
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Psychiatry , 03 September 2020
Sec. Mood Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fpsyt.2020.00837
This article is part of the Research Topic Stress Induced Neural Changes in Emotional Disorders View all 19 articles
 Wei Liu1†
Wei Liu1† Qian Li1†
Qian Li1† Binglu Ye1†
Binglu Ye1† Hang Cao1Fuyi Shen1
Hang Cao1Fuyi Shen1 Zhendong Xu1
Zhendong Xu1 Weijia Du1
Weijia Du1 Fei Guo2Jinqi Liu3
Fei Guo2Jinqi Liu3 Tianyu Li2
Tianyu Li2 Bing Zhang1,4*
Bing Zhang1,4* Zhiqiang Liu1,5*
Zhiqiang Liu1,5*Clinical studies have demonstrated that exposure to the inhalational general anesthetic nitrous oxide (N2O) produces antidepressant effects in depressed patients. However, the mechanisms underlying the antidepressant effects of N2O remain largely unknown. Neuronal nitric oxide synthase (nNOS)–mediated nitric oxide (NO) synthesis is essential for brain function and underlies the molecular mechanisms of many neuromodulators. We hypothesized that activation of the nNOS/NO pathway in the medial prefrontal cortex (mPFC) might mediate the antidepressant effects of N2O. In this study, we revealed that repeated N2O exposure produced antidepressant-like responses in mice. Our mechanistic exploration showed that repeated N2O exposure increased burst firing activity and that the expression levels of BDNF with nNOS activation were dependent in the mPFC. In particular, the antidepressant-like effects of N2O were also antagonized by local nNOS inhibition in the mPFC. In summary, our results indicated that N2O exposure enhances BDNF expression levels and burst firing rates in an nNOS activation dependent manner, which might underlie the pharmacological mechanism of the antidepressant-like effects of N2O exposure. The present study appears to provide further mechanistic evidence supporting the antidepressant effects of N2O.
Major depressive disorder (MDD) is one of the most severe mental disorders in the world and accounts for most of the nonfatal burden of mental and substance use disorders (1). The currently the available antidepressants mostly target the monoaminergic system, but these classic antidepressants are associated with low remission rates and a lag in the onset of antidepressant action obtained (2–4), highlighting an urgent and clear need for identifying a more efficacious and faster-acting therapeutic agents.
In recent decades, studies on the antidepressant effects of anesthetics have attracted considerable attention. Recent studies have suggested that some anesthetics show promise as therapeutics against MDD. Ketamine has attracted keen interest due to its remarkably rapid and sustained antidepressant effects (5–7). However, when administrated as a single intravenous dose, ketamine is associated with some unwanted side effects, including dissociation, headache, dizziness, elevated blood pressure, and blurred vision (8). Thus, the safety concerns of ketamine should be carefully considered, particularly after repeated dosing.
Nitrous oxide (N2O) is a widely used inhalational general anesthetic (9). Recent clinical studies have shown that N2O exerts rapid and marked antidepressant effects in patients with treatment-resistant major depression (TRMD), and the improvements can last for a full week (10). Importantly, compared with ketamine, N2O does not have psychotomimetic or cognitive side effects (11). Hence, N2O might be an attractive alternative for the development of mood disorder therapeutic. Because the improvements in TRMD obtained with N2O are associated with relatively few safety concerns, studies of the mechanism of the N2O-induced antidepressant effects will support a better understanding of the pharmacological treatment of depression.
N2O influences brain functions through multiple mechanisms of action, and this agent is commonly used in dentistry and obstetrics because it is an effective analgesic and anxiolytic agent. The anxiolytic effect might involve the activation of GABAA receptors through the benzodiazepine-binding site (12). N2O is also an antagonist of the N-methyl-D-aspartate (NMDA) receptor, which is a therapeutic target that might exert antidepressant effects (10, 11) similar to those obtained with ketamine. In addition, this agent has a wide range of other potential therapeutic targets, but the precise mechanism underlying the antidepressant effects of N2O has been less well addressed and might involve other molecular and receptor systems.
Nitric oxide (NO), a widespread signaling molecule that can be regulated by N2O, has diverse biological effects in the central nervous system (CNS) (13, 14). However, it remains unclear whether an increase in NO as a result of N2O is part of the antidepressant mechanism of N2O. Neuronal nitric oxide synthase (nNOS) is the key enzyme that mediates the NO signal transduction pathway in the brain (15), this enzyme participates in the regulation of learning, memory, and pathological conditions (16–19). Many shreds of evidence suggest that an imbalance of nNOS activity in the CNS implicated in the pathophysiology of MDD, and the nNOS system is proposed to be a potential therapeutic target for MDD (20). Thus, we hypothesized that the antidepressant effects of N2O might be mediated by the activation of the nNOS/NO pathway in the mPFC.
In this study, we observed antidepressant-like effects in mice after repeated N2O exposure. To address the underlying mechanism, we investigated the neuronal activity in the mPFC and further identified the functional roles of nNOS in the antidepressant-like effects of N2O.
Adult male CD-1 mice [weighing 25–35 g, postnatal day (PD) 42] were used in our experiments. The animals were housed in a limited-access rodent facility with four to five mice per cage in a room with a 12-h light/12-h dark cycle (cage size: 320 × 210 × 160 millimeters, lights on from 7:00 a.m. to 7:00 p.m.) and a constant temperature (22 ± 2°C). Sterilized drinking water and standard food were provided ad libitum. Before the experiments, the mice were habituated to these conditions for 2–3 days. In all experiments, animals were divided randomly into groups. Briefly, we numbered all animals and used Excel’s random number generator to randomize the animals to groups. All the animal studies and experimental procedures were approved by the Animal Care Committees of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences, and the experiments were performed in accordance with EU Directive 2010/63/EU on the protection of animals used for scientific purposes.
Adult CD-1 mice (aged 6–8 weeks) were anesthetized with pentobarbital sodium (80 mg/kg, i.p.) and fixed on an animal stereotaxic frame (RWD, 68016). Two 29-gauge guide cannulas were bilaterally placed 1 mm above the mPFC (anterior/posterior (A/P): +2.10 mm; medial/lateral (M/L): ± 0.35 mm; dorsal/ventral (D/V): -2.0 mm; Supplementary Figure 3A). A 39-gauge needle with a plastic cap on top was inserted into the guide cannula to prevent clogging during the recovery period. The cannulas were fixed to the skull using dental cement. After surgery, the mice were then allowed to recover for at least 1 week before the subsequent experiments. Drugs were microinjected with a 39-gauge injector cannula that was inserted into the guide cannula. One microliter of drug was infused (0.1 µl/min, 5 min total) into each side with a microsyringe. The injector cannulas were kept in place for an additional 5 min to minimize spread of the drug along the injection track. Finally, to confirm the accurate drug injection site and the spreading region of injected fluid, we have injected an equal volume of Chicago Sky Blue solution (2M NaCl and 0.5% Chicago Sky Blue) at the same coordinates in the mPFC (A/P: +2.10 mm; M/L: ± 0.35 mm; D/V: -3.0 mm). After behavioral tests, mice were sacrificed and each brain was sliced to examine the injection sites. Data were excluded where the injection site and the diffusion region were not located in the prelimbic subregion (PrL) of the mPFC.
Mice were randomly divided into four groups. (i) Mice in the control group were exposed to air in the chamber to exclude the effects of gas exposures. In the vehicle (ii) and experimental groups (iii), mice were exposed to the 50% N2 + 50% O2 mixture and 50% N2O + 50% O2 mixture for one 2-h session per day for three consecutive days, respectively. The mice in the positive control (iv) group were exposed to ketamine (20 mg/kg) 24 h before behavioral test. The dose of 20 mg/kg is an available concentration to elicit antidepressant effects in rodents that was confirmed by previous reports (21, 22). For drug administration, the mice were temporarily placed in a transparent airtight chamber (35 cm × 25 cm × 15 cm) at room temperature for 10 min of acclimatization before the gas exposure sessions. The concentration of N2O was monitored in real time. The gas flow was maintained at 2–3 L/min. At the end of the drug administration period, the mice were returned to their home cages, and 24 h after drug administration, the behavioral experiments of the mice were tested.
For local mPFC administration, the mice were fixed with gauze and allowed to recover for 1 week. The intracerebral drug deliveries were given in awake animals. The NOS inhibitor L-NG-nitroarginine methyl ester (L-NAME, Abcam, ab120136) was dissolved in saline to a concentration of 10 μg/μL. Local L-NAME administration was performed 30 min prior to gas exposure. L-NAME was infused into the mPFC (0.1 µl/min, 5 min total).
For the electrophysiological experiments, L-NAME (20 mg/kg) was dissolved in saline and administered to the mice 30 min before every gas exposure through an intraperitoneal injection (i.p.).
The apparatus used for the FST was a transparent plastic cylinder with size of 35 cm height × 10 cm diameter. During the FST, mice were individually placed in a cylinder of water with the depth of 20–26 cm to prevent the mice from touching the bottom with their limbs. The behavior tests were performed 24 h after the final N2O exposure. The water temperature was maintained at 24–25°C. The mice were allowed to swim for 10 min at least 1 day before the normal experiments were performed. Twenty-four hours after drug administration, the mice were placed in a quiet behavioral testing room and left undisturbed for at least 1 h before initiation of the experiment. The behavioral test was performed under subdued light (the same condition controlled during the tail suspension and open field tests). During the experiments, the mice were allowed to swim for 6 min, and their activity was videotaped. The duration of immobility, which was defined as the time during which the mice were floating or remained motionless, was assessed during the last 4 min.
In the TST, the mice were suspended by affixing their tails to the edge of a shelf at a height of 80 cm above the floor, and each animal was subjected to a 6-min suspension session. The activity of the mice was videotaped. The duration of immobility, which was defined as the time during which the mice remained motionless, was assessed during the last 4 min.
The OFT is a standard method for profiling the exploratory behavior and general activity of mice. The apparatus was composed of opaque black plastic (35 cm × 35 cm × 40 cm) and was placed in a soundproof room. The mice were randomly placed in the center of the field and were allowed to explore the box for 10 min, and their activities were monitored online. The path and velocity of each animal were calculated offline.
Twenty-four hours after drug administration, the mice were anesthetized with pentobarbital sodium (80 mg/kg, i.p.) and euthanized by decapitation. The bilateral mPFC regions were dissected immediately and homogenized in RIPA Lysis Buffer (Beyotime, P0013B) with 1 mM PMSF (Sigma, P7626), and dissociated by sonic disruption. The homogenate was centrifuged at 14,000 g for 15 min, and the total protein concentration was assessed. The primary and secondary antibodies used in the Western blotting assay were anti-nNOS (1:500, Abcam, ab76067), anti-iNOS (1:500, Abcam, ab15323), anti-BDNF (1:1,000, Abcam, ab108319), anti-β-tubulin (1:1,000, Cell Signaling Technology, 2128), and goat anti-rabbit IgG (1:5,000, Abcam, ab6721) antibodies. An enhanced chemiluminescence (ECL, Thermo, 32109) reaction solution was added. The samples were then recorded on X-ray film (Carestream, 8294985) for visualization, and the immune reactivity was quantified using ImageJ software.
The mice were anesthetized using pentobarbital sodium (80 mg/kg, i.p.) 24 h after drug administration, and then the electrophysiological experiment was performed. The recording lasted for 3–5 h, and during the recording period, the mice were maintained under anesthesia. The anesthetized mice were then mounted on a stereotaxic apparatus (Narishige, Japan) for extracellular recording. The body temperature was maintained at 36–37°C using a thermostatically controlled heating pad (ATC1000; WPI). In our study, we mainly recorded the spontaneous neuronal activities in the mPFC. The glass electrodes were filled with 2 M NaCl (Sigma, St Louis, MO, USA) and 0.5% Chicago sky blue (Sigma, C8679) solution. The electrodes were placed in the mPFC (coordinates: A/P: + 2.0 to +2.9 mm; M/L: 0 to ±0.90 mm; D/V: -2.5 to -3.5 mm) by using an electric-microdrive (Narishige). Neuronal firing was recorded by a preamplifier and bandpass filtered (Axon clamp, 900A). The data were analyzed with an oscilloscope (Nicolet, Model 2090-I, USA) and stored in a computer equipped with a Clampfit (10.2 Axon, USA) analysis system. To obtain stable firing, the first 2 min of firing were not included in the analysis, and the cells that fired for less than 5 min were excluded. After the recording, the location of the recorded neurons was histologically confirmed in the mPFC.
The data analysis was performed in double-blinded manner. The data are expressed as the means ± SEMs. SPSS software (version 19 for Windows) was used for the statistical analyses. The statistical parameters, including the exact value of n, precision measures (means ± SEMs) and statistical significance, are presented in the figures. In single drug treatment experimental design (Figures 1, 2, 3A, B, and Supplementary Figures 1 and 3), the data were evaluated by one-way ANOVA with a post hoc least significant difference (LSD) test. In the experimental design with an added NOS inhibitor (L-NAME, Figures 3C, D–5 and Supplementary Figure 2), the data were analyzed using two -way ANOVA and post hoc LSD tests. p < 0.05 indicated statistical significance.
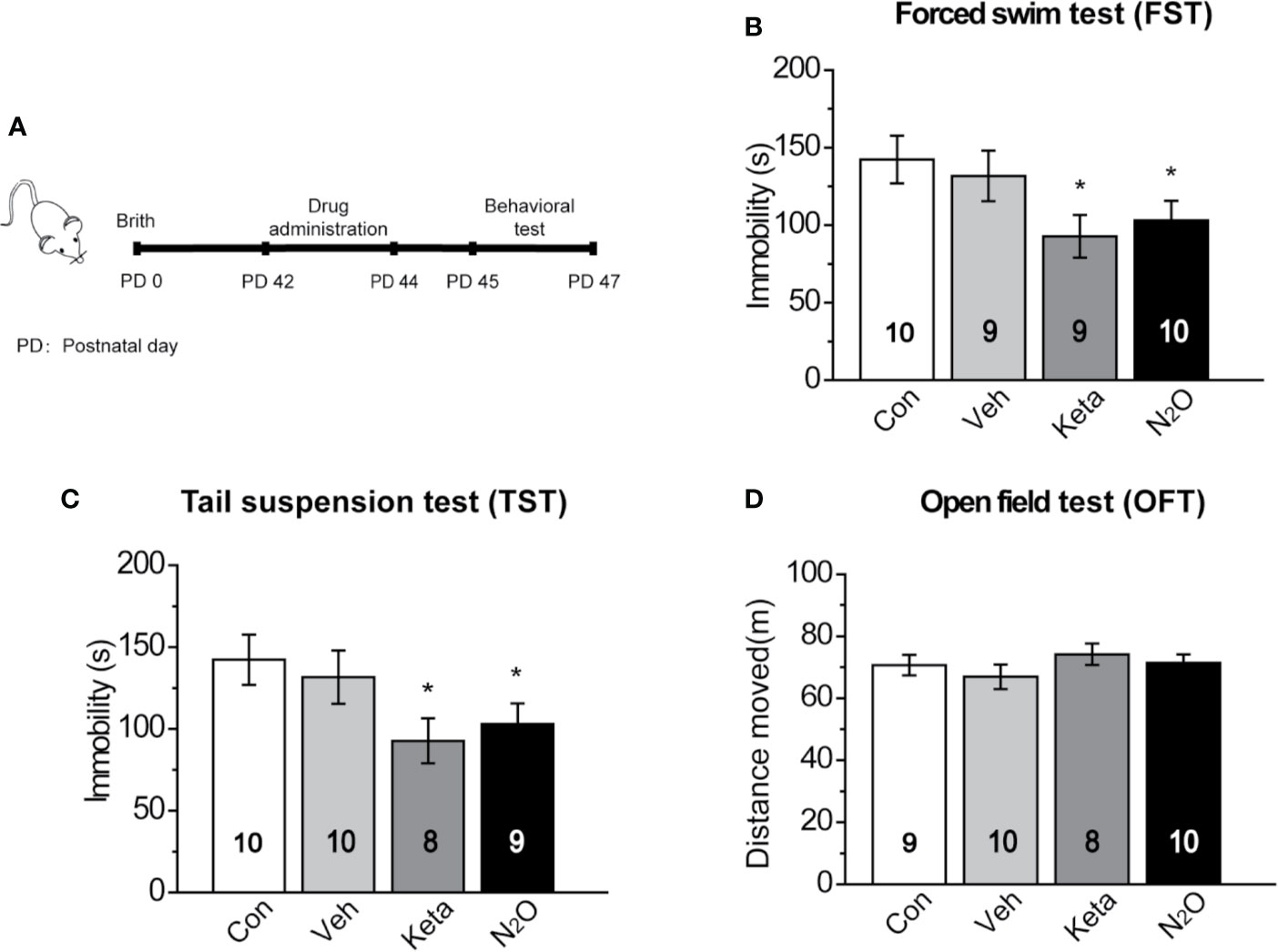
Figure 1 Antidepressant-like effects induced by repeated N2O exposure. (A) The timeline of the experiments and treatments is presented. Twenty-four hours after the final drug administration, the mice were subjected to forced swim test (FST) (B), tail suspension test (TST) (C), and open field test (OFT) (D). The group details as follows: Control (Con, air exposure); Vehicle (Veh, 50% N2 + 50% O2 exposure); Ketamine (Keta, 20 mg/kg, i.p.); N2O (50% N2O + 50% O2 exposure). All gas exposures were performed by administering a single dose for 2 h per day for three consecutive days. Ketamine was injected only once at the last day of drug exposure. The immobility time of the mice in the FST and TST was measured, and the total distance that the mice moved in the OFT was measured. The inserted number represents the number of animals in each group. *p < 0.05 compared with the control group.
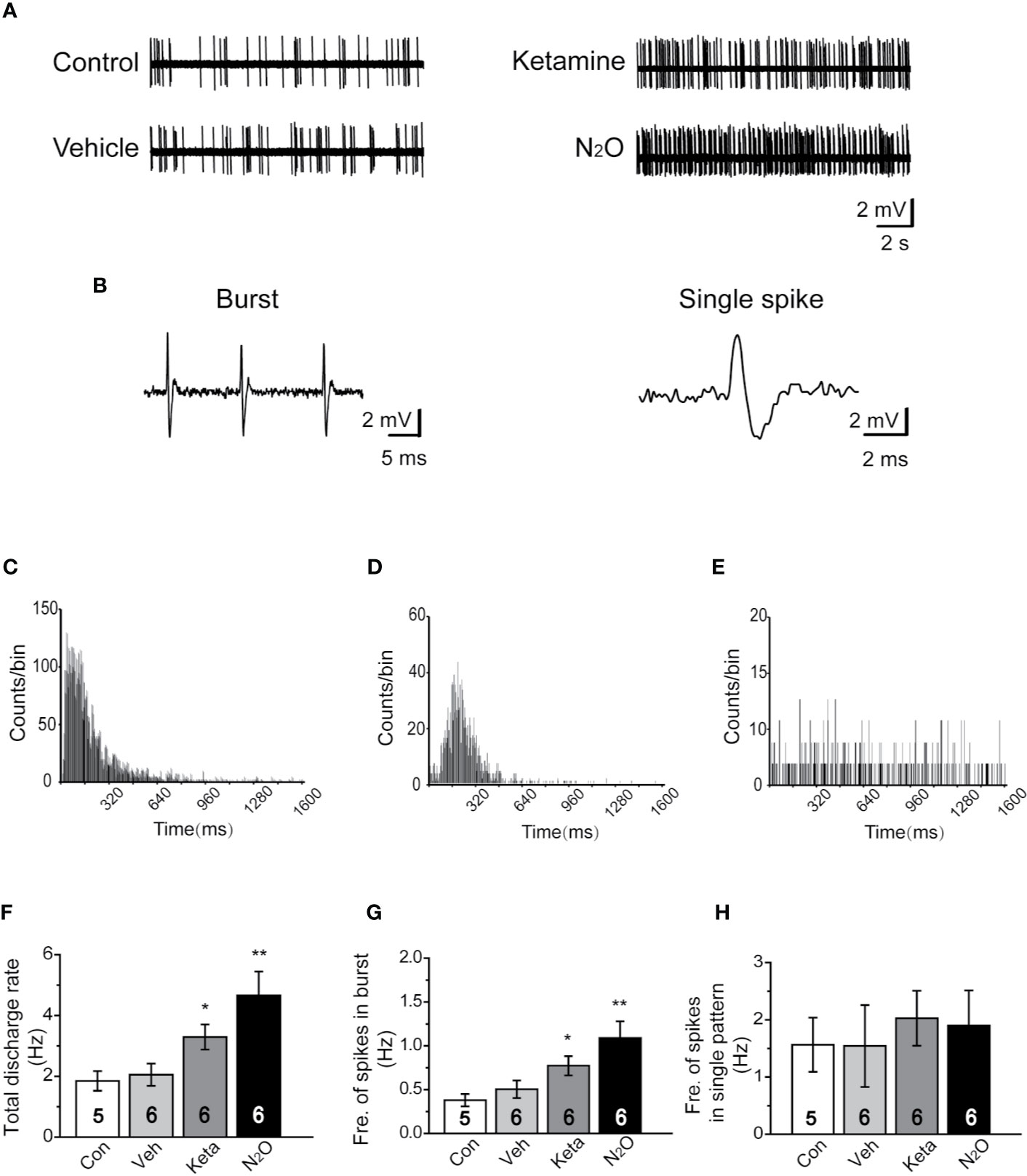
Figure 2 N2O enhanced the neuronal activity in the mPFC. In vivo extracellular electrophysiological recordings were performed 24 h after the final drug treatment. (A) Representative samples of the spontaneous firing activities of each group are shown. (B) Representative traces of the burst firing pattern (left) and single firing pattern (right) are shown. (C) The burst firing pattern is represented by a positive-skewed distribution of the inter-spike interval histogram (ISIH). The single firing patterns are represented by a nearly symmetrical ISIH (D), and the other is a relatively straight ISIH (E). The numbers of burst and single spikes were counted in the discharge rate. (F) The total discharge rates of the neurons in the mPFC of the different groups were counted. (G) The frequency of spikes in the burst firing patterns was shown for the different groups. (H) The frequency of spikes in the single firing patterns was shown for the different groups. The inserted number was the number of animals used for the electrophysiological recording. The group info is consistent with that in Figure 1. *p < 0.05 and **p < 0.01 compared with the control group.
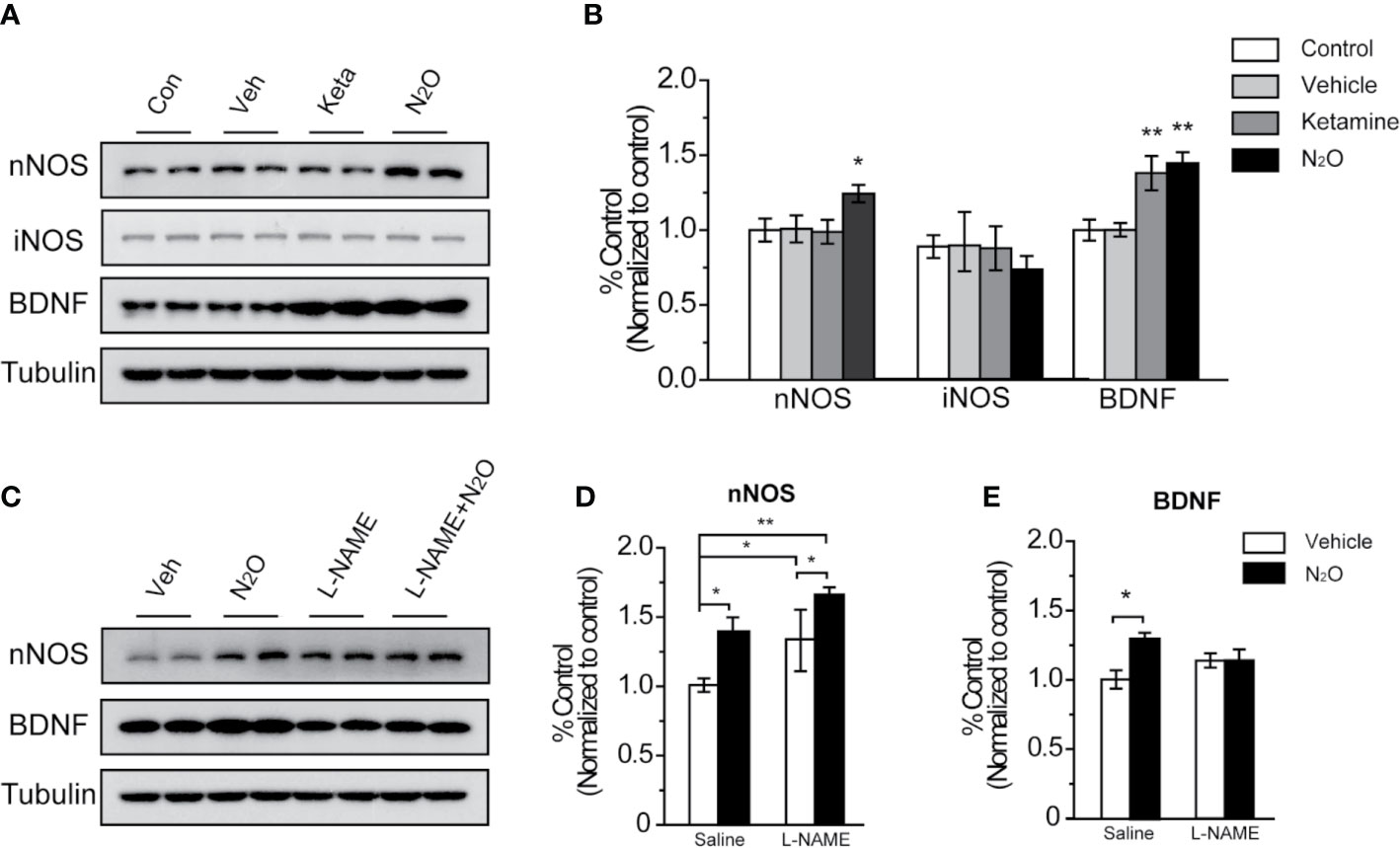
Figure 3 N2O enhanced the expression levels of nNOS and BDNF in the mPFC. All the mPFC tissues were extracted 24 h after the final drug administration. (A, C) Western blot images and (B, D, E) quantification analysis of nNOS, iNOS and BDNF expression levels in the mPFC. The β-tubulin was measured as the reference protein. For panels (A, B), the group info is consistent with that in Figures 1 and 2. *p < 0.05 and **p < 0.01 compared with the control group. For panels (C–E), the mouse mPFCs were pre-infused with saline (5 μL/side) or L-NAME (5 μg/side) 30 min before every gas exposure. The groups were as follows: Vehicle (saline or L-NAME mPFC infusion before 50% N2 + 50% O2 exposure) and N2O (saline or L-NAME mPFC infusion before 50% N2O + 50% O2 exposure). *p < 0.05 and **p < 0.01 compared with the vehicle group.
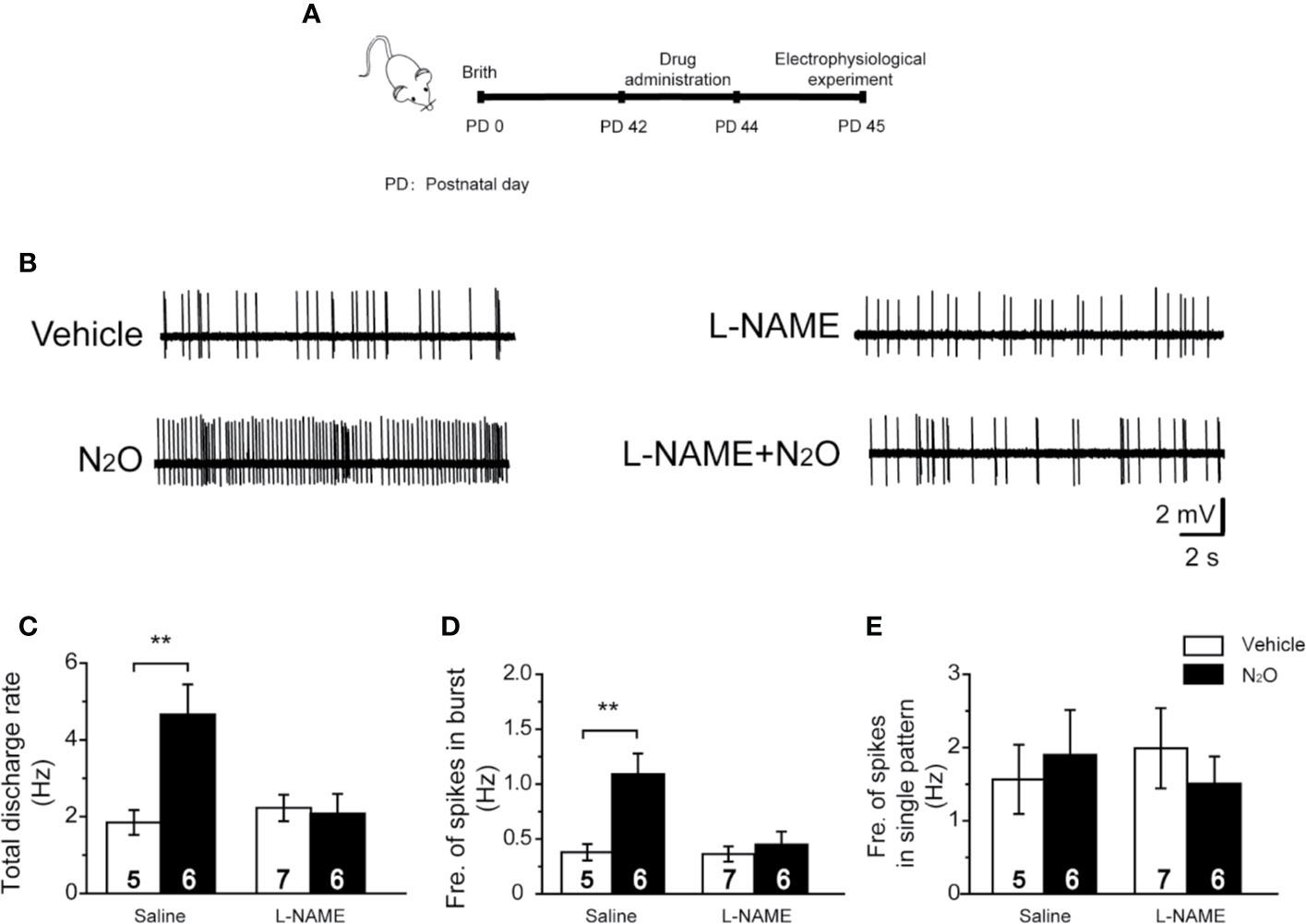
Figure 4 A NOS inhibitor reversed the action of N2O on the neuronal activity in the mPFC. (A) The timeline of the experimental process. In vivo extracellular electrophysiological recordings were performed 24 h after the final drug treatment. (B) Representative samples of the spontaneous firing activities of each group are shown. The numbers of burst and single spikes were both counted in the discharge rate. (C) The total discharge rates of the neurons in the mPFC were counted for the different groups. (D) The frequency of spikes in the burst firing patterns was shown for the different groups. (E) The frequency of spikes in the single firing patterns was shown for the different groups. The mice were pretreated with saline (10 mL/kg, i.p.) or L-NAME (20 mg/kg, i.p.) by intraperitoneal injection 30 min before the gas exposure. The groups were as follows: Vehicle (saline or L-NAME intraperitoneal injection before 50% N2 + 50% O2 exposure); N2O (saline or L-NAME intraperitoneal injection before 50% N2O + 50% O2 exposure). The inserted number was the number of animals used for the electrophysiological recording. **p < 0.01 compared with the vehicle group.
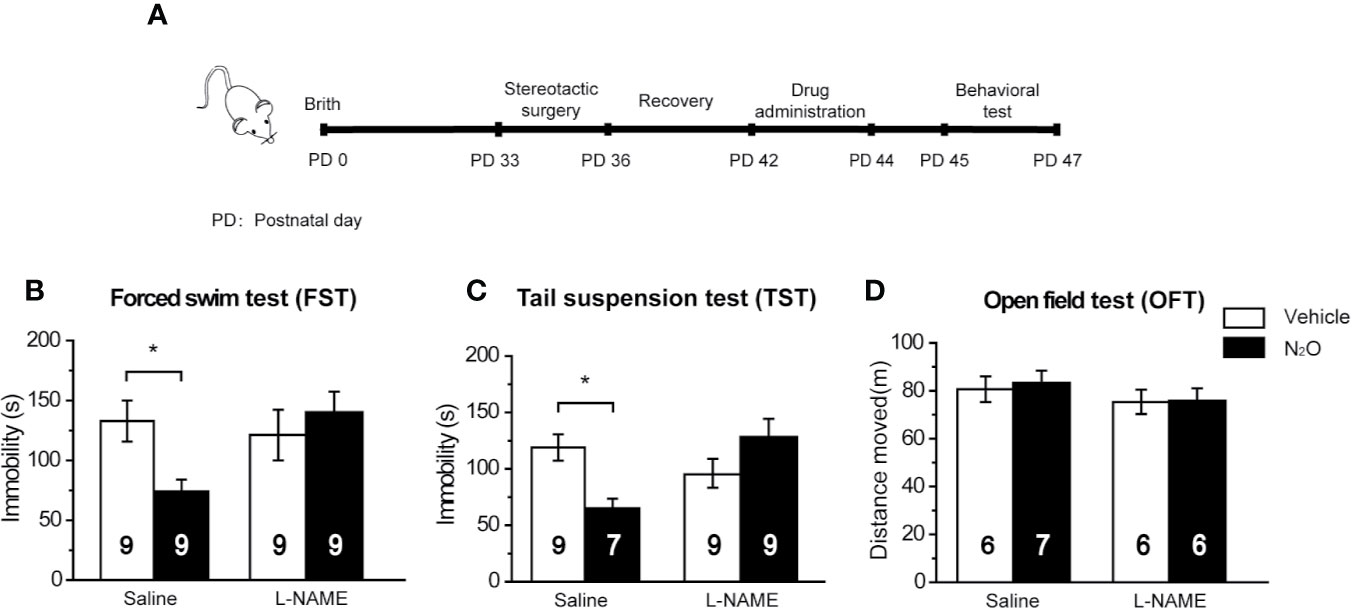
Figure 5 The antidepressant-like effects of N2O were blocked by a NOS inhibitor. (A) The timeline of the experimental process. All the experiments were performed 24 h after the final drug administration, and the mice were then subjected to FST (B), TST (C) and OFT (D). The mPFC of the mice was pre-infused with saline (5 μL/side) or the NOS inhibitor L-NAME (5 μg/side) 30 min before the gas exposure. The group info is consistent with that in Figures 3C–E. The immobility time of the mice in the FST and TST was measured, and the total distance that the mice moved in the OFT was measured. The inserted number represents the number of animals in each group. *p < 0.05 compared with the vehicle group.
In this study, 50% N2O mixed with 50% O2 mixture was used based on the clinical study by Nagele et al. (10). First, we assessed whether N2O exerted antidepressant-like effects in the male mice. A single dose of the N2O mixture was administered for 2 h per day, and this exposure was repeated on three consecutive days (Supplementary Figure 1). To avoid stress response, three behaviors (FST, TST, and OFT) were tested once a day. The FST, TST, and OFT were performed 24, 48, and 72 h after the last exposure to N2O, respectively. The timeline of the experiments and treatments is presented in Figure 1A. The results showed that both repeated N2O exposure, and ketamine treatments significantly reduced the immobility duration in the FST and TST compared with those of the control and vehicle groups [Figure 1B, one-way ANOVA, F (3, 34) = 2.896, p = 0.049; Figure 1C, one-way ANOVA, F (3, 33) = 2.923, p = 0.048]. Total distance in the OFT was not significantly altered in the N2O- and ketamine-treated groups [Figure 1D, one-way ANOVA, F (3, 33) = 0.455, p = 0.715], and this finding excluded the possibility that N2O or ketamine-induced psychomotor changes and yielded false-positive results. Collectively, the results indicated that repeated exposure to N2O induces antidepressant-like responses in mice.
To understand whether N2O can affect neuronal activity, the spontaneous neuronal activity was assessed through in vivo extracellular electrophysiological recordings. The neural activity of the prelimbic subregion of the mPFC (PrL, A/P: +2.0 to +2.9 mm; M/L: 0 to ± 0.9 mm; D/V: -2.5 to -3.5 mm; Supplementary Figure 3B) was recorded. Representative samples of the spontaneous firing activities of different groups are shown (Figure 2A). A positive-skewed distribution in the inter-spike interval histogram (ISIH) (Figure 2C) indicated a burst firing pattern (Figure 2B, left). A single firing pattern was observed as the firing activity of neurons fired in irregular single spikes (Figure 2B, right). We recorded two discharge single firing patterns: one pattern was represented by a nearly symmetrical ISIH (Figure 2D), and the other pattern was represented by a relatively straight ISIH (Figure 2E).
Because neurons often showed burst and single-spike firing activities within a single-unit recording, we calculated the discharge rate, which included the burst and single spikes. The results showed that N2O can elicit a significant change in the discharge rate of neurons in the mPFC [Figure 2F, one-way ANOVA, F (3, 64) = 7.637, p = 0.0001]. Besides, the burst activity, including the frequency of spikes in the burst, obtained with N2O was higher than that of the vehicle group [Figure 2G, one-way ANOVA, F (3, 41) =10.192, p = 0.00009]. Furthermore, N2O significantly increased the overall firing rate and shifted the cumulative frequency of inter-spike intervals (ISIs) toward those observed in a burst pattern. Thus, burst firing in the mPFC might contribute to the antidepressant-like effects of N2O. We counted and analyzed the burst firing and single firing patterns, respectively, and found that approximately 65% of the recorded neurons exhibited burst firing and the remaining 35% of neurons showed single firing patterns. However, we found that single firing patterns did not cause significant changes in discharge in the different groups [Figure 2H, one-way ANOVA, F (3, 19) =0.173, p = 0.913]. The increase in burst firing obtained after N2O administration made major contributions to the enhancement in neuronal activities in the mPFC, which suggested that the burst firing pattern can be regarded as a predictor of the N2O treatment response.
To further examine the possible mechanisms underlying the antidepressant-like effects of N2O treatment, we assessed the changes in protein expression by Western blotting.
NOS has three isoforms, namely, neuronal NOS (nNOS), inducible NOS (iNOS) and endothelial NOS (eNOS), and functional deficits in nNOS or iNOS had been reported to associated with MDD. However, in our study, we found that repeated N2O exposure increased the expression level of nNOS but not iNOS in the mPFC [Figures 3A, B, one-way ANOVA, F (3, 20) = 5.803, p = 0.005 for BDNF, one-way ANOVA, F (3, 20) = 3.241, p = 0.044 or nNOS; and one-way ANOVA, F (3,12) = 1.551, p = 0.252 for iNOS]. Furthermore, the level of BDNF, a critical neurotropic factor for neurogenesis and neuroplasticity, was also increased by the administration of N2O (Figures 3A, B). BDNF plays important an role in neural development and the regulation of synaptic plasticity (23), which is diminished in depressive disorders (24).
To confirm whether BDNF expression is dependent on the activation of nNOS after N2O administration, we applied NOS inhibitor, L-NAME (5 μg/side), which we locally infused into the mPFC before drug treatment to specifically block nNOS activity in the mPFC. The results showed that N2O exposure didn’t further increase BDNF levels in the mPFC when that region was pre-treated with L-NAME [Figures 3C–E, two-way ANOVA, F (1, 19) = 3.766, p = 0.067 for BDNF, and two-way ANOVA, F (1, 20) = 1.325, p = 0.263 for nNOS]. In addition, we unexpectedly observed that local L-NAME treatment increased nNOS expression levels without affecting BDNF expression levels (Figures 3C–E). Because BDNF is a major growth factor in the brain that might represent a potential therapeutic target in MDD, the increase nNOS activation dependent increase in BDNF might be involved in the antidepressant-like effects of N2O.
The above results demonstrated that repeated N2O exposure increased BDNF signaling in a nNOS activation-dependent manner. Therefore, we hypothesized that nNOS might play a key role in the N2O-induced enhancement of neuronal activity in the mPFC.
To verify this hypothesis, we injected the mice with L-NAME (20 mg/kg, i.p.) in the mPFC 30 min before N2O exposure. Twenty-four hours after the final N2O exposure, extracellular electrophysiologyical recording was performed in the mPFC (Figure 4A). Interestingly, we found that the systemic injection of L-NAME could block the antidepressant-like effects induced by repeated N2O exposure [Supplementary Figure 2, two-way ANOVA, F (1, 35) = 3.259, p = 0.080 for FST; two-way ANOVA, F (1, 33) = 2.983, p = 0.93 for TST; two-way ANOVA, F (1,30) = 1.867, p = 0.182 for OFT]. We further examined whether mPFC neuronal activity could also be inhibited by pretreatment with L-NAME. As expected, the N2O-induced increases in the discharge rate and burst firing were blocked by pretreatment with L-NAME [Figures 4B, C, two-way ANOVA, F (1, 42) = 3.902, p = 0.055; Figure 4D; two-way ANOVA, F (1, 42) = 3.434, p = 0.071]. The irregular firing did not significantly change after drug administration [Figure 4E, two-way ANOVA, F (1, 24) = 1.485, p = 0.235], as described above. These results suggested that the antidepressant-like effects of N2O might be due to increased mPFC excitability followed by nNOS activation.
Based on the above results, we confirmed that N2O increased neuronal activity and BDNF expression in the mPFC, and these processes were dependent on the activation of nNOS. Therefore, we further asked whether the antidepressant-like effects of N2O were dependent on nNOS activation in the mPFC. To block the activity of nNOS in the mPFC, we pretreated the mPFC with L-NAME 30 min before N2O exposure. The behavioral tests were performed 24 h after the final exposure of N2O (Figure 5A). The results showed that repeated N2O exposure decreased immobility duration in the FST and TST was reversed by pretreatment with L-NAME (5 μg/side) in the mPFC (PrL). L-NAME alone showed no significant effects on the FST and TST [Figure 5B, two-way ANOVA, F (1, 32) = 1.768, p = 0.193; Figure 5C, two-way ANOVA, F (1, 29) = 0.391, p = 0.537]. The total distance traveled in the open field did not show significant changes between the groups, which indicated that L-NAME and/or N2O exposure did not affect the locomotor activity of the mice [Figure 5D, two-way ANOVA, F (1, 21) = 0.092, p = 0.765]. In summary, these results suggested that the antidepressant-like effects induced by repeated N2O exposure were dependent on the activation of nNOS in the mPFC (PrL).
In our study, we investigated the antidepressant-like properties of N2O through multiple behavioral tests. To address the underlying mechanism, we explored the neuronal activities in the mPFC and intracellular signal transduction pathways. The results showed that repeated N2O exposure increased the neuronal burst-firing activity and upregulated the nNOS and BDNF expression levels in the mPFC. Furthermore, the antidepressant-like effects and enhanced neuronal activities induced by N2O were blocked by the inhibition of nNOS. The results suggested that repeated N2O exposure enhances BDNF expression level and burst firing rate in an nNOS activation-dependent manner in the mPFC, and these effects might underpin the molecular mechanisms underlying the antidepressant-like effects of N2O.
The concentration of general anesthetics is a critical impact factor for CNS function, and the affected sites of the CNS by general anesthetics are concentration dependent (25). The binding preference of general anesthetics for subunits of excitatory and inhibitory neurotransmitter receptors, such as NMDA receptors and GABAA receptors, is also concentration dependent (26). In addition, substantial studies have shown that subanesthetic concentrations prompt neuronal stem cell survival and neurogenesis, but high concentrations may cause neurotoxicity in the CNS (27). For N2O, a clinical study by Nagele and colleagues applied short-term subanesthetic N2O exposure (50% N2O + 50% O2 inhaled for 1 h in a single session), which showed antidepressant effects in TRMD. The subanesthetic dose of N2O was selected based on the routine use for analgesia and mild sedation in anesthesiology and dentistry (10, 28). In the present study, the 50% N2O concentration was selected, but unlike appearances in humans, repeated 2 h rather than a single 1-h subanesthetic N2O exposure triggered antidepressant-like effects in mice. However, the underlying mechanism of this phenomenon is unclear. The optimal antidepressant dose of N2O should be further addressed in future studies.
NO is a gaseous neurotransmitter that governs multiple physiological functions in the CNS. NO is synthesized by NOS, which has three isoforms, namely, nNOS, iNOS, and eNOS, and all three NOS isoforms exist in the CNS and affect cell signaling in brain (29). Evidence has shown that deficits in NOS activity are associated with the neurobiology of MDD (30, 31). Especially for nNOS, studies have shown that dysfunction of nNOS in the paraventricular hypothalamic nucleus, locus coeruleus, mPFC, and hippocampus, is related to MDD (29, 32, 33). Besides, iNOS also is involved in stress-triggered depression, as iNOS derived NO and iNOS mRNA levels increased in the cortices of depression animal model (34). However, a lower number of studies reported the relationship between eNOS and depression and genetic studies suggested there is no correlation between eNOS and MDD (35). In the present study, we demonstrated that N2O exposure significantly increased the expression level of nNOS rather than iNOS. As the major route for the production of NO in the brain is mediated by nNOS catalysis (29), our results suggest that nNOS might be a dominant molecule in mediating the antidepressant-like effects of N2O.
Although abundant reports have emphasized the critical role of nNOS in the neuropathology of MDD, the conclusion is controversial (29). Some studies pointed to an increased activity of nNOS in the corticolimbic system in MDD, and NOS inhibitors showed antidepressant effects through indirect regulation of the monoaminergic system (29, 36). However, several other studies showed that stress exposure diminished nNOS activity in the hippocampus which further caused a deficit in learning and memory processes in animals (37, 38). Postmortem studies also revealed a decrease in nNOS expression in the anterior cingulate cortex (ACC) of MDD patients, which indicated weakened nNOS activity in the process of MDD (39). Therefore, there exists an imbalance but not simply an increase or decrease in nNOS activity in the pathophysiology of MDD. In the present study, we unexpectedly observed that local NOS inhibitor (L-NAME) treatment increased nNOS expression levels without affecting BDNF levels in the mPFC or the depressive behaviors of animals. For this paradoxical phenomenon, we infer that there may exist a feedback mechanism by which L-NAME, as a NOS inhibitor, inhibits the activity of nNOS and further elevates the expression level of nNOS as a result. Because the real activity of nNOS was low (inhibited by L-NAME), L-NAME induced higher expression levels of nNOS could not further activate BDNF signaling and trigger antidepressant-like effects. The concrete mechanisms need to be studied in the future.
BDNF is a developmentally expressed growth factor that regulates plasticity in the adult brain. Preclinical studies have shown that the function of BDNF could be decreased by several forms of stresses Chronic treatments with antidepressants activate BDNF-mediated signaling (40). nNOS-derived NO could modulate BDNF signaling in stress adaptation, which further affects synaptic plasticity in emotionally related brain regions such as cerebral cortex, amygdala, hippocampus and striatum (29, 41). However, the interplay between NO and BDNF signaling has been elusive. Some reports point to that NO appears to negatively modulate BDNF function, because BDNF signaling is augmented by the NOS inhibitor L-NAME (42). However, evidence from cultured hippocampal neurons demonstrated that endogenous low levels of NO could facilitate BDNF signaling, indicating that BDNF signaling is regulated by NO in a concentration-dependent manner (43). In the present study, we observed that the BDNF expression level in the mPFC was upregulated by N2O exposure, and the increase of BDNF was blocked by pre-administration of the NOS inhibitor L-NAME. Based on the above information, we suggest that N2O exposure with a subanesthetic dose might increase intracellular NO, which activates BDNF-dependent synaptic plasticity. Our findings enhance the understanding of the role of nNOS activation induced increase of BDNF signaling in the antidepressant-like responses and support a promising research direction of N2O as a potential antidepressant for clinical use.
Although N2O and ketamine have similar antidepressant actions and mechanisms, the antidepressant efficacy of N2O might not be robust as that of ketamine. Moreover, previous studies have indicated that N2O exposure is associated with many unfavorable side-effects (e.g., euphoria, altered body awareness and image, altered time and perception, and dreamy, detached experiential states) (44) and prolonged N2O administration interferes with the activity of vitamin B12, which results in sensory ataxia problems (11, 45). Therefore, the efficacy, safety and tolerability of N2O should be carefully evaluated in the future. More work needs to be done to investigate the effective initial dose and maintenance dose, the therapeutic drug duration and the potential risks of N2O when given repeatedly over time (44).
A valid animal model of depression is necessary to identify the biological mechanisms of MDD and the features of antidepressants. Currently, stress-induced models are the most commonly used depression model, which might be based on the fact that initial episodes of depression are precipitated by adversity. In addition, the etiology-based animal model of depression that directly targeting the underlying biological causations, including the hypothalamus-pituitary-adrenal (HPA) axis, the neuroinflammation system and the neurotransmission, also recapitulating specific dysfunctions of depression (46). MDD is a heterogeneous disease, and diverse animal models are believed to recapitulate different aspects of symptomatic dimensions and neuropathology associated with depression (47). The antidepressant efficacies and pharmacology mechanisms of N2O should be carefully detected in the future study.
Mood disorders are characterized by profound deficits in reward circuits of brain. Mesocorticolimbic dopaminergic system, as the most important reward circuits, is constructed by the ventral tegmental area (VTA) originated dopaminergic projections to many brain areas such as the mPFC, nucleus accumbens (NAc), amygdala, and hippocampus. Through highly complex inter-projections, these brain regions have been shown to be involved in emotion-related behaviors, especially in the encoding of stressful events (48). The antidepressants might have broad influences on the reward circuit system. According to literatures, ketamine rapidly increases the activity of reward-related brain regions, including the orbitofrontal cortex, ventral striatum, VTA, amygdala, insula, and the ACC, which accompanied with the fast remission of depressive symptoms by ketamine (49). Moreover, ketamine also have positive impacts on dopaminergic transmission (50, 51). In the present study, we depicted a specific antidepressant mechanism of N2O by focusing on the mPFC. We believe that N2O might also have diverse influences on other reward-related brain regions that similar to the effects of ketamine, which should be addressed in the future studies.
In conclusion, we verified that repeated N2O exposure exerts antidepressant-like effects in animals. Our preliminary results suggested that repeated N2O exposure can enhance BDNF expression level and burst firing rates in an nNOS activation dependent manner in the mPFC, which might underpin the pharmacological mechanism underlying the antidepressant-like effects of N2O exposure. Collectively, the results obtained in the present study might provide some theoretical basis for developing a relatively promising and safe method for the treatment of MDD.
The datasets generated for this study are available on request to the corresponding authors.
The animal study was reviewed and approved by All the animal studies and experimental procedures were approved by the Animal Care Committees of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences, and the experiments were performed in accordance with EU Directive 2010/63/EU on the protection of animals used for scientific purposes.
WL, QL, and BY conducted the majority of the experiments, including stereotactic surgery, electrophysiological part and behavioral tests. HC, JL, and WD conducted the western blotting and stereotactic surgery. ZX, JL, and FS carried out the animal behavioral tests and electrophysiological part. TL and FG conducted the data analysis and brain regions detection. WL, QL, BY, BZ, and ZL. prepared the manuscript. WL, BZ, and ZL conceived and supervised the project. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation (grant numbers 81771188, 31671049, 81971418, and 81901376), Science and Technology Commission of Shanghai Municipality (grant numbers 14411966700 and 16411967400), the China Postdoctoral Science Foundation Funded Project (grant number 2017M621535), the Ministry of Science and Technology (grant number 2013CB91060101), Shanghai Municipal Commission of Health and Family Planning (grant number 201740072), the Fundamental Research Funds for the Central Universities (grant number 22120180534), and “Personalized Medicines-Molecular Signature-based Drug Discovery and Development”, Strategic Priority Research Program of the Chinese Academy of Sciences (grant numbers XDA12040302 and XDA120402140).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2020.00837/full#supplementary-material
1. Whiteford HA, Degenhardt L, Rehm J, Baxter AJ, Ferrari AJ, Erskine HE, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet (2013) 382:1575–86. doi: 10.1016/s0140-6736(13)61611-6
2. Hirschfeld RMA. History and evolution of the monoamine hypothesis of depression. J Clin Psychiat (2000) 61:4–6.
3. Wang PS, Insel TR. NIMH-funded pragmatic trials: moving on. Neuropsychopharmacology (2010) 35:2489–90. doi: 10.1038/npp.2010.161
4. Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: Implications for clinical practice. Am J Psychiat (2006) 163:28–40. doi: 10.1176/appi.ajp.163.1.28
5. Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiat (2000) 47:351–4. doi: 10.1016/S0006-3223(99)00230-9
6. Duman RS, Aghajanian GK. Synaptic Dysfunction in Depression: Potential Therapeutic Targets. Science (2012) 338:68–72. doi: 10.1126/science.1222939
7. Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiat (2006) 63:856–64. doi: 10.1001/archpsyc.63.8.856
8. Short B, Fong J, Galvez V, Shelker W, Loo CK. Side-effects associated with ketamine use in depression: a systematic review. Lancet Psychiatry (2018) 5:65–78. doi: 10.1016/S2215-0366(17)30272-9
9. West JB. Humphry Davy, nitrous oxide, the Pneumatic Institution, and the Royal Institution. Am J Physiol Lung Cell Mol Physiol (2014) 307:L661–667. doi: 10.1152/ajplung.00206.2014
10. Nagele P, Duma A, Kopec M, Gebara MA, Parsoei A, Walker M, et al. Nitrous Oxide for Treatment-Resistant Major Depression: A Proof-of-Concept Trial. Biol Psychiatry (2015) 78:10–8. doi: 10.1016/j.biopsych.2014.11.016
11. Zorumski CF, Nagele P, Mennerick S, Conway CR. Treatment-Resistant Major Depression: Rationale for NMDA Receptors as Targets and Nitrous Oxide as Therapy. Front Psychiatry (2015) 6:172:172. doi: 10.3389/fpsyt.2015.00172
12. Sanders RD, Weimann J, Maze M. Biologic effects of nitrous oxide: a mechanistic and toxicologic review. Anesthesiology (2008) 109:707–22. doi: 10.1097/ALN.0b013e3181870a17
13. Bredt DS, Snyder SH. Nitric-Oxide Mediates Glutamate-Linked Enhancement of Cgmp Levels in the Cerebellum. P Natl Acad Sci USA (1989) 86:9030–3. doi: 10.1073/pnas.86.22.9030
14. de Sousa RT, Zanetti MV, Busatto GF, Mouro MG, Zarate CA Jr., Gattaz WF, et al. Lithium increases nitric oxide levels in subjects with bipolar disorder during depressive episodes. J Psychiatr Res (2014) 55:96–100. doi: 10.1016/j.jpsychires.2014.03.023
15. Forstermann U, Schmidt HHHW, Pollock JS, Sheng H, Mitchell JA, Warner TD, et al. Isoforms of Nitric-Oxide Synthase - Characterization and Purification from Different Cell-Types. Biochem Pharmacol (1991) 42:1849–57. doi: 10.1016/0006-2952(91)90581-O
16. Bohme GA, Bon C, Lemaire M, Reibaud M, Piot O, Stutzmann JM, et al. Altered Synaptic Plasticity and Memory Formation in Nitric-Oxide Synthase Inhibitor-Treated Rats. P Natl Acad Sci USA (1993) 90:9191–4. doi: 10.1073/pnas.90.19.9191
17. Schuman EM, Madison DV. A Requirement for the Intercellular Messenger Nitric-Oxide in Long-Term Potentiation. Science (1991) 254:1503–6. doi: 10.1126/science.1720572
18. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J (2012) 33:829–+. doi: 10.1093/eurheartj/ehr304
19. Zhou L, Zhu DY. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide Biol Ch (2009) 20:223–30. doi: 10.1016/j.niox.2009.03.001
20. Wegener G, Volke V. Nitric Oxide Synthase Inhibitors as Antidepressants. Pharmaceut (Basel) (2010) 3:273–99. doi: 10.3390/ph3010273
21. Shirayama Y, Hashimoto K. Lack of Antidepressant Effects of (2R,6R)-Hydroxynorketamine in a Rat Learned Helplessness Model: Comparison with (R)-Ketamine. Int J Neuropsychopharmacol (2018) 21:84–8. doi: 10.1093/ijnp/pyx108
22. Yang Y, Cui Y, Sang K, Dong Y, Ni Z, Ma S, et al. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature (2018) 554:317–22. doi: 10.1038/nature25509
23. Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci (2001) 2:24–32. doi: 10.1038/35049004
24. Castren E, Rantamaki T. The Role of BDNF and Its Receptors in Depression and Antidepressant Drug Action: Reactivation of Developmental Plasticity. Dev Neurobiol (2010) 70:289–97. doi: 10.1002/dneu.20758
25. Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci (2004) 5:709–20. doi: 10.1038/nrn1496
26. Rudolph U, Mohler H. GABAA receptor subtypes: Therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism. Annu Rev Pharmacol Toxicol (2014) 54:483–507. doi: 10.1146/annurev-pharmtox-011613-135947
27. Vutskits L, Xie Z. Lasting impact of general anaesthesia on the brain: mechanisms and relevance. Nat Rev Neurosci (2016) 17:705–17. doi: 10.1038/nrn.2016.128
28. Nagele P, Zorumski CF, Conway C. Exploring Nitrous Oxide as Treatment of Mood Disorders: Basic Concepts. J Clin Psychopharmacol (2018) 38:144–8. doi: 10.1097/JCP.0000000000000837
29. Joca SRL, Sartim AG, Roncalho AL, Diniz CFA, Wegener G. Nitric oxide signalling and antidepressant action revisited. Cell Tissue Res (2019) 377:45–58. doi: 10.1007/s00441-018-02987-4
30. Dhir A, Kulkarni SK. Nitric oxide and major depression. Nitric Oxide (2011) 24:125–31. doi: 10.1016/j.niox.2011.02.002
31. Ghasemi M, Claunch J, Niu K. Pathologic role of nitrergic neurotransmission in mood disorders. Prog Neurobiol (2019) 173:54–87. doi: 10.1016/j.pneurobio.2018.06.002
32. Bernstein HG, Stanarius A, Baumann B, Henning H, Krell D, Danos P, et al. Nitric oxide synthase-containing neurons in the human hypothalamus: reduced number of immunoreactive cells in the paraventricular nucleus of depressive patients and schizophrenics. Neuroscience (1998) 83:867–75. doi: 10.1016/s0306-4522(97)00461-2
33. Bernstein HG, Heinemann A, Krell D, Mawrin C, Bielau H, Danos P, et al. Further immunohistochemical evidence for impaired NO signaling in the hypothalamus of depressed patients. Ann N Y Acad Sci (2002) 973:91–3. doi: 10.1111/j.1749-6632.2002.tb04613.x
34. Peng YL, Liu YN, Liu L, Wang X, Jiang CL, Wang YX. Inducible nitric oxide synthase is involved in the modulation of depressive behaviors induced by unpredictable chronic mild stress. J Neuroinflam (2012) 9:75. doi: 10.1186/1742-2094-9-75
35. Zeman M, Jachymova M, Jirak R, Vecka M, Tvrzicka E, Stankova B, et al. Polymorphisms of genes for brain-derived neurotrophic factor, methylenetetrahydrofolate reductase, tyrosine hydroxylase, and endothelial nitric oxide synthase in depression and metabolic syndrome. Folia Biol (Praha) (2010) 56:19–26.
36. Silva M, Aguiar DC, Diniz CR, Guimaraes FS, Joca SR. Neuronal NOS inhibitor and conventional antidepressant drugs attenuate stress-induced fos expression in overlapping brain regions. Cell Mol Neurobiol (2012) 32:443–53. doi: 10.1007/s10571-011-9775-1
37. Steinert JR, Chernova T, Forsythe ID. Nitric Oxide Signaling in Brain Function, Dysfunction, and Dementia. Neuroscientist (2010) 16:435–52. doi: 10.1177/1073858410366481
38. Paul V, Ekambaram P. Involvement of nitric oxide in learning & memory processes. Indian J Med Res (2011) 133:471–8.
39. Gao SF, Qi XR, Zhao J, Balesar R, Bao AM, Swaab DF. Decreased NOS1 Expression in the Anterior Cingulate Cortex in Depression. Cereb Cortex (2013) 23:2956–64. doi: 10.1093/cercor/bhs285
40. Li J, Zhou Y, Liu B-B, Liu Q, Geng D, Weng L-J, et al. Nobiletin Ameliorates the Deficits in Hippocampal BDNF, TrkB, and Synapsin I Induced by Chronic Unpredictable Mild Stress. Evidence-Based Complement Altern Med (2013) 2013:11. doi: 10.1155/2013/359682
41. Prast H, Philippu A. Nitric oxide as modulator of neuronal function. Prog Neurobiol (2001) 64:51–68. doi: 10.1016/S0301-0082(00)00044-7
42. Biojone C, Casarotto PC, Joca SR, Castren E. Interplay Between Nitric Oxide and Brain-Derived Neurotrophic Factor in Neuronal Plasticity. CNS Neurol Disord Drug Targets (2015) 14:979–87. doi: 10.2174/1871527314666150909113727
43. Kolarow R, Kuhlmann CR, Munsch T, Zehendner C, Brigadski T, Luhmann HJ, et al. BDNF-induced nitric oxide signals in cultured rat hippocampal neurons: time course, mechanism of generation, and effect on neurotrophin secretion. Front Cell Neurosci (2014) 8:323:323. doi: 10.3389/fncel.2014.00323
44. Zarate CA Jr., Machado-Vieira R. Potential Pathways Involved in the Rapid Antidepressant Effects of Nitrous Oxide. Biol Psychiatry (2015) 78:2–4. doi: 10.1016/j.biopsych.2015.04.007
45. Massey TH, Pickersgill TT, K JP. Nitrous oxide misuse and vitamin B12 deficiency. BMJ Case Rep (2016) 2016. doi: 10.1136/bcr-2016-215728
46. Gururajan A, Reif A, Cryan JF, Slattery DA. The future of rodent models in depression research. Nat Rev Neurosci (2019) 20:686–701. doi: 10.1038/s41583-019-0221-6
47. Planchez B, Surget A, Belzung C. Animal models of major depression: drawbacks and challenges. J Neural Transm (Vienna) (2019) 126:1383–408. doi: 10.1007/s00702-019-02084-y
48. Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci (2013) 14:609–25. doi: 10.1038/nrn3381
49. Sterpenich V, Vidal S, Hofmeister J, Michalopoulos G, Bancila V, Warrot D, et al. Increased Reactivity of the Mesolimbic Reward System after Ketamine Injection in Patients with Treatment-resistant Major Depressive Disorder. Anesthesiology (2019) 130:923–35. doi: 10.1097/Aln.0000000000002667
50. Hunt MJ, Kessal K, Garcia R. Ketamine induces dopamine-dependent depression of evoked hippocampal activity in the nucleus accumbens in freely moving rats. J Neurosci (2005) 25:524–31. doi: 10.1523/Jneurosci.3800-04.2005
Keywords: nitrous oxide (N2O), depression, mPFC, burst firing, neuronal nitric oxide synthase (nNOS), BDNF
Citation: Liu W, Li Q, Ye B, Cao H, Shen F, Xu Z, Du W, Guo F, Liu J, Li T, Zhang B and Liu Z (2020) Repeated Nitrous Oxide Exposure Exerts Antidepressant-Like Effects Through Neuronal Nitric Oxide Synthase Activation in the Medial Prefrontal Cortex. Front. Psychiatry 11:837. doi: 10.3389/fpsyt.2020.00837
Received: 04 February 2020; Accepted: 31 July 2020;
Published: 03 September 2020.
Edited by:
Fushun Wang, Nanjing University of Chinese Medicine, ChinaReviewed by:
Chiara Fabbri, King’s College London, United KingdomCopyright © 2020 Liu, Li, Ye, Cao, Shen, Xu, Du, Guo, Liu, Li, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bing Zhang, YmluZ296enpAMTI2LmNvbQ==; Zhiqiang Liu, ZHJsaXV6aGlxaWFuZ0AxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.