 Susan F. Sonnenschein
Susan F. Sonnenschein Felipe V. Gomes
Felipe V. Gomes Anthony A. Grace
Anthony A. Grace
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 30 June 2020
Sec. Schizophrenia
Volume 11 - 2020 | https://doi.org/10.3389/fpsyt.2020.00613
This article is part of the Research Topic Dopaminergic Alterations in Schizophrenia View all 11 articles
Dysregulation of the dopamine system is central to many models of the pathophysiology of psychosis in schizophrenia. However, emerging evidence suggests that this dysregulation is driven by the disruption of upstream circuits that provide afferent control of midbrain dopamine neurons. Furthermore, stress can profoundly disrupt this regulatory circuit, particularly when it is presented at critical vulnerable prepubertal time points. This review will discuss the dopamine system and the circuits that regulate it, focusing on the hippocampus, medial prefrontal cortex, thalamic nuclei, and medial septum, and the impact of stress. A greater understanding of the regulation of the dopamine system and its disruption in schizophrenia may provide a more complete neurobiological framework to interpret clinical findings and develop novel treatments.
Dopamine (DA) modulates circuit reactivity based on environmental stimuli and prior experience and thus plays a central role in functions including reward processing, reinforcement, and habit formation (1–3). Midbrain DA neurons have also been shown respond to novel or aversive stimuli in the absence of reward (4) and it has been proposed that DA signaling may more generally influence sensory processing, such as weighting the salience (5) or certainty (6) of perceived stimuli. Dysregulation of the DA system has been fundamental to many models of the pathophysiology of schizophrenia (7, 8). It is implicated particularly in psychotic symptoms, which involve profound perceptual disturbances (hallucinations) and fixed beliefs resistant to contradictory evidence (delusions). Hallucinations and delusions tend to co-occur and are thus proposed to manifest due to a common pathophysiological mechanism (6, 9). Psychotic symptoms can be attenuated by D2 receptor blocking drugs (10, 11) that reduce the abnormal increased DA neuron activity (12–14), but the underlying cognitive processes likely involve complex connections between numerous brain regions that remain dysfunctional. This article will discuss some of the circuits that regulate DA neuron activity and how dysfunction in these upstream circuits may influence the DA system in schizophrenia.
Clinical imaging studies have provided strong support for the DA hypothesis of schizophrenia. Imaging studies that measured radioligand displacement from DA receptors as a measure of DA activity have shown that patients with schizophrenia display increased DA release in response to low-dose amphetamine, compared to healthy controls (15–17), which correlates with transient worsening of psychotic symptoms (17). Patients also demonstrate increased baseline levels of synaptic DA in the striatum, measured in a DA depletion paradigm (18), which has been shown to correlate with their amphetamine-induced DA release (19). Both measures are observed in antipsychotic drug-naive patients and drug-free patients with prior APD treatment, and both predict treatment response of psychosis to antipsychotic drugs (18–20). Elevated striatal DA synthesis capacity, measured by fluorodopa uptake into DA terminals, is also consistently observed in patients and shown to correlate with psychotic severity (21). Numerous studies have found increased response capacity of the DA system in individuals at clinical high risk (CHR) for psychosis, which correlates with greater severity of prodromal symptoms (22–25). Longitudinal studies have further shown that there is a progressive increase in striatal DA function as CHR patients transition to full syndrome expression (24), which has been shown to predict conversion to psychosis (23, 25). Elevated DA synthesis capacity is a less consistent finding in chronic patients in remission, shown to be significantly elevated compared to healthy controls in some studies (26–29), though not all (30–32), suggesting that increased DA function most clearly signals active psychosis. The elevation in DA is limited to striatal projections (33, 34). In contrast, mesocortical projections, particularly to the dorsolateral PFC, display reduced DA release compared to healthy controls, which may contribute to impaired prefrontal-dependent cognitive processes (35). It is currently unknown what accounts for these coexisting differences in DA regulation. Together, these findings point to dysregulation of the DA system as central to the development and expression of psychotic symptoms.
Midbrain DA neurons can be subdivided with respect to their location, projection target, and functional significance (36, 37). The striatum is one of the primary targets of DA signaling and receives dense projections from DA neurons following a topological gradient. In rodents, more medial DA neurons of the ventral tegmental area (VTA) innervate more reward-related ventral striatal regions, including the nucleus accumbens (38). More lateral DA neurons of the substantia nigra project to the dorsomedial and dorsolateral striatum, which are relevant to habit formation and motor function, respectively (39, 40). DA neurons that are located at the transition from lateral VTA to substantia nigra project to the rostral caudate, or associative striatum, which is most implicated in measures of increased presynaptic DA function and demonstrates the strongest correlation to psychotic symptoms in patients with schizophrenia (33, 34). In primates, the relative position of VTA DA neurons shift, but their topological organization is retained. Whereas rodents have a prominent VTA that is located medial to the substantia nigra, in the primate the DA neurons are shifted, with the rodent VTA projection to limbic and associative striatum now becoming the dorsal tier of the substantia nigra and the rodent substantia nigra that projects to the dorsal striatum now comprising the primate ventral tier substantia nigra neurons (41, 42).
DA neurons exhibit two patterns of activity, known as tonic and phasic states, that have different functional implications and are regulated by distinct afferent systems. In vitro in the absence of inputs, DA neurons maintain a basal activity state through the generation of a pacemaker conductance (43–45). However, in vivo recordings in normal rats have shown that not all DA neurons are showing spontaneous activity; instead, approximately half of midbrain DA neurons are not spontaneously active, and instead exist in a hyperpolarized state (45–47) due to inhibitory input from the ventral pallidum (48), an area that is regulated by a pathway that arises from the ventral subiculum of the hippocampus (vHipp). When the vHipp is activated, it provides a glutamatergic drive of GABAergic projection neurons in the nucleus accumbens, which in turn inhibits the ventral pallidum and increases the proportion of VTA DA neurons that are spontaneously active (i.e. “population activity”; Figure 1).
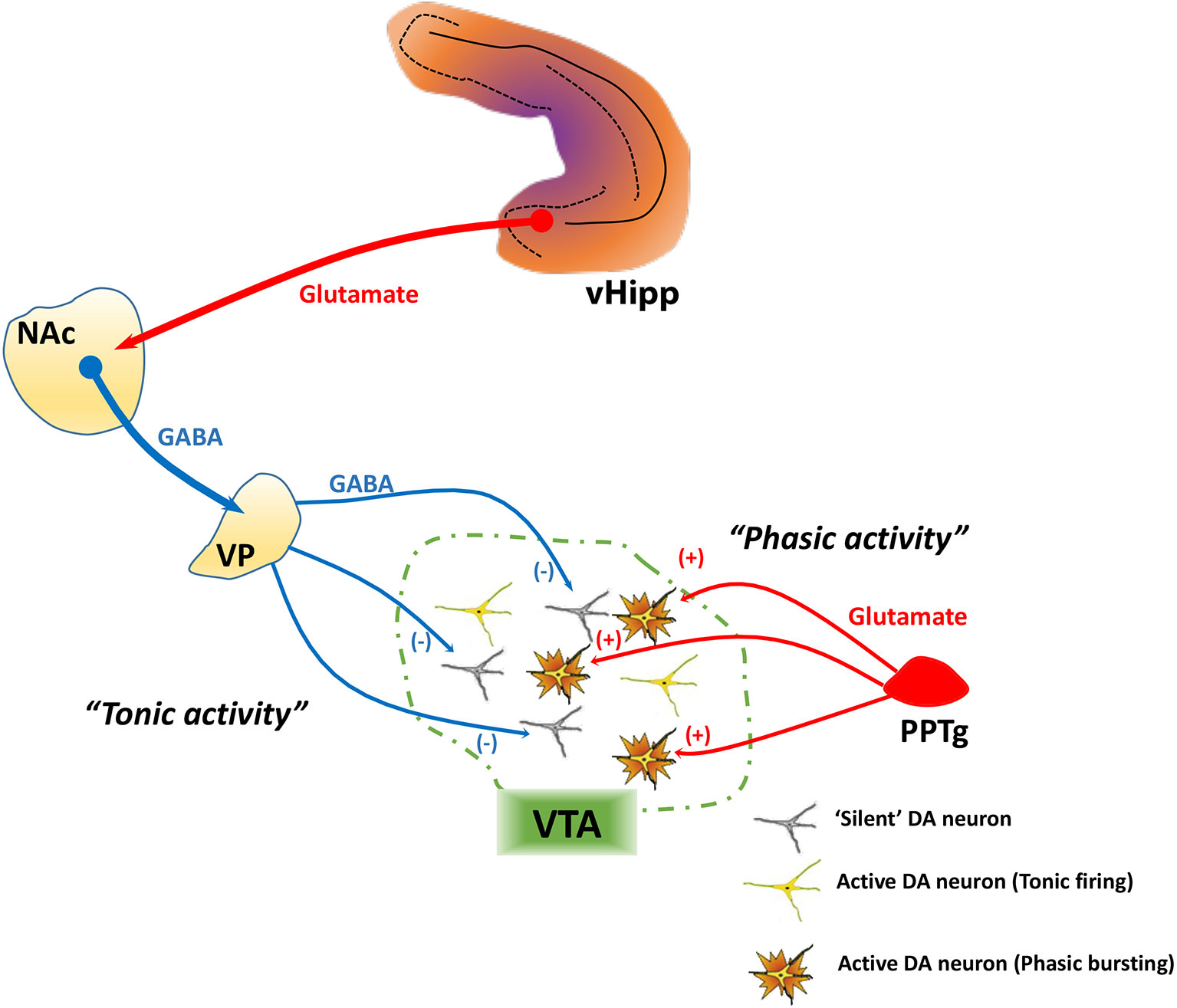
Figure 1 Tonic and phasic dopamine (DA) neuron activity are regulated by distinct afferent systems. DA neurons generate their own activity through a pacemaker conductance. However, a substantial population of DA neurons is not firing spontaneously, being held in a hyperpolarized state by a GABA-mediated inhibitory input from the ventral pallidum (VP). The VP, in turn, is controlled by a pathway originating from the ventral hippocampus (vHipp). The vHipp projects to the nucleus accumbens (NAc), which inhibits the VP. By contrast, phasic burst firing is driven by glutamatergic inputs arising from several areas, primary among these being the pedunculopontine tegmentum (PPTg). This afferent system regulates firing states within the population of spontaneously active DA neurons, because only neurons that are firing spontaneously can burst fire—NMDA channels on hyperpolarized (“silent”) DA neurons are under magnesium block and won't change state. Therefore, the PPTg provides the rapid, behaviorally salient phasic signal, whereas the VP, by controlling the number of DA neurons firing, determines the gain of the phasic signal.
Spontaneously active DA neurons, in vivo, can display an irregular tonic firing pattern and rapid, phasic burst firing (45, 49, 50). Burst firing is dependent on glutamatergic afferents from the pedunculopontine tegmentum via activation of NMDA receptors (Figure 1) (48, 51). In DA neurons that are nonfiring, NMDA fails to activate NMDA receptors due to a magnesium block that is present at hyperpolarized membrane potentials (52). Thus, only DA neurons that are depolarized (spontaneously active) have the potential to exhibit burst firing. DA neurons exhibit burst firing when exposed to a behaviorally salient stimuli, such as a potential threat or reward (53, 54). Therefore, the number of neurons firing can control the amplitude of the behaviorally salient phasic burst response; when there are more DA neurons firing (i.e., greater population activity), NMDA will cause a greater number to exhibit phasic bursts, thus amplifying the phasic response to stimuli (45, 55). In other words, the vHipp-nucleus accumbens-ventral pallidum (vHipp-NAc-VP) circuit allows the baseline level of responsivity of the DA system, which is dependent on population activity, to be adjusted based on the context in which the stimuli are presented.
Elevated DA system activity in schizophrenia results from dysfunction in a larger hippocampal-midbrain-striatal circuit, with a primary locus of pathophysiology that appears to develop in the vHipp. Deficits in the structure and function of the hippocampus are consistently observed in imaging and post-mortem studies of schizophrenia patients (56). Imaging studies show that the anterior hippocampus, which is homologous to the limbic vHipp in rodents (57), is hyperactive in individuals with schizophrenia (58). Most studies report increased hippocampal glutamate levels in both first-episode and chronic patients, independent of medication status (59), and changes in hippocampal metabolism and blood flow are associated with more severe psychotic symptoms in patients (60–62) and those at CHR (63, 64). Increased cerebral blood volume (CBV) has been reported specifically in the CA1 and subiculum of the hippocampus in patients with schizophrenia (65). Increased CBV is also present during the prodromal stage and predicts conversion to psychosis (66, 67) and hippocampal atrophy (68). Multiple lines of evidence have suggested that the hippocampal hypermetabolism is due to reduced parvalbumin (PV)+ GABA interneuron regulation of pyramidal neuron activity, secondary to excitotoxic degeneration of PV+ interneurons (69, 70). NMDA receptor antagonists, such as PCP and ketamine, may similarly exacerbate or mimic psychosis by blocking NMDA receptors on PV+ interneurons and thus disinhibiting pyramidal neurons (71, 72). This can lead to increased levels of glutamate and loss of PV+ interneurons following chronic NMDA receptor antagonist administration (68, 73–75).
One can induce an analogous disruption of hippocampal physiology in animal models based on developmental disruption, including the methylazoxymethanol acetate (MAM) neurodevelopmental rat model (76, 77). The MAM model involves administration of the mitotoxin MAM to pregnant dams on gestational day 17, which correlates with the vulnerable timepoint of the 2nd trimester in humans to adverse events such as maternal infection (78). The offspring of MAM-treated dams (“MAM rats”) develop region-specific disruption of neuronal maturation that results in adult phenotypes relevant to schizophrenia, in contrast to the offspring of dams that receive a saline injection, (“SAL rats”) (76, 79, 80). Adult MAM rats display loss of PV+ interneurons in the vHipp (81), resulting in a baseline hyperactive state from loss of inhibitory control of pyramidal cell activity (82). The increased vHipp drive results in an increase in DA neuron population activity through the vHipp-NAc-VP circuit and inactivation of vHipp in MAM rats can normalize the DA neuron activity and related aberrant behavior (48, 82). Taken together, these data suggest a that a loss of PV+ interneurons in the hippocampus leads to increased DA neuron population activity and a hyper-responsive DA state, in line with clinical evidence of increased presynaptic DA function (21, 42).
Several brain regions can enhance VTA DA system activity through interactions with the vHipp-NAc-VP pathway. Here we discuss evidence indicating the involvement of the medial prefrontal cortex (mPFC), thalamic nuclei, and medial septum on the VTA DA system and how changes in the activity of these regions may lead to a hyperdopaminergic state as seen in schizophrenia.
Dysfunction within the mPFC plays a central role in the pathophysiology of several psychiatric illnesses, including schizophrenia. For instance, contrary to the increased presynaptic striatal DA synthesis and release (83), it has been found that DA transmission is decreased in the PFC of schizophrenia patients (35). This cortical hypodopaminergic state is thought to be associated with impairments in cognitive and executive function in schizophrenia (35, 84). Also, a reduced PFC activity has been associated with elevated striatal DA function in schizophrenia patients and at-risk individuals (28, 85).
The mPFC is thought to be a major regulator of the DA system but with the outcome, either inhibitory and excitatory responses, reflecting the specific anatomy of mPFC afferents to the VTA. Two major mPFC subdivisions, the infralimbic (ilPFC) and the prelimbic (plPFC) cortices, send direct projections to the VTA (86) as well as to other regions linked with control of the midbrain DA system, such as the NAc (87). The ilPFC, in particular, seems to regulate the DA system activity through its modulation of the activity of the vHipp and basolateral amygdala (BLA). It was showed that the ilPFC exerts a bidirectional control over VTA DA system via the BLA and vHipp. Whereas the inactivation of the ilPFC increases VTA DA neuron population activity in a vHipp-dependent manner, the activation of the ilPFC decreased VTA DA neuron population activity (88). Compared with the ilPFC, the inactivation of the plPFC produced opposite effects on VTA DA neurons. Whereas the activation of the plPFC had no effect, the plPFC inactivation decreased VTA DA neuron population activity (88). This is consistent with the opposite manner that the ilPFC and plPFC impacts behavioral responses (89, 90). The mechanism by which the plPFC affects VTA DA system is still not completely understood, but it may involve the removal of plPFC attenuation of vHipp activity and/or removal of the inhibitory influence of the plPFC over the ilPFC.
Whereas vHipp activation upregulates DA responsivity, the amygdala decreases tonic DA neuron firing. Activation of the BLA has been shown decrease DA neuron population activity in the medial affect-related regions of the rat VTA, which is proposed to be due to a glutamatergic projection to the ventral pallidum, because blocking glutamate in the ventral pallidum prevents BLA activation-dependent down-regulation of DA neuron firing (91). Furthermore, the decrease in DA neuron population activity observed following activation of the ilPFC depends on an intact amygdala (88). Therefore, the opposing modulatory actions of the vHipp and the amygdala are determined by ilPFC activity.
It is worth noting that the mPFC does not project directly to the vHipp (92). Thus, the effect of ilPFC inactivation on increases in VTA DA neuron population activity, that was prevented by removal of the vHipp influence, may involve other brain regions such as the entorhinal cortex and thalamic nucleus reuniens (93, 94) since both receive direct excitatory projection from the ilPFC (92) and in turn provide powerful excitatory influence over the vHipp (92, 95, 96). Therefore, both the entorhinal cortex and nucleus reuniens could be a relay between the ilPFC and the vHipp that could potentially affect activity of DA neurons in the VTA.
The thalamus has long been implicated as a potential node of dysfunction in schizophrenia (97) mainly due to its heavily reciprocal connectivity with the hippocampus and prefrontal cortex (98–100), thus serving as a critical mediator of communication between these brain regions. Reductions in resting-state functional connectivity between the thalamus and the hippocampus and prefrontal cortex have been reported at both the chronic and early stages of schizophrenia. It has also been reported in at-risk individuals and may predict conversion to psychosis in this group (101–104). Thalamic dysconnectivity patterns consistent with those seen in schizophrenia were also observed in healthy individuals after receiving ketamine to model psychosis (105). Also, a reduction in sleep spindles, which are non-rapid eye movement sleep oscillations generated by the thalamic reticular nucleus, has been consistently reported in schizophrenia patients, and the magnitude of this reduction was inversely correlated with the severity of psychotic symptoms (106, 107). These findings suggest that the thalamus may serve as a hub of wide-scale network dysfunction in schizophrenia.
Recent rodent studies have similarly indicated circuit abnormalities underlying and resulting from thalamic dysfunction (108). The thalamus is composed of multiple nuclei, each with their distinct afferent and efferent projections (109). Our group has focused on the nucleus reuniens, a thalamic midline nucleus, since it is bidirectionally connected to the hippocampus and prefrontal cortex (98, 99, 110). The nucleus reuniens, in rodents, forms the primary route of communication between the prefrontal cortex and vHipp and is essential for behaviors involving coordinated action of these two regions, such as spatial navigation and fear memory (111–113). Regarding corticothalamic projection to nucleus reuniens, pyramidal neurons from layers 5 and 6 of the medial prefrontal cortex send direct projections to the nucleus reuniens (114, 115) and some neurons from layer 6 of the ilPFC neurons send collaterals to the antero-medial portion of the thalamic reticular nucleus (116), the same subregion of the thalamic reticular nucleus that projects to reuniens (117).
We showed that activation of the nucleus reuniens increases DA neuron population activity in the VTA via its projection to the vHipp, since it was prevented by vHipp inactivation (93). Also, as described above, the inactivation of the ilPFC increases DA neuron population activity in the VTA, an effect that was dependent on the vHipp (88). The mPFC, however, does not send direct projections to the vHipp (92). Besides sending dense projections to the vHipp (96), the nucleus reuniens drives vHipp activity (118). The ilPFC inactivation enhances VTA DA system activity via vHipp likely by disinhibiting the nucleus reuniens since the inactivation of the nucleus reuniens prevented these changes (88). These findings suggest that 1) the ilPFC potently regulates the vHipp via nucleus reuniens and 2) the ilPFC inhibition leads to disinhibition of nucleus reuniens, likely due to deactivation of the thalamic reticular nucleus, which in turn, via its excitatory projections to the vHipp, enhances VTA DA system activity. The ilPFC was found to modulate several aspects of the firing pattern of neurons in the nucleus reuniens (119). Thus, the nucleus reuniens may mediate the regulation of the VTA DA system activity by the ilPFC. Overall, these findings suggest that a loss of top-down prefrontal regulation via disruption of corticothalamic communication, as has been observed in schizophrenia, could contribute to hippocampal overdrive and, consequently, to the hyperdopaminergic state characteristic of the disorder (Figure 2).
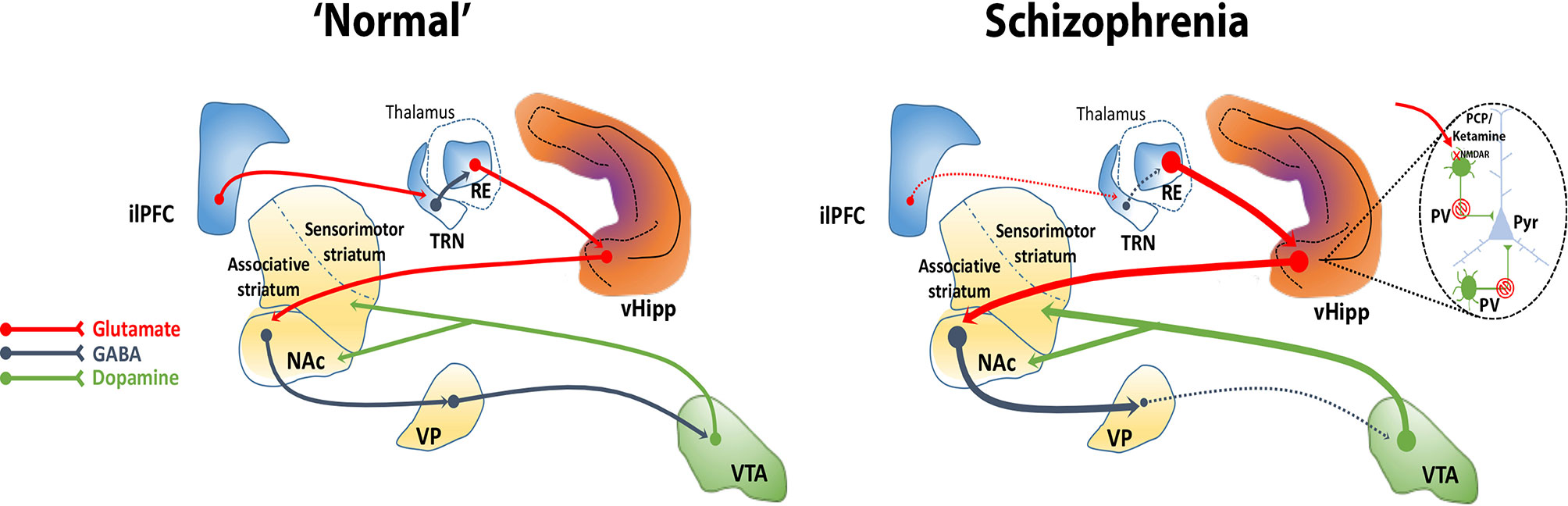
Figure 2 The ventral hippocampus (vHipp) regulates midbrain DA system activity through a polysynaptic circuit. The vHipp excites neurons in the nucleus accumbens (NAc) that, in turn, inhibit ventral pallidal (VP) activity. Given that the VP provides an inhibitory tone to VTA DA neurons, activation of the vHipp results in an enhance VTA DA neuron activity. In schizophrenia, a PV+ interneuron cell loss combined with a disruption of corticothalamic projections contributes to the hyperactivity of glutamatergic pyramidal (Pyr) neurons in the vHipp that drives an increase in active DA neurons projecting to the associative striatum that underlies the emergence of psychotic symptoms in schizophrenia. NMDA receptor antagonists, such as PCP and ketamine, may similarly exacerbate or mimic psychosis by blocking NMDA receptors on PV+ interneurons and thus disinhibiting Pyr neurons. Corticothalamic-hippocampal abnormal interactions can induce a hyperdopaminergic state, for instance, through a dysfunction of the medial prefrontal cortex (mPFC) that could disinhibit the nucleus reuniens (RE), possibly via loss of feedforward inhibition from the reticular nucleus of the thalamus (TRN).
Another thalamic nucleus recently implicated in the regulation of the VTA DA system is the paraventricular nucleus of the thalamus. It was observed that the pharmacological activation of the paraventricular nucleus of the thalamus enhanced VTA DA neuron population activity, which was completely prevented by the inactivation of either vHipp or NAc (120). Both the paraventricular nucleus of the thalamus and vHipp send extensive glutamatergic innervation to the NAc (121, 122). Interestingly, the inactivation of the paraventricular nucleus of the thalamus attenuated the increased VTA DA neuron population activity induced by the vHipp activation (120). Moreover, this regulation seems to simultaneously require activity in both the vHipp and paraventricular nucleus of the thalamus. The inactivation of the paraventricular nucleus of the thalamus reverses vHipp-induced increases in VTA DA neuron population activity. Similarly, vHipp inactivation reverses the paraventricular nucleus of the thalamus-induced increases (120). Together, these findings suggest that convergent glutamatergic inputs from the vHipp and paraventricular nucleus of the thalamus to the NAc work in concert to regulate VTA DA neuron activity. In addition, the inactivation of the paraventricular nucleus of the thalamus reverses the abnormal increase in VTA DA neuron population activity exhibited by MAM rats (120), similar to what is observed after the inactivation of the vHipp in MAM rats (81). These findings indicate that aberrant thalamic activity may contribute substantially to the hyperdopaminergic state seen in schizophrenia.
Another brain region that may influence vHipp activity and, in turn, regulate midbrain DA system activity is the medial septum. The medial septum sends dense cholinergic and GABAergic projections to several hippocampal regions (123, 124), including the vHipp (125). These projections are critical for hippocampal theta oscillation (126, 127), a major operational mode of the hippocampus, which is thought to be indicative of cognitive processing of environmental information (128).
The GABAergic projections from the medial septum synapse primarily on PV+ interneurons in the hippocampus (124, 126, 129), which is the interneuron subtype associated with the hippocampal hyperactivity and downstream hyperdopaminergic state present in schizophrenia (8, 42). On the other hand, the cholinergic projections provide slow depolarization of their target pyramidal neurons (126). Thus, the GABAergic and cholinergic projections from the medial septum can differently impact the excitatory-inhibitory balance in the vHipp which could ultimately lead to changes in the VTA DA system. In this context, our group recently found that pharmacological activation of the medial septum by a local infusion of NMDA increased the number of spontaneously active DA neurons in the VTA (130). An opposite effect was found in the substantia nigra. These effects induced by medial septum activation on both the VTA and substantia nigra depend on the vHipp since they were prevented by the inactivation of this brain region (130). Moreover, the effects of medial septum activation on VTA DA neuron population activity were also prevented by the infusion of the muscarinic receptor antagonist scopolamine into the vHipp, suggesting that medial septum cholinergic inputs to the vHipp may be involved in these effects (130). In addition, the inactivation of the anterior portion of the VP blocked the increased VTA DA neuron population activity induced by medial septum activation (130). On the other hand, inactivation of the posterior portion of the VP blocked the suppression of substantia nigra DA neuron population activity by medial septum activation. This suggests that there are topographically organized parallel circuits by which medial septum activity can bi-directionally affect DA neurons. Also, these findings indicate that medial septum seems to modulate midbrain DA system activity via the vHipp-NAc-VP pathway.
These opposite actions on VTA and substantia nigra DA neurons mediated by medial septum activation were recently associated with an enhancement of cognitive flexibility (131), a process profoundly attenuated in schizophrenia (132). The concept is that activation of the VTA causes the subject to think about the action, while attenuation of the substantia nigra prevents action until after weighing options. Interestingly, the regulation of the midbrain DA system activity by the medial septum in the MAM model of schizophrenia is different from that observed in normal rats (133). Whereas medial septum activation increases VTA DA neuron population activity and inhibits the substantia nigra in the normal rat (130, 133), an activation of the substantia nigra and a reduction of the abnormal increased VTA DA neuron population activity in MAM rats to baseline levels was observed (133). A possible explanation for these findings is that, in MAM rats, medial septum activation leads to an increase in the pyramidal neuron inhibition which would mitigate the vHipp hyperactivity (81). For example, the medial septum activation in normal rats leads to the release of GABA from the medial septum GABAergic projections to vHipp (123, 125). Since interneurons tend to be more sensitive to GABA than pyramidal neurons (134, 135), the released GABA would activate GABAA receptors on interneurons in the vHipp. This would inhibit interneurons, which in turn leads to the disinhibition of pyramidal neurons. On the other hand, in MAM rats, GABA released in the vHipp induced by the medial septum activation would be more likely to reach pyramidal neurons due to the loss of interneurons in the vHipp. These changes combined with the loss of cholinergic activation of a parallel set of GABAergic projections to vHipp pyramidal neurons that impact the substantia nigra (123, 124) would increase vHipp drive of the VTA while increasing vHipp inhibition of the substantia nigra. Therefore, in contrast to the control condition, in the MAM rats the excitation of the substantia nigra combined with inhibition of the VTA would cause the subject to act before thinking, or causing impulsive behavior (133). Overall, the findings observed in MAM rats indicate that the medial septum-vHipp pathway as a potential target to reverse the hyperdopaminergic state in schizophrenia patients.
A diathesis-stress model proposes that schizophrenia develops due to stress exposure acting on a pre-existing vulnerability (136). Indeed, a large body of work highlights the importance of stress as a risk factor in the development of schizophrenia (43, 137–139). Early life stress and chronic social stressors, in particular, have been shown to increase the risk of schizophrenia (140, 141). Acute stress can trigger psychotic symptoms (142) and impaired stress tolerance is associated with prodromal symptoms (143). The correlation between early life stress and severity of positive symptoms (144) may partially be due to the interaction between stress, the hippocampus, and the DA system (145, 146).
The vHipp, which is integral in regulating context-dependent responses (147, 148), also shows marked vulnerability to stress across many psychiatric conditions. This may in part be due to a high expression of glucocorticoid receptors to respond to activation of the hypothalamic-pituitary-adrenal (HPA) axis (149). While an elevation in glucocorticoids is essential to respond to perceived threat, chronic elevation can result in impaired function and hippocampal atrophy (150, 151). This would be exacerbated by stress-induced activation of amygdala-hippocampal glutamatergic projections that target PV+ interneurons (152). Prolonged stressors can lead to dendritic shrinkage and neuronal loss in the hippocampus (149), including a loss of PV+ interneurons (153). It has thus been hypothesized that vHipp dysfunction may contribute to the diathesis in prodromal patients that puts them at risk for developing psychosis in response to stress (154).
Both CHR individuals and schizophrenia patients demonstrate elevated DA release in response to stress compared to healthy controls (155, 156). In adult rats, prolonged stressors, such as restraint stress (157) or repeated footshock (158), increase DA neuron population activity and the level of DA in nucleus accumbens (159). The increase in DA neuron activity can be normalized by inhibiting the vHipp (157, 158). However, at later timepoints, there is a compensatory reduction in DA neuron population activity, referred to as an opponent process (160), and shown to be dependent on the BLA (146, 161). In contrast, during puberty, prolonged stress exposure in rats has been shown to result in a long lasting increase DA neuron activity in adulthood, suggesting that stress before or during puberty is particularly impactful to the responsivity of the DA system (162, 163).
Heightened stress responsivity, insufficient prefrontal inhibition activity in the amygdala (152, 164, 165), and general loss of corticothalamic communication, may contribute to vHipp dysfunction and the emergent hyperdopaminergic state. Extreme stress, or a failure of the PFC to mitigate the impact of stress, could lead to loss of PV+ interneurons in the hippocampus in late adolescence or early adulthood. This in turn would lead to hippocampal hyperactivity and DA system dysregulation. We have shown previously that peripubertal administration of the benzodiazepine diazepam, can prevent the increased anxiety-like behavior and BLA hyperactivity, and normalize hyperdopaminergic activity typically present in adult MAM rats (166–168). These studies suggest that increased stress responsivity, particularly at crucial developmental stages, could lead to the emergence of psychosis in adults and that decreasing stress or other means of reducing vHipp activity during peripubertal period has the potential to circumvent the pathological processes that leads to DA system dysregulation (8). Evidence from animal studies indicate that sex differences should be taken into account since female rodents appear to show greater resilience to schizophrenia-like traits resulting from developmental stress (169). These findings may be associated with the delayed onset and lesser severity of schizophrenia in females (170, 171).
The DA system has long been implicated in the expression and treatment of psychotic symptoms in schizophrenia. Study of the circuits that drive DA dysfunction can provide greater and more integrative understanding of a system-wide pathophysiology. Disruption of these circuits through developmental insults and pathological stressors can lead to DA system dysregulation. Ultimately, a greater understanding of the circuits that drive DA system dysfunction in schizophrenia can provide a neurobiological basis for interpreting clinical studies and potential targets for the treatment and prevention of schizophrenia and related psychotic disorders.
All authors contributed to the article and approved the submitted version.
Research activity of the authors is supported by grants from US National Institutes of Health (MH57440 to AG) and Sao Paulo Research Foundation (FAPESP Young Investigators Grant - 2018/17597-3 to FG).
AAG has received consulting fees from Alkermes, Lundbeck, Takeda, Roche, Lyra, Concert, and research funding from Lundbeck.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. (1997) 275(5306):1593–9. doi: 10.1126/science.275.5306.1593
2. Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Rev (1998) 28(3):309–69. doi: 10.1016/S0165-0173(98)00019-8
3. Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci (2004) 5(6):483–94. doi: 10.1038/nrn1406
4. Matsumoto M, Hikosaka O. Two types of dopamine neuron distinctly convey positive and negative motivational signals. Nature. (2009) 459(7248):837. doi: 10.1038/nature08028
5. Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry (2003) 160(1):13–23. doi: 10.1176/appi.ajp.160.1.13
6. Horga G, Abi-Dargham A. An integrative framework for perceptual disturbances in psychosis. Nat Rev Neurosci (2019) 20:763–78. doi: 10.1038/s41583-019-0234-1
7. McCutcheon RA, Abi-Dargham A, Howes OD. Schizophrenia, dopamine and the striatum: from biology to symptoms. Trends Neurosci (2019) 42(3):205–20. doi: 10.1016/j.tins.2018.12.004
8. Grace AA, Gomes FV. The Circuitry of Dopamine System Regulation and its Disruption in Schizophrenia: Insights Into Treatment and Prevention. Schizophr Bull (2019) 45(1):148–57. doi: 10.1093/schbul/sbx199
9. Emsley R, Rabinowitz J, Torreman M, Group R-I-EPGW. The factor structure for the Positive and Negative Syndrome Scale (PANSS) in recent-onset psychosis. Schizophr Res (2003) 61(1):47–57. doi: 10.1016/S0920-9964(02)00302-X
10. Kapur S, Remington G. Dopamine D 2 receptors and their role in atypical antipsychotic action: still necessary and may even be sufficient. Biol Psychiatry (2001) 50(11):873–83. doi: 10.1016/S0006-3223(01)01251-3
11. Leucht S, Cipriani A, Spineli L, Mavridis D, Örey D, Richter F, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet (2013) 382(9896):951–62. doi: 10.1016/S0140-6736(13)60733-3
12. Valenti O, Cifelli P, Gill KM, Grace AA. Antipsychotic drugs rapidly induce dopamine neuron depolarization block in a developmental rat model of schizophrenia. J Neurosci (2011) 31(34):12330–8. doi: 10.1523/JNEUROSCI.2808-11.2011
13. Sonnenschein SF, Gill KM, Grace AA. State-dependent effects of the D 2 partial agonist aripiprazole on dopamine neuron activity in the MAM neurodevelopmental model of schizophrenia. Neuropsychopharmacology (2019) 44(3):572. doi: 10.1038/s41386-018-0219-1
14. Grace AA, Bunney BS, Moore H, Todd CL. Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci (1997) 20(1):31–7. doi: 10.1016/S0166-2236(96)10064-3
15. Laruelle M, Abi-Dargham A, Van Dyck CH, Gil R, D'Souza CD, Erdos J, et al. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci (1996) 93(17):9235–40. doi: 10.1073/pnas.93.17.9235
16. Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, et al. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry (1998) 155(6):761–7. doi: 10.1176/ajp.155.6.761
17. Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry (1999) 46(1):56–72. doi: 10.1016/S0006-3223(99)00067-0
18. Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci (2000) 97(14):8104–9. doi: 10.1073/pnas.97.14.8104
19. Abi-Dargham A, van de Giessen E, Slifstein M, Kegeles LS, Laruelle M. Baseline and amphetamine-stimulated dopamine activity are related in drug-naive schizophrenic subjects. Biol Psychiatry (2009) 65(12):1091–3. doi: 10.1016/j.biopsych.2008.12.007
20. Demjaha A, Murray RM, McGuire PK, Kapur S, Howes OD. Dopamine synthesis capacity in patients with treatment-resistant schizophrenia. Am J Psychiatry (2012) 169(11):1203–10. doi: 10.1176/appi.ajp.2012.12010144
21. Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment: meta-analysis of imaging studies. Arch Gen Psychiatry (2012) 69(8):776–86. doi: 10.1001/archgenpsychiatry.2012.169
22. Howes OD, Bose SK, Turkheimer F, Valli I, Egerton A, Valmaggia LR, et al. Dopamine synthesis capacity before onset of psychosis: a prospective [18F]-DOPA PET imaging study. Am J Psychiatry (2011) 168(12):1311–7. doi: 10.1176/appi.ajp.2011.11010160
23. Howes OD, Montgomery AJ, Asselin M-C, Murray RM, Valli I, Tabraham P, et al. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry (2009) 66(1):13–20. doi: 10.1001/archgenpsychiatry.2008.514
24. Howes O, Bose S, Turkheimer F, Valli I, Egerton A, Stahl D, et al. Progressive increase in striatal dopamine synthesis capacity as patients develop psychosis: a PET study. Mol Psychiatry (2011) 16(9):885. doi: 10.1038/mp.2011.20
25. Egerton A, Chaddock CA, Winton-Brown TT, Bloomfield MA, Bhattacharyya S, Allen P, et al. Presynaptic striatal dopamine dysfunction in people at ultra-high risk for psychosis: findings in a second cohort. Biol Psychiatry (2013) 74(2):106–12. doi: 10.1016/j.biopsych.2012.11.017
26. Howes OD, Williams M, Ibrahim K, Leung G, Egerton A, McGuire PK, et al. Midbrain dopamine function in schizophrenia and depression: a post-mortem and positron emission tomographic imaging study. Brain (2013) 136(11):3242–51. doi: 10.1093/brain/awt264
27. McGowan S, Lawrence AD, Sales T, Quested D, Grasby P. Presynaptic dopaminergic dysfunction in schizophrenia: a positron emission tomographic [18F] fluorodopa study. Arch Gen Psychiatry (2004) 61(2):134–42. doi: 10.1001/archpsyc.61.2.134
28. Meyer-Lindenberg A, Miletich RS, Kohn PD, Esposito G, Carson RE, Quarantelli M, et al. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci (2002) 5(3):267. doi: 10.1038/nn804
29. Reith J, Benkelfat C, Sherwin A, Yasuhara Y, Kuwabara H, Andermann F, et al. Elevated dopa decarboxylase activity in living brain of patients with psychosis. Proc Natl Acad Sci (1994) 91(24):11651–4. doi: 10.1073/pnas.91.24.11651
30. Dao-Castellana M-H, Paillère-Martinot M-L, Hantraye P, Attar-Lévy D, Rémy P, Crouzel C, et al. Presynaptic dopaminergic function in the striatum of schizophrenic patients. Schizophr Res (1997) 23(2):167–74. doi: 10.1016/S0920-9964(96)00102-8
31. Elkashef AM, Doudet D, Bryant T, Cohen RM, Li S-H, Wyatt RJ. 6-18F-DOPA PET study in patients with schizophrenia. Psychiatry Res: Neuroimag (2000) 100(1):1–11. doi: 10.1016/S0925-4927(00)00064-0
32. Shotbolt P, Stokes P, Owens S, Toulopoulou T, Picchioni M, Bose S, et al. Striatal dopamine synthesis capacity in twins discordant for schizophrenia. psychol Med (2011) 41(11):2331–8. doi: 10.1017/S0033291711000341
33. Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, et al. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry (2010) 67(3):231–9. doi: 10.1001/archgenpsychiatry.2010.10
34. McCutcheon R, Beck K, Jauhar S, Howes OD. Defining the locus of dopaminergic dysfunction in schizophrenia: a meta-analysis and test of the mesolimbic hypothesis. Schizophr Bull (2017) 44(6):1301–11. doi: 10.1093/schbul/sbx180
35. Slifstein M, van de Giessen E, Van Snellenberg J, Thompson JL, Narendran R, Gil R, et al. Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry (2015) 72(4):316–24. doi: 10.1001/jamapsychiatry.2014.2414
36. Menegas W, Bergan JF, Ogawa SK, Isogai Y, Venkataraju KU, Osten P, et al. Dopamine neurons projecting to the posterior striatum form an anatomically distinct subclass. Elife (2015) 4:e10032. doi: 10.7554/eLife.10032
37. Sesack SR, Grace AA. Cortico-basal ganglia reward network: microcircuitry. Neuropsychopharmacology. (2010) 35(1):27. doi: 10.1038/npp.2009.93
38. Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev (2007) 56(1):27–78. doi: 10.1016/j.brainresrev.2007.05.004
39. Lerner TN, Shilyansky C, Davidson TJ, Evans KE, Beier KT, Zalocusky KA, et al. Intact-brain analyses reveal distinct information carried by SNc dopamine subcircuits. Cell (2015) 162(3):635–47. doi: 10.1016/j.cell.2015.07.014
40. Menegas W, Babayan BM, Uchida N, Watabe-Uchida M. Opposite initialization to novel cues in dopamine signaling in ventral and posterior striatum in mice. Elife. (2017) 6:e21886. doi: 10.7554/eLife.21886
41. Lynd-Balta E, Haber S. Primate striatonigral projections: a comparison of the sensorimotor-related striatum and the ventral striatum. J Comp Neurol (1994) 345(4):562–78. doi: 10.1002/cne.903450407
42. Grace AA. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci (2016) 17(8):524–32. doi: 10.1038/nrn.2016.57
43. Grace A, Bunney B. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons—1. Identification and characterization. Neuroscience. (1983) 10(2):301–15. doi: 10.1016/0306-4522(83)90135-5
44. Grace AA, Onn S-P. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci (1989) 9(10):3463–81. doi: 10.1523/JNEUROSCI.09-10-03463.1989
45. Grace A, Bunney B. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci: Off J Soc Neurosci (1984) 4(11):2866. doi: 10.1523/JNEUROSCI.04-11-02866.1984
46. Freeman AS, Bunney BS. Activity of A9 and A10 dopaminergic neurons in unrestrained rats: further characterization and effects of apomorphine and cholecystokinin. Brain Res (1987) 405(1):46–55. doi: 10.1016/0006-8993(87)90988-7
47. Freeman AS, Meltzer LT, Bunney BS. Firing properties of substantia nigra dopaminergic neurons in freely moving rats. Life Sci (1985) 36(20):1983–94. doi: 10.1016/0024-3205(85)90448-5
48. Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci (2003) 6(9):968. doi: 10.1038/nn1103
49. Grace AA, Bunney BS. Opposing effects of striatonigral feedback pathways on midbrain dopamine cell activity. Brain Res (1985) 333(2):271–84. doi: 10.1016/0006-8993(85)91581-1
50. Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci (1984) 4(11):2877–90. doi: 10.1523/JNEUROSCI.04-11-02877.1984
51. Lodge D, Grace A. The laterodorsal tegmentum is essential for burst firing of ventral tegmental area dopamine neurons. Proc Natl Acad Sci U S A (2006) 103(13):5167–72. doi: 10.1073/pnas.0510715103
52. Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature (1984) 309(5965):261–3. doi: 10.1038/309261a0
53. Schultz W. Dopamine reward prediction-error signalling: a two-component response. Nat Rev Neurosci (2016) 17(3):183. doi: 10.1038/nrn.2015.26
54. Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron. (2011) 70(5):855–62. doi: 10.1016/j.neuron.2011.03.025
55. Chergui K, Charlety PJ, Akaoka H, Saunier CF, Brunet JL, Buda M, et al. Tonic activation of NMDA receptors causes spontaneous burst discharge of rat midbrain dopamine neurons in vivo. Eur J Neurosci (1993) 5(2):137–44.
56. Tamminga CA, Stan AD, Wagner AD. The hippocampal formation in schizophrenia. Am J Psychiatry (2010) 167(10):1178–93. doi: 10.1176/appi.ajp.2010.09081187
57. Heilbronner SR, Rodriguez-Romaguera J, Quirk GJ, Groenewegen HJ, Haber SN. Circuit-based corticostriatal homologies between rat and primate. Biol Psychiatry (2016) 80(7):509–21. doi: 10.1016/j.biopsych.2016.05.012
58. Heckers S, Konradi C. Hippocampal Pathology in Schizophrenia. In: Swerdlow N editor. Behavioral Neurobiology of Schizophrenia and Its Treatment. Current Topics in Behavioral Neurosciences. Vol. 4. Berlin, Heidelberg: Springer (2010).
59. Poels EM, Kegeles LS, Kantrowitz JT, Javitt DC, Lieberman JA, Abi-Dargham A, et al. Glutamatergic abnormalities in schizophrenia: a review of proton MRS findings. Schizophr Res (2014) 152(2–3):325–32. doi: 10.1016/j.schres.2013.12.013
60. Gur RE, Mozley PD, Resnick SM, Mozley LH, Shtasel DL, Gallacher F, et al. Resting cerebral glucose metabolism in first-episode and previously treated patients with schizophrenia relates to clinical features. Arch Gen Psychiatry (1995) 52(8):657–67. doi: 10.1001/archpsyc.1995.03950200047013
61. Liddle P, Friston K, Frith C, Hirsch S, Jones T, Frackowiak R. Patterns of cerebral blood flow in schizophrenia. Br J Psychiatry (1992) 160(2):179–86. doi: 10.1192/bjp.160.2.179
62. Friston K, Liddle P, Frith C, Hirsch S, Frackowiak R. The left medial temporal region and schizophrenia: a PET study. Brain (1992) 115(2):367–82. doi: 10.1093/brain/115.2.367
63. Allen P, Azis M, Modinos G, Bossong MG, Bonoldi I, Samson C, et al. Increased resting hippocampal and basal ganglia perfusion in people at ultra high risk for psychosis: replication in a second cohort. Schizophr Bull (2017) 44(6):1323–31. doi: 10.1093/schbul/sbx169
64. Allen P, Chaddock CA, Egerton A, Howes OD, Bonoldi I, Zelaya F, et al. Resting hyperperfusion of the hippocampus, midbrain, and basal ganglia in people at high risk for psychosis. Am J Psychiatry (2015) 173(4):392–9. doi: 10.1176/appi.ajp.2015.15040485
65. Talati P, Rane S, Kose S, Blackford JU, Gore J, Donahue MJ, et al. Increased hippocampal CA1 cerebral blood volume in schizophrenia. NeuroImage: Clin (2014) 5:359–64. doi: 10.1016/j.nicl.2014.07.004
66. Harrison PJ. The hippocampus in schizophrenia: a review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology (2004) 174(1):151–62. doi: 10.1007/s00213-003-1761-y
67. Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, et al. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry (2009) 66(9):938–46. doi: 10.1001/archgenpsychiatry.2009.115
68. Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron (2013) 78(1):81–93. doi: 10.1016/j.neuron.2013.02.011
69. Benes FM. Emerging principles of altered neural circuitry in schizophrenia. Brain Res Rev (2000) 31(2–3):251–69. doi: 10.1016/S0165-0173(99)00041-7
70. Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology (2001) 25(1):1–27. doi: 10.1016/S0893-133X(01)00225-1
71. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci (2007) 27(43):11496–500. doi: 10.1523/JNEUROSCI.2213-07.2007
72. Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry (1994) 51(3):199–214. doi: 10.1001/archpsyc.1994.03950030035004
73. Braun I, Genius J, Grunze H, Bender A, Möller H-J, Rujescu D. Alterations of hippocampal and prefrontal GABAergic interneurons in an animal model of psychosis induced by NMDA receptor antagonism. Schizophr Res (2007) 97(1–3):254–63. doi: 10.1016/j.schres.2007.05.005
74. Rujescu D, Bender A, Keck M, Hartmann AM, Ohl F, Raeder H, et al. A pharmacological model for psychosis based on N-methyl-D-aspartate receptor hypofunction: molecular, cellular, functional and behavioral abnormalities. Biol Psychiatry (2006) 59(8):721–9. doi: 10.1016/j.biopsych.2005.08.029
75. Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci (1997) 17(8):2921–7. doi: 10.1523/JNEUROSCI.17-08-02921.1997
76. Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry (2006) 60(3):253–64. doi: 10.1016/j.biopsych.2006.01.003
77. Johnston M, Carman A, Coyle J. Effects of fetal treatment with methylazoxymethanol acetate at various gestational dates on the neurochemistry of the adult neocortex of the rat. J Neurochem (1981) 36(1):124–8. doi: 10.1111/j.1471-4159.1981.tb02386.x
78. Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophr Bull (2006) 32(2):200–2. doi: 10.1093/schbul/sbj052
79. Modinos G, Allen P, Grace AA, McGuire P. Translating the MAM model of psychosis to humans. Trends Neurosci (2015) 38(3):129–38. doi: 10.1016/j.tins.2014.12.005
80. Gomes FV, Rincon-Cortes M, Grace AA. Adolescence as a period of vulnerability and intervention in schizophrenia: Insights from the MAM model. Neurosci Biobehav Rev (2016) 70:260–70. doi: 10.1016/j.neubiorev.2016.05.030
81. Lodge DJ, Behrens MM, Grace AA. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci (2009) 29(8):2344–54. doi: 10.1523/JNEUROSCI.5419-08.2009
82. Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci (2007) 27(42):11424–30. doi: 10.1523/JNEUROSCI.2847-07.2007
83. McCutcheon RA, Abi-Dargham A, Howes OD. Schizophrenia, Dopamine and the Striatum: From Biology to Symptoms. Trends Neurosci (2019) 42(3):205–20. doi: 10.1016/j.tins.2018.12.004
84. Weinberger DR, Berman KF, Daniel DG. Mesoprefrontal cortical dopaminergic activity and prefrontal hypofunction in schizophrenia. Clin Neuropharmacol (1992) 15 Suppl 1 Pt A:568A–9A. doi: 10.1097/00002826-199201001-00296
85. Fusar-Poli P, Howes OD, Allen P, Broome M, Valli I, Asselin MC, et al. Abnormal prefrontal activation directly related to pre-synaptic striatal dopamine dysfunction in people at clinical high risk for psychosis. Mol Psychiatry (2011) 16(1):67–75. doi: 10.1038/mp.2009.108
86. Sesack SR, Carr DB. Selective prefrontal cortex inputs to dopamine cells: implications for schizophrenia. Physiol Behav (2002) 77(4–5):513–7. doi: 10.1016/S0031-9384(02)00931-9
87. Sesack SR, Deutch AY, Roth RH, Bunney BS. Topographical organization of the efferent projections of the medial prefrontal cortex in the rat: an anterograde tract-tracing study with Phaseolus vulgaris leucoagglutinin. J Comp Neurol (1989) 290(2):213–42. doi: 10.1002/cne.902900205
88. Patton MH, Bizup BT, Grace AA. The infralimbic cortex bidirectionally modulates mesolimbic dopamine neuron activity via distinct neural pathways. J Neurosci (2013) 33(43):16865–73. doi: 10.1523/JNEUROSCI.2449-13.2013
89. Vidal-Gonzalez I, Vidal-Gonzalez B, Rauch SL, Quirk GJ. Microstimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learn Mem (2006) 13(6):728–33. doi: 10.1101/lm.306106
90. Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. (2011) 36(2):529–38. doi: 10.1038/npp.2010.184
91. Chang CH, Grace AA. Amygdala-ventral pallidum pathway decreases dopamine activity after chronic mild stress in rats. Eur J Neurosci (2014) 76(3):223–30.
92. Vertes RP, Hoover WB, Do Valle AC, Sherman A, Rodriguez JJ. Efferent projections of reuniens and rhomboid nuclei of the thalamus in the rat. J Comp Neurol (2006) 499(5):768–96. doi: 10.1002/cne.21135
93. Zimmerman EC, Grace AA. The Nucleus Reuniens of the Midline Thalamus Gates Prefrontal-Hippocampal Modulation of Ventral Tegmental Area Dopamine Neuron Activity. J Neurosci (2016) 36(34):8977–84. doi: 10.1523/JNEUROSCI.1402-16.2016
94. Todd CL, Grace AA. Modulation of ventral tegmental area dopamine cell activity by the ventral subiculum and entorhinal cortex. Ann N Y Acad Sci (1999) 877:688–90. doi: 10.1111/j.1749-6632.1999.tb09302.x
95. van Groen T, Miettinen P, Kadish I. The entorhinal cortex of the mouse: organization of the projection to the hippocampal formation. Hippocampus (2003) 13(1):133–49. doi: 10.1002/hipo.10037
96. Wouterlood FG, Saldana E, Witter MP. Projection from the nucleus reuniens thalami to the hippocampal region: light and electron microscopic tracing study in the rat with the anterograde tracer Phaseolus vulgaris-leucoagglutinin. J Comp Neurol (1990) 296(2):179–203. doi: 10.1002/cne.902960202
97. McGhie A, Chapman J. Disorders of attention and perception in early schizophrenia. Br J Med Psychol (1961) 34:103–16. doi: 10.1111/j.2044-8341.1961.tb00936.x
98. Amaral DG, Cowan WM. Subcortical afferents to the hippocampal formation in the monkey. J Comp Neurol (1980) 189(4):573–91. doi: 10.1002/cne.901890402
99. Vertes RP, Linley SB, Hoover WB. Limbic circuitry of the midline thalamus. Neurosci Biobehav Rev (2015) 54:89–107. doi: 10.1016/j.neubiorev.2015.01.014
100. Mitchell AS, Chakraborty S. What does the mediodorsal thalamus do? Front Syst Neurosci (2013) 7:37. doi: 10.3389/fnsys.2013.00037
101. Woodward ND, Heckers S. Mapping Thalamocortical Functional Connectivity in Chronic and Early Stages of Psychotic Disorders. Biol Psychiatry (2016) 79(12):1016–25. doi: 10.1016/j.biopsych.2015.06.026
102. Samudra N, Ivleva EI, Hubbard NA, Rypma B, Sweeney JA, Clementz BA, et al. Alterations in hippocampal connectivity across the psychosis dimension. Psychiatry Res (2015) 233(2):148–57. doi: 10.1016/j.pscychresns.2015.06.004
103. Anticevic A, Cole MW, Repovs G, Murray JD, Brumbaugh MS, Winkler AM, et al. Characterizing thalamo-cortical disturbances in schizophrenia and bipolar illness. Cereb Cortex (2014) 24(12):3116–30. doi: 10.1093/cercor/bht165
104. Anticevic A, Haut K, Murray JD, Repovs G, Yang GJ, Diehl C, et al. Association of Thalamic Dysconnectivity and Conversion to Psychosis in Youth and Young Adults at Elevated Clinical Risk. JAMA Psychiatry (2015) 72(9):882–91. doi: 10.1001/jamapsychiatry.2015.0566
105. Rivolta D, Heidegger T, Scheller B, Sauer A, Schaum M, Birkner K, et al. Ketamine Dysregulates the Amplitude and Connectivity of High-Frequency Oscillations in Cortical-Subcortical Networks in Humans: Evidence From Resting-State Magnetoencephalography-Recordings. Schizophr Bull (2015) 41(5):1105–14. doi: 10.1093/schbul/sbv051
106. Ferrarelli F, Tononi G. The thalamic reticular nucleus and schizophrenia. Schizophr Bull (2011) 37(2):306–15. doi: 10.1093/schbul/sbq142
107. Ferrarelli F, Tononi G. Reduced sleep spindle activity point to a TRN-MD thalamus-PFC circuit dysfunction in schizophrenia. Schizophr Res (2017) 180:36–43. doi: 10.1016/j.schres.2016.05.023
108. Kupferschmidt DA, Gordon JA. The dynamics of disordered dialogue: Prefrontal, hippocampal and thalamic miscommunication underlying working memory deficits in schizophrenia. Brain Neurosci Adv (2018) 2:1–15. doi: 10.1177/2398212818771821
109. Lambert C, Simon H, Colman J, Barrick TR. Defining thalamic nuclei and topographic connectivity gradients in vivo. Neuroimage. (2017) 158:466–79. doi: 10.1016/j.neuroimage.2016.08.028
110. Reagh ZM, Murray EA, Yassa MA. Repetition reveals ups and downs of hippocampal, thalamic, and neocortical engagement during mnemonic decisions. Hippocampus (2017) 27(2):169–83. doi: 10.1002/hipo.22681
111. Xu W, Sudhof TC. A neural circuit for memory specificity and generalization. Science (2013) 339(6125):1290–5. doi: 10.1126/science.1229534
112. Griffin AL. Role of the thalamic nucleus reuniens in mediating interactions between the hippocampus and medial prefrontal cortex during spatial working memory. Front Syst Neurosci (2015) 9:29. doi: 10.3389/fnsys.2015.00029
113. Vetere G, Kenney JW, Tran LM, Xia F, Steadman PE, Parkinson J, et al. Chemogenetic Interrogation of a Brain-wide Fear Memory Network in Mice. Neuron (2017) 94(2):363–74 e4. doi: 10.1016/j.neuron.2017.03.037
114. McKenna JT, Vertes RP. Afferent projections to nucleus reuniens of the thalamus. J Comp Neurol (2004) 480(2):115–42. doi: 10.1002/cne.20342
115. Varela C, Kumar S, Yang JY, Wilson MA. Anatomical substrates for direct interactions between hippocampus, medial prefrontal cortex, and the thalamic nucleus reuniens. Brain Struct Funct (2014) 219(3):911–29. doi: 10.1007/s00429-013-0543-5
116. Cornwall J, Cooper JD, Phillipson OT. Projections to the rostral reticular thalamic nucleus in the rat. Exp Brain Res (1990) 80(1):157–71. doi: 10.1007/BF00228857
117. Kolmac CI, Mitrofanis J. Organisation of the reticular thalamic projection to the intralaminar and midline nuclei in rats. J Comp Neurol (1997) 377(2):165–78. doi: 10.1002/(SICI)1096-9861(19970113)377:2<165::AID-CNE2>3.0.CO;2-1
118. Bertram EH, Zhang DX. Thalamic excitation of hippocampal CA1 neurons: a comparison with the effects of CA3 stimulation. Neuroscience (1999) 92(1):15–26. doi: 10.1016/S0306-4522(98)00712-X
119. Zimmerman EC, Grace AA. Prefrontal cortex modulates firing pattern in the nucleus reuniens of the midline thalamus via distinct corticothalamic pathways. Eur J Neurosci (2018) 48(10):3255–72. doi: 10.1111/ejn.14111
120. Perez SM, Lodge DJ. Convergent Inputs from the Hippocampus and Thalamus to the Nucleus Accumbens Regulate Dopamine Neuron Activity. J Neurosci (2018) 38(50):10607–18. doi: 10.1523/JNEUROSCI.2629-16.2018
121. Su HS, Bentivoglio M. Thalamic midline cell populations projecting to the nucleus accumbens, amygdala, and hippocampus in the rat. J Comp Neurol (1990) 297(4):582–93. doi: 10.1002/cne.902970410
122. Dong X, Li S, Kirouac GJ. Collateralization of projections from the paraventricular nucleus of the thalamus to the nucleus accumbens, bed nucleus of the stria terminalis, and central nucleus of the amygdala. Brain Struct Funct (2017) 222(9):3927–43. doi: 10.1007/s00429-017-1445-8
123. Sotty F, Danik M, Manseau F, Laplante F, Quirion R, Williams S. Distinct electrophysiological properties of glutamatergic, cholinergic and GABAergic rat septohippocampal neurons: novel implications for hippocampal rhythmicity. J Physiol (2003) 551(Pt 3):927–43. doi: 10.1113/jphysiol.2003.046847
124. Dutar P, Bassant MH, Senut MC, Lamour Y. The septohippocampal pathway: structure and function of a central cholinergic system. Physiol Rev (1995) 75(2):393–427. doi: 10.1152/physrev.1995.75.2.393
125. Milner TA, Amaral DG. Evidence for a ventral septal projection to the hippocampal formation of the rat. Exp Brain Res (1984) 55(3):579–85. doi: 10.1007/BF00235290
126. Buzsaki G. Theta oscillations in the hippocampus. Neuron (2002) 33(3):325–40. doi: 10.1016/S0896-6273(02)00586-X
127. Yoder RM, Pang KC. Involvement of GABAergic and cholinergic medial septal neurons in hippocampal theta rhythm. Hippocampus (2005) 15(3):381–92. doi: 10.1002/hipo.20062
128. Buzsaki G, Moser EI. Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nat Neurosci (2013) 16(2):130–8. doi: 10.1038/nn.3304
129. Freund TF, Antal M. GABA-containing neurons in the septum control inhibitory interneurons in the hippocampus. Nature (1988) 336(6195):170–3. doi: 10.1038/336170a0
130. Bortz DM, Grace AA. Medial septum differentially regulates dopamine neuron activity in the rat ventral tegmental area and substantia nigra via distinct pathways. Neuropsychopharmacology (2018) 43(10):2093–100. doi: 10.1038/s41386-018-0048-2
131. Bortz DM, Gazo KL, Grace AA. The medial septum enhances reversal learning via opposing actions on ventral tegmental area and substantia nigra dopamine neurons. Neuropsychopharmacology (2019) 44(13):2186–94. doi: 10.1038/s41386-019-0453-1
132. Waltz JA. The neural underpinnings of cognitive flexibility and their disruption in psychotic illness. Neuroscience (2017) 345:203–17. doi: 10.1016/j.neuroscience.2016.06.005
133. Bortz DM, Grace AA. Medial septum activation produces opposite effects on dopamine neuron activity in the ventral tegmental area and substantia nigra in MAM vs. normal rats. NPJ Schizophr (2018) 4(1):17. doi: 10.1038/s41537-018-0059-3
134. Grace AA, Bunney BS. Paradoxical GABA excitation of nigral dopaminergic cells: indirect mediation through reticulata inhibitory neurons. Eur J Pharmacol (1979) 59(3-4):211–8. doi: 10.1016/0014-2999(79)90283-8
135. Xiang Z, Huguenard JR, Prince DA. GABAA receptor-mediated currents in interneurons and pyramidal cells of rat visual cortex. J Physiol (1998) 506( Pt 3):715–30. doi: 10.1111/j.1469-7793.1998.715bv.x
136. Thompson JL, Pogue-Geile MF, Grace AA. Developmental pathology, dopamine, and stress: a model for the age of onset of schizophrenia symptoms. Schizophr Bull (2004) 30(4):875–900. doi: 10.1093/oxfordjournals.schbul.a007139
137. Holtzman C, Trotman H, Goulding S, Ryan A, Macdonald A, Shapiro D, et al. Stress and neurodevelopmental processes in the emergence of psychosis. Neuroscience (2013) 249:172–91. doi: 10.1016/j.neuroscience.2012.12.017
138. van Os J, Kenis G, Rutten BP. The environment and schizophrenia. Nature. (2010) 468(7321):203. doi: 10.1038/nature09563
139. Gomes FV, Grace AA. Adolescent stress as a driving factor for schizophrenia development—a basic science perspective. Schizophr Bull (2017) 43(3):486–9. doi: 10.1093/schbul/sbx033
140. Varese F, Smeets F, Drukker M, Lieverse R, Lataster T, Viechtbauer W, et al. Childhood adversities increase the risk of psychosis: a meta-analysis of patient-control, prospective-and cross-sectional cohort studies. Schizophr Bull (2012) 38(4):661–71. doi: 10.1093/schbul/sbs050
141. McGrath J, Saha S, Welham J, El Saadi O, MacCauley C, Chant D. A systematic review of the incidence of schizophrenia: the distribution of rates and the influence of sex, urbanicity, migrant status and methodology. BMC Med (2004) 2(1):13. doi: 10.1186/1741-7015-2-13
142. Day R, Nielsen J, Korten A, Ernberg G, Dube K, Gebhart J, et al. Stressful life events preceding the acute onset of schizophrenia: a cross-national study from the World Health Organization. Cult Med Psychiatry (1987) 11(2):123–205. doi: 10.1007/BF00122563
143. Reininghaus U, Kempton MJ, Valmaggia L, Craig TK, Garety P, Onyejiaka A, et al. Stress sensitivity, aberrant salience, and threat anticipation in early psychosis: an experience sampling study. Schizophr Bull (2016) 42(3):712–22. doi: 10.1093/schbul/sbv190
144. Ruby E, Rothman K, Corcoran C, Goetz RR, Malaspina D. Influence of early trauma on features of schizophrenia. Early Int Psychiatry (2017) 11(4):322–33. doi: 10.1111/eip.12239
145. Howes OD, McCutcheon R, Owen MJ, Murray RM. The role of genes, stress, and dopamine in the development of schizophrenia. Biol Psychiatry (2017) 81(1):9–20. doi: 10.1016/j.biopsych.2016.07.014
146. Belujon P, Grace AA. Regulation of dopamine system responsivity and its adaptive and pathological response to stress. Proc R Soc London B: Biol Sci (2015) 282(1805):20142516. doi: 10.1098/rspb.2014.2516
147. Fanselow MS. Contextual fear, gestalt memories, and the hippocampus. Behav Brain Res (2000) 110(1–2):73–81. doi: 10.1016/S0166-4328(99)00186-2
148. Lisman J, Grace A. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron (2005) 46(5):703. doi: 10.1016/j.neuron.2005.05.002
149. Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry (2000) 57(10):925–35. doi: 10.1001/archpsyc.57.10.925
150. Sapolsky RM, Uno H, Rebert CS, Finch CE. Hippocampal damage associated with prolonged glucocorticoid exposure in primates. J Neurosci (1990) 10(9):2897–902. doi: 10.1523/JNEUROSCI.10-09-02897.1990
151. Sapolsky RM, Krey LC, McEWEN BS. Prolonged glucocorticoid exposure reduces hippocampal neuron number: implications for aging. J Neurosci (1985) 5(5):1222–7. doi: 10.1523/JNEUROSCI.05-05-01222.1985
152. Gomes FV, Zhu X, Grace AA. Stress during critical periods of development and risk for schizophrenia. Schizophr Res (2019) 213:107–13. doi: 10.1016/j.schres.2019.01.030
153. Czeh B, Simon M, Van Der Hart MG, Schmelting B, Hesselink MB, Fuchs E. Chronic stress decreases the number of parvalbumin-immunoreactive interneurons in the hippocampus: prevention by treatment with a substance P receptor (NK 1) antagonist. Neuropsychopharmacology (2005) 30(1):67–79. doi: 10.1038/sj.npp.1300581
154. Corcoran C, Walker E, Huot R, Mittal V, Tessner K, Kestler L, et al. The stress cascade and schizophrenia: etiology and onset. Schizophr Bull (2003) 29(4):671–92. doi: 10.1093/oxfordjournals.schbul.a007038
155. Pruessner JC, Champagne F, Meaney MJ, Dagher A. Dopamine release in response to a psychological stress in humans and its relationship to early life maternal care: a positron emission tomography study using [11C] raclopride. J Neurosci (2004) 24(11):2825–31. doi: 10.1523/JNEUROSCI.3422-03.2004
156. Mizrahi R, Addington J, Rusjan PM, Suridjan I, Ng A, Boileau I, et al. Increased stress-induced dopamine release in psychosis. Biol Psychiatry (2012) 71(6):561–7. doi: 10.1016/j.biopsych.2011.10.009
157. Valenti O, Lodge DJ, Grace AA. Aversive stimuli alter ventral tegmental area dopamine neuron activity via a common action in the ventral hippocampus. J Neurosci (2011) 31(11):4280–9. doi: 10.1523/JNEUROSCI.5310-10.2011
158. Valenti O, Gill KM, Grace AA. Different stressors produce excitation or inhibition of mesolimbic dopamine neuron activity: response alteration by stress pre-exposure. Eur J Neurosci (2012) 35(8):1312–21. doi: 10.1111/j.1460-9568.2012.08038.x
159. Kalivas PW, Duffy P. Selective activation of dopamine transmission in the shell of the nucleus accumbens by stress. Brain Res (1995) 675(1-2):325–8. doi: 10.1016/0006-8993(95)00013-G
160. Koob GF, Le Moal M. Neurobiological mechanisms for opponent motivational processes in addiction. Philos Trans R Soc B: Biol Sci (2008) 363(1507):3113–23. doi: 10.1098/rstb.2008.0094
161. Belujon P, Jakobowski NL, Dollish HK, Grace AA. Withdrawal from acute amphetamine induces an amygdala-driven attenuation of dopamine neuron activity: reversal by ketamine. Neuropsychopharmacology (2016) 41(2):619. doi: 10.1038/npp.2015.191
162. Gomes FV, Grace AA. Prefrontal cortex dysfunction increases susceptibility to schizophrenia-like changes induced by adolescent stress exposure. Schizophr Bull (2016) 43(3):592–600. doi: 10.1093/schbul/sbw156
163. Gomes FV, Zhu X, Grace AA. The pathophysiological impact of stress on the dopamine system is dependent on the state of the critical period of vulnerability. Mol Psychiatry (2019) 213:107–13. doi: 10.1038/s41380-019-0527-9
164. Rosenkranz JA, Grace AA. Dopamine attenuates prefrontal cortical suppression of sensory inputs to the basolateral amygdala of rats. J Neurosci (2001) 21(11):4090–103. doi: 10.1523/JNEUROSCI.21-11-04090.2001
165. Rosenkranz JA, Grace AA. Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo. J Neurosci (2002) 22(1):324–37. doi: 10.1523/JNEUROSCI.22-01-00324.2002
166. Du Y, Grace AA. Peripubertal diazepam administration prevents the emergence of dopamine system hyperresponsivity in the MAM developmental disruption model of schizophrenia. Neuropsychopharmacology (2013) 38(10):1881. doi: 10.1038/npp.2013.101
167. Du Y, Grace AA. Loss of parvalbumin in the hippocampus of MAM schizophrenia model rats is attenuated by peripubertal diazepam. Int J Neuropsychopharmacol (2016) 19(11):1–5. doi: 10.1093/ijnp/pyw065
168. Du Y, Grace AA. Amygdala hyperactivity in MAM model of schizophrenia is normalized by peripubertal diazepam administration. Neuropsychopharmacology. (2016) 41(10):2455–62. doi: 10.1038/npp.2016.42
169. Klinger K, Gomes FV, Rincon-Cortes M, Grace AA. Female rats are resistant to the long-lasting neurobehavioral changes induced by adolescent stress exposure. Eur Neuropsychopharmacol (2019) 29(10):1127–37. doi: 10.1016/j.euroneuro.2019.07.134
170. Seeman MV, Lang M. The role of estrogens in schizophrenia gender differences. Schizophr Bull (1990) 16(2):185–94. doi: 10.1093/schbul/16.2.185
Keywords: dopamine, ventral tegmental area, hippocampus, amygdala, thalamus, prefrontal cortex, medial septum
Citation: Sonnenschein SF, Gomes FV and Grace AA (2020) Dysregulation of Midbrain Dopamine System and the Pathophysiology of Schizophrenia. Front. Psychiatry 11:613. doi: 10.3389/fpsyt.2020.00613
Received: 19 April 2020; Accepted: 12 June 2020;
Published: 30 June 2020.
Edited by:
Carol A. Tamminga, University of Texas Southwestern Medical Center, United StatesReviewed by:
Didier Pinault, INSERM U1114 Neuropsychologie Cognitive et Physiophatologie de la Schizophrénie, FranceCopyright © 2020 Sonnenschein, Gomes and Grace. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony A. Grace, R3JhY2VBQUBwaXR0LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.