron. (2002) 36:989–91. doi: 10.1016/S0896-6273(02)01136-4
88. Zafeiriou DI, Ververi A, Dafoulis V, Kalyva E, Vargiami E. Autism spectrum disorders: the quest for genetic syndromes. Am J Med Genet B Neuropsychiatr Genet. (2013) 162B:327–66. doi: 10.1002/ajmg.b.32152
89. Port RG, Oberman LM, Roberts TP. Revisiting the excitation/inhibition imbalance hypothesis of ASD through a clinical lens. Br J Radiol. (2019) 92:20180944. doi: 10.1259/bjr.20180944
90. Culotta L, Penzes P. Exploring the mechanisms underlying excitation/inhibition imbalance in human iPSC-derived models of ASD. Mol Autism. (2020) 11:32. doi: 10.1186/s13229-020-00339-0
91. Bosl W, Tierney A, Tager-Flusberg H, Nelson C. EEG complexity as a biomarker for autism spectrum disorder risk. BMC Med. (2011) 9:18. doi: 10.1186/1741-7015-9-18
Keywords: autistic spectrum disorder, electroencephalography, epilepsy, epileptiform discharges, excitation/inhibition imbalance
Citation: Milovanovic M and Grujicic R (2021) Electroencephalography in Assessment of Autism Spectrum Disorders: A Review. Front. Psychiatry 12:686021. doi: 10.3389/fpsyt.2021.686021
Received: 26 March 2021; Accepted: 30 August 2021;
Published: 29 September 2021.
Edited by:
Marija Raleva, Saints Cyril and Methodius University of Skopje, North MacedoniaReviewed by:
Elena Dmitrievna Belousova, Pirogov Russian National Research Medical University, RussiaCopyright © 2021 Milovanovic and Grujicic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maja Milovanovic, bWFqYS5taWxvdmFub3ZpY0BpbWgub3JnLnJz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.