
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 26 February 2020
Sec. Schizophrenia
Volume 11 - 2020 | https://doi.org/10.3389/fpsyt.2020.00072
This article is part of the Research Topic The Role of Inflammation in the Etiology and Treatment of Schizophrenia View all 12 articles
 Adrianna P. Kępińska1
Adrianna P. Kępińska1 Conrad O. Iyegbe1
Conrad O. Iyegbe1 Anthony C. Vernon2,3
Anthony C. Vernon2,3 Robert Yolken4
Robert Yolken4 Robin M. Murray1
Robin M. Murray1 Thomas A. Pollak1*
Thomas A. Pollak1*Associations between influenza infection and psychosis have been reported since the eighteenth century, with acute “psychoses of influenza” documented during multiple pandemics. In the late 20th century, reports of a season-of-birth effect in schizophrenia were supported by large-scale ecological and sero-epidemiological studies suggesting that maternal influenza infection increases the risk of psychosis in offspring. We examine the evidence for the association between influenza infection and schizophrenia risk, before reviewing possible mechanisms via which this risk may be conferred. Maternal immune activation models implicate placental dysfunction, disruption of cytokine networks, and subsequent microglial activation as potentially important pathogenic processes. More recent neuroimmunological advances focusing on neuronal autoimmunity following infection provide the basis for a model of infection-induced psychosis, potentially implicating autoimmunity to schizophrenia-relevant protein targets including the N-methyl-D-aspartate receptor. Finally, we outline areas for future research and relevant experimental approaches and consider whether the current evidence provides a basis for the rational development of strategies to prevent schizophrenia.
Schizophrenia risk is associated with a variety of environmental and genetic factors (1), including those associated with immunity and inflammation (2). Genome-wide association studies (GWAS) implicate loci at the major histocompatibility complex (MHC) which encodes multiple genes involved in immunity such as the human leukocyte antigen (HLA) genes (3–6) and complement component 4 (C4) (7), among others, and enhancers related to B-lymphocyte lineages (CD19 and CD20 lines) involved in acquired/adaptive immunity (8). Overall there is strong evidence supporting the involvement of specific immune variants in schizophrenia risk (7), some evidence of convergence across genomics, transcriptomic, and methylomic processes (9), but conflicting evidence for both (i) enrichment of specific immune cell types or pathways (10) and (ii) for genetic overlap between SZ and specific immune diseases (11, 12).
Substantial epidemiological evidence exists suggesting that maternal, perinatal, childhood, and adult infection may all increase the risk of schizophrenia diagnosis (13–19). While many organisms and infection types have been implicated in schizophrenia risk, the influenza virus has special status: not only is maternal influenza infection the most well-replicated infective risk factor for schizophrenia, but the history of schizophrenia research has been shaped at crucial points by observations concerning the apparent, sometimes surprising, role of influenza as an exposure. This review aims to present the current state of knowledge on mechanisms by which influenza infection may confer schizophrenia risk, along with the implications of this understanding for future research, prevention, and treatment.
Before the focus of this review moves to schizophrenia and related psychotic disorders, it should be noted that some of the associations that will be discussed are now thought not to be specific to schizophrenia risk. The late winter/spring season of birth effect has also been reported in bipolar disorder (BD) (20), but the evidence for a link between BD and influenza is somewhat mixed (21) and addressed in limited studies (22). Influenza (including serologically documented infection) has been reported as a risk factor for BD with psychotic features but not nonpsychotic BD [reviewed in (22–24)]. Furthermore, some evidence suggests an association between maternal infection and autism spectrum disorders (ASD) [reviewed in (25)]. A substantial body of work from Scandinavian (largely Danish) health register studies supports the notion that clinically diagnosed maternal, childhood, or adulthood infection is a pluripotent risk factor for the subsequent development of psychiatric disorder, with effects observed across diagnostic boundaries (13–16, 26, 27). Therefore, while the focus of this review is on schizophrenia and psychosis, the potential transdiagnostic relevance of some of the mechanisms reviewed here should not be ignored.
Currently, influenza is regarded predominantly as a respiratory illness, but before the last century a far broader conceptualisation existed. As early as a 1732 epidemic, clinicians made note of the nervous sequelae of infection, with manifestations including neurasthenia, melancholy, hysteria, mental prostration, and insanity (28). According to the historian of medicine Mark Honigsbaum: “in the mid-1890s British medical journals were full of tales of Victorian professionals driven to the brink of madness and beyond by the nervous sequelae of influenza… for some 30 years, from the first epidemics of Russian influenza in the 1890s through to the ‘Spanish’ influenza of 1918–19, the ‘psychoses of influenza' were a widely recognised psychiatric phenomenon” (29). In 1919, Karl Menninger published a now-classic paper reporting the characteristics of 100 patients with mental disturbances associated with influenza infection admitted in a 3-month period to the Boston Psychopathic Hospital. Of 80 on whom full data were available, 16 were diagnosed with delirium, 25 with “dementia praecox,” 23 with “other psychoses,” and 16 were unclassified (30). Interestingly, two-thirds of the “dementia praecox” patients were reported to have fully recovered at 5-year follow up (31). A further historically important strand of evidence came from von Economo's (32) research on encephalitis lethargica (EL), a still poorly understood inflammatory CNS condition featuring psychotic and catatonic symptoms, which was broadly contemporaneous with and potentially aetiologically related to the 1918-1919 Spanish influenza pandemic. Table 1 provides an overview of historical influenza pandemics that have been linked to the occurrence of psychosis.
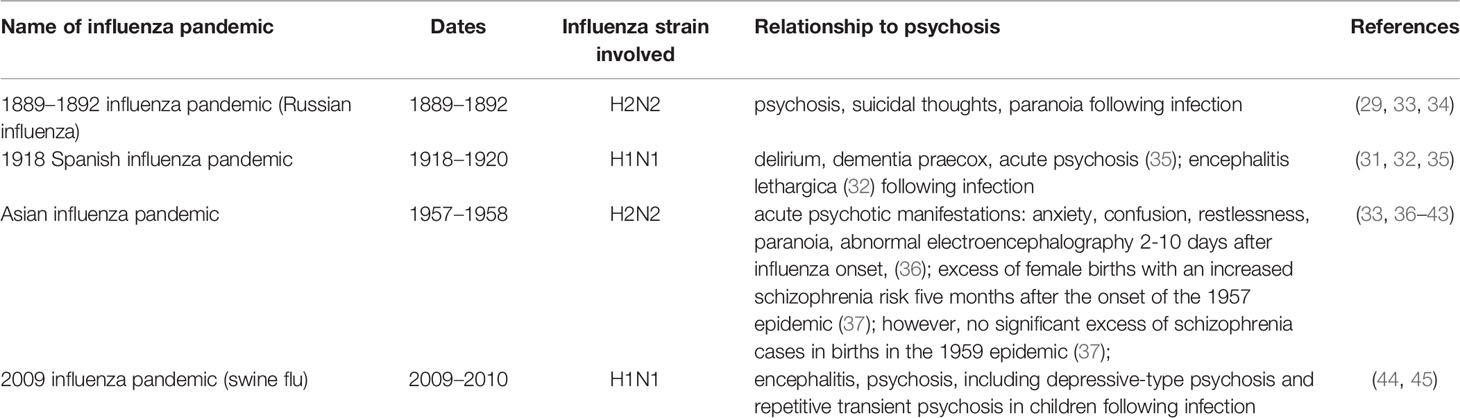
Table 1 Influenza pandemics and their relationships to psychosis.
While suggestive, these reports do not provide evidence of a causal link between influenza infection and psychotic disorders. Renewed interest in the second half of the 20th century shifted focus towards maternal infection, following consistent findings of an increased risk for schizophrenia in late winter/spring season births (20, 46), raising the possibility of winter-borne infection as a plausible mechanism. Beginning with Mednick et al.'s 1988 study of a Finnish population exposed to the 1957 influenza A2 pandemic, epidemiological studies in the 1980s–1990s, of an ecological nature, described increased risk for schizophrenia in children who were in utero during an influenza epidemic (37–41). These studies are comprehensively reviewed in (47). Frequently, rates were highest for second trimester exposures, although the first trimester appeared also to be a period of increased risk. Some subsequent studies however—often with more accurate case ascertainment and larger samples—were not able to replicate these initial findings [e.g. (48, 49)]. While estimates of risk varied greatly and heterogeneity in methodology somewhat limits generalisability, a 2010 review calculated that maternal influenza exposure increased schizophrenia risk with an odds ratio of 3.0 and a population attributable proportion of 14% (47).
Partly because of the manifold methodological problems involved in imputing precisely who was exposed to influenza, these ecological studies were followed by so-called “sero-epidemiological” studies, in which infection was verified using archived biological specimens: in one such early study first trimester maternal exposure was associated with a sevenfold increase in offspring schizophrenia risk, with threefold increase in risk associated with early-to-mid gestation exposure (50).
Other studies explored whether other viral and bacterial infections are associated with differential schizophrenia risk. A meta-analysis found that childhood viral infection was associated with a nearly twofold increased risk of adult nonaffective psychosis and that of all childhood infections, viral infections in particular, were associated with a nearly twofold increased risk of adult schizophrenia (18). However, bacterial infections were not associated with risk for psychosis, suggesting that risk may be specific for childhood viral infections.
Some controversy persists as to whether the evidence for maternal influenza as a schizophrenia risk factor is sufficient. A recent review of studies of schizophrenia risk in relation to the 1957 influenza pandemic criticised the serological studies for using strain-specific antibody titres that were too low to be specific for recent infection and so were insufficient as proxy measures of recent infection; furthermore, a pooled meta-analysis of eight ecological studies and one serological study found no overall increased risk of schizophrenia in children of influenza-exposed mothers (51). This review was in turn criticised as inappropriate given the heterogeneity of methods used in the pooled studies (52); furthermore it appeared to omit some relevant serological data [e.g. (39, 40)] and as it focuses on the 1957 pandemic only, it does not include studies on other strains of influenza infection and psychosis.
Complicating interpretation of ecological/epidemiological and serological studies is the fact that obstetric complications are more likely following influenza or influenza-like illness, and that obstetric complications are an independent risk factor for the subsequent development of psychotic disorders and/or symptoms (53–55).
The late 1980s and 1990s saw the emergence of neurodevelopmental theories offering mechanistic accounts of how schizophrenia develops. The neurodevelopmental hypothesis (56, 57) posits that schizophrenia results from a pathological disruption of normal brain development which commences many years before schizophrenia onset (58). Infection and other insults could disrupt developmental processes such as cell proliferation, cell migration, arborisation, and myelination (59) with resulting brain structural alterations [e.g. ventricular enlargement, grey matter reductions, and white matter disruption; (60)]; activation of pathologically developed brain systems in adolescence or young adulthood then manifests in schizophrenia symptoms (59).
Amongst other criticisms, the theory fails to account for later-onset schizophrenia [45 years or older; (61)] and postadolescence changes (62). Extended neurodevelopmental models posited further “hits,” e.g., genetic and environmental factors first predisposing to schizophrenia prenatally and then later in life [“three-hit” model, (63); multiple hit theory, (64)]. Infection is a possible “hit”; for instance, human endogenous retrovirus infections, activated by viruses including influenza, were suggested as late “hits” (64). This theory is consistent with evidence that maternal infection contributes to later increased offspring risk for childhood infections, which in turn contribute to schizophrenia development (65).
There is no clear evidence that genetic liability to schizophrenia increases the likelihood of influenza infection or predisposes to a disrupted immune response to influenza, or that influenza genetic risk loci are implicated in schizophrenia. In terms of genetic risk for influenza infection, while significant genetic effects accounting for the antibody level in influenza A and B (66, 67) have been reported with h2 (heritability) range of 0.20–0.27 and c2 (shared environnment) = 0.19 for influenza A and B (66), results for discrete serostatus (seropositive/seronegative) were significant for influenza B only. However, another GWAS of IgG response to viruses identified HLA class II residues as causal variants and found an overlap between variants affecting the humoral response to influenza A and variants linked to influenza-related autoimmune disorders including narcolepsy (68). Neither of these studies nor any others to date have directly addressed the issue of overlap between genetic risk for schizophrenia and specific risk for influenza infection, although this has been explored for other pathogens (69–71). A UK population-based cohort study of 7,921 mothers found no association between schizophrenia polygenic risk score (PRS) and perinatal infection (using a single “any infection” category) (72). Similarly, a case-control study by Benros et al. explored an association between schizophrenia PRS and a history of hospital contacts for viral infections, including influenza infection: PRS for schizophrenia did not account for the association between hospitalisation for infection and subsequent schizophrenia risk, indicating that schizophrenia risk does not increase proneness to such severe infections (73).
The influenza virus is an enveloped RNA virus from the family Orthomyxoviridae, with three genera, influenza A, B, and C (74). Given that influenza type A is responsible for pandemics (75) historically linked to schizophrenia and psychotic symptoms (see Table 1), we will focus on this alone. Influenza A viruses are classified into subtypes based on the antigenic properties of their envelope glycoproteins (see Figure 1), hemagglutinin, and neuraminidase. The viral envelope is a lipid membrane derived from plasma membrane of an infected host cell. Influenza strain targets also differ. Notably, the H5N1 virus and other avian-derived strains are neurotropic while H1N1 is thought not to be (76–78).
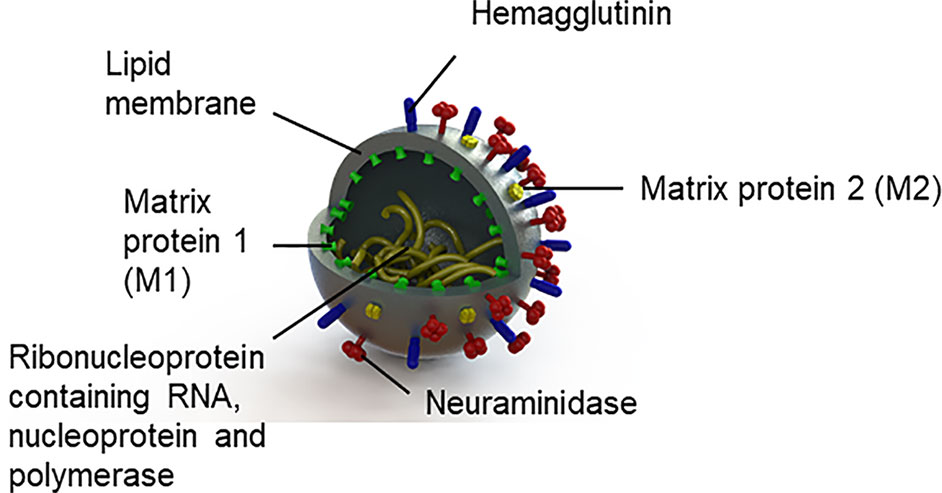
Figure 1 Structure of the influenza virus.
For infection to be successful, hemagglutinin binds the influenza virus to its receptors, sialyloligosaccharides, on the host cell surface. The viral envelope and the host cell membrane fuse giving the viral RNA access to the host cell (74, 79). Neuraminidase facilitates virus release (74, 75). Following the production of viral particles in the nucleus of the host cell, the host cell lyses and dies (75). Protective immune responses from the cell occur; the viral hemagglutinin, neuraminidase, and matrix 2 (M2) proteins are targeted by antibodies; matrix 1 (M1) proteins are targeted by T cells (80); and nucleoproteins are targeted by T cells (80) and nonneutralizing antibodies (81).
While maternal infection is reported to be a risk factor for schizophrenia, controversy remains concerning which biological processes underlie this risk. There is scant evidence for transplacental passage and persistence of the influenza virus in the offspring brain (82). More likely to be relevant are the effects of infection-induced maternal immune activation (MIA) on the developing brain (83).
MIA cannot be easily modelled in humans and longitudinal, prospective research on effects of infection during pregnancy on human development is scarce (84). Hence, translational animal models of MIA have been developed: these models have been critical in providing causality to the epidemiological data and are starting to provide clues as to the cellular and molecular mechanisms that may underlie the associations (83). Rodents in gestational periods are exposed either directly to a pathogen such as influenza, or more commonly to nonvirulent immune-activating agents such as the viral mimetic polyriboinosinic-polyribocytidilic acid (poly(I:C)) or the bacterial endotoxin lipopolysaccharide (LPS), the inflammatory agent turpentine, or specific proinflammatory cytokines (83, 85). These animal models of MIA provide evidence for behavioral, neurochemical, neuroanatomic, and neurophysiologic disruptions in the offspring which map onto endophenotypes observed across human psychiatric disorders with a neurodevelopmental onset (83, 86). Such translational research complements the insights from human epidemiology by establishing causal relationships, identifying cellular and molecular mechanisms and offering the potential to explore therapeutic interventions (85, 86). Frequently, these aforementioned deficits in the MIA model demonstrate a maturational delay, such that they are not evident before young adulthood, and many studies have sought to mitigate these deficits with treatments (e.g., pharmacological, immunological, behavioral) (83). Another important etiological advance for such animal models is to recapitulate a “two-hit” approach, in which pathology becomes evident in MIA-exposed offspring only after a second hit, such as unpredictable psychological stress (87).
MIA may lead straightforwardly to damage to the foetal brain during the early stages of neurodevelopment (82), but may also provide entry into a deviant trajectory of neural development which predisposes offspring to behavioral deficits depending on the intensity of the infection and when in gestation it occurs [early vs. late—and potentially as late as the lactation stage (88)]. MIA-associated abnormalities have been described, sometimes inconsistently, for multiple brain cell types, all of which are implicated across psychiatric disorders from postmortem data and genetic studies to a greater or lesser extent: Schwann cells (89), astrocytes and microglia (90, 91), hippocampal GABAergic cells (92, 93), dopaminergic neurons (94), and parvalbumin interneurons (95, 96).
Notably, most rodent (and primate) MIA models use a dose of poly(I:C) which models a high intensity, acute and transient (<24 h) infection, the physiological relevance of which could be questioned. Furthermore, factors such as the source, molecular weight, and endotoxin contamination of experimental poly(I:C) may be unrecognised sources of variability in foetal outcomes (97). Although MIA models specifically using pathogens as the immune activating stimulus have become rarer in recent years, primarily due to increasingly stringent safety frameworks around the use of potentially virulent pathogens (98), a series of MIA studies using human H1N1 influenza infection by the group of S. Hossein Fatemi are particularly valuable in elucidating potential mechanisms of psychosis risk. Broadly, maternal human H1N1 infection has been demonstrated to cause abnormalities, within the offspring, of the following [summarized in (99) and (59)]:
Gene Expression: the breadth of gene expression changes was greater the later in embryonic development infection occurred; for example embryonic day 16 or 18 (E16 or E18) infection disrupted more genes, across more brain regions, than did E7 infection. Furthermore, infection at later embryonic stages disrupted expression of genes involved in myelination and implicated in schizophrenia risk.
Protein Expression: increase in production of potentially harmful neuronal nitric oxide synthase (nNOS), reduction of reelin expression indicating abnormal neuronal migration and decreased synaptic plasticity, and downregulation of myelin basic protein.
Brain Structure: reduced cerebral cortical volume; increased total brain volume after early embryonic infection, decreased total brain volume after late infection.
Behavior: decreased prepulse inhibition (PPI), increased head twitch response.
Neurotransmitter Levels: reduced serotonin and taurine levels.
Placental Development: increased cytoarchitectural disorganisation, increased presence of immune cells, presence of variously sized thrombi, and dysregulation of placental gene expression.
Additional selected studies, focused on models of infection with influenza virus, are presented in Table 2.
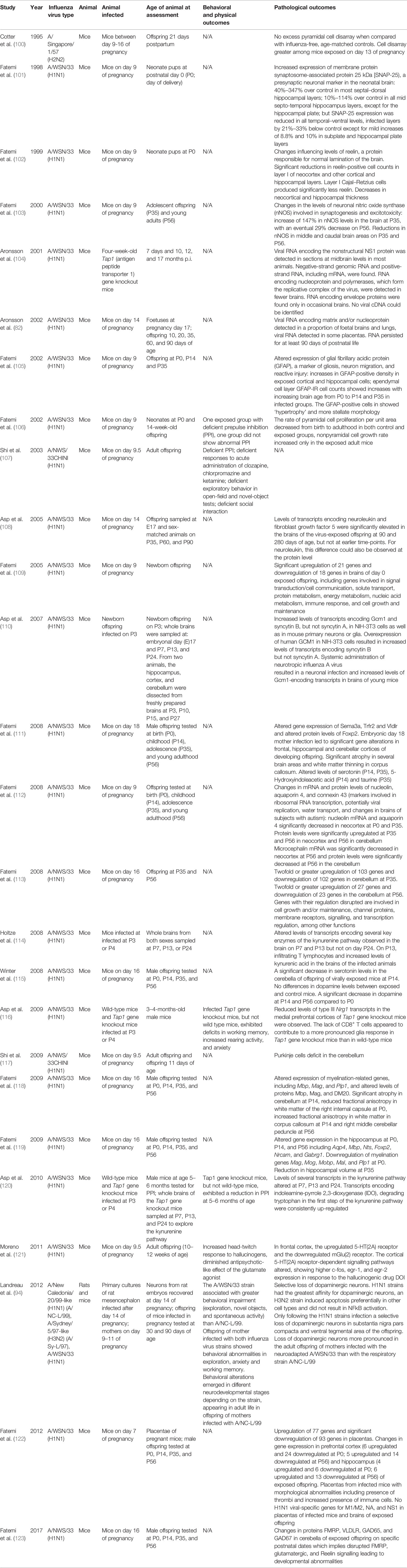
Table 2 Summary of selected behavioral and pathological outcomes following influenza infection in rodents.
Other animal studies have demonstrated associations between maternal influenza infection and schizophrenia-related neurotransmitter dysfunction including elevated serotonin 5-HT2A receptor expression in the frontal cortex (121), reductions of cerebellar serotonin levels at postpartum days (P) 14 and P35 (111, 115), downregulation of the metabotropic glutamate receptor 2 in the frontal cortex (121), and decrease in dopamine levels at P14 and P56 (115). Changes following poly(I:C) MIA exposure included subtle metabolic perturbations of postnatal prefrontal cortex maturation (124), and dynamic changes in volumes of multiple brain structures (125), including adult changes which can be prevented by periadolescent administration of antipsychotic medication (at nonantipsychotic dose equivalents) (126). Supporting the translational relevance of these studies, there is an emerging parallel literature in humans suggesting that early immune activation affects subsequent brain development and behavior: for example, maternal IL-6 levels during pregnancy predicted greater neonatal amygdala volumes and connectivity, which in turn predicted poorer impulse control at two years of age (127); complementary results for amygdala connectivity and internalizing behaviors have been reported for maternal cortisol levels (128).
An important mediator of the maternal immune response to infection is likely to be disruption of cytokines regulating brain development. Notably maternal infection could dysregulate cytokine networks either by direct transplacental transfer of cytokines to the foetus, by placental cytokine production or by increased foetal production of cytokines, including within the CNS (129). Cytokine dysregulation can result in perturbations of both proinflammatory and antiinflammatory cytokines. The deleterious or protective effects of any individual cytokine are likely determined by its context within a network of proinflammatory and antiinflammatory mediators, dynamically responding to external and endogenous challenges with differential expression in different brain regions over time (130). For example, macrophage-driven expression of antiinflammatory IL-10 in a mouse model can attenuate the long-term effects of prenatal viral infection, but in the absence of inflammatory stimulus, IL-10 itself precipitates offspring behavioral abnormalities (131).
Cytokines are induced in response to inflammation by neurons, astrocytes, and microglia, with the role of activated microglia in schizophrenia pathogenesis being the object of much recent attention (132). MIA exposure leads to alterations of the microglial transcriptome, with an initial shift to a more reactive state proximal to the MIA insult, followed by a delay in the maturation of brain microglia, as compared to controls (133). Alterations in the microglial transcriptome also lead to phagocytic function abnormalities and behavioral abnormalities in the adult MIA offspring (134). Results from the MIA literature are heterogenous with regards to microglial activation in offspring (91). Some studies report increased microglial density [e.g. (135)], morphology [e.g. (136)] or expression of activation markers [e.g. (137)], while other studies using late MIA have failed to demonstrate long-term changes in microglia density, morphology, or activation (138). Inference regarding the translational relevance of these findings, too, has been limited by a lack of clarity around the utility of the putative human markers of microglial activation such as translocator protein (TSPO), bound by ligands in Positron Emission Tomography (PET) studies, and of microglial markers used in post-mortem studies; notably, in addition to microglia, both astrocytes and vascular endothelial cells show dynamic changes in TSPO expression in response to inflammatory stimuli, and in a mouse model schizophrenia-relevant behavioral abnormalities and increased inflammatory cytokine expression were associated with reduced, rather than increased, prefrontal TSPO levels (139).
Both neurotropic and nonneurotropic influenza strains can cause microglial activation and potentially contribute to inflammation (76, 77). Innate immune training against influenza confers protection against infection with antiviral interferon-stimulated defence genes, including MXA (prevents nuclear import of the virus), IFITM3 and other IFITM proteins (block host-virus cell membrane fusion), and viperin [blocks influenza virus release; (140)]. Innate immune training also promotes disease tolerance of host tissues (140) and previous activation primes microglia to respond strongly to a new stimulus (141). Previous neuropathology potentially attunes microglia to respond more strongly to systemic inflammation (142), including inflammation by chronic mild stress in periadolesence following MIA (86). Consequently, infection could prime microglia towards heightened activation, potentially increasing the risk of developing psychotic symptoms (143); alternatively, the opposite could be true, i.e., that the microglia become tolerant and as such cannot respond flexibly to new stimuli.
In terms of potential downstream consequences of such foetal microglial activation, there is an emerging literature implicating the role of microglia in shaping brain development, including the potential for synaptic pruning via the full or partial engulfment [phago- or trogocytosis (144)] and putative degradation of synaptic inputs, a process mediated at least in the rodent visual thalamus in a complement and activity-dependent manner (145), which may also involve other molecular mediators such as TREM2 for example or the fractalkine receptor [CX3CR1; (146)] [potential mechanisms are reviewed in (147)]. Given the well-replicated finding of reduced dendritic spine number in schizophrenia (148), and evidence that patient-derived microglia-like cells are capable of synapse elimination at least in vitro (149), it is plausible also that maternal infection-induced foetal microglial activation could lead to later psychopathology through upregulation of synaptic pruning mechanisms (or conversely, loss of them: for example, early in development, microglia contact of neurons actually stimulates dendritic spine formation (150) suggesting the underexplored possibility that different pathologies play out in a potentially time- and region-specific manner).
Additionally, influenza infection may lead to placental abnormalities that result in hypoxia and/or nutritional deficiency or foetal brain growth restriction (122). Brain abnormalities may also stem from the maternal immune response whereby maternal autoantibodies are transported via the placenta and interact with foetal brain antigens to disrupt brain development [the teratogenic antibody hypothesis of schizophrenia; (151–153)]. The concept of infection-induced brain autoimmunity is explored in the next section.
Research on autoimmune disorders and schizophrenia dates to the 1950s, when schizophrenia was found to be protective against the development of rheumatoid arthritis (154, 155). Subsequently, the cooccurrence of schizophrenia with celiac disease was noted [e.g. (156)]. Recent meta-analysis suggests a positive association between nonneurological autoimmune disorders and psychosis (157). The risk for developing schizophrenia in people with autoimmune disorders was found to increase in association with increasing number of hospitalisations for infections (158), suggesting a synergistic effect. Drawing on the clinical observation that patients with autoantibody-mediated encephalitis frequently presented with psychosis (159), serological research has reported the presence of these same autoantibodies against cell surface neuronal antigens in some patients with schizophrenia [e.g. (160, 161)]. An extensive literature also focuses on markers of previous infection (usually IgG antibodies to specific pathogens, including influenza) in adults with psychotic disorders, with exposure to several organisms associated with increased schizophrenia risk or risk for a specific psychosis symptom profile (e.g. impaired cognition) (17, 162).
Autoimmune encephalitis (AE) frequently presents with acute psychosis in adults (163, 164). Autoantibodies to a variety of CNS cell surface antigens (neuronal surface autoantibodies; NSAbs) have been implicated in AE, including the NMDAR and more rarely LGI1, CASPR2, AMPAR, GABAAR, GABABR, D2R, DPPX, mGluR5, and GlyR (165–169). Of the autoimmune encephalitides associated with the above antigens, NMDAR encephalitis presents most frequently with psychosis. The typical pattern includes prodromal malaise, or influenza-like symptoms, before the emergence of psychiatric symptoms. 4% of patients show isolated psychotic episodes at presentation or relapse (170) and behavioral and cognitive impairments including psychosis are predominant early symptoms (171). The psychosis reported in anti-NMDAR encephalitis is distinctive, polymorphic (with significant affective elements) and does not correspond clearly to currently existing categories of psychotic disorder in mainstream psychiatric use (172). Anti-NMDAR encephalitis is caused by IgG antibodies directed against an epitope on the N-terminal domain of the NR1 subunit of the NMDA glutamate receptor (173, 174), with intrathecal antibody production by B lymphocyte descendants thought to be essential for pathogenesis.
Anti-NMDAR encephalitis is associated with ovarian teratoma in under a third of cases. An intriguing association between infection and the development of the disorder became apparent when it was observed that a number of patients experiencing “relapses” following herpes simplex virus (HSV) encephalitis had cerebrospinal fluid (CSF) NMDAR antibodies, suggesting that these “relapses” were in fact a postinfectious AE, rather than the result of reinfection or viral reactivation (175). Subsequent work has established that NMDAR antibody production can occur following HSV encephalitis even in the absence of clear “relapse” or encephalopathy (176), and that nonencephalitic HSV infection is also more common in patients with anti-NMDAR encephalitis (177). Other viral pathogens—including Epstein Barr Virus, Human Herpesvirus 6, cytomegalovirus, adenovirus and HIV—have been implicated in this and other autoimmune encephalitides [including those characterized by antibodies to the GABAA and GABAB receptors, the AMPA receptor and the dopamine D2 receptor; reviewed in (178)].
A potential association between anti-NMDAR encephalitis and influenza is supported by reports of at least five patients who developed the disorder following influenza vaccination (179–182)—although in none of these cases can causation be proven. A phylogenetic relationship has been suggested between microRNAs related to anti-NMDAR encephalitis and the H1N1 influenza virus, with some authors suggesting a theoretical basis for the possibility that anti-NMDAR encephalitis could be induced by influenza vaccination [(183); see also next section].
Anti-NMDAR encephalitis shares clinical features with EL in children, and indeed NMDAR antibodies have been reported in children with contemporary EL (184). When considered in the light of classic research on EL following the 1918-1919 Spanish influenza pandemic (32), this suggests a potential relationship between influenza infection, NSAbs and psychosis in anti-NMDAR encephalitis. [The association between influenza and EL is, however, highly controversial, not least because of temporal and geographical discrepancies between the start of the pandemic and the first recorded EL cases, as well as studies on post-mortem tissue which have frequently failed to find evidence of influenza virus; but given historical issues with case ascertainment and storage of biological samples potentially undermining efforts at viral detection, the association remains a plausible hypothesis for some authors (185)].
This interpretation is supported by findings that anti-NMDAR encephalitis may be seasonal, with a peak in incidence during winter (186), potentially converging with seasonality of influenza. Recent research found that Māori and Pacific Island populations have higher incidence and potentially more severe outcomes of anti-NMDAR encephalitis, a finding of significance given that population's apparent increased susceptibility to severe influenza infection (187, 188).
NMDAR antibodies are of interest in schizophrenia because of their links to the glutamate/NMDAR hypofunction hypothesis of psychotic disorders: NMDAR antibodies found in patients with schizophrenia can disrupt NMDAR dynamics in vivo (189, 190) providing prima facie support for the NMDAR hypofunction hypothesis. Crucially, Hammer et al. (191) reported the presence of influenza virus A or B IgG was significantly associated with NMDAR antibody seropositivity in a large cohort of adult patients with psychotic disorders and disease and healthy controls, a finding that was subsequently replicated in an independent cohort (192).
As described above, infection-induced neuronal autoimmunity may have relevance for some acute psychoses. However neuronal autoimmunity also has relevance in the context of maternal transmission. Maternal-foetal transfer of pathogenic antibodies has long been proposed as a potential mechanism in the development of ASD and, to a lesser extent, for schizophrenia also (153). Although not formally regarded as part of the MIA paradigm, recent animal models have had some successes in recapitulating neurodevelopmental phenotypes in immunisation paradigms whereby maternal antibodies are transferred to the offspring, resulting in neuropathological and behavioral abnormalities (193–195). Two of these studies used CASPR2 antibodies, cell surface IgG antibodies which have been implicated in encephalitis and a variety of peripheral nerve manifestations. Intriguingly, a study by Coutinho et al. found that NMDAR antibodies were more frequent in mothers of children with neurodevelopmental disorders, who themselves (i.e. the mothers) subsequently developed psychosis. This finding was not replicated in another cohort in which the mothers did not go on to develop psychosis, but clearly mandates attempts at replication (196). A recent animal study has shown that maternal-foetal transfer of recombinant NMDAR NR1 antibodies—at levels that did not affect the behavior of the pregnant mother—resulted in impaired neurodevelopmental reflexes, reduced anxiety, motor hyperactivity, and impaired sensorimotor gating, the latter two of which were regarded as psychosis-like phenotypes (197) (but see section “Experimental paradigms of maternal infection” for transdiagnostic relevance of these behaviors).
The association between influenza infection and NMDAR autoantibody status may have structural molecular basis. The influenza A M2 channel and NMDAR share a ligand, the antiviral compound amantadine (198), suggesting putative structural homology which could form the basis for NMDAR autoimmunity occurring after infection. In molecular mimicry, there is sharing of sequences, such as linear amino acid sequences, by molecules from dissimilar genes or their protein products. In infection, if the virus shares cross-reactive epitopes for B or T cells with the host, the host immune cells can target both the infecting agent and the host itself, potentially inducing autoimmune disease (199). The processes involved include Tc cells damaging self-tissue by lysis or Th cells releasing cytokines. Cytokines in turn activate macrophages or stimulate secretion of antibodies, and antibodies bind to cross-reactive epitopes on the surface of tissues, triggering further cytokine production by macrophages (200). Damaged tissues can also release new self-epitopes which activate autoantigen-reactive T and B cells, recognising those self-epitopes [epitope spreading; (201)].
There are multiple strands of evidence that influenza infection may have an aetiological role in systemic autoimmunity, including in Henoch-Schonlein purpura, type 1 diabetes mellitus and antiphospholipid syndrome [reviewed in (202)]. In one study, influenza vaccination induced autoimmunity (primarily antiphospholipid antibodies) in apparently healthy volunteers (203). H1N1 infection in rabbits has also been shown to induce brain-reactive antibodies, including to a 37kDa target also present in humans (204). Precedent for the role of influenza exposure initiating neurological disorder, potentially via molecular mimicry, exists for Guillain-Barre syndrome (205) and narcolepsy, in which hypocretin-producing neurons could be an autoimmune target due to molecular mimicry between H1N1 virus-derived antigen and a neuronal autoantigen in HLA-DQB1*06:02 positive patients (206, 207); see (208) for an example model of narcolepsy. This association is supported by epidemiological findings of an increased risk of narcolepsy in children following the H1N1 vaccination, Pandemrix (209, 210), and by serological findings that antibodies to influenza nucleoprotein might cross-react with hypocretin receptor 2 in patients with Pandemrix vaccination [(211), although see (212–214)].
Further evidence for molecular mimicry as a bridging link between influenza infection, the adaptive immune response and neurodevelopmental risk for schizophrenia comes from gene sequence overlap between the H5N1 virus and genes abnormally regulated in schizophrenia (215). Furthermore, the H1N1 influenza antiviral protein hemagglutinin was found to share peptide structure with a variety of human axon guidance proteins; the majority of proteins identified as containing homologous sequences are involved in processes which, if disrupted, could lead to deviant neurodevelopmental trajectories. The observed peptide matches were conserved across influenza strains and frequently involved experimentally validated hemagglutinin epitopes (216). Finally, the NMDAR 2A subunit was found to share peptides with several pathogens, including the influenza A virus (217). The findings suggest that anti-pathogen immune responses to the influenza A virus may cross-react with multiple schizophrenia-related proteins. This reaction could potentially trigger processes which may ultimately lead to schizophrenia. Work from our group has confirmed the higher-than-expected overlap between the influenza proteome and schizophrenia-relevant proteins, additionally identifying hemagglutinin as contributing, amongst influenza proteins, the most extensive peptide sharing [Kepinska et al., in submission; see also (218)].
Converging evidence demonstrates that infection with the influenza virus has a multiplicity of effects on prenatal and postnatal processes which, when disrupted, could result in increased risk of the development of schizophrenia or acute psychoses in adulthood. Figure 2 outlines potential prenatal and postnatal pathogenic contributions. Nonetheless, it is important to emphasise that infection has been linked with increased risk of several psychiatric disorders (see Introduction). It is therefore not clear to what extent the mechanisms discussed in this review are schizophrenia-specific, or whether, as is highly likely, other factors may shape the clinical expression of disease.
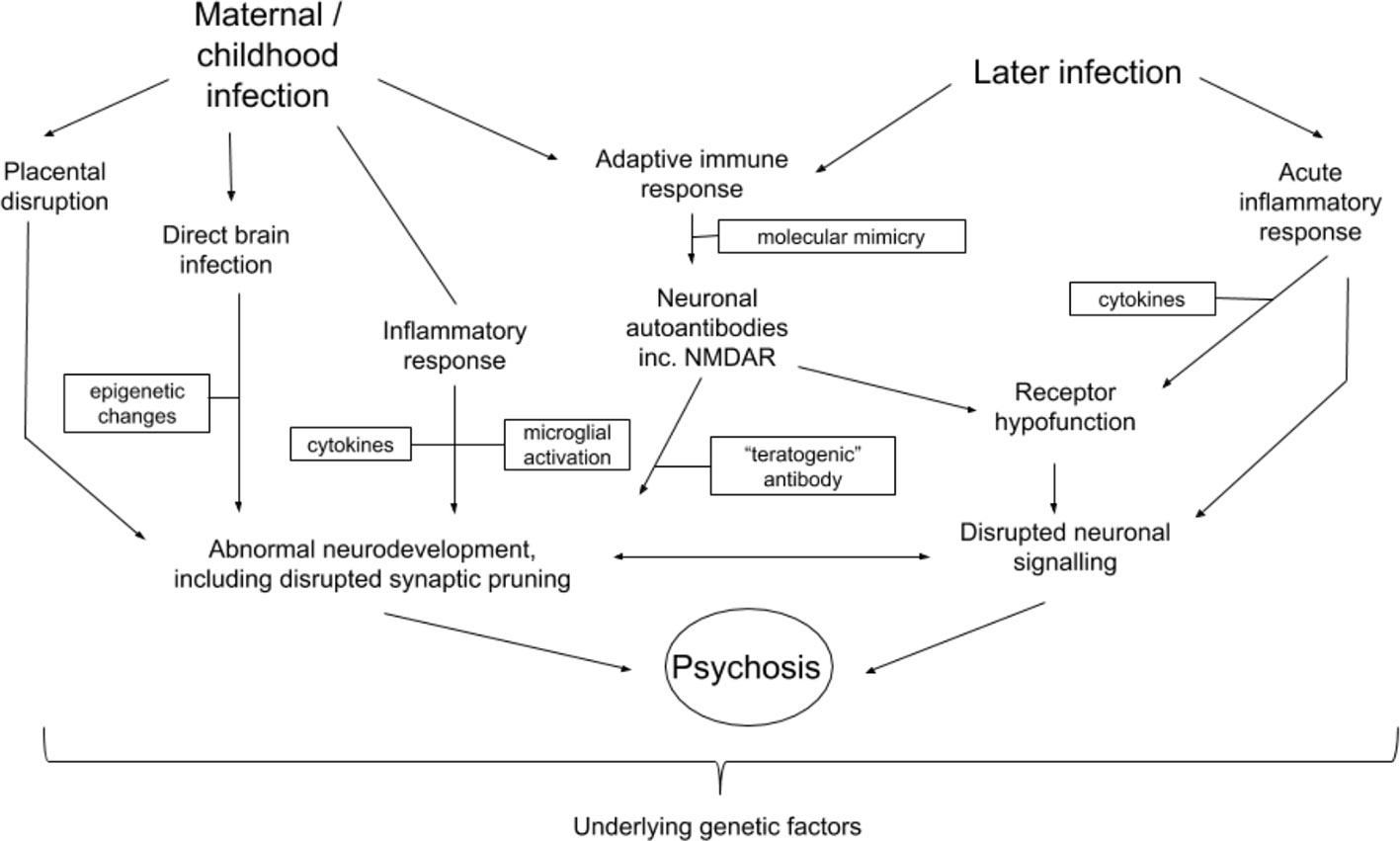
Figure 2 Potential interactions between mechanisms related to influenza infection and development of schizophrenia or other psychotic disorders. Arrows indicate possible directions of interaction. Boxes represent different factors or changes which mediate processes possibly leading to the development of psychosis or schizophrenia.
Outstanding questions and possible future experimental approaches are summarized in Box A.
Box A | Outstanding questions for future research.
- Do influenza MIA models, or models of adulthood influenza infection, demonstrate an antibody response to brain antigens including, for example, the NMDAR?
- Is there an epidemiological association between influenza vaccination (maternal, childhood or adulthood; seasonal or pandemic) with differential risk of subsequent development of psychotic disorder?
- Is antiviral use in pregnancy associated with a reduced risk of psychotic disorders in offspring?
- Is the acute response to influenza infection or vaccination in healthy individuals instructive for understanding the development of psychosis? Relevant approaches could include neuroimaging and behavioral testing following vaccination, similar to paradigms using LPS or typhoid vaccine administration (219–221).
- Will next generation viral metagenomic sequencing reveal differential presence of virus in biofluids from patients with psychotic disorders?
- Can an in silico approach be used to assess the plausibility of the molecular mimicry hypothesis, potentially assessing linear or structural overlap between viral proteome and schizophrenia-relevant proteins?
Future immunity-focused research on schizophrenia and influenza should further explore the relationship between infection and the innate and adaptive immune response in schizophrenia using animal models and large-scale serological studies in patients at different stages of disease. To date, MIA models typically include very little deep immunophenotyping, and discussion of the adaptive immune response in these models has been almost entirely lacking. Standardised and more sensitive testing technologies are required, including improved noninvasive methods to assess central neuroinflammation in humans and nonhuman animals (222, 223).Recent developments in stem cell technology suggest the possibility of using induced pluripotent stem-cell (iPSC) microglia-like cells [as per (149)] to assess how influenza infection affects the phenotype of these cells. Potentially, iPSC-derived cerebral organoids [so-called ‘mini brains’ (224)] could offer a window into the effects of influenza infection on relevant aspects of neurodevelopment.
While this paper reviews limited case studies and series indicating that in some instances influenza vaccination has been linked to CNS-directed autoimmunity, there is currently no evidence demonstrating a clear association between influenza vaccination and the development of schizophrenia or other psychotic disorders. The limited reported cases constitute a weight of evidence which is far weaker than the many epidemiological studies supporting the association between maternal influenza infection and schizophrenia. Influenza vaccination—both pandemic and seasonal—has saved and continues to save countless millions of lives worldwide, with an overwhelming evidence base supporting its efficacy. Within this context, influenza vaccination may nonetheless represent an as-yet underutilised opportunity for epidemiological and mechanistic explorations of potential influenza-psychosis associations. For example, healthy volunteers having the vaccination could be assessed using immunophenotyping, brain imaging, and behavioral measures to further characterize the acute response to influenza exposure [analogous to similar human studies of the acute response to LPS or typhoid vaccine administration (219–221)].
From the perspective of prevention of psychosis, consideration has been given to the potential use of antiviral medication in at-risk pregnant women. Although human studies are lacking, pilot studies in mice suggest that giving oseltamivir to pregnant mice can prevent some influenza-induced changes in the offspring (99). And while oseltamivir is regarded as having a favourable profile in pregnancy, there are no data on the long-term effects on neurodevelopment in human children.
Consideration has also been given to the potential role of influenza vaccines prior to, or during, pregnancy as a preventive measure to limit the prenatal teratogenic influence of viruses (225–227). The seasonal influenza vaccine has established efficacy in preventing maternal infection, as well as partially preventing the infant through passive immunity, and its administration remains best practice for protection of mother and child, with the World Health Organisation recognising pregnant women as a priority vulnerable group. In addition, educating pregnant women to contact their healthcare provide if they have a fever is recommended in order to expedite administration of antiviral medication and supportive care (228). Some authors consider the fact that influenza vaccination is not recommended in the first trimester in some countries as cause for concern, leaving women and the developing foetus vulnerable during a critical neurodevelopmental window (228). Epidemiologically, first trimester (or any other trimester) pandemic influenza vaccination does not appear to be associated with increased childhood morbidity (229). Although neurodevelopmental outcome data are largely lacking, some mouse models suggested that influenza vaccination early in pregnancy can indeed promote behavioral function and neurogenesis in the offspring, and confer protection from the effects of MIA with LPS (230, 231). One note of caution has been raised by a cohort study of nearly 200,000 children in California which reported a small but statistically significantly increased risk of ASD following first trimester vaccination (232); unsurprisingly the report was controversial, with ensuing disagreement concerning interpretation of the findings and whether the correct statistical measures were used (233, 234).
Given that the vast majority of children of mothers who experience an infection do not develop psychiatric disease, recent consideration has been given to maternal and foetal mechanisms of resilience to perinatal infection and inflammation: these include maternal nutritional status, the microbiome, and a variety of postnatal environmental factors (235). In terms of interventions within the MIA paradigm that have potential widespread relevance, dietary supplementation with omega-3 polyunsaturated fatty acids (PUFAs) may represent an attractive preventative strategy (236, 237).
An increase in our understanding of neuro-immune interactions has enabled a fuller understanding of the mechanistic underpinnings of the neurodevelopmental hypothesis of schizophrenia and have contributed to a more nuanced picture of schizophrenia pathogenesis which can accommodate the influence of influenza infections after the perinatal period. Our understanding of both influenza and schizophrenia has changed immensely since the 1918-1919 pandemic. The development of next-generation genetic, immunological and bioinformatic technologies may bring a resolution of the centuries-old puzzle of the relationship between influenza and psychosis.
TP, RM, CI and AK developed the initial idea for the review. AK and TP wrote the first draft of the manuscript. AK, CI, AV, RY, RM and TP contributed to revision and editing of the manuscript.
TP was supported in a Clinical Research Training Fellowship by the Wellcome Trust (grant number: 105758/Z/14/Z) and in a clinical lectureship by the National Institute for Health Research (NIHR). AK is funded by the NIHR Maudsley Biomedical Research Centre at South London and Maudsley Foundation Trust and King's College London. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. AV acknowledges financial support from the Medical Research Council (New Investigator Research Grant (NIRG), MR/N025377/1). The work (at King’s College, London) was also supported by the Medical Research Council (MRC) Centre grant (MR/N026063/1). AV has received research funding from F. Hoffman La Roche Ltd and UBC Biopharma SPRL as part of a research programme on early life immune activation. The views expressed are those of the authors and not necessarily those of F. Hoffman La Roche or UCB Biopharma SPRL. These funders had no influence on the decision to publish this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past collaboration with the authors TP and RY.
1. Owen MJ, Sawa A, Mortensen PB. Schizophrenia. Lancet (2016) 388(10039):86–97. doi: 10.1016/S0140-6736(15)01121-6
2. Khandaker GM, Cousins L, Deakin J, Lennox BR, Yolken R, Jones PB. Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry (2015) 2(3):258–70. doi: 10.1016/S2215-0366(14)00122-9
3. Corvin A, Morris DW. Genome-wide association studies: findings at the major histocompatibility complex locus in psychosis. Biol Psychiatry (2014) 75(4):276–83. doi: 10.1016/j.biopsych.2013.09.018
4. Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature (2009) 460(7256):748–52. doi: 10.1038/nature08185
5. McGuffin P, Power RA. Schizophrenia as a human leukocyte antigen-associated disease revisited. Am J Psychiatry (2013) 170(8):821–3. doi: 10.1176/appi.ajp.2013.13030336
6. Consortium, S.G.o.t.P.G. Biological insights from 108 schizophrenia-associated genetic loci. Nature (2014) 511(7510):421–7. doi: 10.1038/nature13595
7. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature (2016) 530(7589):177–83. doi: 10.1038/nature16549
8. Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature (2014) 511(7510):421–7. doi: 10.1038/nature13595
9. van Mierlo HC, Schot A, Boks MPM, de Witte ,LD. The association between schizophrenia and the immune system: review of the evidence from unbiased 'omic-studies'. Schizophr Res (2019) in press. doi: 10.1016/j.schres.2019.05.028
10. Network and Pathway Analysis Subgroup of Psychiatric Genomics, C. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci (2015) 18(2):199–209. doi: 10.1038/nn.3922
11. Pouget JG. The emerging immunogenetic architecture of schizophrenia. Schizophr Bull (2018) 44(5):993–1004. doi: 10.1093/schbul/sby038
12. Pouget JG, Schizophrenia Working Group of the Psychiatric Genomics Consortium, Han B, Wu Y, Mignot E, Ollila HM, et al. Cross-disorder analysis of schizophrenia and 19 immune-mediated diseases identifies shared genetic risk. Hum Mol Genet (2019) 28(20):3498–513. doi: 10.1093/hmg/ddz145
13. Benros ME, Mortensen PB, Eaton WW. Autoimmune diseases and infections as risk factors for schizophrenia. Ann N Y Acad Sci (2012) 1262:56–66. doi: 10.1111/j.1749-6632.2012.06638.x
14. Benros ME, Nielsen PR, Nordentoft M, Eaton WW, Dalton SO, Mortensen PB. Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30-year population-based register study. Am J Psychiatry (2011) 168(12):1303–10. doi: 10.1176/appi.ajp.2011.11030516
15. Kohler O, Petersen L, Mors O, Mortensen PB, Yolken RH, Gasse C, et al. Infections and exposure to anti-infective agents and the risk of severe mental disorders: a nationwide study. Acta Psychiatr Scand (2017) 135(2):97–105. doi: 10.1111/acps.12671
16. Lydholm CN, Kohler-Forsberg O, Nordentoft M, Yolken RH, Mortensen PB, Petersen L, et al. Parental infections before, during, and after pregnancy as risk factors for mental disorders in childhood and adolescence: a Nationwide danish study. Biol Psychiatry (2019) 85(4):317–25. doi: 10.1016/j.biopsych.2018.09.013
17. Yolken RH, Torrey EF. Are some cases of psychosis caused by microbial agents? a review of the evidence. Mol Psychiatry (2008) 13(5):470–9. doi: 10.1038/mp.2008.5
18. Khandaker GM, Zimbron J, Dalman C, Lewis G, Jones PB. Childhood infection and adult schizophrenia: a meta-analysis of population-based studies. Schizophr Res (2012) 139(1-3):161–8. doi: 10.1016/j.schres.2012.05.023
19. Khandaker GM, Zimbron J, Lewis G, Jones PB. Prenatal maternal infection, neurodevelopment and adult schizophrenia: a systematic review of population-based studies. Psychol Med (2013) 43(2):239–57. doi: 10.1017/S0033291712000736
20. Torrey EF, Miller J, Rawlings R, Yolken RH. Seasonality of births in schizophrenia and bipolar disorder: a review of the literature. Schizophr Res (1997) 28(1):1–38. doi: 10.1016/S0920-9964(97)00092-3
21. Barichello T, Badawy M, Pitcher MR, Saigal P, Generoso JS, Goularte JA, et al. Exposure to perinatal infections and bipolar disorder: a systematic review. Curr Mol Med (2016) 16(2):106–18. doi: 10.2174/1566524016666160126143741
22. Marangoni C, Hernandez M, Faedda GL. The role of environmental exposures as risk factors for bipolar disorder: a systematic review of longitudinal studies. J Affect Disord (2016) 193:165–74. doi: 10.1016/j.jad.2015.12.055
23. Brown AS. The Kraepelinian dichotomy from the perspective of prenatal infectious and immunologic insults. Schizophr Bull (2015) 41(4):786–91. doi: 10.1093/schbul/sbv063
24. Canetta SE, Bao Y, Co MD, Ennis FA, Cruz J, Terajima M, et al. Serological documentation of maternal influenza exposure and bipolar disorder in adult offspring. Am J Psychiatry (2014) 171(5):557–63. doi: 10.1176/appi.ajp.2013.13070943
25. Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol (2012) 72(10):1272–6. doi: 10.1002/dneu.22024
26. Kohler-Forsberg O, Petersen L, Gasse C, Mortensen PB, Dalsgaard S, Yolken RH, et al. A nationwide study in Denmark of the association between treated infections and the subsequent risk of treated mental disorders in children and adolescents. JAMA Psychiatry (2019) 76(3):271–9. doi: 10.1001/jamapsychiatry.2018.3428
27. Benros ME, Waltoft BL, Nordentoft M, Ostergaard SD, Eaton WW, Krogh J, et al. Autoimmune diseases and severe infections as risk factors for mood disorders: a nationwide study. JAMA Psychiatry (2013) 70(8):812–20. doi: 10.1001/jamapsychiatry.2013.1111
28. Honigsbaum M. A History of the Great Influenza Pandemics: Death, Panic and Hysteria. London: I.B. Tauris (2014) p. 1830–920.
29. Honigsbaum M. "An inexpressible dread": psychoses of influenza at fin-de-siecle. Lancet (2013) 381(9871):988–9. doi: 10.1016/S0140-6736(13)60701-1
30. Menninger KA. Psychoses associated with influenza: i. general data: statistical analysis. JAMA (1919) 72(4):235–41. doi: 10.1001/jama.1919.02610040001001
31. Menninger KA. Influenza and schizophrenia. An analysis of post-influenzal "dementia precox," as of 1918, and five years later further studies of the psychiatric aspects of influenza 1926. Am J Psychiatry (1994) 151(6 Suppl):182–7. doi: 10.1176/ajp.151.6.182
32. von Economo C. Encephalitis lethargica: its sequelae and treatment. Oxford, England: Oxford Univ. Press (1931). xvi, 200.
33. Hilleman MR. Realities and enigmas of human viral influenza: pathogenesis, epidemiology and control. Vaccine (2002) 20(25-26):3068–87. doi: 10.1016/S0264-410X(02)00254-2
34. Kraepelin E. Ueber psychosen nach influenza. Dtsch Med Wochenschr (1890) 16(11):209–12. doi: 10.1055/s-0029-1207083
35. Menninger KA. Psychoses associated with influenza - I - general data statistical analysis. J Am Med Assoc (1919) 72:235–41. doi: 10.1001/jama.1919.02610040001001
36. Bental E. Acute psychoses due to encephalitis following Asian influenza. Lancet (1958) 2(Jul5):18–20. doi: 10.1016/S0140-6736(58)90005-9
37. McGrath JJ, Pemberton MR, Welham JL, Murray RM. Schizophrenia and the influenza epidemics of 1954, 1957 and 1959 - a southern-hemisphere study. Schizophr Res (1994) 14(1):1–8. doi: 10.1016/0920-9964(94)90002-7
38. Mednick SA, Machon RA, Huttunen MO, Bonett D. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry (1988) 45(2):189–92. doi: 10.1001/archpsyc.1988.01800260109013
39. Kunugi H, Nanko S, Takei N, Saito K, Hayashi N, Kazamatsuri H. Schizophrenia following in utero exposure to the 1957 influenza epidemics in Japan. Am J Psychiatry (1995) 152(3):450–2. doi: 10.1176/ajp.152.3.450
40. Adams W, Kendell RE, Hare EH, Munk-Jorgensen P. Epidemiological evidence that maternal influenza contributes to the aetiology of schizophrenia. an analysis of Scottish, English, and Danish data. Br J Psychiatry (1993) 163:522–34. doi: 10.1192/bjp.163.4.522
41. O'Callaghan E, Sham P, Takei N, Murray RM, Glover G. Schizophrenia after prenatal exposure to 1957 A2 influenza epidemic. Lancet (1991) 337(8752):1248–50. doi: 10.1016/0140-6736(91)92919-S
42. Still RM. Psychosis following Asian influenza in Barbados. Lancet (1958) 2(7036):20–1. doi: 10.1016/S0140-6736(58)90006-0
43. Selten JP, Frissen A, Lensvelt-Mulders G, Morgan VA. Schizophrenia and 1957 pandemic of influenza: meta-analysis. Schizophr Bull (2010) 36(2):219–28. doi: 10.1093/schbul/sbp147
44. Khandaker G, Zurynski Y, Buttery J, Marshall H, Richmond PC, Dale RC, et al. Neurologic complications of influenza A(H1N1)pdm09: surveillance in 6 pediatric hospitals. Neurology (2012) 79(14):1474–81. doi: 10.1212/WNL.0b013e31826d5ea7
45. Chang CH, Chang YC, Tzang RF. Childhood psychosis after H1N1 influenza. J Neuropsychiatry Clin Neurosci (2015) 27(1):e87–9. doi: 10.1176/appi.neuropsych.14010012
46. Davies G, Welham J, Chant D, Torrey EF, McGrath J. A systematic review and meta-analysis of Northern Hemisphere season of birth studies in schizophrenia. Schizophr Bull (2003) 29(3):587–93. doi: 10.1093/oxfordjournals.schbul.a007030
47. Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry (2010) 167(3):261–80. doi: 10.1176/appi.ajp.2009.09030361
48. Erlenmeyer-Kimling L, Folnegovic Z, Hrabak-Zerjavic V, Borcic B, Folnegovic-Smalc V, Susser E. Schizophrenia and prenatal exposure to the 1957 A2 influenza epidemic in Croatia. Am J Psychiatry (1994) 151(10):1496–8. doi: 10.1176/ajp.151.10.1496
49. Susser E, Lin SP, Brown AS, Lumey LH, Erlenmeyer-Kimling L. No relation between risk of schizophrenia and prenatal exposure to influenza in Holland. Am J Psychiatry (1994) 151(6):922–4. doi: 10.1176/ajp.151.6.922
50. Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, et al. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry (2004) 61(8):774–80. doi: 10.1001/archpsyc.61.8.774
51. Selten JP, Termorshuizen F. The serological evidence for maternal influenza as risk factor for psychosis in offspring is insufficient: critical review and meta-analysis. Schizophr Res (2017) 183:2–9. doi: 10.1016/j.schres.2016.11.006
52. Brown AS, Meyer U. Comment on: “The serological evidence for maternal influenza as risk factor for psychosis in offspring is insufficient: critical review and meta-analysis”. Schizophr Res (2017) 189:223–4. doi: 10.1016/j.schres.2017.02.023
53. Wright P, Takei N, Rifkin L, Murray RM. Maternal influenza, obstetric complications, and schizophrenia. Am J Psychiatry (1995) 152(12):1714–20. doi: 10.1176/ajp.152.12.1714
54. Hultman CM, Sparen P, Takei N, Murray RM, Cnattingius S. Prenatal and perinatal risk factors for schizophrenia, affective psychosis, and reactive psychosis of early onset: case-control study. BMJ (1999) 318(7181):421–6. doi: 10.1136/bmj.318.7181.421
55. Dreier JW, Berg-Beckhoff G, Andersen AMN, Susser E, Nordentoft M, Strandberg-Larsen K. Fever and infections during pregnancy and psychosis-like experiences in the offspring at age 11. A prospective study within the Danish National Birth Cohort. Psychol Med (2018) 48(3):426–36. doi: 10.1017/S0033291717001805
56. Murray RM, Lewis SW. Is schizophrenia a neurodevelopmental disorder? Br Med J (Clin Res ed.) (1987) 295(6600):681–2. doi: 10.1136/bmj.295.6600.681
57. Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry (1987) 44(7):660–9. doi: 10.1001/archpsyc.1987.01800190080012
58. Fatemi SH, Folsom TD. The Neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr Bull (2009) 35(3):528–48. doi: 10.1093/schbul/sbn187
59. Rapoport JL, Giedd JN, Gogtay N. Neurodevelopmental model of schizophrenia: update 2012. Mol Psychiatry (2012) 17(12):1228–38. doi: 10.1038/mp.2012.23
60. Howes OD, Murray RM. Schizophrenia: an integrated sociodevelopmental-cognitive model. Lancet (2014) 383(9929):1677–87. doi: 10.1016/S0140-6736(13)62036-X
61. Folsom DP, Lebowitz BD, Lindamer LA, Palmer BW, Patterson TL, Jeste DV. Schizophrenia in late life: emerging issues. Dialog Clin Neurosci (2006) 8(1):45–52.
62. Kochunov P, Hong LE. Neurodevelopmental and neurodegenerative models of schizophrenia: white matter at the center stage. Schizophr Bull (2014) 40(4):721–8. doi: 10.1093/schbul/sbu070
63. Keshavan MS. Development, disease and degeneration in schizophrenia: a unitary pathophysiological model. J Psychiatr Res (1999) 33(6):513–21. doi: 10.1016/S0022-3956(99)00033-3
64. Davis J, Eyre H, Jacka FN, Dodd S, Dean O, McEwen S, et al. A review of vulnerability and risks for schizophrenia: Beyond the two hit hypothesis. Neurosci Biobehav Rev (2016) 65:185–94. doi: 10.1016/j.neubiorev.2016.03.017
65. Blomström Å., Karlsson H, Gardner R, Jorgensen L, Magnusson C, Dalman C. Associations between maternal infection during pregnancy, childhood infections, and the risk of subsequent psychotic disorder–a Swedish cohort study of nearly 2 million individuals. Schizophr Bull (2016) 42(1):125–33. doi: 10.1093/schbul/sbv112
66. Rubicz R, Leach CT, Kraig E, Dhurandhar NV, Duggirala R, Blangero J, et al. Genetic factors influence serological measures of common infections. Hum Hered (2011) 72(2):133–41. doi: 10.1159/000331220
67. Rubicz R, Yolken R, Drigalenko E, Carless MA, Dyer TD, Kent J Jr., et al. Genome-wide genetic investigation of serological measures of common infections. Eur J Hum Genet (2015) 23(11):1544–8. doi: 10.1038/ejhg.2015.24
68. Hammer C, Begemann M, McLaren PJ, Bartha I, Michel A, Klose B, et al. Amino acid variation in HLA Class II proteins is a major determinant of humoral response to common viruses. Am J Hum Genet (2015) 97(5):738–43. doi: 10.1016/j.ajhg.2015.09.008
69. Avramopoulos D, Pearce BD, McGrath J, Wolyniec P, Wang R, Eckart N, et al. Infection and inflammation in schizophrenia and bipolar disorder: a genome wide study for interactions with genetic variation. PloS One (2015) 10(3):e0116696. doi: 10.1371/journal.pone.0116696
70. Wang AW, Avramopoulos D, Lori A, Mulle J, Conneely K, Powers A, et al. Genome-wide association study in two populations to determine genetic variants associated with Toxoplasma gondii infection and relationship to schizophrenia risk. Prog Neuropsychopharmacol Biol Psychiatry (2019) 92:133–47. doi: 10.1016/j.pnpbp.2018.12.019
71. Grove J, Borglum AD, Pearce BD. GWAS, cytomegalovirus infection, and schizophrenia. Curr Behav Neurosci Rep (2014) 1(4):215–23. doi: 10.1007/s40473-014-0022-1
72. Leppert B, Havdahl A, Riglin L, Jones HJ, Zheng J, Davey Smith G, et al. Association of maternal neurodevelopmental risk alleles with early-life exposures. JAMA Psychiatry (2019) 76(8):834–42. doi: 10.1001/jamapsychiatry.2019.0774
73. Benros ME, Trabjerg BB, Meier S, Mattheisen M, Mortensen PB, Mors O, et al. Influence of Polygenic risk scores on the association between infections and schizophrenia. Biol Psychiatry (2016) 80(8):609–16. doi: 10.1016/j.biopsych.2016.04.008
74. Shaw M, Palese P. Orthomyxoviridae. In: Knipe DM, Howley PM, editors. Fields virology. Philadelphia, PA: Lippincott Williams and Wilkins (2013). p. 1151–84.
75. Treanor J. Influenza (including avian influenza and swine influenza). In: D.R. Bennett JE, Blaser MJ, editors. Mandell, Douglas, and Bennett"s Principles and Practice of Infectious Diseases, 8th ed. Philadelphia, PA: Elsevier Saunders (2015). p. 2000–24.
76. Sadasivan S, Zanin M, O'Brien K, Schultz-Cherry S, Smeyne RJ. Induction of microglia activation after infection with the non-neurotropic A/CA/04/2009 H1N1 influenza virus. PloS One (2015) 10(4):e0124047. doi: 10.1371/journal.pone.0124047
77. Wang G, Zhang J, Li W, Xin G, Su Y, Gao Y, et al. Apoptosis and proinflammatory cytokine responses of primary mouse microglia and astrocytes induced by human H1N1 and avian H5N1 influenza viruses. Cell Mol Immunol (2008) 5(2):113–20. doi: 10.1038/cmi.2008.14
78. Jang HM, Boltz D, Sturm-Ramirez K, Shepherd KR, Jiao Y, Webster R, et al. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci United States America (2009) 106(33):14063–8. doi: 10.1073/pnas.0900096106
79. Kash JC, Taubenberger JK. The role of viral, host, and secondary bacterial factors in influenza pathogenesis. Am J Pathol (2015) 185(6):1528–36. doi: 10.1016/j.ajpath.2014.08.030
80. Kreijtz JH, Fouchier RA, Rimmelzwaan GF. Immune responses to influenza virus infection. Virus Res (2011) 162(1-2):19–30. doi: 10.1016/j.virusres.2011.09.022
81. Carragher DM, Kaminski DA, Moquin A, Hartson L, Randall TD. A novel role for non-neutralizing antibodies against nucleoprotein in facilitating resistance to influenza virus. J Immunol (2008) 181(6):4168–76. doi: 10.4049/jimmunol.181.6.4168
82. Aronsson F, Lannebo C, Paucar M, Brask J, Kristensson K, Karlsson H. Persistence of viral RNA in the brain of offspring to mice infected with influenza A/WSN/33 virus during pregnancy. J Neurovirol (2002) 8(4):353–7. doi: 10.1080/13550280290100480
83. Brown AS, Meyer U. Maternal immune activation and neuropsychiatric illness: a translational research perspective. Am J Psychiatry (2018) 175(11):1073–83. doi: 10.1176/appi.ajp.2018.17121311
84. Estes ML, McAllister AK. Maternal immune activation: implications for neuropsychiatric disorders. Science (2016) 353(6301):772–7. doi: 10.1126/science.aag3194
85. Meyer U. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry (2014) 75(4):307–15. doi: 10.1016/j.biopsych.2013.07.011
86. Harvey L, Boksa P. Prenatal and postnatal animal models of immune activation: relevance to a range of neurodevelopmental disorders. Dev Neurobiol (2012) 72(10):1335–48. doi: 10.1002/dneu.22043
87. Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R, et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science (2013) 339(6123):1095–9. doi: 10.1126/science.1228261
88. Arad M, Piontkewitz Y, Albelda N, Shaashua L, Weiner I. Immune activation in lactating dams alters sucklings' brain cytokines and produces non-overlapping behavioral deficits in adult female and male offspring: a novel neurodevelopmental model of sex-specific psychopathology. Brain Behav Immun (2017) 63:35–49. doi: 10.1016/j.bbi.2017.01.015
89. Levine J, Buchman CA, Fregien N. Influenza A virus infection of human Schwann cells in vitro. Acta Oto-Laryngologica (2003) 123(1):41–5. doi: 10.1080/0036554021000028092
90. Nakai Y, Itoh M, Mizuguchi M, Ozawa H, Okazaki E, Kobayashi Y, et al. Apoptosis and microglial activation in influenza encephalopathy. Acta Neuropathol (2003) 105(3):233–9. doi: 10.1007/s00401-002-0605-x
91. Smolders S, Notter T, Smolders SMT, Rigo JM, Brone B. Controversies and prospects about microglia in maternal immune activation models for neurodevelopmental disorders. Brain Behav Immun (2018) 73:51–65. doi: 10.1016/j.bbi.2018.06.001
92. Brask J, Owe-Larsson B, Hill RH, Kristensson K. Changes in calcium currents and GABAergic spontaneous activity in cultured rat hippocampal neurons after a neurotropic influenza A virus infection. Brain Res Bull (2001) 55(3):421–9. doi: 10.1016/S0361-9230(01)00536-6
93. Pearce BD, Valadi NM, Po CL, Miller AH. Viral infection of developing GABAergic neurons in a model of hippocampal disinhibition. Neuroreport (2000) 11(11):2433–8. doi: 10.1097/00001756-200008030-00019
94. Landreau F, Galeano P, Caltana LR, Masciotra L, Chertcoff A, Pontoriero A, et al. Effects of two commonly found strains of influenza A virus on developing dopaminergic neurons, in relation to the pathophysiology of schizophrenia. PloS One (2012) 7(12):e51068. doi: 10.1371/journal.pone.0051068
95. Canetta S, Bolkan S, Padilla-Coreano N, Song LJ, Sahn R, Harrison NL, et al. Maternal immune activation leads to selective functional deficits in offspring parvalbumin interneurons. Mol Psychiatry (2016) 21(7):956–68. doi: 10.1038/mp.2015.222
96. Thion MS, Mosser CA, Ferezou I, Grisel P, Baptista S, Low D, et al. Biphasic impact of prenatal inflammation and macrophage depletion on the wiring of neocortical inhibitory circuits. Cell Rep (2019) 28(5):1119–1126 e4. doi: 10.1016/j.celrep.2019.06.086
97. Kowash HM, Potter HG, Edye ME, Prinssen EP, Bandinelli S, Neill JC, et al. Poly(I:C) source, molecular weight and endotoxin contamination affect dam and prenatal outcomes, implications for models of maternal immune activation. Brain Behav Immun (2019) 82:160–6. doi: 10.1016/j.bbi.2019.08.006
98. Meyer U, Feldon J. To poly(I:C) or not to poly(I:C): advancing preclinical schizophrenia research through the use of prenatal immune activation models. Neuropharmacology (2012) 62(3):1308–21. doi: 10.1016/j.neuropharm.2011.01.009
99. Kneeland RE, Fatemi SH. Viral infection, inflammation and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2013) 42:35–48. doi: 10.1016/j.pnpbp.2012.02.001
100. Cotter D, Takei N, Farrell M, Sham P, Quinn P, Larkin C, et al. Does prenatal exposure to influenza in mice induce pyramidal cell disarray in the dorsal hippocampus? Schizophr Res (1995) 16(3):233–41. doi: 10.1016/0920-9964(94)E0082-I
101. Fatemi SH, Sidwell R, Kist D, Akhter P, Meltzer HY, Bailey K, et al. Differential expression of synaptosome-associated protein 25 kDa [SNAP-25] in hippocampi of neonatal mice following exposure to human influenza virus in utero. Brain Res (1998) 800(1):1–9. doi: 10.1016/S0006-8993(98)00450-8
102. Fatemi SH, Emamian ES, Kist D, Sidwell RW, Nakajima K, Akhter P, et al. Defective corticogenesis and reduction in Reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol Psychiatry (1999) 4(2):145–54. doi: 10.1038/sj.mp.4000520
103. Fatemi SH, Cuadra AE, El-Fakahany EE, Sidwell RW, Thuras P. Prenatal viral infection causes alterations in nNOS expression in developing mouse brains. Neuroreport (2000) 11(7):1493–6. doi: 10.1097/00001756-200005150-00027
104. Aronsson F, Karlsson H, Ljunggren H-G, Kristensson K. Persistence of the influenza A/WSN/33 virus RNA at midbrain levels of immunodefective mice. J NeuroViro (2001) 7(2):117–24. doi: 10.1080/13550280152058771
105. Fatemi SH, Emamian ES, Sidwell RW, Kist DA, Stary JM, Earle JA, et al. Human influenza viral infection in utero alters glial fibrillary acidic protein immunoreactivity in the developing brains of neonatal mice. Mol Psychiatry (2002) 7(6):633–40. doi: 10.1038/sj.mp.4001046
106. Fatemi SH, Earle J, Kanodia R, Kist D, Emamian ES, Patterson PH, et al. Prenatal viral infection leads to pyramidal cell atrophy and macrocephaly in adulthood: implications for genesis of autism and schizophrenia. Cell Mol Neurobiol (2002) 22(1):25–33. doi: 10.1023/A:1015337611258
107. Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci (2003) 23(1):297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003
108. Asp L, Beraki S, Aronsson F, Rosvall L, Ogren SO, Kristensson K, et al. Gene expression changes in brains of mice exposed to a maternal virus infection. Neuroreport (2005) 16(10):1111–5. doi: 10.1097/00001756-200507130-00016
109. Fatemi SH, Pearce DA, Brooks AI, Sidwell RW. Prenatal viral infection in mouse causes differential expression of genes in brains of mouse progeny: a potential animal model for schizophrenia and autism. Synapse (2005) 57(2):91–9. doi: 10.1002/syn.20162
110. Asp L, Nellaker C, Karlsson H. Influenza A virus transactivates the mouse envelope gene encoding syncytin B and its regulator, glial cells missing 1. J Neurovirol (2007) 13(1):29–37. doi: 10.1080/13550280601103125
111. Fatemi SH, Reutiman TJ, Folsom TD, Huang H, Oishi K, Mori S, et al. Maternal infection leads to abnormal gene regulation and brain atrophy in mouse offspring: implications for genesis of neurodevelopmental disorders. Schizophr Res (2008) 99(1-3):56–70. doi: 10.1016/j.schres.2007.11.018
112. Fatemi SH, Folsom TD, Reutiman TJ, Sidwell RW. Viral regulation of aquaporin 4, connexin 43, microcephalin and nucleolin. Schizophr Res (2008) 98(1-3):163–77. doi: 10.1016/j.schres.2007.09.031
113. Fatemi SH, Reutiman TJ, Folsom TD, Sidwell RW. The role of cerebellar genes in pathology of autism and schizophrenia. Cerebellum (2008) 7(3):279–94. doi: 10.1007/s12311-008-0017-0
114. Holtze M, Asp L, Schwieler L, Engberg G, Karlsson H. Induction of the kynurenine pathway by neurotropic influenza A virus infection. J Neurosci Res (2008) 86(16):3674–83. doi: 10.1002/jnr.21799
115. Winter C, Reutiman TJ, Folsom TD, Sohr R, Wolf RJ, Juckel G, et al. Dopamine and serotonin levels following prenatal viral infection in mouse–implications for psychiatric disorders such as schizophrenia and autism. Eur Neuropsychopharmacol (2008) 18(10):712–6. doi: 10.1016/j.euroneuro.2008.06.001
116. Asp L, Beraki S, Kristensson K, Ogren SO, Karlsson H. Neonatal infection with neurotropic influenza A virus affects working memory and expression of type III Nrg1 in adult mice. Brain Behav Immun (2009) 23(6):733–41. doi: 10.1016/j.bbi.2009.04.004
117. Shi L, Smith SE, Malkova N, Tse D, Su Y, Patterson PH. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav Immun (2009) 23(1):116–23. doi: 10.1016/j.bbi.2008.07.012
118. Fatemi SH, Folsom TD, Reutiman TJ, Abu-Odeh D, Mori S, Huang H, et al. Abnormal expression of myelination genes and alterations in white matter fractional anisotropy following prenatal viral influenza infection at E16 in mice. Schizophr Res (2009) 112(1-3):46–53. doi: 10.1016/j.schres.2009.04.014
119. Fatemi SH, Folsom TD, Reutiman TJ, Huang H, Oishi K, Mori S. Prenatal viral infection of mice at E16 causes changes in gene expression in hippocampi of the offspring. Eur Neuropsychopharmacol (2009) 19(9):648–53. doi: 10.1016/j.euroneuro.2009.03.004
120. Asp L, Holtze M, Powell SB, Karlsson H, Erhardt S. Neonatal infection with neurotropic influenza A virus induces the kynurenine pathway in early life and disrupts sensorimotor gating in adult Tap1-/- mice. Int J Neuropsychopharmacol (2010) 13(4):475–85. doi: 10.1017/S1461145709990253
121. Moreno JL, Kurita M, Holloway T, Lopez J, Cadagan R, Martinez-Sobrido L, et al. Maternal influenza viral infection causes schizophrenia-like alterations of 5-HT(2)A and mGlu(2) receptors in the adult offspring. J Neurosci (2011) 31(5):1863–72. doi: 10.1523/JNEUROSCI.4230-10.2011
122. Fatemi SH, Folsom TD, Rooney RJ, Mori S, Kornfield TE, Reutiman TJ, et al. The viral theory of schizophrenia revisited: abnormal placental gene expression and structural changes with lack of evidence for H1N1 viral presence in placentae of infected mice or brains of exposed offspring. Neuropharmacology (2012) 62(3):1290–8. doi: 10.1016/j.neuropharm.2011.01.011
123. Fatemi SH, Folsom TD, Liesch SB, Kneeland RE, Karkhane Yousefi M, Thuras PD. The effects of prenatal H1N1 infection at E16 on FMRP, glutamate, GABA, and reelin signaling systems in developing murine cerebellum. J Neurosci Res (2017) 95(5):1110–22. doi: 10.1002/jnr.23949
124. Vernon AC, So PW, Lythgoe DJ, Chege W, Cooper JD, Williams SC, et al. Longitudinal in vivo maturational changes of metabolites in the prefrontal cortex of rats exposed to polyinosinic-polycytidylic acid in utero. Eur Neuropsychopharmacol (2015) 25(12):2210–20. doi: 10.1016/j.euroneuro.2015.09.022
125. Crum WR, Sawiak SJ, Chege W, Cooper JD, Williams SCR, Vernon AC. Evolution of structural abnormalities in the rat brain following in utero exposure to maternal immune activation: a longitudinal in vivo MRI study. Brain Behav Immun (2017) 63:50–9. doi: 10.1016/j.bbi.2016.12.008
126. Piontkewitz Y, Arad M, Weiner I. Risperidone administered during asymptomatic period of adolescence prevents the emergence of brain structural pathology and behavioral abnormalities in an animal model of schizophrenia. Schizophr Bull (2011) 37(6):1257–69. doi: 10.1093/schbul/sbq040
127. Graham AM, Rasmussen JM, Rudolph MD, Heim CM, Gilmore JH, Styner M, et al. Maternal systemic interleukin-6 during pregnancy is associated with newborn amygdala phenotypes and subsequent behavior at 2 years of age. Biol Psychiatry (2018) 83(2):109–19. doi: 10.1016/j.biopsych.2017.05.027
128. Graham AM, Rasmussen JM, Entringer S, Ben Ward E, Rudolph MD, Gilmore JH, et al. Maternal cortisol concentrations during pregnancy and sex-specific associations with neonatal amygdala connectivity and emerging internalizing behaviors. Biol Psychiatry (2019) 85(2):172–81. doi: 10.1016/j.biopsych.2018.06.023
129. Urakubo A, Jarskog LF, Lieberman JA, Gilmore JH. Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophr Res (2001) 47(1):27–36. doi: 10.1016/S0920-9964(00)00032-3
130. Garay PA, Hsiao EY, Patterson PH, McAllister AK. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav Immun (2013) 31:54–68. doi: 10.1016/j.bbi.2012.07.008
131. Meyer U, Murray PJ, Urwyler A, Yee BK, Schedlowski M, Feldon J. Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol Psychiatry (2008) 13(2):208–21. doi: 10.1038/sj.mp.4002042
132. Mondelli V, Vernon AC, Turkheimer F, Dazzan P, Pariante CM. Brain microglia in psychiatric disorders. Lancet Psychiatry (2017) 4(7):563–72. doi: 10.1016/S2215-0366(17)30101-3
133. Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science (2016) 353(6301):aad8670. doi: 10.1126/science.aad8670
134. Mattei D, Ivanov A, Ferrai C, Jordan P, Guneykaya D, Buonfiglioli A, et al. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl Psychiatry (2017) 7(5):e1120. doi: 10.1038/tp.2017.80
135. Li WY, Chang YC, Lee LJ, Lee LJ. Prenatal infection affects the neuronal architecture and cognitive function in adult mice. Dev Neurosci (2014) 36(5):359–70. doi: 10.1159/000362383
136. Juckel G, Manitz MP, Brune M, Friebe A, Heneka MT, Wolf RJ. Microglial activation in a neuroinflammational animal model of schizophrenia–a pilot study. Schizophr Res (2011) 131(1-3):96–100. doi: 10.1016/j.schres.2011.06.018
137. Van den Eynde K, Missault S, Fransen E, Raeymaekers L, Willems R, Drinkenburg W, et al. Hypolocomotive behaviour associated with increased microglia in a prenatal immune activation model with relevance to schizophrenia. Behav Brain Res (2014) 258:179–86. doi: 10.1016/j.bbr.2013.10.005
138. Giovanoli S, Notter T, Richetto J, Labouesse MA, Vuillermot S, Riva MA, et al. Late prenatal immune activation causes hippocampal deficits in the absence of persistent inflammation across aging. J Neuroinflamm (2015) 12:221. doi: 10.1186/s12974-015-0437-y
139. Notter T, Coughlin JM, Gschwind T, Weber-Stadlbauer U, Wang Y, Kassiou M, et al. Translational evaluation of translocator protein as a marker of neuroinflammation in schizophrenia. Mol Psychiatry (2017) 23(2):323–4. doi: 10.1038/mp.2016.248
140. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol (2014) 14:315. doi: 10.1038/nri3665
141. Schroder K, Sweet MJ, Hume DA. Signal integration between IFNgamma and TLR signalling pathways in macrophages. Immunobiology (2006) 211(6-8):511–24. doi: 10.1016/j.imbio.2006.05.007
142. Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol (2010) 6(4):193–201. doi: 10.1038/nrneurol.2010.17
143. Salam AP, Borsini A, Zunszain PA. Trained innate immunity: a salient factor in the pathogenesis of neuroimmune psychiatric disorders. Mol Psychiatry (2018) 23(2):170–6. doi: 10.1038/mp.2017.186
144. Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun (2018) 9(1):1228. doi: 10.1038/s41467-018-03566-5
145. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron (2012) 74(4):691–705. doi: 10.1016/j.neuron.2012.03.026
146. Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci (2014) 17(3):400–6. doi: 10.1038/nn.3641
147. Wilton DK, Dissing-Olesen L, Stevens B. Neuron-Glia signaling in synapse elimination. Annu Rev Neurosci (2019) 42:107–27. doi: 10.1146/annurev-neuro-070918-050306
148. Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry (2000) 57(1):65–73. doi: 10.1001/archpsyc.57.1.65
149. Sellgren CM, Gracias J, Watmuff B, Biag JD, Thanos JM, Whittredge PB, et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat Neurosci (2019) 22(3):374–85. doi: 10.1038/s41593-018-0334-7
150. Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, et al. Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun (2016) 7:12540. doi: 10.1038/ncomms12540
151. Wright P, Laing P, Donaldson PT, Murray RM. Schizophrenia: the teratogenic antibody hypothesis. In: Müller N, editor. Psychiatry Psychoimmunology and Viruses. Key Topics in Brain Research. Vienna: Springer (1999). p. 89–99.
152. Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, et al. Serologic-evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry (2004) 61(8):774–80. doi: 10.1001/archpsyc.61.8.774
153. Wright P, Murray RM. Schizophrenia: prenatal influenza and autoimmunity. Ann Med (1993) 25(5):497–502. doi: 10.3109/07853899309147318
154. Trevathan RD, Tatum JC. Rarity of concurrence of psychosis and rheumatoid arthritis in individual patients; report of a case. J Nerv Ment Dis (1954) 120(1-2):83–4.
155. Pilkington TL. The coincidence of rheumatoid-arthritis and schizophrenia. J Nerv Ment Dis (1956) 124(6):604–6. doi: 10.1097/00005053-195612000-00007
156. Graff H, Handford A. Celiac syndrome in the case histories of five schizophrenics. Psychiatr Q (1961) 35:306–13. doi: 10.1007/BF01566581
157. Cullen AE, Holmes S, Pollak TA, Blackman G, Joyce DW, Kempton MJ, et al. Associations between non-neurological autoimmune disorders and psychosis: a meta-analysis. Biol Psychiatry (2019) 85(1):35–48. doi: 10.1016/j.biopsych.2018.06.016
158. Benros ME, Nielsen PR, Nordentoft M, Eaton WW, Dalton SO, Mortensen PB. Autoimmune Diseases and Severe Infections as Risk Factors for Schizophrenia: A 30-Year Population-Based Register Study. Am J Psychiatry (2011) 168(12):1303–10. doi: 10.1176/appi.ajp.2011.11030516
159. Graus F, Saiz A, Dalmau J. Antibodies and neuronal autoimmune disorders of the CNS. J Neurol (2010) 257(4):509–17. doi: 10.1007/s00415-009-5431-9
160. Steiner J, Walter M, Glanz W, Sarnyai Z, Bernstein HG, Vielhaber S, et al. Increased prevalence of diverse N-methyl-D-aspartate glutamate receptor antibodies in patients with an initial diagnosis of schizophrenia: specific relevance of IgG NR1a antibodies for distinction from N-methyl-D-aspartate glutamate receptor encephalitis. JAMA Psychiatry (2013) 70(3):271–8. doi: 10.1001/2013.jamapsychiatry.86
161. Zandi MS, Irani SR, Lang B, Waters P, Jones PB, McKenna P, et al. Disease-relevant autoantibodies in first episode schizophrenia. J Neurol (2011) 258(4):686–8. doi: 10.1007/s00415-010-5788-9
162. Pollak TA, Rogers JP, Nagele RG, Peakman M, Stone JM, David AS, et al. Antibodies in the diagnosis, prognosis, and prediction of psychotic disorders. Schizophr Bull (2019) 45(1):233–46. doi: 10.1093/schbul/sby021
163. Pollak TA, McCormack R, Peakman M, Nicholson TR, David AS. Prevalence of anti-N-methyl-D-aspartate (NMDA) receptor [corrected] antibodies in patients with schizophrenia and related psychoses: a systematic review and meta-analysis. Psychol Med (2014) 44(12):2475–87. doi: 10.1017/S003329171300295X
164. Al-Diwani A, Pollak TA, Langford AE, Lennox BR. Synaptic and neuronal autoantibody-associated psychiatric syndromes: controversies and hypotheses. Front Psychiatry (2017) 8:13. doi: 10.3389/fpsyt.2017.00013
165. Coutinho E, Harrison P, Vincent A. Do neuronal autoantibodies cause psychosis? a neuroimmunological perspective. Biol Psychiatry (2014) 75(4):269–75. doi: 10.1016/j.biopsych.2013.07.040
166. Deakin J, Lennox BR, Zandi MS. Antibodies to the N-methyl-D-aspartate receptor and other synaptic proteins in psychosis. Biol Psychiatry (2014) 75(4):284–91. doi: 10.1016/j.biopsych.2013.07.018
167. Höftberger R, van Sonderen A, Leypoldt F, Houghton D, Geschwind M, Gelfand J, et al. Encephalitis and AMPA receptor antibodies Novel findings in a case series of 22 patients. Neurology (2015) 84(24):2403–12. doi: 10.1212/WNL.0000000000001682
168. Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr Res (2016) 176(1):36–40. doi: 10.1016/j.schres.2014.10.007
169. Pollak TA, Beck K, Irani SR, Howes OD, David AS, McGuire PK. Autoantibodies to central nervous system neuronal surface antigens: psychiatric symptoms and psychopharmacological implications. Psychopharmacol (Berl) (2016) 233(9):1605–21. doi: 10.1007/s00213-015-4156-y
170. Kayser MS, Titulaer MJ, Gresa-Arribas N, Dalmau J. Frequency and characteristics of isolated psychiatric episodes in anti-N-methyl-d-aspartate receptor encephalitis. JAMA Neurol (2013) 70(9):1133–9. doi: 10.1001/jamaneurol.2013.3216
171. Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol (2013) 12(2):157–65. doi: 10.1016/S1474-4422(12)70310-1
172. Al-Diwani A, Handel A, Townsend L, Pollak T, Leite MI, Harrison PJ, et al. The psychopathology of NMDAR-antibody encephalitis in adults: a systematic review and phenotypic analysis of individual patient data. Lancet Psychiatry (2019) 6(3):235–46. doi: 10.1016/S2215-0366(19)30001-X
173. Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol (2008) 7(12):1091–8. doi: 10.1016/S1474-4422(08)70224-2
174. Dalmau J, Tuzun E, Wu HY, Masjuan J, Rossi JE, Voloschin A, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol (2007) 61(1):25–36. doi: 10.1002/ana.21050
175. Armangue T, Leypoldt F, Malaga I, Raspall-Chaure M, Marti I, Nichter C, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol (2014) 75(2):317–23. doi: 10.1002/ana.24083
176. Westman G, Studahl M, Ahlm C, Eriksson BM, Persson B, Ronnelid J, et al. N-Methyl-D-Aspartate receptor autoimmunity affects cognitive performance in herpes simplex encephalitis. Clin Microbiol Infect (2016) 22(11):934–40. doi: 10.1016/j.cmi.2016.07.028
177. Salovin A, Glanzman J, Roslin K, Armangue T, Lynch DR, Panzer JA. Anti-NMDA receptor encephalitis and nonencephalitic HSV-1 infection. Neurol Neuroimmunol Neuroinflamm (2018) 5(4):e458. doi: 10.1212/NXI.0000000000000458
178. Pruss H. Postviral autoimmune encephalitis: manifestations in children and adults. Curr Opin Neurol (2017) 30(3):327–33. doi: 10.1097/WCO.0000000000000445
179. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol (2011) 10(1):63–74. doi: 10.1016/S1474-4422(10)70253-2
180. Baltagi SA, Shoykhet M, Felmet K, Kochanek PM, Bell MJ. Neurological sequelae of 2009 influenza A (H1N1) in children: a case series observed during a pandemic. Pediatr Crit Care Med (2010) 11(2):179–84. doi: 10.1097/PCC.0b013e3181cf4652
181. Cartisano T, Kicker J. Anti-N-methyl-D-aspartate receptor encephalitis in 7-month old infant following influenza vaccination (P5.136). Neurology (2016) 86(16 Supplement). P5.136.
182. Hung TY, Foo NH, Lai MC. Anti-N-methyl-d-aspartate receptor encephalitis. Pediatr Neonatol (2011) 52(6):361–4. doi: 10.1016/j.pedneo.2011.08.012
183. Wang H. Anti-NMDA receptor encephalitis and vaccination. Int J Mol Sci (2017) 18(1):193. doi: 10.3390/ijms18010193
184. Dale RC, Irani SR, Brilot F, Pillai S, Webster R, Gill D, et al. N-methyl-D-aspartate receptor antibodies in pediatric dyskinetic encephalitis lethargica. Ann Neurol (2009) 66(5):704–9. doi: 10.1002/ana.21807
185. McCall S, Vilensky JA, Gilman S, Taubenberger JK. The relationship between encephalitis lethargica and influenza: a critical analysis. J Neurovirol (2008) 14(3):177–85. doi: 10.1080/13550280801995445
186. Pillai SC, Hacohen Y, Tantsis E, Prelog K, Merheb V, Kesson A, et al. Infectious and autoantibody-associated encephalitis: clinical features and long-term outcome. Pediatrics (2015) 135(4):e974–84. doi: 10.1542/peds.2014-2702
187. Wilson N, Barnard LT, Summers JA, Shanks GD, Baker MG. Differential mortality rates by ethnicity in 3 influenza pandemics over a century, New Zealand. Emerg Infect Dis (2012) 18(1):71–7. doi: 10.3201/eid1801.110035
188. Jones HF, Mohammad SS, Reed PW, Dunn PPJ, Steele RH, Dale RC, et al. Anti-N-methyl-d-aspartate receptor encephalitis in Maori and Pacific Island children in New Zealand. Dev Med Child Neurol (2017) 59(7):719–24. doi: 10.1111/dmcn.13420
189. Jezequel J, Rogemond V, Pollak T, Lepleux M, Jacobson L, Grea H, et al. Cell- and single molecule-based methods to detect anti-N-methyl-D-aspartate receptor autoantibodies in patients with first-episode psychosis from the OPTiMiSE project. Biol Psychiatry (2017) 82(10):766–72. doi: 10.1016/j.biopsych.2017.06.015
190. Jezequel J, Johansson EM, Dupuis JP, Rogemond V, Grea H, Kellermayer B, et al. Dynamic disorganization of synaptic NMDA receptors triggered by autoantibodies from psychotic patients. Nat Commun (2017) 8(1):1791. doi: 10.1038/s41467-017-01700-3
191. Hammer C, Stepniak B, Schneider A, Papiol S, Tantra M, Begemann M, et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol Psychiatry (2014) 19(10):1143–9. doi: 10.1038/mp.2013.110
192. Castillo-Gomez E, Kastner A, Steiner J, Schneider A, Hettling B, Poggi G, et al. The brain as immunoprecipitator of serum autoantibodies against N-Methyl-D-aspartate receptor subunit NR1. Ann Neurol (2016) 79(1):144–51. doi: 10.1002/ana.24545
193. Brimberg L, Mader S, Jeganathan V, Berlin R, Coleman TR, Gregersen PK, et al. Caspr2-reactive antibody cloned from a mother of an ASD child mediates an ASD-like phenotype in mice. Mol Psychiatry (2016) 21(12):1663–71. doi: 10.1038/mp.2016.165
194. Coutinho E, Menassa DA, Jacobson L, West SJ, Domingos J, Moloney TC, et al. Persistent microglial activation and synaptic loss with behavioral abnormalities in mouse offspring exposed to CASPR2-antibodies in utero. Acta Neuropathol (2017) 134(4):567–83. doi: 10.1007/s00401-017-1751-5
195. Jones KL, Pride MC, Edmiston E, Yang M, Silverman JL, Crawley JN, et al. Autism-specific maternal autoantibodies produce behavioral abnormalities in an endogenous antigen-driven mouse model of autism. Mol Psychiatry (2018) in press. doi: 10.1038/s41380-018-0126-1
196. Coutinho E, Jacobson L, Pedersen MG, Benros ME, Norgaard-Pedersen B, Mortensen PB, et al. CASPR2 autoantibodies are raised during pregnancy in mothers of children with mental retardation and disorders of psychological development but not autism. J Neurol Neurosurg Psychiatry (2017) 88(9):718–21. doi: 10.1136/jnnp-2016-315251
197. Jurek B, Chayka M, Kreye J, Lang K, Kraus L, Fidzinski P, et al. Human gestational NMDAR autoantibodies impair neonatal murine brain function. Ann Neurol (2019) 86(5):656–70. doi: 10.1002/ana.25552
198. Blanpied TA, Clarke RJ, Johnson JW. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J Neurosci (2005) 25(13):3312–22. doi: 10.1523/JNEUROSCI.4262-04.2005
199. Kohm AP, Fuller KG, Miller SD. Mimicking the way to autoimmunity: an evolving theory of sequence and structural homology. Trends In Microbiol (2003) 11(3):101–5. doi: 10.1016/S0966-842X(03)00006-4
200. Ercolini AM, Miller SD. The role of infections in autoimmune disease. Clin Exp Immunol (2009) 155(1):1–15. doi: 10.1111/j.1365-2249.2008.03834.x
201. Munz C, Lunemann JD, Getts MT, Miller SD. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol (2009) 9(4):246–58. doi: 10.1038/nri2527
202. Toplak N, Avcin T. Influenza and autoimmunity. Ann N Y Acad Sci (2009) 1173:619–26. doi: 10.1111/j.1749-6632.2009.04759.x
203. Toplak N, Kveder T, Trampus-Bakija A, Subelj V, Cucnik S, Avcin T. Autoimmune response following annual influenza vaccination in 92 apparently healthy adults. Autoimmun Rev (2008) 8(2):134–8. doi: 10.1016/j.autrev.2008.07.008
204. Laing P, Knight JG, Hill JM, Harris AG, Oxford JS, Webster RG, et al. Influenza viruses induce autoantibodies to a brain-specific 37-kDa protein in rabbit. Proc Natl Acad Sci U S A (1989) 86(6):1998–2002. doi: 10.1073/pnas.86.6.1998
205. Vellozzi C, Iqbal S, Broder K. Guillain-Barre syndrome, influenza, and influenza vaccination: the epidemiologic evidence. Clin Infect Dis (2014) 58(8):1149–55. doi: 10.1093/cid/ciu005
206. Kornum BR, Faraco J, Mignot E. Narcolepsy with hypocretin/orexin deficiency, infections and autoimmunity of the brain. Curr Opin In Neurobiol (2011) 21(6):897–903. doi: 10.1016/j.conb.2011.09.003
207. Singh AK, Mahlios J, Mignot E. Genetic association, seasonal infections and autoimmune basis of narcolepsy. J Autoimmun (2013) 43:26–31. doi: 10.1016/j.jaut.2013.02.003
208. Partinen M, Kornum BR, Plazzi G, Jennum P, Julkunen I, Vaarala O. Narcolepsy as an autoimmune disease: the role of H1N1 infection and vaccination. Lancet Neurol (2014) 13(6):600–13. doi: 10.1016/S1474-4422(14)70075-4
209. Miller E, Andrews N, Stellitano L, Stowe J, Winstone AM, Shneerson J, et al. Risk of narcolepsy in children and young people receiving AS03 adjuvanted pandemic A/H1N1 2009 influenza vaccine: retrospective analysis. BMJ (2013) 346:f794. doi: 10.1136/bmj.f794
210. Partinen M, Saarenpaa-Heikkila O, Ilveskoski I, Hublin C, Linna M, Olsen P, et al. Increased incidence and clinical picture of childhood narcolepsy following the 2009 H1N1 pandemic vaccination campaign in Finland. PloS One (2012) 7(3):e33723. doi: 10.1371/journal.pone.0033723
211. Ahmed SS, Volkmuth W, Duca J, Corti L, Pallaoro M, Pezzicoli A, et al. Antibodies to influenza nucleoprotein cross-react with human hypocretin receptor 2. Sci Transl Med (2015) 7(294):294ra105. doi: 10.1126/scitranslmed.aab2354
212. Vassalli A, Li S, Tafti M. Comment on “Antibodies to influenza nucleoprotein cross-react with human hypocretin receptor 2”. Sci Transl Med (2015) 7(314):314le2. doi: 10.1126/scitranslmed.aad2353
213. Luo G, Lin L, Jacob L, Bonvalet M, Ambati A, Plazzi G, et al. Absence of anti-hypocretin receptor 2 autoantibodies in post pandemrix narcolepsy cases. PloS One (2017) 12(12):e0187305. doi: 10.1371/journal.pone.0187305
214. Luo G, Ambati A, Lin L, Bonvalet M, Partinen M, Ji X, et al. Autoimmunity to hypocretin and molecular mimicry to flu in type 1 narcolepsy. Proc Natl Acad Sci U S A (2018) 115(52):E12323–32. doi: 10.1073/pnas.1818150116
215. Kanduc D. Describing the hexapeptide identity platform between the influenza A H5N1 and Homo sapiens proteomes. Biologics : Targets Ther (2010) 4:245–61. doi: 10.2147/BTT.S12097
216. Lucchese G, Capone G, Kanduc D. Peptide sharing between influenza a H1N1 hemagglutinin and human axon guidance Proteins. Schizophr Bull (2014) 40(2):362–75. doi: 10.1093/schbul/sbs197
217. Lucchese G. Understanding neuropsychiatric diseases, analyzing the peptide sharing between infectious agents and the language-associated NMDA 2A protein. Front In Psychiatry (2016) 7:60. doi: 10.3389/fpsyt.2016.00060
218. Kepinska A, Pollak T, Iyegbe C, Murray R. T189. Peptide sharing between schizophrenia-related proteins and the influenza a virus may offer a window into the immune aetiology of psychotic disorders. Schizophr Bull (2018) 44(suppl_1):S189–9. doi: 10.1093/schbul/sby016.465
219. Harrison NA, Cercignani M, Voon V, Critchley HD. Effects of inflammation on hippocampus and substantia nigra responses to novelty in healthy human participants. Neuropsychopharmacology (2015) 40(4):831–8. doi: 10.1038/npp.2014.222
220. Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD. Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry (2009) 66(5):407–14. doi: 10.1016/j.biopsych.2009.03.015
221. Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Dolan RJ, et al. Neural origins of human sickness in interoceptive responses to inflammation. Biol Psychiatry (2009) 66(5):415–22. doi: 10.1016/j.biopsych.2009.03.007
222. Marques TR, Ashok AH, Pillinger T, Veronese M, Turkheimer FE, Dazzan P, et al. Neuroinflammation in schizophrenia: meta-analysis of in vivo microglial imaging studies. Psychol Med (2019) 49(13), 2186–96. doi: 10.1017/S0033291718003057
223. Holmes SE, Hinz R, Drake RJ, Gregory CJ, Conen S, Matthews JC, et al. In vivo imaging of brain microglial activity in antipsychotic-free and medicated schizophrenia: a [(11)C](R)-PK11195 positron emission tomography study. Mol Psychiatry (2016) 21(12):1672–9. doi: 10.1038/mp.2016.180
224. Quadrato G, Brown J, Arlotta P. The promises and challenges of human brain organoids as models of neuropsychiatric disease. Nat Med (2016) 22(11):1220–8. doi: 10.1038/nm.4214
225. Cono J, Cragan JD, Jamieson DJ, Rasmussen SA. Prophylaxis and treatment of pregnant women for emerging infections and bioterrorism emergencies. Emerg Infect Dis (2006) 12(11):1631–7. doi: 10.3201/eid1211.060618
226. Yudin MH. Optimizing knowledge of antiviral medications for prophylaxis and treatment of influenza during pregnancy. Expert Rev Respir Med (2011) 5(4):495–501. doi: 10.1586/ers.11.37
227. Bednarczyk RA, Adjaye-Gbewonyo D, Omer SB. Safety of influenza immunization during pregnancy for the fetus and the neonate. Am J Obstet Gynecol (2012) 207(3 Suppl):S38–46. doi: 10.1016/j.ajog.2012.07.002
228. Al-Haddad BJS, Oler E, Armistead B, Elsayed NA, Weinberger DR, Bernier R, et al. The fetal origins of mental illness. Am J Obstet Gynecol (2019) 221(6): 549–62. doi: 10.1016/j.ajog.2019.06.013
229. Hviid A, Svanstrom H, Molgaard-Nielsen D, Lambach P. Association between pandemic influenza A(H1N1) vaccination in pregnancy and early childhood morbidity in offspring. JAMA Pediatr (2017) 171(3):239–48. doi: 10.1001/jamapediatrics.2016.4023
230. Xia Y, Qi F, Zou J, Yao Z. Influenza A(H1N1) vaccination during early pregnancy transiently promotes hippocampal neurogenesis and working memory. involvement of Th1/Th2 balance. Brain Res (2014) 1592:34–43. doi: 10.1016/j.brainres.2014.09.076
231. Xia Y, Qi F, Zou J, Yang J, Yao Z. Influenza vaccination during early pregnancy contributes to neurogenesis and behavioral function in offspring. Brain Behav Immun (2014) 42:212–21. doi: 10.1016/j.bbi.2014.06.202
232. Zerbo O, Qian Y, Yoshida C, Fireman BH, Klein NP, Croen LA. Association between influenza infection and vaccination during pregnancy and risk of autism spectrum disorder. JAMA Pediatr (2017) 171(1):e163609. doi: 10.1001/jamapediatrics.2016.3609
233. Sandy K, Gandham S, Nanan R. Influenza a infections in infants of vaccinated and unvaccinated mothers. JAMA Pediatr (2017) 171(10):1014–5. doi: 10.1001/jamapediatrics.2017.2391
234. Hviid A, Svanstrom H. Influenza a infections in infants of vaccinated and unvaccinated mothers-reply. JAMA Pediatr (2017) 171(10):1015. doi: 10.1001/jamapediatrics.2017.2394
235. Meyer U. Neurodevelopmental resilience and susceptibility to maternal immune activation. Trends Neurosci (2019) 42(11):793–806. doi: 10.1016/j.tins.2019.08.001
236. Li Q, Leung YO, Zhou I, Ho LC, Kong W, Basil P, et al. Dietary supplementation with n-3 fatty acids from weaning limits brain biochemistry and behavioural changes elicited by prenatal exposure to maternal inflammation in the mouse model. Transl Psychiatry (2015) 5:e641. doi: 10.1038/tp.2015.126
Keywords: infection, epidemiology, autoimmunity, neurodevelopment, maternal immune activation (MIA), influenza, schizophrenia
Citation: Kępińska AP, Iyegbe CO, Vernon AC, Yolken R, Murray RM and Pollak TA (2020) Schizophrenia and Influenza at the Centenary of the 1918-1919 Spanish Influenza Pandemic: Mechanisms of Psychosis Risk. Front. Psychiatry 11:72. doi: 10.3389/fpsyt.2020.00072
Received: 07 November 2019; Accepted: 28 January 2020;
Published: 26 February 2020.
Edited by:
Belinda Lennox, University of Oxford, United KingdomReviewed by:
Sabina Berretta, McLean Hospital, United StatesCopyright © 2020 Kępińska, Iyegbe, Vernon, Yolken, Murray and Pollak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas A. Pollak, dGhvbWFzLnBvbGxha0BrY2wuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.