 Veronika Andrashko
Veronika Andrashko Tomas Novak
Tomas Novak Martin Brunovsky
Martin Brunovsky Monika Klirova
Monika Klirova Peter Sos1
Peter Sos1 Jiri Horacek
Jiri Horacek- 1Clinical Research of Mental Disorders, National Institute of Mental Health, Klecany, Czechia
- 2Third Faculty of Medicine, Charles University, Prague, Czechia
The rapid antidepressant effect of ketamine has become a breakthrough in the research and treatment of depression. Although predictive and modulating factors of the response to ketamine are broadly studied, little is known about optimal concurrent medication protocols. Concerning gamma-aminobutyric acid neurotransmission being a shared target for both ketamine and benzodiazepines (BZD), we evaluated the influence of BZD on the antidepressant effect of a single ketamine infusion in depressed patients. Data from 47 patients (27 females) with major depression (MADRS ≥ 20, ≥ 1 prior nonresponse to antidepressant treatment in current episode) who participated in two previous studies (EudraCT Number: 2009-010625-39 and 2013-000952-17) entered the analysis. All of the subjects were given an infusion of a subanesthetic dose of racemic ketamine (0.54 mg per kg) as an add-on medication to ongoing antidepressant treatment. Thirteen patients (28%) reached ≥ 50% reduction in MADRS within one week after ketamine administration. Nineteen (40%) patients took concomitant benzodiazepines on a daily basis. The doses of BZDs were significantly higher in nonresponders (p=0.007). ROC analysis distinguished responders from nonresponders by a criterion of >8mg of diazepam equivalent dose (DZ equivalent) with a sensitivity of 80% and a specificity of 85% (p<0.001). RM-ANOVA revealed a different time pattern of response to ketamine between the BZD+ (>8mg of DZ equivalent) and BZD− (≤8mg of DZ equivalent) groups, with a significantly worse outcome in BZD+ on day 3 (p=0.04) and day 7 (p=0.02). The results of the study indicate that concomitant benzodiazepine treatment in higher doses may attenuate ketamine’s antidepressant effect. The pathophysiological, clinical and methodological implications of this finding should be considered in future research and ketamine treatment.
Introduction
The rapid antidepressant effect of ketamine, first published in 2000 (1) and further abundantly replicated (2), has become a breakthrough in the research and treatment of depression, including novel insight in its mechanisms of antidepressant effect (3, 4). Briefly, the rapid improvement of the depressive mood induced by ketamine may be mediated by the induction of the multiple processes involved in neuroplasticity, with synaptogenesis being a reputable explanation for its antidepressant effect (5–8). These mechanisms are allegedly triggered through ketamine’s antagonism of N-methyl-d-aspartate (NMDA) receptors on gamma-aminobutyric acid (GABA) interneurons and consequent activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (6, 9). Ketamine in subanesthetic doses is considered an auspicious treatment option, particularly in treatment-resistant depression (10).
However, several knowledge gaps are still to be remediated, including the precise methodology of treatment timing, frequency of administration and the effect of concomitant medication (11). In particular, the question of benzodiazepine (BZD) medication in depressed patients receiving ketamine is important for several reasons. Firstly, BZDs represent very commonly prescribed psychotropics (12, 13) and, although not recommended by guidelines (14, 15), are often added to antidepressants in initial depression treatment. In a cohort study, up to 12.5% of depressed patients initiating antidepressant treatment simultaneously started BZD therapy, with 12.3% of them becoming long-term BZD users later on (16). Several other studies have proven that several patients treated for depression fail to withdraw from BZD even after the acute treatment period (17–19), despite the fact that they may contribute to treatment resistance per se in these patients (20). Secondly, BZDs act as GABA-A receptor agonists, allosterically increasing the inhibitory tone of GABA-interneurons (21, 22) and may, thereby, interfere with the therapeutic effect of ketamine in light of ketamine’s blockage of NMDA receptors on the identical population of GABAergic interneurons (3, 9).
This interaction was documented in animal experimental studies, contributing to the understanding of both its pharmacodynamics and the behavioral effects of ketamine. Specifically, diazepam selectively blocked ketamine’s influence on the limbic system metabolism (23) and inhibited ketamine-induced hyperlocomotion (24) in rodents. Moreover, the more than five decades of experience in anesthesiology has documented that BZDs constrict ketamine’s psychotomimetic side effects during general anesthesia (25, 26). These clinical experiences together with the opposite effects of ketamine and BZD on GABAergic interneurons suggest that BZD could negatively interfere with ketamine’s antidepressant effect and the treatment outcome. This assumption has been supported by one case report (27) and two case series suggesting the modulatory influence of BZDs on ketamine’s antidepressant effect (28, 29).
In this study, we aimed to assess the influence of BZD on the antidepressant effect of a single ketamine infusion on a large sample of patients with depression, and to elucidate the eventual dose-dependence of this interaction.
Materials and Methods
Subjects and Study Design
We included 47 inpatients from two consecutive single-site, randomized, placebo-controlled, double-blind, cross-over trials between 2010 and 2015 and regarding the research question and purpose of this analysis, we utilized the data from the active branch only. The results of the first study have been previously published (30). All of the subjects were 18 to 65 years old inpatients, diagnosed with recurrent or a single episode of Major Depressive Disorder (MDD) according to DSM-IV criteria (31), confirmed also by The Mini-International Neuropsychiatric Interview—M.I.N.I. (32), Czech version 5.0.0. The inclusion criteria were as follows: Montgomery-Åsberg Depression Rating Scale (MADRS) (33) score of 20 and more, at least one prior nonresponse to adequate antidepressant treatment in a current major depressive episode (MDE), and being on a stable dose of antidepressants (monotherapy or combination) for a minimum of four weeks prior to ketamine administration (and 7 days after ketamine infusion). The exclusion criteria were as follows: any suicide risk assessed by clinical examination, current psychiatric comorbidity on Axis I and II (less than 6 months prior to enrolment in the study), serious unstable medical or neurological illness, monoamine oxidase inhibitors and treatment augmentation by lamotrigine, lithium, antipsychotics and a lifetime history of a psychotic disorder or symptoms in first- or second-degree relatives. All of the patients underwent a physical examination, routine blood tests, electrocardiogram, urinalysis, and urine toxicology before inclusion in the study.
The patients taking BZDs before the study were allowed to remain on an unchanged dose during the trial. For emergency purposes (severe anxiety, tension, or agitation), an increased dose of ongoing BZDs or a single dose of BZDs (in BZD nonusers) up to 20 mg of oxazepam per day or its equivalent could be administered. The protocol also had presupposed the administration of parenteral BZDs as emergency medication in the event of extreme anxiety or agitation as an adverse reaction during the infusion of ketamine.
Patients using BZDs regularly (regardless of dose) during the study period (7 days before the infusion and for 7 days after ketamine infusion) were defined as BZD users and patients without regular anxiolytic medication or receiving BZDs not more than twice per week, were defined as BZD nonusers. For the purpose of the study, the dosage of BZDs was adjusted to estimated diazepam equivalent (DZ equivalent) as a reference (e.g. 10 mg DZ equivalent = 20 mg of oxazepam or 0.75 mg of alprazolam/clonazepam) (34).
All of the patients provided an informed written consent. Both source studies were registered in the European Clinical Trials Database (EudraCT Number: 2009-010625-39 and 2013-000952-17), approved by the Ethical committee of Prague Psychiatric Centre/National Institute of Mental Health, Czech Republic and performed in accordance with the ethical standards from the Declaration of Helsinki 1975, revised Hong Kong 1989.
Treatment and Assessment
All of the patients received an intravenous infusion of racemic ketamine hydrochloride (Calypsol, Gedeon Richter Plc., Czech Republic), delivered via an infusion pump (ID 20/50, Polymed medical CZ Ltd) at fixed time from 8 a.m. to 8:30 a.m. Ketamine was administered as a loading dose of 0.27 mg/kg for the first 10 min, followed by a maintenance infusion of 0.27 mg/kg within 20 min. This dosing schedule was calculated with respect to the pharmacokinetics of ketamine (35, 36) in order to produce stable ketamine blood levels and the total dose applied was the same as in the majority of antidepressant trials of intravenous ketamine (2).
The severity of depressive symptoms was assessed by the MADRS at the baseline (day before the infusion), and 24 h, 72 h, and 7 days after the infusion. Subjective scales (Beck Depression Inventory (BDI) and Beck Anxiety Inventory (BAI) were administered at the same time points. Response to treatment was defined as a MADRS reduction of at least 50% from the baseline at any visit during the 1-week follow-up period.
Blood sampling was performed in order to evaluate serum levels of ketamine and its first metabolite norketamine (2 ml of venous blood 5 min before the infusion, 10 and 30 min after the beginning of the infusion). For the assessment of ketamine and norketamine serum levels, we used gas chromatography–mass spectrometry (GS-MS), developed and validated according to international standards (37). The analytical standards of norketamine, ketamine and deuterated ketamine (ketamine-D4) supplied as hydrochlorides from Cerilliant, USA were used. For quantitation, the internal standard method was applied using ketamine-D4. Isolation of analytes from blood serum samples was performed using SPECDAU disks and the analyses were performed with acetyl derivatives using a HP 6890–5973 instrument (Agilent, Germany) operating in THE electron impact single ion monitoring (SIM) mode. The lower limit of quantification (LLOQ) for ketamine was 50 ng/ml and for norketamine 8 ng/ml. The limit of detection (LOD) for ketamine was 20 ng/ml and for norketamine 1 ng/ml (37).
Statistical Methods
Demographic, clinical, and treatment characteristics including ketamine and norketamine serum levels, fluoxetine dose equivalent of ongoing antidepressants (38), DZ equivalent (34), and proportion of BZD users (using BZDs on a daily basis regardless of dose), between responders (≥50% MADRS score reduction from the baseline at any visit during the one-week follow-up period) and nonresponders were compared using the unpaired t test, Mann-Whitney U test and Fisher’s exact test as appropriate. As the BZD doses were allowed to be adjusted during the study, DZ equivalent was calculated as the mean daily dose from one day before ketamine administration to the end of the follow-up (day −1 to day 7). In a second step, to address the possibility of different treatment outcomes between zero-to-low-dose and high-dose BZD users (rather than between clear-cut defined BZD users and nonusers), we applied a receiver operating characteristic (ROC) curve to determine the cut-off BZD daily dose with the highest combined sensitivity and specificity for distinguishing responders and nonresponders. BZD+ (BZD high-dose users) and BZD– (BZD zero-to-low-dose users) were compared across demographic and clinical parameters using the unpaired t test, Mann-Whitney U test and Fisher’s exact test and then entered into a logistic regression model as a categorical variable to predict the treatment outcome (response), adjusted for baseline depression and anxiety severity (MADRS, BAI) as possible confounders. Repeated measures analysis of variance (RM-ANOVA) with BZD+ and BZD− as a group variable and MADRS scores at four time points (baseline to day 7) as a within subject repeated variable with the degree of freedom corrected for a lack of sphericity, was performed to find a different pattern of response to ketamine within the study period, if present. In the event of a significant group x time interaction, Bonferroni post hoc tests was used. All of the tests were two-sided and an exact significance level of 0.05 was adopted. The analyses were performed using Statistica, version 12 [StatSoft, Inc. (2013)] and MedCalc, version 15.11.4 (MedCalc Software, Ostend, Belgium).
Results
The data obtained from 47 patients (27 (57%) females) aged between 31 and 52 years (mean 43.0 ± 12.3) were entered into the analyses. The mean baseline depression severity measured by MADRS was 24.0 ± 6.3. The mean duration of depression diagnosis was 10.0 ± 10.1 years, whereas the mean duration of current depression episode was 19.3 ± 26.7 weeks. Thirteen patients (28%) were on AD monotherapy and 34 (72%) had a combination of two or more antidepressants. Sertraline and escitalopram (together 36%), venlafaxine (28%), and mirtazapine (17%) were the most frequently used antidepressants in the study patients. The mean AD daily dosage was 54.1 ± 24.5 mg of fluoxetine equivalent (38). Thirteen patients (28%) responded to a single dose of ketamine (≥ 50% MADRS reduction anytime within one week). The responders had significantly lower baseline anxiety (p=0.04) and depression severity levels (p=0.052), otherwise both groups were comparable (Table 1).
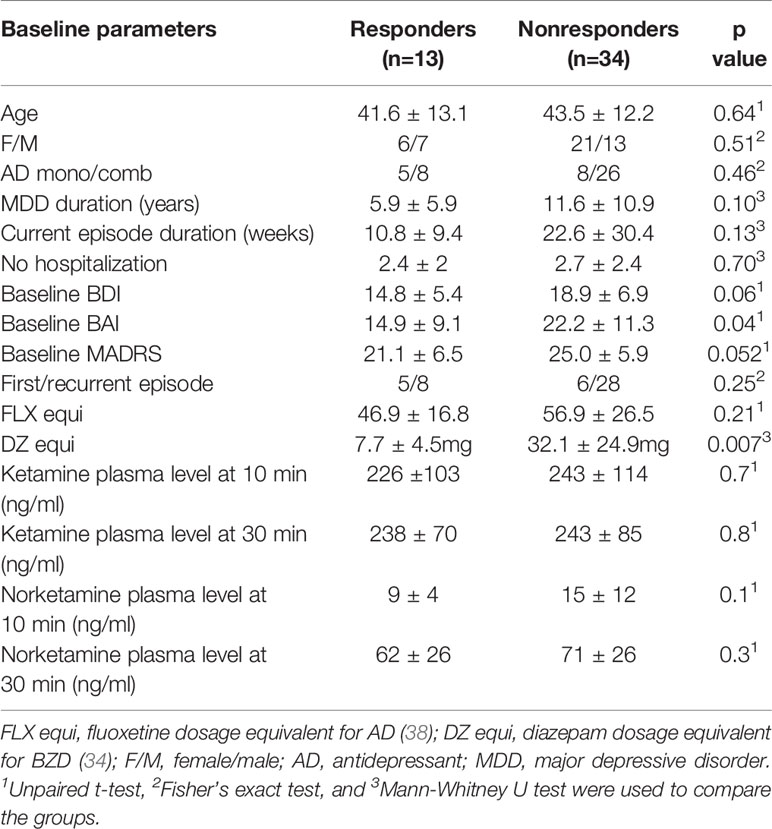
Table 1 Baseline characteristics of responders vs. nonresponders.
Twenty-one patients have used at least one dose of BZDs during the study period. Two of them required just a single BZD dose due to worsening anxiety (one patient 24 h before and second one day after ketamine administration). Nineteen patients (40% of sample) were taking concomitant BZDs on daily basis upon admission to the study (BZD users) with doses varying from 3.8 to 94.0 mg of DZ equivalent per day. Thirteen (68%) patients were taking clonazepam, five (26%) took alprazolam and one patient had a combination of clonazepam and midazolam. While concomitant BZD medication (of any dose) was equally distributed in the responders (38%) and nonresponders (42%), both groups markedly differed in the BZD doses with significantly higher doses in nonresponders (7.7 ± 4.5 vs. 32.1 ± 24.9 mg of DZ equivalent; p=0.007, Table 1).
The ROC analysis distinguished nonresponders from responders by a criterion of >8mg of DZ equivalent with a sensitivity of 80% and a specificity of 84.6% (AUC=0.91, 95%CI 0.68-0.99, p=0.001, Figure 1). After dividing the patients into high-dose BZDs users (BZD+, >8mg of DZ equivalent, N=13) and none-to-low-dose BZDs users (BZD-, 0–8mg of DZ equivalent, i.e., up to 0.5mg of clonazepam or alprazolam; N=34), only one patient from the BZD+ group belonged to the responders, while the other responders were from the BZD- group (response rate 8% in BZD+ vs. 35% in BZD-; Fisher’s exact test, p=0.07). Additional logistic regression with a response as a dependent variable adjusted for baseline depression (MADRS score) and anxiety (BAI score) severity as possible confounding factors indicated a higher chance of being a nonresponder in patients on high-dose BZDs after ketamine infusion (OR=9.7, 95%CI 1.2–69.7, p=0.03). Moreover, when MADRS scores between the BZD+ and BZD- group were compared from the baseline to day 7, a different time pattern of response to ketamine was revealed (RM-ANOVA; group x time interaction: F(3,135)=2.98, p=0.03), with a comparable decrease of depressive symptoms 24 h after infusion (Bonferroni post hoc test, p=0.53), but a significantly worse outcome in BZD+ on day 3 (p=0.04, Hedges´ g= 0.67) and on day 7 (p=0.02, g= 0.78) (Figure 2). Otherwise, the BZD+ and BZD- groups did not differ either in demographic or clinical variables, or in ketamine and its metabolite norketamine plasma levels (Table 2).
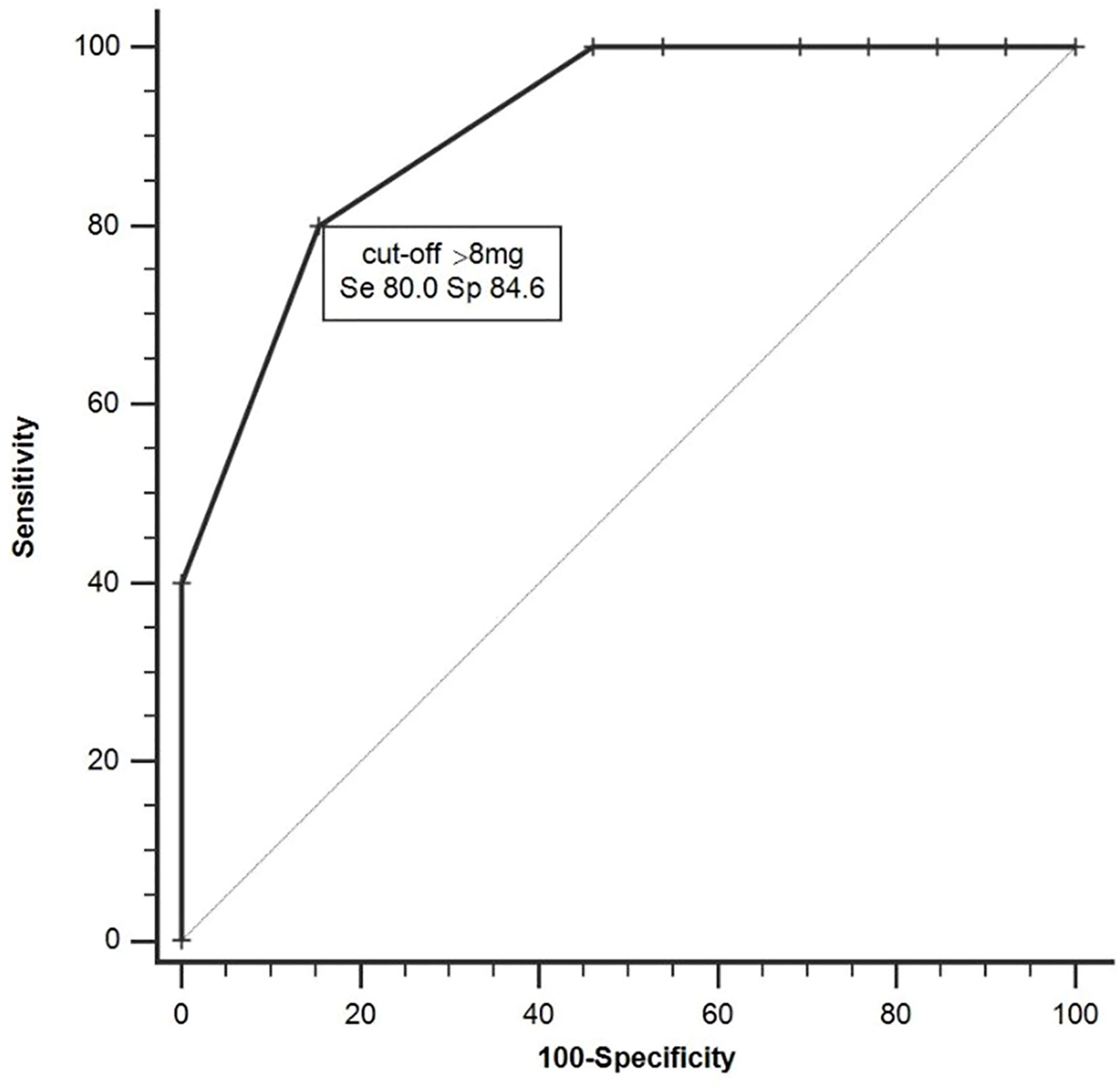
Figure 1 Receiver operating characteristic (ROC) analysis distinguishing responders and nonresponders (AUC=0.91, 95%CI 0.68–0.99, p < 0.001).
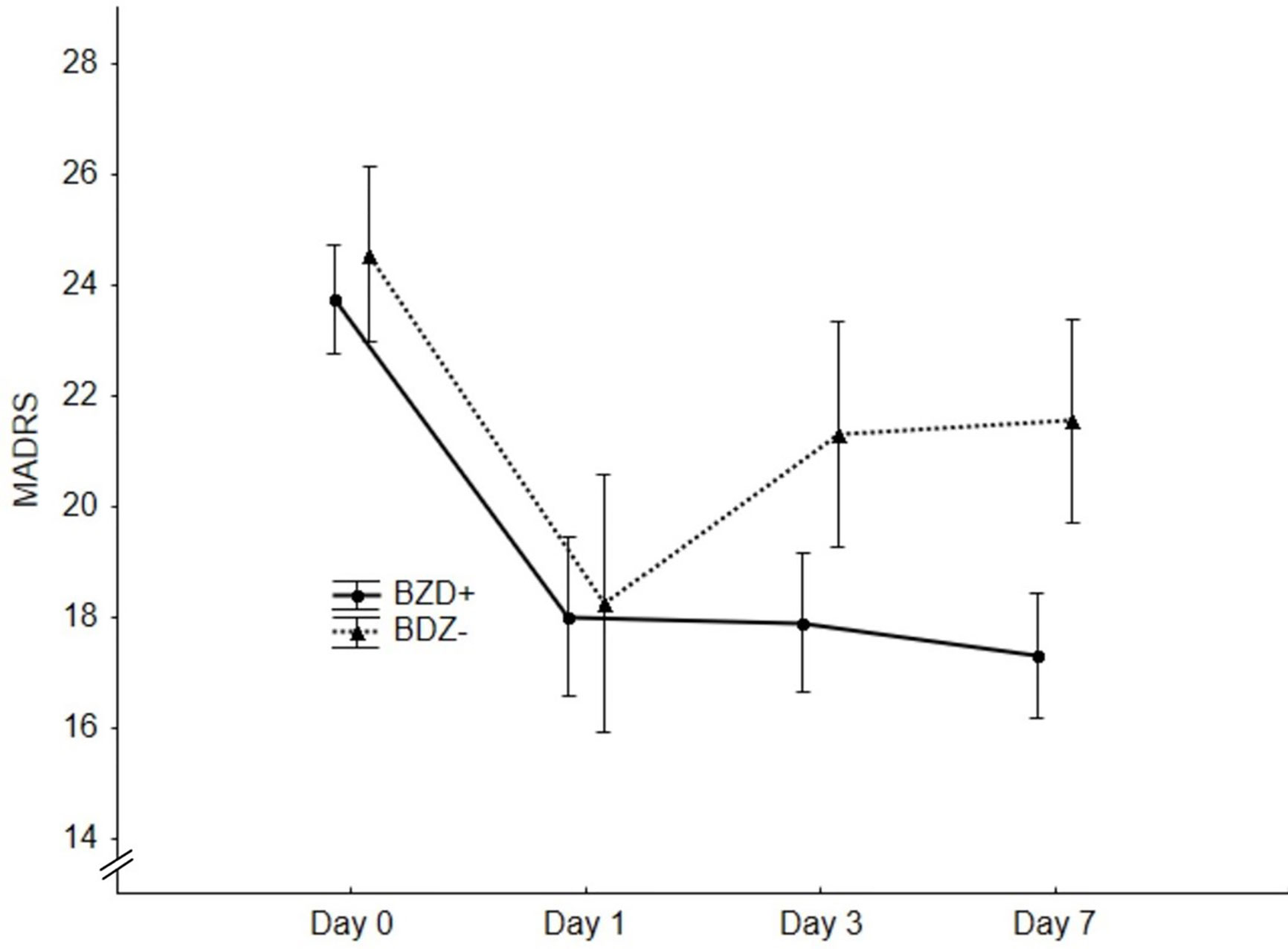
Figure 2 Significantly better outcome in the benzodiazepines (BZD−) group on day 3 (p = 0.04) and day 7 (p = 0.02) revealed by repeated measures analysis of variance (RM-ANOVA).
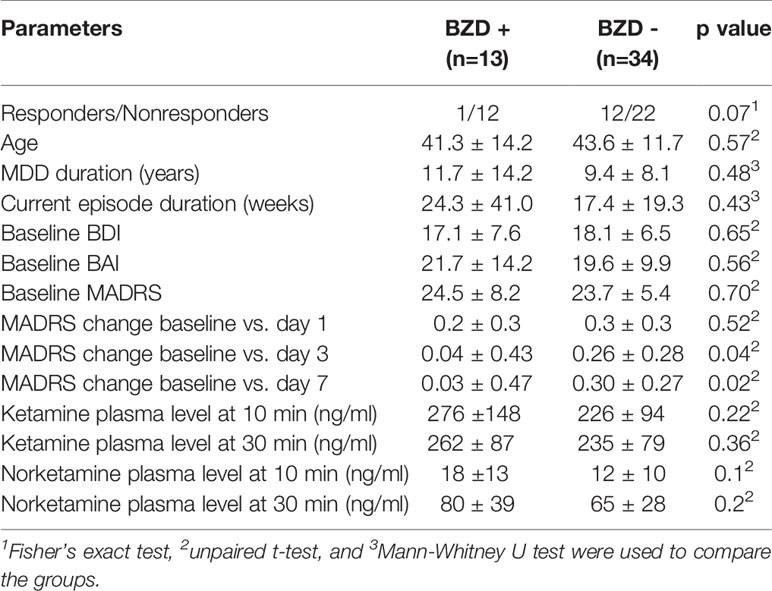
Table 2 Characteristics of high-dose benzodiazepines (BZD) users (BZD+; >8mg of DE) and none-to-low-dose BZD users (BZD-; 0–8mg of DE) during the follow-up period.
Discussion
The main finding of this study is that BZDs attenuate ketamine’s antidepressant effect in patients with MDD. This effect occurred in patients taking higher BZD doses and remained present even when adjusted to baseline anxiety and depression scores and was also not affected by plasma levels of ketamine and its first metabolite norketamine measured during or after the infusion.
Our findings are congruent with two previous case-series reports [pointing lower daily dose of BZDs in ketamine responders among 10 depressed patients (28) or accelerated loss of therapeutic effect in 4 out of 13 subjects (29)] and a case report of a patient with attenuated response to ketamine with concurrent lorazepam (27)] and have pathophysiological and substantial clinical and methodological implications. Firstly, we speculated that BZDs increase the inhibition of pyramidal neurons and, thereby, counteract the inhibition of GABA-interneurons mediated by ketamine-induced antagonism of extrasynaptic NMDA receptors, located foremost at these interneurons (3). This interpretation is in line with the fact that BZDs effectively inhibit the psychotomimetic effect of ketamine during or after general anesthesia (25, 26), especially considering the correlation between the intensity of psychotomimetic or dissociative symptoms during ketamine infusion and its antidepressant effect, documented in our previous study (30), and replicated for an independent sample (39). Interestingly, our results show that BZDs only interfered with a delayed response to ketamine, observed on day 3 and day 7 after the administration, but not in the first 24 h. This fact suggests that BZD-mediated inhibition of ketamine’s antidepressant effect may be related to attenuation of neuroplastic processes, emerging subsequently after the acute effect and after ketamine and its active metabolites are eliminated from the blood. Preclinical data suggested that a ketamine-induced elevation of synaptic proteins (such as synapsin I, PSD95 and GluR1) could be initially observed 2 h after the application and remained increased up to 7 days (8). This observation corresponds with the duration of ketamine’s antidepressant effect in humans (2). Together, a subanesthetic dose of ketamine acutely induces dissociative/psychotomimetic effects by blocking NMDA receptors, followed by an extracellular release of glutamate. However, glutamate then preferentially activates glutamatergic AMPA receptors, inducing their phosphorylation (40, 41) and subsequently triggering activation of protein kinases such as mTOR (mammalian target of rapamycin), promoting an expression of neurotrophic proteins that regulate synaptic growth and remodellation (39, 42). Our data suggest that BZDs may interfere with these downstream signaling steps and, therefore, relate to a sustained antidepressant effect. This assumption should be elucidated by future research addressing the temporal dynamics of BDNF (brain-derived neurotrophic factor) and other neurotrophins in patients with concomitant BZDs.
The fact that BZDs did not influence ketamine’s antidepressant effect in the first 24 h (similar improvement in both groups) may be interpreted by possible initial symptom relief due to a robust acute psychological effect (not related to the antidepressant effect per se) linked to psychotropic effect of ketamine infusion. This psychotropic effect of ketamine limits effective study blinding (43, 44), especially when an inactive comparator is used.
Clinically, our results raise discussion on the negative therapeutic impact of concomitant BZD medication in depressed patients, whereas concurrent medication is indeed a modifiable factor, unlike other (e.g. demographic or pharmacokinetic) characteristics in subjects receiving ketamine. Anxious depression has been shown to be associated with a poorer response to antidepressant treatment per se (45). Moreover, BZDs have been identified as a possible causative factor for treatment resistance in depressed patients (20). With ketamine being a novel option in treatment-resistant depression, it is likely that a substantial number of patients could be on BZDs at the time of infusion. This should be taken into account especially since the U.S. Food and Drug Administration has recently approved intranasal esketamine as an additional treatment for resistant depression, where the influence of BZD’s may be similar, albeit to our knowledge there is yet no published data examining this consideration.
In addition, ketamine’s effect may also be influenced by antiglutamatergic anticonvulsive drugs used in bipolar depression or as adjunctive therapy in resistant unipolar depression (46). It has been documented that pre-treatment with antiglutamatergic anticonvulsant lamotrigine attenuates ketamine’s acute neuropsychiatric effects (47, 48) as well as its neuroimaging correlates in pharmacological fMRI studies (48, 49). Nevertheless, the interaction between antiglutamatergic anticonvulsants and the antidepressant effect of ketamine was not yet systematically evaluated in MDD patients, apart from an anecdotal report where no difference was found in glutamatergic medication between ketamine responders and nonresponders among 10 patients (28).
Our results should be interpreted in the context of several limitations. First, we utilized retrospective analysis of previously existing data, thus were disabled in designing and controlling the preceding experiment. Second, the study was performed on a relatively small sample size. Third, due to retrospective character of the study, blood levels of BZDs were not available for the analysis and thus we were not able to objectively assess their dose-dependent impact taking into account their pharmacokinetics. Last, the responders and nonresponders in our study differed in baseline anxiety scores (BAI, Table 1) with the difference in baseline depression being close to significant. Despite the elimination of these confounding factors by performing additional logistic regression, the small sample size and post hoc design may enervate the magnitude of our consideration about BZDs being a solitary determinant. The severity of symptomatology remains relevant especially in the scope of recent studies focusing on the differences in cortical excitation and inhibition in regard to glutamatergic (50) and GABAergic (51, 52) neurotransmission, showing that higher severity and/or resistance of depressive symptomatology is linked to impaired GABAergic functions, especially regarding GABA neurotransmission as a junction target for ketamine and benzodiazepines.
Our findings evoke a substantial methodological implication for future studies with ketamine, suggesting that: a) the results should be replicated together with blood levels of BZDs and b) concomitant medication should be controlled and comprised as an independent factor in effectiveness studies and meta-analyses. Regarding the kinetics of different BZDs, further studies should also address the time period for withdrawal before ketamine infusion.
To conclude, our study indicates that concomitant BZD treatment attenuates ketamine’s antidepressant effect. This fact should be considered in the clinical management of ketamine treatment and future research design.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethical committee of Prague Psychiatric Centre/National Institute of Mental Health, Czech Republic. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
PS designed and directed the studies and performed the clinical experiments together with MK. JH encouraged VA to investigate the topic. VA wrote the manuscript with input from TN, who performed the statistical calculations. TN, JH, MB, and VA contributed to the interpretation of the results. JH supervised the project and the process of manuscript preparation.
Funding
This work was supported by: Czech Health Research Council grant number NV18-04-00260, ERDF/ESF project PharmaBrain, Reg. No. CZ.02.1.01/0.0/0.0/16_025/0007444, by grant number LO1611 from the Ministry of Education, Youth and Sports of the Czech Republic under the NPU I program and by MH CZ - DRO (“National Institute of Mental Helth – NIMH, IN: 00023752).
Conflict of Interest
TN reports that he has received honoraria from KRKA ČR s.r.o. in the past three years.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry (2000) 47:351–4. doi: 10.1016/S0006-3223(99)00230-9
2. Xu Y, Hackett M, Carter G, Loo C, Gálvez V, Glozier N, et al. Effects of low-dose and very low-dose ketamine among patients with major depression: a systematic review and meta-analysis. IJNP (2016) 19(4):1–15. doi: 10.1093/ijnp/pyv124
3. Abdallah CG, Sanacora G, Duman RS, Krystal JH. Ketamine and rapid-acting antidepressants: a window into a new neurobiology for mood disorder therapeutics. Annu Rev Med (2015) 66:509–23. doi: 10.1146/annurev-med-053013-062946
4. Starsburger SE, Bhimani PM, Kaabe JH, Krysiak JT, Nanchanatt DL, Nguyen TN, et al. What is the mechanism of ketamine’s rapid-onset antidepressant effect? A concise overview of the surprisingly large of possibilities. J Clin Pharm Ther (2017) 42(2):147–54. doi: 10.1111/jcpt.12497
5. Zarate CA, Manji HK. The role of AMPA receptor modulation in the treatment of neuropsychiatric diseases. Exp Neurol (2008) 211(1):7–10. doi: 10.1016/j.expneurol.2008.01.011
6. Koike H, Iijima M, Chaki S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res (2011) 224(1):107–11. doi: 10.1016/j.bbr.2011.05.035
7. Garcia LS, Comim CM, Valvassori SS, Reus GZ, Barbosa LM, Andreazza AC, et al. Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry (2008) 32(1):140–4. doi: 10.1016/j.pnpbp.2007.07.027
8. Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science (2010) 329:959–64. doi: 10.1126/science.1190287
9. Blier P. Exploiting N-Methyl-D-aspartate channel blockade for a rapid antidepressant response in major depressive disorder. Biol Psychiatry (2013) 74(4):238–9. doi: 10.1016/j.biopsych.2013.05.029
10. Wilkinson ST, Sanacora G. Consideration on the off-label use of ketamine as a treatment for mood disorders. JAMA (2017) 318(9):793–4. doi: 10.1001/jama.2017.10697
11. Sanacora G, Katz R. Ketamine: a review for clinicians. Focus (Am Psychiatr Publ) (2018) 16(3):243–50. doi: 10.1176/appi.focus.20180012
12. Fassaert T, Dorn T, Spreeuwenberg PM, van Dongen MC, van Gool CJ, Yzermans CJ. Prescription of benzodiazepines in general practice in the context of a man-made disaster: a longitudinal study. Eur J Public Health (2007) 17:612–7. doi: 10.1093/eurpub/ckm020
13. Olfson M, King M, Schoenbaum M. Benzodiazepine use in the United States. JAMA Psychiatry (2015) 72(2):136–42. doi: 10.1001/jamapsychiatry.2014.1763
14. American Psychiatric Association. Treatment of patients with major depression and practical guideline. American Psychiatric Association (2010). [cited 2018 Jan 28]. Available at: http://www.psychiatryonline.com/pracGuide/pracGuideTopic_7.aspx
15. Kennedy SH, Lam RW, Mclntyre RS, Tourjman SV, Bhat V, Blier P, et al. Canadian network for mood and anxiety treatments (CANMAT) 2016 clinical guidelines for the management of adults with major depressive disorder. Section 3: pharmacological treatments. Can J Psychiatry (2016) 61(9):540–60. doi: 10.1177/0706743716659417
16. Bushnell GA, Sturmer T, Gaynes BN, Pate V, Miller M. Simultaneous antidepressant and benzodiazepine new use and subsequent long-term benzodiazepine use in adults with depression, United States, 2001-2014. JAMA Psychiatry (2017) 74(7):747–55. doi: 10.1001/jamapsychiatry.2017.1273
17. Sjostedt C, Ohlsson H, Xinjun L, Sundquist K. Socio-demographic factors and long-term use of benzodiazepines in patients with depression, anxiety or insomnia. Psych Res (2017) 249:221–5. doi: 10.1016/j.psychres.2017.01.046
18. Valenstein M, Taylor KK, Austin K, Kales HC, McCarthy JF, Blow FC. : Benzodiazepine use among depressed patients treated in mental health settings. Am J Psychiatry (2004) 161:654–61. doi: 10.1176/appi.ajp.161.4.654
19. Sanyal C, Asbridge M, Kisely S, Sketris I, Andreou P. The utilization of antidepressants and benzodiazepines among people with major depression in Canada. Can J Psychiatry (2011) 56:667–76. doi: 10.1177/070674371105601105
20. Parker GB, Graham RK. Determinants of treatment-resistant depression. The salience of benzodiazepines. J Nerv Ment Dis (2015) 203(9):659–63. doi: 10.1097/NMD.0000000000000348
21. Costa E, Guidotti A, Mao CC. Evidence for involvement of GABA in the action of benzodiazepines: studies on rat cerebellum. Adv Biochem Psychopharmacol (1975) 14:113–30.
22. Downing SS, Lee YT, Farb DH, Gibbs TT. Benzodiazepine modulation of partial agonist efficacy and spontaneously active GABA(A) receptors supports an allosteric model of modulation. Br J Pharmacol (2005) 145:894–906. doi: 10.1038/sj.bjp.0706251
23. Eintrei C, Sokoloff L, Smith CB. Effects of diazepam and ketamine administered individually or in combination on regional rates of glucose utilization on rat brain. Br J Anaest (1999) 82:596–602. doi: 10.1093/bja/82.4.596
24. Irifune M, Sato T, Kamata Y, Nishikawa T, Nomoto M, Fukuda T, et al. Inhibition by diazepam of ketamine-induced hyperlocomotion and dopamine turnover in mice. Can J Anaesth (1998) 45(5):471–8. doi: 10.1007/BF03012584
25. Catwright PD, Pingel SM. Midazolam and diazepam in ketamine anaesthesia. Anaesthesia (1984) 39(5):439–42. doi: 10.1111/j.1365-2044.1984.tb07312.x
26. Freuchen I, Ostergaard J, Kuhl JB, Mikkelsen BO. Reduction of psychotomimetic side effects of Ketalar (ketamine) by Rohypnol (flunitrazepam). A randomized, double-blind trial. Acta Anaesth Scand (1976) 20(2):97–103. doi: 10.1111/j.1399-6576.1976.tb05015.x
27. Ford N, Ludbrook G, Galletly Ch. Benzodiazepines may reduce the effectiveness of ketamine in the treatment of depression. Aust N Z J Psychiatry (2015) 49(12):1227. doi: 10.1177/0004867415590631
28. Frye MA, Blier P, Tye SJ. Concomitant benzodiazepine use attenuates ketamine response: implications for large scale study design and clinical development. J Clin Psychofarmacol (2015) 35(3):334–6. doi: 10.1097/JCP.0000000000000316
29. Albott CS, Shiroma PR, Cullen KR, Johns B, Thuras P, Wels J, et al. The antidepressant effect of repeat dose intravenous ketamin is delayed by concurrent benzodiazepine use. J Clin Psychiatry (2017) 78(3):e308–209. doi: 10.4088/JCP.16l11277
30. Sos P, Klirova M, Novak T, Kohutova B, Horacek J, Palenicek T. Relationship of ketamine’s antidepressant and psychotomimetic effects in unipolar depression. Neuro Endocrinol Lett (2013) 34(4):287–93.
31. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: American Psychiatric Publishing (2009).
32. Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry (1998) 59:22–33. doi: 10.1037/t18597-000
33. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatr (1979) 134:382–9. doi: 10.1192/bjp.134.4.382
34. Kane SP. Equivalent Benzodiazepine Canculator (2017). Available from: http://clincalc.com/Benzodiazepine/
35. Hetem LA, Danion JM, Diemunsch P, Brandt C. Effect of a subanesthetic dose of ketamine on memory and conscious awareness in healthy volunteers. Psychopharmacology (2000) 152:283–8. doi: 10.1007/s002130000511
36. Horacek J, Brunovsky M, Novak T, Tislerova B, Palenicek T, Bubenikova-Valesova V, et al. Subanesthetic dose of ketamine decreases prefrontal theta cordance in healthy volunteers: implications for antidepressant effect. Psychol Med (2010) 40:1443–51. doi: 10.1017/S0033291709991619
37. Penders J, Verstraete A. Laboratory guidelines and standards in clinical and forensic toxicology. Accred Qual Assur (2006) 11:284–90. doi: 10.1007/s00769-006-0131-y
38. Hayasaka Y, Purgato M, Magni LR, Ogawa Y, Takeshima N, Cipriani A, et al. Dose equivalents of antidepressants: Evidence-based recommendations from randomized controlled trials. J Affect Disord (2015) 180:179–84. doi: 10.1016/j.jad.2015.03.021
39. Luckenbaugh DA, Niciu MJ, Ionescu DF, Nolan NM, Richards EM, Brutsche EM, et al. Do the dissociative side effects of ketamine mediate its antidepressant effects? J Affect Disord (2014) 159:56–61. doi: 10.1016/j.jad.2014.02.017
40. Du J, Machado-Vieira R, Maeng S, Martinowich K, Manji HK, Zarate CA , Jr. Enhancing AMPA to NMDA throughput as a convergent mechanism for antidepressant action. Drug Discovery Today Ther Strated (2006) 3(4):519–26. doi: 10.1016/j.ddstr.2006.11.012
41. Maeng S, Zarate CA Jr, Du J, Schloesser RJ, McCammon J, Chen G, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-mathylisoxazole-4-propionic acid receptors. Biol Psychiatry (2008) 63(4):349–52. doi: 10.1016/j.biopsych.2007.05.028
42. Cavalleri L, Merlo Pich E, Millan MJ, Chiamulera C, Kunath T, Spano PF, et al. Ketamine enhances structural plasticity in mouse mesencephalic and human iPSC-derived dopaminergic neurons via AMPAR-driven BDNF and mTOR signalling. Mol Psychiatry (2018) 23(4):812–23. doi: 10.1038/mp.2017.241
43. Sanderson C, Hardy J, Odette S, Currow DC. Placebo and nocebo effects in randomized controlled trials: the implications for research and practice. J Pain Symptom Manage (2013) 46(5):722–30. doi: 10.1016/j.jpainsymman.2012.12.005
44. Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner D, et al. Subanestetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive and neuroendocrine responses. Arch Gen Psychiatry (1994) 51(3):199–214. doi: 10.1001/archpsyc.1994.03950030035004
45. Fava M, Rush AJ, Alpert JE, Balasubramani GK, Wisniewski SR, Carmin CN, et al. Difference in treatment outcome in outpatients with anxious versus nonanxious depression: a STAR*D report. Am J Psychiatry (2008) 165(3):342–51. doi: 10.1176/appi.ajp.2007.06111868
46. Vigo DV, Baldessarini RJ. Anticonvulsants in the treatment of major depressive disorder: an overview. Har Rev Psychiatry (2009) 17(4):231–41. doi: 10.1080/10673220903129814
47. Anand A, Charney DS, Oren DA, Berman RM, Hu XS, Cappiello A, et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of N-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry (2000) 57(3):270–6. doi: 10.1001/archpsyc.57.3.270
48. Deakin JF, Lees J, McKie S, Hallak JE, Williams SR, Dursun SM. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry (2008) 65(2):154–64. doi: 10.1001/archgenpsychiatry.2007.37
49. Doyle OM, De Simoni S, Schwarz AJ, Brittain C, O’Daly OG, Williams SC, et al. Quantifying the attenuation of the ketamine pharmacological magnetic resonance imaging response in humans: a validation using antipsychotic and glutamatergic agents. J Pharmacol Exp Ther (2013) 345(1):151–60. doi: 10.1124/jpet.112.201665
50. Li CT, Yang KC, Lin WC. Glutamatergic dysfunction and glutamatergic compounds for major psychiatric disorders: evidence from clinical neuroimaging studies. Front Psychiatry (2018) 9:767. doi: 10.3389/fpsyt.2018.00767
51. Bajbouj M, Lisanby SH, Lang UE, Danker-Hopfe H, Heuser I, Neu P. Evidence for impaired cortical inhibition in patients with unipolar major depression. Biol Psychiatry (2006) 59(5):395–400. doi: 10.1016/j.biopsych.2005.07.036
Keywords: ketamine, depression, clinical effect, antidepressant, concomitant medication, benzodiazepines
Citation: Andrashko V, Novak T, Brunovsky M, Klirova M, Sos P and Horacek J (2020) The Antidepressant Effect of Ketamine Is Dampened by Concomitant Benzodiazepine Medication. Front. Psychiatry 11:844. doi: 10.3389/fpsyt.2020.00844
Received: 29 February 2020; Accepted: 03 August 2020;
Published: 28 August 2020.
Edited by:
Marijn Lijffijt, Baylor College of Medicine, United StatesReviewed by:
Casimiro Cabrera Abreu, Queens University, CanadaCheng-Ta Li, Taipei Veterans General Hospital, Taiwan
Copyright © 2020 Andrashko, Novak, Brunovsky, Klirova, Sos and Horacek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Veronika Andrashko, dmVyb25pa2EuYW5kcmFzaGtvQG51ZHouY3o=