 Swapna Mahurkar-Joshi
Swapna Mahurkar-Joshi Lin Chang
Lin Chang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 14 August 2020
Sec. Psychological Therapy and Psychosomatics
Volume 11 - 2020 | https://doi.org/10.3389/fpsyt.2020.00805
This article is part of the Research Topic Neurogastroenterology – Focus on the Gut-Brain Axis View all 19 articles
Irritable bowel syndrome (IBS) is a brain-gut axis disorder characterized by abdominal pain and altered bowel habits. IBS is a multifactorial, stress-sensitive disorder with evidence for familial clustering attributed to genetic or shared environmental factors. However, there are weak genetic associations reported with IBS and a lack of evidence to suggest that major genetic factor(s) contribute to IBS pathophysiology. Studies on animal models of stress, including early life stress, suggest a role for environmental factors, specifically, stress associated with dysregulation of corticotropin releasing factor and hypothalamus-pituitary-adrenal (HPA) axis pathways in the pathophysiology of IBS. Recent evidence suggests that epigenetic mechanisms, which constitute molecular changes not driven by a change in gene sequence, can mediate environmental effects on central and peripheral function. Epigenetic alterations including DNA methylation changes, histone modifications, and differential expression of non-coding RNAs (microRNA [miRNA] and long non-coding RNA) have been associated with several diseases. The objective of this review is to elucidate the molecular factors in the pathophysiology of IBS with an emphasis on epigenetic mechanisms. Emerging evidence for epigenetic changes in IBS includes changes in DNA methylation in animal models of IBS and patients with IBS, and various miRNAs that have been associated with IBS and endophenotypes, such as increased visceral sensitivity and intestinal permeability. DNA methylation, in particular, is an emerging field in the realm of complex diseases and a promising mechanism which can provide important insights into IBS pathogenesis and identify potential targets for treatment.
Irritable bowel syndrome (IBS) is a complex condition characterized by alterations of bidirectional brain-gut interactions affecting gastrointestinal (GI) function. It is a widely prevalent disorder affecting about 5% to 11% of general population and occurs in children and adults and in men and women although it is considered a female-predominant condition. Hallmark symptoms include the presence of chronic or recurrent abdominal pain associated with altered bowel habits without underlying structural abnormalities (1–4). IBS has been subdivided on the basis of predominant bowel habits into diarrhea-predominant (IBS-D), constipation-predominant (IBS-C), or a mix of diarrhea and constipation (IBS-M) subtypes (3). IBS can coexist with other GI disorders including gastroesophageal reflux disease and functional dyspepsia, as well as somatic syndromes including fibromyalgia, interstitial cystitis, migraine headaches, and psychologic disorders (5). Due to its high prevalence, recurrent nature of symptoms and a negative impact on health-related quality of life (6), IBS is associated with substantial cost to patients, the health care system, and society (7).
IBS is considered to be a multi-factorial disorder, however, its pathophysiology is not completely understood. IBS and other functional GI disorders have more recently been redefined by experts as “disorders of gut-brain interactions (DGBI) classified by GI symptoms related to any combination of the following: motility disturbance, visceral hypersensitivity, altered mucosal and immune function, altered gut microbiota, and altered central nervous system (CNS) processing” (4). The presence of emotional and psychological factors and food intolerance contribute to the clinical presentation and can exacerbate IBS symptoms (8, 9).
Studies have shown that genetic factors have a modest effect in IBS (10). In addition, there is increasing evidence of a strong influence of environmental factors such as stress in its pathogenesis. A number of studies have found that IBS patients have a higher prevalence of stressful events including early adverse life events (EALs), or traumatic experiences during childhood, as well as current stressful life events in adulthood (11–13). The mechanisms underlying long-term effects of stress and EALs may result from epigenetic programing (14). Epigenetic changes refer to molecular alterations that potentially lead to altered gene expression resulting in a change in phenotype in absence of alteration in the underlying gene sequence.
In this review, we summarize the genetic factors associated with IBS and describe the role of epigenetic factors including DNA methylation and histone modifications as links between genes and environmental factors (e.g., stress) in the etiopathology of IBS. We review the current knowledge of epigenetic modifications associated with IBS in patients as well as in early life stress animal models of IBS, and those associated with IBS endophenotypes (defined as intermediate phenotypes of subclinical traits) including stress and hypothalamic–pituitary–adrenal (HPA) axis function, visceral hypersensitivity and abdominal pain, and GI motility. Further, we briefly outline the role of other epigenetic factors including non-coding RNAs (long non-coding RNAs [lncRNAs] and microRNAs [miRNAs]) in IBS. Finally, we will present a schematic model of our current understanding of factors associated with IBS pathogenesis. A better understanding of the epigenetic mechanisms in IBS can open new avenues for the identification of novel therapeutic targets.
IBS is often associated with familial clustering in which patients report a family history of IBS (15–17). However, the strength of the genetic association varies between studies. One study reported familial aggregation in IBS but found no evidence of association in spouses, suggesting either a possible genetic etiology or an exposure to a shared household environmental factor early in life as an underlying cause of IBS (18). Additional evidence in favor of both a genetic and environmental etiology of IBS comes from twin studies. Twin studies by Morris-Yates et al. (19) and Svedberg et al. (20) provided evidence for genetic basis of IBS in Australian and Swedish populations. In two large studies on 281 twin pairs in the United States (21) and 3334 twin pairs in Norway (22), Levy et al. and Bengtson et al. showed a higher concordance rate among monozygotic twins than in dizygotic twins for IBS. However, one study by Mohammed et al. (23), failed to replicate the differences in the concordance rates between the monozygotic and dizygotic twin groups. Interestingly, Levy et al. also reported that the presence of IBS in the mother was a strong predictor of having IBS. The proportion of twins who had mothers with IBS was 15.2% which was significantly higher than the 6.7% of twins with IBS who had a co-twin with IBS. Since dizygotic twins share about the same number of genes with each other as each twin shares with their mother, this study suggested that in addition to heredity, social learning, and behavior may contribute to the development of IBS (24).
IBS has been associated with genetic variants in a number of candidate genes. Genes associated with IBS in various studies are listed in Table 1. These include single nucleotide polymorphisms (SNPs) in genes related to signaling systems important in the control of gut motility or sensation in IBS, which includes serotoninergic (5-HT) system including tryptophan hydroxylase (TPH), serotonin reuptake transporter (SERT), a, cholecystokinin (CCK), voltage-gated sodium channels (Nav), Catechol-O-methyltransferase (COMT), cannabinoids, and ion channels, such as transient receptor potential (TRP) channels (TRPV1). Immune related SNPs have been of particular interest in IBS based on accumulating evidence showing immune activation in IBS (25). However, findings have been variable across studies and association of genes such as tumor necrosis factor (TNFα) and IL-10 have not been consistent (26). A recent meta-analysis, which included 12 published case-control studies found no significant association with IBS with polymorphisms in genes such as IL-4, IL-6, IL-8, IL-10, TNFA, IL-1R1, and IL-23R. However, SNP rs4263839 which encodes for TNFSF15 was only moderately associated with IBS, in particular with IBS-C (25). Candidate gene association studies in IBS have been comprehensively reviewed by Cheung et al. (27), Camilleri (28), and Gazouli et al. (29).
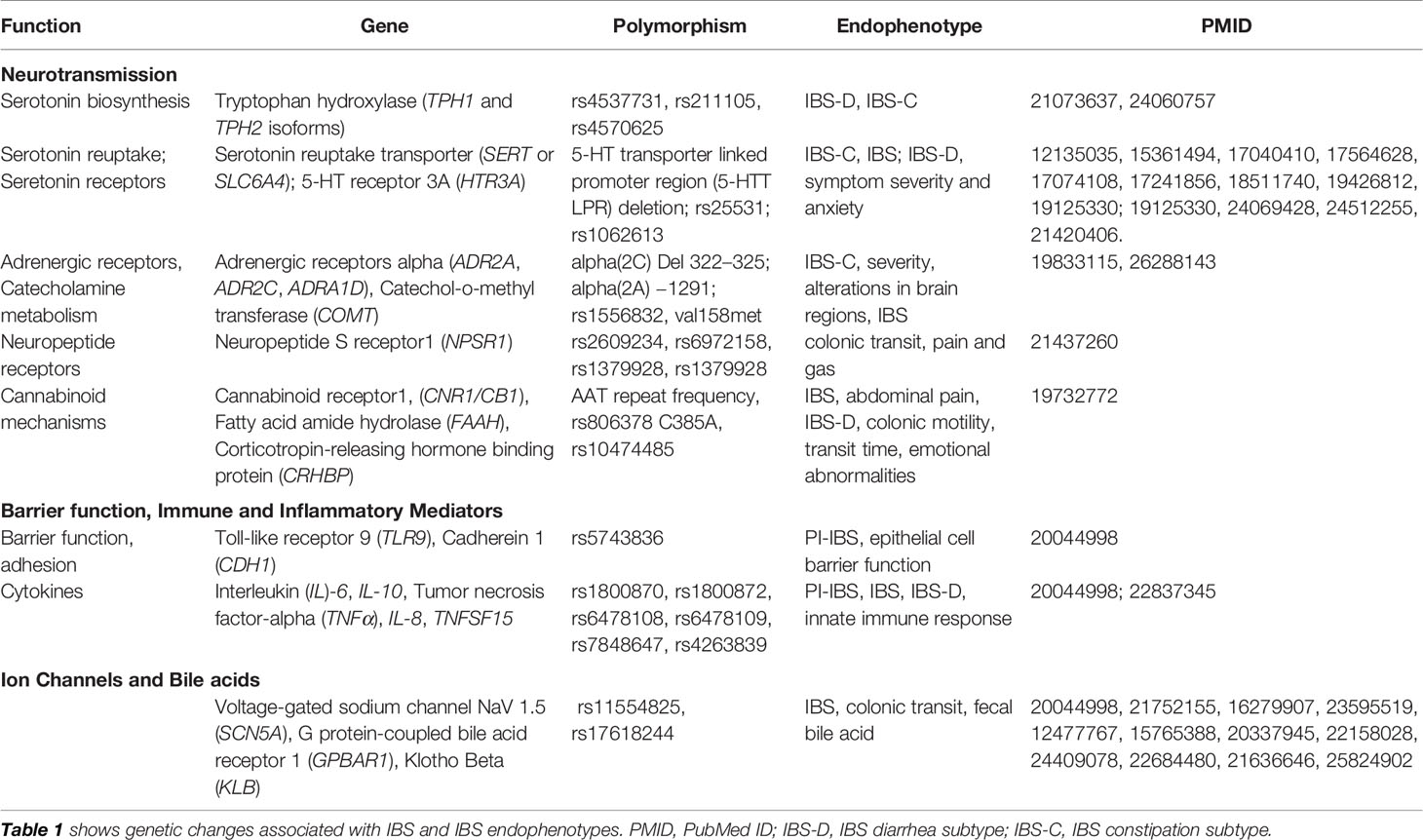
Table 1 Genetic changes associated with irritable bowel syndrome (IBS).
Despite these genetic associations, it is not entirely unexpected that the effects of an individual polymorphism on the overall phenotype are modest because IBS is a complex, multifactorial condition. Moreover, the development of disease likely involves more than the presence of just a moderately associated common variant. While SNPs of these genes alone may not be sufficient to cause IBS or other complex chronic pain conditions, they may interact with other genes and environmental factors including EALs and contribute to the disease etiology. Therefore, an alternative approach has been to evaluate an association of gene variants with specific IBS subtypes (IBS-D, IBS-C, and IBS-M) as well as endophenotypes. For example, we found that the SNP rs1556832 in the catecholaminergic gene, adrenoceptor alpha 1D (ADRA1D), was associated with IBS symptom severity and morphological changes in brain regions that modulate sensory processing (30). In another study, we demonstrated that the presence of IBS was significantly associated with SNPs in corticotropin releasing hormone receptor 1 (CRH-R1) gene. These SNPs were associated with increased GI symptom-related anxiety and acoustic startle response to threat in IBS patients, suggesting that that CRH-R1 is involved in altered stress responsiveness in IBS (30).
Considering the challenges of identifying individual risk alleles in case-control studies and the difficulty of defining significant gene association with IBS, a GWAS using large samples has been proposed as an alternative approach in an attempt to increase sample size and homogeneity. Ek et al. reported a GWAS study in IBS comprising of 534 IBS patients and 4,932 healthy controls, followed by six independent clinical case-control replication studies from different countries (31) where they identified variants in KDLER2 and GRIP2IP (chromosome 7p22.1) genes to be associated with IBS. KDLER2 codes for a family of integral membrane protein with seven transmembrane domains involved in intracellular signaling of bacterial toxins (32), potentially relevant to the role of microbiota in IBS. The GRID2IP gene encodes for a protein (delphilin) expressed on fiber-Purkinje cell synapses in the brain involved in glutamatergic neurotransmission, potentially relevant to pain signaling (31, 33). Another GWAS with a smaller sample size (172 IBS cases and 1,398 controls) conducted in an Australian cohort found an association of protocadherin 15 (PCDH15) gene, encoding an integral membrane protein that mediates calcium-dependent cell–cell adhesion (P~9 × 10−9).
GWAS studies have also evaluated other SNP associations in IBS. TNFSF15 was found to be only nominally significant in the GWAS study, contrasting with prior reports as mentioned previously. Similar nominal associations were detected for other genes such as Cell Division Cycle 42 (CDC42), Neurexophilin 1 (NXPH1) (34), 5-HT Receptor 3E (HTR3E) (35), Klothoβ (KLB) (36) and Sodium Voltage-Gated Channel Alpha Subunit 5 (SCN5A). Interestingly, SCN5A encodes the α-subunit of the voltage-gated sodium channel NaV1.5. About 2% of patients with IBS were found to carry mutations in SCN5A, most of which were loss-of-function mutations that disrupted NaV1.5 channel function (37). Additionally, in a GWAS study on self-reported IBS patients and controls, Bonfiglio et al. identified variants at 9q31.2 locus that were associated with IBS in women suggesting a role for sex hormones in IBS (38). However, most genes associated with IBS thus far represent non-validated findings and therefore their role in IBS needs to be cautiously interpreted. Moreover, such discrepancies are believed to arise from multifactorial nature of the disease, phenotype heterogeneity (including variability in endophenotypes) and/or sample sizes, among others.
Additionally, the mechanisms involved in pain sensitization and altered motility are likely multifactorial as demonstrated in multiple clinical and animal studies in the past decades (39). These functional alterations are mediated through cellular and molecular changes mediated by genetic and epigenetic alterations (40) detailed in the following sections. At the CNS level, proposed mechanisms include plasticity of the endogenous pain modulation system and structural changes in the brain (41, 42). An important step towards understanding the complex pathogenesis of IBS lies in the ability to discover the interface between genetic pathways and epigenetic regulation mediated by gene-environment interaction at peripheral (gut) and central (CNS) levels.
IBS is associated with various environmental factors including chronic stress in early life and/or adulthood, diet (43–45), and gastrointestinal infections (46, 47). Chronic stress can increase an individual’s vulnerability to developing IBS and/or can trigger or exacerbate the symptoms of IBS (48, 49). Stress is the body’s reaction to a physical or psychological stimulus that disturbs the homeostasis of an organism. Stress has wide-spread effects on gut physiology, including changes in intestinal motility, mucosal transport, and gut barrier function leading to changes in permeability, and visceral perception. The biological effects of stress are mediated by the sympathetic nervous system and corticotropin releasing factor (CRF)/HPA axis pathways. Glucocorticoids, which are major effector molecules of the HPA axis, bind to their intracellular receptors and regulate the physiological adaptations to stress (50, 51). Glucocorticoids including cortisol/corticosterone initiates negative feedback control via binding to glucocorticoid receptors (GR) and mineralocorticoid receptors (MR) in brain regions including hippocampus, paraventricular nucleus (PVN), and anterior pituitary gland (52). However, in response to chronic and uncontrollable stressors, maladaptive changes can be elicited resulting in malfunctioning of stress systems affecting the brain structure and function (53, 54).
Many studies support an important role for stress in the IBS pathophysiology and symptoms (8). The stress-induced activation or augmentation of the CRF and HPA axis systems has been associated with visceral hypersensitivity, an important feature of IBS, in animal models (55–58). IBS patients have a greater reactivity to stress compared to healthy subjects, as manifested by a dysregulated HPA axis response, enhanced visceral perception and gut motility, among other findings (59–61). IBS has been associated with increased prevalence of EALs and a growing body of evidence from both animal and human studies supports the hypothesis that chronic stress, including EALs, represent an important mechanism leading to changes in glucocorticoid receptor (GR) expression, thereby increasing responsiveness of the HPA axis (62). The HPA axis response is regulated by a negative feedback though binding of cortisol to GRs at multiple levels including the hypothalamus and hippocampus. Impairment of this negative feedback mechanism can lead to a dysregulation of the HPA axis, specifically an enhanced HPA axis response due to reduced negative feedback from reduced expression of GRs. The importance of an early life and adulthood stress on this IBS phenotype was demonstrated in the maternal separation (MS) animal model, where pups that were maternally separated in early life and later subjected to psychologic stress as an adult displayed post‐stress visceral hypersensitivity, increased corticosterone levels, and reduced expression of GRs in the hippocampus (11, 12). Additionally, stress-induced visceral hyperalgesia has been investigated in repeated water avoidance stress (WAS), a validated rat model of psychological stress that demonstrates many human IBS-like traits. A knockdown of GRs has also been shown to increase visceromotor response to colonic distention in animal models (63). Additionally, a neonatal inflammation rat model suggested a role for inflammatory insult in early life, which upregulates vasoactive intestinal peptide (Vip) in the colon muscularis externa contributing to altered motility and diarrhea-like symptoms as seen in IBS-D patients (64–66).
We found that GR expression was decreased in peripheral blood mononuclear cells (PBMCs) in IBS patients in comparison to healthy controls and that GR expression levels negatively correlated with pituitary responsiveness (ACTH levels) to CRF stimulation (67). That is, reduced GR expression was associated with an enhanced HPA axis response. HPA axis function was assessed in PBMCs because they are accessible and feasible to study. Although GRs regulate HPA axis via negative feedback in the CNS, changes in GRs on PBMCs have been reported in psychiatric diseases, including changes in the number and sensitivity of GRs (68, 69) and GR promoter methylation status and mRNA expression (67, 70–72). Furthermore, the transcriptome of peripheral blood has been shown to share >80% homology with genes expressed in the brain, heart, liver, spleen, colon, kidney, prostate, and stomach, and that there is a broad movement of leukocyte subsets to and from the gut at steady state, suggesting that PBMCs can reflect the molecular events at the central and peripheral locations (73).
Various animal models representing different stress paradigms (e.g. restraint stress, WAS, neonatal MS, etc.) as well as studies in human subjects have demonstrated an impairment in mucosal barrier function, the enteric nervous system (ENS), and immune system (74–76). These stress-induced changes result in alterations in GI functions including increased intestinal permeability, altered ion transport and hypersecretion, and mucus secretion and are mediated by neuro-immune mechanisms including the CRF system, which consists of CRF, urocortins 1–3 (Ucn) and their receptors CRF-1R and CRF-2R (77, 78). Barrier dysfunction may also occur early in IBS and is hypothesized to contribute to low-grade intestinal immune activation and increased visceral perception (79), specifically in IBS-D patients (80, 81) and post-infection IBS (PI-IBS) (82). Additionally, an increase in paracellular permeability has been correlated with the magnitude of visceral pain in IBS-D patients (81). Furthermore, an exaggerated response to CRH infusion in IBS patients was associated with an increase in cytokine levels suggesting a correlation between stress and increased cytokine levels (83). This is hypothesized to be mediated by glucocorticoid-related epigenetic changes leading to inadequate suppression of proinflammatory cytokines (40). It conceivable that this contributes, at least in part, to the higher plasma levels of cytokines reported in some IBS patients (84, 85).
Both local (intestinal) and systemic factors contribute to the altered epithelial barrier function. Recent data indicate that soluble mediators from fecal supernatants and mucosal homogenates of IBS patients affect the epithelial integrity, thereby increasing colonic permeability (86, 87). These studies showed that the impairment of barrier integrity may be mediated via the release of various mediators such as serine proteases or histamine. Additionally, molecular alterations such as expression of genes involved in barrier function (88) can mediate permeability changes, which can in turn lead to permeation of bacteria and their products.
Epigenetic mechanisms alter gene expression without alterations of underlying DNA sequence (89) and are key to the normal development, cellular function, and differentiation into specific lineages (90). These mechanisms broadly include DNA methylation, histone modification, and non-coding RNA mediated gene regulation as shown in Figure 1. Epigenetic mechanisms play a role in synaptic plasticity, learning, and memory (91), as well as in various neuropsychiatric conditions including depression and pain (92). Long-lasting epigenetic changes have been linked to early stress, childhood trauma or abuse. Epigenetic changes are amenable to exogenous influences and involve complex and dynamic interaction between the DNA sequence, DNA and histone modifications and environmental factors, all of which combine to produce the phenotype, thus providing an important link between environment and phenotype (93).
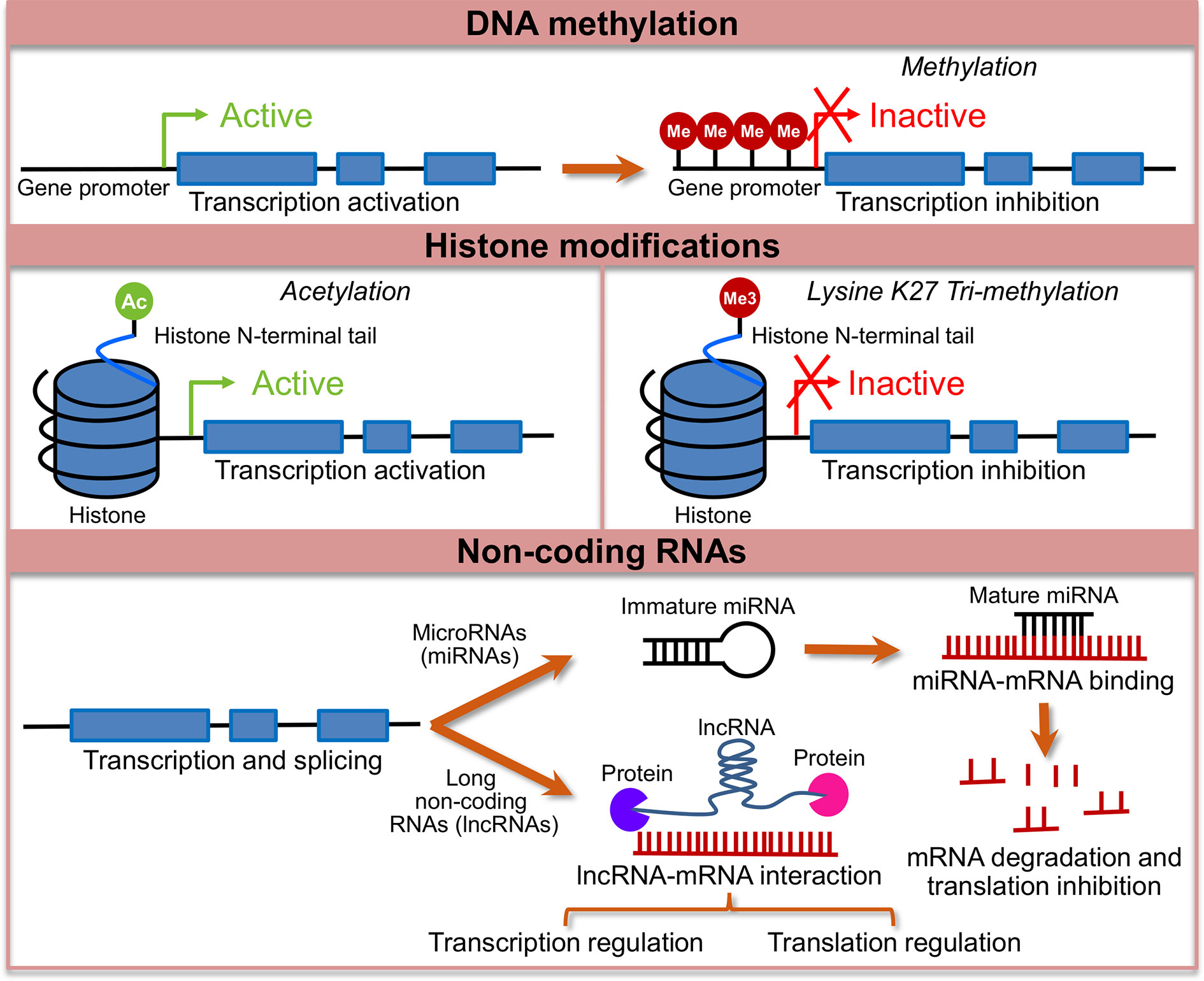
Figure 1 Major epigenetic changes studied in the context of irritable bowel syndrome (IBS). Shows a conceptual model of major epigenetic changes studied in the context of IBS. Lines with blue boxes represent genes with promoter regions. Blue boxes represent exons, lines before exon 1 represents promoter region and the lines between exons represent introns. The top panel shows active transcription in the unmethylated state of the gene, which when methylated (Me) at the promoter region leads to transcription inactivation. Middle panel shows two representative histone modifications, histone acetylation at the N-terminal tail, which is usually associated with activation of transcription and histone methylation, specifically, addition of a tri-methyl group (Me3) at 27th lysine (K) on the N-terminal tail, which is associated with transcription repression. The bottom panel shows mechanism of transcription regulation by non-coding RNAs. MicroRNA genes are transcribed to immature precursor miRNAs that are processed to form mature miRNAs, which bind to miRNAs either leading to mRNA degradation or inhibition of translation. Long non-coding RNAs regulate transcription and translation, and function at the level of chromatin via interaction with RNA binding proteins.
In vertebrates, DNA methylation occurs mostly in the context of CpG dinucleotides by a covalent attachment of a methyl group to the C5 position of cytosine (89). CpG islands (CGIs) are short interspersed DNA sequences (usually 1000 base-pairs) with a high concentration of CpG residues, which are normally non-methylated in contrast to the rest of the genome, which is globally methylated. CGIs typically occur at or near the transcription start site of genes (94) and when a CGI in the promoter region of a gene is methylated, expression of the gene is repressed. The exact mechanism of DNA methylation mediated repression of gene expression has begun to be elucidated in recent years. DNA methylation results in binding of methyl-binding-domain (MBD) proteins, which are associated with large protein complexes that contain histone deacetylases (HDACs) and recruit histone methyl transferases (HMTs) leading to chromatin remodeling (95). Both DNA methylation and the proteins associated with MDBs are being investigated as promising therapeutic targets (96). Additionally, recent studies have demonstrated that methylation of CpG sites in the gene body are positively correlated with gene expression and is a potential therapeutic target in cancer (97). The quantification of DNA methylation in diseased or environmentally impacted cells could provide useful information for detection and treatment of the disease.
DNA methylation changes, in particular, have been studied in various chronic conditions including cancer (98), chronic pain (99), and psychiatric diseases (100). Stress and other environmental factors including EALs, diet and gut microbial metabolites can potentially trigger epigenetic alterations (101, 102). For example, studies have demonstrated that maternal care influences HPA axis function through epigenetic programming of GR (coded by Nuclear Receptor Subfamily 3 Group C Member 1, or NR3C1) expression and that environment-induced remodeling of the epigenome, or during chronic stress, can result in long-term changes in gene expression (103–105). Table 2A lists the epigenetic modifications reported in association with IBS or animal models of IBS. The role of central epigenetic regulatory mechanisms in stress-induced visceral hypersensitivity has been demonstrated in MS and WAS rat models. While MS animal models mimic the early life stress, WAS simulates both acute and chronic effects of a psychological stressor on colonic sensitivity, which have been extensively reviewed by Greenwood-Van Meerveld et al. (106). Stress-induced visceral hypersensitivity has been associated with an increase in DNA methylation in the GR gene promoter and a decreased expression of the GR gene in the amygdala of WAS rats (107, 108). Additionally, the study identified a decrease in DNA methylation and increased expression of the CRF gene associate with visceral hypersensitivity in the amygdala of the stressed rats. Hong et al. demonstrated that chronic stress increased methylation of genes that regulate visceral pain sensation in the peripheral nervous system of rats. They reported that chronic stress resulted in increased promoter methylation and reduced expression of the NR3C1 (or GR) gene in L6-S2 dorsal root ganglia (109). In human subjects, DNA methylation in brains of suicide victims with a history of childhood abuse was associated with increased methylation and decreased expression of GR gene compared to suicide victims with no history of childhood maltreatment (104). However, no clear consensus exists regarding DNA methylation of the GR gene in IBS patients.
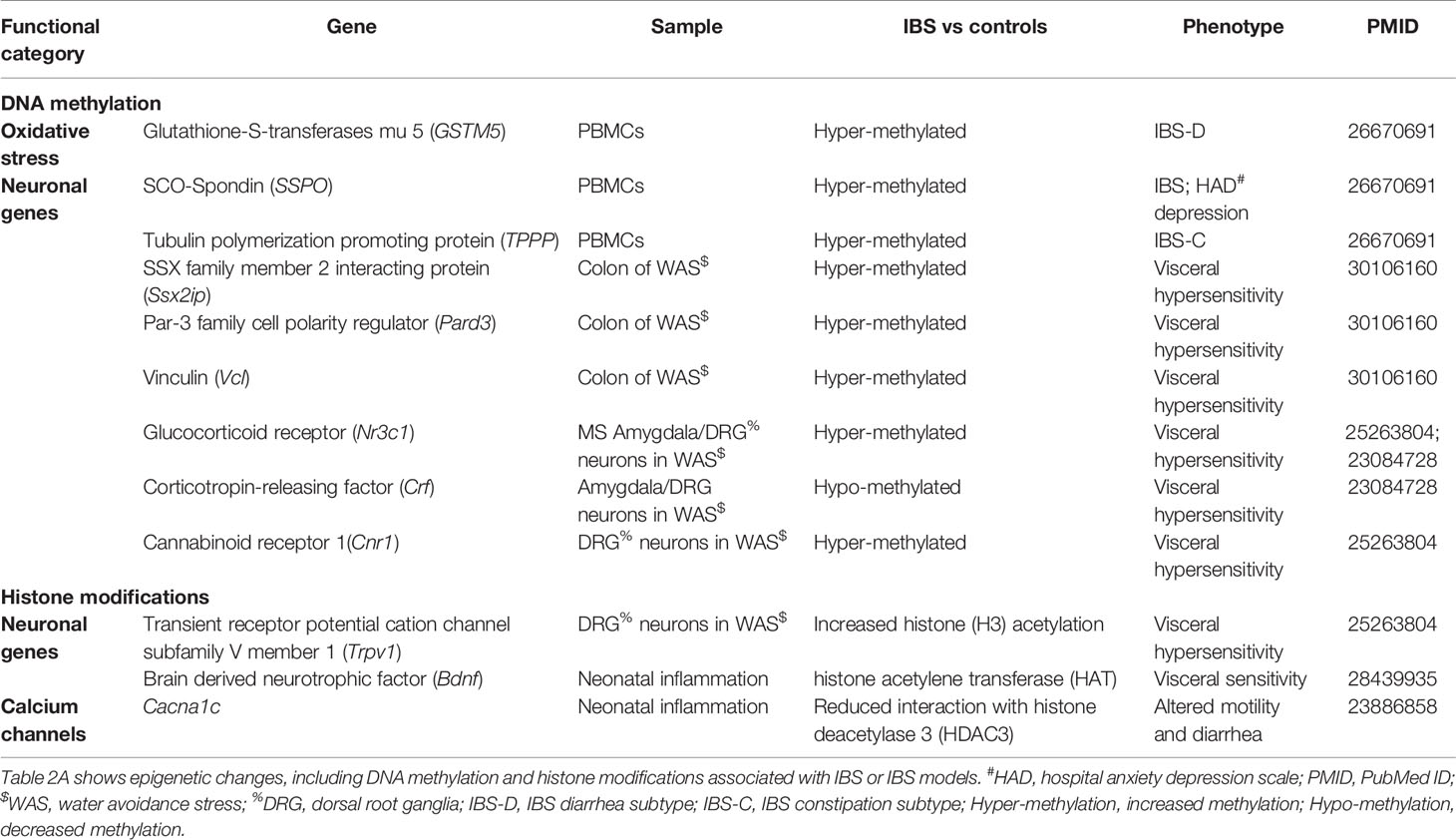
Table 2A Epigenetic changes associated with irritable bowel syndrome (IBS).
In a genome-wide methylation scan followed by targeted sequencing, we previously demonstrated an association of DNA methylation of several CpG sites in PBMCs in IBS patients compared to healthy controls (110). We reported an increase in DNA methylation in genes including sub-commissural organ (SCO)-Spondin (SSPO), glutathione-S-transferases mu 5 (GSTM5) and tubulin polymerization promoting protein (TPPP) in IBS patients compared to healthy controls. SSPO is associated with neuronal function (111) and has been suggested to play a role in depression and evidence suggests that SCO secretory activity is regulated by the serotonin system, which plays an important role in stress-related pathways and in IBS (112). Additionally, an increased methylation of GSTM5, a gene that codes an enzyme that plays an important role in antioxidant defense was associated with decreased gene expression compared to controls. Although a role for oxidative stress and the significance of epigenetic silencing of GSTM5 in IBS is not known, DNA methylation mediated repression of GSTM5 gene expression has been shown in other conditions (113). Although larger independent studies may be required to confirm the functional role of the associated genes, these studies highlight the importance of epigenetic changes in IBS. DNA methylation changes in blood cells can provide insights into systemic changes associated with IBS and can serve as important diagnostic and prognostic biomarkers (114).
Epigenetic changes in the gut mucosa can provide important insights into the peripheral mechanisms of IBS. A recent study investigated the genome-wide methylation predominantly in promoter regions of genes, and gene expression in the colon of rat WAS model and suggested an association of Notch signaling and focal adhesion pathways with psychological stress (115). In a recent study that included a relatively large cohort of IBS subjects and healthy controls (n=102 and 36, respectively), we found several DNA methylation changes in PBMCs as well as colonic mucosa that were associated with IBS. There was increased methylation of stress-related genes such as NR3C1, CRHR1, brain-derived neurotrophic factor (BDNF) in PBMCs and/or colon (116). In the colonic mucosa of IBS patients, we identified distinct clusters of DNA methylation patterns highlighting the heterogeneity in the epigenetic profiles of colonic mucosa of IBS patients. A hyper-methylated cluster was associated with higher symptom severity and abdominal pain compared to clusters with lower methylation levels and included genes such as protocadherins (PCDHs), cadherins (CDHs), VIP, TRPV4, and Guanylate Cyclase 1, Soluble, Beta 3 (GUCY1B3) which were significant after correcting for multiple comparisons. Thus, these studies suggest that DNA methylation changes are important pathophysiologic mechanisms in IBS and should be further evaluated.
In eukaryotic cells, genes complex with histone and other chromosomal proteins to form a chromatin scaffold. Histone modifications play an important role in regulation of gene expression. The histone tails undergo a variety of covalent modifications, that include lysine acetylation, methylation, ubiquitination, and sumoylation, among others (117) (Figure 1). Acetylation and methylation are some of the most studied histone modifications so far. In general, acetylation of core histone tails leads to open chromatin structure to allow transcription and the histone deacetylases (HDACs) oppose the effects of histone acetylases and are predominantly transcriptional repressors (118). Histone methylation is more complex and can occur on a specific lysine or arginine residue. Depending on the residues being methylated and the number of methylation molecules added (each methylated lysine residue can exist in a mono-, di-, or tri-methylated state), histone methylation may be associated with either an active or a silent state of chromatin. For example, H3K27me3 is associated with transcription repression whereas, H3K4me3 is generally associated with active transcription.
Recent studies have highlighted antinociceptive effects of histone acetylation and lysine tri-methylation in inflammatory and neuropathic pain models (119, 120). In the partial sciatic nerve ligation model of neuropathic pain, an increase in the expression levels of monocyte chemotactic protein-3 (MCP3), a pro-inflammatory cytokine was associated with reduced levels of repressive histone methylation, H3K27me3 (121). A role for histone acetylation has been suggested in the pathophysiology of visceral hypersensitivity induced by early-life stress in the MS animal model of IBS (122). Moloney et al. showed that HDAC inhibitor, suberoylanilide hydroxamic acid (SAHA), reversed visceral hypersensitivity, and the effects of stress on fecal pellet output in animal models of early life stress highlighting the importance of histone acetylation in stress-related conditions (123). Hong et al. demonstrated an increased expression of histone acetyltransferase EP300, which induced acetylation of histone H3 of promoter of nociceptive endovanilloid TRPV1 gene in the chronic WAS model of IBS. Moreover, they demonstrated that siRNA mediated knockdown of EP300 prevented visceral hyperalgesia (124).
Animal models suggest that neonatal inflammation may contribute to altered gut motility via histone modification. In rats subjected to neonatal inflammation, Vip levels increased, which reduced the interaction of histone deacetylase 3 (HDAC3) with α1C-subunit of Cav1.2b channel (Cacna1c or α1C1b). This resulted in increased acetylation of histone H3 lysine 9 (H3K9) in the promoter region inducing the transcription of α1C1b which may result in gut dysmotility and diarrhea (65). Similarly, neonatal immune challenge led to an upregulation of tyrosine hydroxylase in the locus coeruleus, mediated by epigenetic programming (125). The study showed a cascade of events involving upregulation of norepinephrine, activation of adrenergic receptors, and involvement of enhanced pCREB binding to the cAMP response element, which resulted in recruitment of histone acetylene transferase (HAT) to the brain derived neurotrophic factor (BDNF) gene. This led to an enhanced expression of the BDNF and aggravated visceromotor response to colorectal distension.
MiRNAs are endogenous noncoding RNAs of small size (18–25 nucleotides) that have been characterized as important gene expression regulators via binding through complementary sequence homology to the 3′-untranslated region (UTR) of target mRNAs thereby causing repression of translation or mRNA degradation (126) (Figure 1). Involvement of miRNA in cancer is well established and emerging research indicates a role of miRNA in the regulation of genes that play a role in nociceptive circuits (127). It has been suggested that miRNAs interacting with nervous and immune systems may act as “master switches” regulating a network of genes orchestrating the pain response and may be targeted for therapeutic purposes contrasting with the current strategy focusing on single targets (127). This approach is highly relevant to the GI tract where neuroimmune interactions are key contributors to the control of GI functions.
Recent translational studies in IBS have identified several miRNAs (Table 2B) that appear to be important in regulating the expression of genes involved in visceral pain response or intestinal permeability. In a study conducted in two independent cohorts of IBS-D women in in the UK and Germany, there was an association between the c.*76G>A variant in the 3′UTR of the serotonin receptor 3 subunit gene (HTR3E), leading to increased expression of the 5HT3E subunit, and (129). Using luciferase assays, this variation was located in the binding element sequence of miR-510 suggesting a functional implication of the HTR3E variation in the ability of miR-510 to regulate its gene expression.
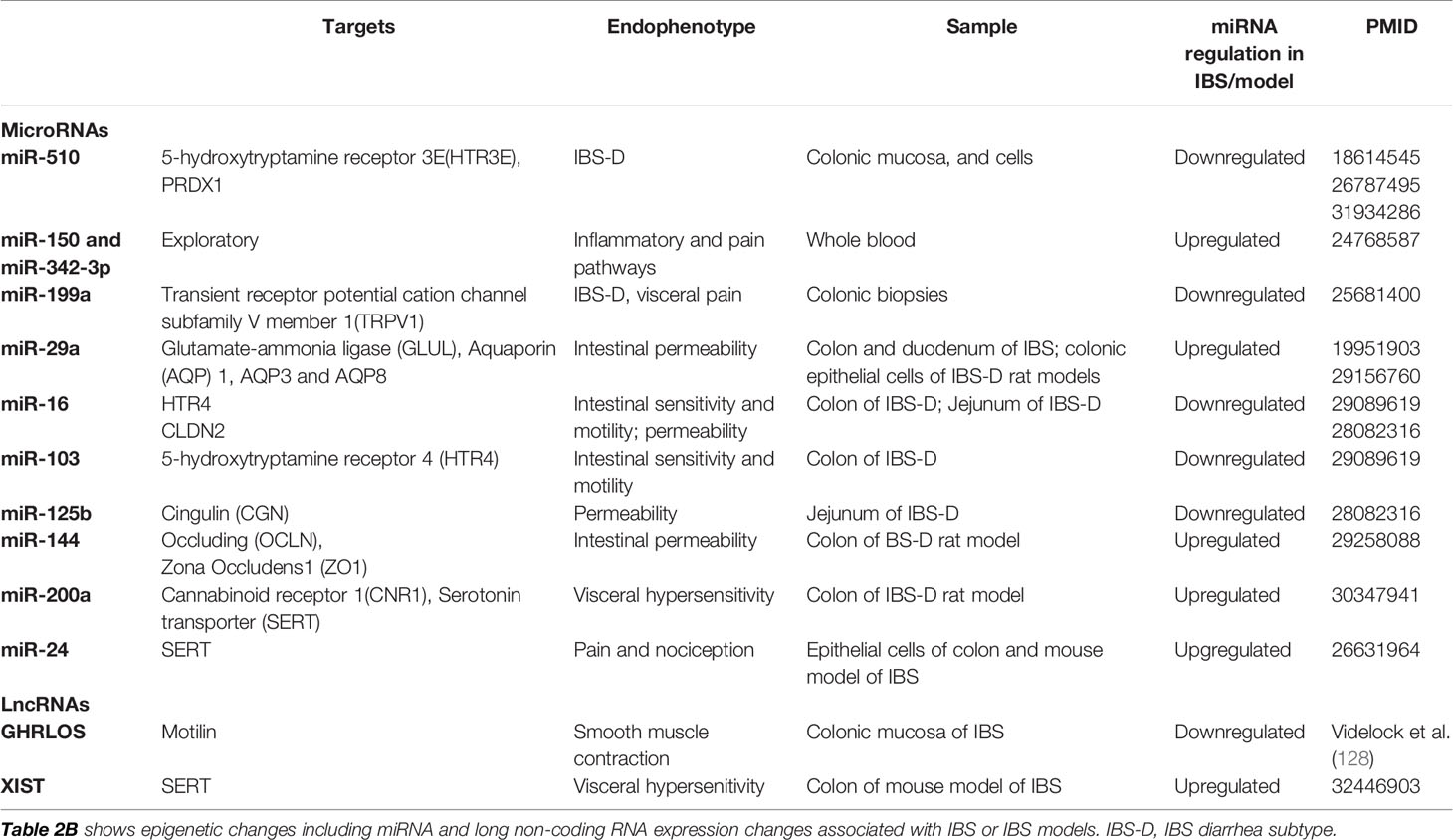
Table 2B Non-coding RNAs associated with irritable bowel syndrome (IBS).
Fourie et al. investigated whether circulating miRNAs are differentially expressed in a small number of IBS patients compared to healthy controls (130). This study found an upregulation of miR-150 and miR-342-3p, which are involved in inflammatory (131) and pain pathways, in IBS patients compared to healthy controls (132). Subsequent studies from Zhou et al, using a miRNA microarray approach, revealed increased expression of miR-29a in blood microvesicles, small bowel and colonic biopsies from IBS-D patients compared to healthy controls, and it was associated with increased intestinal permeability (133). Glutamine synthetase was confirmed as a target of miR-29A and was significantly reduced in the small bowel mucosa in IBS patients suggesting a relationship between miR-29a, glutamine dependent signaling pathways and intestinal permeability in IBS patients. In a randomized placebo-controlled trial, glutamate was shown to safely and effectively reduce IBS symptoms in post-infection IBS-D patients with increased intestinal permeability (134). Subsequently, Zhou et al. showed increased levels of mir-29A/B and reduced expression of NFKB Repressing Factor (NKRF) and Claudin 1 (CLDN1) genes in intestinal tissue from IBS-D patients as well as TNBS colitis and WAS rat models of IBS (135). Additionally, they showed that miR-199a was significantly decreased in IBS-D patients compared to controls and an upregulation in animal models decreased visceral pain via inhibition of TRPV1 signaling.
Subsequently, the role of other miRNAs has been identified in IBS. CGN and CLDN2, associated with barrier function were shown to be the targets of hsa-miR-125b-5p and hsa-miR-16, which were downregulated in jejunal mucosal samples of IBS-D (136). Similarly, occludin (OCLN) and zonula occludens 1 (ZO1/TJP1), which are associated with intestinal permeability, were identified as direct targets of miR-144 in the colon of IBS-D rat models (137). In addition, the role of miRNAs in visceral hyperalgesia has been suggested by altered levels of miRNAs, including miR-200a which targets cannabinoid receptor 1 (CNR1) and serotonin transporter (SERT) (138), miR-214 which targets SERT (139), and miR-16 and miR-103 which target HTR4 (140) in a rat model of IBS-D and human IBS-D colonic epithelial cells.
These studies have led to an increased understanding of the molecular mechanisms underlying some of the endophenotypes of IBS. Thus, they may be explored as diagnostic tools and have potential to form a basis for the therapeutic interventions being proposed in IBS (133). However, further studies examining their exact mechanisms in IBS and that can reproduce previous findings in a larger population are needed. These translational discoveries have prompted growing interest in miRNA-based therapy for IBS, although delivering drugs targeting miRNA to the intestinal tissue currently stands as a major obstacle and is being actively investigated (141).
LncRNAs are transcripts that measure more than 200 nucleotides in length and are processed similar to protein-coding mRNAs (142). Although the functional mechanisms of most lncRNAs are not fully understood, they are known to exhibit diverse functional roles, including the gene regulation by chromatin remodeling, modulation of gene expression, localization, and stability (143) (Figure 1). Recently, Videlock et al. investigated the entire colonic mucosal transcriptome and found that a lncRNA, GREHLOS, which regulates the expression of motilin involved in smooth muscle contraction, was downregulated in IBS patients compared to healthy controls (128). Recently, increased expression of a lncRNA, X inactivate-specific transcript (XIST) was associated with decrease SERT transcription and increased visceral hypersensitivity in mouse model of IBS-D. The study suggested a role for XIST in recruiting DNA methyl transferases, DNMT1, DNMT3A, and DNMT3B to reduce SERT transcription via promoter methylation.
Recent studies are starting to investigate an interaction of microbiome, diet, and epigenetics defined as “microbiota-nutrient metabolism-epigenetics axis” in complex diseases (144). Evidence suggests that epigenetic events are dynamic and responsive to changing nutrient availability and microbiome (102, 144, 145). Although the role of microbes and their metabolites on epigenetic machinery in the manifestation of IBS symptoms has not been investigated, there is indirect evidence for the role of microbial products involved in epigenetic modifications in eliciting visceral hypersensitivity (146). These interactions may be mediated by metabolites synthesized by commensal bacteria including neurotransmitters or short-chain fatty acids (SCFAs) (147). SCFAs, including butyrate, propionate, and acetate produced by the fermentation of host dietary polysaccharides, have neuroactive properties (148) and may play an important role in the brain-gut microbiome axis in IBS (149). SFCAs have been shown to regulate post-translational modifications of histones by inhibiting histone deacetylases, promoting active chromatin state and thereby promoting transcription (150, 151).
Nutrigenomics, the study of interaction of diet and genomic factors is an emerging topic in the context of IBS (152, 153). The majority of patients with IBS report meal-related symptoms and dietary modifications is an increasing treatment intervention used in IBS. For example, a low FODMAP (fermentable oligo-, di-, and mono-saccharides and polyols) diet has been associated with alleviation of IBS symptoms (154–156). Additionally, nutrition or diet can affect the epigenomic state. The role of diet in regulating epigenetic pathways is highlighted by a recent study, which showed that calorie restriction changes gene expression and DNA methylation profile of subcutaneous adipose tissue (145). It is suggested that diet and microbial metabolites influence the epigenome by impacting the pool of compounds or enzymes involved in epigenetic pathways (102). In particular, dietary components, co-factors, and vitamins including, S-adenosyl methionine (SAM), folate, vitamin B12, vitamin B6, acetyl-CoA have been shown to play a role in regulating histone modifications or DNA methylation levels (144). Therefore, investigating interactions between diet, microbiome and epigenetic factors may be important in understanding the etiology of IBS and developing personalized therapy for IBS.
Genetic, epigenetic, and other factors associated with IBS have been summarized in a schematic figure (Figure 2). IBS is a multifactorial disorder of gut-brain interactions. In addition to stress, diet, and other environmental factors, changes at molecular level including genetic and epigenetic factors may contribute to pain modulation at the CNS level and/or periphery, and affect immune function, oxidative stress, mucosal barrier function, and GI motor and secretory function at the peripheral level in IBS. Moreover, gut microbiota and their metabolites likely contribute to this integrated system and play a major role in the pathogenesis of IBS. Given that IBS is a complex, multifactorial disorder, we propose that epigenomic mechanisms imprint dynamic environmental effects on the fixed genome resulting in alterations in phenotype leading to a disease state. These changes are potentially reversible and can be powerful diagnostic and prognostic markers and therapeutic targets (157, 158).
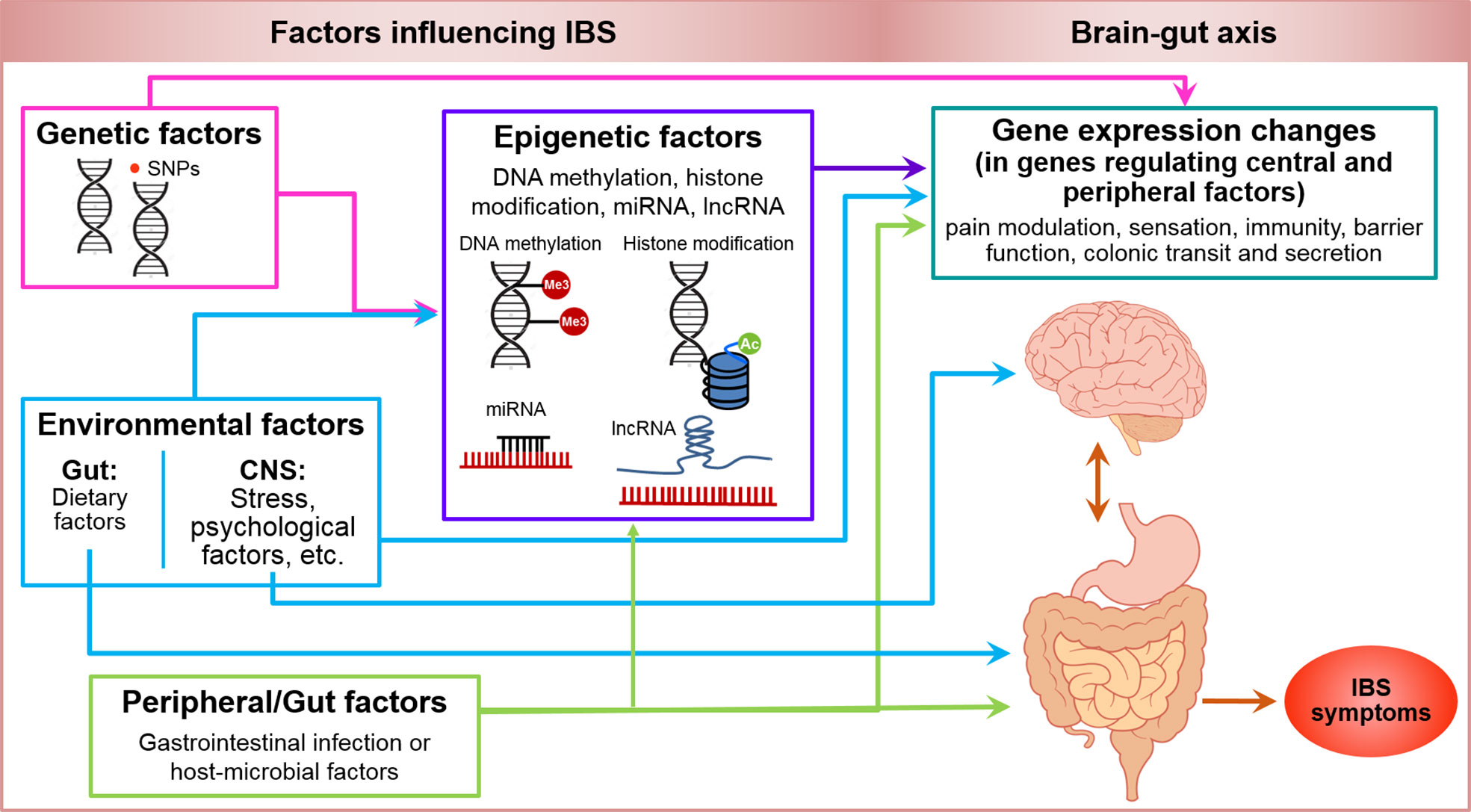
Figure 2 Genetic, epigenetic, environmental and peripheral factors in irritable bowel syndrome. Shows a schematic model of genetic and epigenetic factors influencing IBS. Pink arrows illustrate that genetic factors including SNPs can influence the gene expression either directly or mediated by epigenetic factors including DNA methylation, histone modifications, miRNA and lncRNA expression (purple arrow). Environmental factors including stress and psychological factors at CNS level and dietary factors at gastrointestinal level can induce changes in gene expression mediated by epigenetic or non-genetic/epigenetic factors, and can have a direct influence on CNS and gut function (blue arrows). Peripheral or gut factors including GI infection or other host or microbial factors, can potentially modify the function of genes mediated by epigenetic or non-epigenetic factors, and influence the CNS and gut function (green arrows) such as, pain modulation, sensation, immunity, barrier function, colonic transit and secretion to manifest the symptoms of IBS (orange-red arrow).
Understanding the role of neuroimmune, genetic, epigenetic, and microbial underpinnings in IBS is crucial to understanding the pathophysiology of IBS. The mechanisms of visceral pain and neuro-motor dysfunction, resulting in the symptoms of IBS are influenced by several factors including stress, genetic, epigenetic as well as microbiota. An in-depth investigation of these factors independently, as well as integratively, in a sufficiently large, well-characterized patient and control populations is crucial in understanding the etio-pathology of IBS and in identifying reliable and validated diagnostic biomarkers and therapeutic targets in IBS.
SM-J reviewed the literature and wrote the manuscript. LC reviewed the literature, wrote and edited the manuscript and provided the resources.
NIH/NIDDK grants P50 DK64539, R21 DK104078, P30 DK 41301, UL1TR000124.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Cathy Liu for her assistance in the creation of the figures.
1. Lovell RM, Ford AC. Global prevalence of and risk factors for irritable bowel syndrome: a meta-analysis. Clin Gastroenterol Hepatol. (2012) 10:712–721.e4. doi: 10.1016/j.cgh.2012.02.029
2. Heitkemper M, Jarrett M, Bond EF, Chang L. Impact of sex and gender on irritable bowel syndrome. Biol Res Nurs (2003) 5:56–65. doi: 10.1177/1099800403005001006
3. Longstreth GF, Thompson WG, Chey WD, Houghton LA, Mearin F, Spiller RC. Functional bowel disorders. Gastroenterology (2006) 130:1480–91. doi: 10.1053/j.gastro.2005.11.061
4. Drossman DA, Hasler WL. Rome IV-Functional GI Disorders: Disorders of Gut-Brain Interaction. Gastroenterology (2016) 150:1257–61. doi: 10.1053/j.gastro.2016.03.035
5. Kim SE, Chang L. Overlap between functional GI disorders and other functional syndromes: what are the underlying mechanisms? Neurogastroenterol. Motil (2012) 24:895–913. doi: 10.1111/j.1365-2982.2012.01993.x
6. Gralnek IM, Hays RD, Kilbourne A, Naliboff B, Mayer EA. The impact of irritable bowel syndrome on health-related quality of life. Gastroenterology (2000) 119:654–60. doi: 10.1053/gast.2000.16484
7. Canavan C, West J, Card T. Review article: the economic impact of the irritable bowel syndrome. Aliment. Pharmacol Ther (2014) 40:1023–34. doi: 10.1111/apt.12938
8. Chang L. The role of stress on physiologic responses and clinical symptoms in irritable bowel syndrome. Gastroenterology (2011) 140:761–5. doi: 10.1053/j.gastro.2011.01.032
9. Monsbakken KW, Vandvik PO, Farup PG. Perceived food intolerance in subjects with irritable bowel syndrome– etiology, prevalence and consequences. Eur J Clin Nutr (2006) 60:667–72. doi: 10.1038/sj.ejcn.1602367
10. Saito YA, Mitra N, Mayer EA. Genetic approaches to functional gastrointestinal disorders. Gastroenterology (2010) 138:1276–85. doi: 10.1053/j.gastro.2010.02.037
11. Bradford K, Shih W, Videlock EJ, Presson AP, Naliboff BD, Mayer EA, et al. Association between early adverse life events and irritable bowel syndrome. Clin Gastroenterol Hepatol. (2012) 10:385–390.e1–3. doi: 10.1016/j.cgh.2011.12.018
12. Park SH, Videlock EJ, Shih W, Presson AP, Mayer EA, Chang L. Adverse childhood experiences are associated with irritable bowel syndrome and gastrointestinal symptom severity. Neurogastroenterol. Motil (2016) 28:1252–60. doi: 10.1111/nmo.12826
13. Parker CH, Naliboff BD, Shih W, Presson AP, Videlock EJ, Mayer EA, et al. Negative Events During Adulthood Are Associated With Symptom Severity and Altered Stress Response in Patients With Irritable Bowel Syndrome. Clin Gastroenterol Hepatol. (2019) 17:2245–52. doi: 10.1016/j.cgh.2018.12.029
14. Meaney MJ, Szyf M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci (2005) 7:103–23.
15. Whorwell PJ, McCallum M, Creed FH, Roberts CT. Non-colonic features of irritable bowel syndrome. Gut (1986) 27:37–40. doi: 10.1136/gut.27.1.37
16. Levy RL, Whitehead WE, Von Korff MR, Feld AD. Intergenerational transmission of gastrointestinal illness behavior. Am J Gastroenterol (2000) 95:451–6. doi: 10.1111/j.1572-0241.2000.01766.x
17. Locke GR, Zinsmeister AR, Talley NJ, Fett SL, Melton LJ. Familial association in adults with functional gastrointestinal disorders. Mayo Clin Proc (2000) 75:907–12. doi: 10.4065/75.9.907
18. Saito YA, Petersen GM, Larson JJ, Atkinson EJ, Fridley BL, de Andrade M, et al. Familial Aggregation of Irritable Bowel Syndrome: A Family Case–Control Study. Am J Gastroenterol (2010) 105:833–41. doi: 10.1038/ajg.2010.116
19. Morris-Yates A, Talley NJ, Boyce PM, Nandurkar S, Andrews G. Evidence of a genetic contribution to functional bowel disorder. Am J Gastroenterol (1998) 93:1311–7. doi: 10.1111/j.1572-0241.1998.440_j.x
20. Svedberg P, Johansson S, Wallander M-A, Hamelin B, Pedersen NL. Extra-intestinal manifestations associated with irritable bowel syndrome: a twin study. Aliment. Pharmacol Ther (2002) 16:975–83. doi: 10.1046/j.1365-2036.2002.01254.x
21. Levy RL, Jones KR, Whitehead WE, Feld SI, Talley NJ, Corey LA. Irritable bowel syndrome in twins: heredity and social learning both contribute to etiology. Gastroenterology (2001) 121:799–804. doi: 10.1053/gast.2001.27995
22. Bengtson M-B, Rønning T, Vatn MH, Harris JR. Irritable bowel syndrome in twins: genes and environment. Gut (2006) 55:1754–9. doi: 10.1136/gut.2006.097287
23. Mohammed I, Cherkas LF, Riley SA, Spector TD, Trudgill NJ. Genetic influences in irritable bowel syndrome: a twin study. Am J Gastroenterol (2005) 100:1340–4. doi: 10.1111/j.1572-0241.2005.41700.x
24. Levy RL, Jones KR, Whitehead WE, Feld SI, Talley NJ, Corey LA. Irritable bowel syndrome in twins: heredity and social learning both contribute to etiology. Gastroenterology (2001) 121:799–804. doi: 10.1053/gast.2001.27995
25. Czogalla B, Schmitteckert S, Houghton LA, Sayuk GS, Camilleri M, Olivo-Diaz A, et al. A meta-analysis of immunogenetic Case-Control Association Studies in irritable bowel syndrome. Neurogastroenterol. Motil (2015) 27:717–27. doi: 10.1111/nmo.12548
26. Bashashati M, Rezaei N, Shafieyoun A, McKernan DP, Chang L, Öhman L, et al. Cytokine imbalance in irritable bowel syndrome: a systematic review and meta-analysis. Neurogastroenterol. Motil (2014) 26:1036–48. doi: 10.1111/nmo.12358
27. Cheung CKY, Wu JCY. Genetic polymorphism in pathogenesis of irritable bowel syndrome. World J Gastroenterol (2014) 20:17693–8. doi: 10.3748/wjg.v20.i47.17693
28. Camilleri M. Genetics of Human Gastrointestinal Sensation. Neurogastroenterol. Motil (2013) 25:458–66. doi: 10.1111/nmo.12132
29. Gazouli M, Wouters MM, Kapur-Pojskić L, Bengtson M-B, Friedman E, Nikčević G, et al. Lessons learned — resolving the enigma of genetic factors in IBS. Nat Rev Gastroenterol. Hepatol. (2016) 13:77–87. doi: 10.1038/nrgastro.2015.206
30. Orand A, Gupta A, Shih W, Presson AP, Hammer C, Niesler B, et al. Catecholaminergic Gene Polymorphisms Are Associated with GI Symptoms and Morphological Brain Changes in Irritable Bowel Syndrome. PLoS One (2015) 10:e0135910. doi: 10.1371/journal.pone.0135910
31. Ek WE, Reznichenko A, Ripke S, Niesler B, Zucchelli M, Rivera NV, et al. Exploring the genetics of irritable bowel syndrome: a GWA study in the general population and replication in multinational case-control cohorts. Gut (2015) 64:1774–82. doi: 10.1136/gutjnl-2014-307997
32. Kreitman RJ, Pastan I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem J (1995) 307(Pt 1):29–37. doi: 10.1042/bj3070029
33. Miyagi Y, Yamashita T, Fukaya M, Sonoda T, Okuno T, Yamada K, et al. Delphilin: a novel PDZ and formin homology domain-containing protein that synaptically colocalizes and interacts with glutamate receptor delta 2 subunit. J Neurosci (2002) 22:803–14. doi: 10.1523/JNEUROSCI.22-03-00803.2002
34. Wouters MM, Lambrechts D, Knapp M, Cleynen I, Whorwell P, Agréus L, et al. Genetic variants in CDC42 and NXPH1 as susceptibility factors for constipation and diarrhoea predominant irritable bowel syndrome. Gut (2014) 63:1103–11. doi: 10.1136/gutjnl-2013-304570
35. Gu Q-Y, Zhang J, Feng Y-C, Dai G-R, Du W-P. Association of genetic polymorphisms in HTR3A and HTR3E with diarrhea predominant irritable bowel syndrome. Int J Clin Exp Med (2015) 8:4581–5.
36. Wong BS, Camilleri M, Carlson PJ, Guicciardi ME, Burton D, McKinzie S, et al. Gores GJ. A Klothoβ variant mediates protein stability and associates with colon transit in irritable bowel syndrome with diarrhea. Gastroenterology (2011) 140:1934–42. doi: 10.1053/j.gastro.2011.02.063
37. Beyder A, Mazzone A, Strege PR, Tester DJ, Saito YA, Bernard CE, et al. Loss-of-function of the voltage-gated sodium channel NaV1.5 (channelopathies) in patients with irritable bowel syndrome. Gastroenterology (2014) 146:1659–68. doi: 10.1053/j.gastro.2014.02.054
38. Bonfiglio F, Zheng T, Garcia-Etxebarria K, Hadizadeh F, Bujanda L, Bresso F, et al. Female-Specific Association Between Variants on Chromosome 9 and Self-Reported Diagnosis of Irritable Bowel Syndrome. Gastroenterology (2018) 155:168–79. doi: 10.1053/j.gastro.2018.03.064
39. Lee YJ, Park KS. Irritable bowel syndrome: emerging paradigm in pathophysiology. World J Gastroenterol (2014) 20:2456–69. doi: 10.3748/wjg.v20.i10.2456
40. Dinan TG, Cryan J, Shanahan F, Keeling PWN, Quigley EMM. IBS: An epigenetic perspective. Nat Rev Gastroenterol Hepatol. (2010) 7:465–71. doi: 10.1038/nrgastro.2010.99
41. Mayer EA, Tillisch K. The brain-gut axis in abdominal pain syndromes. Annu Rev Med (2011) 62:381–96. doi: 10.1146/annurev-med-012309-103958
42. Seminowicz DA, Labus JS, Bueller JA, Tillisch K, Naliboff BD, Bushnell MC, et al. Regional gray matter density changes in brains of patients with irritable bowel syndrome. Gastroenterology (2010) 139:48–57.e2. doi: 10.1053/j.gastro.2010.03.049
43. Singh R, Salem A, Nanavati J, Mullin GE. The Role of Diet in the Treatment of Irritable Bowel Syndrome: A Systematic Review. Gastroenterol Clin North Am (2018) 47:107–37. doi: 10.1016/j.gtc.2017.10.003
44. Hayes PA, Fraher MH, Quigley EMM. Irritable bowel syndrome: the role of food in pathogenesis and management. Gastroenterol Hepatol. (N. Y.) (2014) 10:164–74.
45. Dimidi E, Rossi M, Whelan K. Irritable bowel syndrome and diet: where are we in 2018? Curr Opin Clin Nutr Metab Care (2017) 20:456–63. doi: 10.1097/MCO.0000000000000416
46. Koloski NA, Jones M, Weltman M, Kalantar J, Bone C, Gowryshankar A, et al. Identification of early environmental risk factors for irritable bowel syndrome and dyspepsia. Neurogastroenterol. Motil (2015) 27:1317–25. doi: 10.1111/nmo.12626
47. Barbara G, Grover M, Bercik P, Corsetti M, Ghoshal UC, Ohman L, et al. Rome Foundation Working Team Report on Post-Infection Irritable Bowel Syndrome. Gastroenterology (2019) 156:46–58.e7. doi: 10.1053/j.gastro.2018.07.011
48. Lackner JM, Brasel AM, Quigley BM, Keefer L, Krasner SS, Powell C, et al. The ties that bind: perceived social support, stress, and IBS in severely affected patients. Neurogastroenterol. Motil (2010) 22:893–900. doi: 10.1111/j.1365-2982.2010.01516.x
49. Bennett EJ, Tennant CC, Piesse C, Badcock CA, Kellow JE. Level of chronic life stress predicts clinical outcome in irritable bowel syndrome. Gut (1998) 43:256–61. doi: 10.1136/gut.43.2.256
50. Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev (1984) 5:25–44. doi: 10.1210/edrv-5-1-25
51. Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev (1996) 17:245–61. doi: 10.1210/edrv-17-3-245
52. Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci (1997) 20:78–84. doi: 10.1016/s0166-2236(96)10069-2
53. Nutt DJ, Malizia AL. Structural and functional brain changes in posttraumatic stress disorder. J Clin Psychiatry (2004) 65 Suppl 1:11–7.
54. Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci (2009) 10:434–45. doi: 10.1038/nrn2639
55. Venkova K, Johnson AC, Myers B, Greenwood-Van Meerveld B. Exposure of the amygdala to elevated levels of corticosterone alters colonic motility in response to acute psychological stress. Neuropharmacology (2010) 58:1161–7. doi: 10.1016/j.neuropharm.2010.02.012
56. Greenwood-Van Meerveld B, Moloney RD, Johnson AC, Vicario M. Mechanisms of Stress-Induced Visceral Pain: Implications in Irritable Bowel Syndrome. J Neuroendocrinol. (2016) 28. doi: 10.1111/jne.12361
57. Taché Y, Million M. Role of Corticotropin-releasing Factor Signaling in Stress-related Alterations of Colonic Motility and Hyperalgesia. J Neurogastroenterol. Motil (2015) 21:8–24. doi: 10.5056/jnm14162
58. Larauche M, Moussaoui N, Biraud M, Bae WK, Duboc H, Million M, et al. Brain corticotropin-releasing factor signaling: Involvement in acute stress-induced visceral analgesia in male rats. Neurogastroenterol. Motil (2019) 31:e13489. doi: 10.1111/nmo.13489
59. Welgan P, Meshkinpour H, Beeler M. Effect of anger on colon motor and myoelectric activity in irritable bowel syndrome. Gastroenterology (1988) 94:1150–6. doi: 10.1016/0016-5085(88)90006-6
60. Posserud I, Agerforz P, Ekman R, Björnsson ES, Abrahamsson H, Simrén M. Altered visceral perceptual and neuroendocrine response in patients with irritable bowel syndrome during mental stress. Gut (2004) 53:1102–8. doi: 10.1136/gut.2003.017962
61. Dickhaus B, Mayer EA, Firooz N, Stains J, Conde F, Olivas TI, et al. Irritable bowel syndrome patients show enhanced modulation of visceral perception by auditory stress. Am J Gastroenterol (2003) 98:135–43. doi: 10.1111/j.1572-0241.2003.07156.x
62. Videlock EJ, Adeyemo M, Licudine A, Hirano M, Ohning G, Mayer M, et al. Childhood trauma is associated with hypothalamic-pituitary-adrenal axis responsiveness in irritable bowel syndrome. Gastroenterology (2009) 137:1954–62. doi: 10.1053/j.gastro.2009.08.058
63. Winston JH, Xu G-Y, Sarna SK. Adrenergic stimulation mediates visceral hypersensitivity to colorectal distension following heterotypic chronic stress. Gastroenterology (2010) 138:294–304.e3. doi: 10.1053/j.gastro.2009.09.054
64. Sarna SK. Colonic Motility: From Bench Side to Bedside (2010). San Rafael (CA: Morgan & Claypool Life Sciences. Available at: http://www.ncbi.nlm.nih.gov/books/NBK53477/ (Accessed July 13, 2020).
65. Li Q, Winston JH, Sarna SK. Developmental origins of colon smooth muscle dysfunction in IBS-like rats. Am J Physiol Gastrointest. Liver Physiol (2013) 305:G503–512. doi: 10.1152/ajpgi.00160.2013
66. Choudhury BK, Shi X-Z, Sarna SK. Gene plasticity in colonic circular smooth muscle cells underlies motility dysfunction in a model of postinfective IBS. Am J Physiol Gastrointest. Liver Physiol (2009) 296:G632–642. doi: 10.1152/ajpgi.90673.2008
67. Videlock EJ, Shih W, Adeyemo M, Mahurkar-Joshi S, Presson AP, Polytarchou C, et al. The effect of sex and irritable bowel syndrome on HPA axis response and peripheral glucocorticoid receptor expression. Psychoneuroendocrinology (2016) 69:67–76. doi: 10.1016/j.psyneuen.2016.03.016
68. de Kloet CS, Vermetten E, Bikker A, Meulman E, Geuze E, Kavelaars A, et al. Leukocyte glucocorticoid receptor expression and immunoregulation in veterans with and without post-traumatic stress disorder. Mol Psychiatry (2007) 12:443–53. doi: 10.1038/sj.mp.4001934
69. Yehuda R, Golier JA, Yang R-K, Tischler L. Enhanced sensitivity to glucocorticoids in peripheral mononuclear leukocytes in posttraumatic stress disorder. Biol Psychiatry (2004) 55:1110–6. doi: 10.1016/j.biopsych.2004.02.010
70. Yehuda R, Flory JD, Bierer LM, Henn-Haase C, Lehrner A, Desarnaud F, et al. Lower methylation of glucocorticoid receptor gene promoter 1F in peripheral blood of veterans with posttraumatic stress disorder. Biol Psychiatry (2015) 77:356–64. doi: 10.1016/j.biopsych.2014.02.006
71. Gola H, Engler A, Morath J, Adenauer H, Elbert T, Kolassa I-T, et al. Reduced peripheral expression of the glucocorticoid receptor α isoform in individuals with posttraumatic stress disorder: a cumulative effect of trauma burden. PLoS One (2014) 9:e86333. doi: 10.1371/journal.pone.0086333
72. Hepgul N, Cattaneo A, Zunszain PA, Pariante CM. Depression pathogenesis and treatment: what can we learn from blood mRNA expression? BMC Med (2013) 11:28. doi: 10.1186/1741-7015-11-28
73. Liew C-C, Ma J, Tang H-C, Zheng R, Dempsey AA. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med (2006) 147:126–32. doi: 10.1016/j.lab.2005.10.005
74. Lennon EM, Maharshak N, Elloumi H, Borst L, Plevy SE, Moeser AJ. Early life stress triggers persistent colonic barrier dysfunction and exacerbates colitis in adult IL-10-/- mice. Inflammation Bowel Dis (2013) 19:712–9. doi: 10.1097/MIB.0b013e3182802a4e
75. Santos J, Benjamin M, Yang PC, Prior T, Perdue MH. Chronic stress impairs rat growth and jejunal epithelial barrier function: role of mast cells. Am J Physiol Gastrointest. Liver Physiol (2000) 278:G847–854. doi: 10.1152/ajpgi.2000.278.6.G847
76. Castagliuolo I, Lamont JT, Qiu B, Fleming SM, Bhaskar KR, Nikulasson ST, et al. Acute stress causes mucin release from rat colon: role of corticotropin releasing factor and mast cells. Am J Physiol (1996) 271:G884–892. doi: 10.1152/ajpgi.1996.271.5.G884
77. Hoffman JM, Baritaki S, Ruiz JJ, Sideri A, Pothoulakis C. Corticotropin-Releasing Hormone Receptor 2 Signaling Promotes Mucosal Repair Responses after Colitis. Am J Pathol (2016) 186:134–44. doi: 10.1016/j.ajpath.2015.09.013
78. Moss AC, Anton P, Savidge T, Newman P, Cheifetz AS, Gay J, et al. Urocortin II mediates pro-inflammatory effects in human colonocytes via corticotropin-releasing hormone receptor 2alpha. Gut (2007) 56:1210–7. doi: 10.1136/gut.2006.110668
79. Bertiaux-Vandaële N, Youmba SB, Belmonte L, Lecleire S, Antonietti M, Gourcerol G, et al. The expression and the cellular distribution of the tight junction proteins are altered in irritable bowel syndrome patients with differences according to the disease subtype. Am J Gastroenterol (2011) 106:2165–73. doi: 10.1038/ajg.2011.257
80. Dunlop SP, Hebden J, Campbell E, Naesdal J, Olbe L, Perkins AC, et al. Abnormal intestinal permeability in subgroups of diarrhea-predominant irritable bowel syndromes. Am J Gastroenterol (2006) 101:1288–94. doi: 10.1111/j.1572-0241.2006.00672.x
81. Zhou Q, Zhang B, Verne GN. Intestinal membrane permeability and hypersensitivity in the irritable bowel syndrome. Pain (2009) 146:41–6. doi: 10.1016/j.pain.2009.06.017
82. Marshall JK, Thabane M, Garg AX, Clark W, Meddings J, Collins SM, et al. Intestinal permeability in patients with irritable bowel syndrome after a waterborne outbreak of acute gastroenteritis in Walkerton, Ontario. Aliment. Pharmacol Ther (2004) 20:1317–22. doi: 10.1111/j.1365-2036.2004.02284.x
83. O’Mahony L, McCarthy J, Kelly P, Hurley G, Luo F, Chen K, et al. Lactobacillus and bifidobacterium in irritable bowel syndrome: symptom responses and relationship to cytokine profiles. Gastroenterology (2005) 128:541–51. doi: 10.1053/j.gastro.2004.11.050
84. Dinan TG, Quigley EMM, Ahmed SMM, Scully P, O’Brien S, O’Mahony L, et al. Hypothalamic-Pituitary-Gut Axis Dysregulation in Irritable Bowel Syndrome: Plasma Cytokines as a Potential Biomarker? Gastroenterology (2006) 130:304–11. doi: 10.1053/j.gastro.2005.11.033
85. Clarke G, Quigley EMM, Cryan JF, Dinan TG. Irritable bowel syndrome: towards biomarker identification. Trends Mol Med (2009) 15:478–89. doi: 10.1016/j.molmed.2009.08.001
86. Gecse K, Róka R, Ferrier L, Leveque M, Eutamene H, Cartier C, et al. Increased faecal serine protease activity in diarrhoeic IBS patients: a colonic lumenal factor impairing colonic permeability and sensitivity. Gut (2008) 57:591–9. doi: 10.1136/gut.2007.140210
87. Barbara G, Wang B, Stanghellini V, de Giorgio R, Cremon C, Di Nardo G, et al. Mast cell-dependent excitation of visceral-nociceptive sensory neurons in irritable bowel syndrome. Gastroenterology (2007) 132:26–37. doi: 10.1053/j.gastro.2006.11.039
88. Piche T, Barbara G, Aubert P, Bruley des Varannes S, Dainese R, Nano JL, et al. Impaired intestinal barrier integrity in the colon of patients with irritable bowel syndrome: involvement of soluble mediators. Gut (2009) 58:196–201. doi: 10.1136/gut.2007.140806
89. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev (2002) 16:6–21. doi: 10.1101/gad.947102
91. Géranton SM, Fratto V, Tochiki KK, Hunt SP. Descending serotonergic controls regulate inflammation-induced mechanical sensitivity and methyl-CpG-binding protein 2 phosphorylation in the rat superficial dorsal horn. Mol Pain (2008) 4:35. doi: 10.1186/1744-8069-4-35
92. Géranton SM, Morenilla-Palao C, Hunt SP. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J Neurosci (2007) 27:6163–73. doi: 10.1523/JNEUROSCI.1306-07.2007
93. McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonté B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci (2009) 12:342–8. doi: 10.1038/nn.2270
94. Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev (2011) 25:1010–22. doi: 10.1101/gad.2037511
95. Teodoridis JM, Strathdee G, Brown R. Epigenetic silencing mediated by CpG island methylation: potential as a therapeutic target and as a biomarker. Drug Resist Update (2004) 7:267–78. doi: 10.1016/j.drup.2004.06.005
96. Patnaik S. Anupriya null. Drugs Targeting Epigenetic Modifications and Plausible Therapeutic Strategies Against Colorectal Cancer. Front Pharmacol (2019) 10:588. doi: 10.3389/fphar.2019.00588
97. Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell (2014) 26:577–90. doi: 10.1016/j.ccr.2014.07.028
99. Denk F, McMahon SB. Chronic pain: emerging evidence for the involvement of epigenetics. Neuron (2012) 73:435–44. doi: 10.1016/j.neuron.2012.01.012
100. Radley JJ, Kabbaj M, Jacobson L, Heydendael W, Yehuda R, Herman JP. Stress risk factors and stress-related pathology: neuroplasticity, epigenetics and endophenotypes. Stress (2011) 14:481–97. doi: 10.3109/10253890.2011.604751
101. Vaiserman AM. Epigenetic programming by early-life stress: Evidence from human populations. Dev Dyn. (2015) 244:254–65. doi: 10.1002/dvdy.24211
102. Hullar MAJ, Fu BC. Diet, the gut microbiome, and epigenetics. Cancer J (2014) 20:170–5. doi: 10.1097/PPO.0000000000000053
103. Weaver ICG, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci (2004) 7:847–54. doi: 10.1038/nn1276
104. Labonte B, Yerko V, Gross J, Mechawar N, Meaney MJ, Szyf M, et al. Differential glucocorticoid receptor exon 1(B), 1(C), and 1(H) expression and methylation in suicide completers with a history of childhood abuse. Biol Psychiatry (2012) 72:41–8. doi: 10.1016/j.biopsych.2012.01.034
105. Watkeys OJ, Kremerskothen K, Quidé Y, Fullerton JM, Green MJ. Glucocorticoid receptor gene (NR3C1) DNA methylation in association with trauma, psychopathology, transcript expression, or genotypic variation: A systematic review. Neurosci Biobehav Rev (2018) 95:85–122. doi: 10.1016/j.neubiorev.2018.08.017
106. Greenwood-Van Meerveld B, Johnson AC. Stress-Induced Chronic Visceral Pain of Gastrointestinal Origin. Front Syst Neurosci (2017) 11:86. doi: 10.3389/fnsys.2017.00086
107. Perroud N, Paoloni-Giacobino A, Prada P, Olié E, Salzmann A, Nicastro R, et al. Increased methylation of glucocorticoid receptor gene (NR3C1) in adults with a history of childhood maltreatment: a link with the severity and type of trauma. Transl Psychiatry (2011) 1:e59. doi: 10.1038/tp.2011.60
108. Tran L, Chaloner A, Sawalha AH, Greenwood Van-Meerveld B. Importance of epigenetic mechanisms in visceral pain induced by chronic water avoidance stress. Psychoneuroendocrinology (2013) 38:898–906. doi: 10.1016/j.psyneuen.2012.09.016
109. Hong S, Zheng G, Wiley JW. Epigenetic regulation of genes that modulate chronic stress-induced visceral pain in the peripheral nervous system. Gastroenterology (2015) 148:148–157.e7. doi: 10.1053/j.gastro.2014.09.032
110. Mahurkar S, Polytarchou C, Iliopoulos D, Pothoulakis C, Mayer EA, Chang L. Genome-wide DNA methylation profiling of peripheral blood mononuclear cells in irritable bowel syndrome. Neurogastroenterol. Motil (2016) 28:410–22. doi: 10.1111/nmo.12741
111. Grondona JM, Hoyo-Becerra C, Visser R, Fernández-Llebrez P, López-Ávalos MD. The subcommissural organ and the development of the posterior commissure. Int Rev Cell Mol Biol (2012) 296:63–137. doi: 10.1016/B978-0-12-394307-1.00002-3
112. Richter HG, Tomé MM, Yulis CR, Vío KJ, Jiménez AJ, Pérez-Fígares JM, et al. Transcription of SCO-spondin in the subcommissural organ: evidence for down-regulation mediated by serotonin. Brain Res Mol Brain Res (2004) 129:151–62. doi: 10.1016/j.molbrainres.2004.07.003
113. Hunter A, Spechler PA, Cwanger A, Song Y, Zhang Z, Ying G, et al. DNA Methylation Is Associated with Altered Gene Expression in AMD. Invest Ophthalmol Vis Sci (2012) 53:2089–105. doi: 10.1167/iovs.11-8449
114. Leygo C, Williams M, Jin HC, Chan MWY, Chu WK, Grusch M, et al. DNA Methylation as a Noninvasive Epigenetic Biomarker for the Detection of Cancer. Dis Markers (2017) 2017:3726595. doi: 10.1155/2017/3726595
115. Zhu S, Min L, Guo Q, Li H, Yu Y, Zong Y, et al. Transcriptome and methylome profiling in a rat model of irritable bowel syndrome induced by stress. Int J Mol Med (2018) 42:2641–9. doi: 10.3892/ijmm.2018.3823
116. Mahurkar-Joshi S, Videlock EJ, Iliopoulos D, Pothoulakis C, Mayer EA, Chang L. Epigenetic Changes in Blood Cells and Colonic Mucosa are Associated with Irritable Bowel Syndrome (IBS). Gastroenterology (2018) 154:S–214. doi: 10.1016/S0016-5085(18)31105-3
117. Hake SB, Xiao A, Allis CD. Linking the epigenetic “language” of covalent histone modifications to cancer. Br J Cancer (2004) 90:761–9. doi: 10.1038/sj.bjc.6601575
118. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res (2011) 21:381–95. doi: 10.1038/cr.2011.22
119. Bai G, Wei D, Zou S, Ren K, Dubner R. Inhibition of class II histone deacetylases in the spinal cord attenuates inflammatory hyperalgesia. Mol Pain (2010) 6:51. doi: 10.1186/1744-8069-6-51
120. Imai S, Ikegami D, Yamashita A, Shimizu T, Narita M, Niikura K, et al. Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long-lasting neuropathic pain. Brain (2013) 136:828–43. doi: 10.1093/brain/aws330
121. Imai S, Ikegami D, Yamashita A, Shimizu T, Narita M, Niikura K, et al. Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long-lasting neuropathic pain. Brain (2013) 136:828–43. doi: 10.1093/brain/aws330
122. Moloney RD, Stilling RM, Dinan TG, Cryan JF. Early-life stress-induced visceral hypersensitivity and anxiety behavior is reversed by histone deacetylase inhibition. Neurogastroenterol. Motil (2015) 27:1831–6. doi: 10.1111/nmo.12675
123. Moloney RD, Johnson AC, O’Mahony SM, Dinan TG, Greenwood-Van Meerveld B, Cryan JF. Stress and the Microbiota-Gut-Brain Axis in Visceral Pain: Relevance to Irritable Bowel Syndrome. CNS Neurosci Ther (2016) 22:102–17. doi: 10.1111/cns.12490
124. Hong S, Zheng G, Wiley JW. Epigenetic regulation of genes that modulate chronic stress-induced visceral pain in the peripheral nervous system. Gastroenterology (2015) 148:148–157.e7. doi: 10.1053/j.gastro.2014.09.032
125. Aguirre JE, Winston JH, Sarna SK. Neonatal immune challenge followed by adult immune challenge induces epigenetic-susceptibility to aggravated visceral hypersensitivity. Neurogastroenterol. Motil (2017) 29:125. doi: 10.1111/nmo.13081
126. Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet (2008) 9:102–14. doi: 10.1038/nrg2290
127. Kress M, Hüttenhofer A, Landry M, Kuner R, Favereaux A, Greenberg D, et al. microRNAs in nociceptive circuits as predictors of future clinical applications. Front Mol Neurosci (2013) 6:33. doi: 10.3389/fnmol.2013.00033
128. Videlock EJ, Mahurkar-Joshi S, Iliopoulos D, Pothoulakis C, Meyer EA, Chang L. Dysregulation of the long-noncoding RNA, GHRLOS, in irritable bowel syndrome. Gastroenterology (2017) 152:S722. doi: 10.1016/S0016-5085(17)32511-8
129. Kapeller J, Houghton LA, Mönnikes H, Walstab J, Möller D, Bönisch H, et al. First evidence for an association of a functional variant in the microRNA-510 target site of the serotonin receptor-type 3E gene with diarrhea predominant irritable bowel syndrome. Hum Mol Genet (2008) 17:2967–77. doi: 10.1093/hmg/ddn195
130. Fourie NH, Peace RM, Abey SK, Sherwin LB, Rahim-Williams B, Smyser PA, et al. Elevated circulating miR-150 and miR-342-3p in patients with irritable bowel syndrome. Exp Mol Pathol (2014) 96:422–5. doi: 10.1016/j.yexmp.2014.04.009
131. Gheinani AH, Burkhard FC, Monastyrskaya K. Deciphering microRNA code in pain and inflammation: lessons from bladder pain syndrome. Cell Mol Life Sci (2013) 70:3773–89. doi: 10.1007/s00018-013-1275-7
132. Pekow JR, Kwon JH. MicroRNAs in inflammatory bowel disease. Inflammation Bowel Dis (2012) 18:187–93. doi: 10.1002/ibd.21691
133. Zhou Q, Souba WW, Croce CM, Verne GN. MicroRNA-29a regulates intestinal membrane permeability in patients with irritable bowel syndrome. Gut (2010) 59:775–84. doi: 10.1136/gut.2009.181834
134. Randomised placebo-controlled trial of dietary glutamine supplements for postinfectious irritable bowel syndrome. Gut. Available at: https://gut.bmj.com/content/68/6/996 (Accessed December 20, 2019).
135. Zhou Q, Costinean S, Croce CM, Brasier AR, Merwat S, Larson SA, et al. MicroRNA 29 targets nuclear factor-κB-repressing factor and Claudin 1 to increase intestinal permeability. Gastroenterology (2015) 148:158–169.e8. doi: 10.1053/j.gastro.2014.09.037
136. Martinez C, Rodino-Janeiro BK, Lobo B, Stanifer ML, Klaus B, Granzow M, et al. miR-16 and miR-125b are involved in barrier function dysregulation through the modulation of claudin-2 and cingulin expression in the jejunum in IBS with diarrhoea. Gut (2017) 66:1537–8. doi: 10.1136/gutjnl-2016-311477
137. Hou Q, Huang Y, Zhu S, Li P, Chen X, Hou Z, et al. MiR-144 Increases Intestinal Permeability in IBS-D Rats by Targeting OCLN and ZO1. Cell Physiol Biochem (2017) 44:2256–68. doi: 10.1159/000486059
138. Hou Q, Huang Y, Zhang C, Zhu S, Li P, Chen X, et al. MicroRNA-200a Targets Cannabinoid Receptor 1 and Serotonin Transporter to Increase Visceral Hyperalgesia in Diarrhea-predominant Irritable Bowel Syndrome Rats. J Neurogastroenterol. Motil (2018) 24:656–68. doi: 10.5056/jnm18037
139. Liao X-J, Mao W-M, Wang Q, Yang G-G, Wu W-J, Shao S-X. MicroRNA-24 inhibits serotonin reuptake transporter expression and aggravates irritable bowel syndrome. Biochem Biophys Res Commun (2016) 469:288–93. doi: 10.1016/j.bbrc.2015.11.102
140. Wohlfarth C, Schmitteckert S, Härtle JD, Houghton LA, Dweep H, Fortea M, et al. miR-16 and miR-103 impact 5-HT 4 receptor signalling and correlate with symptom profile in irritable bowel syndrome. Sci Rep (2017) 7:1–14. doi: 10.1038/s41598-017-13982-0
141. Merhautova J, Demlova R, Slaby O. MicroRNA-Based Therapy in Animal Models of Selected Gastrointestinal Cancers. Front Pharmacol (2016) 7:329. doi: 10.3389/fphar.2016.00329
142. Chen L-L. Linking Long Noncoding RNA Localization and Function. Trends Biochem Sci (2016) 41:761–72. doi: 10.1016/j.tibs.2016.07.003
143. Yarani R, Mirza AH, Kaur S, Pociot F. The emerging role of lncRNAs in inflammatory bowel disease. Exp Mol Med (2018) 50. doi: 10.1038/s12276-018-0188-9
144. Miro-Blanch J, Yanes O. Epigenetic Regulation at the Interplay Between Gut Microbiota and Host Metabolism. Front Genet (2019) 10:638. doi: 10.3389/fgene.2019.00638
145. Bouchard L, Rabasa-Lhoret R, Faraj M, Lavoie M-E, Mill J, Pérusse L, et al. Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction. Am J Clin Nutr (2010) 91:309–20. doi: 10.3945/ajcn.2009.28085
146. Rea K, O’Mahony SM, Dinan TG, Cryan JF. The Role of the Gastrointestinal Microbiota in Visceral Pain. In: Greenwood-Van Meerveld B, editor. Gastrointestinal Pharmacology Handbook of Experimental Pharmacology. Cham: Springer International Publishing (2017). p. 269–87. doi: 10.1007/164_2016_115
147. Stilling RM, Dinan TG, Cryan JF. Microbial genes, brain & behaviour – epigenetic regulation of the gut–brain axis. Genes Brain Behav (2014) 13:69–86. doi: 10.1111/gbb.12109
148. Russell WR, Hoyles L, Flint HJ, Dumas M-E. Colonic bacterial metabolites and human health. Curr Opin Microbiol. (2013) 16:246–54. doi: 10.1016/j.mib.2013.07.002
149. Martin CR, Osadchiy V, Kalani A, Mayer EA. The Brain-Gut-Microbiome Axis. Cell Mol Gastroenterol. Hepatol. (2018) 6:133–48. doi: 10.1016/j.jcmgh.2018.04.003
150. Maslowski KM, Mackay CR. Diet, gut microbiota and immune responses. Nat Immunol (2011) 12:5–9. doi: 10.1038/ni0111-5
151. Krautkramer KA, Rey FE, Denu JM. Chemical signaling between gut microbiota and host chromatin: What is your gut really saying? J Biol Chem (2017) 292:8582–93. doi: 10.1074/jbc.R116.761577
152. Vaiopoulou A, Karamanolis G, Psaltopoulou T, Karatzias G, Gazouli M. Molecular basis of the irritable bowel syndrome. World J Gastroenterol (2014) 20:376–83. doi: 10.3748/wjg.v20.i2.376
153. DeBusk RM, Fogarty CP, Ordovas JM, Kornman KS. Nutritional genomics in practice: where do we begin? J Am Diet Assoc (2005) 105:589–98. doi: 10.1016/j.jada.2005.01.002
154. de Roest RH, Dobbs BR, Chapman BA, Batman B, O’Brien LA, Leeper JA, et al. The low FODMAP diet improves gastrointestinal symptoms in patients with irritable bowel syndrome: a prospective study. Int J Clin Pract (2013) 67:895–903. doi: 10.1111/ijcp.12128
155. Halmos EP, Power VA, Shepherd SJ, Gibson PR, Muir JG. A diet low in FODMAPs reduces symptoms of irritable bowel syndrome. Gastroenterology (2014) 146:67–75.e5. doi: 10.1053/j.gastro.2013.09.046
156. Coutinho SV, Plotsky PM, Sablad M, Miller JC, Zhou H, Bayati AI, et al. Neonatal maternal separation alters stress-induced responses to viscerosomatic nociceptive stimuli in rat. Am J Physiol Gastrointest. Liver Physiol (2002) 282:G307–316. doi: 10.1152/ajpgi.00240.2001
157. Kelly TK, De Carvalho DD, Jones PA. Epigenetic Modifications as Therapeutic Targets. Nat Biotechnol (2010) 28:1069–78. doi: 10.1038/nbt.1678
Keywords: irritable bowel syndrome, IBS, epigenetics, visceral hypersensitivity, DNA methylation, microRNA, histone modifications, long non-coding RNA
Citation: Mahurkar-Joshi S and Chang L (2020) Epigenetic Mechanisms in Irritable Bowel Syndrome. Front. Psychiatry 11:805. doi: 10.3389/fpsyt.2020.00805
Received: 06 January 2020; Accepted: 27 July 2020;
Published: 14 August 2020.
Edited by:
Guillaume Gourcerol, Université de Rouen, FranceReviewed by:
Miriam Goebel-Stengel, HELIOS Klinik Rottweil, GermanyCopyright © 2020 Mahurkar-Joshi and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Chang, bGluY2hhbmdAbWVkbmV0LnVjbGEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.