 Nina Vanessa Kraguljac
Nina Vanessa Kraguljac Adrienne Carol Lahti
Adrienne Carol Lahti
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 11 March 2021
Sec. Schizophrenia
Volume 12 - 2021 | https://doi.org/10.3389/fpsyt.2021.613764
This article is part of the Research Topic Trajectories of Brain Abnormalities in Early Schizophrenia View all 14 articles
Schizophrenia is a complex neuropsychiatric disorder with a diverse clinical phenotype that has a substantial personal and public health burden. To advance the mechanistic understanding of the illness, neuroimaging can be utilized to capture different aspects of brain pathology in vivo, including brain structural integrity deficits, functional dysconnectivity, and altered neurotransmitter systems. In this review, we consider a number of key scientific questions relevant in the context of neuroimaging studies aimed at unraveling the pathophysiology of schizophrenia and take the opportunity to reflect on our progress toward advancing the mechanistic understanding of the illness. Our data is congruent with the idea that the brain is fundamentally affected in the illness, where widespread structural gray and white matter involvement, functionally abnormal cortical and subcortical information processing, and neurometabolic dysregulation are present in patients. Importantly, certain brain circuits appear preferentially affected and subtle abnormalities are already evident in first episode psychosis patients. We also demonstrated that brain circuitry alterations are clinically relevant by showing that these pathological signatures can be leveraged for predicting subsequent response to antipsychotic treatment. Interestingly, dopamine D2 receptor blockers alleviate neural abnormalities to some extent. Taken together, it is highly unlikely that the pathogenesis of schizophrenia is uniform, it is more plausible that there may be multiple different etiologies that converge to the behavioral phenotype of schizophrenia. Our data underscore that mechanistically oriented neuroimaging studies must take non-specific factors such as antipsychotic drug exposure or illness chronicity into consideration when interpreting disease signatures, as a clear characterization of primary pathophysiological processes is an imperative prerequisite for rational drug development and for alleviating disease burden in our patients.
Schizophrenia is a complex neuropsychiatric disorder with a diverse clinical phenotype that manifests in variable levels of positive and negative symptoms and cognitive impairment. Even though the course of the illness can be variable, for many patients the disease is chronic and debilitating, resulting in a substantial personal and public health burden (1). Available pharmacological interventions can alleviate positive symptoms, but effective treatments across symptom dimensions are lacking and a cure remains elusive. This can in part be attributed to the gaps in the knowledge of the underlying brain pathology. To close these gaps, tremendous efforts geared toward elucidating key pathophysiological brain signatures and at advancing our mechanistic understanding of the illness are undertaken with the ultimate goal to lower disease burden and improve long-term outcomes for patients.
Neuroimaging offers versatility in terms of capturing different aspects of brain pathology in vivo, including brain structural integrity deficits, functional dysconnectivity, and altered neurotransmitter systems, positioning the field well to contribute to the discovery of clinically relevant biological processes in schizophrenia. The first mechanistic discovery studies utilizing neuroimaging date back to the 1970s and 80s, when ventricular enlargement was described as a potential diagnostic marker (2–4). Since these early studies, the field has grown exponentially; more than 16,000 neuroimaging manuscripts in schizophrenia spectrum disorder patients have been published (Figure 1). Even though major strides toward delineating relevant pathophysiological processes have been made, progress in the field has been impeded by a number of confounding factors that make interpretation of data difficult. This ultimately hampers the development of a comprehensive explanatory model capturing the complex underlying pathophysiology and integrating findings from diverse imaging modalities. Two principal confounding variables need to be considered when interpreting neuroimaging studies. These are antipsychotic medication exposure and illness chronicity, which impact virtually all imaging measures. This has resulted in a debate to what extent brain imaging alterations reported in schizophrenia studies reflect primary disease pathology (5). To make further progress, it is imperative to disentangle confounding effects from illness specific brain signatures. Investigating the impact of antipsychotic medications in longitudinal brain imaging studies and studying patients in the early illness stages who have had no prior antipsychotic medication exposure affords the opportunity to characterize antipsychotic drug action, mitigate confounds and gain a more in depth understanding of the nature of the illness.

Figure 1. Neuroimaging studies in schizophrenia spectrum disorders. A PubMed search was performed on 7/29/20 with the following search term: (Schizophrenia OR Schizoaffective disorder OR Schizophreniform disorder OR Brief psychotic disorder OR Psychosis) AND (Neuroimaging OR PET OR SPECT OR MRI OR Diffusion OR Spectroscopy OR Connectivity). The search was restricted to studies published 1970 or later and to human subjects.
Our group's work has been dedicated to contribute to the efforts in delineating the schizophrenia pathophysiology and in disentangling disease signatures from those of antipsychotic medications and disease chronicity. In the following paragraphs, we discuss a number of key scientific questions and take the opportunity to reflect on our progress toward advancing the mechanistic understanding of the illness.
Antipsychotic medications are the cornerstone of treating patients with schizophrenia spectrum disorders. They principally act on the dopamine D2 receptors (6), but also modulate a number of other neurotransmitter systems (7, 8), affect receptor expression profiles, have downstream effects on protein synthesis (9), and impact brain plasticity (9). Given the complexity of antipsychotic drug action, it is plausible that molecular imaging geared at investigating the dopamine system and other imaging markers are impacted by these medications. Longitudinal brain imaging studies that contrast neural signatures in the same patients before and after they are treated can shed light on modulatory effects of antipsychotic medications on brain structure, function and neurochemistry [for reviews of the larger field, see (10–15)]. We have conducted several studies utilizing different imaging techniques to broadly characterize antipsychotic drug effects on neural signatures (Table 1).
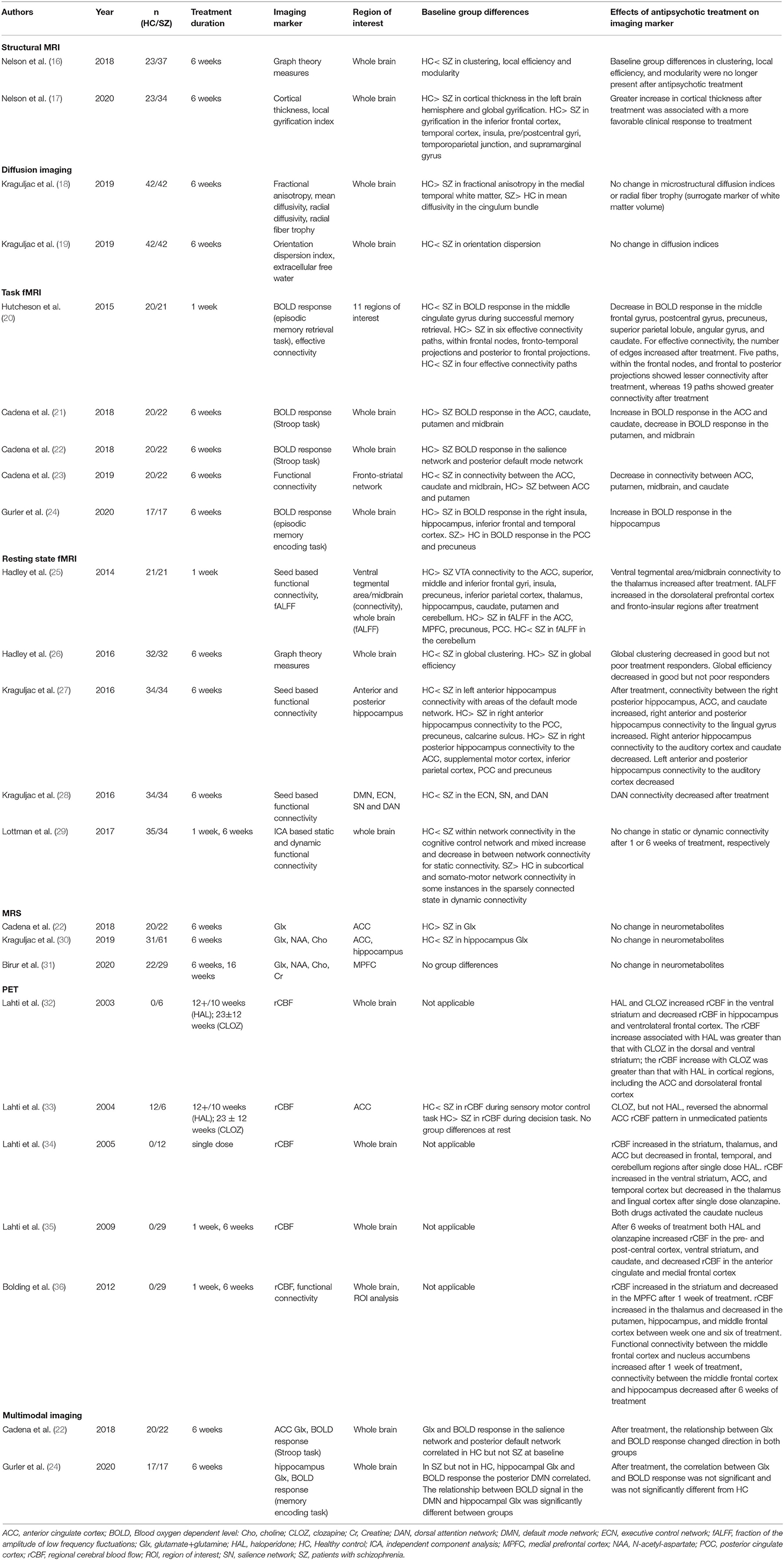
Table 1. Neuroimaging studies of antipsychotic drug effects from by the authors.
Our structural imaging studies show mixed results in terms of antipsychotic medication effects. We observed antipsychotic medication related changes in cortical thickness (17), but not in white matter microstructural integrity or radial fiber trophy (a surrogate marker for white matter volume) (18, 19). The disparity in effects across tissue classes is perhaps not surprising, given the different anatomical, functional and neurochemical makeup of these tissues. For example, brain derived neurotrophic factor (BDNF) is a neurotrophin that regulates synaptic plasticity (37, 38) and spine density (39). The 66met allele has been associated with diminished activity-dependent BDNF secretion (40) and decreased cortical volumes (41) but not with white matter integrity in healthy subjects (42). BDNF also is found to be abnormally low in schizophrenia (43) and can be increased by second generation antipsychotics (44). It is therefore possible that antipsychotic medication related modulation of BDNF results in alleviation of dendritic deficits in the outer layers of the cortex (45), which would be reflected in an increase in cortical thickness, but not white matter integrity. Alternatively, it is possible that the acute gray matter tissue response can be captured at the time scale at which we investigated structural changes, but the duration is not sufficient to demonstrate subtle changes in white matter architecture and microstructure that may become more evident with longer duration of antipsychotic treatment.
Our longitudinal task based and resting state functional neuroimaging studies consistently show changes associated with antipsychotic drugs at different time scales, ranging from a single dose drug administration to several weeks of treatment. Our findings are in agreement with drug challenge studies in healthy subjects demonstrating that dopamine antagonism affects brain function (46) and animal studies showing that antipsychotics can be used to restore cortical synchronization and functional connectivity after disruption with hallucinogenic agents (47, 48). When interpreting our functional data in context of differences between patients and healthy controls, we show that antipsychotic treatment appears to, at least partially, restore abnormal brain function to levels that are more similar to those of healthy controls. We also observed a recurring theme of brain regions that are functionally modulated by antipsychotic treatment. Relevant areas include the anterior cingulate cortex, medial prefrontal cortex, hippocampus, caudate and putamen. This suggests that cortico-striatal circuitry may be functionally remodeled by antipsychotic medications. Interestingly, anterior cingulate and medial frontal cortex function (35, 49–52) and cortico-striatal connectivity have also been identified as critical for antipsychotic drug action by other groups (53, 54). When examining brain function on a more global level, we found that antipsychotic medications affect brain network topology, i.e., global efficiency and clustering, but not dynamic connectivity states (26, 29), which is consistent with the idea that antipsychotic medications partially but not fully restore abnormal brain function.
In our magnetic resonance spectroscopy studies, we unexpectedly did not find changes with antipsychotic medications on glutamate levels. Several lines of evidence describe complex interactions between the dopamine and glutamate systems and identify glutamate receptor complexes as potentially important indirect targets for dopamine D2 receptor blockers (55–57), suggesting antipsychotic medications may affect glutamate levels. A number of prior studies report a decrease of glutamate in the temporal lobe (58), striatum, and the anterior cingulate cortex (59), though the group failed to replicate the latter in a subsequent report (60). It is possible that this discrepancy is due to differential effects of various antipsychotic medications on the glutamate system. Alternatively, risperidone may affect glutamate levels only in a subset of patients, or the shifts in glutamatergic neurotransmission may be too subtle to be captured with magnetic resonance spectroscopy, which measures tissue metabolites rather than compartment specific levels.
Our data also suggest that heterogeneity in the patient population may need to be taken into account when examining antipsychotic medication related changes in neural signatures. We demonstrated in several studies that changes in brain structure and function following antipsychotic treatment may be more prominent in patients with a favorable clinical response, compared to those who show little improvement with medications (17, 26, 27). It is tempting to speculate that the variability in modulatory effects of antipsychotics is reflective of differences in brain pathology. It is possible that patients who respond favorably to medications do so because they still have actively destructive, yet reversible, neurobiological changes that can be ameliorated with antipsychotic treatment. In contrast, poor responders may be beyond the period of active deficit process formation in their brain and the pathological state becomes more permanent, rendering antipsychotic medications ineffective both on a clinical level and in reversing structural and functional brain injury. If that is the case, it should be possible to leverage neuroimaging data to aid in the prediction of response to antipsychotic treatment. Much of the work described here has been performed in patients who had prior antipsychotic medication exposure, and in mixed groups of antipsychotic medication-naïve patients previously medicated patients. Adequately powered longitudinal studies conducted in exclusively medication-naïve patients are direly needed to reduce the possible effects of illness chronicity and prior antipsychotic medication exposure on changes related to antipsychotic medication treatment.
The current strategy for management of psychosis spectrum disorders consists of sequential treatment with different antipsychotic medications based on trial and error. Unfortunately, it is not possible to predict how a patient will respond to treatment based on clinical assessments alone. To make progress in this context, we have conducted a number of studies geared toward characterizing neural signatures in unmedicated and antipsychotic medication-naïve patients that predict a subsequent favorable response to antipsychotic treatment (Table 2). Here, the ultimate goal is to realize the potential of neuroimaging in guiding treatment decisions based on the underlying brain pathology [for further review of the topic, see (63, 64)].
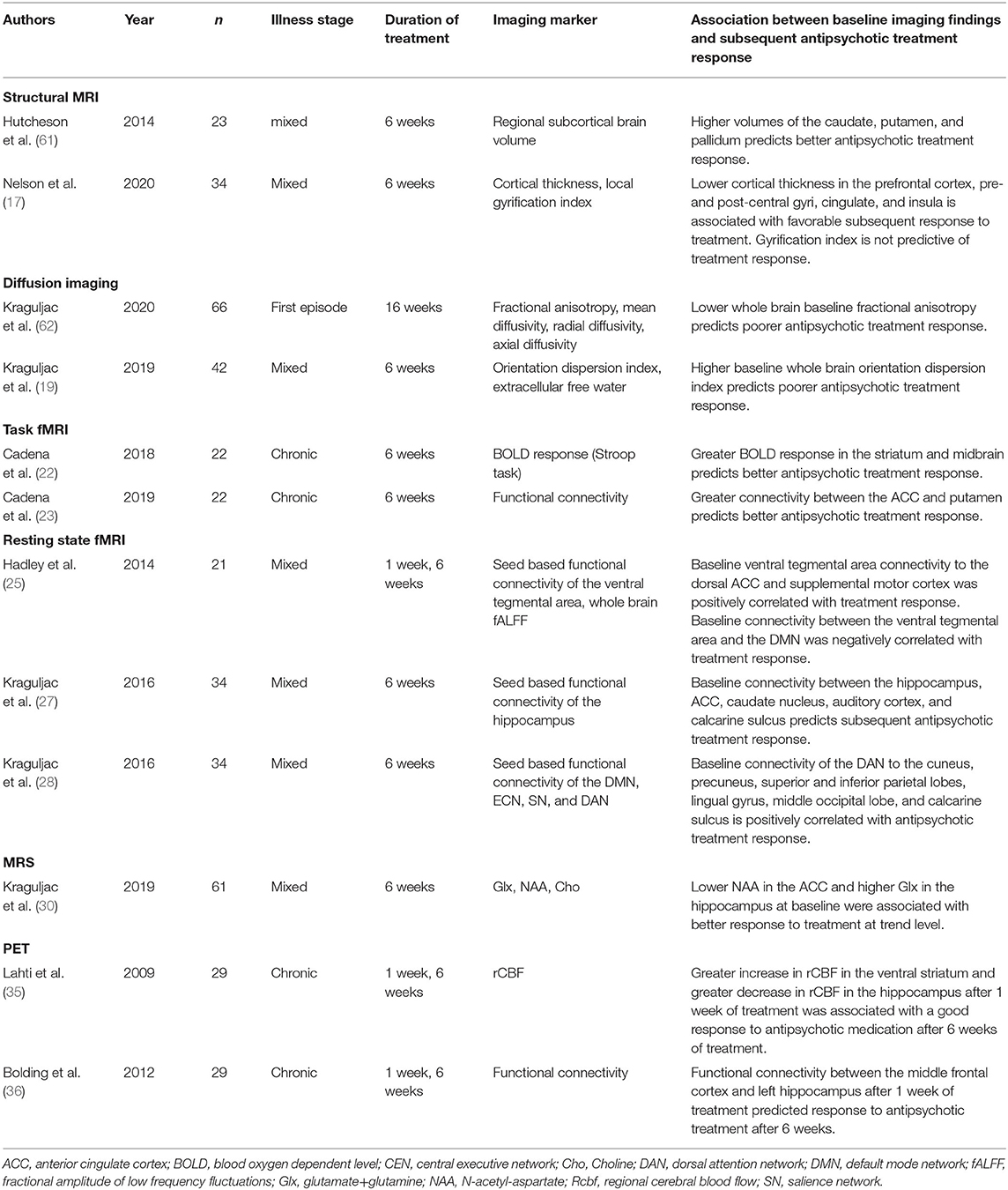
Table 2. Selected baseline neuroimaging markers that are relevant to the prediction of subsequent antipsychotic drug response.
In structural imaging studies, we found that both gray and white matter integrity at baseline are predictive of subsequent response to antipsychotic treatment. Lower cortical thickness and greater basal ganglia volumes [basal ganglia volumes are increased in patients, possibly as a result of antipsychotic drug exposure (65)] are associated with better treatment response (17, 61). This suggests that greater baseline alterations in gray matter are indicative of a favorable clinical outcome. In contrast, greater alterations in whole brain fractional anisotropy, a non-specific marker of white matter integrity, and in whole brain orientation dispersion, a measure of fiber complexity, were associated with subsequent poor treatment response, suggesting that greater alterations in white matter are indicative of poor clinical outcomes (18, 19, 62).
Our functional neuroimaging studies, both positron emission tomography (PET) and functional MRI, found consistent associations between more intact brain function at baseline and a favorable subsequent response to antipsychotic medications. Here again we found a recurring theme of brain regions and networks that were relevant for clinical outcomes, specifically anterior cingulate cortex, hippocampus, thalamus, striatum and caudate, as well as the default mode network and dorsal attention network. Interestingly, we also found that the degree of normalization in brain function after 1 week of antipsychotic medication was predictive of clinical outcomes after 6 weeks of treatment (35, 36). Our data suggest that both baseline function and early functional changes may be important predictors of treatment response.
Our magnetic resonance spectroscopy studies investigated neurometabolite levels in the anterior cingulate cortex and the hippocampus (30). We found that N-acetyl-aspartate (NAA), a marker of neuronal health, in the anterior cingulate cortex and glutamate in the hippocampus at baseline were associated with response to risperidone after 6 weeks of treatment. At first glance, the association between low cortical NAA and good response to treatment appears counter-intuitive. It is striking, however, that greater alterations in both NAA and glutamate are linked to better treatment response. One could speculate that these features point toward an excitation/inhibition imbalance and a deprived neuronal state that is reversible by successful antipsychotic treatment.
Taken together, our studies suggest two groups of neuroimaging markers that are relevant for subsequent antipsychotic treatment response. The first is a set of markers for which greater abnormalities predict subsequent poor response to antipsychotic treatment. These markers are largely centered around the theme of brain connectivity, where greater abnormal functional connectivity and white matter integrity deficits are indicative of worse response to antipsychotic treatment. The second is a set of markers where greater abnormalities predict a favorable response to antipsychotic treatment. These markers include cortical thickness, subcortical brain volumes and neurometabolites levels. Abnormalities in these imaging features may signify a deprived neuronal state or a disequilibrium in the excitation/inhibition balance that may be reversible with successful antipsychotic treatment. Overall, our findings suggest that the brain is structurally and functionally “wired” in a way that does or does not favor response to antipsychotic medication treatment. However, based on these studies, it remains unclear if the neural signatures relevant for antipsychotic treatment response are also core features of the schizophrenia pathophysiology or not. To disentangle core illness features from confounds of antipsychotic medication exposure as well as illness chronicity, it is imperative to characterize neuroimaging signatures in antipsychotic medication-naïve first-episode psychosis patients.
There is a growing sense that progressive brain changes occur beyond the first psychotic episode and that illness chronicity may fundamentally affect the brain (66). It is difficult to pinpoint the extent to which abnormalities can be attributed to antipsychotic medication effects or to ongoing disease progression. Studying patients at later illness stages makes it problematic to discern downstream effects of pathology and prior treatment attempts from primary pathology. Even though this patient population is difficult to recruit (67), studying antipsychotic medication-naïve first episode psychosis patients can mitigate these issues. Our group has conducted studies in this population using different imaging modalities in an effort to characterize relevant neural signatures (Table 3).
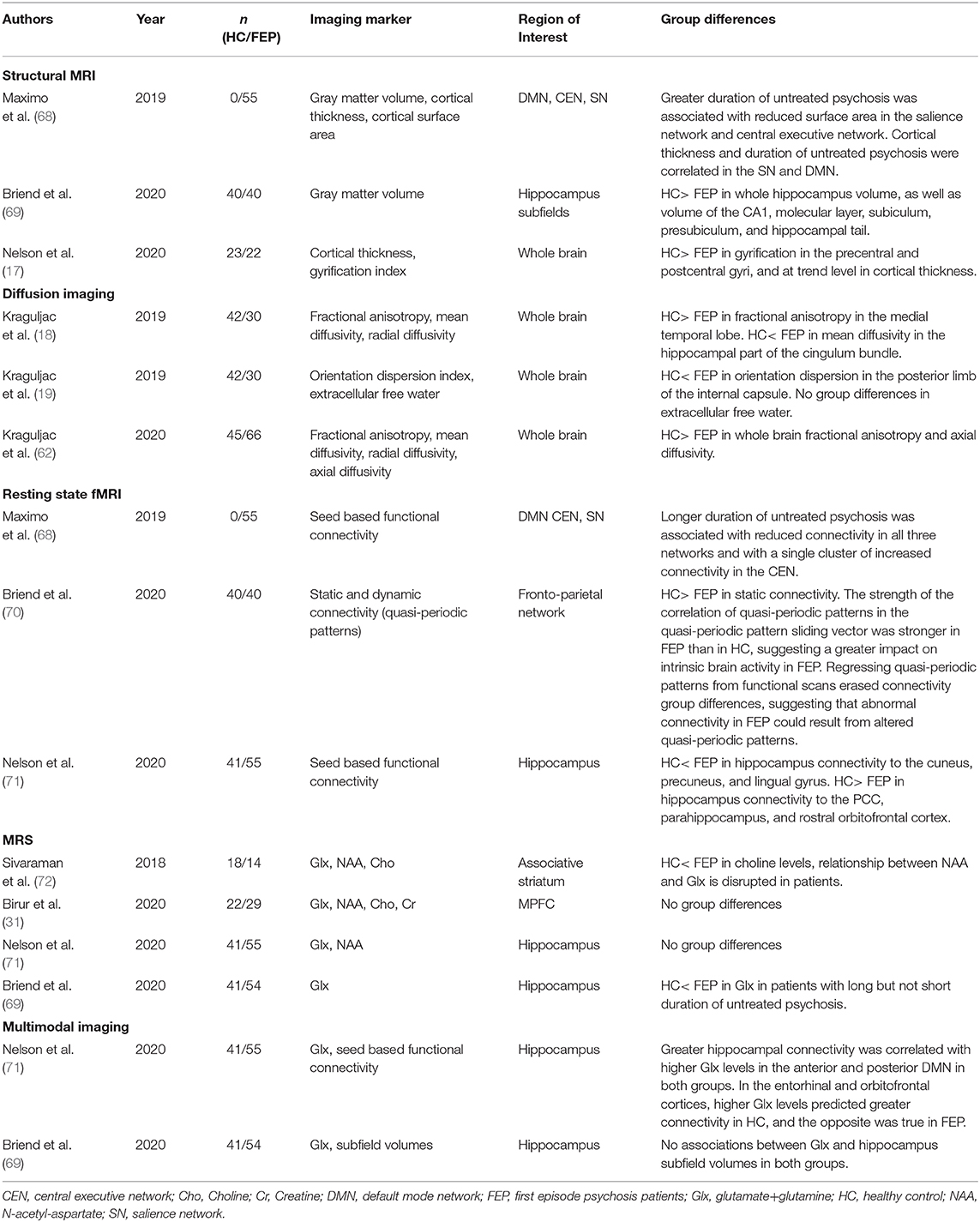
Table 3. Neuroimaging studies of antipsychotic medication-naïve first episode psychosis patients from our group.
Using structural neuroimaging techniques, we found that a number of alterations are already present in medication-naive patients. Alterations include decreased gyrification in the pre- and post-central gyri, decreased total hippocampus and hippocampus subfield volumes, a trend level decrease in cortical thickness, as well as a decrease in whole brain fractional anisotropy and axial diffusivity (17, 18, 62, 69). Additionally, an increase in mean diffusivity in the hippocampal part of the cingulum, which is often interpreted as evidence of neuroinflammation, is already evident in first episode patients (18). Importantly, the duration of untreated psychosis, which is the time between first onset of psychotic symptoms and the first antipsychotic treatment, seems to be an important modulatory factor for structural integrity, as a longer duration of untreated psychosis is associated with greater gray and white matter alterations (62, 68, 69).
We saw similar results in our functional imaging studies where greater duration of untreated psychosis was associated with greater disruptions in functional connectivity in large-scale brain networks supporting higher order cognition (68). In the overall group of antipsychotic medication-naïve patients, we note that hippocampus connectivity is disrupted. Spatial patterns of dysconnectivity resemble those in unmedicated, chronic schizophrenia patients (71). When examining dynamic connectivity, we found that quasi-periodic patterns had a greater impact of fronto-parietal control network connectivity in first episode patients compared to controls, suggesting that brain network dynamics are already altered in this patient group (70). Taken together, connectivity alterations are clearly already present in medication-naïve patients, which is consistent with the hypothesis that brain network dysconnectivity is a core feature of the illness (73–75).
In magnetic resonance spectroscopy studies, we found elevated choline and a disruption of the relationship between NAA and glutamate in the left striatum, indicating possible mitochondrial, membrane, and glial dysfunction (72). However, we reported no neurometabolite abnormalities in the medial prefrontal cortex or the hippocampus when contrasting antipsychotic medication-naïve first episode patients and controls in the overall group (31, 69). It is important to note that glutamate levels in both areas showed a greater variance in patients compared to controls, suggesting heterogeneity in glutamatergic metabolism in first episode psychosis. When the duration of untreated psychosis is taken into consideration, those with a shorter but not those with a longer duration of untreated psychosis did have an elevation in hippocampal glutamate (69).
Collectively, it is evident that functional and structural abnormalities are already present in patients who are in their early illness stages who have no prior exposure to antipsychotic medications. However, wee did not observe group level alterations in NAA or glutamate which was somewhat unexpected. Interestingly, the variance in measurements was greater in patients compared to controls, suggesting that neurometabolite alterations may only be present in a subset of patients, or become more pronounced as the illness progresses. Importantly, we found the duration of untreated psychosis to be an important mediator, such that a longer interval adversely impacts brain structure and function, underscoring the importance of early intervention efforts in this syndrome.
The field is moving from traditional models of schizophrenia focused on the disruption of a single molecule such as dopamine or glutamate, to models of pathway dysregulation as a means of integrating findings from diverse imaging modalities. An individual component of a pathway in this context can be best understood as part of a highly interactive network that may influence other parts of the network and in turn be regulated by a number of other factors (76). An abnormality may represent a primary etiological factor, but could also be present because of a disruption in modulatory inputs and thus reflect a secondary consequence of a pathophysiological process. It is critical to develop disease models with a degree of complexity that accurately reflect these highly interactive patterns. Multimodal imaging allows investigation of brain dysfunction using a range of techniques within the same individual and to test hypotheses about relationships between biological mechanisms. This approach may reveal details about the pathophysiology that would not be detectable when using one modality alone.
For example, glutamatergic hyperactivity is hypothesized to be a key pathological feature in schizophrenia (77, 78). Glutamate neurotransmitter flux, neuronal firing rate, and the blood oxygen level dependent (BOLD) response are tightly coupled (79, 80), and glutamate plays a role in long-range functional connections (81). In preclinical studies, excess glutamate has been shown to be associated with disorganized neuronal activity (82) and may result in increased synapse turnover as well as axonal or glial injury (83–85). If this premise holds true, multimodal neuroimaging studies should detect relationships between glutamate levels and brain function in healthy subjects that are disrupted in patients with schizophrenia. They also should reveal an association between glutamate excess and structural integrity deficits in patients. Our group has conducted a number of such studies to empirically test the relationship between glutamate and brain function the impact of glutamate excess on brain structure.
We used several task paradigms and resting state functional MRI to characterize the relationship between glutamate levels and brain function in healthy subjects and schizophrenia spectrum patients. We reported a positive correlation between hippocampal glutamate and inferior frontal activation during a memory retrieval task in controls that was absent in medicated chronic schizophrenia patients (86). The relationship between hippocampal glutamate and the BOLD response in default mode network regions during memory encoding also differed between unmedicated patients and controls (24). Using a Stroop task, we observed a negative correlation between anterior cingulate cortex glutamate and the BOLD response in the posterior cingulate cortex, precuneus, occipital cortex and cerebellum in controls; this correlation was inverted in medicated first-episode psychosis patients (87). Similarly, we noted a correlation between anterior cingulate cortex glutamate and the BOLD response in the salience network and posterior default network during a Stroop task in healthy controls but not unmedicated patients with schizophrenia (22). Using a reward task, we observed an association between the prediction error related BOLD response in the midbrain and substantia nigra glutamate in healthy controls but not medicated patients with chronic schizophrenia. Interestingly, glutamate levels were elevated in patients, suggesting that glutamatergic dysfunction might contribute to abnormal neural prediction error coding (88). At rest, we initially did not detect a relationship between hippocampal glutamate and aberrant hippocampal connectivity to the precuneus in a small group of unmedicated schizophrenia patients (89). However, after expanding the sample size, we did observe that higher glutamate levels were correlated with higher hippocampus resting state connectivity to the anterior default mode network in healthy controls, but the relationship between measures was inverse in first episode psychosis patients (71). Taken together, our studies consistently demonstrated a link between glutamate levels and brain function, regardless of task condition and even in absence of a task in healthy subjects. Importantly, this relationship was consistently found to be altered or absent in patients across paradigms tapping into various aspects of brain function, which implicates that the disruption in this coupling may be a fundamental feature of the illness. Interestingly, a detectable alteration in glutamate levels does not seem to be a necessary element to reach the threshold of a disruption in brain function, suggesting the presence of subtle abnormalities in glutamatergic neurotransmission that may be below the level of detection with magnetic resonance spectroscopy. Our longitudinal studies further demonstrate that the relationship between glutamate and brain function in patients is modulated by antipsychotic drug treatment, suggesting a potential mechanism of antipsychotic drug action (22, 24).
We also conducted studies to examine the impact of glutamate (excess) on brain structures. In unmedicated chronic schizophrenia patients, we found that higher hippocampal glutamate was associated with lower hippocampal volumes, suggesting that glutamate related excitotoxicity (neurotransmitter excess related increased synapse turnover) might affect brain structure (90). Our finding is in agreement with a later study that reported a negative relationship between glutamate excess and brain volumes in the caudate nucleus in first episode psychosis patients (91). In contrast, we did not observe a linear relationship between hippocampus subfield volumes and glutamate levels in antipsychotic medication-naïve first episode psychosis patients (69). Interestingly though, those with a longer duration of untreated psychosis showed lower hippocampus subfield volumes. While this discrepancy between our studies at first glance does not support the hypothesis that glutamate impacts brain structures as a fundamental mechanism of the illness, it is possible that the amount of time the brain is exposed to altered glutamate is the key factor in adversely affecting brain structures rather than the amount of glutamate present in the brain at any given time.
As demonstrated with the above examples, multimodal neuroimaging clearly can give important insights into the pathophysiology of the illness, and allows testing of hypotheses about the relationships between biological mechanisms which is not possible with a single imaging modality. However, because different aspects of the pathophysiology are typically assessed at the same time in multimodal imaging, interpretations are limited to discovery of associations but not causality. In other words, it is possible to detect a relationship between two different imaging markers, but it is not feasible to discern if one abnormality is caused by the other or vice versa.
A complementary line of research, pharmacological challenge studies, which are designed to model aspects of the pathophysiology, can inform inferences drawn about causality for neuroimaging alterations seen in schizophrenia spectrum patients. Our group has done several pharmacological challenge studies using subanesthetic ketamine, a non-competitive N-methyl-d-aspartate (NMDA) receptor blocker that transiently induces a behavioral phenotype similar to that seen in the illness (92–96), to test the hypothesis that experimentally induced NMDA receptor hypofunction causes changes in brain function and neurochemistry that are comparable to neural signatures observed in schizophrenia.
Early imaging studies testing the effects of a pharmacological challenge found that sub-anesthetic ketamine affects regional cerebral blood flow (rCBF). In response to the drug, an increase in psychosis symptom severity was paralleled by an increase in rCBF in the anterior cingulate cortex, both in healthy controls and medicated patients with schizophrenia (97). In addition to a ketamine related increase in rCBF in the anterior cingulate cortex, a reduction in rCBF was found in the hippocampus, lingual gyrus and fusiform gyrus in a different study of healthy controls (98). Interestingly, kinetic analyses suggest that the ketamine induced rCBF response differs across brain areas (99). Using a MRI based rather than PET based technique to quantify blood flow, our group later reported increased rCBF in the prefrontal cortex, cingulate cortex caudate, putamen, thalamus and hippocampus, as well as interregional connectivity alterations in areas of the salience network (100). This again suggests systems level, but not uniform, effects of experimentally induced NMDA receptor hypofunction.
In a combined magnetic resonance spectroscopy and resting state functional connectivity study of the hippocampus (101), we reported that experimentally induced NMDA receptor hypofunction resulted in an increase in glutamate that was similar in magnitude to that we saw in unmedicated schizophrenia patients (90). In parallel, we also found a reduction in hippocampal resting state connectivity to the anterior cingulate cortex, medial prefrontal cortex, insula, hippocampus, precuneus, posterior cingulate cortex and lingual gyrus. Spatial patterns of abnormalities here closely resemble those we observed in both unmedicated schizophrenia patients (27, 71) and antipsychotic medication-naïve first episode psychosis patients (71).
Taken together, our findings from ketamine challenge studies lend empirical support to the putative link between NMDA receptor hypofunction, disruption in brain function and glutamate excess and provide a theoretical framework for the interpretation of abnormal brain signatures in schizophrenia in the context of NMDA receptor hypofunction. It is important to note that this type of framework can also be leveraged to test target engagement for putative novel pharmacological agents as recently demonstrated a multi-center proof of mechanism study of pomaglumetad (102), which will ideally translate into accelerated development of novel drugs.
Postmortem studies reporting abnormalities in γ-Aminobutyric-acid (GABA)ergic interneurons (103, 104) and glutamatergic signaling (105) provided the impetus for the measurement of glutamate and GABA using MR spectroscopy, as well as the measurement of brain oscillations using EEG/MEG. The fast-spiking GABA-interneurons play a fundamental role in controlling of the synchrony of cortical pyramidal neurons by producing rhythmic inhibitory postsynaptic potentials. Consequently, these interneurons appear to be key to the generation of gamma oscillations (106). In a group of first episode psychosis patients scanned using an ultra-high field magnet [7 Tesla (T)] we reported a decrease in glutamate, but not GABA levels, in the anterior cingulate cortex (107). Interestingly, in a larger group of first episode psychosis patients also scanned at 7T, Wang reported both a decrease in anterior cingulate glutamate and GABA levels (108). In the same group of first episode psychosis patients in which we found a glutamate decrease (107), we did not find abnormalities in gamma range oscillations using MEG (109), although others, also in first episode patients, have reported such alterations (110–112). All three of these studies used EEG, and it has been shown that there can be differences in the cortical auditory evoked response between MEG and EEG (113). Inspired by imaging studies reporting decrease in N-acetyl-aspartate, a marker of neuronal integrity, in the anterior cingulate cortex in schizophrenia (114), Roberts (115) used electron microscopy in anterior cingulate cortex postmortem brain samples and identified a decrease in the number of excitatory synaptic connections, as well as a decrease in the number of mitochondria per neuronal somata, suggesting a decrease in cortical efficiency in schizophrenia.
Our work has contributed to the extensive efforts in studying the structural and functional neuroanatomy of schizophrenia by mapping relevant neural signatures in psychosis spectrum patients. Our data is congruent with the idea that the brain is fundamentally affected in the illness, where widespread structural gray and white matter involvement, functionally abnormal cortical and subcortical information processing, and neurometabolic dysregulation are present in patients. Importantly, data indicate that pathology is not merely diffusely distributed across the entire brain, rather it appears that certain brain circuits are preferentially affected. The evidence is compelling that many of the subtle abnormalities described in chronic schizophrenia are already evident in first episode psychosis patients, highlighting that these brain signatures are likely to be relevant to the core pathology and not a just be a consequence of other non-specific factors associated with the illness (116). We also demonstrated that these brain circuitry alterations are clinically relevant by showing that these pathological signatures can be leveraged for predicting subsequent response to antipsychotic treatment, further underscoring that they are key features of the illness.
Despite these successes in delineating disease signatures, it remains challenging to identify casual factors leading to these alterations through cross-sectional unimodal mapping alone (117). To gain further insights, our group has leveraged different approaches. First, we used various pharmacological challenges to characterize the effects of active modulation of major neurotransmitter systems on neural signatures. We clearly demonstrated that dopamine D2 receptor blockers alleviate neural abnormalities to some extent, and that experimentally induced NMDA receptor blockage results in alterations that resemble those seen schizophrenia, underscoring the pathophysiological relevance of these neurotransmitter systems. Second, we combined findings from different imaging modalities to gain additional insights into the role of glutamatergic neurotransmission for the modulation of functional brain networks. Our studies consistently demonstrated a disruption in the link between glutamate levels and brain function in patients across paradigms tapping into various aspects of brain function, which implicates that the disruption in this coupling may be a fundamental feature of the illness. Third, postmortem studies can also inform mechanistic understanding. For example, decrease in mitochondrial function observed in schizophrenia postmortem samples (115) suggest abnormalities in brain bioenergetics.
Taken together, it is highly unlikely that the pathogenesis of schizophrenia is uniform (118), it is more plausible that there may be multiple different etiologies that converge to the behavioral phenotype of schizophrenia (119). Our data underscore that mechanistically oriented neuroimaging studies must take non-specific factors such as antipsychotic drug exposure or illness chronicity into consideration when interpreting disease signatures, as a clear characterization of primary pathophysiological processes is an imperative prerequisite for rational drug development and for alleviating disease burden in our patients.
AL and NK wrote the manuscript. Both authors contributed to and approved the final version of this manuscript.
This work as supported by the National Institute of mental Health [Grant Nos: K23MH06683 and R01MH118484 (NK), R01MH081014, R01MH102951, and R01MH113800 [AL)].
NK serves as consultant for Neurocrine Biosciences, Inc.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Charlson FJ, Ferrari AJ, Santomauro DF, Diminic S, Stockings E, Scott JG, et al. Global epidemiology and burden of schizophrenia: findings from the global burden of disease study 2016. Schizophr Bull. (2018) 44:1195–203. doi: 10.1093/schbul/sby058
2. Johnstone EC, Crow TJ, Frith CD, Husband J, Kreel L. Cerebral ventricular size and cognitive impairment in chronic schizophrenia. Lancet. (1976) 2:924–6. doi: 10.1016/S0140-6736(76)90890-4
3. Reveley AM, Reveley MA, Clifford CA, Murray RM. Cerebral ventricular size in twins discordant for schizophrenia. Lancet. (1982) 1:540–1. doi: 10.1016/S0140-6736(82)92047-5
4. Andreasen NC, Olsen SA, Dennert JW, Smith MR. Ventricular enlargement in schizophrenia: relationship to positive and negative symptoms. Am J Psychiatry. (1982) 139:297–302. doi: 10.1176/ajp.139.3.297
5. Tomelleri L, Jogia J, Perlini C, Bellani M, Ferro A, Rambaldelli G, et al. Neuroimaging Network of the, Brain structural changes associated with chronicity and antipsychotic treatment in schizophrenia. Eur Neuropsychopharmacol. (2009) 19:835–40. doi: 10.1016/j.euroneuro.2009.07.007
6. Seeman P, Kapur S. Schizophrenia: more dopamine, more D2 receptors. Proc Natl Acad Sci USA. (2000) 97:7673–5. doi: 10.1073/pnas.97.14.7673
7. Ananth J, Burgoyne KS, Gadasalli R, Aquino S. How do the atypical antipsychotics work? J Psychiatry Neurosci. (2001) 26:385–94.
8. Reynolds GP. Antipsychotic drug mechanisms and neurotransmitter systems in schizophrenia. Acta Psychiatr Scand Suppl. (1994) 380:36–40. doi: 10.1111/j.1600-0447.1994.tb05830.x
9. Morrison PD, Murray RM. The antipsychotic landscape: dopamine and beyond. Ther Adv Psychopharmacol. (2018) 8:127–35. doi: 10.1177/2045125317752915
10. Davis CE, Jeste DV, Eyler LT. Review of longitudinal functional neuroimaging studies of drug treatments in patients with schizophrenia. Schizophr Res. (2005) 78:45–60. doi: 10.1016/j.schres.2005.05.009
11. Tarcijonas G, Sarpal DK. Neuroimaging markers of antipsychotic treatment response in schizophrenia: an overview of magnetic resonance imaging studies. Neurobiol Dis. (2019) 131:104209. doi: 10.1016/j.nbd.2018.06.021
12. Egerton A, Stone JM, Chaddock CA, Barker GJ, Bonoldi I, Howard RM, et al. Relationship between brain glutamate levels and clinical outcome in individuals at ultra high risk of psychosis. Neuropsychopharmacology. (2014) 39:2891–9. doi: 10.1038/npp.2014.143
13. Gonzalez-Vivas C, Soldevila-Matias P, Sparano O, Garcia-Marti G, Marti-Bonmati L, Crespo-Facorro B, et al. Longitudinal studies of functional magnetic resonance imaging in first-episode psychosis: a systematic review. Eur Psychiatry. (2019) 59:60–9. doi: 10.1016/j.eurpsy.2019.04.009
14. Garcia GJ, Chagas MH, Silva CH, Machado-de-Sousa JP, Crippa JA, Hallak JE. Structural and functional neuroimaging findings associated with the use of clozapine in schizophrenia: a systematic review. Braz J Psychiatry. (2015) 37:71–9. doi: 10.1590/1516-4446-2014-1387
15. Roiz-Santianez R, Suarez-Pinilla P, Crespo-Facorro B. Brain structural effects of antipsychotic treatment in schizophrenia: a systematic review. Curr Neuropharmacol. (2015) 13:422–34. doi: 10.2174/1570159X13666150429002536
16. Nelson EA, White DM, Kraguljac NV, Lahti AC. Gyrification connectomes in unmedicated patients with schizophrenia and following a short course of antipsychotic drug treatment. Front Psychiatry. (2018) 9:699. doi: 10.3389/fpsyt.2018.00699
17. Nelson EA, Kraguljac NV, White DM, Jindal RD, Shin AL, Lahti AC. A prospective longitudinal investigation of cortical thickness and gyrification in schizophrenia. Can J Psychiatry. (2020) 65:381–91. doi: 10.1177/0706743720904598
18. Kraguljac NV, Anthony T, Skidmore FM, Marstrander J, Morgan CJ, Reid MA, et al. Micro- and macrostructural white matter integrity in never-treated and currently unmedicated patients with schizophrenia and effects of short-term antipsychotic treatment. Biol Psychiatry Cogn Neurosci Neuroimaging. (2019) 4:462–71. doi: 10.1016/j.bpsc.2019.01.002
19. Kraguljac NV, Anthony T, Monroe WS, Skidmore FM, Morgan CJ, White DM, et al. A longitudinal neurite and free water imaging study in patients with a schizophrenia spectrum disorder. Neuropsychopharmacology. (2019) 44:1932–9. doi: 10.1038/s41386-019-0427-3
20. Hutcheson NL, Sreenivasan KR, Deshpande G, Reid MA, Hadley J, White DM, et al. Effective connectivity during episodic memory retrieval in schizophrenia participants before and after antipsychotic medication. Hum Brain Mapp. (2015) 36:1442–57. doi: 10.1002/hbm.22714
21. Cadena EJ, White DM, Kraguljac NV, Reid MA, Lahti AC. Evaluation of fronto-striatal networks during cognitive control in unmedicated patients with schizophrenia and the effect of antipsychotic medication. NPJ Schizophr. (2018) 4:8. doi: 10.1038/s41537-018-0051-y
22. Cadena EJ, White DM, Kraguljac NV, Reid MA, Maximo JO, Nelson EA, et al. A longitudinal multimodal neuroimaging study to examine relationships between resting state glutamate and task related BOLD response in schizophrenia. Front Psychiatry. (2018) 9:632. doi: 10.3389/fpsyt.2018.00632
23. Cadena EJ, White DM, Kraguljac NV, Reid MA, Jindal R, Pixley RM, et al. Cognitive control network dysconnectivity and response to antipsychotic treatment in schizophrenia. Schizophr Res. (2019) 204:262–70. doi: 10.1016/j.schres.2018.07.045
24. Gurler D, White DM, Kraguljac NV, Ver Hoef L, Martin C, Tennant B, et al. Neural signatures of memory encoding in schizophrenia are modulated by antipsychotic treatment. Neuropsychobiology. (2021) 80:12–24. doi: 10.1159/000506402
25. Hadley JA, Nenert R, Kraguljac NV, Bolding MS, White DM, Skidmore FM, et al. Ventral tegmental area/midbrain functional connectivity and response to antipsychotic medication in schizophrenia. Neuropsychopharmacology. (2014) 39:1020–30. doi: 10.1038/npp.2013.305
26. Hadley JA, Kraguljac NV, White DM, Ver Hoef L, Tabora J, Lahti AC. Change in brain network topology as a function of treatment response in schizophrenia: a longitudinal resting-state fMRI study using graph theory. NPJ Schizophr. (2016) 2:16014. doi: 10.1038/npjschz.2016.14
27. Kraguljac NV, White DM, Hadley N, Hadley JA, Ver Hoef L, Davis E, et al. Aberrant hippocampal connectivity in unmedicated patients with schizophrenia and effects of antipsychotic medication: a longitudinal resting state functional MRI study. Schizophr Bull. (2016) 42:1046–55. doi: 10.1093/schbul/sbv228
28. Kraguljac NV, White DM, Hadley JA, Visscher K, Knight D, ver Hoef L, et al. Abnormalities in large scale functional networks in unmedicated patients with schizophrenia and effects of risperidone. Neuroimage Clin. (2016) 10:146–58. doi: 10.1016/j.nicl.2015.11.015
29. Lottman KK, Kraguljac NV, White DM, Morgan CJ, Calhoun VD, Butt A, et al. Risperidone effects on brain dynamic connectivity-a prospective resting-state fMRI study in Schizophrenia. Front Psychiatry. (2017) 8:14. doi: 10.3389/fpsyt.2017.00014
30. Kraguljac NV, Morgan CJ, Reid MA, White DM, Jindal RD, Sivaraman S, et al. A longitudinal magnetic resonance spectroscopy study investigating effects of risperidone in the anterior cingulate cortex and hippocampus in schizophrenia. Schizophr Res. (2019) 210:239–44. doi: 10.1016/j.schres.2018.12.028
31. Birur B, Kraguljac NV, VerHoef L, Morgan CJ, Jindal RD, Reid MA, et al. Neurometabolic correlates of 6 and 16 weeks of treatment with risperidone in medication-naive first-episode psychosis patients. Transl Psychiatry. (2020) 10:15. doi: 10.1038/s41398-020-0700-6
32. Lahti AC, Holcomb HH, Weiler MA, Medoff DR, Tamminga CA. Functional effects of antipsychotic drugs: comparing clozapine with haloperidol. Biol Psychiatry. (2003) 53:601–8. doi: 10.1016/S0006-3223(02)01602-5
33. Lahti AC, Holcomb HH, Weiler MA, Medoff DR, Frey KN, Hardin M, et al. Clozapine but not haloperidol Re-establishes normal task-activated rCBF patterns in schizophrenia within the anterior cingulate cortex. Neuropsychopharmacology. (2004) 29:171–8. doi: 10.1038/sj.npp.1300312
34. Lahti AC, Weiler MA, Medoff DR, Tamminga CA, Holcomb HH. Functional effects of single dose first- and second-generation antipsychotic administration in subjects with schizophrenia. Psychiatry Res. (2005) 139:19–30. doi: 10.1016/j.pscychresns.2005.02.006
35. Lahti AC, Weiler MA, Holcomb HH, Tamminga CA, Cropsey KL. Modulation of limbic circuitry predicts treatment response to antipsychotic medication: a functional imaging study in schizophrenia. Neuropsychopharmacology. (2009) 34:2675–90. doi: 10.1038/npp.2009.94
36. Bolding MS, White DM, Hadley JA, Weiler M, Holcomb HH, Lahti AC. Antipsychotic drugs alter functional connectivity between the medial frontal cortex, hippocampus, and nucleus accumbens as measured by H215O PET. Front Psychiatry. (2012) 3:105. doi: 10.3389/fpsyt.2012.00105
37. Kuipers SD, Bramham CR. Brain-derived neurotrophic factor mechanisms and function in adult synaptic plasticity: new insights and implications for therapy. Curr Opin Drug Discov Devel. (2006) 9:580–586.
38. Pandya CD, Kutiyanawalla A, Pillai A. BDNF-TrkB signaling and neuroprotection in schizophrenia. Asian J Psychiatry. (2013) 6:22–8. doi: 10.1016/j.ajp.2012.08.010
39. Tyler WJ, Pozzo-Miller L. Miniature synaptic transmission and BDNF modulate dendritic spine growth and form in rat CA1 neurones. J Physiol. (2003) 553:497–509. doi: 10.1113/jphysiol.2003.052639
40. Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. (2003) 112:257–69. doi: 10.1016/S0092-8674(03)00035-7
41. Pezawas L, Verchinski BA, Mattay VS, Callicott JH, Kolachana BS, Straub RE, et al. The brain-derived neurotrophic factor val66met polymorphism and variation in human cortical morphology. J Neurosci. (2004) 24:10099–102. doi: 10.1523/JNEUROSCI.2680-04.2004
42. Montag C, Schoene-Bake JC, Faber J, Reuter M, Weber B. Genetic variation on the BDNF gene is not associated with differences in white matter tracts in healthy humans measured by tract-based spatial statistics. Genes Brain Behav. (2010) 9:886–91. doi: 10.1111/j.1601-183X.2010.00626.x
43. Fernandes BS, Steiner J, Berk M, Molendijk ML, Gonzalez-Pinto A, Turck CW, et al. Peripheral brain-derived neurotrophic factor in schizophrenia and the role of antipsychotics: meta-analysis and implications. Mol Psychiatry. (2015) 20:1108–19. doi: 10.1038/mp.2014.117
44. Park SW, Lee CH, Cho HY, Seo MK, Lee JG, Lee BJ, et al. Effects of antipsychotic drugs on the expression of synaptic proteins and dendritic outgrowth in hippocampal neuronal cultures. Synapse. (2013) 67:224–234. doi: 10.1002/syn.21634
45. Lieberman JA, Bymaster FP, Meltzer HY, Deutch AY, Duncan GE, Marx CE, et al. Antipsychotic drugs : comparison in animal models of efficacy, neurotransmitter regulation, and neuroprotection. Pharmacol Rev. (2008) 60:358–403. doi: 10.1124/pr.107.00107
46. Cole DM, Beckmann CF, Oei NY, Both S, van Gerven JM, Rombouts SA. Differential and distributed effects of dopamine neuromodulations on resting-state network connectivity. NeuroImage. (2013) 78:59–67. doi: 10.1016/j.neuroimage.2013.04.034
47. Kargieman L, Riga MS, Artigas F, Celada P. Clozapine reverses phencyclidine-induced desynchronization of prefrontal cortex through a 5-HT1A receptor-dependent mechanism. Neuropsychopharmacology. (2012) 37:723–33. doi: 10.1038/npp.2011.249
48. Celada P, Puig MV, Díaz-Mataix L, Artigas F. The hallucinogen DOI reduces low-frequency oscillations in rat prefrontal cortex: reversal by antipsychotic drugs. Biol Psychiatry. (2008) 64:392–400. doi: 10.1016/j.biopsych.2008.03.013
49. Ngan ET, Lane CJ, Ruth TJ, Liddle PF. Immediate and delayed effects of risperidone on cerebral metabolism in neuroleptic naive schizophrenic patients: correlations with symptom change. J Neurol Neurosurg Psychiatry. (2002) 72:106–10. doi: 10.1136/jnnp.72.1.106
50. Holcomb HH, Cascella NG, Thaker GK, Medoff DR, Dannals RF, Tamminga CA. Functional sites of neuroleptic drug action in the human brain: PET/FDG studies with and without haloperidol. Am J Psychiatry. (1996) 153:41–9. doi: 10.1176/ajp.153.1.41
51. Miller DD, Andreasen NC, O'Leary DS, Rezai K, Watkins GL, Ponto LL, et al. Effect of antipsychotics on regional cerebral blood flow measured with positron emission tomography. Neuropsychopharmacology. (1997) 17:230–40. doi: 10.1016/S0893-133X(97)00042-0
52. Snitz BE, MacDonald A, 3rd, Cohen JD, Cho RY, Becker T, Carter CS. Lateral and medial hypofrontality in first-episode schizophrenia: functional activity in a medication-naive state and effects of short-term atypical antipsychotic treatment. Am J Psychiatry. (2005) 162:2322–9. doi: 10.1176/appi.ajp.162.12.2322
53. Sarpal DK, Argyelan M, Robinson DG, Szeszko PR, Karlsgodt KH, John M, et al. Baseline striatal functional connectivity as a predictor of response to antipsychotic drug treatment. Am J Psychiatry. (2016) 173:69–77. doi: 10.1176/appi.ajp.2015.14121571
54. Sarpal DK, Robinson DG, Lencz T, Argyelan M, Ikuta T, Karlsgodt K, et al. Antipsychotic treatment and functional connectivity of the striatum in first-episode schizophrenia. JAMA Psychiatry. (2015) 72:5–13. doi: 10.1001/jamapsychiatry.2014.1734
55. Krzystanek M, Bogus K, Palasz A, Krzystanek E, Worthington JJ, Wiaderkiewicz R. Effects of long-term treatment with the neuroleptics haloperidol, clozapine and olanzapine on immunoexpression of NMDA receptor subunits NR1, NR2A and NR2B in the rat hippocampus. Pharmacol Reports. (2015) 67:965–9. doi: 10.1016/j.pharep.2015.01.017
56. Hanaoka T, Toyoda H, Mizuno T, Kikuyama H, Morimoto K, Takahata R, et al. Alterations in NMDA receptor subunit levels in the brain regions of rats chronically administered typical or atypical antipsychotic drugs. Neurochem Res. (2003) 28:919–24. doi: 10.1023/A:1023231611616
57. Del'guidice T, Beaulieu JM. Messing up with traffic: different effects of antipsychotic agents on glutamate receptor complexes in vivo. Mol Pharmacol. (2008) 73:1339–42. doi: 10.1124/mol.108.046540
58. Szulc A, Galinska B, Tarasow E, Waszkiewicz N, Konarzewska B, Poplawska R, et al. Proton magnetic resonance spectroscopy study of brain metabolite changes after antipsychotic treatment. Pharmacopsychiatry. (2011) 44:148–57. doi: 10.1055/s-0031-1279739
59. Egerton A, Broberg BV, Van Haren N, Merritt K, Barker GJ, Lythgoe DJ, et al. Response to initial antipsychotic treatment in first episode psychosis is related to anterior cingulate glutamate levels: a multicentre (1)H-MRS study (OPTiMiSE). Mol Psychiatry. (2018) 23:2145–55. doi: 10.1038/s41380-018-0082-9
60. Merritt K, Perez-Iglesias R, Sendt KV, Goozee R, Jauhar S, Pepper F, et al. Remission from antipsychotic treatment in first episode psychosis related to longitudinal changes in brain glutamate. NPJ Schizophr. (2019) 5:12. doi: 10.1038/s41537-019-0080-1
61. Hutcheson NL, Clark DG, Bolding MS, White DM, Lahti AC. Basal ganglia volume in unmedicated patients with schizophrenia is associated with treatment response to antipsychotic medication. Psychiatry Res. (2014) 221:6–12. doi: 10.1016/j.pscychresns.2013.10.002
62. Kraguljac NV, Anthony T, Morgan CJ, Jindal RD, Burger MS, Lahti AC. White matter integrity, duration of untreated psychosis, and antipsychotic treatment response in medication-naive first-episode psychosis patients. Mol Psychiatry. (2020). doi: 10.1038/s41380-020-0765-x
63. Kraguljac NV, McDonald WM, Widge AS, Rodriguez CI, Tohen M, Nemeroff CB. Neuroimaging biomarkers in schizophrenia. Am J Psychiatry. (2021). doi: 10.1176/appi.ajp.2020.20030340. [Epub ahead of print].
64. Dazzan P. Neuroimaging biomarkers to predict treatment response in schizophrenia: the end of 30 years of solitude? Dialogues Clin Neurosci. (2014) 16:491–503. doi: 10.31887/DCNS.2014.16.4/pdazzan
65. Heckers S, Neuropathology of schizophrenia: cortex, thalamus, basal ganglia, and neurotransmitter-specific projection systems. Schizophr Bull. (1997) 23:403-21. doi: 10.1093/schbul/23.3.403
66. Hulshoff Pol HE, Kahn RS. What happens after the first episode? A review of progressive brain changes in chronically ill patients with schizophrenia. Schizophr Bull. (2008) 34:354–66. doi: 10.1093/schbul/sbm168
67. Kempton MJ, McGuire P. How can neuroimaging facilitate the diagnosis and stratification of patients with psychosis? Eur Neuropsychopharmacol. (2015) 25:725–32. doi: 10.1016/j.euroneuro.2014.07.006
68. Maximo JO, Nelson EA, Armstrong WP, Kraguljac NV, Lahti AC. Duration of untreated psychosis correlates with brain connectivity and morphology in medication-naive patients with first-episode psychosis. Biol Psychiatry Cogn Neurosci Neuroimaging. (2020) 5:231–8. doi: 10.1016/j.bpsc.2019.10.014
69. Briend F, Nelson EA, Maximo O, Armstrong WP, Kraguljac NV, Lahti AC. Hippocampal glutamate and hippocampus subfield volumes in antipsychotic-naive first episode psychosis subjects and relationships to duration of untreated psychosis. Transl Psychiatry. (2020) 10:137. doi: 10.1038/s41398-020-0812-z
70. Briend F, Armstrong WP, Kraguljac NV, Keilhloz SD, Lahti AC. Aberrant static and dynamic functional patterns of frontoparietal control network in antipsychotic-naive first-episode psychosis subjects. Hum Brain Mapp. (2020) 41:2999–3008. doi: 10.1002/hbm.24992
71. Nelson EA, Kraguljac NV, Maximo JO, Briend F, Armstrong W, Ver Hoef LW, et al. Hippocampal dysconnectivity and altered glutamatergic modulation of the default mode network: a combined resting-state connectivity and magnetic resonance spectroscopy study in schizophrenia. Biol Psychiatry Cogn Neurosci Neuroimaging. (2020). doi: 10.1016/j.bpsc.2020.04.014. [Epub ahead of print].
72. Sivaraman S, Kraguljac NV, White DM, Morgan CJ, Gonzales SS, Lahti AC. Neurometabolic abnormalities in the associative striatum in antipsychotic-naive first episode psychosis patients. Psychiatry Res Neuroimaging. (2018) 281:101–6. doi: 10.1016/j.pscychresns.2018.06.003
73. Pettersson-Yeo W, Allen P, Benetti S, McGuire P, Mechelli A. Dysconnectivity in schizophrenia: where are we now? Neurosci Biobehav Rev. (2011) 35:1110–24. doi: 10.1016/j.neubiorev.2010.11.004
74. M.P. van den Heuvel, Fornito A. Brain networks in schizophrenia. Neuropsychol Rev. (2014) 24:32–48. doi: 10.1007/s11065-014-9248-7
75. Baker JT, Holmes AJ, Masters GA, Yeo BT, Krienen F, Buckner RL, et al. Disruption of cortical association networks in schizophrenia and psychotic bipolar disorder. JAMA Psychiatry. (2014) 71:109–18. doi: 10.1001/jamapsychiatry.2013.3469
76. Goff DC, Romero K, Paul J, M. Mercedes Perez-Rodriguez, Crandall D, Potkin SG. Biomarkers for drug development in early psychosis: current issues and promising directions. Eur Neuropsychopharmacol. (2016) 26:923–37. doi: 10.1016/j.euroneuro.2016.01.009
77. Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. (1995) 52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004
78. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. (1991) 148:1301–8. doi: 10.1176/ajp.148.10.1301
79. Hyder F, Rothman DL, Shulman RG, Total neuroenergetics support localized brain activity: implications for the interpretation of fMRI. Proc Natl Acad Sci USA. (2002) 99:10771–6. doi: 10.1073/pnas.132272299
80. Smith AJ, Blumenfeld H, Behar KL, Rothman DL, Shulman RG, Hyder F, et al. Cerebral energetics and spiking frequency: the neurophysiological basis of fMRI. Proc Natl Acad Sci USA. (2002) 99:10765–70. doi: 10.1073/pnas.132272199
81. Duncan NW, Wiebking C, Northoff G. Associations of regional GABA and glutamate with intrinsic and extrinsic neural activity in humans-a review of multimodal imaging studies. Neurosci Biobehav Rev. (2014) 47:36–52. doi: 10.1016/j.neubiorev.2014.07.016
82. Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. (2012) 37:4–15. doi: 10.1038/npp.2011.181
83. Matute C, Alberdi E, Domercq M, Sanchez-Gomez MV, Perez-Samartin A, Rodriguez-Antiguedad A, et al. Excitotoxic damage to white matter. J Anat. (2007) 210:693–702. doi: 10.1111/j.1469-7580.2007.00733.x
84. Hassel B, Boldingh KA, Narvesen C, Iversen EG, Skrede KK. Glutamate transport, glutamine synthetase and phosphate-activated glutaminase in rat CNS white matter. A quantitative study. J Neurochem. (2003) 87:230–7. doi: 10.1046/j.1471-4159.2003.01984.x
85. Karadottir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. (2005) 438:1162–6. doi: 10.1038/nature04302
86. Hutcheson NL, Reid MA, White DM, Kraguljac NV, Avsar KB, Bolding MS, et al. Multimodal analysis of the hippocampus in schizophrenia using proton magnetic resonance spectroscopy and functional magnetic resonance imaging. Schizophr Res. (2012) 140:136–42. doi: 10.1016/j.schres.2012.06.039
87. Overbeek G, Gawne TJ, Reid MA, Salibi N, Kraguljac NV, White DM, et al. Relationship between cortical excitation and inhibition and task-induced activation and deactivation: a combined magnetic resonance spectroscopy and functional magnetic resonance imaging study at 7T in first-episode psychosis. Biol Psychiatry Cogn Neurosci Neuroimaging. (2019) 4:121–30. doi: 10.1016/j.bpsc.2018.10.002
88. White DM, Kraguljac NV, Reid MA, Lahti AC. Contribution of substantia nigra glutamate to prediction error signals in schizophrenia: a combined magnetic resonance spectroscopy/functional imaging study. NPJ Schizophr. (2015) 1:14001. doi: 10.1038/npjschz.2014.1
89. Kraguljac NV, White DM, Hadley J, Reid MA, Lahti AC. Hippocampal-parietal dysconnectivity and glutamate abnormalities in unmedicated patients with schizophrenia. Hippocampus. (2014) 24:1524–32. doi: 10.1002/hipo.22332
90. Kraguljac NV, White DM, Reid MA, Lahti AC. Increased hippocampal glutamate and volumetric deficits in unmedicated patients with schizophrenia. JAMA Psychiatry. (2013) 70:1294–302. doi: 10.1001/jamapsychiatry.2013.2437
91. Plitman E, Patel R, Chung JK, Pipitone J, Chavez S, Reyes-Madrigal F, et al. Glutamatergic metabolites, volume and cortical thickness in antipsychotic-naive patients with first-episode psychosis: implications for excitotoxicity. Neuropsychopharmacology. (2016) 41:2606–13. doi: 10.1038/npp.2016.84
92. Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology. (2001) 25:455–67. doi: 10.1016/S0893-133X(01)00243-3
93. Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. (1995) 13:9–19. doi: 10.1016/0893-133X(94)00131-I
94. Kraguljac NV, Carle M, Frolich MA, Tran S, Yassa MA, White DM, et al. Mnemonic discrimination deficits in first-episode psychosis and a ketamine model suggests dentate gyrus pathology linked to N-Methyl-D-Aspartate receptor hypofunction. Biol Psychiatry Cogn Neurosci Neuroimaging. (2018) 3:231–8. doi: 10.1016/j.bpsc.2017.02.005
95. Parwani A, Weiler MA, Blaxton TA, Warfel D, Hardin M, Frey K, et al. The effects of a subanesthetic dose of ketamine on verbal memory in normal volunteers. Psychopharmacology. (2005) 183:265–74. doi: 10.1007/s00213-005-0177-2
96. Weiler MA, Thaker GK, Lahti AC, Tamminga CA. Ketamine effects on eye movements. Neuropsychopharmacology. (2000) 23:645–53. doi: 10.1016/S0893-133X(00)00156-1
97. Holcomb HH, Lahti AC, Medoff DR, Cullen T, Tamminga CA. Effects of noncompetitive NMDA receptor blockade on anterior cingulate cerebral blood flow in volunteers with schizophrenia. Neuropsychopharmacology. (2005) 30:2275–82. doi: 10.1038/sj.npp.1300824
98. Lahti AC, Holcomb HH, Medoff DR, Tamminga CA. Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport. (1995) 6:869–72. doi: 10.1097/00001756-199504190-00011
99. Holcomb HH, Lahti AC, Medoff DR, Weiler M, Tamminga CA. Sequential regional cerebral blood flow brain scans using PET with H2(15)O demonstrate ketamine actions in CNS dynamically. Neuropsychopharmacology. (2001) 25:165–72. doi: 10.1016/S0893-133X(01)00229-9
100. Bryant JE, Frolich M, Tran S, Reid MA, Lahti AC, Kraguljac NV. Ketamine induced changes in regional cerebral blood flow, interregional connectivity patterns, and glutamate metabolism. J Psychiatr Res. (2019) 117:108–15. doi: 10.1016/j.jpsychires.2019.07.008
101. Kraguljac NV, Frolich MA, Tran S, White DM, Nichols N, Barton-McArdle A, et al. Ketamine modulates hippocampal neurochemistry and functional connectivity: a combined magnetic resonance spectroscopy and resting-state fMRI study in healthy volunteers. Mol Psychiatry. (2017) 22:562–9. doi: 10.1038/mp.2016.122
102. Kantrowitz JT, Grinband J, Goff DC, Lahti AC, Marder SR, Kegeles LS, et al. Proof of mechanism and target engagement of glutamatergic drugs for the treatment of schizophrenia: RCTs of pomaglumetad and TS-134 on ketamine-induced psychotic symptoms and pharmacoBOLD in healthy volunteers. Neuropsychopharmacology. (2020) 45:1842–50. doi: 10.1101/2020.03.09.20029827
103. de Jonge JC, Vinkers CH, Hulshoff Pol HE, Marsman A. GABAergic mechanisms in schizophrenia: linking postmortem and in vivo studies. Front Psychiatry. (2017) 8:118. doi: 10.3389/fpsyt.2017.00118
104. Glausier JR, Lewis DA. GABA and schizophrenia: where we stand and where we need to go. Schizophr Res. (2017) 181:2–3. doi: 10.1016/j.schres.2017.01.050
105. Hu W, MacDonald ML, Elswick DE, Sweet RA. The glutamate hypothesis of schizophrenia: evidence from human brain tissue studies. Ann N Y Acad Sci. (2015) 1338:38–57. doi: 10.1111/nyas.12547
106. Whittington MA, Traub RD, Jefferys JG. Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation. Nature. (1995) 373:612–5. doi: 10.1038/373612a0
107. Reid MA, Salibi N, White DM, Gawne TJ, Denney TS, and Lahti AC. 7T proton magnetic resonance spectroscopy of the anterior cingulate cortex in first-episode schizophrenia. Schizophr Bull. (2018) 45:180–9. doi: 10.1093/schbul/sbx190
108. Wang AM, Pradhan S, Coughlin JM, Trivedi A, DuBois SL, Crawford JL, et al. Assessing brain metabolism with 7-T proton magnetic resonance spectroscopy in patients with first-episode psychosis. JAMA Psychiatry. (2019) 76:314–23. doi: 10.1001/jamapsychiatry.2018.3637
109. Gawne TJ, Overbeek G, Killen JF, Kraguljac NV, White DM, Reid MA, et al. A multimodal magnetoencephalography, 7T fMRI stroop and 7T proton MR spectroscopy study in first episode psychosis. (2020) 6:23. doi: 10.1093/schbul/sbx023.070
110. Spencer KM, Niznikiewicz MA, Nestor PG, Shenton ME, McCarley RW. Left auditory cortex gamma synchronization and auditory hallucination symptoms in schizophrenia. BMC Neurosci. (2009) 10:85. doi: 10.1186/1471-2202-10-85
111. Tada M, Nagai T, Kirihara K, Koike S, Suga M, Araki T, et al. Differential alterations of auditory gamma oscillatory responses between pre-onset high-risk individuals and first-episode schizophrenia. Cereb Cortex. (2016) 26:1027–35. doi: 10.1093/cercor/bhu278
112. Wang J, Tang Y, Curtin A, Chan CKR, Wang Y, Li H, et al. Abnormal auditory-evoked gamma band oscillations in first-episode schizophrenia during both eye open and eye close states. Prog Neuropsychopharmacol Biol Psychiatry. (2018) 86:279–86. doi: 10.1016/j.pnpbp.2018.04.016
113. Edgar JC, Fisk CI, Chen YH, Stone-Howell B, Hunter MA, Huang M, et al. By our bootstraps: comparing methods for measuring auditory 40 Hz steady-state neural activity. Psychophysiology. (2017) 54:1110–27. doi: 10.1111/psyp.12876
114. Reid MA, Stoeckel LE, White DM, Avsar KB, Bolding MS, Akella NS, et al. Assessments of function and biochemistry of the anterior cingulate cortex in schizophrenia. Biol Psychiatry. (2010) 68:625–33. doi: 10.1016/j.biopsych.2010.04.013
115. Roberts RC, McCollum LA, Schoonover KE, Mabry SJ, Roche JK, Lahti AC. Ultrastructural evidence for glutamatergic dysregulation in schizophrenia. Schizophr Res. (2020). doi: 10.1016/j.schres.2020.01.016. [Epub ahead of print].
116. Kalin NH. Gaining ground on schizophrenia: conceptualizing how to use neuroimaging and genomics in its diagnosis and treatment. Am J Psychiatry. (2019) 176:771–3. doi: 10.1176/appi.ajp.2019.19080828
117. Meyer-Lindenberg A. From maps to mechanisms through neuroimaging of schizophrenia. Nature. (2010) 468:194–202. doi: 10.1038/nature09569
118. Kahn RS, Sommer IE. The neurobiology and treatment of first-episode schizophrenia. Mol Psychiatry. (2015) 20:84–97. doi: 10.1038/mp.2014.66
Keywords: pharmacological challenge, treatment response, duration of untreated psychosis, functional MRI, magnetic resonance spectroscopy, diffusion weighted imaging, first episode psychosis, antipsychotic naïve
Citation: Kraguljac NV and Lahti AC (2021) Neuroimaging as a Window Into the Pathophysiological Mechanisms of Schizophrenia. Front. Psychiatry 12:613764. doi: 10.3389/fpsyt.2021.613764
Received: 03 October 2020; Accepted: 15 February 2021;
Published: 11 March 2021.
Edited by:
Luca De Peri, Cantonal Sociopsychological Organization, SwitzerlandReviewed by:
Jessica A. Turner, Georgia State University, United StatesCopyright © 2021 Kraguljac and Lahti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrienne Carol Lahti, YWxhaHRpQHVhYm1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.