 Heidi N. Boyda1Amanzo A. Ho1Lurdes Tse1Ric M. Procyshyn2
Heidi N. Boyda1Amanzo A. Ho1Lurdes Tse1Ric M. Procyshyn2 Jessica W. Y. Yuen2
Jessica W. Y. Yuen2 David D. Kim1William G. Honer2
David D. Kim1William G. Honer2 Alasdair M. Barr1*
Alasdair M. Barr1*- 1Department of Anesthesiology, Pharmacology and Therapeutics, Faculty of Medicine, University of British Columbia, Vancouver, BC, Canada
- 2Department of Psychiatry, Faculty of Medicine, University of British Columbia, Vancouver, BC, Canada
Antipsychotic drugs represent the most effective treatment for chronic psychotic disorders. The newer second generation drugs offer the advantage of fewer neurological side-effects compared to prior drugs, but many cause serious metabolic side-effects. The underlying physiology of these side-effects is not well-understood, but evidence exists to indicate that the sympathetic nervous system may play an important role. In order to examine this possibility further, we treated separate groups of adult female rats acutely with either the first generation antipsychotic drug haloperidol (0.1 or 1 mg/kg) or the second generation drugs risperidone (0.25 or 2.5 mg/kg), clozapine (2 or 20 mg/kg), olanzapine (3 or 15 mg/kg) or vehicle by intraperitoneal injection. Blood samples were collected prior to drug and then 30, 60, 120, and 180 mins after treatment. Plasma samples were assayed by HPLC-ED for levels of norepinephrine, epinephrine, and dopamine. Results confirmed that all antipsychotics increased peripheral catecholamines, although this was drug and dose dependent. For norepinephrine, haloperidol caused the smallest maximum increase (+158%], followed by risperidone (+793%), olanzapine (+952%) and clozapine (+1,684%). A similar pattern was observed for increases in epinephrine levels by haloperidol (+143%], olanzapine (+529%), risperidone (+617%) then clozapine (+806%). Dopamine levels increased moderately with olanzapine [+174%], risperidone [+271%], and clozapine [+430%]. Interestingly, levels of the catecholamines did not correlate strongly with each other prior to treatment at baseline, but were increasingly correlated after treatment as time proceeded. The results demonstrate antipsychotics can potently regulate peripheral catecholamines, in a manner consistent with their metabolic liability.
Introduction
The second generation antipsychotic drugs (also known as the “atypical” antipsychotics, to differentiate them from the original first generation, “typical,” antipsychotics) represent the most effective pharmacological treatment for chronic psychotic illnesses, which include the schizophrenia spectrum disorders (1). This class of drugs is also increasingly being used to treat additional psychiatric indications, such as bipolar disorder, mood, and anxiety disorders (2–7). The widespread preference for the use of the second over the first generation antipsychotics is largely driven by the lower incidence of neurological side-effects in the former, which include extrapyramidal symptoms and tardive dyskinesia (8), as well as lower rates of neuroendocrine abnormalities such as hyperprolactinemia (9, 10). The second generation drugs, however, have been linked to their own serious side-effects as well. These most commonly include cardiometabolic side-effects, which significantly increase the risk of developing cardiometabolic disorders such as Type 2 diabetes mellitus (DM) and cardiovascular disease (11–19). Typically, Type 2 DM is preceded by the onset of the metabolic syndrome, which is characterized by weight gain, hypertension, hyperlipidemia, hyperglycemia, glucose intolerance, and insulin resistance (20–22). It is important to note, though, that these metabolic side-effects vary considerably between the different second generation antipsychotics, with some drugs having more severe metabolic effects than others (23, 24).
Currently, the physiological substrates that mediate the metabolic side-effects of the second generation antipsychotic drugs remain incompletely understood. As it is likely that invasive procedures will be required to fully elucidate the biochemical pathways involved, which may not be appropriate for application in humans, there is a key need to develop translational animal models of these drug side-effects. The use of preclinical paradigms offers the additional benefit of helping to disentangle the multifactorial causes of metabolic dysregulation in psychiatric illness, where variables such as diet, exercise, and drug use can all contribute to cardiometabolic dysregulation (25–27). Fortunately, the past 10–15 years has seen significant advances in animal models of the metabolic side-effects of antipsychotic drugs (28), which have demonstrated that many of these compounds cause acute glucose dysregulation and insulin resistance independent of weight gain (29–39).
The biochemical substrates involved are under examination, and important advances have been made. But the present perspective suggests a complex picture, as both central and peripheral mechanisms may be involved (40). It is also possible that multiple biochemical pathways may be affected, upstream, and downstream of each other. One physiological network that spans both central and peripheral systems, and that has been implicated in the metabolic side-effect of antipsychotic drugs in both animal and human studies, is the sympathetic nervous system. Under physiological conditions, an overactive sympathetic nervous system predicts an increase in metabolic abnormalities over time (41). Increased sympathetic activity; as evident by elevated plasma catecholamines, is a consistently reported effect of high metabolic-risk second generation antipsychotic drugs such as clozapine, whereas minimal effects on catecholamine levels are observed in patients treated with lower metabolic-risk drugs (42–44).
While animal studies also suggest a role for peripheral catecholamines (45, 46), a limitation of the literature is the absence of data on the impact of concurrent multiple different antipsychotics, including both first and second generation drugs (to capture the spectrum of metabolic liability), on peripheral catecholamine levels over repeated time points. To address this gap in the literature, we performed a head-to-head comparison of the effects of treatment with the first generation antipsychotic drug haloperidol—with low metabolic liability—against the second generation drugs risperidone, olanzapine and clozapine—which exert increasing metabolic effects. To increase the validity of the study, multiple doses of each drug were used, based on known metabolic effects reported by our laboratory previously (47–51). Animals were unanesthetized and freely moving, handled by a staff well-trained in stress-free phlebotomy, and catecholamine levels were measured with multiple blood draws over a 3-h period. Catecholamines were measured with a sensitive HPLC-ECD assay.
Materials and Methods
Animals
One hundred twenty female, adult Sprague-Dawley rats (225–250 g) were obtained from the animal supplier (Charles River, Montreal, QC) and allowed to habituate in the UBC animal colony for at least 1 week before all experiments commenced. Females are used by our laboratory and many other groups because they exhibit more consistent metabolic abnormalities than males following antipsychotic drug treatment (28, 33, 52–54). Animals were housed in groups of 3–4 in large polycarbonate cages and given ad libitum access to food (Purina rat chow) and tap water. All rats were maintained on a 12-h light-dark cycle (lights on at 07:00 h) in a temperature-controlled colony at 22 ± 1°C. Experimental procedures were conducted during the light cycle, and in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The University of British Columbia's Animal Care and Use Committee approved all experimental methods.
Pharmaceutical Agents and Solutions
Doses of antipsychotic drugs were based on previous studies of the metabolic side-effects of antipsychotic drugs which we and others have demonstrated previously. A lower and higher dose were selected for each antipsychotic, spanning a 3–10-fold range. Doses for the present study included risperidone (0.25; 2.5 mg/kg), haloperidol (0.1; 1.0 mg/kg), clozapine (2; 20 mg/kg), and olanzapine (3; 15 mg/kg) [purchased from Toronto Research Chemicals Inc, Toronto, ON, Canada]. Dosing solutions were prepared fresh daily: risperidone, clozapine, and olanzapine were formulated in a vehicle composed of 50% polyethylene glycol 400, 40% distilled water, and 10% ethanol. Haloperidol was formulated in a vehicle of 0.3% tartaric acid. All other chemicals were commercially available and of reagent grade. Each rat received a 1 ml/kg intraperitoneal (IP) injection of the vehicle control or antipsychotic formulation. Animals were all experimentally naïve, and randomized to treatment group.
Treatment and Plasma Collection
Fasted animals [n = 10 per group] were weighed and allowed 1 h to acclimate. We used fasted animals, as this is the condition under which we typically assess the metabolic effects of antipsychotic drugs. Rats were then given a saphenous blood draw (200 μL) prior to drug treatment to obtain a baseline measure of plasma catecholamines. Immediately following, animals were treated with their assigned antipsychotic drug or vehicle. Additional blood samples were then collected at 30, 60, 120, and 180 mins after drug treatment. Blood samples were centrifuged (10,000 RPM, 10 mins, 4°C) and stored at −80°C until analysis.
Determination of Plasma Catecholamine Concentration
Standard solutions of epinephrine, norepinephrine and dopamine (50–25,600 pg/ml) were prepared by dilution of a stock 1 mg/ml solution with standard diluent (ThermoFisher, Sunnyvale, CA) to generate a standard curve. The internal standard (IS) solution consisted of 20 ng/ml 3,4-dihydroxybenzylamine (DHBA). Samples were prepared by adding 90 μL of 3 M Tris-5% EDTA buffer, 10 μL of DHBA, and 50 μL of plasma or standard to a 0.6 ml centrifuge tubes containing 5.0 mg activated alumina oxide (Sigma Aldrich, St. Louis, MO). Samples were vortexed and placed on a rotary mixer for 10 mins at 4°C, followed by centrifugation and removal of supernatant. Addition of 400 μl of ultra-pure water followed by aspiration was performed three consecutive times, followed by centrifugation. Addition of 50 μl of 0.1 M perchloric acid was added and samples were mixed on the rotary mixer for 10 mins. Samples were vortexed, centrifuged, and the remaining supernatant was injected into the HPLC system.
HPLC-ECD
Catecholamine levels were analyzed by HPLC coupled to electrochemical detection (ECD). A Shimadzu series HPLC system, including an ESA Coulochem III electrochemical detector, separated analytes on a ESA HR-80 column (80 × 4.6 mm, 3 μm). Mobile phase contained 0.7% sodium phosphate, 3% sodium citrate, 0.02% EDTA, 0.2% diethylamine HCl, 0.1% 1-octanesulfonic acid, 5% acetonitrile, 0.2% dimethylacetamide and water, pH–adjusted to 3.1. A 20 μl injection was loaded at 0.4 ml/min. The ECD system contained an ESA 5020 guard cell (200 mV) and an ESA 5011 analytical cell (E1 = −150 mV; E2 = 225 mV) for acquisition of epinephrine, norepinephrine, dopamine, and IS plasma levels. Data were processed blind to treatment condition, with chromatogram peaks analyzed by the LCsolution software package.
Statistical Analysis
Data obtained included plasma levels of norepinephrine, epinephrine, and dopamine. All data were subjected to a repeated-measures analysis of variance (ANOVA), with drug treatment and dose as between group factors, and time as the within-subjects factor. Main effects or interactions were followed up with the LSD post-hoc test. Correlations were conducted using the Pearson correlation coefficient (IBM SPSS Statistics for Windows, Version 24.0. Armonk, NY: IBM Corp).
Results
Blood samples were successfully collected for all animals at all time points. Observation of the chromatograms demonstrated that peaks for norepinephrine, epinephrine, and dopamine (as well as the IS) were all well-separated and with no overlap (Figure 1). We also identified an unknown peak at ~8–9 mins (peak “X,” Figure 1), which we confirmed was not serotonin, and may represent a catecholamine metabolite.
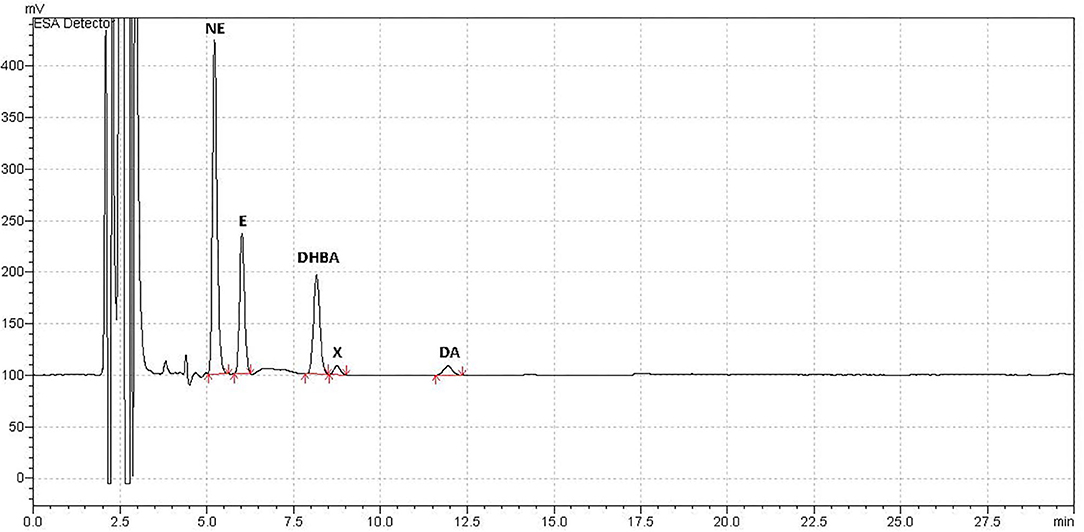
Figure 1. Example Chromatograph. Example of typical chromatograph obtained using HPLC-ED for analysis of plasma catecholamine concentrations in a sample from adult female rats treated with an antipsychotic drug or vehicle. Catecholamine peaks were all well-separated with no peak overlap. Catecholamines measured were norepinephrine (NE), epinephrine (E), and dopamine (DA). The internal standard, added after plasma collection, was 3,4-dihydroxybenzylamine (DHBA). An unidentified peak (“X”) occurred at ~8–8.5 mins, which correlated with catecholamine levels, and may have been a metabolite.
Norepinephrine
When reviewing the data on norepinephrine levels, an analysis for outliers determined that two samples were strong outliers based on SPSS criteria (>3 × the interquartile range) and so were excluded from analysis and replaced with next observation carried backward (55). Analyzing each drug individually, risperidone demonstrated a significant effect of dose [F(2,27) = 31.44, p < 0.001], time [F(4,108) = 29.23, p < 0.001] and a dose × time interaction [F(8,108) = 13.71, p < 0.001]. Post-hoc analysis revealed that both doses of risperidone caused a significant increase in norepinephrine levels by 30 mins compared to controls, but by 60 mins the lower dose of risperidone did not differ from controls, for the duration of the test (Figure 2A). By contrast, norepinephrine levels remained significantly elevated at all time points after treatment with the higher dose of risperidone compared to both the low dose of risperidone and controls.
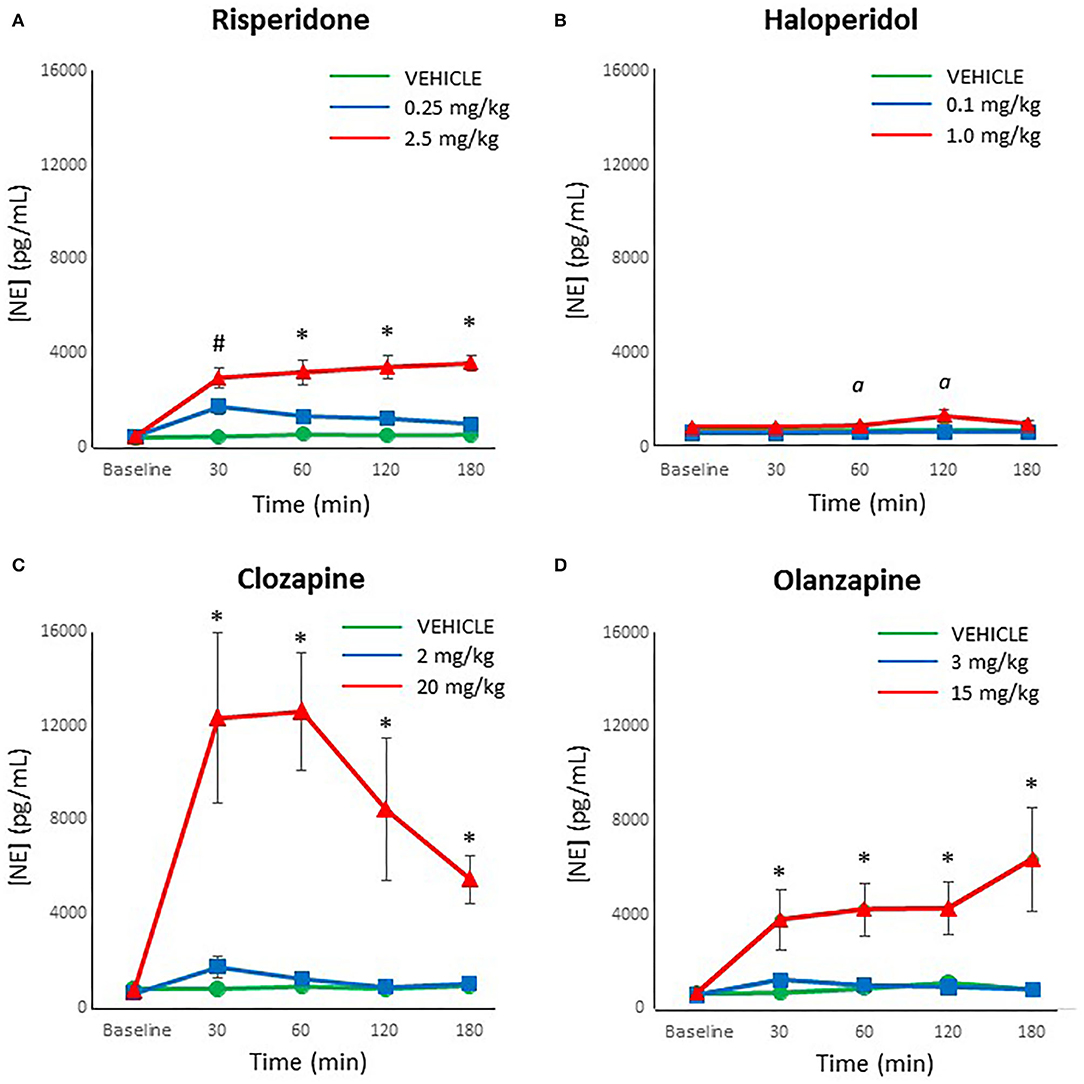
Figure 2. Acute changes in plasma norepinephrine (NE) concentrations (pg/ml) after antipsychotic drug treatment in adult female rats. Animals (n = 10 per group) were given a single IP injection of (A) risperidone (0.25 or 2.5 mg/kg) or (B) haloperidol (0.1 or 1.0 mg/kg) or (C) clozapine (2 or 20 mg/kg) or (D) olanzapine (3 or 15 mg/kg) or vehicle. Values are expressed in group means ± SEM. Plasma samples were collected immediately prior to drug treatment (Baseline) and at 30, 60, 120, and 180 mins after treatment. #Statistically significant difference between both drug treated groups and vehicle, p < 0.05; *Statistically significant difference between high dose group and both other groups, p < 0.05; α Statistically significant difference between high dose group and vehicle, p < 0.05.
For haloperidol, the ANOVA indicated a significant effect of dose [F(2,27) = 7.31, p < 0.005], time [F(4,108) = 5.80, p < 0.001] and a dose × time interaction [F(8,108) = 3.01, p < 0.005]. Post-hoc analysis confirmed that levels of norepinephrine with the low dose of haloperidol did not differ at any time point from controls (Figure 2B). In contrast, the high dose of haloperidol was associated with a trend for increased norepinephrine levels compared to controls at 30 mins after treatment (p = 0.06), and then significantly higher between 60 and 120 mins (p < 0.05), but no longer at 180 mins (p = 0.07).
With clozapine, the ANOVA indicated a significant effect of dose [F(2,27) = 18.60, p < 0.001], time [F(4,108) = 6.82, p < 0.001] and a dose × time interaction [F(8,108) = 5.56, p < 0.001]. Post-hoc analysis demonstrated that levels of norepinephrine with the low dose of clozapine did not differ at any time point from controls (Figure 2C). However, the high dose of clozapine evinced a large, highly significant increase in norepinephrine levels compared to both the controls and the low dose of clozapine at all time points after treatment (p < 0.001).
For olanzapine, analysis of the data by the ANOVA revealed a significant effect of dose [F(2,27) = 11.07, p < 0.001], time [F(4,108) = 4.00, p = 0.005] and a dose × time interaction [F(8,108) = 3.08, p < 0.005]. Similarly to clozapine, the post-hoc analysis indicated that levels of norepinephrine with the low dose of olanzapine did not differ at any time point from controls (Figure 2D). In contrast, the high dose of olanzapine caused a large, significant increase in norepinephrine levels compared to both the controls and the low dose of clozapine at all time points after treatment (p < 0.01).
Epinephrine
Epinephrine levels were reliably measured with the HPLC-ED, with no samples below the limit of detection. Looking at each drug individually, the ANOVA noted that risperidone demonstrated a significant effect of dose [F(2,27) = 20.17, p < 0.001], time [F(4,108) = 17.11, p < 0.001] and a dose × time interaction [F(8,108) = 7.97, p < 0.001]. The post-hoc analysis revealed that both doses of risperidone caused a significant increase in epinephrine levels by 30 mins compared to controls (p ≤ 0.01), but by 60 mins the lower dose of risperidone did not differ significantly from controls, for the duration of the test (Figure 3A). By contrast, epinephrine levels remained significantly elevated at all time points after treatment with the higher dose of risperidone compared to both the low dose of risperidone and controls (p ≤ 0.005).
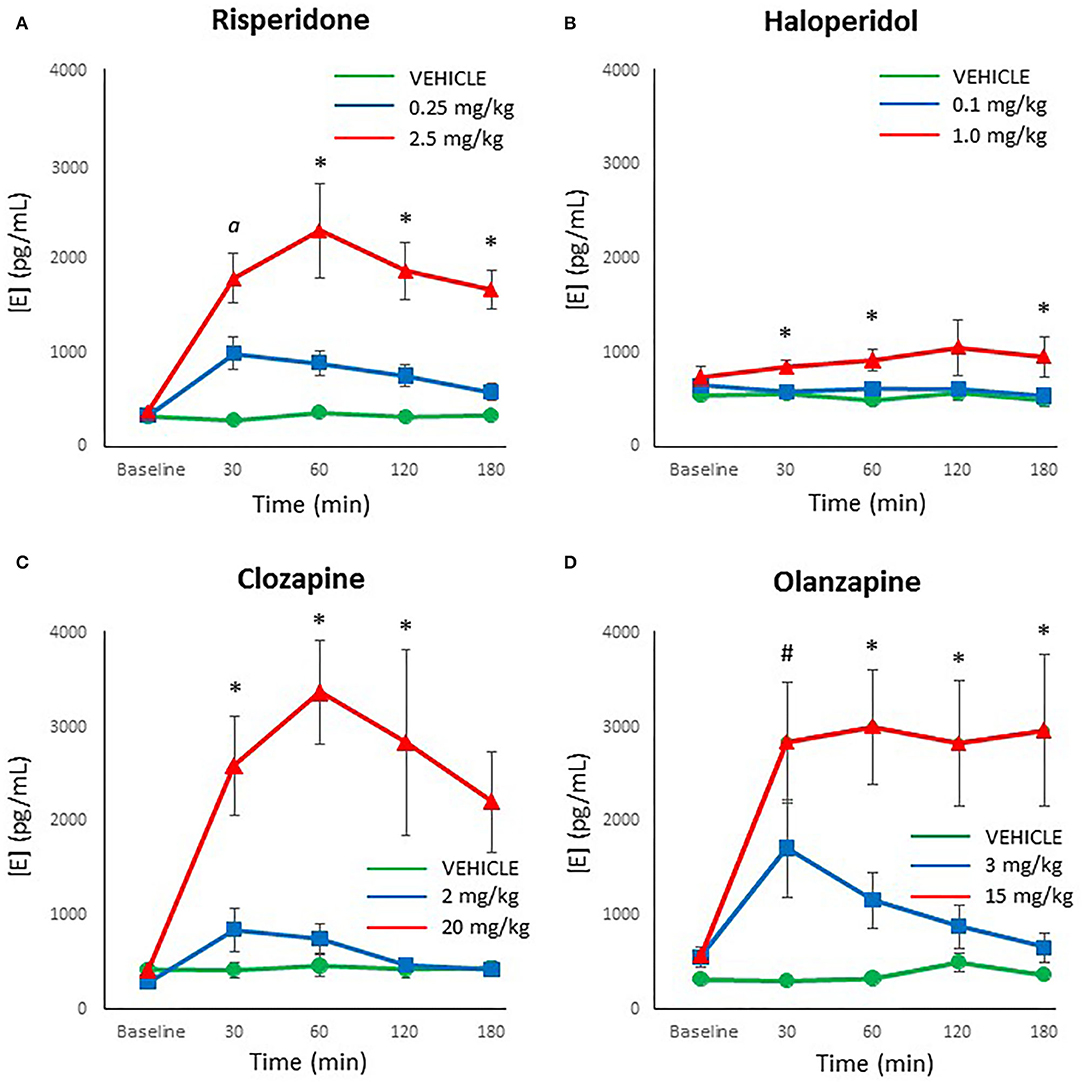
Figure 3. Acute changes in plasma epinephrine (E) concentrations (pg/ml) after antipsychotic drug treatment in adult female rats. Animals (n = 10 per group) were given a single IP injection of (A) risperidone (0.25 or 2.5 mg/kg) or (B) haloperidol (0.1 or 1.0 mg/kg) or (C) clozapine (2 or 20 mg/kg) or (D) olanzapine (3 or 15 mg/kg) or vehicle. Values are expressed in group means ± SEM. Plasma samples were collected immediately prior to drug treatment (Baseline) and at 30, 60, 120, and 180 mins after treatment. α Statistically significant difference between all three groups, p < 0.05; *Statistically significant difference between high dose group and both other groups, p < 0.05; #Statistically significant difference between both drug treated groups and vehicle, p < 0.05.
Analysis of the results with haloperidol by ANOVA indicated a significant effect of dose [F(2,27) = 4.05, p < 0.05], but no main effect of time or dose × time interaction. Post-hoc analysis of the dose effect showed that the low dose of haloperidol and controls did not differ at any time point (Figure 3B), but the high dose of haloperidol caused a significant increase in epinephrine levels compared to both other groups at 30, 60, and 180 mins (p < 0.05).
For clozapine, ANOVA indicated a significant effect of dose [F(2,27) = 14.15, p < 0.001], time [F(4,108) = 8.14, p < 0.001] and a dose × time interaction [F(8,108) = 5.27, p < 0.001]. The post-hoc analysis confirmed that showed that the low dose of clozapine and controls did not differ at any time point in epinephrine levels (Figure 3C), but the high dose of clozapine had higher epinephrine levels than both other groups at all time points after drug treatment (p < 0.01).
With olanzapine, the analysis showed significant main effects of dose [F(2,27) = 14.05, p < 0.001], time [F(4,108) = 6.51, p < 0.001] and a dose × time interaction [F(8,108) = 3.77, p = 0.001]. Follow-up post-hoc tests revealed that both doses of olanzapine caused a significant increase in epinephrine levels by 30 mins compared to controls (p ≤ 0.01) (Figure 3D), but by 60 mins the lower dose of olanzapine did not differ from controls, for the duration of the test. Epinephrine levels with the higher dose of olanzapine remained significantly higher than both other groups from 60 through to 180 mins (p < 0.005).
Dopamine
Although dopamine levels were substantially lower than both norepinephrine and epinephrine, dopamine was reliably detected and above the limit of detection in all processed plasma samples. An analysis for outliers determined that three samples were strong outliers (> 3 × the interquartile range) and so were excluded from analysis and replaced with next observation carried backward (55). ANOVA indicated a main effect of dose of risperidone on dopamine levels [F(2,27) = 19.60, p < 0.001], time [F(4,108) = 4.93, p = 0.001], and a dose × time interaction [F(8,108) = 3.97, p = 0.001]. Follow-up post-hoc tests showed that while low dose risperidone and vehicle-treated rats did not differ at any time point in dopamine levels, the levels were significantly higher in the high dose group compared to the low dose group at 30 and 60 mins (p < 0.05) (Figure 4A), with a strong trend vs. controls (p = 0.057 and 0.072, respectively). By 120–180 mins, the high dose exhibited higher levels of dopamine than both other groups (p ≤ 0.001).
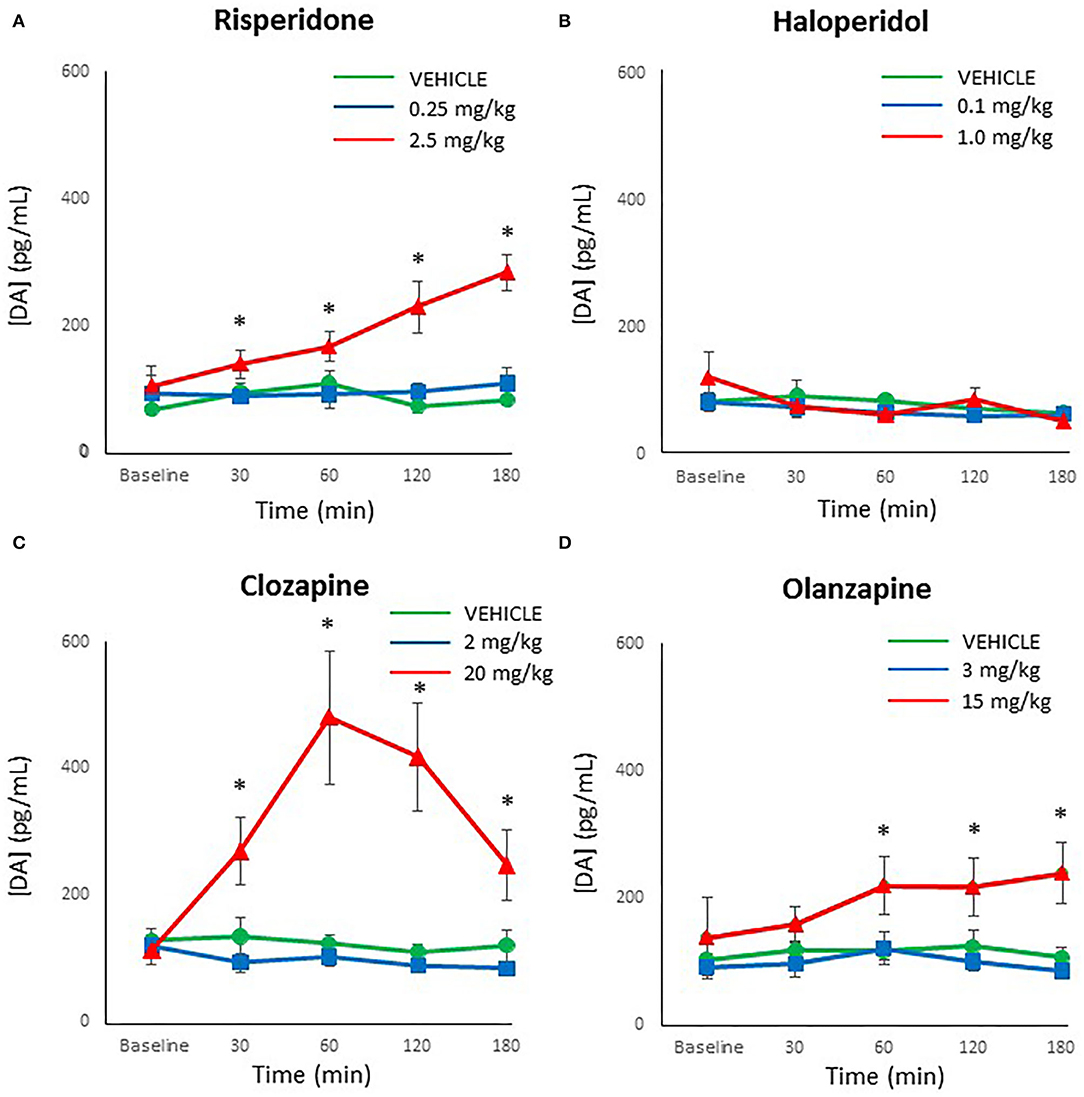
Figure 4. Acute changes in plasma dopamine (DA) concentrations (pg/ml) after antipsychotic drug treatment in adult female rats. Animals (n = 10 per group) were given a single IP injection of (A) risperidone (0.25 or 2.5 mg/kg) or (B) haloperidol (0.1 or 1.0 mg/kg) or (C) clozapine (2 or 20 mg/kg) or (D) olanzapine (3 or 15 mg/kg) or vehicle. Values are expressed in group means ± SEM. Plasma samples were collected immediately prior to drug treatment (Baseline) and at 30, 60, 120, and 180 mins after treatment. *Statistically significant difference between high dose group and both other groups, p < 0.05.
For haloperidol, there was a main effect of time [F(4,108) = 2.51, p < 0.05] but no effect of dose or dose × time interaction. This time effect reflected a gradual reduction of dopamine levels in all groups over time (Figure 4B).
With clozapine, the ANOVA indicated a main effect of dose [F(2,27) = 11.48, p < 0.001], time [F(4,108) = 6.79, p < 0.001] and a dose × time interaction [F(8,108) = 8.44, p < 0.001]. Follow-up post-hoc tests revealed that low dose clozapine and controls did not differ at any time point (Figure 4C), but high dose clozapine demonstrated higher dopamine levels than both other groups at all timepoints after treatment (p < 0.05).
Analysis of olanzapine data by ANOVA indicated a main effect of dose [F(2,27) = 7.50, p < 0.001] but no effect of time or dose × time interaction. Dopamine levels were higher in the high dose olanzapine group than both other groups from 60–180 mins (p < 0.05) (Figure 4D).
Comparison Between Drugs
As the low doses of antipsychotics did not strongly affect catecholamine levels, we limited our head-to-head comparison of different drugs to the high doses. Baseline norepinephrine levels were included as a covariate for post-treatment norepinephrine levels. The ANOVA indicated a main effect of drug [F(3,36) = 10.02, p < 0.001], no effect of time, and a drug × time interaction [F(9,108) = 2.75, p < 0.01]. Follow-up analysis revealed that norepinephrine levels were higher in the clozapine group than all others from 30 to 60 mins (p < 0.05), and higher than haloperidol from 120 to 180 mins. Olanzapine had marginally higher levels than haloperidol at 180 mins (p = 0.055). For epinephrine, ANOVA indicated a main effect of drug [F(3,36) = 4.65, p < 0.01] and no effect of time or drug × time interaction. Post-hoc analyses demonstrated that risperidone, clozapine, and risperidone had higher epinephrine levels than haloperidol at all points after drug treatment. With dopamine, there were main effects of drug [F(3,36) = 8.34, p < 0.001], time [F(3,108) = 6.12, p = 0.001], and a dose × time interaction [F(9,108) = 5.54, p < 0.001], whereby clozapine-treated animals had higher dopamine levels than all other groups from 30 to 120 mins, and all groups had higher levels than haloperidol at 180 mins.
Relationship Between Catecholamines
As an exploratory analysis, we examined the relationship between the three catecholamines at each time point (all treatment groups were combined). At pre-treatment baseline, there was a moderate correlation between norepinephrine and the other two catecholamines (r = 0.31–0.36, p < 0.001) while epinephrine and dopamine were not correlated (r = 0.02, NS). Following drug treatment at 30 mins, all catecholamine levels showed a large increase in their correlation with each other (r = 0.50–0.73, p < 0.001), which increased further at 60 mins (r = 0.67–0.88, p < 0.001), at 120 mins (r = 0.59–0.79, p < 0.001) and at 180 mins (r = 0.70–0.91, p < 0.001).
Discussion
In the present study, we compared treatment in freely moving rats with multiple doses of four different antipsychotic drugs, over a 180-mins period, on levels of peripheral catecholamines. The drugs included the first generation antipsychotic drug haloperidol, as well as the second generation drugs risperidone, olanzapine and clozapine. Metabolic abnormalities in patients are most common with the latter two drugs (21, 56), although can occur with all antipsychotic medications, including first generation drugs (57). The key results of the study were that all antipsychotics caused an increase in plasma catecholamine levels. For norepinephrine, this varied considerably by drug, and maximal increases relative to baseline for risperidone were [low dose: +382%; high dose: +793%], haloperidol [low dose: no change; high dose: +158%], clozapine [low dose: +273%; high dose: +1,684%] and olanzapine [low dose: +212%; high dose: +952%]. A similar pattern was observed with epinephrine, where maximal changes relative to baseline for risperidone were [low dose: +299%; high dose: +617%], haloperidol [low dose: no change; high dose: +143%], clozapine [low dose: +293%; high dose: +806%] and olanzapine [low dose: +307%; high dose: +529%]. Increases in peripheral dopamine were smaller, showing no change with the low dose of each drug, and only moderate increases relative to baseline with the high dose for risperidone [+271%], haloperidol [no change], clozapine [+430%] and olanzapine [+174%]. Interestingly, levels of the catecholamines did not correlate strongly with each other prior to treatment at baseline, but were increasingly correlated after treatment as time proceeded.
Overall, these findings clearly demonstrate that antipsychotic drugs can increase peripheral plasma catecholamines in a dose- and drug-dependent manner. The smallest increases in catecholamines were caused by haloperidol, followed by risperidone/olanzapine, and largest by clozapine. These findings are in general agreement with the limited literature on the effects of antipsychotic drugs on peripheral catecholamine levels in animals and humans. An earlier study observed that intravenous treatment of rats with the first generation antipsychotic chlorpromazine resulted in a dose-dependent elevation of both norepinephrine and epinephrine to levels comparable to those observed in the present study with the second generation drugs (58). Of interest, chlorpromazine has been characterized as the first generation drug most likely to cause metabolic syndrome (59). More recently, a pair of studies by the same research group noted that intravenous olanzapine and clozapine both caused significant increases in plasma epinephrine, to a comparable degree to the present study (46, 60); norepinephrine and dopamine were not measured in their studies. Indirect evidence for elevated peripheral catecholamines following treatment with clozapine and chlorpromazine was provided by Savoy et al. (45), who demonstrated that the hyperglycemia caused by these two drugs could be decreased by treatment with the ganglionic blocker hexamethonium, which presumably worked by preventing the release of peripheral norepinephrine and epinephrine. Clinical studies of patients treated with antipsychotic drugs have also reliably observed increases in peripheral catecholamines with higher metabolic liability drugs, such as clozapine (42, 44, 61, 62). For example, plasma norepinephrine levels were ~three times higher in patients treated with clozapine than in controls or patients treated with the first generation antipsychotics haloperidol and fluphenazine (42, 44). We are not aware of any studies that have measured the effects of antipsychotic drugs on peripheral dopamine, which may be due to the greater sensitivity of the assay needed.
While the link between elevated peripheral catecholamines and the metabolic effects of antipsychotics was not determined in this study (as we did not measure metabolic indices), norepinephrine and epinephrine are well-established as two of the most potent hormones involved in the regulation of blood glucose levels. They tightly control insulin release, glucagon secretion, hepatic gluconeogenesis, and glycogenolysis (41, 63), and elevated levels of these catecholamines are related to metabolic dysregulation and metabolic syndrome (64). As such, the observed increased in catecholamines provide a possible pathway to hyperglycemia that requires further study. However, the catecholamines may represent only part of the story. The present doses of drugs were based on prior studies by our laboratory, that observed effects on glucose intolerance and insulin resistance (32, 34, 47, 48). These doses were selected to produce metabolic effects comparable in magnitude to metabolic effects observed in humans treated with the same drugs (65, 66). Some of those doses, such as the high dose (1 mg/kg) of haloperidol and the low dose of clozapine (2 mg/kg), previously exerted pronounced effects in the glucose tolerance test (47), yet those same doses currently only produced very modest increases in catecholamines. Whether these are sufficient to produce metabolic dysregulation will require additional experimentation. The role of peripheral dopamine also requires further evaluation; the hormone has diverse functions peripherally, and is mainly co-released from sympathetic nerve fibers with norepinephrine (67). There is considerable evidence to show that peripheral dopamine regulates body weight and glucose homeostasis via insulin release (68, 69), and so may represent an unexplored contributor to the metabolic effects of antipsychotics.
Importantly, the observed effects were all determined in fasted animals, as this is the condition under which we and many other groups typically assess the metabolic effects of antipsychotic drugs (28). Fasting is associated with decreased sympathetic activity (70), whereas carbohydrate and fat feeding causes increased sympathetic activity (71, 72). The present use of fasting animals therefore likely maximized the capacity to observe increases in catecholamines and reduced pre-treatment variability in catecholamine levels between animals.
The mechanism by which antipsychotics increase peripheral catecholamines remains unknown. One potential substrate is the α2-adrenoceptor, which acts as an autoreceptor to decrease catecholamine release from nerve terminals (41, 73). Many antipsychotics have a strong affinity for the α2-adrenoceptor, and clozapine (which caused the greatest increase of peripheral catecholamines) has the highest α2 receptor load (74). However, high α2 receptor loads are also associated with antipsychotics with minimal metabolic liability, such as asenapine (32, 74), suggesting that this may not be the main mechanism. We would posit that antipsychotic drugs may be acting centrally in brain regions such as the locus coeruleus (75) or the lateral and paraventricular nuclei of the hypothalamus (76), which serve to activate the sympathetic nervous system and have been implicated in the metabolic effects of antipsychotics (77). The possibility certainly remains, though, that antipsychotics could be exerting peripheral effects on the sympathetic nervous system, such as through their affinity for the monoamine transporters (78).
The current study has a number of limitations. The first of these is that antipsychotic drugs were only administered to female rats, and not to males. The basis for this decision was that female rats typically produce more reliable and robust metabolic effects than do male rats when treated with antipsychotic drugs (28, 52, 53), and so better model the human condition. However, the physiological basis for these sex differences remains incompletely understood, and could feasibly be related to sex differences in catecholamine release (79). This straightforward experiment therefore represents a logical next step in following up the present results. Another limitation of the present study is the acute nature of drug treatment, as animals were only given a single injection of the drug. We would predict that increases in catecholamine levels would not change with repeated treatment, as we have found the metabolic effects of antipsychotics to be stable in rats over time in longitudinal studies (34, 50), and reports of elevated catecholamines in antipsychotic-treated patients include subjects who have also been administered the drugs chronically (42, 44, 61, 62). Nevertheless, the current effects should be determined in rats treated over an extended period with antipsychotic drugs to confirm that the findings remain consistent, and thus better model clinical populations. It would also be informative to assess plasma antipsychotic drug levels in animals treated with the current doses of drugs. Finally, the present study did not concurrently measure both catecholamine release and metabolic dysregulation to confirm, at an individual level, the link between elevated catecholamines and glucose dysregulation. While this would have been informative, there was a conscious decision to progress in a step-by-step manner, which partly comes from a legitimate concern that additional blood draws, injections, and a glucose challenge could significantly have affected stress levels and thus modified catecholamine release. However, future studies will clearly be needed to determine whether these increased catecholamines are causal to metabolic dysregulation using well-designed experimental protocols.
In conclusion, the present results provide strong evidence that antipsychotic drugs exert potent stimulatory effects on peripheral catecholamines, which may be relevant to drug metabolic side-effects. The observation that even the first generation antipsychotic drug haloperidol can significantly increase catecholamine levels with a sufficient dose is consistent with the clinical observation that many first generation drugs are not metabolically neutral and may be associated with metabolic dysregulation (22, 80), albeit milder than most second generation drugs. To our knowledge, the present preclinical study is the first to compare multiple antipsychotics head-to-head, and is also the first to report effects on all three peripheral catecholamine levels. Time-series data collected from unrestrained, unanesthetized animals by a team experienced in stress-free phlebotomy increase the validity of the data. However, many questions remain regarding the direct relationship of these catecholamines to metabolic dysregulation, as well as the physiological pathways involved in catecholamine release, and so significant further research in required on this topic, including assessment of complementary indices of sympathetic activity.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by UBC Animal Care Committee.
Author Contributions
HB, AH, and LT collected and processed data. AB designed and supervised the study. All authors contributed to the writing and final draft of this manuscript and approve it for publication.
Funding
This work was supported by a Natural Sciences and Engineering Research Council of Canada grant to AB and a British Columbia Provincial Health Services Authority grant to AB and RP. WH was supported by the Jack Bell Chair in Schizophrenia.
Conflict of Interest
WH has received consulting fees or sat on paid advisory boards for Otsuka/Lundbeck, Newron, AlphaSights, Guidepoint, Translational Life Sciences and holds shares in Translational Life Sciences and Eli Lilly. He was additionally supported by the Jack Bell Chair in Schizophrenia. RP has been a member of the following advisory boards in the past 3 years: Janssen, Lundbeck, and Otsuka; a member of the following speaker's bureaus in the past 3 years: Janssen, Lundbeck, and Otsuka; and received grants from the Canadian Institutes of Health Research.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Helen Jin, June Lam, Jenny Li, and Daniel Wong for their assistance with collecting and processing blood samples.
References
1. Honer WG, Thornton AE, Sherwood M, MacEwan GW, Ehmann TS, Williams R, et al. Conceptual and methodological issues in the design of clinical trials of antipsychotics for the treatment of schizophrenia. CNS Drugs. (2007) 21:699–714. doi: 10.2165/00023210-200721090-00001
2. Procyshyn RM, Honer WG, Wu TK, Ko RW, McIsaac SA, Young AH, et al. Persistent antipsychotic polypharmacy and excessive dosing in the community psychiatric treatment setting: a review of medication profiles in 435 Canadian outpatients. J Clin Psychiatry. (2010) 71:566–73. doi: 10.4088/JCP.08m04912gre
3. Vasudev A, Chaudhari S, Sethi R, Fu R, Sandieson RM, Forester BP. A review of the pharmacological and clinical profile of newer atypical antipsychotics as treatments for bipolar disorder: considerations for use in older patients. Drugs Aging. (2018) 35:887–95. doi: 10.1007/s40266-018-0579-6
4. Del Casale A, Sorice S, Padovano A, Simmaco M, Ferracuti S, Lamis DA, et al. Psychopharmacological treatment of Obsessive-Compulsive Disorder (OCD). Curr Neuropharmacol. (2019) 17:710–36. doi: 10.2174/1570159X16666180813155017
5. Hershenberg R, Gros DF, Brawman-Mintzer O. Role of atypical antipsychotics in the treatment of generalized anxiety disorder. CNS Drugs. (2014) 28:519–33. doi: 10.1007/s40263-014-0162-6
6. Mela M, Okpalauwaekwe U, Anderson T, Eng J, Nomani S, Ahmed A, et al. The utility of psychotropic drugs on patients with Fetal Alcohol Spectrum Disorder (FASD): a systematic review. Psychiatry Clin Psychopharmacol. (2018) 28:436–45. doi: 10.1080/24750573.2018.1458429
7. Linton D, Barr AM, Honer WG, Procyshyn RM. Antipsychotic and psychostimulant drug combination therapy in attention deficit/hyperactivity and disruptive behavior disorders: a systematic review of efficacy and tolerability. Curr Psychiatry Rep. (2013) 15:355. doi: 10.1007/s11920-013-0355-6
8. Rummel-Kluge C, Komossa K, Schwarz S, Hunger H, Schmid F, Kissling W, et al. Second-generation antipsychotic drugs and extrapyramidal side effects: a systematic review and meta-analysis of head-to-head comparisons. Schizophr Bull. (2010) 38:167–77. doi: 10.1093/schbul/sbq042
9. Haddad PM, Wieck A. Antipsychotic-induced hyperprolactinaemia: mechanisms, clinical features and management. Drugs. (2004) 64:2291–314. doi: 10.2165/00003495-200464200-00003
10. Tandon R, Halbreich U. The second-generation “atypical” antipsychotics: similar improved efficacy but different neuroendocrine side effects. Psychoneuroendocrinology. (2003) 28(Suppl. 1):1–7. doi: 10.1016/S0306-4530(02)00109-9
11. Kessing LV, Thomsen AF, Mogensen UB, Andersen PK. Treatment with antipsychotics and the risk of diabetes in clinical practice. Br J Psychiatry. (2010) 197:266–71. doi: 10.1192/bjp.bp.109.076935
12. Newcomer JW. Antipsychotic medications: metabolic and cardiovascular risk. J Clin Psychiatry. (2007) 68(Suppl. 4):8–13. doi: 10.4088/JCP.0307e07
13. Tumiel E, Wichniak A, Jarema M, Lew-Starowicz M. Nonpharmacological interventions for the treatment of cardiometabolic risk factors in people with schizophrenia-a systematic review. Front Psychiatry. (2019) 10:566. doi: 10.3389/fpsyt.2019.00566
14. Yuen JWY, Kim DD, Procyshyn RM, White RF, Honer WG, Barr AM. Clozapine-induced cardiovascular side effects and autonomic dysfunction: a systematic review. Front Neurosci. (2018) 12:203. doi: 10.3389/fnins.2018.00203
15. Bozymski KM, Whitten JA, Blair ME, Overley AM, Ott CA. Monitoring and treating metabolic abnormalities in patients with early psychosis initiated on antipsychotic medications. Community Mental Health J. (2018) 54:717–24. doi: 10.1007/s10597-017-0203-y
16. Kim DD, Barr AM, Fredrikson DH, Honer WG, Procyshyn RM. Association between serum lipids and antipsychotic response in schizophrenia. Curr Neuropharmacol. (2019) 17:852–60. doi: 10.2174/1570159X17666190228113348
17. Tse L, Procyshyn RM, Fredrikson DH, Boyda HN, Honer WG, Barr AM. Pharmacological treatment of antipsychotic-induced dyslipidemia and hypertension. Int Clin Psychopharmacol. (2014) 29:125–37. doi: 10.1097/YIC.0000000000000014
18. Whitney Z, Procyshyn RM, Fredrikson DH, Barr AM. Treatment of clozapine-associated weight gain: a systematic review. Eur J Clin Pharmacol. (2015) 71:389–401. doi: 10.1007/s00228-015-1807-1
19. Fredrikson DH, Boyda HN, Tse L, Whitney Z, Pattison MA, Ott FJ, et al. Improving metabolic and cardiovascular health at an early psychosis intervention program in Vancouver, Canada. Front Psychiatry. (2014) 5:105. doi: 10.3389/fpsyt.2014.00105
20. Hasnain M, Fredrickson SK, Vieweg WV, Pandurangi AK. Metabolic syndrome associated with schizophrenia and atypical antipsychotics. Curr Diab Rep. (2010) 10:209–16. doi: 10.1007/s11892-010-0112-8
21. Mitchell AJ, Vancampfort D, Sweers K, van Winkel R, Yu W, De Hert M. Prevalence of metabolic syndrome and metabolic abnormalities in schizophrenia and related disorders–a systematic review and meta-analysis. Schizophr Bull. (2013) 39:306–18. doi: 10.1093/schbul/sbr148
22. Deng C. Effects of antipsychotic medications on appetite, weight, and insulin resistance. Endocrinol Metab Clin North Am. (2013) 42:545–63. doi: 10.1016/j.ecl.2013.05.006
23. Rummel-Kluge C, Komossa K, Schwarz S, Hunger H, Schmid F, Lobos CA, et al. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res. (2010) 123:225–33. doi: 10.1016/j.schres.2010.07.012
24. Meyer JM, Davis VG, Goff DC, McEvoy JP, Nasrallah HA, Davis SM, et al. Change in metabolic syndrome parameters with antipsychotic treatment in the CATIE schizophrenia trial: prospective data from phase 1. Schizophr Res. (2008) 101:273–86. doi: 10.1016/j.schres.2007.12.487
25. Lang DJ, Barr AM, Procyshyn RM. Management of medication-related cardiometabolic risk in patients with severe mental illness. Curr Cardiovasc Risk Rep. (2013) 7:283–7. doi: 10.1007/s12170-013-0321-1
26. Laursen TM, Munk-Olsen T, Vestergaard M. Life expectancy and cardiovascular mortality in persons with schizophrenia. Curr Opin Psychiatry. (2012) 25:83–8. doi: 10.1097/YCO.0b013e32835035ca
27. Marteene W, Winckel K, Hollingworth S, Kisely S, Gallagher E, Hahn M, et al. Strategies to counter antipsychotic-associated weight gain in patients with schizophrenia. Expert Opin Drug Saf. (2019) 18:1149–60. doi: 10.1080/14740338.2019.1674809
28. Boyda HN, Tse L, Procyshyn RM, Honer WG, Barr AM. Preclinical models of antipsychotic drug-induced metabolic side effects. Trends Pharmacol Sci. (2010) 31:484–97. doi: 10.1016/j.tips.2010.07.002
29. Chintoh AF, Mann SW, Lam L, Giacca A, Fletcher P, Nobrega J, et al. Insulin resistance and secretion in vivo: effects of different antipsychotics in an animal model. Schizophr Res. (2009) 108:127–33. doi: 10.1016/j.schres.2008.12.012
30. Kowalchuk C, Teo C, Wilson V, Chintoh A, Lam L, Agarwal SM, et al. In male rats, the ability of central insulin to suppress glucose production is impaired by olanzapine, whereas glucose uptake is left intact. J Psychiatry Neurosci. (2017) 42:424–31. doi: 10.1503/jpn.170092
31. Girault EM, Alkemade A, Foppen E, Ackermans MT, Fliers E, Kalsbeek A. Acute peripheral but not central administration of olanzapine induces hyperglycemia associated with hepatic and extra-hepatic insulin resistance. PLoS ONE. (2012) 7:e43244. doi: 10.1371/journal.pone.0043244
32. Boyda HN, Procyshyn RM, Pang CC, Hawkes E, Wong D, Jin CH, et al. Metabolic side-effects of the novel second-generation antipsychotic drugs asenapine and iloperidone: a comparison with olanzapine. PLoS ONE. (2013) 8:e53459. doi: 10.1371/journal.pone.0053459
33. Barr AM, Wu CH, Wong C, Hercher C, Topfer E, Boyda HN, et al. Effects of chronic exercise and treatment with the antipsychotic drug olanzapine on hippocampal volume in adult female rats. Neuroscience. (2013) 255:147–57. doi: 10.1016/j.neuroscience.2013.10.010
34. Boyda HN, Procyshyn RM, Tse L, Wong D, Pang CC, Honer WG, et al. Intermittent treatment with olanzapine causes sensitization of the metabolic side-effects in rats. Neuropharmacology. (2012) 62:1391–400. doi: 10.1016/j.neuropharm.2011.02.019
35. Park S, Hong SM, Ahn IL, Kim da S, Kim SH. Estrogen replacement reverses olanzapine-induced weight gain and hepatic insulin resistance in ovariectomized diabetic rats. Neuropsychobiology. (2010) 61:148–61. doi: 10.1159/000285780
36. Bush ND, Townsend LK, Wright DC. AICAR prevents acute olanzapine-induced disturbances in glucose homeostasis. J Pharmacol Exp Ther. (2018) 365:526–35. doi: 10.1124/jpet.118.248393
37. Shamshoum H, Medak KD, Townsend LK, Ashworth KE, Bush ND, Hahn MK, et al. AMPK β1 activation suppresses antipsychotic-induced hyperglycemia in mice. FASEB J. (2019) 33:14010–21. doi: 10.1096/fj.201901820R
38. Weston-Green K, Huang XF, Deng C. Second generation antipsychotic-induced type 2 diabetes: a role for the muscarinic M3 receptor. CNS Drugs. (2013) 27:1069–80. doi: 10.1007/s40263-013-0115-5
39. Babic I, Gorak A, Engel M, Sellers D, Else P, Osborne AL, et al. Liraglutide prevents metabolic side-effects and improves recognition and working memory during antipsychotic treatment in rats. J Psychopharmacol. (2018) 32:578–90. doi: 10.1177/0269881118756061
40. Kowalchuk C, Castellani LN, Chintoh A, Remington G, Giacca A, Hahn MK. Antipsychotics and glucose metabolism: how brain and body collide. Am J Physiol Endocrinol Metab. (2019) 316:e1–15. doi: 10.1152/ajpendo.00164.2018
41. Boyda HN, Procyshyn RM, Pang CC, Barr AM. Peripheral adrenoceptors: the impetus behind glucose dysregulation and insulin resistance. J Neuroendocrinol. (2013) 25:217–28. doi: 10.1111/jne.12002
42. Elman I, Goldstein DS, Eisenhofer G, Folio J, Malhotra AK, Adler CM, et al. Mechanism of peripheral noradrenergic stimulation by clozapine. Neuropsychopharmacology. (1999) 20:29–34. doi: 10.1016/S0893-133X(98)00047-5
43. Spivak B, Roitman S, Vered Y, Mester R, Graff E, Talmon Y, et al. Diminished suicidal and aggressive behavior, high plasma norepinephrine levels, and serum triglyceride levels in chronic neuroleptic-resistant schizophrenic patients maintained on clozapine. Clin Neuropharmacol. (1998) 21:245–50.
44. Brown AS, Gewirtz G, Harkavy-Friedman J, Cooper T, Brebion G, Amador XF, et al. Effects of clozapine on plasma catecholamines and relation to treatment response in schizophrenia: a within-subject comparison with haloperidol. Neuropsychopharmacology. (1997) 17:317–25. doi: 10.1016/S0893-133X(97)00073-0
45. Savoy YE, Ashton MA, Miller MW, Nedza FM, Spracklin DK, Hawthorn MH, et al. Differential effects of various typical and atypical antipsychotics on plasma glucose and insulin levels in the mouse: evidence for the involvement of sympathetic regulation. Schizophr Bull. (2010) 36:410–8. doi: 10.1093/schbul/sbn104
46. Nagata M, Nakajima M, Ishiwata Y, Takahashi Y, Takahashi H, Negishi K, et al. Mechanism underlying induction of hyperglycemia in rats by single administration of olanzapine. Biol Pharmaceut Bull. (2016) 39:754–61. doi: 10.1248/bpb.b15-00842
47. Boyda HN, Tse L, Procyshyn RM, Wong D, Wu TK, Pang CC, et al. A parametric study of the acute effects of antipsychotic drugs on glucose sensitivity in an animal model. Prog Neuropsychopharmacol Biol Psychiatry. (2010) 34:945–54. doi: 10.1016/j.pnpbp.2010.04.024
48. Boyda HN, Procyshyn RM, Tse L, Xu J, Jin CH, Wong D, et al. Antipsychotic polypharmacy increases metabolic dysregulation in female rats. Exp Clin Psychopharmacol. (2013) 21:164–71. doi: 10.1037/a0031228
49. Yuen JWY, Wu C, Wang CK, Kim DD, Procyshyn RM, Honer WG, et al. A comparison of the effects of clozapine and its metabolite norclozapine on metabolic dysregulation in rodent models. Neuropharmacology. (2020) 175:107717. doi: 10.1016/j.neuropharm.2019.107717
50. Boyda HN, Ramos-Miguel A, Procyshyn RM, Topfer E, Lant N, Choy HH, et al. Routine exercise ameliorates the metabolic side-effects of treatment with the atypical antipsychotic drug olanzapine in rats. Int J Neuropsychopharmacol. (2014) 17:77–90. doi: 10.1017/S1461145713000795
51. Boyda HN, Procyshyn RM, Tse L, Hawkes E, Jin CH, Pang CC, et al. Differential effects of 3 classes of antidiabetic drugs on olanzapine-induced glucose dysregulation and insulin resistance in female rats. J Psychiatry Neurosci. (2012) 37:407–15. doi: 10.1503/jpn.110140
52. Weston-Green K, Huang XF, Deng C. Sensitivity of the female rat to olanzapine-induced weight gain–far from the clinic? Schizophr Res. (2010) 116:299–300. doi: 10.1016/j.schres.2009.09.034
53. Davey KJ, O'Mahony SM, Schellekens H, O'Sullivan O, Bienenstock J, Cotter PD, et al. Gender-dependent consequences of chronic olanzapine in the rat: effects on body weight, inflammatory, metabolic and microbiota parameters. Psychopharmacology. (2012) 221:155–69. doi: 10.1007/s00213-011-2555-2
54. Boyda HN, Procyshyn RM, Asiri Y, Wu C, Wang CK, Lo R, et al. Antidiabetic-drug combination treatment for glucose intolerance in adult female rats treated acutely with olanzapine. Prog Neuropsychopharmacol Biol Psychiatry. (2014) 48:170–6. doi: 10.1016/j.pnpbp.2013.10.006
55. Blankers M, Koeter MW, Schippers GM. Missing data approaches in eHealth research: simulation study and a tutorial for nonmathematically inclined researchers. J Med Internet Res. (2010) 12:e54. doi: 10.2196/jmir.1448
56. De Hert M, Detraux J, van Winkel R, Yu W, Correll CU. Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat Rev Endocrinol. (2011) 8:114–26. doi: 10.1038/nrendo.2011.156
57. Allison DB, Mentore JL, Heo M, Chandler LP, Cappelleri JC, Infante MC, et al. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry. (1999) 156:1686–96.
58. Sato S, Mizuno S, Hatanaka T, Ohshima K, Endoh M, Katayama K, et al. A kinetic study of chlorpromazine on the hyperglycemic response in rats. I. effect of chlorpromazine on plasma catecholamines. J Pharmacobio Dynam. (1988) 11:486–91. doi: 10.1248/bpb1978.11.486
59. Solmi M, Murru A, Pacchiarotti I, Undurraga J, Veronese N, Fornaro M, et al. Safety, tolerability, and risks associated with first- and second-generation antipsychotics: a state-of-the-art clinical review. Ther Clin Risk Manag. (2017) 13:757–77. doi: 10.2147/TCRM.S117321
60. Nagata M, Kimura Y, Ishiwata Y, Takahashi H, Yasuhara M. Clozapine-induced acute hyperglycemia is accompanied with elevated serum concentrations of adrenaline and glucagon in rats. Biol Pharm Bull. (2018) 41:1286–90. doi: 10.1248/bpb.b18-00195
61. Green AI, Alam MY, Sobieraj JT, Pappalardo KM, Waternaux C, Salzman C, et al. Clozapine response and plasma catecholamines and their metabolites. Psychiatry Res. (1993) 46:139–49. doi: 10.1016/0165-1781(93)90016-A
62. Schulz E, Fleischhaker C, Clement HW, Remschmidt H. Blood biogenic amines during clozapine treatment of early-onset schizophrenia. J Neural Transm. (1997) 104:1077–89. doi: 10.1007/BF01273320
63. Barth E, Albuszies G, Baumgart K, Matejovic M, Wachter U, Vogt J, et al. Glucose metabolism and catecholamines. Crit Care Med. (2007) 35(Suppl. 9):S508–18. doi: 10.1097/01.CCM.0000278047.06965.20
64. Mancia G, Bousquet P, Elghozi JL, Esler M, Grassi G, Julius S, et al. The sympathetic nervous system and the metabolic syndrome. J Hypertension. (2007) 25:909–20. doi: 10.1097/HJH.0b013e328048d004
65. Wu C, Yuen J, Boyda HN, Procyshyn RM, Wang CK, Asiri YI, et al. An evaluation of the effects of the novel antipsychotic drug lurasidone on glucose tolerance and insulin resistance: a comparison with olanzapine. PLoS ONE. (2014) 9:e107116. doi: 10.1371/journal.pone.0107116
66. Albaugh VL, Singareddy R, Mauger D, Lynch CJ. A double blind, placebo-controlled, randomized crossover study of the acute metabolic effects of olanzapine in healthy volunteers. PLoS ONE. (2011) 6:e22662. doi: 10.1371/journal.pone.0022662
67. Goldstein DS, Holmes C. Neuronal source of plasma dopamine. Clin Chem. (2008) 54:1864–71. doi: 10.1373/clinchem.2008.107193
68. Rubí B, Maechler P. Minireview: new roles for peripheral dopamine on metabolic control and tumor growth: let's seek the balance. Endocrinology. (2010) 151:5570–81. doi: 10.1210/en.2010-0745
69. Schreiner A, Aadamsoo K, Altamura AC, Franco M, Gorwood P, Neznanov NG, et al. Paliperidone palmitate versus oral antipsychotics in recently diagnosed schizophrenia. Schizophr Res. (2015) 169:393–9. doi: 10.1016/j.schres.2015.08.015
70. Young JB, Landsberg L. Suppression of sympathetic nervous system during fasting. Science. (1977) 196:1473–5. doi: 10.1126/science.867049
71. Schwartz JH, Young JB, Landsberg L. Effect of dietary fat on sympathetic nervous system activity in the rat. J Clin Invest. (1983) 72:361–70. doi: 10.1172/JCI110976
72. Young JB, Landsberg L. Stimulation of the sympathetic nervous system during sucrose feeding. Nature. (1977) 269:615–7. doi: 10.1038/269615a0
73. Brede M, Philipp M, Knaus A, Muthig V, Hein L. alpha2-adrenergic receptor subtypes - novel functions uncovered in gene-targeted mouse models. Biol Cell. (2004) 96:343–8. doi: 10.1111/j.1768-322X.2004.tb01424.x
74. Minzenberg MJ, Yoon JH. An index of relative central α-adrenergic receptor antagonism by antipsychotic medications. Exp Clin Psychopharmacol. (2011) 19:31–9. doi: 10.1037/a0022258
75. Ramirez OA, Wang RY. Locus coeruleus norepinephrine-containing neurons: effects produced by acute and subchronic treatment with antipsychotic drugs and amphetamine. Brain Res. (1986) 362:165–70. doi: 10.1016/0006-8993(86)91411-3
76. de Diego AM, Gandía L, García AG. A physiological view of the central and peripheral mechanisms that regulate the release of catecholamines at the adrenal medulla. Acta Physiol. (2008) 192:287–301. doi: 10.1111/j.1748-1716.2007.01807.x
77. Kowalchuk C, Kanagasundaram P, McIntyre WB, Belsham DD, Hahn MK. Direct effects of antipsychotic drugs on insulin, energy sensing and inflammatory pathways in hypothalamic mouse neurons. Psychoneuroendocrinology. (2019) 109:104400. doi: 10.1016/j.psyneuen.2019.104400
78. Tatsumi M, Jansen K, Blakely RD, Richelson E. Pharmacological profile of neuroleptics at human monoamine transporters. Eur J Pharmacol. (1999) 368:277–83. doi: 10.1016/S0014-2999(99)00005-9
79. Gomes HL, Graceli JB, Gonçalves WL, dos Santos RL, Abreu GR, Bissoli NS, et al. Influence of gender and estrous cycle on plasma and renal catecholamine levels in rats. Can J Physiol Pharmacol. (2012) 90:75–82. doi: 10.1139/y11-102
Keywords: antipsychotic, norepinephrine, epinephrine, dopamine, catecholamine, rat, metabolic side effects
Citation: Boyda HN, Ho AA, Tse L, Procyshyn RM, Yuen JWY, Kim DD, Honer WG and Barr AM (2020) Differential Effects of Acute Treatment With Antipsychotic Drugs on Peripheral Catecholamines. Front. Psychiatry 11:617428. doi: 10.3389/fpsyt.2020.617428
Received: 14 October 2020; Accepted: 13 November 2020;
Published: 01 December 2020.
Edited by:
Margaret K. Hahn, University of Toronto, CanadaReviewed by:
Chao Deng, University of Wollongong, AustraliaSridhar Natesan, King's College London, United Kingdom
Copyright © 2020 Boyda, Ho, Tse, Procyshyn, Yuen, Kim, Honer and Barr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alasdair M. Barr, YWwuYmFyckB1YmMuY2E=