 André C. Tonon
André C. Tonon Luísa K. Pilz
Luísa K. Pilz Regina P. Markus
Regina P. Markus Maria Paz Hidalgo1,2
Maria Paz Hidalgo1,2 Elaine Elisabetsky
Elaine Elisabetsky- 1Laboratório de Cronobiologia e Sono, Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil
- 2Graduate Program in Psychiatry and Behavioral Sciences, Federal University of Rio Grande do Sul, Porto Alegre, Brazil
- 3Laboratório de Cronofarmacologia, Departamento de Fisiologia, Instituto de Biociência, Universidade de São Paulo, São Paulo, Brazil
- 4Programa de Pós-Graduação em Ciências Biológicas-Bioquímica, Departamento de Bioquímica, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
Daily rhythm of melatonin synchronizes the body to the light/dark environmental cycle. Several hypotheses have been raised to understand the intersections between melatonin and depression, in which changes in rest-activity and sleep patterns are prominent. This review describes key experimental and clinical evidence that link melatonin with the etiopathology and symptomatology of depressive states, its role in the follow up of therapeutic response to antidepressants, as well as the clinical evidence of melatonin as MDD treatment. Melatonin, as an internal temporal cue contributing to circadian organization and best studied in the context of circadian misalignment, is also implicated in neuroplasticity. The monoaminergic systems that underly MDD and melatonin production overlap. In addition, the urinary metabolite 6-sulfatoxymelatonin (aMT6) has been proposed as biomarker for antidepressant responders, by revealing whether the blockage of noradrenaline uptake has taken place within 24 h from the first antidepressant dose. Even though animal models show benefits from melatonin supplementation on depressive-like behavior, clinical evidence is inconsistent vis-à-vis prophylactic or therapeutic benefits of melatonin or melatonin agonists in depression. We argue that the study of melatonin in MDD or other psychiatric disorders must take into account the specificities of melatonin as an integrating molecule, inextricably linked to entrainment, metabolism, immunity, neurotransmission, and cell homeostasis.
Introduction
Melatonin (N-acetyl-5-methoxytryptamine) is an amphiphilic indoleamine conserved from unicellular organisms to plants and animals. In mammals, pineal and extra-pineal synthesis were described (1). Darkness at night triggers a polysynaptic pathway that begins with the retino-hypothalamic tract projecting to the suprachiasmatic nuclei (SCN) of the hypothalamus and ends as a sympathetic input to the pineal gland. Melatonin synthesis is prompted by norepinephrine and requires serotonin as a precursor (2). Pineal melatonin, a primary output of the circadian pacemaker (SCN), transduces light-dark information to the whole body, coordinating daily physiological functions and behaviors.
Biological rhythms and melatonin represent recent frameworks in the investigation of the etiology, prognostic improvement, and treatment approach in the clinical course of depression. The somatic, cognitive, and affective symptoms of depressive states that occur in association with neurochemical/hormonal imbalance, may result, among other factors, from the misalignment of biological rhythms (3–5). A summary of the physiological regulation and organization of biological rhythms is shown in Figure 1.
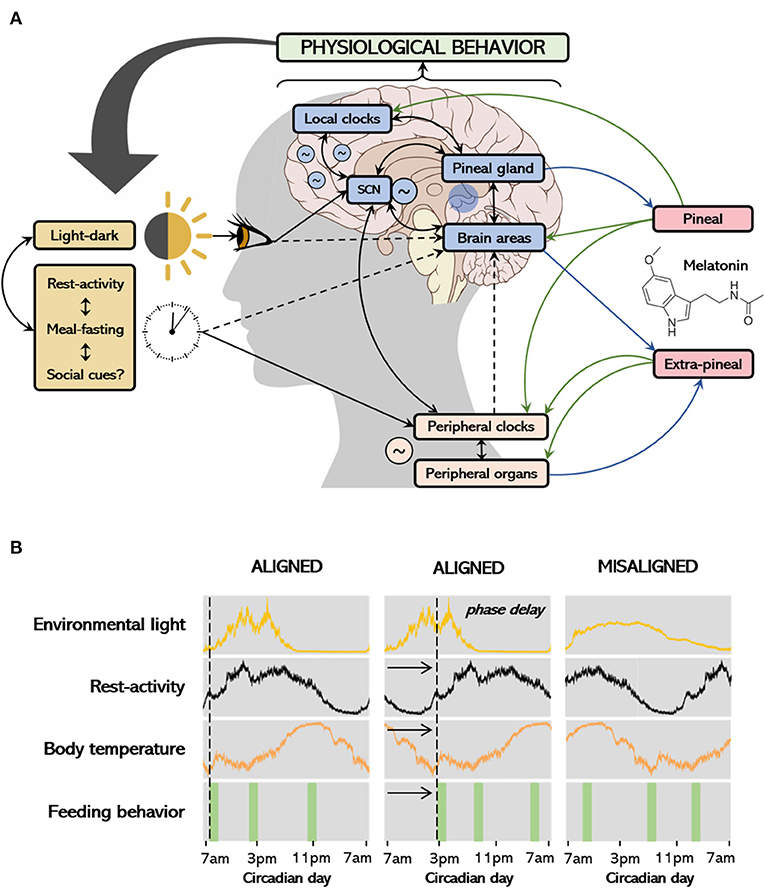
Figure 1. Environmental and internal regulation of biological rhythms, melatonin production and associations with behavior (A); the process of entrainment and alignment (B). The main environmental cue that synchronizes the circadian system is the light-dark cycle. The suprachiasmatic nucleus (SCN) receives photic information and projects to several structures in the body, coordinating peripheral tissues by modulating other oscillators according to light-dark transitions. Other stimuli of importance include feeding schedules, rest-activity rhythm and social cues. Peripheral tissues respond to timing neural and endocrine signals modulated by the SCN, including the release of melatonin to the bloodstream, as well as from feeding. On the other hand, endogenous circadian rhythms also influence daily patterns in behavior/exposure to environmental factors and might also influence melatonin production (see details in text). In (A), black arrows connect the environmental stimuli to the central and peripheral clocks and tissues and show the interconnection among systems. Continuous arrows represent higher levels of evidence and dashed arrows represent lower levels of evidence. Melatonin is produced in the pineal gland, as a result of darkness or on-demand via other pathways (see details in text), acting locally or transducing the dark signal to peripheral tissues (green arrows). Extra-pineal sources of melatonin include the skin, guts, and lungs, which only act locally with no known chronobiotic effect. While the physiological actions of melatonin potentially impact behavior, individual behavior might also determine the timing of light exposure, rest-activity rhythms and feeding behavior, thus regulating melatonin secretion. The first column at (B) shows rhythms of activity, temperature, and feeding behavior aligned with the light signal; the second depicts rhythms which are phase delayed. The rhythms at the third column are no longer entrained to the light signal and are not synchronous within themselves, determining a misalignment.
Considering the cyclic nature of mood disorders, the shared monoaminergic involvement, the data from seasonal depression, and the marked sleep and circadian changes in depressive disorders, several hypotheses have been raised to understand the intersections between melatonin with depression. Experimental and clinical studies were designed to understand possible pathophysiological connections and devise therapeutic strategies to optimize the pharmacological management of depressive patients. This review aims to discuss the interconnections of melatonin production pathways and its physiological actions with the etiopathology and clinical approaches to depression. We will initially describe the framework of depression relevant for the understanding its association with biological rhythms and melatonin. We proceed by discussing the role of melatonin as an internal temporal cue, its production and physiological actions, and its role as a neurohormone sharing common neuro-endocrine inputs with the pathways associated with depression. Finally, we describe the current knowledge from clinical research in the field, including the use of exogenous melatonin and urinary 6-sulfatoxymelatonin as treatment and response biomarker, respectively.
Depression and Biological Rhythms
What Do we Mean by Depression?
Depression is a term with multiple meanings. As a state, a depressive mood may be an adaptive stress response (6). Depressed mood states manifest as typical behavioral phenomena (e.g., anhedonia, psychomotor disturbance) and dysregulation in neurovegetative functions (e.g., altered appetite, disturbed sleep), which may be a result of a great variety of physiological events. As part of a mental disturbance, depressive symptoms can manifest in several conditions such as unipolar or bipolar mood disorder, substance abuse, and general medical disease. On the other hand, Major Depressive Disorder (MDD), as defined by international guidelines such as the Diagnostic and Statistical Manual of Mental Disorders (DSM) and the International Classification of Diseases ICD, is a syndromic construct assembled as a distinct clinical condition (7, 8). According to the fifth edition of the DSM, at least one of the two cardinal symptoms must be manifested to reach a positive diagnosis: depressed mood and/or loss of interest or pleasure in daily activities. Of the nine cognitive (e.g., impaired concentration), emotional (e.g., feelings of worthlessness) and somatic symptoms (e.g., psychomotor agitation or retardation, changes in appetite, sleep disturbances) listed as diagnostic criteria, at least five must be present during the same 2-week period, with the symptoms representing a change from previous baseline. Of note, other medical conditions that can cause the same symptoms must be excluded. MDD is one of the most burdensome disorders and the leading cause of disability worldwide (9, 10). Therefore, the assessment of the severity of the depressive syndrome (as a continuous feature) in clinical and research settings is fundamental, which typically implies the use of validated self-reported instruments and structured interviews of cognitive, physical, and psychomotor changes. Though sadness and anhedonia are the cardinal symptoms of MDD, the multivariate features of depression, together with its multifactorial etiology and the lack of a reliable biomarker, can explain the individual differences in pharmacological response.
Clinical response is usually noticeable after 2–5 weeks (11–13). The poor or absent response to antidepressants that is associated with the morbidity and mortality of MDD justifies the search for improvement in diagnosis, while searching for subtypes of depression that show treatment idiosyncrasies. In addition, preventing the relapse and recurrence of clinically significant symptoms is essential for new treatment approaches and/or the identification of markers of response to established treatments. In fact, several studies document a vast heterogeneity of clinical features of depressive disorders, indicating that specific symptoms represent different physiological phenomena, boosting the search for specific diagnostic and treatment biomarkers and unraveling new therapeutic strategies (14). In this context, despite inherent limitations, animal models have been instrumental in the investigation of subjacent neuronal mechanisms associated with the genesis, trigger factors, and maintenance of depressive states, as well as potential treatments.
The difficulties in modeling depression and neuropsychiatric disorders in experimental non-human species may, in part, be attributed to the variability of depressive symptoms (across and within individuals), and the sheer impossibility to reproduce in non-human animals some important features, such as guilt and suicidality. This somehow mirrors the challenges of Psychiatry as a field in categorizing debilitating disorders (with overlapping symptoms and varied underlying mechanisms) as opposed to normal behavior. Chronic stress, maternal deprivation, and olfactory bulbectomy are among the models that present higher degrees of face validity (i.e., animal behavior correlates with MDD symptomatology), predictive validity (i.e., good chances of replicating the positive or negative results in human patients), and construct validity (i.e., consistent underlying pathology biomarkers, triggering factors, time course, and response to antidepressants). The majority of animal models only replicate anhedonia (with varying degrees of face value), which is certainly one of the core symptoms of depression. The widely used tail suspension and forced swimming tests have no pathophysiological or undisputed behavioral manifestations equivalent to those seen in depression but possess some degree of translation predictability. It is of utmost importance to select the model appropriate for a given question, and interpret results considering specific limitations.
Are Biological Rhythms Related to Depression?
Although the challenges of assessing behavior pose inherent limitations to both experimental and clinical studies, the relationships between depressive-like behavior and biological rhythms have been abundantly documented. Nile grass rats (Arvicanthis niloticus), a diurnal rodent, exposed to short photoperiod (5 h light/19 h dark) for 6 weeks presented reduced saccharin preference, higher immobility time in the forced swim test, and no changes on time spent in the light side of the dark/light box in comparison to controls (15). The same species submitted to low light intensity during the daytime (cycles of dim light of 50 lux during 12 h/dark during 12 h) for 4 weeks showed increased immobility and decreased climbing in the forced swim test; again, no effects were observed in the light/dark test or locomotion in the open field compared to bright light controls. Nile grass rats also show lower dopaminergic and somatostatin neurons in the hypothalamus under both short photoperiods and dim light/dark cycle (16). In nocturnal mice, dim light at night [16 h of light (~150 lux) and 8 h of darkness (5 lux)] for 4 weeks lead to depression-like behaviors such as increased immobility in the forced swim test and reduced sucrose preference (17). In addition, nocturnal rodents under constant light for 3–4 weeks, a light regimen known to render rats behaviorally arrhythmic (18), exhibited significant higher number of grooming events during the open field test, and diminished sucrose preference throughout the study (19). Under forced desynchrony (light/dark cycles of 22 h), a protocol known to uncouple neuronal oscillators in the suprachiasmatic nucleus (SCN; see Figure 1), rats develop into depressive-like phenotypes (i.e., anhedonia, sexual dysfunction, increased immobility in the forced swim test) (3).
Landgraf et al. (4) showed that knocking down Bmal1 (one of the essential clock genes that participate in the maintenance of the circadian periodicity at the cellular level) in mice suprachiasmatic nucleus (SCN) led to increased escape latencies and number of escape failures in the learned helplessness paradigm, induced higher immobility time in the tail suspension test, and decreased the time spent in the light compartment of the light/dark test. These results indicate that disrupting central SCN rhythms causes helplessness, behavioral despair, and anxiety-like behavior. The experiment supports a causal relationship because environmental light-dark cycles and light input pathways were unchanged. Therefore, results cannot be explained by light affecting depressive-like behavior through other pathways [i.e., without disturbing sleep or circadian rhythmicity, or through pathways unrelated to the SCN (20)]. Unlike in global knockouts in which mice may also present altered behavior related to pleiotropic functions of the clock genes, the model has the advantage of selectively inducing disturbed SCN rhythms while avoiding other neural damages caused by SCN lesions. Moreover, hedonic behavior measured by the sucrose preference test, spatial preference in the open field test, and aversion to eating in a novel environment were unchanged, suggesting that only specific aspects of depressive-like behavior are influenced by the disruption of the central clock (4). Of note, neurotransmitter systems altered in depression show blood levels circadian oscillations (21, 22), though it is unclear whether or how mood states could affect this circadian pattern.
Clinical evidence supports that dysfunctions of the circadian timekeeping system are present in a variety of morbid clinical conditions, such as obesity, diabetes, hypercholesterolemia, cardiovascular diseases, and cancer (23, 24). Mood alterations have also been extensively studied in models of circadian disruption or misalignment (see Figure 1). Of relevance to this discussion, depressive symptomatology can assume seasonal variation (i.e., seasonal affective disorder), and is associated with non-respiratory sleep disturbances and distinguishable daily motor activity patterns (i.e., comparing depressed individuals with non-depressed individuals and among depressive subtypes). Diurnal variations in mood have been seen in naturalistic conditions (25) and experimental results suggested mood to be affected by an interaction between circadian phase and duration of prior wakefulness (26). A few initiatives arouse in the past decade aiming to elucidated whether altered patterns of diurnal variations in mood are associated with depressive disorders. These reports show that individuals with depression are more likely to report diurnal variations in mood (27, 28).
Acute or chronic dysfunctions of the circadian timekeeping system (e.g., Eastbound travels across time-zones and rotating night shift workers, respectively) impact the organism with consequences at different biological scales, i.e., cellular, tissue, and systemic level. In the light of the above, circadian misalignment can result from: (1) damage to cerebral structures like the retina, the retino-hypothalamic tract or the SCN; (2) genetic manipulation or variations in clock and clock-controlled genes; (3) a response to external factors (zeitgebers) in situations of rotating night shift work, travel across time zones, lack of light exposure during daytime or the excessive exposure during the night. All these potentially impact the production of pineal melatonin, an important temporal cue of biological rhythms.
Melatonin: The Neuroendocrine Timing Signal
The main environmental cue that synchronizes biological rhythms with the environment is the light-dark cycle. The SCN, a small collection of hypothalamic cells just above the optic chiasm, receives environmental photic information collected by intrinsically photosensitive ganglion cells (ipRGC) in the retina. The ipRGC expresses the photopigment melanopsin, which transduces light wavelengths into neural input through the retino-hypothalamic tract to the SCN. The SCN projects to several structures, including the paraventricular hypothalamic nucleus that communicate with the intermediolateral spinal column (29). The light/dark information reaches the pineal gland through sympathetic postsynaptic fibers of the superior cervical ganglion (30), and melatonin is simultaneously released to the peripheral circulation and to the cerebrospinal fluid (CSF) (31). It is the daily pattern of melatonin secretion that carries information for circadian and seasonal temporal organization (32). Melatonin is also synthesized by the skin, guts, and lungs in a constitutive manner, and on-demand by activated immune-competent cells, such as monocyte-derived and resident macrophages, microglia and lymphocytes (1, 33, 34).
It is currently impossible to monitor the central clock (SCN activity) directly in humans. Since the timing of melatonin secretion is strongly regulated by the SCN, the onset of melatonin secretion under dim light (dim light melatonin onset; DLMO) has been considered a gold standard to assess the central clock's phase and whether it is misaligned in relation to other internal rhythms or environmental cycles. A fair number of studies have investigated the relationship between depressive symptoms and the phase angle difference between DLMO and the timing of other rhythmic functions, including sleep (35, 36). Results are heterogeneous, which may reflect the multifactorial nature of depression and the multiple actions and regulatory systems related to melatonin. Aside from phase shifts, lower nocturnal melatonin levels are often, though not always, reported in depressed individuals (37–39).
Even though the molecular and mechanistic underpinnings are yet to be unveiled, the way the clock, sleep and behavior co-exist and co-affect each other seems crucial to balance health and disease [see (40) for discussion]. In the context of mood disorders, we still need to identify sub-optimal phase relationships and other altered circadian states that may affect the susceptibility to develop specific depressive-like behaviors. Melatonin—as the important phase marker and eigen-zeitgeber it is—is likely to remain a central player in this framework.
Melatonin Production and Physiological Actions
What Is the Pathway of Melatonin Production?
In mammals, the biosynthesis of melatonin starts with the conversion of tryptophan to l-5-hydroxytryptophan, which is converted to serotonin [5-hydroxytryptamine(5-HT)] by the aromatic l-amino acid decarboxylase (AADC). Serotonin is acetylated by the phosphorylated arylalkylamine N-acetyltransferase (P-AANAT), forming N-acetylserotonin (NAS), which is converted to melatonin by N-acetylserotonin O-methyltransferase (ASMT). The melatonin pineal daily rhythm is determined by the conversion of 5HT to NAS under sympathetic control (Figure 2). Environmental light modifies the structure of melanopsin in the retinal ganglion cells triggering glutamate excitation at the retino-hypothalamic tract that projects to the SCN. The SCN inhibits the hypothalamic paraventricular nucleus (PVN) via GABAergic projection. In the absence of light, the PVN activates the ganglion cervical nuclei (via the intermediolateral column of the medulla) activating noradrenergic fibers that innervate the pineal gland, ultimately releasing the co-transmitters noradrenaline and ATP (41). Consequently, the production of melatonin depends on the integrity of the brain monoaminergic system (2). Beta-1 adrenergic receptor activation leads to increase in cAMP and activation of protein kinase A, which promotes the phosphorylation of the cyclic AMP regulating element (CREB) and the phosphorylation of AANAT (P-AANAT). Phospho-CREB induces the transcription of the gene that codifies AANAT (the native form of the enzyme), which is immediately degraded by the proteasome. In nocturnal animals both the control of transcription and activation of AANAT plays an important role in melatonin synthesis, while in diurnal animals the transcription of the gene is constitutive (1); thus, nocturnal melatonin surge is delayed in nocturnal rodents when compared to humans. Circulating melatonin levels in the bloodstream accurately reflects pineal synthesis (2). At dark-night the pineal melatonin plasma levels show a 10–20-fold increase in normally entrained individuals. When light is turned on in the middle of the dark night, the pineal melatonin synthesis stops, and melatonin concentration in the blood is abruptly reduced due to liver metabolization (first-pass effect). In the liver, melatonin is metabolized to 6-sulfatoxymelatonin (aMT6s), excreted in the urine, which is well-correlated with plasma melatonin (Figure 2) (42).
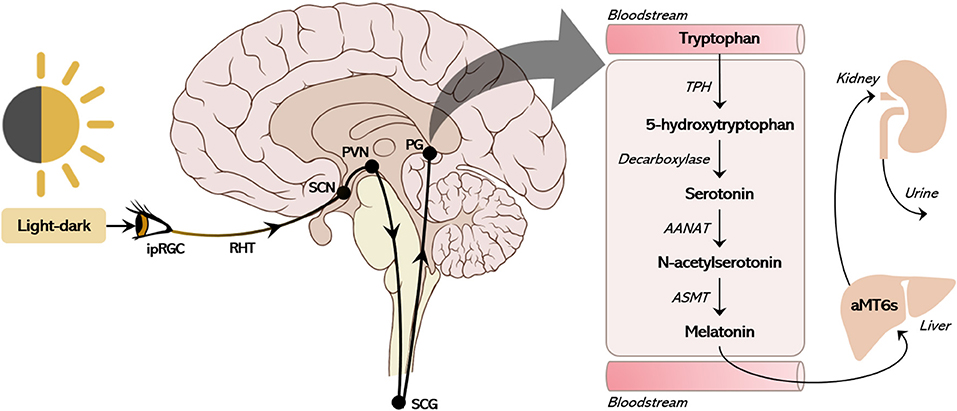
Figure 2. The canonical pathway of pineal melatonin production. The suprachiasmatic nucleus (SCN) receives environmental photic information collected by intrinsically photosensitive ganglion cells (ipRGC) in the retina. The ipRGCs express the photopigment melanopsin, which transduces light wavelengths into neural input through the retinohypothalamic tract (RHT) to the SCN. The SCN constitutively inhibits the hypothalamic paraventricular nucleus (PVN) via GABAergic projection. In the absence of light, the PVN activates the ganglion cervical nuclei (SCG, via the intermediolateral column of the medulla) triggering noradrenergic fibers that innervate the pineal gland (PG), ultimately releasing the co-transmitters noradrenaline and ATP. This sympathetic stimulus triggers the action of arylalkylamine N-acetyltransferase (AANAT) converting serotonin (5TH) into N-acetylserotonin (NAS) within the pinealocyte. With the constitutive action of N-acetylserotonin O-methyltransferase (ASMT), NAS is then converted to melatonin and immediately released into the cerebrospinal fluid and bloodstream. Through first-pass metabolism in the liver, melatonin is converted to 6-sulfatoxymelatonin (aMT6s), which is then excreted in the urine.
How Is Melatonin Production Regulated?
The pineal gland is a circumventricular organ that monitors the level of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) in the cerebrospinal fluid (CSF) and peripheral circulation. In pinealocytes, the activation of the NFκB pathway by toll-like receptors and TNFRs inhibits AANAT-gene transcription both in diurnal and nocturnal animals. Cortisol or corticosterone at concentrations compatible with arousal (but not the ones found in models of chronic unpredictable mild stress) potentiate melatonin synthesis, while concentrations compatible with immune suppression blocks it. In immune-competent cells PAMPs and DAMPs promote the synthesis of melatonin by activating the transcription of the gene that codifies AANAT. This process is also dependent on the binding of NFkB dimers to the gene promoter, but it is not an immediate effect. Activation of PAMPs or DAMPs receptors promote the nuclear translocation of the NFkB dimer (p50/p65) that leads to the synthesis of the subunit (cRel) necessary for activating AANAT transcription. The induction of melatonin synthesis in rodents and human macrophages by the transcription factor is a hallmark in inflammatory responses and inflammation (43–46). The fact that stressful conditions increase extra-pineal melatonin synthesis independently of environmental light strongly suggests that the amplitude of daily melatonin rhythm, classically attributed to reduction of nocturnal melatonin synthesis, may also result from an increase of the output from extra-pineal sources (1).
During the mounting of an innate immune response melatonin plays various roles: the reduction of nocturnal melatonin leads to the mobilization of leukocytes from the bone marrow to the blood, and from the blood to the site of lesion; the synthesis of melatonin by macrophages/microglia increases its phagocytic ability and reduces the oxidative stress participating in the recovery phase of the inflammatory response (1). The return of macrophages/microglia/phagocytes counts to baseline requires the return of mediators' levels active at the recovery phase, including melatonin, to basal values. When the recovery from the acute inflammatory response is mediated only by pineal activity return to baseline, without restoration of macrophage melatonin synthesis to baseline, the result is a resilient state (low-inflammatory grade).
Over the past decades, studies reported neuroinflammatory responses in a series of neurodegenerative and psychiatric disorders. Intense research has been undertaken to determine the critical elements of such responses and putative therapies for their suppression. Some reports associate MDD to increased levels of cytokines including TNFα, IL-6, and IL-1β, as well as a reduction in complement C3 (47–49). Increased immune-inflammation, with high oxidative and nitrosative stress leading to changes in neuronal regulating tryptophan catabolites (TRYCATs) and mitochondrial dysfunction, have been documented in the progressive course of MDD (50, 51). In animal models, melatonin treatment significantly abolished the effects of LPS and reduced NF-κB in the cortex and the hippocampus, both effects resulting in an improvement of depressive-like behaviors (52). These results point to the possibility of an “antidepressant effect” of melatonin via the interplay with the immune system. Furthermore, as described above, cortisol functioning and its circadian fluctuation is essential for the adequate melatonin surge. Blunted cortisol rhythms (i.e., lower morning cortisol peak and higher daytime values) have been demonstrated in association with depressive symptoms in humans (49, 53, 54).
How Does Melatonin Exert Its Physiological Actions?
Melatonin effects triggered by a large range of concentrations (pM to mM) are mediated by mechanisms of action with different grades of sensitivity. The effects of exogenous vs. endogenous melatonin, as well as the concentration reached by intracrine/autocrine, paracrine and hormonal sources, depend on the melatonin bioavailability and the mechanisms that mediate each production source. In a concentration range compatible with the nocturnal surge (pM to nM), melatonin orchestrates a plethora of physiological functions by activating GPCRs receptors (MT1 and MT2) localized in plasmatic, nuclei and mitochondrial membranes. At much higher concentrations, melatonin may act as an electron donor, promoting a receptor free non-specific antioxidant response (55). Other non-receptor mediated actions include the regulation of clock genes expression (by directly inhibiting the proteasome), and inhibition of the ubiquitin–proteasome system that ultimately controls protein degradation.
Melatonin receptors MT1 and MT2 couple to Gi/o and beta-arrestins and depending on the context also couple to Gq (55). The second messenger immediate responses result in an increase/decrease of cAMP or increase in intracellular or intramitochondrial free calcium (56). The internalization of the receptors upon stimulation leads to the activation of ERK pathways, which modulate intranuclear responses. MT1/MT2 receptors form heterodimers with pharmacological properties different from MT2 homodimers. The heterodimerization of the MT1 receptor with the orphan receptor GPR50 impairs its interaction with melatonin (57). Thus, another source of variability of melatonin response are the changes in the expression and the dimerization of melatonin receptors induced by pathophysiological and pathological states.
Pineal melatonin exerts a wide range of physiological actions due to its release into the cerebrospinal fluid (CSF) and bloodstream. As a hormone that gains the bloodstream at night in the dark, melatonin is an internal temporal cue that has immediate interactions with its molecular effectors as well as prospective effects, since it primes physiological responses that will take place hours after its peak. The duration, timing, and cyclic-nature of pineal melatonin production, as well as its seasonal variation according to photoperiod changes, contribute to the temporal organization of physiological phenomena into circadian and circannual timescales (58). Finally, transgenerational effects have been described, as maternal melatonin reaches the fetus via placenta and constitutes the only fetal source of intrauterine melatonin. As previously stated, extra-pineal melatonin shows immediate effects in the cells and tissues where it is produced, and the current knowledge does not support any eigen-zeitgeber action (i.e., being an internal temporal cue) or seasonal effect resulting from these sources. A detailed review on the physiological actions of melatonin was composed by Cipolla-Neto and Amaral (59).
Melatonin and Depression: Is There a Physiological Overlap?
How Does Melatonin Relate to the Monoamine and Neurotrophic Hypotheses of Depression?
Since the report of two pharmacological agents in the late 1950s, the pathophysiology of MDD has been linked to a depletion in monoaminergic neurotransmitters, such as noradrenaline, serotonin and dopamine (5, 60). Initial studies showed that severe depressive episodes could be effectively treated with monoamine oxidase inhibitors (MAOi) and tricyclic antidepressants (TCA), which share increased availability of brain catecholamines as a common pathway. The involvement of other monoamines has been studied, with growing evidence to the role of serotonin with the introduction of Selective Serotonin Reuptake Inhibitors (SSRIs) in the 1960s.
Since melatonin production occurs from serotonin after stimulation of adrenoreceptors (see section What Is the Pathway of Melatonin Production? above), it has been hypothesized that a disturbance in melatonin secretion could be present in the acute phase of depressive illness, and possibly be related to its pathophysiology. Moreover, melatonin controls dopamine signaling in the forebrain, hypothalamic and hippocampal areas (61–63). Of note, dopamine is not only the precursor of noradrenaline, but also has been implicated in the circadian regulation of melatonin production (64), via activation of dopamine D4 receptors by noradrenergic signals (65).
Several clinical studies tried to identify alterations in absolute levels of melatonin as a possible maker of depressive states; considering the above detailed physiological regulation of melatonin, this was a misleading proposition, and the evidence is at best controversial. While some earlier studies reported lower nocturnal serum or saliva levels of melatonin in depressed individuals, others reported no differences, or even higher levels compared to controls (39). One study (38) reported that individuals with levels of melatonin in the two lowest quartiles had higher risk of severe symptomatology; nevertheless, the authors described similar levels of nocturnal melatonin in depressed patients and controls. Studies that investigated the nocturnal production of 6-sulfatoxymelatonin (aMT6s) found no difference between depressed individuals and controls before treatment (66, 67), suggesting the absence of a linear relationship between depressive behavior and melatonin levels. One study (37) found lower bedtime levels of plasma melatonin in depressed individuals with melancholic features; others (68, 69) indicated that subgroups of depressed individuals who do not suppress morning cortisol after dexamethasone stimulation test are more likely to present lower nocturnal melatonin. These data indicate that the lack of agreement in findings on melatonin levels in depressed individuals may be a result of (1) the diversity of behavioral and etiological phenomena in depressive states [as previously commented (14)]; (2) methodological heterogeneity, like inconsistency in the methods for melatonin assessment, difficulties in complying with the protocol of melatonin collection, and the distinctions among study designs; and (3) the erroneous premise that (lack of or excessive) melatonin could contribute or counteract (as antidepressant) depression needs to evolve to the understanding that melatonin is an integrative molecule which potentially affects mood through regulation of physiology and behavior in several levels.
Though the immediate mechanisms of action of current antidepressants are well-understood, the delay in clinical outcome conveys the gap of knowledge on the complete pathways that ultimately determine the clinical response. This delayed response shifted theories toward the neuroplastic hypothesis, whereby reduced levels of neurotrophic factors result in atrophic morphological changes, especially at the synaptic level (5). Animal studies that examine the neurobiological changes induced by experimental models that lead to depressive-like behaviors highlighted the relevance of the neuroplasticity phenomena in depression (70). The activation of cellular signaling pathways by antidepressant strategies would eventually elevate the levels of growth factors (e.g., BDNF, VEGF, VGF), promote proliferation on hippocampal progenitor cells, and ultimately restore monoaminergic synapses (70–72).
Melatonin exerts neuroprotective actions by counteracting the NMDA-mediated excitotoxic effects of glutamate, including the impaired BDNF signaling and cell death (73). Exogenous melatonin can differentially modulate glutamate release in CNS structures in rodents (74), suggesting it might correct the imbalance in the glutamatergic system in patients with mood disorders. This body of evidence suggests that melatonin may increase neuroplasticity in the CNS acting on the glutamatergic system.
Can Urinary 6-Sulfatoxymelatonin (aMT6S) Be a Treatment Biomarker?
Biomarkers are easily accessible molecules that discriminate the presence or absence of a disease or predict treatment response. They are based on genomic, proteomic, functional, and structural features that characterize the disorders. “Diagnostic biomarkers” identifiable at any stage of the illness are often measured before treatment. The term “treatment biomarkers” refers either to baseline values that predict how effective treatments may be and guide therapeutic choice, or short-term variation between baseline and an early phase in the course of therapy (75).
A promising group of “treatment biomarkers” for SNRI/SSRI search for consistent changes in monoamines neuronal reuptake inhibition associated with subsequent mood changes (usually after 4–6 weeks). To measure the blockage of neuronal monoamine transporters directly, it would be necessary to assess changes in an immediate molecular product on the postsynaptic level that directly results from the neurotransmitter release (see section What Is the Pathway of Melatonin Production?). We proposed the assessment of 6-sulfatoxymelatonin (aMT6s, Figure 2) to detect whether the blockage of noradrenaline neuronal reuptake on the first day of medication took place. This can be done by using the relationship between the morning aMT6s content 1 day before and 1 day after the installment of the therapy. This procedure is not expected to evaluate the implication of melatonin or the pineal gland in the mechanism of action of the antidepressants, but rather to produce a proxy of the immediate events that follow the consequences of antidepressants at synaptic level. The same protocol could theoretically be used for both SNRIs and SSRIs, as the pinealocyte function depends on the uptake of serotonin, which is mediated by the transporter found in neuronal membranes.
Because the difference in aMT6s urine concentration in the light and dark phases reflects pineal activity, it has been suggested that an increase in the nighttime production of aMT6s after the first dose of antidepressant medication could work as a fingerprint of the integrity of this system; thus, it could predict a later improvement in depressive symptoms (76). A first study to test that hypothesis evaluated the association of a “aMT6-SNRI biomarker” and the improvement of depressive symptoms in MDD patients taking placebo (n = 12) or antidepressants (n = 22; fluoxetine, duloxetine or Hypericum perforatum) for 8 weeks. Both placebo and SNRIs groups showed improvement in depressive symptoms but only the group treated with the drug showed a significant increase in aMT6 urinary excretion in the first urine of the day (77). In patients taking clomipramine, the fraction of aMT6s excreted from 24:00 to 06:00 relative to the total amount excreted in 24 h was significantly higher 24 h after the first dose than at baseline (before treatment) in responders in comparison to non-responders (78). The same pattern was seen in 22 women taking nortriptyline (79), and 20 women taking fluoxetine (80): only responders had a significant increase in aMT6s levels. Altogether these studies suggest that the “aMT6 urinary biomarker” reveals whether the blockage of neuronal uptake was effective on the first day of administration, thus representing that aMT6 reflects the integrity of the monoaminergic systems that are required for antidepressant action. The advantage of this method over those that measure changes in the content of monoamines in leukocytes, platelets, or even plasma is the direct estimation of the increase of neurotransmission efficiency that is the inductor of neuronal plasticity.
Do Exogenous Melatonin and Melatonin Agonists Have Therapeutic and/or Prophylactic Effects on Depression?
In a series of elegant experiments, Nagy et al. (81) showed that de 24-h pattern of the dorsal raphe 5-HT reuptake in C57BL/6J mice under shortened photoperiods may be altered at the transcriptional level by specifically timed melatonin. The data suggest that daily melatonin treatment can induce and sustain receptor 5HT1A mRNA expression throughout the light phase. In the same line, Otsuka et al. (82), using the same mice, showed that daily melatonin treatments 2 h before the end of the light phase can restore the amplitude of the daily rhythm of 5-HT contents in the amygdala. In models with higher face and construct validities, such as the chronic unpredictable mild stress (CUMS), melatonin at high concentrations (10 mg/kg) showed antidepressant-like effects, preventing the CUMS-induced decrease in norepinephrine content and the expression of tyrosine hydroxylase, dopamine-b-hydroxylase and norepinephrine transporter in the adrenal medulla (83), as well as depressive-like behaviors, such as impaired sucrose intake, physical coat deterioration, and decreased grooming (84, 85).
Overall, there is substantial heterogeneity in the body of clinical evidence regarding the prophylactic or therapeutic use of exogenous melatonin or melatonin agonists in depression, complicating a conclusive assessment. Different methodological approaches and small convenience samples precludes robust comparisons among studies, as there is no consensus on dosing and timing. Moreover, it is known that the secretion of pineal melatonin shows a wide interindividual difference as well as significant dependence on the exposure to dark each night (86); thus, therapeutic doses or an optimal serum levels are yet to be determined.
A recent systematic review (87) included eight randomized double-blind controlled trials using exogenous melatonin as an augmentation strategy in major depression, bipolar disorder or seasonal affective disorder in comparison with placebo. The dosage of exogenous melatonin ranged from 0.125 to 10 mg. Among the three studies that evaluated melatonin in the context of depression, one showed that melatonin improved subjective sleep quality but not depressive symptoms [(88), using 5–10 mg slow release melatonin], the second showed no effect [(89), using 6 mg] and the third compared the use of 3 mg slow-release melatonin plus 15 mg buspirone vs. 15 mg buspirone and placebo, showing a significant antidepressant effect of the melatonin combination (90). Of the four studies using melatonin for SAD, one (91) showed significant antidepressant effect (using 0.125 mg twice daily), while the other three did not show significant effects (92–94). Another systematic review (95) pooled results of clinical trials testing the prophylactic or therapeutic effect of melatonin for depression in adults, including comorbid conditions. Among the three studies that tested prophylactic melatonin, one study with older adults with sleep complaints showed lower depressive scores after supplementation with 5 mg melatonin at bedtime. Two other studies were in individuals with irritable bowel syndrome and found no antidepressant effect of 3 mg melatonin. Of the studies testing melatonin as a treatment for depression, one showed a decrease in depressive scores in individuals with Delayed Sleep Phase Syndrome treated with melatonin 5 mg between 19:00 and 21:00; the other six found no significant antidepressant effects. Adverse effects reported in these studies include mild sleepiness, headache, poor sleep, vivid dreams, daytime sleepiness, and fuzzy feeling.
Following the initial evidence on the potential antidepressant effect of melatonin, efforts have been directed toward the development of antidepressants with melatonin agonist activity. Among them, agomelatine (S20098, N-[2-(7-methoxynaphth-1-yl)ethyl]acetamide) first reported in 1992, is by far the best studied. This synthetic molecule has high affinity for MT1 and MT2 receptors, but also present moderate affinity for the serotonin receptor 5HT2C. There is few clinical evidence of the effect of agomelatine in reducing overall depressive symptoms: results show agomelatine's higher response and lower remission rate compared to placebo (96, 97) and its similar efficacy compared to other antidepressants (i.e., paroxetine, fluoxetine, sertraline, escitalopram, and venlafaxine) (97, 98). However, most studies did not show superior or additional effects. Agomelatine seems to be better tolerated than other antidepressant agents, as no significant adverse events have been reported; furthermore, it did not show significant discontinuation symptoms. Some studies suggest agomelatine offers benefits for initial insomnia, improvement of sleep quality and efficiency, reduced daytime sleepiness, but no changes in sleep architecture of depressed patients (97). In one study (99), agomelatine was used in difficult-to-treat and refractory patients showing a significant improvement after 12 weeks. Of note, samples are overall composed of mild to moderate cases of depression. In addition, most studies compared agomelatine with low doses of other antidepressants. Finally, the evidence on melatonin or agomelatine efficacy in preventing or treating SAD is highly controversial (100), with no solid support for its prescription.
The knowledge of clinical use of melatonin is growing, with evidence supporting the prescription of exogenous melatonin for a few clinical situations. In the case of adult chronic insomnia, a few studies show modest benefits for sleep onset, sleep latency and total sleep time, although clinical significance is still questionable (101, 102). For this recommendation, typical doses are in the range of 1–5 mg. Melatonin is also currently recommended as an adjuvant treatment of insomnia in children and adolescents, particularly in those with comorbid ADHD or autism (103). In this case, the prescribed dose should be initially of 0.2–0.5 mg, 3–4 h prior to bedtime, with progressive increase up to 3 mg for children or 5 mg for adolescents (104).
Circadian Rhythm Sleep-Wake Disorders are a set of clinical conditions to which melatonin are classically indicated. For sleep disturbances in shift workers, the readjustment to nighttime sleep following a night work shift or the promotion of desired daytime sleep can be reached with doses of no more than 1–3 mg about 30 min prior to the desired sleep onset (105, 106). In the case of jet lag syndrome of eastward trips, exogenous melatonin can promote the adjustment of sleep phase when taken at the desired destination bedtime (107, 108). For individuals with Delayed Sleep-Wake Phase Disorder, 0.5 up to 5 mg melatonin scheduled at ~1.5–2 h prior to habitual bedtime significantly advanced sleep onset (109–111). Finally, 0.5 mg of melatonin either 1 h prior to a preferred bedtime or at a fixed time can hasten synchronization of individuals with 24-h Sleep-Wake Rhythm Disorder (107).
In conclusion, the evidence supporting the clinical use of melatonin should be analyzed very carefully. A potential therapeutic effect of melatonin for mood disorders can only be expected if compatible with the physiological regulation of melatonin detailed in this review. Pineal melatonin is a neuro-endocrine message of darkness that is physiologically secreted in humans after a few minutes in the dark, peaking shortly after, serving as an internal temporal cue to cells, tissues, and systems. Extra-pineal production of melatonin happens in a constitutive manner, and on-demand production or suppression may also be regulated by levels of glucocorticoids, and different stages of an inflammatory response triggered by immune-competent cells. Hence, the optimization of potential therapeutic uses of melatonin in psychiatric disorders must encompass the specificities of an integrative molecule inextricably linked to entrainment, metabolism, immunity, neurotransmission, and cell homeostasis.
Conclusion and Perspectives
Localizing phenomena, dissecting underlying mechanisms, and decomposing systems to understand their functioning have been helpful strategies in building knowledge in Biology. The downside of tackling questions in such a manner is that one might overlook how interconnected systems are, and thus lose perspective of the whole. Therein lies the challenge of decomposing complex regulatory structures like the timekeeping and melatonergic systems.
Pineal melatonin is synthesized in the absence of light (typically at night) under sympathetic control. Its production relies on the neurotransmitter norepinephrine and requires serotonin as precursor, monoamines linked to depression. The rhythmic production of pineal melatonin is a message of darkness to the body, which aids in synchronizing oscillations in physiological functions and behaviors. Nevertheless, melatonin may be implicated in the pathophysiology of depressive mood for its chronobiotic effects and the various immunological processes in which it is involved. A growing number of studies highlight the relevance of exploring the potential of melatonin extra-pineal synthesis, as well as the regulation of pineal melatonin production apart from the circadian light stimuli. Therefore, the study of melatonin in the context of depression must acknowledge its regulatory effect via chronobiotic (endocrine), paracrine and autocrine functions. It is crucial to acknowledge that detectable circulating levels of melatonin are a reflection of its amplitude and phase of secretion.
Studies of the pharmacodynamics of exogenous melatonin or melatonin agonists should always anticipate distinct clinical outputs depending on the route of administration, dosage, and timing, as these substances would potentially represent (or interfere with) different physiological actions. The pleiotropic actions through which melatonin might affect mood include the role of melatonin as an internal temporal cue, as a neurohormone in close relation with the monoaminergic system, and its indirect effects on depression via the immune and stress response systems. In this context, the resulting effects of melatonin actions on mood must be understood as a more complex and multifactorial pattern of systemic regulation, rather than an “antidepressant” effect per se.
Author Contributions
AT and EE reviewed and edited the final manuscript. MH was responsible for funding acquisition. All authors participated in the conceptualization and the writing of the original draft.
Funding
The authors wish to acknowledge funding the Research Support Fund at Hospital de Clínicas de Porto Alegre (FIPE-HCPA) MH, Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2013/13691-1) RM. The following received fellowships: AT and LP from Higher Education Personnel Improvement Coordination (CAPES); 305331/2012-4 PQ/CNPq to MH; senior fellowship to RM from CNPq.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Markus RP, Fernandes PA, Kinker GS, Cruz-Machado SdS, Marçola M. Immune-pineal axis – acute inflammatory responses coordinate melatonin synthesis by pinealocytes and phagocytes. Br J Pharmacol. (2018) 175:3239–50. doi: 10.1111/bph.14083
2. Simonneaux V, Ribelayga C. Generation of the melatonin endocrine message in mammals: a review of the complex regulation of melatonin synthesis by norepinephrine, peptides, and other pineal transmitters. Pharmacol Rev. (2003) 55:325–95. doi: 10.1124/pr.55.2.2
3. Ben-Hamo M, Larson TA, Duge LS, Sikkema C, Wilkinson CW, de la Iglesia HO, et al. Circadian forced desynchrony of the master clock leads to phenotypic manifestation of depression in rats. eNeuro. (2016) 3. doi: 10.1523/ENEURO.0237-16.2016
4. Landgraf D, Long JE, Proulx CD, Barandas R, Malinow R, Welsh DK. Genetic disruption of circadian rhythms in the suprachiasmatic nucleus causes helplessness, behavioral despair, and anxiety-like behavior in mice. Biol Psychiatry. (2016) 80:827–35. doi: 10.1016/j.biopsych.2016.03.1050
5. Hasler G. Pathophysiology of depression: do we have any solid evidence of interest to clinicians? World Psychiatry. (2010) 9:155–61. doi: 10.1002/j.2051-5545.2010.tb00298.x
6. Institute of Medicine (US) Committee on Nervous System Disorders in Developing Countries. Neurological, Psychiatric, and Developmental Disorders: Meeting the Challenge in the Developing World. Washington, DC: National Academies Press (US) (2001). Available online at: http://www.ncbi.nlm.nih.gov/books/NBK223475/ (accessed February 18, 2021).
7. ICD-11. Available online at: https://icd.who.int/en (accessed December 7, 2020).
8. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington DC: American Psychiatric Association (2013).
9. GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. (2016) 388:1545–602. doi: 10.1016/S0140-6736(16)31678-6
10. Mathers C Fat DM Boerma JT World Health Organization eds. The Global Burden of Disease: 2004 Update. Geneva, Switzerland: World Health Organization (2008).
11. Fava M. Diagnosis and definition of treatment-resistant depression. Biol Psychiatry. (2003) 53:649–59. doi: 10.1016/S0006-3223(03)00231-2
12. Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. AJP. (2006) 163:1905–17. doi: 10.1176/ajp.2006.163.11.1905
13. Kennedy SH, Lam RW, McIntyre RS, Tourjman SV, Bhat V, Blier P, et al. Canadian network for mood and anxiety treatments (CANMAT) 2016 clinical guidelines for the management of adults with major depressive disorder: section 3. Pharmacological treatments. Can J Psychiatry. (2016) 61:540–60. doi: 10.1177/0706743716659417
14. Fried EI, Nesse RM. Depression sum-scores don't add up: why analyzing specific depression symptoms is essential. BMC Med. (2015) 13:72. doi: 10.1186/s12916-015-0325-4
15. Ashkenazy-Frolinger T, Kronfeld-Schor N, Juetten J, Einat H. It is darkness and not light: depression-like behaviors of diurnal unstriped Nile grass rats maintained under a short photoperiod schedule. J Neurosci Methods. (2010) 186:165–70. doi: 10.1016/j.jneumeth.2009.11.013
16. Deats SP, Adidharma W, Yan L. Hypothalamic dopaminergic neurons in an animal model of seasonal affective disorder. Neurosci Lett. (2015) 602:17–21. doi: 10.1016/j.neulet.2015.06.038
17. Fonken LK, Nelson RJ. Dim light at night increases depressive-like responses in male C3H/HeNHsd mice. Behav Brain Res. (2013) 243:74–8. doi: 10.1016/j.bbr.2012.12.046
18. Ohta H, Yamazaki S, McMahon DG. Constant light desynchronizes mammalian clock neurons. Nat Neurosci. (2005) 8:267–9. doi: 10.1038/nn1395
19. Tapia-Osorio A, Salgado-Delgado R, Angeles-Castellanos M, Escobar C. Disruption of circadian rhythms due to chronic constant light leads to depressive and anxiety-like behaviors in the rat. Behav Brain Res. (2013) 252:1–9. doi: 10.1016/j.bbr.2013.05.028
20. LeGates TA, Altimus CM, Wang H, Lee H-K, Yang S, Zhao H, et al. Aberrant light directly impairs mood and learning through melanopsin-expressing neurons. Nature. (2012) 491:594–8. doi: 10.1038/nature11673
21. Hampp G, Ripperger JA, Houben T, Schmutz I, Blex C, Perreau-Lenz S, et al. Regulation of monoamine oxidase A by circadian-clock components implies clock influence on mood. Curr Biol. (2008) 18:678–83. doi: 10.1016/j.cub.2008.04.012
22. Hampp G, Albrecht U. The circadian clock and mood-related behavior. Commun Integr Biol. (2008) 1:1–3. doi: 10.4161/cib.1.1.6286
23. Panda S. Circadian physiology of metabolism. Science. (2016) 354:1008–15. doi: 10.1126/science.aah4967
24. Sulli G, Lam MTY, Panda S. Interplay between circadian clock and cancer: new Frontiers for cancer treatment. Trends Cancer. (2019) 5:475–94. doi: 10.1016/j.trecan.2019.07.002
25. Stone AA, Schwartz JE, Schkade D, Schwarz N, Krueger A, Kahneman D. A population approach to the study of emotion: diurnal rhythms of a working day examined with the day reconstruction method. Emotion. (2006) 6:139–49. doi: 10.1037/1528-3542.6.1.139
26. Boivin DB, Czeisler CA, Dijk D-J, Duffy JF, Folkard S, Minors DS, et al. Complex interaction of the sleep-wake cycle and circadian phase modulates mood in healthy subjects. Arch Gen Psychiatry. (1997) 54:145–52. doi: 10.1001/archpsyc.1997.01830140055010
27. Oliveira MAB, Epifano K, Mathur S, Carvalho FG, Scop M, Carissimi A, et al. Validation of the English version of the mood rhythm instrument. BMC Psychol. (2020) 8:35. doi: 10.1186/s40359-020-00397-2
28. Pilz LK, Carissimi A, Oliveira MAB, Francisco AP, Fabris RC, Medeiros MS, et al. Rhythmicity of mood symptoms in individuals at risk for psychiatric disorders. Sci Rep. (2018) 8:11402. doi: 10.1038/s41598-018-29348-z
29. Lazzerini Ospri L, Prusky G, Hattar S. Mood, the circadian system, and melanopsin retinal ganglion cells. Ann Rev Neurosci. (2017) 40:539–56. doi: 10.1146/annurev-neuro-072116-031324
30. Reiter RJ. The pineal gland: an intermediary between the environment and the endocrine system. Psychoneuroendocrinology. (1983) 8:31–40. doi: 10.1016/0306-4530(83)90039-2
31. Reiter RJ, Tan DX, Kim SJ, Cruz MHC. Delivery of pineal melatonin to the brain and SCN: role of canaliculi, cerebrospinal fluid, tanycytes and Virchow–Robin perivascular spaces. Brain Struct Funct. (2014) 219:1873–87. doi: 10.1007/s00429-014-0719-7
32. Pévet P. The internal time-giver role of melatonin. A key for our health. Rev Neurol. (2014) 170:646–52. doi: 10.1016/j.neurol.2014.05.008
33. Carvalho-Sousa CE, Pereira EP, Kinker GS, Veras M, Ferreira ZS, Barbosa-Nunes FP, et al. Immune-pineal axis protects rat lungs exposed to polluted air. J Pineal Res. (2020) 68:e12636. doi: 10.1111/jpi.12636
34. Fernandes PA, Kinker GS, Navarro BV, Jardim VC, Ribeiro-Paz ED, Córdoba-Moreno MO, et al. Melatonin-Index as a biomarker for predicting the distribution of presymptomatic and asymptomatic SARS-CoV-2 carriers. Melatonin Res. (2021) 4:189–205. doi: 10.32794/mr11250090
35. Murray JM, Sletten TL, Magee M, Gordon C, Lovato N, Bartlett DJ, et al. Prevalence of circadian misalignment and its association with depressive symptoms in delayed sleep phase disorder. Sleep. (2017) 40:1–10. doi: 10.1093/sleep/zsw002
36. Robillard R, Carpenter JS, Rogers NL, Fares S, Grierson AB, Hermens DF, et al. Circadian rhythms and psychiatric profiles in young adults with unipolar depressive disorders. Transl Psychiatry. (2018) 8:1–8. doi: 10.1038/s41398-018-0255-y
37. Nair NPV, Hariharasubramanian N, Pilapil C. Circadian rhythm of plasma melatonin in endogenous depression. Prog Neuropsychopharmacol Biol Psychiatry. (1984) 8:715–8. doi: 10.1016/0278-5846(84)90044-7
38. Sundberg I, Ramklint M, Stridsberg M, Papadopoulos FC, Ekselius L, Cunningham JL. Salivary melatonin in relation to depressive symptom severity in young adults. PLOS ONE. (2016) 11:e0152814. doi: 10.1371/journal.pone.0152814
39. Ogłodek EA, Just MJ, Szromek AR, Araszkiewicz A. Melatonin and neurotrophins NT-3, BDNF, NGF in patients with varying levels of depression severity. Pharmacol Rep. (2016) 68:945–951. doi: 10.1016/j.pharep.2016.04.003
40. Roenneberg T, Merrow M. The circadian clock and human health. Curr Biol. (2016) 26:R432–43. doi: 10.1016/j.cub.2016.04.011
41. Møller M, Baeres FM. The anatomy and innervation of the mammalian pineal gland. Cell Tissue Res. (2002) 309:139–50. doi: 10.1007/s00441-002-0580-5
42. Arendt J. Melatonin and human rhythms. Chronobiol Int. (2006) 23:21–37. doi: 10.1080/07420520500464361
43. Cruz-Machado S, da S, Pinato L, Tamura EK, Carvalho-Sousa CE, Markus RP. Glia-Pinealocyte network: the paracrine modulation of melatonin synthesis by tumor necrosis factor (TNF). PLoS ONE. (2012) 7:e40142. doi: 10.1371/journal.pone.0040142
44. Carvalho-Sousa CE, da Silveira Cruz-Machado S, Tamura EK, Fernandes PACM, Pinato L, Muxel SM, et al. Molecular basis for defining the pineal gland and pinealocytes as targets for tumor necrosis factor. Front Endocrinol. (2011) 2:10. doi: 10.3389/fendo.2011.00010
45. Cruz-Machado SDS, Carvalho-Sousa CE, Tamura EK, Pinato L, Cecon E, Fernandes PACM, et al. TLR4 and CD14 receptors expressed in rat pineal gland trigger NFKB pathway. J Pineal Res. (2010) 49:183–92. doi: 10.1111/j.1600-079X.2010.00785.x
46. Cecon E, Fernandes PA, Pinato L, Ferreira ZS, Markus RP. Daily variation of constitutively activated nuclear factor kappa B (NFKB) in rat pineal gland. Chronobiol Int. (2010) 27:52–67. doi: 10.3109/07420521003661615
47. Antonioli M, Rybka J, Carvalho LA. Neuroimmune endocrine effects of antidepressants. Neuropsychiatr Dis Treat. (2012) 8:65–83. doi: 10.2147/NDT.S16409
48. Tao H, Chen X, Zhou H, Fu J, Yu Q, Liu Y. Changes of serum melatonin, interleukin-6, homocysteine, and complement C3 and C4 levels in patients with depression. Front Psychol. (2020) 11:1271. doi: 10.3389/fpsyg.2020.01271
49. Peacock BN, Scheiderer DJ, Kellermann GH. Biomolecular aspects of depression: a retrospective analysis. Compreh Psychiatry. (2017) 73:168–80. doi: 10.1016/j.comppsych.2016.11.002
50. Maes GA M. Oxidative/nitrosative stress and immuno-inflammatory pathways in depression: treatment implications. Curr Pharm Des. (2014) 20:3812–47. doi: 10.2174/13816128113196660738
51. Anderson G. Linking the biological underpinnings of depression: role of mitochondria interactions with melatonin, inflammation, sirtuins, tryptophan catabolites, DNA repair and oxidative and nitrosative stress, with consequences for classification and cognition. Prog Neuropsychopharmacol Biol Psychiatry. (2018) 80:255–66. doi: 10.1016/j.pnpbp.2017.04.022
52. Ali T, Rahman SU, Hao Q, Li W, Liu Z, Shah FA, et al. Melatonin prevents neuroinflammation and relieves depression by attenuating autophagy impairment through FOXO3a regulation. J Pineal Res. (2020) 69:e12667. doi: 10.1111/jpi.12667
53. Adam EK, Quinn ME, Tavernier R, McQuillan MT, Dahlke KA, Gilbert KE. Diurnal cortisol slopes and mental and physical health outcomes: a systematic review and meta-analysis. Psychoneuroendocrinology. (2017) 83:25–41. doi: 10.1016/j.psyneuen.2017.05.018
54. Tonon AC, Carissimi A, Schimitt RL, de Lima LS, Pereira FDS, Hidalgo MP. How do stress, sleep quality, and chronotype associate with clinically significant depressive symptoms? A study of young male military recruits in compulsory service. Braz J Psychiatry. (2020) 42:54–62. doi: 10.1590/1516-4446-2018-0286
55. Jockers R, Delagrange P, Dubocovich ML, Markus RP, Renault N, Tosini G, et al. Update on melatonin receptors: IUPHAR review 20. Br J Pharmacol. (2016) 173:2702–25. doi: 10.1111/bph.13536
56. Suofu Y, Li W, Jean-Alphonse FG, Jia J, Khattar NK, Li J, et al. Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc Natl Acad Sci USA. (2017) 114:E7997–8006. doi: 10.1073/pnas.1705768114
57. Levoye A, Dam J, Ayoub MA, Guillaume J-L, Couturier C, Delagrange P, et al. The orphan GPR50 receptor specifically inhibits MT1 melatonin receptor function through heterodimerization. EMBO J. (2006) 25:3012–23. doi: 10.1038/sj.emboj.7601193
58. Amaral FG do, Cipolla-Neto J. A brief review about melatonin, a pineal hormone. Arch Endocrinol Metabol. (2018) 62:472–9. doi: 10.20945/2359-3997000000066
59. Cipolla-Neto J, Amaral FG do. Melatonin as a hormone: new physiological and clinical insights. Endocr Rev. (2018) 39:990–1028. doi: 10.1210/er.2018-00084
60. Mulinari S. Monoamine theories of depression: historical impact on biomedical research. J History Neurosci. (2012) 21:366–92. doi: 10.1080/0964704X.2011.623917
61. Alexiuk NAM, Uddin M, Vriend J. Melatonin increases the in situ activity of tyrosine hydroxylase in the mediobasal hypothalamus of male syrian hamsters. Life Sci. (1996) 59:687–94. doi: 10.1016/0024-3205(96)00350-5
62. Di Chiara G, Morelli M, Consolo S. Modulatory functions of neurotransmitters in the striatum: ACh/dopamine/NMDA interactions. Trends Neurosci. (1994) 17:228–33. doi: 10.1016/0166-2236(94)90005-1
63. Zisapel N, Egozi Y, Laudon M. Inhibition of dopamine release by melatonin: regional distribution in the rat brain. Brain Res. (1982) 246:161–3. doi: 10.1016/0006-8993(82)90157-3
64. González S, Moreno-Delgado D, Moreno E, Pérez-Capote K, Franco R, Mallol J, et al. Circadian-related heteromerization of adrenergic and dopamine D4 receptors modulates melatonin synthesis and release in the pineal gland. PLoS Biol. (2012) 10:e1001347. doi: 10.1371/journal.pbio.1001347
65. Bailey MJ, Coon SL, Carter DA, Humphries A, Kim J, Shi Q, et al. Night/day changes in pineal expression of >600 genes central role of adrenergic/cAMP signaling. J Biol Chem. (2009) 284:7606–22. doi: 10.1074/jbc.M808394200
66. Waterman GS, Ryan ND, Perel JM, Dahl RE, Birmaher B, Williamson DE, et al. Nocturnal urinary excretion of 6-hydroxymelatonin sulfate in prepubertal major depressive disorder. Biol Psychiatry. (1992) 31:582–90. doi: 10.1016/0006-3223(92)90244-T
67. Carvalho LA, Gorenstein C, Moreno RA, Markus RP. Melatonin levels in drug-free patients with major depression from the southern hemisphere. Psychoneuroendocrinology. (2006) 31:761–8. doi: 10.1016/j.psyneuen.2006.02.010
68. Steiner M, Brown GM, Goldman S. Nocturnal melatonin and cortisol secretion in newly admitted psychiatric inpatients. Eur Arch Psychiatry Clin Nuerosci. (1990) 240:21–7. doi: 10.1007/BF02190088
69. Beck-Friis J, Kjellman BF, Aperia B, Undén F, von Rosen D, Ljunggren JG, et al. Serum melatonin in relation to clinical variables in patients with major depressive disorder and a hypothesis of a low melatonin syndrome. Acta Psychiatr Scand. (1985) 71:319–30. doi: 10.1111/j.1600-0447.1985.tb02531.x
70. Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. (2008) 455:894–902. doi: 10.1038/nature07455
71. Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. (2008) 33:88–109. doi: 10.1038/sj.npp.1301574
72. Krishnan V, Nestler EJ. Animal models of depression: molecular perspectives. In: Hagan JJ, editor. Molecular Functional Models in Neuropsychiatry Current Topics in Behavioral Neurosciences. Berlin, Heidelberg: Springer (2011) p. 121–47.
73. Bavithra S, Sugantha Priya E, Selvakumar K, Krishnamoorthy G, Arunakaran J. Effect of melatonin on glutamate: BDNF signaling in the cerebral cortex of polychlorinated biphenyls (PCBs)-exposed adult male rats. Neurochem Res. (2015) 40:1858–69. doi: 10.1007/s11064-015-1677-z
74. Evely KM, Hudson RL, Dubocovich ML, Haj-Dahmane S. Melatonin receptor activation increases glutamatergic synaptic transmission in the rat medial lateral habenula. Synapse. (2016) 70:181–6. doi: 10.1002/syn.21892
75. Jentsch MC, Van Buel EM, Bosker FJ, Gladkevich AV, Klein HC, Oude Voshaar RC, et al. Biomarker approaches in major depressive disorder evaluated in the context of current hypotheses. Biomark Med. (2015) 9:277–97. doi: 10.2217/bmm.14.114
76. Lane EA, Moss HB. Pharmacokinetics of melatonin in man: first pass hepatic metabolism. J Clin Endocrinol Metab. (1985) 61:1214–6. doi: 10.1210/jcem-61-6-1214
77. Carvalho LA, Gorenstein C, Moreno R, Pariante C, Markus RP. Effect of antidepressants on melatonin metabolite in depressed patients. J Psychopharmacol. (2009) 23:315–21. doi: 10.1177/0269881108089871
78. Markus RP, Franco DG, Carvalho LA, Gentil V, Gorenstein C. Acute increase in urinary 6-sulfatoximelatonin after clomipramine, as a predictive measure for emotional improvement. J Psychopharmacol. (2010) 24:855–60. doi: 10.1177/0269881109102542
79. Hidalgo MPL, Caumo W, Dantas G, Franco DG, Torres IL, da S, Pezzi J, et al. 6-Sulfatoxymelatonin as a predictor of clinical outcome in depressive patients. Hum Psychopharmacol Clin Exp. (2011) 26:252–7. doi: 10.1002/hup.1204
80. Jury Freitas J, Bertuol Xavier N, Comiran Tonon A, Carissimi A, Timm Pizutti L, Vieira Ilgenfritz CA, et al. 6-Sulfatoxymelatonin predicts treatment response to fluoxetine in major depressive disorder. Ther Adv Psychopharmacol. (2019) 9:2045125319881927. doi: 10.1177/2045125319881927
81. Nagy AD, Iwamoto A, Kawai M, Goda R, Matsuo H, Otsuka T, et al. Melatonin adjusts the expression pattern of clock genes in the suprachiasmatic nucleus and induces antidepressant-like effect in a mouse model of seasonal affective disorder. Chronobiol Int. (2015) 32:447–57. doi: 10.3109/07420528.2014.992525
82. Otsuka T, Kawai M, Togo Y, Goda R, Kawase T, Matsuo H, et al. Photoperiodic responses of depression-like behavior, the brain serotonergic system, and peripheral metabolism in laboratory mice. Psychoneuroendocrinology. (2014) 40:37–47. doi: 10.1016/j.psyneuen.2013.10.013
83. Stefanovic B, Spasojevic N, Jovanovic P, Dronjak S. Melatonin treatment affects changes in adrenal gene expression of catecholamine biosynthesizing enzymes and norepinephrine transporter in the rat model of chronic-stress-induced depression. Can J Physiol Pharmacol. (2019) 97:685–90. doi: 10.1139/cjpp-2018-0612
84. Vega-Rivera NM, Ortiz-López L, Granados-Juárez A, Estrada-Camarena EM, Ramírez-Rodríguez GB. Melatonin reverses the depression-associated behaviour and regulates microglia, fractalkine expression and neurogenesis in adult mice exposed to chronic mild stress. Neuroscience. (2020) 440:316–36. doi: 10.1016/j.neuroscience.2020.05.014
85. Detanico BC, Piato AL, Freitas JJ, Lhullier FL, Hidalgo MP, Caumo W, et al. Antidepressant-like effects of melatonin in the mouse chronic mild stress model. Eur J Pharmacol. (2009) 607:121–5. doi: 10.1016/j.ejphar.2009.02.037
86. Burgess HJ, Fogg LF. Individual differences in the amount and timing of salivary melatonin secretion. PLoS ONE. (2008) 3:e3055. doi: 10.1371/journal.pone.0003055
87. Crescenzo FD, Lennox A, Gibson JC, Cordey JH, Stockton S, Cowen PJ, et al. Melatonin as a treatment for mood disorders: a systematic review. Acta Psychiatr Scand. (2017) 136:549–58. doi: 10.1111/acps.12755
88. Dolberg OT, Hirschmann S, Grunhaus L. Melatonin for the treatment of sleep disturbances in major depressive disorder. Am J Psychiatry. (1998) 155:1119–21. doi: 10.1176/ajp.155.8.1119
89. Serfaty MA, Osborne D, Buszewicz MJ, Blizard R, Raven PW. A randomized double-blind placebo-controlled trial of treatment as usual plus exogenous slow-release melatonin (6 mg) or placebo for sleep disturbance and depressed mood. Int Clin Psychopharmacol. (2010) 25:132–42. doi: 10.1097/YIC.0b013e32832c260b
90. Fava M, Targum SD, Nierenberg AA, Bleicher LS, Carter TA, Wedel PC, et al. An exploratory study of combination buspirone and melatonin SR in major depressive disorder (MDD): a possible role for neurogenesis in drug discovery. J Psychiatr Res. (2012) 46:1553–63. doi: 10.1016/j.jpsychires.2012.08.013
91. Lewy AJ, Bauer VK, Cutler NL, Sack RL. Melatonin treatment of winter depression: a pilot study. Psychiatry Res. (1998) 77:57–61. doi: 10.1016/S0165-1781(97)00128-5
92. Sherer MA, Weingartner H, James SP, Rosenthal NE. Effects of melatonin on performance testing in patients with seasonal affective disorder. Neurosci Lett. (1985) 58:277–82. doi: 10.1016/0304-3940(85)90066-7
93. Danilenko KV, Putilov AA. Melatonin treatment of winter depression following total sleep deprivation: waking EEG and mood correlates. Neuropsychopharmacology. (2005) 30:1345–52. doi: 10.1038/sj.npp.1300698
94. Lewy AJ, Lefler BJ, Emens JS, Bauer VK. The circadian basis of winter depression. Proc Natl Acad Sci USA. (2006) 103:7414–9. doi: 10.1073/pnas.0602425103
95. Hansen MV, Danielsen AK, Hageman I, Rosenberg J, Gögenur I. The therapeutic or prophylactic effect of exogenous melatonin against depression and depressive symptoms: a systematic review and meta-analysis. Eur Neuropsychopharmacol. (2014) 24:1719–28. doi: 10.1016/j.euroneuro.2014.08.008
96. Krause M, Gutsmiedl K, Bighelli I, Schneider-Thoma J, Chaimani A, Leucht S. Efficacy and tolerability of pharmacological and non-pharmacological interventions in older patients with major depressive disorder: a systematic review, pairwise and network meta-analysis. Eur Neuropsychopharmacol. (2019) 29:1003–22. doi: 10.1016/j.euroneuro.2019.07.130
97. De Berardis D, Marini S, Fornaro M, Srinivasan V, Iasevoli F, Tomasetti C, et al. The melatonergic system in mood and anxiety disorders and the role of agomelatine: implications for clinical practice. Int J Mol Sci. (2013) 14:12458–83. doi: 10.3390/ijms140612458
98. Guaiana G, Gupta S, Chiodo D, Davies SJ, Haederle K, Koesters M. Agomelatine versus other antidepressive agents for major depression. Cochrane Database Syst Rev. (2013) 12:CD008851. doi: 10.1002/14651858.CD008851.pub2
99. Sparshatt A, Williams RHM, Baldwin DS, Haddad PM, Bazire S, Weston E, et al. A naturalistic evaluation and audit database of agomelatine: clinical outcome at 12 weeks. Acta Psychiatr Scand. (2013) 128:203–11. doi: 10.1111/acps.12044
100. Nussbaumer-Streit B, Greenblatt A, Kaminski-Hartenthaler A, Noord MGV, Forneris CA, Morgan LC, et al. Melatonin and agomelatine for preventing seasonal affective disorder. Cochrane Database Syst Rev. (2019) 6:CD011271. doi: 10.1002/14651858.CD011271.pub3
101. Brzezinski A, Vangel MG, Wurtman RJ, Norrie G, Zhdanova I, Ben-Shushan A, et al. Effects of exogenous melatonin on sleep: a meta-analysis. Sleep Med Rev. (2005) 9:41–50. doi: 10.1016/j.smrv.2004.06.004
102. Low TL, Choo FN, Tan SM. The efficacy of melatonin and melatonin agonists in insomnia - an umbrella review. J Psychiatr Res. (2020) 121:10–23. doi: 10.1016/j.jpsychires.2019.10.022
103. Buckley AW, Hirtz D, Oskoui M, Armstrong MJ, Batra A, Bridgemohan C, et al. Practice guideline: treatment for insomnia and disrupted sleep behavior in children and adolescents with autism spectrum disorder: report of the guideline development, dissemination, and implementation subcommittee of the American Academy of Neurology. Neurology. (2020) 94:392–404. doi: 10.1212/WNL.0000000000009033
104. Bruni O, Alonso-Alconada D, Besag F, Biran V, Braam W, Cortese S, et al. Current role of melatonin in pediatric neurology: clinical recommendations. Eur J Paediatr Neurol. (2015) 19:122–33. doi: 10.1016/j.ejpn.2014.12.007
105. Liira J, Verbeek JH, Costa G, Driscoll TR, Sallinen M, Isotalo LK, et al. Pharmacological interventions for sleepiness and sleep disturbances caused by shift work. Cochrane Database Syst Rev. (2014) 8:CD009776. doi: 10.1002/14651858.CD009776.pub2
106. Morgenthaler TI, Lee-Chiong T, Alessi C, Friedman L, Aurora RN, Boehlecke B, et al. Practice parameters for the clinical evaluation and treatment of circadian rhythm sleep disorders. An American Academy of Sleep Medicine report. Sleep. (2007) 30:1445–59. doi: 10.1093/sleep/30.11.1445
107. Auger RR, Burgess HJ, Emens JS, Deriy LV, Thomas SM, Sharkey KM. Clinical practice guideline for the treatment of intrinsic circadian rhythm sleep-wake disorders: advanced sleep-wake phase disorder (ASWPD), delayed sleep-wake phase disorder (DSWPD), non-24-hour sleep-wake rhythm disorder (N24SWD), and irregular sleep-wake rhythm disorder (ISWRD). An Update for 2015: An American Academy of Sleep Medicine Clinical Practice Guideline. J Clin Sleep Med. (2015) 11:1199–236. doi: 10.5664/jcsm.5100
108. Lewy A. Clinical implications of the melatonin phase response curve. J Clin Endocrinol Metab. (2010) 95:3158–60. doi: 10.1210/jc.2010-1031
109. Sletten TL, Magee M, Murray JM, Gordon CJ, Lovato N, Kennaway DJ, et al. Efficacy of melatonin with behavioural sleep-wake scheduling for delayed sleep-wake phase disorder: a double-blind, randomised clinical trial. PLoS Med. (2018) 15:e1002587. doi: 10.1371/journal.pmed.1002587
110. Mundey K, Benloucif S, Harsanyi K, Dubocovich ML, Zee PC. Phase-dependent treatment of delayed sleep phase syndrome with melatonin. Sleep. (2005) 28:1271–8. doi: 10.1093/sleep/28.10.1271
Keywords: psychiatry, mood disorder, neuropsychaitric disorders, behavior, biological rhythms, chronobiology, 6-sulfatoxymelatonin (aMT6s), biomarker
Citation: Tonon AC, Pilz LK, Markus RP, Hidalgo MP and Elisabetsky E (2021) Melatonin and Depression: A Translational Perspective From Animal Models to Clinical Studies. Front. Psychiatry 12:638981. doi: 10.3389/fpsyt.2021.638981
Received: 08 December 2020; Accepted: 15 March 2021;
Published: 08 April 2021.
Edited by:
Mirko Manchia, University of Cagliari, ItalyReviewed by:
Anna Brancato, University of Palermo, ItalyElena Martín-García, Pompeu Fabra University, Spain
Copyright © 2021 Tonon, Pilz, Markus, Hidalgo and Elisabetsky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: André C. Tonon, bGFiY3Jvbm9lc29ub0BoY3BhLmVkdS5iciA=