
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Psychiatry , 06 August 2020
Sec. Schizophrenia
Volume 11 - 2020 | https://doi.org/10.3389/fpsyt.2020.00627
This article is part of the Research Topic The Role of Inflammation in the Etiology and Treatment of Schizophrenia View all 12 articles
 Dominique Endres1,2*
Dominique Endres1,2* Sebastian Rauer3
Sebastian Rauer3 Alexander Pschibul4
Alexander Pschibul4 Patrick Süß5
Patrick Süß5 Nils Venhoff6
Nils Venhoff6 Kimon Runge1,2
Kimon Runge1,2 Bernd Feige1,2Dominik Denzel1,2
Bernd Feige1,2Dominik Denzel1,2 Kathrin Nickel1,2
Kathrin Nickel1,2 Tina Schweizer1,2
Tina Schweizer1,2 Simon Maier1,2
Simon Maier1,2 Karl Egger7
Karl Egger7 Katharina Domschke2,8
Katharina Domschke2,8 Philipp T. Meyer9
Philipp T. Meyer9 Harald Prüss10,11†
Harald Prüss10,11† Ludger Tebartz van Elst1,2†
Ludger Tebartz van Elst1,2†Background: Autoimmune encephalitis, such as anti-NMDA-receptor encephalitis, typically presenting with subacute onset of neuropsychiatric symptoms, can be detected by antineuronal autoantibodies or inflammatory changes in the cerebrospinal fluid (CSF), as well as pathological alterations in electroencephalography (EEG), magnetic resonance imaging (MRI), or [18F]fluorodeoxyglucose positron emission tomography (FDG PET). For patients with predominant psychotic symptoms, the term autoimmune psychosis was proposed. Here, the authors present the case of a patient with probable autoimmune psychosis associated with unknown antineuronal antibodies.
Case Presentation: A 18-year-old male patient with preexisting autism spectrum disorder developed a severe catatonic syndrome over 2.5 years. The MRI showed normal findings, the EEG depicted intermittent slowing, and the independent component analyses showed additional sharp spikes. However, FDG PET, the basic laboratory analysis and testing of the serum/CSF for well-characterized antineuronal autoantibodies were unsuspicious. The serum and CSF “tissue-based assay” using indirect immunofluorescence on unfixed murine brain tissue revealed antineuronal autoantibodies against an unknown epitope in granule cells in the cerebellum and to neurites of hippocampal interneurons with a somatodendritic staining pattern. The immunosuppressive treatment with high-dose glucocorticoids, plasma exchange, and rituximab led to partial improvement.
Conclusion: The patient probably suffered from autoantibody-associated autoimmune psychosis. The special features of the case were that the patient (1) presented with mostly inconspicuous basic diagnostics, except for the altered EEG in combination with the detection of CSF autoantibodies directed against a currently unknown epitope, (2) experienced an isolated and long-lasting psychotic course, and (3) had pre-existing autism spectrum disorder. The detection of a probable autoimmune pathophysiology in such cases seems important, as it offers new and more causal immunosuppressive treatment alternatives.
Autoimmune encephalitis, such as anti-NMDA-receptor encephalitis, is typically associated with a subacute onset of neuropsychiatric symptoms, and can often be identified by the presence of antineuronal autoantibodies or inflammatory changes in the cerebrospinal fluid (CSF), electroencephalography (EEG) pathologies, and evidence of encephalitis in magnetic resonance imaging (MRI) or [18F]fluorodeoxyglucose positron emission tomography (FDG PET; 1–5). Until now, a series of antineuronal autoantibodies against surface (e.g., NMDA-R, LGI1, CASPR2, AMPA1/2-R, GABA-B-R, DPPX) or intracellular antigens (e.g., Yo, Hu, CV2/CRMP5, Ri, Ma1/2, SOX1, GAD65, amphiphysin), or “potentially antineuronal” systemic antibodies (e.g., antinuclear antibodies [ANAs] against double-stranded [ds]-DNA or gliadin autoantibodies) (3–5) are known. The associated neuropsychiatric syndromes are classically characterized by a combination of neurological (e.g., seizures, movement disorders, focal-neurological deficits, or reduced consciousness) and psychiatric (e.g., psychosis, mania, or catatonia) symptoms (3, 6, 7). However, there are also variants with milder isolated or predominant psychiatric symptoms described in the literature associated with some of the autoantibodies (8–11). Catatonic syndromes are typically reported in anti-NMDA-receptor encephalitis (12). Some authors have suggested the term “autoimmune psychosis” to describe that subgroup of patients with predominant psychotic symptoms (13–15). New methods (e.g., “tissue-based assays” via indirect immunofluorescence on unfixed murine brain tissue) can be used to increase sensitivity in discovering also novel autoantibodies against so far unknown epitopes on the cell surface (16). Previously “seronegative” autoimmune neurological syndromes have already been confirmed by such positive tests (17). The role of tissue-based assays in the detection of autoimmune psychoses is largely unclear. The rationale of the current case report is to present a patient with probable autoimmune psychosis associated with antineuronal autoantibodies directed against a currently unknown antigen detected by a tissue-based assay.
Here, the authors present the case of a 18-year-old male German pupil who developed a severe catatonic syndrome over 2.5 years. The catatonic syndrome presented with symptoms of mutism (the patient did not speak at all during the course of the disease), catalepsy and rigor, echo phenomena, and intermittent states of psychomotor agitation (the patient would run up and down the hallway in an excited manner). Further, he developed fecal and urinary incontinence, displayed impaired perception and formal thought disorder, exhibited severe concentration and attention deficits, and reported delusions (e.g., he thought he was telepathically controlled) and auditory hallucinations hearing commenting voices (e.g., he heard songs and the voice of a classmate that would tell him funny things). The symptoms initially began with cognitive deficits and seizure-like states with laughing. An epileptic examination by telemetry did not reveal any evidence of gelastic seizures. Auditory hallucinations probably existed for about 1.5 years. In addition, the patient developed an increased muscle tone and cramps of the tongue for half a year. These were interpreted as tardive dyskinesias after risperidone intake. At that time, due to severe abulia and akinetic mutism he had to be fed via stomach tube and in retrospect probably also suffered from headache.
The diagnostic examinations were carried out at first presentation in our tertiary care hospital, approximately 2.5 years after symptom onset. The structural brain MRI was normal (Figure 1). There were no white matter or contrast-enhanced lesions. A routine EEG depicted intermittent, generalized, but frontally accentuated slowing. The independent component analyses (ICA) revealed sharp waves (component 4) and intermittent rhythmic delta activity (IRDA, component 5 + 6, Figure 1). In the serum, increased ANA titers (1:800) without specificity against a defined cluster of extractable nuclear antigens (ENA) including double-stranded (ds)-DNA were found. The patient’s basic CSF analytic results (white blood cell count, albumin quotient, IgG-index, and oligoclonal bands) were normal. No antibodies against the intracellular (synaptic) antigens Yo, Hu, CV2/CRMP5, Ri, Ma1/2, Tr, Zic4, GAD65, and amphiphysin were found. Initially, anti-Sox1 autoantibodies were once weakly positive, in the course no more. Antibodies against various well-characterized neuronal cell surface antigens (NMDA-R, LGI1, CASPR2, AMPA1/2-R, GABA-B-R, DPPX) were also negative. However, in his serum and CSF, autoantibody binding to granule cells in the granule cell layer of the cerebellum and to neurites of hippocampal interneurons with a somatodendritic staining pattern against an unknown neuronal epitope were found in addition to an ANA pattern (Figure 2). An FDG PET examination of his brain showed no relevant regional hypermetabolism (suggestive of active inflammation) or hypometabolism (as a possible sequel of inflammation or degeneration) aside from mild relative striatal hypermetabolism, compatible with ongoing neuroleptic medication (Figure 1). A whole-body FDG PET/computer tomography (CT) detected no metabolic or structural pathologies suggestive of malignancy or inflammation. All diagnostic investigations and findings are summarized in Table 1.
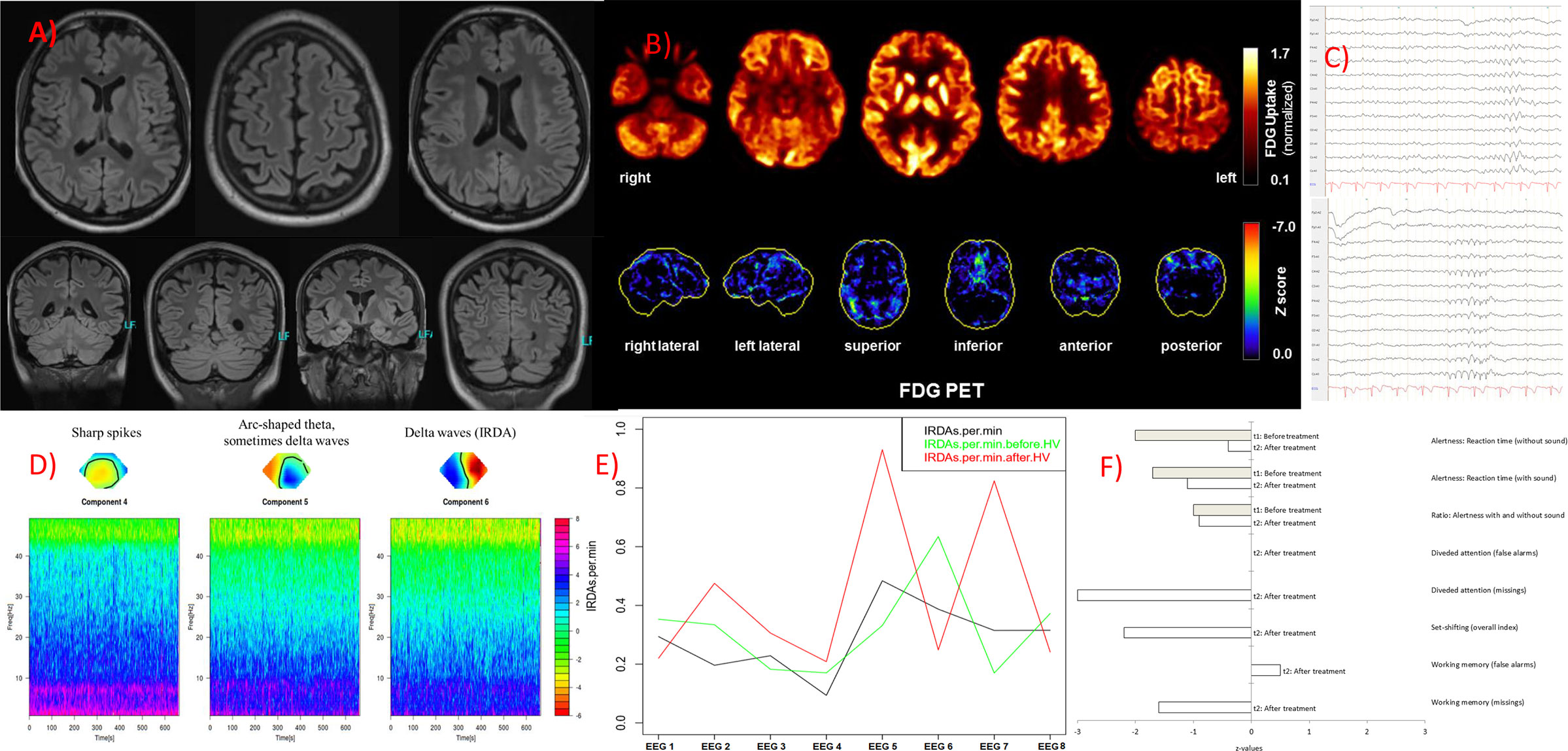
Figure 1 Imaging, electroencephalography (EEG) and neuropsychological findings. (A) The magnetic resonance imaging produced unsuspicious findings. (B) An [18F]fluorodeoxyglucose positron emission tomography (FDG PET) examination at baseline revealed no relevant regional metabolic abnormality suggestive of inflammation. Upper row: Transaxial FDG PET images (voxel-wise FDG uptake normalized to whole brain uptake), lower row: 3D surface projections of regions with decreased FDG uptake (colour-coded Z-score, compared to age-matched healthy controls; minor, linear-shaped areas of hypometabolism were judged to be non-specific partial volume effects due to atrophy; 18). The cerebral FDG PET scan was performed as part of the whole-body PET/CT scan 50 min after injection of 340 MBq FDG (Vereos Digital PET/CT, Philips Healthcare, The Netherlands). (C) The routine EEG revealed marginal intermittent, rhythmic, and generalized slowing. (D) The independent component analyses showed sharp spikes in component 4, arc-shaped theta (sometimes delta waves) in component 5, and intermittent rhythmic, generalized delta activity (IRDAs) in component 6. (E) Rate of IRDAs during initial treatment: Under benzodiazepines (EEG 1, 3 and 4) the IRDA activity appeared to be low, after the steroid pulse (5) the IRDA rate increased significantly, during plasma exchange (6) they were reduced again, in the two follow-up EEGs directly after initial treatment (7) and half a year later under rituximab (8) the IRDA activity decreased overall, whereas without valproate the IRDAs were increased again after hyperventilation (7). EEG 1 was performed with a medication of lorazepam, amisulpride, and olanzapine; EEG 2 with valproate (lamotrigine in low dose) and amisulpride; EEG 3 with clobazam, valproate (lamotrigine in low dose), and amisulpride; EEG 4 with clobazam (low dose), valproate (lamotrigine in low dose), and amisulpride; EEG 5 with valproate (lamotrigine in low dose), amisulpride; after steroid pulse treatment, with still 40 mg methylprednisolone orally; EEG 6 with valproate (reduced), lamotrigine, amisulpride, still 10 mg methylprednisolone and after 4 cycles of plasma exchange; EEG 7 with valproate (low dose), lamotrigine, amisulpride, with still 10 mg methylprednisolone orally; EEG 8 follow up (a half year later) with lamotrigine and amisulpride. (F) Measures of attentional performance during the course of the disease (t2 was performed at six-months follow-up).
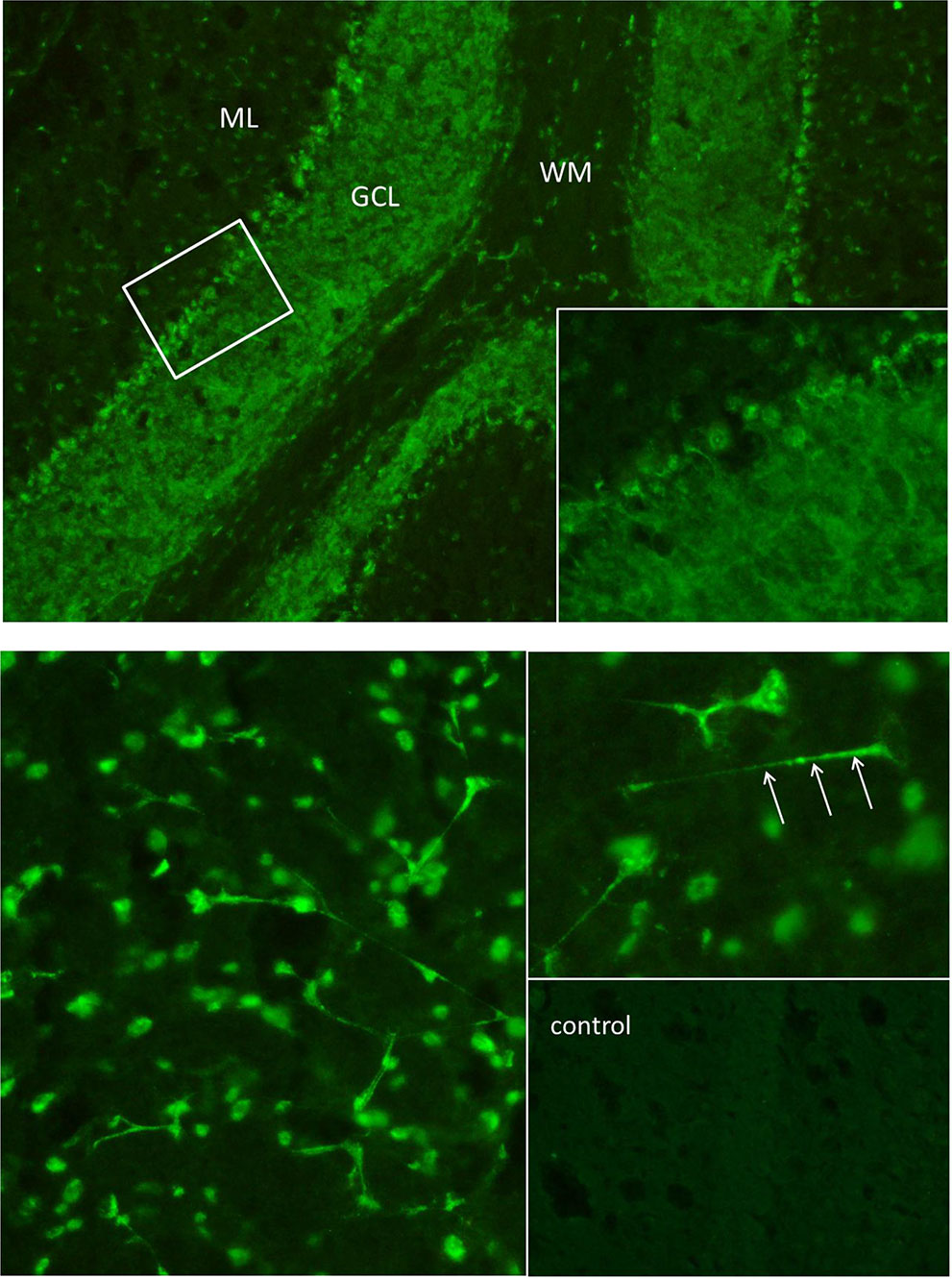
Figure 2 The “tissue-based assay” using indirect immunofluorescence screening on unfixed brain sections of rodents identified antibody binding to granule cells in the granule cell layer of the cerebellum (above) and to neurites of hippocampal interneurons (below). Autoantibodies in the cerebrospinal fluid showed a “somatodendritic staining pattern” (arrows point to dendritic processes on hippocampal interneurons in stratum lacunosum moleculare), but also to cell nuclei, reflecting antinuclear antibodies (ANAs). In a control cerebrospinal fluid sample of the hippocampus (downright) there was no comparable signal. The cerebrospinal fluid material was used undiluted. ML, molecular layer; GCL, granule cell layer; WM, white matter.

Table 1 Diagnostic findings (approximately 2.5 years after symptom onset).
Past medical history was negative for in-utero or birth complications, febrile convulsions or epileptic seizures, inflammatory brain diseases, relevant systemic infections, or craniocerebral traumata with unconsciousness. Since the first decade, he suffered from autism spectrum disorder with difficulties in social cognition, communication and interaction with peers like playing with other kids, ritualized repetition of everyday situations/dialogues, and special interests for automatic machines and trains. He had no history of autoimmune disease, infection, cancer, or other relevant somatic disorders. His family history (for parents and grandparents, he has no siblings) was devoid of any diagnosed psychiatric disorders. His mother suffers from psoriasis. The paternal grandfather died of a glioblastoma, the maternal grandfather had an unclear tumor disease of the eye.
The classical neuroleptic treatment with risperidone, olanzapine, and amisulpride, as well as treatment with lorazepam, did not lead to relevant improvement. An increase in extrapyramidal motor symptoms was observed with both risperidone and amisulpride application in a dose-related extent. Under valproate treatment, the mutism improved slightly (so that basic communication became possible). Of the three different IRDA components visible before valproate (Figure 1), only one single left frontal component was still visible in the ICA after valproate. Due to an increase in weight as a probable side effect of valproate, the medication was changed to lamotrigine. Because of the refractory disease course and the suspected autoimmune origin, the diagnosis of a probable autoimmune psychosis was made, and immunosuppressive treatment was initiated. Treatment consisted of high-dose glucocorticoid pulse therapy (1,000 mg methylprednisolone daily for five days), followed by oral treatment and stepwise glucocorticoid tapering. Plasma exchange (eight cycles) was used to rapidly decrease autoantibody concentrations. For maintenance treatment the B-cell depleting anti-CD20 antibody rituximab was given intravenously in a dose of 2x1,000 mg within 14 days and subsequently 1,000 mg every six months. The treatment steps were carried out in succession. Following high-dose glucocorticoid treatment and plasma exchange, slight improvements were observed. His incontinence improved and he made considerable progress in his ability to communicate. At the first six-month follow-up (the admission was made for the third rituximab treatment), the patient had gone back to school to gain structure in his daily routine (initially 3 h, later the whole day). A complete neuropsychological testing was possible for the first-time half a year after initiation of immunosuppressive treatment, showing ongoing deficits in alertness and working memory and particularly in divided attention and set shifting (Figure 1, t2). His formal thinking and cognitive functions were still compromised; intermittently there were long response latencies. Auditory hallucinations remained, however, on a reduced level, delusions did not occur any more. In summary, there was a relevant clinical improvement, but the patient was still significantly limited in his ability to function in everyday life. The follow-up MRI was unchanged, and CSF autoantibody testing showed reduced titers of the anti-neuronal autoantibody, although still clearly detectable. Figure 1 illustrates the course of the EEG slowing (IRDAs) under treatment and after a 6-months follow-up period.
The present case report describes a male patient suffering from a probable autoantibody-associated psychotic syndrome presenting with severe catatonic and paranoid-hallucinatory symptoms. Striking aspects of the presented case are: (1) the mostly non-specific basic diagnostics in combination with the detection of antineuronal autoantibodies showing a “somatodendritic staining pattern” against an unknown neuronal epitope, (2) the long-lasting psychiatric course, (3) the slight improvement under anticonvulsants and clear improvement under immunosuppressive treatment, and (4) the preexisting autism spectrum disorder.
The initial comprehensive diagnostic procedure revealed no clear pathological findings indicating an immunological pathophysiology. The basic CSF diagnostics and the biochip assay tests on fixed cells for detecting established antineuronal autoantibodies, the MRI of the brain, and the FDG PET failed to identify any relevant abnormality. The EEG showed non-specific changes in the form of an intermittent frontally accentuated slowing. The ICA helped to demonstrate electrophysiological instability with sharp spikes and IRDAs. Even with this comprehensive diagnostic procedure, the identification of an autoimmune psychosis would not have been possible. In the presented case, the critical clues were only obtained from a tissue-based assay via indirect immunofluorescence on unfixed murine brain tissue carried out in a specialized CSF laboratory. Antineuronal autoantibody binding to granule cells in the granule cell layer of the cerebellum and to neurites of hippocampal interneurons against an unknown epitope was found. The immunofluorescence pattern was a “somatodendritic staining pattern”, increasing the likelihood of functional relevance of the autoantibody (19). If or not the absence of encephalitis signs in the CSF or FDG PET are related to the long development or the isolated psychiatric course of the disease remains unclear. For some other well-characterized antineuronal antibodies such as LGI1 or IgLON5, an inconspicuous CSF is also common. However, based on present conceptualization, this case can most likely be taken as an example of a probable autoimmune psychosis compatible with a “mild encephalitis” concept (20), even though it would not fulfill the current consensus criteria of probable autoimmune encephalitis (3).
From a clinical point of view, it was interesting to observe that the catatonic symptoms and the EEG pathologies already improved slightly following initial treatment with valproate. Patients with confirmed autoimmune encephalitis (e.g., anti-NMDA-R encephalitis) also often improve initially under “symptomatic” treatment with neuroleptics. The response to psychotropic drugs such as neuroleptics or, in this case, anticonvulsants, is therefore not a contradiction to an autoimmune genesis. For the presented case, it can be speculated that neuronal network instability caused by the detected unspecified autoantibodies improved under anticonvulsive (i.e., network-stabilizing) medication (see Figure 1). This idea would be well explained by the Local Area Network Inhibition (LANI) hypothesis. Following the LANI model, the excitatory spikes and IRDAs could lead to a counterregulatory hyperinhibition of neuronal networks and associated symptoms (e.g., cognitive dysfunction; 21). Finally, in our case report, the link to autism is interesting. In their clinical work, the authors of this paper repeatedly observed patients with developmental disorders who later developed autoimmune encephalitis. It would be interesting to analyze whether autism spectrum diseases with their well-known link to immunological abnormalities (e.g., autoantibodies against brain tissue, microglia activation, inflammatory cytokines; 22) are associated with an increased risk for the subsequent development of autoimmune encephalitis. In the present case, an immunological predisposition with increased titers of antinuclear antibodies in a male patient and a positive family history for immunological disorders (mother suffers from psoriasis) was present. Initial studies in children even showed that a small subgroup of children with autism displayed evidence of autoimmune encephalitis (23).
A pathophysiological significance of the serum/CSF autoantibodies is probable due to the antineuronal pattern in combination with intermittent EEG slowing and sharp spikes. The response to immunosuppressants was only partial and without full remission of symptoms; this could be due to the autoantibody itself or to the long course of the disease. Precise titer determination of the detected antineuronal antibodies and confirmation tests with immunohistochemistry on hippocampal neurons were not performed in the presenting patient; in similar cases in the future, the antibody findings should be analyzed in more detail (see 24). Also, CSF may be subjected to single-cell antibody repertoire analysis, which allows identification of disease-related monoclonal human autoantibodies and their targets, and seems to become the gold-standard also for determining pathogenicity of neuropsychiatric autoantibodies (25, 26). A better identification of the target epitope would help to identify the underlying pathophysiological processes more clearly. Another limiting factor in the current case is that the anti-GABA-A-R antibodies also were not analyzed using a cell-based assay. However, the lack of laminar neuropil binding in the tissue-based assay, as would be seen in autoimmune encephalitis with anti-GABA-A-R antibodies (or anti-NMDA-R or anti-GABA-B-R antibodies), argues against the presence of these antibodies. Larger patient numbers in future studies are required to determine if the hypothetical clinical characteristics discussed here with regard to the LANI hypothesis and the association with autism are correct.
This case report shows an association of a psychotic syndrome with predominant catatonic symptoms and antineuronal autoantibodies against an unknown epitope detected by a tissue-based assay. The basic diagnostics were mostly unremarkable with the exception of a conspicuous EEG. The application of tissue-based assays for the detection of so far unknown autoantibodies might also be helpful in other psychiatric patients with suspected autoimmune pathophysiology.
All datasets necessary for this case study are included in the article.
The patient and his parents have given their signed written informed consent for this case report, including the presented images, to be published.
DE, PS, SR, AP, and LT treated the patient. DE performed the data research and wrote the paper. SR, AP, and HP performed the neurological interpretation. SR performed the CSF basic analyses. HP performed the tissue-based testing on unfixed brain sections. BF and LT performed the EEG analyses. NV performed the rheumatological tests and immunological interpretation. TS performed the neuropsychological testing and interpretation. PM performed the nuclear medicine investigations and interpretation. KE, KN, and SM performed and interpreted the MRIs. AP performed the rituximab treatment. KR, BF, DD, and KD supported the clinical and laboratory interpretation. All authors were critically involved in the theoretical discussion and composition of the manuscript. All authors contributed to the article and approved the submitted version.
The article processing charge was funded by the German Research Foundation (DFG) and the University of Freiburg in the funding program Open Access Publishing.
SR: Receiving consulting and lecture fees, grant and research support from Bayer Vital, Biogen, Merck Serono, Novartis, Sanofi-Aventis, Genzyme, Roche and Teva. Furthermore, SR indicates that he is a founding executive board member of ravo Diagnostika GmbH Freiburg. NV: Advisory boards, lectures and travel grants from Roche. KD: Steering Committee Neurosciences, Janssen. PM: Advisory boards and lectures within the last three years: GE, Philips, OPASCA, Curium, Desitin and Medac. LT: Advisory boards, lectures, or travel grants within the last three years: Roche, Eli Lilly, Janssen-Cilag, Novartis, Shire, UCB, GSK, Servier, Janssen and Cyberonics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the patient for agreeing to the case report. DE was funded by the Berta-Ottenstein-Programme for Advanced Clinician Scientists, Faculty of Medicine, University of Freiburg. PS was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - 270949263/GRK2162 and PS is supported by the Interdisciplinary Center for Clinical Research (IZKF) Erlangen, Clinician Scientist Program.
ANA, antinuclear antibody; CSF, cerebrospinal fluid; CT, computer tomography; ds, double-stranded; EEG, electroencephalography; ENA, extractable nuclear antigens; FDG-PET, [18F]fluorodeoxyglucose positron emission tomography; ICA, independent component analyses; IRDA, intermittent rhythmic delta activity; LANI, Local Area Network Inhibition; MRI, magnetic resonance imaging.
1. Leypoldt F, Armangue T, Dalmau J. Autoimmune encephalopathies. Ann N. Y. Acad Sci (2015) 1338:94–114. doi: 10.1111/nyas.12553
2. Heine J, Prüss H, Bartsch T, Ploner CJ, Paul F, Finke C. Imaging of autoimmune encephalitis–Relevance for clinical practice and hippocampal function. Neuroscience (2015) 309:68–83. doi: 10.1016/j.neuroscience.2015.05.037
3. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol (2016) 15(4):391–404. doi: 10.1016/S1474-4422(15)00401-9
4. Endres D, Bechter K, Prüss H, Hasan A, Steiner J, Leypoldt F, et al. [Autoantibody-associated schizophreniform psychoses: clinical symptomatology]. Nervenarzt (2019) 90(5):547–63. doi: 10.1007/s00115-019-0700-z
5. Tebartz van Elst L, Bechter K, Prüss H, Hasan A, Steiner J, Leypoldt F, et al. Autoantikörper-assoziierte schizophreniforme Psychosen: Pathophysiologie, Diagnostik und Therapie. Nervenarzt (2019) 90(7):745–61. doi: 10.1007/s00115-019-0735-1
6. Dalmau J, Geis C, Graus F. Autoantibodies to Synaptic Receptors and Neuronal Cell Surface Proteins in Autoimmune Diseases of the Central Nervous System. Physiol Rev (2017) 97(2):839–87. doi: 10.1152/physrev.00010.2016
7. Dalmau J, Graus F. Antibody-Mediated Encephalitis. N. Engl J Med (2018) 378(9):840–51. doi: 10.1056/NEJMra1708712
8. van Elst LT, Klöppel S, Rauer S. Voltage-gated potassium channel/LGI1 antibody-associated encephalopathy may cause brief psychotic disorder. J Clin Psychiatry (2011) 72(5):722–3. doi: 10.4088/JCP.10l06510
9. Kayser MS, Titulaer MJ, Gresa-Arribas N, Dalmau J. Frequency and characteristics of isolated psychiatric episodes in anti–N-methyl-d-aspartate receptor encephalitis. JAMA Neurol (2013) 70(9):1133–9. doi: 10.1001/jamaneurol.2013.3216
10. Endres D, Perlov E, Stich O, Meyer PT, Lützen N, Tebartz van Elst L. Case report: low-titre anti-Yo reactivity in a female patient with psychotic syndrome and frontoparieto-cerebellar atrophy. BMC Psychiatry (2015) 15:112. doi: 10.1186/s12888-015-0486-x
11. Endres D, Perlov E, Stich O, Rauer S, Maier S, Waldkircher Z, et al. Hypoglutamatergic state is associated with reduced cerebral glucose metabolism in anti-NMDA receptor encephalitis: a case report. BMC Psychiatry (2015) 15:186. doi: 10.1186/s12888-015-0552-4
12. Rogers JP, Pollak TA, Blackman G, David AS. Catatonia and the immune system: a review. Lancet Psychiatry (2019) 6(7):620–30. doi: 10.1016/S2215-0366(19)30190-7
13. Ellul P, Groc L, Tamouza R, Leboyer M. The Clinical Challenge of Autoimmune Psychosis: Learning from Anti-NMDA Receptor Autoantibodies. Front Psychiatry (2017) 8:54. doi: 10.3389/fpsyt.2017.00054
14. Najjar S, Steiner J, Najjar A, Bechter K. A clinical approach to new-onset psychosis. associated with immune dysregulation: the concept of autoimmune psychosis. J Neuroinflamm. (2018) 15(1):40. doi: 10.1186/s12974-018-1067-y
15. Pollak TA, Lennox BR, Müller S, Benros ME, Prüss H, Tebartz van Elst L, et al. Autoimmune psychosis: an international consensus on an approach to the diagnos is and management of psychosis of suspected autoimmune origin. Lancet Psychiatry (2020) 7(1):93–108. doi: 10.1016/S2215-0366(19)30290-1
16. Kreye J, Wenke NK, Chayka M, Leubner J, Murugan R, Maier N, et al. Human cerebrospinal fluid monoclonal N-methyl-D-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain (2016) 139(Pt 10):2641–52. doi: 10.1093/brain/aww208
17. Edwin Thanarajah S, Prüss H, Warnke C, Barbe MT, Schroeter M, Fink GR, et al. Atypical Autoimmune Encephalitis With Neuropil Antibodies Against a Yet Unknown Epitope. Front Neurol (2019) 10:175. doi: 10.3389/fneur.2019.00175
18. Minoshima S, Frey KA, Koeppe RA, Foster NL, Kuhl DE. A diagnostic approach in Alzheimer’s disease using three-dimensional stereotactic surface projections of fluorine-18-FDG PET. J Nucl. Med (1995) 36(7):1238–48.
19. Lewerenz J, Jarius S, Wildemann B, Wandinger KP, Leypoldt F. Autoantibody-associated autoimmune encephalitis and cerebellitis: Clinical presentation, diagnostic work-up and treatment. Nervenarzt (2016) 87(12):1293–9. doi: 10.1007/s00115-016-0235-5
20. Bechter K. Encephalitis, Mild Encephalitis, Neuroprogression, or Encephalopathy-Not Merely a Question of Terminology. Front Psychiatry (2019) 9:782. doi: 10.3389/fpsyt.2018.00782
21. Tebartz van Elst L, Krishnamoorthy ES, Schulze-Bonhage A, Altenmüller DM, Richter H, Ebert D, et al. Local area network inhibition: a model of a potentially important paraepileptic pathomechanism in neuropsychiatric disorders. Epilep. Behav (2011) 22(2):231–9. doi: 10.1016/j.yebeh.2011.06.016
22. Hughes HK, Mills Ko E, Rose D, Ashwood P. Immune Dysfunction and Autoimmunity as Pathological Mechanisms in Autism Spectrum Disorders. Front Cell Neurosci (2018) 12:405. doi: 10.3389/fncel.2018.00405
23. Connery K, Tippett M, Delhey LM, Rose S, Slattery JC, Kahler SG, et al. Intravenous immunoglobulin for the treatment of autoimmune encephalopathy in children with autism. Transl Psychiatry (2018) 8(1):148. doi: 10.1038/s41398-018-0214-7
24. Vitaliani R, Mason W, Ances B, Zwerdling T, Jiang Z, Dalmau J. Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol (2005) 58(4):594–604. doi: 10.1002/ana.20614
25. Kornau HC, Kreye J, Stumpf A, Fukata Y, Parthier D, Sammons RP, et al. Human Cerebrospinal Fluid Monoclonal LGI1 Autoantibodies Increase Neuronal Excitability. Ann Neurol (2020) 87(3):405–18. doi: 10.1002/ana.25666
Keywords: autoimmune encephalitis, encephalopathy, autoimmune psychosis, antibodies, schizophrenia, catatonia
Citation: Endres D, Rauer S, Pschibul A, Süß P, Venhoff N, Runge K, Feige B, Denzel D, Nickel K, Schweizer T, Maier S, Egger K, Domschke K, Meyer PT, Prüss H and Tebartz van Elst L (2020) Novel Antineuronal Autoantibodies With Somatodendritic Staining Pattern in a Patient With Autoimmune Psychosis. Front. Psychiatry 11:627. doi: 10.3389/fpsyt.2020.00627
Received: 25 October 2019; Accepted: 15 June 2020;
Published: 06 August 2020.
Edited by:
Norbert Müller, Ludwig Maximilian University of Munich, GermanyReviewed by:
Massimo Tusconi, University of Cagliari, ItalyCopyright © 2020 Endres, Rauer, Pschibul, Süß, Venhoff, Runge, Feige, Denzel, Nickel, Schweizer, Maier, Egger, Domschke, Meyer, Prüss and Tebartz van Elst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominique Endres, ZG9taW5pcXVlLmVuZHJlc0B1bmlrbGluaWstZnJlaWJ1cmcuZGU=
†These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.