 Davide Amato
Davide Amato Anna Kruyer1
Anna Kruyer1 Andreas Heinz
Andreas Heinz- 1Department of Neuroscience, Medical University of South Carolina, Charleston, SC, United States
- 2Department of Pharmacology and Physiology, Faculty of Medicine, Université de Montréal, Montreal, QC, Canada
- 3Department of Psychiatry, Charité University Medicine Berlin, Campus Charité Mitte, Berlin, Germany
Antipsychotic treatment resistance in schizophrenia remains a major issue in psychiatry. Nearly 30% of patients with schizophrenia do not respond to antipsychotic treatment, yet the underlying neurobiological causes are unknown. All effective antipsychotic medications are thought to achieve their efficacy by targeting the dopaminergic system. Here we review early literature describing the fundamental mechanisms of antipsychotic drug efficacy, highlighting mechanistic concepts that have persisted over time. We then reconsider the original framework for understanding antipsychotic efficacy in light of recent advances in our scientific understanding of the dopaminergic effects of antipsychotics. Based on these new insights, we describe a role for the dopamine transporter in the genesis of both antipsychotic therapeutic response and primary resistance. We believe that this discussion will help delineate the dopaminergic nature of antipsychotic treatment-resistant schizophrenia.
Introduction
Schizophrenia is a psychiatric condition often involving a complex genetic predisposition (1–3) as well as vulnerability to certain environmental factors (4), eventually culminating in symptoms clinically defined as positive (emergent symptoms, including hallucinations and delusions) or negative (characterized by loss of a particular function, including apathy and lack of motivation) (5–7). Additionally, a proportion of patients with schizophrenia are impaired on standard neurocognitive tasks (8), and this is considered an important correlate of disease severity (9–12). The fundamental neurobiological maladaptations underlying the symptoms of schizophrenia are not completely understood. Regardless, sub-chronic blockade of a proportion (60–80%) of dopamine D2/3 receptors (which we will refer to as “D2”) is considered to underlie treatment efficacy in schizophrenia (13). Previous and recent literature supports the effectiveness of D2 antagonism compared to any alternative pharmacological intervention (14–17). However, blocking dopamine receptors is not an effective therapeutic mechanism for all individuals with schizophrenia (18–24). For example, some patients with first-episode psychosis do not respond to antipsychotic treatment (25). Lack of response to antipsychotic treatment can also be “acquired” and can develop over time with long-term treatment regimens (23, 26) or can develop after a period of treatment abstinence, such as that which occurs during medication nonadherence (27–30). In many of these cases, patients unresponsive to first-line antipsychotic treatments are instead responsive to clozapine (18, 31, 32). Furthermore, there exists an additional group of patients with schizophrenia who will not respond to clozapine or to any other antipsychotic drug. This category of patients is defined as “ultra-resistant” (18, 33).
Whether all instances of antipsychotic resistance share a common neurobiological mechanism is not clear (10, 24, 34–39), nor is there a precise behavioral signature indicating its clinical manifestation, since criteria to define resistance to antipsychotic treatment were standardized only recently (40). It is not within the scope of this review to contribute to the behavioral definition of treatment resistance in schizophrenia. Rather the focus here is narrowed onto the putative role of dopamine clearance in the expression of primary antipsychotic-resistant schizophrenia (i.e., patients with first episode psychosis who never responded to treatment). We do not exclude the possibility that alterations in other neurotransmitter systems might also be involved, nor do we exclude that the dopaminergic mechanisms described here will also apply to other forms of antipsychotic resistance. Simply, we focus on dopamine, because clinical observations emphasize the importance of this neurotransmitter in the pathophysiology of psychosis (41–43) and its treatment (44). Our attention on dopamine clearance is motivated by recent data from ex vivo and in vivo studies with animal models demonstrating that antipsychotic failure is accompanied by tolerance to antipsychotic-induced increases in basal dopamine and dopamine turnover, and that the dopamine transporter (DAT) is a key moderator of both extracellular dopamine and antipsychotic response (35, 38, 45). The link between preserved, or slightly elevated, dopaminergic tone and antipsychotic responsiveness has also been observed in humans with schizophrenia (46). Recent interpretations of these data suggest that a preserved extracellular dopaminergic tone might have an important pharmacological role in the therapeutic efficacy of antipsychotics (24). These observations have been directly and indirectly supported by independent studies (38, 47–50). Due to space limitations, we will only briefly outline dopaminergic biomarkers described in the literature that appear relevant to understanding antipsychotic responsiveness. We will then conclude with the suggestion that DAT could be a more powerful moderator of antipsychotic efficacy and failure than currently recognized. Changes in DAT expression and/or function alone can alter the expected response to antipsychotic medications, making DAT a highly relevant protein when considering the dopaminergic nature of antipsychotic-resistant schizophrenia.
Dopaminergic Dysregulation in Schizophrenia
Before discussing dopaminergic mechanisms of antipsychotic efficacy, it is important to describe the dopaminergic signaling abnormalities in schizophrenia that are targeted by antipsychotic drugs. As described in the Introduction, the underlying etiology and neuropathology of schizophrenia symptoms are still unclear. Genetic studies point to associations with genes regulating neurodevelopment, the immune system, and dopaminergic and glutamatergic transmission (2, 51), while other studies demonstrate a potential role for disruption of multiple intracellular signaling pathways in schizophrenia (52). Furthermore, environmental factors linked to schizophrenia such as migration or obstetric infection can change dopamine neurotransmission (4), in addition to other neurobiological systems (53–58). Despite the many factors that appear to contribute to schizophrenia, treatment has focused on correcting a dysregulated dopaminergic system by inhibiting dopaminergic transmission. However, it should be noted that the efficacy of pharmacologically targeting the dopaminergic system in schizophrenia does not definitively prove a dopaminergic dysregulation. Dopamine has a powerful neuromodulatory role in the brain and in the basal ganglia in particular and it can regulate motor activity as well as motivation and cognition. Since all of these functions are impacted in schizophrenia, it should not be surprising that many antidopaminergic drugs are effective (or deleterious) for schizophrenia symptoms, even though the observable symptoms may have some other underlying cause(s). Thus, the dopaminergic system should be seen as a treatment pathway capable of affecting behavioral features that appear to be disrupted in schizophrenia, but that may be caused by alterations in other neurotransmitter systems.
Mechanisms of Antipsychotic Responsiveness
Brain dopamine receptor blockade has been embraced as a mechanism for the therapeutic efficacy of antipsychotic drugs for over 60 years (9, 59). Thus, very frequently, researchers have focused on the interactions between molecule(s) and receptor(s) to describe antipsychotic mechanisms. Although this approach is correct in principle, practically it may be too simplistic. Receptors do not act in isolation. Receptors on neurons are connected via synapses and organized into networks within neuronal circuitries. Receptors are also functionally linked with intracellular molecular networks that control membrane excitability, as well as neurotransmitter synthesis, release, and metabolism, and by these mechanisms, neurons can regulate their own activity. Due to the nature of neural signaling, changes in the inactivation or activation of neural receptors with antipsychotic drugs, or with any other compound, which cause local intracellular changes, will affect other cell populations through signal propagation along neural pathways. Thus, antipsychotic medications can impact neurotransmitter synthesis, release, and metabolism not only in neurons that directly interact with antipsychotics but also in those neurons that are part of the same neural circuitry. Therefore, a proper understanding of the mechanisms underlying antipsychotic responsiveness should not simply describe the chemical interactions between antipsychotic drugs and their target receptors, but should consider modifications induced by antipsychotics at the cellular and circuit levels. We will focus on neuroadaptations occurring at the cellular level that link receptors to synthesis, release, and uptake of extracellular dopamine.
Striatal D2 Receptor Blockade in Treatment-Responsive Schizophrenia
Striatal D2 receptor blockade is considered the most effective mechanism to reduce psychotic symptoms in schizophrenia (60, 61). Extra-striatal mechanisms of antipsychotics have been debated previously (62) and will not be discussed here. The general theory of the therapeutic efficacy of antipsychotics builds on two main observations. First, clinical potency of antipsychotics, including clozapine, is directly related to their affinity for the dopamine D2 receptor in vitro (14, 15). This is substantiated by evidence that therapeutic concentrations of antipsychotics in the plasma or in the spinal fluid accurately match the antipsychotic dissociation constant (Kd) at D2 receptors (63). Secondly, therapeutic concentrations of all antipsychotics (typical and atypical) produce a similar D2 receptor occupancy (13, 59, 64). Although this observation does not strictly apply for clozapine (21) or quetiapine (65), it has been shown that D2 receptor occupancy in the human brain ranges between 70% and 80% within 2 h of treatment and remains elevated for over 24 h for both typical and atypical antipsychotics (21, 66, 67). D2 receptor occupancy with clozapine (20) and quetiapine (68, 69), on the other hand, decreases significantly within 24 h. Based on these findings, Seeman and Tallerico (63) suggested that the main difference between typical and atypical antipsychotics is the temporal decay of antipsychotic binding to the D2 receptor when challenged by endogenous dopamine. In fact, antipsychotics compete with endogenous dopamine within the synaptic space and the presence of dopamine would theoretically affect the concentration of antipsychotic required to reach a particular range of D2 receptor occupancy. Subsequently, it was observed that the dissociation rate constant, koff (rather than association rate constant, kon), largely accounts for the difference in binding affinity when comparing typical and atypical antipsychotics (70). This also implies that measurements of D2 receptor occupancy with antipsychotics can be affected by the chemistry of the radioligands used (i.e., lipid-soluble spiperone, nemonapride versus water-soluble dopamine, raclopride) (71–73). D2 receptor occupancy by atypical antipsychotics such as clozapine and quetiapine will be reduced by (11C)raclopride less so than if lipid-soluble radioligands such as (11C)methylspiperone were used (63, 73, 74). Therefore, differences in D2 receptor occupancy between clozapine, quetiapine, and other antipsychotics could be influenced by the chemistry of the radioligands used (75). This intriguing interpretation, developed using in vitro assays, has not been confirmed functionally. Typical and atypical antipsychotics dissociate with similar temporal kinetics in electrophysiological evaluations, suggesting that the reversal of D2 receptor antagonism by typical and atypical antipsychotics does not differ markedly (76, 77). These contradictory results point to the possibility that mechanisms other than receptor occupancy may also be involved in the outcomes of these assays, although we cannot dismiss the relevance of ligand binding kinetics at D2 receptors for achieving antipsychotic efficacy (24, 38).
Striatal D2 Receptor Density and Blockade in Treatment-Resistant Schizophrenia
As already mentioned above, the blockade (or occupancy) of a proportion of D2 receptors is not a working antipsychotic mechanism for a significant number of patients with schizophrenia (31). In fact, roughly one-third of individuals with schizophrenia are resistant to treatment with first-line antipsychotics despite sufficient D2 receptor occupancy (19). Clozapine, which works at a relatively low (∼40%) striatal D2 receptor occupancy (20, 21, 78, 79), is the most effective antipsychotic in the majority of patients refractory to other antipsychotic medications (18, 32, 80). If we hypothetically accept the suggestion that this outcome is not attributable to D2 receptor binding kinetics (77), we begin to consider other dopaminergic mechanism that may account for this apparent discrepancy. A growing literature supports the idea that additional dopaminergic mechanisms may underlie therapeutic efficacy of antipsychotic drugs (24, 38). Some patients who respond to first-line antipsychotic treatment experience diminished treatment efficacy over time (23), which can lead to treatment non-compliance and relapse (81). Diminished antipsychotic efficacy may also occur despite stable D2 receptor occupancy (82). These dynamics are depicted in Figure 1. The opposite has also been observed with long-term antipsychotic efficacy occurring despite decreasing D2 receptor occupancy (89–85).
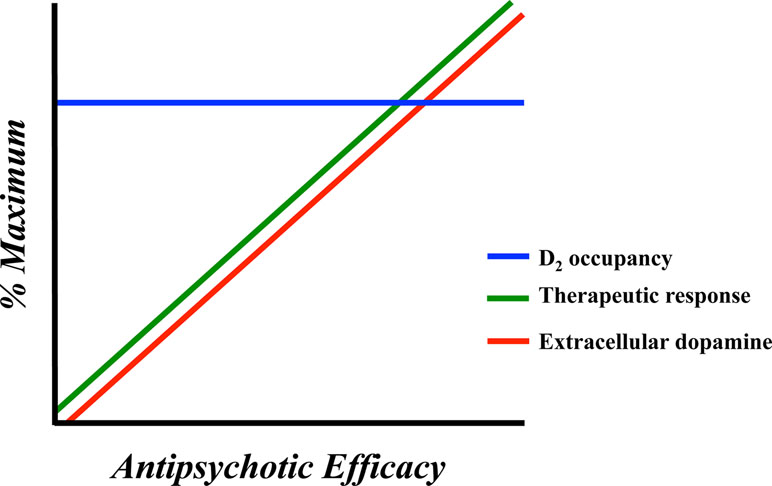
Figure 1 Representation of the neurochemical factors affecting antipsychotic response in humans and animal models. Antipsychotic response is optimal in concert with elevated extracellular dopamine levels. D2 receptor occupancy is less dynamic and appears stable during time periods characterized by both therapeutic efficacy and antipsychotic failure.
Acquired resistance to antipsychotics (tolerance) could involve antipsychotic-induced dopamine receptor supersensitivity, potentially resulting from D2 receptor upregulation, consequent to chronic D2 receptor blockade (34, 86, 87). In patients with schizophrenia, antipsychotic-induced dopamine supersensitivity is thought to impair treatment efficacy, promote relapse to psychosis, and also worsen psychotic symptoms (88–90). In laboratory animals, antipsychotic-induced dopamine supersensitivity produces loss of antipsychotic efficacy (35, 91, 92) and an exaggerated behavioral response to dopamine agonists (35, 93–95). However, the link to antipsychotic-induced striatal D2 upregulation is complex. Changes in levels of dopamine receptor expression in patients have not been replicated reliably by independent research groups (96, 97). Recent studies using animal models also show tolerance to antipsychotics despite clinically representative levels of striatal D2 receptor blockade, as measured either with in vivo imaging (38) or ex vivo receptor autoradiography (35, 91). Antipsychotic-induced dopamine supersensitivity and tolerance to antipsychotics can also be dissociable from changes in striatal D2 receptor density (35). Thus, changes in striatal D2 receptor expression are not always predictive of either changes in antipsychotic efficacy or the emergence of antipsychotic-induced dopamine supersensitivity (24, 35, 38, 98, 99), although high doses of antipsychotics may upregulate striatal D2 receptors (100).
Beyond changes in striatal D2 receptor density, chronic antipsychotic treatment can also increase D2 receptor function, and this has been linked to dopamine supersensitivity and acquired antipsychotic tolerance. When D2 receptors are coupled to Gi/o proteins, they are in a functional, high affinity state for dopamine (referred to as D2HIGH). When D2 receptors are uncoupled to Gi/o proteins, they are in a functionally inert, low affinity state for dopamine (D2LOW). As such, the proportion of D2HIGH can modulate dopamine signaling via D2 receptors. The link between antipsychotic tolerance and changes in striatal D2HIGH sites comes largely from work in animal models showing that chronic antipsychotic treatment increases striatal D2HIGH levels (35, 91, 101). Antipsychotic treatment regimens that promote behavioral dopamine supersensitivity and antipsychotic treatment tolerance produce an even greater increase in D2HIGH sites (91). D2HIGH receptor elevation and antipsychotic-induced dopamine supersensitivity also follow a similar time course (35). However, D2HIGH sites can increase early in antipsychotic treatment, before any behavioral evidence of dopamine supersensitivity or treatment tolerance (35). In addition, antipsychotic dosing regimens that do not produce dopamine supersensitivity can still increase striatal D2HIGH sites (91, 101). Furthermore, there is no conclusive evidence of elevated D2HIGH receptors in patients with schizophrenia [see (102)]. Thus, there is likely a link between changes in D2HIGH sites and acquired antipsychotic treatment tolerance, but this requires further study.
Dopamine D2 Receptor Isoforms and Schizophrenia
The majority of the cells expressing D2 receptors in the striatum are neurons with medium-sized cell bodies and spiny dendrites (medium spiny neurons, MSNs, about 95% of all cells in this region), which are postsynaptic to dopaminergic terminals projecting from the midbrain, among other regions; for an overview, see Refs. (24, 103). The striatum also contains presynaptic D2 receptors expressed on dopaminergic axon terminals, which represent only a small percentage of the total D2 receptor pool found in the striatum and may have a different molecular structure (104). Accordingly, there are two isoforms of dopamine D2 receptors deriving from alternative splicing of exon 6 to produce the long (D2L) and the short (D2S) forms of the protein (105–107) (Figure 2A–C). Both isoforms appear to regulate dopaminergic firing (108), but only D2S controls Ca2+-mediated autoinhibition (109, 110). Furthermore, post-synaptic D2S, but not D2L, controls MSN excitability in rodents (111) and likely in humans (112), despite its pre-dominant presynaptic localization. These effects are likely a consequence of the distinct molecular mechanisms linked to D2 receptor isoforms (113–116) (Figure 2C).
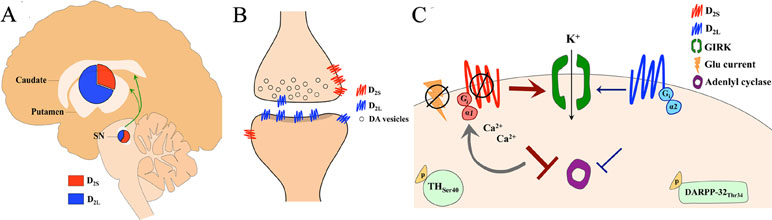
Figure 2 (A) Uneven expression of dopamine D2 receptor isoforms (short, D2S and long, D2L) in the human midbrain (substantia nigra, SN) and striatum (caudate nucleus and putamen). D2L is predominant in the striatum, while D2S is prevalently expressed in the midbrain. This unbalanced D2L/D2S ratio is observed across species. (B) Schematic of a synaptic contact between a dopaminergic terminal projecting from SN and a somatodendritic spine in the striatum shows the unbalanced D2L/D2S ratio on midbrain and striatal neurons. (C) Distinct physiological effects are mediated by the two D2 receptor isoforms. Both D2S and D2L receptors inhibit adenylyl cyclase, though D2L-mediated inhibition is weaker, via Giα1and Giα2, respectively. D2S stimulation leads to phosphorylation of tyrosine hydroxylase (TH) at serine 40 in nigrostriatal dopaminergic neurons, whereas D2L stimulation leads to phosphorylation of dopamine and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) at threonine 34, in medium spiny neurons. D2S, but not D2L, activates G protein-gated inwardly rectifying potassium (GIRK) conductance, which is Ca2+ sensitive. D2S, but not D2L, inhibits excitation in response to glutamate (Glu) currents.
The expression of D2 isoforms in the mammalian brain is distributed unevenly (Figure 2A). Genomic studies of human and rodent D2 mRNA, which share ∼95–99% homology (117), report that while D2L and D2S mRNA are widely expressed in the brain, D2L mRNA is highly expressed in the striatum (i.e., caudate nucleus and putamen) relative to D2S mRNA (117–120). Investigation of D2 protein expression in primates shows that D2L is highly expressed in the striatum and found specifically on MSNs and cholinergic interneurons, while D2S is instead expressed on dopaminergic axons (121). In the cortex and midbrain, D2L is mostly expressed on neuronal somata, while D2S is found on somata, dendrites, and axon terminals (121). Interestingly, high potency antipsychotics (with high affinity for the D2 receptor) appear to selectively bind those receptors expressed in the striatum (a structure with high D2L/D2S ratio) (122, 123), supporting the notion that antipsychotics could bind both D2 isoforms, but that effective antipsychotic doses would bind largely D2L and only a small proportion of total D2S receptors in the brain. Although this possibility is not completely supported from binding studies using cloned D2 receptors in cultured cells (124–127), saturation binding studies and in vivo studies with ED50 antipsychotics using transgenic mice (i.e., D2L receptor knockout mice) appear to confirm an antipsychotic selectivity for D2L (128–131). Consistent with these observations, humans studies have shown that more effective antipsychotics have higher D2 receptor occupancy in the striatum than in the midbrain (SN). (132, 133).
Postmortem studies using brain tissue from patients with schizophrenia that received antipsychotic treatment prior to death demonstrate a significant increase in D2L mRNA in the caudate nucleus (134), arguing in favor of specific adaptations of D2L in response to chronic blockade with antipsychotics. Studies have reported that D2 receptor mRNA adaptations with chronic D2 blockade might (135, 136) or might not (137) associate with membrane receptor expression suggesting that post-transcriptional mechanisms might more robustly control D2 receptor trafficking (138). Other studies instead demonstrate direct links between gene transcription and D2 receptor expression selectively in the striato-pallidal pathway (139). Currently, the precise action of antipsychotics on the D2 receptor isoforms is still inconclusive despite strong evidence from these studies with transgenic rodents.
Dopamine Synthesis, Release, and Uptake
Dopamine levels in schizophrenia are thought to be higher than in healthy individuals especially during psychotic episodes (140) and antipsychotics are intended to reduce this increased dopamine signaling (13). But it is unclear how this could occur when narrowly considering only D2 receptor occupancy (24). D2 receptors are expressed in the dendrites, somata, and terminals of dopaminergic neurons (autoreceptors) and in postsynaptic neurons (heteroreceptors). Dopamine stimulation of D2 autoreceptors at terminals decreases synaptic dopamine release, while stimulation of somatic D2 autoreceptors instead decreases the firing activity of these cells (141). Acute application of antipsychotics with high affinity for the D2 receptor has been found to increase dopamine release in projection areas (142), and this increase in dopamine is only minimally driven by increased dopamine neuron firing (143, 144), since application of antipsychotics directly onto somatic autoreceptors of midbrain dopamine neurons causes only modest dopamine release (145). Also, postsynaptic D2 heteroreceptors can moderately regulate extracellular dopamine in the striatum via GABA transmission, especially if autoreceptors are hypofunctional (131). Altogether, these seminal studies suggest that antipsychotics most effectively control dopamine transmission by targeting receptors in terminals found in the striatum. Interestingly, since most of the striatal receptors are heteroreceptors and only modestly control dopamine release, increases or decreases in extracellular dopamine levels (35, 45) are likely mediated by other mechanisms impacted by antipsychotics (38). These regulatory mechanisms include modifications to dopamine synthesis, release, and uptake.
We previously found that TH expression was not changed by effective doses of typical and atypical antipsychotics in animal models (38). However, TH expression increased when antipsychotics were no longer effective, and this was positively correlated with increased DAT expression (38). Interestingly, although TH expression did not change during antipsychotic efficacy, extracellular dopamine increased and vesicular release of dopamine decreased, suggesting that antipsychotics contributed to modulation of extracellular dopamine via reduced uptake rather than modified synthesis. Thus, changes in extracellular dopamine levels can be independent from the synthesis rate and may rely more on autoinhibition and uptake (38, 164), and/or a compensatory activity of TH (160).
Another possibility as to how antipsychotics regulate striatal dopamine output is through their direct impact on the vesicular exocytosis at active zones linked with Ca2+ channels (170, 171). We previously reported that typical and atypical antipsychotics can accumulate in synaptic vesicles of cultured hippocampal neurons through an acidic trapping mechanism and inhibit Na+ channels upon release. The inhibition of Na+ channels leads to feedback inhibition of Ca2+ influx and reduced vesicular dopamine release (171). We tested this mechanism using antipsychotic treatment regimens reflecting clinically relevant outcomes of antipsychotic efficacy and resistance and found that exocytosis-mediated dopamine release was regulated in distinct ways at different points during haloperidol treatment (38). Specifically, haloperidol inhibited dopamine exocytosis in sub-chronic regimens, i.e., ≤6 days and during treatment efficacy, while dopamine exocytosis was enhanced during chronic antipsychotic treatment associated with loss of behavioral efficacy (38). This distinct regulation of vesicular release of dopamine during sub-chronic versus chronic haloperidol might reflect the involvement of two different mechanisms in which K+ channels mediate the inhibition of vesicular release, while Na+ channels counteract this inhibition (38). Antipsychotics can regulate dopamine release by directly binding the open state of K+ channels (i.e. Kv4.3) during depolarization and accelerating the decay rate of inactivation (172–174). This mechanism of action can regulate dopamine release over time independent of depolarization blockade by modifying the intrinsic excitability of dopaminergic neurons (175). Further, changes in K+ conductance can shunt the effects of innervating signals onto dopaminergic neurons, preventing changes in dopamine release. One additional mechanism through which antipsychotics may impact dopamine release involves elevation in extracellular dopamine as a consequence of antipsychotic-induced DAT blockade (38), which may activate GIRK currents at axon terminals through an interaction between D2 autoreceptors (24, 38) and Kv1 channels (176). We found that K+-mediated release of dopamine is differentially affected during antipsychotic efficacy and failure in freely moving mice undergoing treatment, although it is not yet known if this is due to a direct action of antipsychotics on K+ channels or is instead mediated indirectly by elevated endogenous dopamine. Thus, multiple lines of evidence point to the capacity of antipsychotics to impact dopamine release, even though they may not necessarily impact dopamine synthesis.
Antipsychotic Action on DAT
Previous meta-analytical studies have found no consistent evidence for DAT changes in schizophrenia (186), and autoradiographic studies found no antipsychotic-induced changes in DAT density labeled with [I25I]RTI-121 ([125I]2 beta-carboxylic acid isopropyl ester-3 beta-(4-iodophenyl)tropane) (187, 188). However, other investigations discussed above report that direct blockade of dopamine uptake contributed to the elevated extracellular dopamine in response to acute antipsychotics (146, 147, 165, 189), although the technology at the time did not allow for a clear distinction between release and uptake kinetics. More recent studies using fast scan voltammetry demonstrated that antipsychotics with high affinity for D2 receptors enhanced dopamine half-life by nearly 50% via direct DAT blockade and antagonism of D2 autoreceptors (190–192). Accordingly, a delayed dopamine half-life results from direct inhibition of DAT, since the decay phase of stimulated dopamine overflow entirely depends on uptake (193). In support of this, striatal slice recordings showed that antipsychotics do not enhance dopamine release after the first stimulation (192), contradicting the idea that D2 autoreceptor antagonism by antipsychotics blocks autoinhibition in slices. The direct inhibition of DAT with antipsychotics occurs at low affinity and antipsychotics are less potent than more selective DAT blockers like nomifensine (194–196). This helps us interpret the apparent lack of association between antipsychotics and DAT changes reported by previous studies with low sensitivity methods (187, 188). Since uptake is the main route of elimination of extracellular dopamine (180, 197) and the kinetics of diffusion are independent from release and uptake (177), then DAT blockade by antipsychotics could explain the increase in dopamine and dopamine metabolites observed in previous microdialysis studies (198–202) as well as the prolonged half-life of dopamine stimulated by K+ (189).
Additional findings from ex vivo studies support a direct interaction between antipsychotics and the DAT. Under normal physiological conditions, increased dopamine release rapidly upregulates DAT membrane expression (203, 204). Effective doses of antipsychotics given sub-chronically (i.e., 2–6 days) inhibits the production of DAT mRNA, but does not alter striatal DAT membrane expression (38). These effects are reversed (i.e., upregulation of DAT mRNA and protein) during chronic antipsychotic treatments associated with loss of behavioral efficacy (38). We and others (205) have found similar DAT adaptations in vivo (38). MicroPET imaging using [18F]FP-CMT ([18F] N-3-fluoropropyl-2-beta-carbomethoxy-3-beta-(4’ methylphenyl)) nortropane, with superior properties for imaging the DAT in the living brain (38, 206), was applied to rats at baseline and follow-up (i.e., during loss of antipsychotic efficacy). Rats show an increase in DAT availability (binding potential; BPND) during antipsychotic failure, suggesting the putative relevance of dopamine clearance for achieving antipsychotic therapeutic response, at least in animal models. Interestingly, changes affecting DAT expression and corresponding behavioral responses to antipsychotics are accompanied by a stable and clinically relevant D2 receptor blockade (69%) and by increased or decreased extracellular dopamine in the striatum, during the expression of antipsychotic efficacy and failure, respectively (35, 45). Furthermore, the importance of DAT function in antipsychotic efficacy is supported by genetic studies showing an association between clozapine efficacy and DAT gene polymorphism (207). Regarding the question of where dopamine goes when both presynaptic and postsynaptic D2 receptors are blocked, these studies suggest that it might be captured by DAT, which is upregulated by clinical doses of antipsychotics (38). However, contrary to the obvious theoretical expectation that reduced dopamine would optimize antipsychotic therapeutic response, we found that it coincided with loss of antipsychotic efficacy. This counterintuitive result has been elaborated elsewhere (24, 38), but it will be briefly recapitulated in the next section and discussed in the context of antipsychotic-resistant schizophrenia.
Dopamine Autoinhibition as a Feature of Antipsychotic Responsiveness
We have previously proposed a model of antipsychotic efficacy, based on the potential therapeutic properties of endogenous dopamine, by taking into account a number of factors encountered in the clinic and in experimental studies with humans and animals (24, 38). We suggested that antipsychotic efficacy, as observed in animals treated with continuous doses of antipsychotics reaching clinically relevant D2 receptor blockade, is driven by dynamic interactions between endogenous dopamine and presynaptic D2 receptors. This suggestion is justified by independent but related findings showing that antipsychotic efficacy occurs in conjunction with high striatal extracellular dopamine in humans and animals (35, 38, 45, 46), while only a proportion of the total striatal D2 receptors are blocked with antipsychotics in human patients (13, 62) and animals (35, 38). On the other hand, antipsychotic treatment failure is observed when extracellular dopamine (35, 38, 45), but not D2 receptor blockade (38), is decreased (Figure 1). This fluctuation in extracellular dopamine and antipsychotic response over continuous treatment regimens characterized by stable D2 receptor blockade led us to hypothesize that antipsychotics impact the interaction between endogenous dopamine and the D2 receptor pool available for binding. Under physiological conditions, spontaneous release of dopamine stimulates a greater proportion of D2 than D1 receptors (208, 209) and antipsychotics can bind to all dopamine receptors (24, 210). Therefore, when therapeutic doses of antipsychotics reach the brain, about 70% of D2 receptors will be blocked along with a modest proportion of D1 receptors. As a consequence, endogenous dopamine will interact with spare dopamine receptors and particularly with D2 receptors, since this type, relative to D1 receptors, is stimulated by low levels of dopamine (209). The resulting neuronal response will be dictated by the molecular characteristics of the D2 receptors (i.e., Gi/o inhibitory coupled protein). During phasic dopamine release (i.e., that which would be expected to induce a psychotic episode in schizophrenia), dopamine reaches presynaptic autoreceptors, producing antipsychotic-dependent dopamine-mediated autoinhibition and a corresponding antipsychotic efficacy (24, 38).
This autoinhibition might be mediated by the D2S isoform since the two splice variants have distinct functions and are unevenly distributed within the striatonigral dopaminergic circuitry (Figure 2A, C). Furthermore, antipsychotics appear to preferentially bind dopamine receptors in the striatum (123), a brain structure with predominant expression of D2L as discussed above, and dopamine exhibits higher binding affinity for D2S in transgenic mice (130) and in cell culture (113). Together these data suggest that therapeutic doses of antipsychotics in the brain cause a functional segregation of D2S and D2L, which based on the data available until now could overlap with a functional segregation of pre- and post-synaptic D2 receptors (Figure 2A, C). It should be noted that both isoforms are expressed in pre- and post-synaptic neurons and the functional segregation might also occur within the same cells (Figure 2B). In support of this theory are studies with human schizophrenia patients demonstrating selective reduction in expression of D2S mRNA (211), potentially indicative of a desensitization of the short isoform in response to increased dopamine activity on this receptor. On the other hand, postmortem studies also show that D2L mRNA is upregulated in patients with schizophrenia (212), which may indicate an adaptive response to chronic blockade (119).
Since phasic discharge leads to large extracellular increases in dopamine (213) and is thought to underlie psychotic experiences (9, 41, 46, 140, 214–217), we propose that a therapeutic antipsychotic response is obtained by antipsychotic drugs when an adequate proportion of D2 receptors is blocked and extracellular dopamine levels are sufficiently elevated to trigger autoinhibition. This crucial combination of effects is achieved by the direct blockade of DAT by antipsychotics (38, 146, 147, 165, 189), which allows for an accumulation of synaptic dopamine that reduces the threshold at which phasic dopamine activates homeostatic autoinhibition. The antipsychotic-induced facilitation of dopamine autoinhibition, mediated by DAT blockade and D2 autoreceptor stimulation, which may serve as an antipsychotic mechanism is depicted in Figure 3. Although we have arrived at this hypothesis by analyzing multiple experimental observations, which sometimes lack corresponding human studies, our functional predictions on the association between extracellular dopamine and antipsychotic therapeutic responsiveness in humans and animals have been observed by a number of independent groups (35, 38, 45–48, 49, 218). In the following section, we will provide naturalistic examples of the potential importance of functional DAT to understanding antipsychotic-resistant schizophrenia.
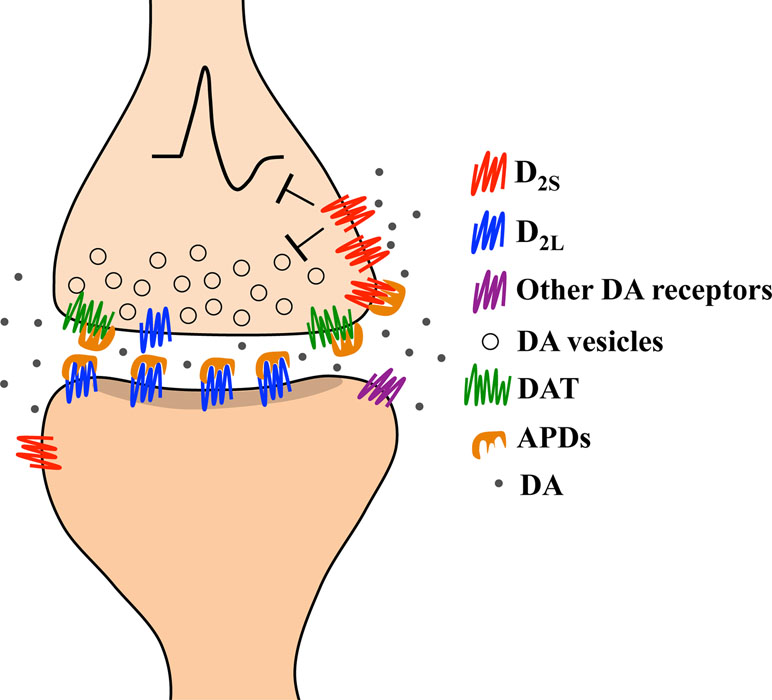
Figure 3 Representation of the hypothesized pharmacological mechanism underlying a therapeutic response in schizophrenia based on human and animal studies. Therapeutic doses of antipsychotic drugs (APDs) block about 70% of striatal D2 receptors. APDs mostly block heteroreceptors, which are more often D2L than D2S, as well as a smaller proportion of autoreceptors (which are more often D2S than D2L). APDs also block the dopamine transporter (DAT). The combined blockade of D2L heteroreceptors and DAT causes synaptic accumulation of dopamine that allows stimulation of spare D2S receptors. Phasic release of dopamine in response to environmental changes will trigger an enduring autoinhibition since extracellular dopamine levels are already elevated. We hypothesize that the autoinhibition triggered by a phasic discharge of dopamine during antipsychotic treatment is the mechanism underlying a therapeutic antipsychotic response.
The Role of DAT in Antipsychotic-Resistant Schizophrenia: Lessons from Aging and Drug Addiction
If extracellular dopamine levels contribute to the generation of a therapeutic antipsychotic response and DAT is the main physiological regulator of extracellular dopamine levels, then DAT should have a role in the expression of antipsychotic-resistant schizophrenia. Furthermore, if DAT activity quickly adapts to changes in extracellular dopamine, then it would be surprising if DAT was unaltered in schizophrenia, a disorder with symptoms attributed to dysregulated dopamine neurotransmission. We have described above how blockade of DAT may be a critical factor in antipsychotic efficacy, since DAT blockade allows accumulation of extracellular dopamine and consequently dopamine-mediated autoinhibition upon phasic transmitter release. We have also described research showing that antipsychotics given to rodents at therapeutic doses induce DAT upregulation during loss of behavioral efficacy (38). The loss of efficacy in this scenario coincides with a reduction in extracellular dopamine, which we predict reduces the capacity of dopaminergic terminals to undergo autoinhibition upon phasic release. On the other hand, we introduce below an additional scenario in which reduced expression of DAT may also prove deleterious in terms of antipsychotic therapeutic efficacy. Although theoretically low DAT expression would allow accumulation of extracellular dopamine, which we hypothesize is essential for therapeutic efficacy (Figure 3), proteins regulating extracellular dopamine levels including DAT, D2 autoreceptors, ion channels, and dopamine synthesis machinery appear to be co-regulated (131, 160–163, 172). Thus, DAT downregulation at the expression level may also negatively impact the capacity of dopaminergic terminals to undergo autoinhibition. We predict that downregulation of proteins regulating physiological dopamine neurotransmission at baseline (i.e., tonic neurotransmission) could be the underlying neurobiology of primary antipsychotic treatment-resistant schizophrenia. Figure 4 depicts a scenario in which the absence of autoinhibition due to ablated DAT expression and autoreceptor co-regulation allows for an enduring stimulation of free unbound post-synaptic receptors, leading to psychosis despite a reduction in dopamine release overall. We can characterize this condition as a form of dopamine supersensitivity driven entirely by presynaptic adaptations. Although DAT expression has been found to change in animal models of antipsychotic responsivity, it cannot be assumed that the same mechanism applies in humans with schizophrenia. Indeed, data may differ across species as already shown with D2 receptor binding (219) and dopamine synthesis (153). Therefore, why should this principle of species incompatibility not also apply for dopamine uptake? We can gain a better understanding of this issue only after testing it in human patients.
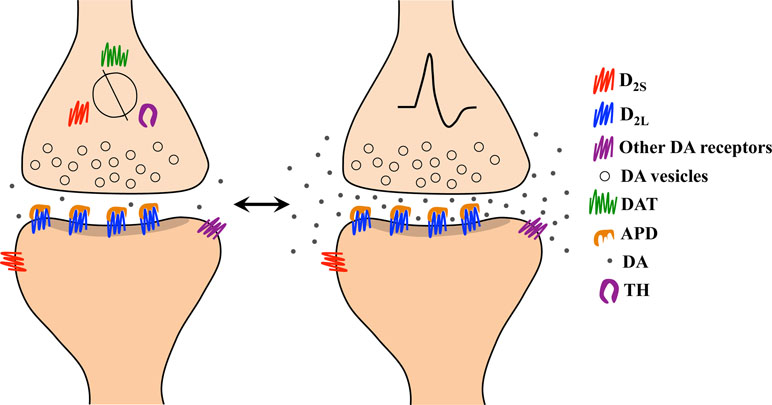
Figure 4 Representation of the pharmacological mechanism underlying the absence of therapeutic response in antipsychotic-resistant schizophrenia based on our model. (Left) Aging and/or addictive drugs consumed before antipsychotic treatment begins (i.e., in first episode psychosis) lead to reduced expression of the dopamine transporter (DAT), D2 autoreceptors, and tyrosine hydroxylase (TH), as these proteins appear to be co-regulated, at least in rodents. (Right) During environmentally evoked phasic dopamine release, impaired capacity for autoinhibition results from low levels of DAT and D2 autoreceptors. The resulting post-synaptic stimulation contributes to psychosis despite a significant blockade of D2 receptors by antipsychotic drugs (APDs).
Aging
Meta-analytical studies report that DAT levels in schizophrenia are mostly decreased, unchanged, and sometimes increased (186). These data were obtained mostly with untreated patients and therefore we hypothesize (24, 38) that the variability of these results was consequent to genetic factors (220–222) and age. For example, DAT density can decrease with age (223). Based on our proposal, reduced DAT expression as a result of aging can decrease autoinhibition mediated by antipsychotics, due to the co-regulation of autoreceptors described above, and thus reduce antipsychotic response. Interestingly, many of the patients that participated in the aforementioned study were ∼40 years old, the age associated with a decline in DAT density (186). It would have been of interest to administer antipsychotics to these individuals and measure their responsiveness. Perhaps, they would have been non-responsive or less responsive than younger individuals and/or those with higher DAT availability. However, these were not the aims of those studies. In support of this suggestion, a previous study (19) showed that the average age of patients with treatment-resistant schizophrenia was 42 years old, while patients responsive to treatment were 25 on average. Interestingly, the treatment with antipsychotics yielded similar levels of D2 receptor occupancy (19).
Aging is an important factor underlying neuropharmacological responsiveness mediated by the dopaminergic system, since D2 receptors and DAT expression decline naturally in healthy aging individuals (224–229). The reduction in D2 receptor and DAT expression is unrelated to dopamine neuron loss (229) and has profound consequences on the antipsychotic therapeutic dosing required to obtain therapeutic responsiveness in schizophrenia (230). Aging can also reveal genetic predisposition to suboptimal DAT and D2 receptor functions affecting cognitive performance in healthy individuals (231), and it can trigger degeneration of dopaminergic neurons through increased nitrative damage resulting from excess cytosolic dopamine due to an imbalance in DAT/VMAT (vesicular monoamine transporter-2) expression (232). This form of toxicity, deriving from an excess of cytosolic dopamine, has relevance to understand some of the extrapyramidal symptoms (232) and the loss of brain tissue in patients with schizophrenia (233). Although it is not clear if DAT changes are a main player in maladaptive functional and structural changes, both are often observed in schizophrenia and might affect antipsychotic response in elderly patients with schizophrenia (234, 235). While aging could explain the expression of antipsychotic treatment resistance in older patients, it is not yet clear why DAT function would affect antipsychotic responsiveness in younger individual with schizophrenia. A theoretical suggestion is provided in the following section.
Drug Addiction in Schizophrenia
Epidemiological studies report that nearly half of patients with schizophrenia also suffer from drug addiction (236, 237). This is about four times more prevalent than in the general population (238). If we consider that the recreational consumption of addictive drugs is common in the general population (i.e., 84% for alcohol consumption), but only a small proportion of individuals exposed to drugs of abuse become drug addicted (239, 240) and that this happens about four times more often in patients with schizophrenia, then it is possible that many of the remaining ∼50% of patients with schizophrenia without formal diagnosis for drug addiction likely consume at least some class of addictive drugs as well. The most commonly consumed drugs in patients with schizophrenia include alcohol, psychostimulants, cannabis, and tobacco (236–238). It has been suggested that patients with schizophrenia may use illicit substances to self-medicate their symptoms (236, 238, 241) as well as the side effects of antipsychotic medications (242), as self-medication with addictive drugs is indeed common in patients with mental illness (243).
All addictive drugs impact the dopaminergic system in the midbrain and in striatal structures (244, 245), a main component of the brain reward circuitry (246), and likely will also impact the DAT (221, 247–253). We theorize that consumption of substances of abuse to medicate pre-psychotic symptoms during the prodromal period is very likely to trigger psychotic episodes, and importantly, to weaken (or blunt) antipsychotic response since repeated exposure to addictive substances (including psychostimulants, cannabis, tobacco, alcohol and heroin) can decrease DAT membrane expression (248–253). This suggestion is based on our model describing the importance of functional DAT to facilitate antipsychotic mediated autoinhibition (Figure 3).
Although reduced DAT expression might be assumed to promote the effectiveness of antipsychotics, since uptake blockade with antipsychotics results in synaptic accumulation of dopamine and facilitates autoinhibition upon phasic dopamine release, receptor desensitization due to a corresponding downregulation (or phosphorylation) of autoreceptors may prevent the occurrence (or reduce the likelihood) of autoinhibition altogether (Figure 4). Not only are the DAT and D2 autoreceptors co-regulated, along with ion channels and the dopamine synthesis machinery (131, 160–163, 172), but reduced DAT, reduced D2 receptor expression, and reduced dopamine release can all be found in human psychostimulant users (254) and are linked to blunted striatal dopaminergic transmission in human patients with co-morbid schizophrenia and drug addiction (255).
It should be noted that the mechanisms described here and depicted in Figure 4 apply to the primary form of antipsychotic-resistant schizophrenia and not to acquired antipsychotic resistance (i.e., tolerance) observed in humans (23) and in animal models (35, 38, 45, 91). This distinction is fundamental since DAT plasticity underlying the acquired resistance to antipsychotics is different than what is described here. In fact, based on our own findings from animal models, chronic antipsychotic treatment up-regulates DAT (38), while other studies with humans and animals show that repeated exposure to addictive drugs reduce DAT (254, 256) and both conditions can lead to lack of antipsychotic response [see Ref. (38) for an expanded discussion of acquired antipsychotic resistance and Figure 4 for a depiction of primary resistance]. The description of several forms of DAT plasticity induced by psychotropic drugs is beyond the scope of this paper, but it should be acknowledged that the reduction of DAT expression with chronic addictive drug use is not absolute and is sensitive to several factors including treatment regimen, drug class, among others, as summarized in these interesting studies (257–259).
Antipsychotic-Resistant Schizophrenia: A Hypothetical Example
A young person who may not be aware of an underlying genetic predisposition to psychosis who becomes exposed to substances of abuse at the same rate as other non-predisposed individuals may risk impacting his or her capacity to buffer excess extracellular dopamine via drug-induced downregulation of DAT expression. This individual may seek medical intervention upon first experience of psychosis, at which time he or she will receive antipsychotic treatment and may already face reduced therapeutic efficacy due to the drug-related changes in DAT expression. On the other hand, if patients have no history of addictive substance consumption before starting antipsychotic treatment and begin using moderate doses of addictive drugs thereafter, we speculate that the effects of antipsychotics and certain categories of addictive substances on the expression and function of the dopaminergic machinery (DAT, TH, D2 receptors) may counterbalance one another (24), producing some therapeutic efficacy for a period of time. This might explain the high rate of smoking and use of illicit substances among patients with schizophrenia.
In summary, we propose that antipsychotic efficacy in patients with schizophrenia and particularly the contribution of DAT expression to antipsychotic response may be influenced by genetic factors as well as environmental factors such as age or history of drug use/abuse. We hypothesize that a history of drug use prior to onset of schizophrenia could be a potential risk factor to becoming antipsychotic treatment resistant, since previous exposure to addictive substances may decrease DAT expression and impair the synaptic machinery required for autoinhibition, which we theorize underlies antipsychotic responsiveness during medical intervention in schizophrenia. Antipsychotic-resistant schizophrenia patients may still respond to clozapine despite reduced DAT expression, because clozapine in particular stimulates serotonin release [for an overview, see Refs. (168, 169)], which suppresses dopaminergic firing (259–262) and thus may compensate for the absence of dopamine-mediated autoinhibition. Though based on a breadth of clinical and bench research, this theoretical suggestion is speculative and requires validation. A more thorough evaluation of this possibility might entail assessment of patient demographics, including history of drug use or abuse, as well as the drug classes used and frequency of use, along with a history of therapeutic responsiveness or resistance when treated with typical or atypical antipsychotics.
Conclusion
Although we acknowledge the genetic and neurobiological complexity of schizophrenia and its relevance for the efficacy of pharmacological treatment, we propose that sufficient DAT expression in the brains of patients with schizophrenia may be necessary for an adequate antipsychotic response in first episode psychosis. Particularly, we suggest that the antipsychotic-mediated reduction in dopamine re-uptake by direct DAT blockade allows accumulation of dopamine in the synaptic cleft, which increases the efficiency by which phasically discharged dopamine triggers presynaptic autoinhibition. Furthermore, given the apparent selectivity of antipsychotics for the D2L isoform and the predominant presynaptic expression of D2S in the midbrain, phasic dopamine is likely to activate D2S, which specifically reduces neuronal excitability. Thus, the functional and spatial segregation of the D2 receptor isoforms within the striatum and midbrain may contribute to the generation of an antipsychotic response. We further propose that consumption of addictive drugs prior to onset of schizophrenia symptoms might reduce expression of both DAT and D2 autoreceptors and will increase the risk of antipsychotic resistance upon treatment. Similarly, since DAT and D2 receptor expression decline with age, aging itself may serve as a risk factor for antipsychotic resistance. Although these hypotheses require further validation, our theory points to the importance of a functional level of membrane DAT expression in patients with schizophrenia in order to gain therapeutic benefit from antipsychotics.
Author Contributions
DA conceptualized the ideas presented and wrote the first draft. DA, AK, A-NS, and AH wrote the final manuscript. AK made the figures. All authors have read and approved the final version of the manuscript.
Funding
DA is supported by the Deutsche Forschungsgemeinschaft (AM 488/1-1) and by the Brain & Behavior Research Foundation (NARSAD Young Investigator Award 2018). AK is supported by the National Institutes of Health (DA044782). A-NS is supported by a salary award from the Fonds de la Recherche du Québec-Santé (28988).
Conflict of Interest Statement
A-NS is a consultant for H. Lundbeck A/S. This had no influence on the manuscript’s content. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature (2014) 511(7510):421–7. doi: 10.1038/nature13595
2. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature (2016) 530(7589):177–83. doi: 10.1038/nature16549
3. Avramopoulos D. Recent advances in the genetics of schizophrenia. Mol Neuropsychiatry (2018) 4(1):35–51. doi: 10.1159/000488679
4. van Os J, Kenis G, Rutten BP. The environment and schizophrenia. Nature (2010) 468(7321):203–12. doi: 10.1038/nature09563
5. Awad AG, Voruganti LN, Heslegrave RJ. A conceptual model of quality of life in schizophrenia: description and preliminary clinical validation. Qual Life Res (1997) 6(1):21–6.
6. Wong AH, Voruganti LN, Heslegrave RJ, Awad AG. Neurocognitive deficits and neurological signs in schizophrenia. Schizophr Res (1997) 23(2):139–46. doi: 10.1016/S0920-9964(96)00095-3
7. Fusar-Poli P, Borgwardt S, Bechdolf A, Addington J, Riecher-Rossler A, Schultze-Lutter F, et al. The psychosis high-risk state: a comprehensive state-of-the-art review. JAMA Psychiatry (2013) 70(1):107–20. doi: 10.1001/jamapsychiatry.2013.269
8. Heinrichs RW, Zakzanis KK. Neurocognitive deficit in schizophrenia: a quantitative review of the evidence. Neuropsychology (1998) 12(3):426–45. doi: 10.1037//0894-4105.12.3.426
9. Heinz A. Dopaminergic dysfunction in alcoholism and schizophrenia—psychopathological and behavioral correlates. Eur Psychiatry (2002) 17(1):9–16. doi: 10.1016/S0924-9338(02)00628-4
10. de Bartolomeis A, Balletta R, Giordano S, Buonaguro EF, Latte G, Iasevoli F. Differential cognitive performances between schizophrenic responders and non-responders to antipsychotics: correlation with course of the illness, psychopathology, attitude to the treatment and antipsychotics doses. Psychiatry Res (2013) 210(2):387–95. doi: 10.1016/j.psychres.2013.06.042
11. Green MF, Harvey PD. Cognition in schizophrenia: past, present, and future. Schizophr Res Cogn (2014) 1(1):e1–e9. doi: 10.1016/j.scog.2014.02.001
12. Radua J, Ramella-Cravaro V, Ioannidis JPA, Reichenberg A, Phiphopthatsanee N, Amir T, et al. What causes psychosis? An umbrella review of risk and protective factors. World Psychiatry (2018) 17(1):49–66. doi: 10.1002/wps.20490
13. Kapur S, Zipursky R, Jones C, Remington G, Houle S. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry (2000a) 157(4):514–20. doi: 10.1176/appi.ajp.157.4.514
14. Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science (1976) 192(4238):481–3. doi: 10.1126/science.3854
15. Seeman P, Lee T, Chau-Wong M, Wong K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature (1976) 261(5562):717–9. doi: 10.1038/261717a0
16. Johnstone EC, Crow TJ, Frith CD, Carney MW, Price JS. Mechanism of the antipsychotic effect in the treatment of acute schizophrenia. Lancet (1978) 1(8069):848–51. doi: 10.1016/S0140-6736(78)90193-9
17. Howes OD, Egerton A, Allan V, McGuire P, Stokes P, Kapur S. Mechanisms underlying psychosis and antipsychotic treatment response in schizophrenia: insights from PET and SPECT imaging. Curr Pharm Des (2009) 15(22):2550–9. doi: 10.2174/138161209788957528
18. Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. Arch Gen Psychiatry (1988) 45(9):789–96. doi: 10.1001/archpsyc.1988.01800330013001
19. Wolkin A, Barouche F, Wolf AP, Rotrosen J, Fowler JS, Shiue CY, et al. Dopamine blockade and clinical response: evidence for two biological subgroups of schizophrenia. Am J Psychiatry (1989) 146(7):905–8. doi: 10.1176/ajp.146.7.905
20. Farde L, Nordstrom AL, Wiesel FA, Pauli S, Halldin C, Sedvall G. Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Arch Gen Psychiatry (1992) 49(7):538–44. doi: 10.1001/archpsyc.1992.01820070032005
21. Pilowsky LS, Costa DC, Ell PJ, Murray RM, Verhoeff NP, Kerwin RW. Clozapine, single photon emission tomography, and the D2 dopamine receptor blockade hypothesis of schizophrenia. Lancet (1992) 340(8813):199–202. doi: 10.1016/0140-6736(92)90467-H
22. Pilowsky LS, Costa DC, Ell PJ, Murray RM, Verhoeff NP, Kerwin RW. Antipsychotic medication, D2 dopamine receptor blockade and clinical response: a 123I IBZM SPET (single photon emission tomography) study. Psychol Med (1993) 23(3):791–7. doi: 10.1017/S0033291700025575
23. Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med (2005) 353(12):1209–23. doi: 10.1056/NEJMoa051688
24. Amato D, Vernon AC, Papaleo F. Dopamine, the antipsychotic molecule: a perspective on mechanisms underlying antipsychotic response variability. Neurosci Biobehav Rev (2017) 85:146–59. doi: 10.1016/j.neubiorev.2017.09.027
25. Demjaha A, Lappin JM, Stahl D, Patel MX, MacCabe JH, Howes OD, et al. Antipsychotic treatment resistance in first-episode psychosis: prevalence, subtypes and predictors. Psychol Med (2017) 47(11):1981–9. doi: 10.1017/S0033291717000435
26. Beasley CM Jr., Stauffer VL, Liu-Seifert H, Taylor CC, Dunayevich E, Davis JM. All-cause treatment discontinuation in schizophrenia during treatment with olanzapine relative to other antipsychotics: an integrated analysis. J Clin Psychopharmacol (2007) 27(3):252–8. doi: 10.1097/JCP.0b013e3180582426
27. Wyatt RJ. Neuroleptics and the natural course of schizophrenia. Schizophr Bull (1991) 17(2):325–51. doi: 10.1093/schbul/17.2.325
28. Sullivan G, Wells KB, Morgenstern H, Leake B. Identifying modifiable risk factors for rehospitalization: a case-control study of seriously mentally ill persons in Mississippi. Am J Psychiatry (1995) 152(12):1749–56. doi: 10.1176/ajp.152.12.1749
29. Morken G, Widen JH, Grawe RW. Non-adherence to antipsychotic medication, relapse and rehospitalisation in recent-onset schizophrenia. BMC Psychiatry (2008) 8:32. doi: 10.1186/1471-244X-8-32
30. Takeuchi H, Siu C, Remington G, Fervaha G, Zipursky RB, Foussias G, et al. Does relapse contribute to treatment resistance? Antipsychotic response in first- vs. Neuropsychopharmacology (2018) 44(6):1036–42. doi: 10.1038/s41386-018-0278-3
31. Meltzer HY. Treatment-resistant schizophrenia—the role of clozapine. Curr Med Res Opin (1997) 14(1):1–20. doi: 10.1185/03007999709113338
32. McEvoy JP, Lieberman JA, Stroup TS, Davis SM, Meltzer HY, Rosenheck RA, et al. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry (2006) 163(4):600–10. doi: 10.1176/appi.ajp.163.4.600
33. Lieberman JA, Safferman AZ, Pollack S, Szymanski S, Johns C, Howard A, et al. Clinical effects of clozapine in chronic schizophrenia: response to treatment and predictors of outcome. Am J Psychiatry (1994) 151(12):1744–52. doi: 10.1176/ajp.151.12.1744
34. Chouinard G, Jones BD. Neuroleptic-induced supersensitivity psychosis: clinical and pharmacologic characteristics. Am J Psychiatry (1980) 137(1):16–21. doi: 10.1176/ajp.137.1.16
35. Samaha AN, Seeman P, Stewart J, Rajabi H, Kapur S. “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci (2007) 27(11):2979–86. doi: 10.1523/JNEUROSCI.5416-06.2007
36. Molina V, Reig S, Sanz J, Palomo T, Benito C, Sarramea F, et al. Differential clinical, structural and P300 parameters in schizophrenia patients resistant to conventional neuroleptics. Prog Neuropsychopharmacol Biol Psychiatry (2008) 32(1):257–66. doi: 10.1016/j.pnpbp.2007.08.017
37. Demjaha A, Egerton A, Murray RM, Kapur S, Howes OD, Stone JM, et al. Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol Psychiatry (2014) 75(5):e11–13. doi: 10.1016/j.biopsych.2013.06.011
38. Amato D, Canneva F, Cumming P, Maschauer S, Groos D, Wrosch JK, et al. A dopaminergic mechanism of antipsychotic drug efficacy, failure, and failure reversal: the role of the dopamine transporter. Molecular Psychiatry (2018). doi: 10.1038/s41380-018-0114-5
39. Iwata Y, Nakajima S, Plitman E, Caravaggio F, Kim J, Shah P, et al. Glutamatergic neurometabolite levels in patients with ultra-treatment-resistant schizophrenia: a cross-sectional 3T proton magnetic resonance spectroscopy study. Biol Psychiatry (2018) 85(7):596–605. doi: 10.1016/j.biopsych.2018.09.009
40. Kane JM, Agid O, Baldwin ML, Howes O, Lindenmayer JP, Marder S, et al. Clinical guidance on the identification and management of treatment-resistant schizophrenia. J Clin Psychiatry (2019) 80(2):e1–9. doi: 10.4088/JCP.18com12123
41. Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry (2003) 160(1):13–23. doi: 10.1176/appi.ajp.160.1.13
42. Abi-Dargham A. From “bedside” to “bench” and back: a translational approach to studying dopamine dysfunction in schizophrenia. Neurosci Biobehav Rev (2018) S0149-7634(18)30314-2. doi: 10.1016/j.neubiorev.2018.12.003
43. Slifstein M, Abi-Dargham A. Is it pre- or postsynaptic? Imaging striatal dopamine excess in schizophrenia. Biol Psychiatry (2018) 83(8):635–7. doi: 10.1016/j.biopsych.2018.02.015
44. Huang E, Zai CC, Lisoway A, Maciukiewicz M, Felsky D, Tiwari AK, et al. Catechol-O-methyltransferase Val158Met polymorphism and clinical response to antipsychotic treatment in schizophrenia and schizo-affective disorder patients: a meta-analysis. Int J Neuropsychopharmacol (2016) 19(5):1–12. doi: 10.1093/ijnp/pyv132
45. Amato D, Natesan S, Yavich L, Kapur S, Muller CP. Dynamic regulation of dopamine and serotonin responses to salient stimuli during chronic haloperidol treatment. Int J Neuropsychopharmacol (2011) 14(10):1327–39. doi: 10.1017/S1461145711000010
46. Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U S A (2000) 97(14):8104–9. doi: 10.1073/pnas.97.14.8104
47. Scheggia D, Mastrogiacomo R, Mereu M, Sannino S, Straub RE, Armando M, et al. Variations in Dysbindin-1 are associated with cognitive response to antipsychotic drug treatment. Nat Commun (2018) 9(1):2265. doi: 10.1038/s41467-018-04711-w
48. Swerdlow NR, Bhakta SG, Talledo JA, Franz DM, Hughes EL, Rana BK, et al. Effects of amphetamine on sensorimotor gating and neurocognition in antipsychotic-medicated schizophrenia patients. Neuropsychopharmacology (2018) 43(4):708–17. doi: 10.1038/npp.2017.285
49. Caravaggio F, Iwata Y, Kim J, Shah P, Gerretsen P, Remington G, et al. What proportion of striatal D2 receptors are occupied by endogenous dopamine at baseline? A meta-analysis with implications for understanding antipsychotic occupancy. Neuropharmacology (2019) S0028-3908(19)30114-5. doi: 10.1016/j.neuropharm.2019.03.034
50. Tamminga CA, Schaffer MH, Smith RC, Davis JM. Schizophrenic symptoms improve with apomorphine. Science (1978) 200(4341):567–8. doi: 10.1126/science.347574
51. Weinberger DR. Thinking about schizophrenia in an era of genomic medicine. Am J Psychiatry (2019) 176(1):12–20. doi: 10.1176/appi.ajp.2018.18111275
52. Karam CS, Ballon JS, Bivens NM, Freyberg Z, Girgis RR, Lizardi-Ortiz JE, et al. Signaling pathways in schizophrenia: emerging targets and therapeutic strategies. Trends Pharmacol Sci (2010) 31(8):381–90. doi: 10.1016/j.tips.2010.05.004
53. Gilmore JH, Jarskog LF. Exposure to infection and brain development: cytokines in the pathogenesis of schizophrenia. Schizophr Res (1997) 24(3):365–7. doi: 10.1016/S0920-9964(96)00123-5
54. Nicodemus KK, Marenco S, Batten AJ, Vakkalanka R, Egan MF, Straub RE, et al. Serious obstetric complications interact with hypoxia-regulated/vascular-expression genes to influence schizophrenia risk. Mol Psychiatry (2008) 13(9):873–7. doi: 10.1038/sj.mp.4002153
55. Moreno JL, Kurita M, Holloway T, Lopez J, Cadagan R, Martinez-Sobrido L, et al. Maternal influenza viral infection causes schizophrenia-like alterations of 5-HT(2)A and mGlu(2) receptors in the adult offspring. J Neurosci (2011) 31(5):1863–72. doi: 10.1523/JNEUROSCI.4230-10.2011
56. Holloway T, Moreno JL, Umali A, Rayannavar V, Hodes GE, Russo SJ, et al. Prenatal stress induces schizophrenia-like alterations of serotonin 2A and metabotropic glutamate 2 receptors in the adult offspring: role of maternal immune system. J Neurosci (2013) 33(3):1088–98. doi: 10.1523/JNEUROSCI.2331-12.2013
57. Jiang Z, Rompala GR, Zhang S, Cowell RM, Nakazawa K. Social isolation exacerbates schizophrenia-like phenotypes via oxidative stress in cortical interneurons. Biol Psychiatry (2013) 73(10):1024–34. doi: 10.1016/j.biopsych.2012.12.004
58. Mizrahi R. Social stress and psychosis risk: common neurochemical substrates? Neuropsychopharmacology (2016) 41(3):666–74. doi: 10.1038/npp.2015.274
59. Kapur S, Mamo D. Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog Neuropsychopharmacol Biol Psychiatry (2003) 27(7):1081–90. doi: 10.1016/j.pnpbp.2003.09.004
60. van Rossum JM. The significance of dopamine-receptor blockade for the mechanism of action of neuroleptic drugs. Arch Int Pharmacodyn Ther (1966) 160(2):492–4.
61. Miyamoto S, Duncan GE, Marx CE, Lieberman JA. Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry (2005) 10(1):79–104. doi: 10.1038/sj.mp.4001556
62. Nord M, Farde L. Antipsychotic occupancy of dopamine receptors in schizophrenia. CNS Neurosci Ther (2011) 17(2):97–103. doi: 10.1111/j.1755-5949.2010.00222.x
63. Seeman P, Tallerico T. Antipsychotic drugs which elicit little or no parkinsonism bind more loosely than dopamine to brain D2 receptors, yet occupy high levels of these receptors. Mol Psychiatry (1998) 3(2):123–34. doi: 10.1038/sj.mp.4000336
64. Suhara T, Okauchi T, Sudo Y, Takano A, Kawabe K, Maeda J, et al. Clozapine can induce high dopamine D(2) receptor occupancy in vivo. Psychopharmacology (Berl) (2002) 160(1):107–12. doi: 10.1007/s00213-001-0967-0
65. Gefvert O, Lundberg T, Wieselgren IM, Bergstrom M, Langstrom B, Wiesel F, et al. D(2) and 5HT(2A) receptor occupancy of different doses of quetiapine in schizophrenia: a PET study. Eur Neuropsychopharmacol (2001) 11(2):105–10. doi: 10.1016/S0924-977X(00)00133-4
66. Farde L, Wiesel FA, Nordstrom AL, Sedvall G. D1- and D2-dopamine receptor occupancy during treatment with conventional and atypical neuroleptics. Psychopharmacology (Berl) (1989) 99:S28–31. doi: 10.1007/BF00442555
67. Nordstrom AL, Farde L, Halldin C. Time course of D2-dopamine receptor occupancy examined by PET after single oral doses of haloperidol. Psychopharmacology (Berl) (1992) 106(4):433–8. doi: 10.1007/BF02244811
68. Gefvert O, Bergstrom M, Langstrom B, Lundberg T, Lindstrom L, Yates R. Time course of central nervous dopamine-D2 and 5-HT2 receptor blockade and plasma drug concentrations after discontinuation of quetiapine (Seroquel) in patients with schizophrenia. Psychopharmacology (Berl) (1998) 135(2):119–26. doi: 10.1007/s002130050492
69. Kapur S, Zipursky R, Jones C, Shammi CS, Remington G, Seeman P. A positron emission tomography study of quetiapine in schizophrenia: a preliminary finding of an antipsychotic effect with only transiently high dopamine D2 receptor occupancy. Arch Gen Psychiatry (2000b) 57(6):553–9. doi: 10.1001/archpsyc.57.6.553
70. Kapur S, Seeman P. Antipsychotic agents differ in how fast they come off the dopamine D2 receptors. J Psychiatry Neurosci (2000) 25(2):161–6.
71. Seeman P, Guan HC, Niznik HB. Endogenous dopamine lowers the dopamine D2 receptor density as measured by [3H]raclopride: implications for positron emission tomography of the human brain. Synapse (1989) 3(1):96–7. doi: 10.1002/syn.890030113
72. Seeman P. Therapeutic receptor-blocking concentrations of neuroleptics. Int Clin Psychopharmacol (1995) 10(Suppl 3):5–13. doi: 10.1097/00004850-199509000-00002
73. Seeman P, Van Tol HH. Deriving the therapeutic concentrations for clozapine and haloperidol: the apparent dissociation constant of a neuroleptic at the dopamine D2 or D4 receptor varies with the affinity of the competing radioligand. Eur J Pharmacol (1995) 291(2):59–66. doi: 10.1016/0922-4106(95)90125-6
74. Seeman P, Tallerico T. Rapid release of antipsychotic drugs from dopamine D2 receptors: an explanation for low receptor occupancy and early clinical relapse upon withdrawal of clozapine or quetiapine. Am J Psychiatry (1999) 156(6):876–84. doi: 10.1176/ajp.156.6.876
75. Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry (2002) 47(1):27–38. doi: 10.1177/070674370204700105
76. Sahlholm K, Marcellino D, Nilsson J, Ogren SO, Fuxe K, Arhem P. Typical and atypical antipsychotics do not differ markedly in their reversibility of antagonism of the dopamine D2 receptor. Int J Neuropsychopharmacol (2014) 17(1):149–55. doi: 10.1017/S1461145713000801
77. Sahlholm K, Zeberg H, Nilsson J, Ogren SO, Fuxe K, Arhem P. The fast-off hypothesis revisited: a functional kinetic study of antipsychotic antagonism of the dopamine D2 receptor. Eur Neuropsychopharmacol (2016) 26(3):467–76. doi: 10.1016/j.euroneuro.2016.01.001
78. Heinz A, Knable MB, Weinberger DR. Dopamine D2 receptor imaging and neuroleptic drug response. J Clin Psychiatry (1996) 57(Suppl 11):84–8; discussion 89-93.
79. Kapur S, Zipursky RB, Remington G. Clinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. Am J Psychiatry (1999) 156(2):286–93. doi: 10.1176/ajp.156.2.286
80. Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet (2013) 382(9896):951–62. doi: 10.1016/S0140-6736(13)60733-3
81. Emsley R, Chiliza B, Asmal L, Harvey BH. The nature of relapse in schizophrenia. BMC Psychiatry (2013) 13:50. doi: 10.1186/1471-244X-13-50
82. Mamo D, Kapur S, Keshavan M, Laruelle M, Taylor CC, Kothare PA, et al. D2 receptor occupancy of olanzapine pamoate depot using positron emission tomography: an open-label study in patients with schizophrenia. Neuropsychopharmacology (2008) 33(2):298–304. doi: 10.1038/sj.npp.1301409
83. Nyberg S, Farde L, Halldin C, Dahl ML, Bertilsson L. D2 dopamine receptor occupancy during low-dose treatment with haloperidol decanoate. Am J Psychiatry (1995) 152(2):173–8. doi: 10.1176/ajp.152.2.173
84. Remington G, Mamo D, Labelle A, Reiss J, Shammi C, Mannaert E, et al. A PET study evaluating dopamine D2 receptor occupancy for long-acting injectable risperidone. Am J Psychiatry (2006) 163(3):396–401. doi: 10.1176/appi.ajp.163.3.396
85. Uchida H, Suzuki T. Dose and dosing frequency of long-acting injectable antipsychotics: a systematic review of PET and SPECT data and clinical implications. J Clin Psychopharmacol (2014) 34(6):728–35. doi: 10.1097/JCP.0000000000000065
86. Silvestri S, Seeman MV, Negrete JC, Houle S, Shammi CM, Remington GJ, et al. Increased dopamine D2 receptor binding after long-term treatment with antipsychotics in humans: a clinical PET study. Psychopharmacology (Berl) (2000) 152(2):174–80. doi: 10.1007/s002130000532
87. Iyo M, Tadokoro S, Kanahara N, Hashimoto T, Niitsu T, Watanabe H, et al. Optimal extent of dopamine D2 receptor occupancy by antipsychotics for treatment of dopamine supersensitivity psychosis and late-onset psychosis. J Clin Psychopharmacol (2013) 33(3):398–404. doi: 10.1097/JCP.0b013e31828ea95c
88. Chouinard G, Jones BD, Annable L. Neuroleptic-induced supersensitivity psychosis. Am J Psychiatry (1978) 135(11):1409–10. doi: 10.1176/ajp.135.11.1409
89. Fallon P, Dursun S, Deakin B. Drug-induced supersensitivity psychosis revisited: characteristics of relapse in treatment-compliant patients. Ther Adv Psychopharmacol (2012) 2(1):13–22. doi: 10.1177/2045125311431105
90. Suzuki T, Kanahara N, Yamanaka H, Takase M, Kimura H, Watanabe H, et al. Dopamine supersensitivity psychosis as a pivotal factor in treatment-resistant schizophrenia. Psychiatry Res (2015) 227(2–3):278–82. doi: 10.1016/j.psychres.2015.02.021
91. Samaha AN, Reckless GE, Seeman P, Diwan M, Nobrega JN, Kapur S. Less is more: antipsychotic drug effects are greater with transient rather than continuous delivery. Biol Psychiatry (2008) 64(2):145–52. doi: 10.1016/j.biopsych.2008.01.010
92. Gill KM, Cook JM, Poe MM, Grace AA. Prior antipsychotic drug treatment prevents response to novel antipsychotic agent in the methylazoxymethanol acetate model of schizophrenia. Schizophr Bull (2014) 40(2):341–50. doi: 10.1093/schbul/sbt236
93. Smith RC, Davis JM. Behavioral supersensitivity to apomorphine and amphetamine after chronic high dose haloperidol treatment. Psychopharmacol Commun (1975) 1(3):285–93.
94. Bedard AM, Maheux J, Levesque D, Samaha AN. Continuous, but not intermittent, antipsychotic drug delivery intensifies the pursuit of reward cues. Neuropsychopharmacology (2011) 36(6):1248–59. doi: 10.1038/npp.2011.10
95. Ericson H, Radesater AC, Servin E, Magnusson O, Mohringe B. Effects of intermittent and continuous subchronic administration of raclopride on motor activity, dopamine turnover and receptor occupancy in the rat. Pharmacol Toxicol (1996) 79(6):277–86. doi: 10.1111/j.1600-0773.1996.tb00009.x
96. Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry (1991) 148(11):1474–86. doi: 10.1176/ajp.148.11.1474
98. Pierce RC, Rowlett JK, Bardo MT, Rebec GV. Chronic ascorbate potentiates the effects of chronic haloperidol on behavioral supersensitivity but not D2 dopamine receptor binding. Neuroscience (1991) 45(2):373–8. doi: 10.1016/0306-4522(91)90234-F
99. Flores G, Barbeau D, Quirion R, Srivastava LK. Decreased binding of dopamine D3 receptors in limbic subregions after neonatal bilateral lesion of rat hippocampus. J Neurosci (1996) 16(6):2020–6. doi: 10.1523/JNEUROSCI.16-06-02020.1996
100. Ginovart N, Wilson AA, Hussey D, Houle S, Kapur S. D2-receptor upregulation is dependent upon temporal course of D2-occupancy: a longitudinal [11C]-raclopride PET study in cats. Neuropsychopharmacology (2009) 34(3):662–71. doi: 10.1038/npp.2008.116
101. Seeman P, Weinshenker D, Quirion R, Srivastava LK, Bhardwaj SK, Grandy DK, et al. Dopamine supersensitivity correlates with D2High states, implying many paths to psychosis. Proc Natl Acad Sci U S A (2005) 102(9):3513–8. doi: 10.1073/pnas.0409766102
102. Graff-Guerrero A, Mizrahi R, Agid O, Marcon H, Barsoum P, Rusjan P, et al. The dopamine D2 receptors in high-affinity state and D3 receptors in schizophrenia: a clinical [11C]-(+)-PHNO PET study. Neuropsychopharmacology (2009) 34(4):1078–86. doi: 10.1038/npp.2008.199
103. Smith AD, Bolam JP. The neural network of the basal ganglia as revealed by the study of synaptic connections of identified neurones. Trends Neurosci (1990) 13(7):259–65. doi: 10.1016/0166-2236(90)90106-K
104. Levey AI, Hersch SM, Rye DB, Sunahara RK, Niznik HB, Kitt CA, et al. Localization of D1 and D2 dopamine receptors in brain with subtype-specific antibodies. Proc Natl Acad Sci U S A (1993) 90(19):8861–65. doi: 10.1073/pnas.90.19.8861
105. Dal Toso R, Sommer B, Ewert M, Herb A, Pritchett DB, Bach A, et al. The dopamine D2 receptor: two molecular forms generated by alternative splicing. EMBO J (1989) 8(13):4025–34. doi: 10.1002/j.1460-2075.1989.tb08585.x
106. Giros B, Sokoloff P, Martres MP, Riou JF, Emorine LJ, Schwartz JC. Alternative splicing directs the expression of two D2 dopamine receptor isoforms. Nature (1989) 342(6252):923–6. doi: 10.1038/342923a0
107. Monsma FJ Jr., McVittie LD, Gerfen CR, Mahan LC, Sibley DR. Multiple D2 dopamine receptors produced by alternative RNA splicing. Nature (1989) 342(6252):926–9. doi: 10.1038/342926a0
108. Jang JY, Jang M, Kim SH, Um KB, Kang YK, Kim HJ, et al. Regulation of dopaminergic neuron firing by heterogeneous dopamine autoreceptors in the substantia nigra pars compacta. J Neurochem (2011) 116(6):966–74. doi: 10.1111/j.1471-4159.2010.07107.x
109. Gantz SC, Robinson BG, Buck DC, Bunzow JR, Neve RL, Williams JT, et al. Distinct regulation of dopamine D2S and D2L autoreceptor signaling by calcium. Elife (2015) 4:1–19. doi: 10.7554/eLife.09358
110. Robinson BG, Condon AF, Radl D, Borrelli E, Williams JT, Neve KA. Cocaine-induced adaptation of dopamine D2S, but not D2L autoreceptors. Elife (2017) 6:1–8. doi: 10.7554/eLife.31924
111. Centonze D, Gubellini P, Usiello A, Rossi S, Tscherter A, Bracci E, et al. Differential contribution of dopamine D2S and D2L receptors in the modulation of glutamate and GABA transmission in the striatum. Neuroscience (2004) 129(1):157–66. doi: 10.1016/j.neuroscience.2004.07.043
112. Zhang Y, Bertolino A, Fazio L, Blasi G, Rampino A, Romano R, et al. Polymorphisms in human dopamine D2 receptor gene affect gene expression, splicing, and neuronal activity during working memory. Proc Natl Acad Sci U S A (2007) 104(51):20552–7. doi: 10.1073/pnas.0707106104
113. Montmayeur JP, Borrelli E. Transcription mediated by a cAMP-responsive promoter element is reduced upon activation of dopamine D2 receptors. Proc Natl Acad Sci U S A (1991) 88(8):3135–9. doi: 10.1073/pnas.88.8.3135
114. Senogles SE. The D2 dopamine receptor isoforms signal through distinct Gi alpha proteins to inhibit adenylyl cyclase. J Biol Chem (1994) 269(37):23120–7. doi: 10.1073/pnas.0730708100
115. Guiramand J, Montmayeur JP, Ceraline J, Bhatia M, Borrelli E. Alternative splicing of the dopamine D2 receptor directs specificity of coupling to G-proteins. J Biol Chem (1995) 270(13):7354–8. doi: 10.1074/jbc.270.13.7354
116. Lindgren N, Usiello A, Goiny M, Haycock J, Erbs E, Greengard P, et al. Distinct roles of dopamine D2L and D2S receptor isoforms in the regulation of protein phosphorylation at presynaptic and postsynaptic sites. Proc Natl Acad Sci U S A (2003) 100(7):4305–9. doi: 10.1073/pnas.0730708100
117. Mack KJ, O’Malley KL, Todd RD. Differential expression of dopaminergic D2 receptor messenger RNAs during development. Brain Res Dev Brain Res (1991) 59(2):249–51. doi: 10.1016/0165-3806(91)90105-R
118. O’Malley KL, Mack KJ, Gandelman KY, Todd RD. Organization and expression of the rat D2A receptor gene: identification of alternative transcripts and a variant donor splice site. Biochemistry (1990) 29(6):1367–71. doi: 10.1021/bi00458a003
119. Neve KA, Neve RL, Fidel S, Janowsky A, Higgins GA. Increased abundance of alternatively spliced forms of D2 dopamine receptor mRNA after denervation. Proc Natl Acad Sci U S A (1991) 88(7):2802–6. doi: 10.1073/pnas.88.7.2802
120. Gandelman KY, Harmon S, Todd RD, O’Malley KL. Analysis of the structure and expression of the human dopamine D2A receptor gene. J Neurochem (1991) 56(3):1024–9. doi: 10.1111/j.1471-4159.1991.tb02024.x
121. Khan ZU, Mrzljak L, Gutierrez A, de la Calle A, Goldman-Rakic PS. Prominence of the dopamine D2 short isoform in dopaminergic pathways. Proc Natl Acad Sci U S A (1998) 95(13):7731–6. doi: 10.1073/pnas.95.13.7731
122. Meador-Woodruff JH, Mansour A, Civelli O, Watson SJ. Distribution of D2 dopamine receptor mRNA in the primate brain. Prog Neuropsychopharmacol Biol Psychiatry (1991) 15(6):885–93. doi: 10.1016/0278-5846(91)90016-T
123. Fishburn CS, David C, Carmon S, Fuchs S. The effect of haloperidol on D2 dopamine receptor subtype mRNA levels in the brain. FEBS Lett (1994) 339(1–2):63–6. doi: 10.1016/0014-5793(94)80385-4
124. Castro SW, Strange PG. Differences in the ligand binding properties of the short and long versions of the D2 dopamine receptor. J Neurochem (1993) 60(1):372–5. doi: 10.1111/j.1471-4159.1993.tb05863.x
125. Malmberg A, Jackson DM, Eriksson A, Mohell N. Unique binding characteristics of antipsychotic agents interacting with human dopamine D2A, D2B, and D3 receptors. Mol Pharmacol (1993) 43(5):749–54.
126. Schotte A, Janssen PF, Gommeren W, Luyten WH, Van Gompel P, Lesage AS, et al. Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology (Berl) (1996) 124(1–2):57–73. doi: 10.1007/BF02245606
127. Kongsamut S, Roehr JE, Cai J, Hartman HB, Weissensee P, Kerman LL, et al. Iloperidone binding to human and rat dopamine and 5-HT receptors. Eur J Pharmacol (1996) 317(2–3):417–23. doi: 10.1016/S0014-2999(96)00840-0
128. Usiello A, Baik JH, Rouge-Pont F, Picetti R, Dierich A, LeMeur M, et al. Distinct functions of the two isoforms of dopamine D2 receptors. Nature (2000) 408(6809):199–203. doi: 10.1038/35041572
129. Wang Y, Xu R, Sasaoka T, Tonegawa S, Kung MP, Sankoorikal EB. Dopamine D2 long receptor-deficient mice display alterations in striatum-dependent functions. J Neurosci (2000) 20(22):8305–14. doi: 10.1523/JNEUROSCI.20-22-08305.2000
130. Xu R, Hranilovic D, Fetsko LA, Bucan M, Wang Y. Dopamine D2S and D2L receptors may differentially contribute to the actions of antipsychotic and psychotic agents in mice. Mol Psychiatry (2002) 7(10):1075–82. doi: 10.1038/sj.mp.4001145
131. Anzalone A, Lizardi-Ortiz JE, Ramos M, De Mei C, Hopf FW, Iaccarino C, et al. Dual control of dopamine synthesis and release by presynaptic and postsynaptic dopamine D2 receptors. J Neurosci (2012) 32(26):9023–34. doi: 10.1523/JNEUROSCI.0918-12.2012
132. Kessler RM , Ansari MS, Riccardi P, Li R, Jayathilake K, Dawant B, et al. Occupancy of striatal and extrastriatal dopamine D2 receptors by clozapine and quetiapine. Neuropsychopharmacology (2006) 31(9):1991–2001. doi: 10.1038/sj.npp.1301108
133. Kessler RM , Ansari MS, Riccardi P, Li R, Jayathilake K, Dawant B, et al. Occupancy of striatal and extrastriatal dopamine D2/D3 receptors by olanzapine and haloperidol. Neuropsychopharmacology (2005) 30:2283–9. doi: 10.1038/sj.npp.1300836
134. Roberts DA, Balderson D, Pickering-Brown SM, Deakin JF, Owen F. The abundance of mRNA for dopamine D2 receptor isoforms in brain tissue from controls and schizophrenics. Brain Res Mol Brain Res (1994) 25(1–2):173–5. doi: 10.1016/0169-328X(94)90296-8
135. Le Moine C, Normand E, Guitteny AF, Fouque B, Teoule R, Bloch B. Dopamine receptor gene expression by enkephalin neurons in rat forebrain. Proc Natl Acad Sci U S A (1990) 87(1):230–4. doi: 10.1073/pnas.87.1.230
136. Angulo JA, Coirini H, Ledoux M, Schumacher M. Regulation by dopaminergic neurotransmission of dopamine D2 mRNA and receptor levels in the striatum and nucleus accumbens of the rat. Brain Res Mol Brain Res (1991) 11(2):161–6. doi: 10.1016/0169-328X(91)90117-G
137. van Tol HH, Riva M, Civelli O, Creese I. Lack of effect of chronic dopamine receptor blockade on D2 dopamine receptor mRNA level. Neurosci Lett (1990) 111(3):303–8. doi: 10.1016/0304-3940(90)90279-I
138. Srivastava LK, Morency MA, Bajwa SB, Mishra RK. Effect of haloperidol on expression of dopamine D2 receptor mRNAs in rat brain. J Mol Neurosci (1990) 2(3):155–61. doi: 10.1007/BF02896840
139. Fox CA, Mansour A, Watson SJ Jr. The effects of haloperidol on dopamine receptor gene expression. Exp Neurol (1994) 130(2):288–303. doi: 10.1006/exnr.1994.1207
140. Laruelle M, Abi-Dargham A. Dopamine as the wind of the psychotic fire: new evidence from brain imaging studies. J Psychopharmacol (1999) 13(4):358–71. doi: 10.1177/026988119901300405
141. Roth RH. CNS dopamine autoreceptors: distribution, pharmacology, and function. Ann N Y Acad Sci (1984) 430:27–53. doi: 10.1111/j.1749-6632.1984.tb14497.x
142. Westerink BH, de Vries JB. On the mechanism of neuroleptic induced increase in striatal dopamine release: brain dialysis provides direct evidence for mediation by autoreceptors localized on nerve terminals. Neurosci Lett (1989) 99(1–2):197–202. doi: 10.1016/0304-3940(89)90289-9
143. Bunney BS, Walters JR, Roth RH, Aghajanian GK. Dopaminergic neurons: effect of antipsychotic drugs and amphetamine on single cell activity. J Pharmacol Exp Ther (1973) 185(3):560–71.
144. Chiodo LA, Bunney BS. Typical and atypical neuroleptics: differential effects of chronic administration on the activity of A9 and A10 midbrain dopaminergic neurons. J Neurosci (1983) 3(8):1607–19. doi: 10.1523/JNEUROSCI.03-08-01607.1983
145. Santiago M, Westerink BH. The regulation of dopamine release from nigrostriatal neurons in conscious rats: the role of somatodendritic autoreceptors. Eur J Pharmacol (1991) 204(1):79–85. doi: 10.1016/0014-2999(91)90838-H
146. Seeman P, Lee T. The dopamine-releasing actions of neuroleptics and ethanol. J Pharmacol Exp Ther (1974) 190(1):131–40.
147. Iversen LL, Rogawski MA, Miller RJ. Comparison of the effects of neuroleptic drugs on pre- and postsynaptic dopaminergic mechanisms in the rat striatum. Mol Pharmacol (1976) 12(2):251–62.
148. Carlsson A, Lindqvist M. Effect of chlorpromazine or haloperidol on formation of 3methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol (Copenh) (1963) 20:140–4. doi: 10.1111/j.1600-0773.1963.tb01730.x
149. Cumming P, Ase A, Laliberte C, Kuwabara H, Gjedde A. In vivo regulation of DOPA decarboxylase by dopamine receptors in rat brain. J Cereb Blood Flow Metab (1997) 17(11):1254–60. doi: 10.1097/00004647-199711000-00014
150. Danielsen EH, Smith D, Hermansen F, Gjedde A, Cumming P. Acute neuroleptic stimulates DOPA decarboxylase in porcine brain in vivo. Synapse (2001) 41(2):172–5. doi: 10.1002/syn.1071
151. Lazar MA, Mefford IN, Barchas JD. Tyrosine hydroxylase activation. Comparison of in vitro phosphorylation and in vivo administration of haloperidol. Biochem Pharmacol (1982) 31(16):2599–607. doi: 10.1016/0006-2952(82)90706-7
152. Hakansson K, Pozzi L, Usiello A, Haycock J, Borrelli E, Fisone G. Regulation of striatal tyrosine hydroxylase phosphorylation by acute and chronic haloperidol. Eur J Neurosci (2004) 20(4):1108–12. doi: 10.1111/j.1460-9568.2004.03547.x
153. Mamo D, Remington G, Nobrega J, Hussey D, Chirakal R, Wilson AA, et al. Effect of acute antipsychotic administration on dopamine synthesis in rodents and human subjects using 6-[18F]-L-m-tyrosine. Synapse (2004) 52(2):153–62. doi: 10.1002/syn.20016
154. Cumming P, Ase A, Kuwabara H, Gjedde A. [3H]DOPA formed from [3H]tyrosine in living rat brain is not committed to dopamine synthesis. J Cereb Blood Flow Metab (1998) 18(5):491–9. doi: 10.1097/00004647-199805000-00004
155. Magelund G, Gerlach J, Casey DE. Neuroleptic-potentiating effect of alpha-methyl-p-tyrosine compared with haloperidol and placebo in a double-blind cross-over trial. Acta Psychiatr Scand (1979) 60(2):185–9. doi: 10.1097/00004647-199805000-00004
156. Ito H, Takano H, Takahashi H, Arakawa R, Miyoshi M, Kodaka F, et al. Effects of the antipsychotic risperidone on dopamine synthesis in human brain measured by positron emission tomography with L-[beta-11C]DOPA: a stabilizing effect for dopaminergic neurotransmission? J Neurosci (2009) 29(43):13730–4. doi: 10.1111/j.1600-0447.1979.tb03587.x
157. Grunder G, Vernaleken I, Muller MJ, Davids E, Heydari N, Buchholz HG, et al. Subchronic haloperidol downregulates dopamine synthesis capacity in the brain of schizophrenic patients in vivo. Neuropsychopharmacology (2003) 28(4):787–94. doi: 10.1523/JNEUROSCI.4172-09.2009
158. Vernaleken I, Kumakura Y, Cumming P, Buchholz HG, Siessmeier T, Stoeter P, et al. Modulation of [18F]fluorodopa (FDOPA) kinetics in the brain of healthy volunteers after acute haloperidol challenge. Neuroimage (2006) 30(4):1332–9. doi: 10.1038/sj.npp.1300103
159. Eisenberg DP, Yankowitz L, Ianni AM, Rubinstein DY, Kohn PD, Hegarty CE, et al. Presynaptic dopamine synthesis capacity in schizophrenia and striatal blood flow change during antipsychotic treatment and medication-free conditions. Neuropsychopharmacology (2017) 42(11):2232–41. doi: 10.1016/j.neuroimage.2005.11.014
160. Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci U S A (1998) 95(7):4029–34. doi: 10.1038/npp.2017.67
161. Rocha BA, Fumagalli F, Gainetdinov RR, Jones SR, Ator R, Giros B, et al. Cocaine self-administration in dopamine-transporter knockout mice. Nat Neurosci (1998) 1(2):132–7. doi: 10.1073/pnas.95.7.4029
162. Dickinson SD, Sabeti J, Larson GA, Giardina K, Rubinstein M, Kelly MA, et al. Dopamine D2 receptor-deficient mice exhibit decreased dopamine transporter function but no changes in dopamine release in dorsal striatum. J Neurochem (1999) 72(1):148–56. doi: 10.1038/381
163. Salvatore MF, Calipari ES, Jones SR. Regulation of tyrosine hydroxylase expression and phosphorylation in dopamine transporter-deficient mice. ACS Chem Neurosci (2016) 7(7):941–51. doi: 10.1046/j.1471-4159.1999.0720148.x
164. Schmitz Y, Schmauss C, Sulzer D. Altered dopamine release and uptake kinetics in mice lacking D2 receptors. J Neurosci (2002) 22(18):8002–9. doi: 10.1021/acschemneuro.6b00064
165. Miller JC, Friedhoff AJ. Effects of haloperidol and apomorphine on the K+-depolarized overflow of [3H] dopamine from rat striatal slices. Biochem Pharmacol (1979) 28(5):688–90. doi: 10.1523/JNEUROSCI.22-18-08002.2002
166. McElvain JS, Schenk JO. Blockade of dopamine autoreceptors by haloperidol and the apparent dynamics of potassium-stimulated endogenous release of dopamine from and reuptake into striatal suspensions in the rat. Neuropharmacology (1992) 31(7):649–59. doi: 10.1016/0006-2952(79)90158-8
167. Seeman P, Staiman A, Lee T, Chau-Wong M. The membrane actions of tranquilizers in relation to neuroleptic-induced parkinsonism and tardive dyskinesia. Adv Biochem Psychopharmacol (1974) 9(0):137–48. doi: 10.1016/0028-3908(92)90143-D
169. Amato D, Beasley CL, Hahn MK, Vernon AC. Neuroadaptations to antipsychotic drugs: insights from pre-clinical and human post-mortem studies. Neurosci Biobehav Rev (2016a) 74(5):830–4. doi: 10.1016/j.bbr.2014.07.025
170. Rayport S, Sulzer D. Visualization of antipsychotic drug binding to living mesolimbic neurons reveals D2 receptor, acidotropic, and lipophilic components. J Neurochem (1995) 65(2):691–703. doi: 10.1016/j.neubiorev.2016.10.004
171. Tischbirek CH, Wenzel EM, Zheng F, Huth T, Amato D, Trapp S, et al. Use-dependent inhibition of synaptic transmission by the secretion of intravesicularly accumulated antipsychotic drugs. Neuron (2012) 74(5):830–44. doi: 10.1046/j.1471-4159.1995.65020691.x
172. Yang SB, Proks P, Ashcroft FM, Rupnik M. Inhibition of ATP-sensitive potassium channels by haloperidol. Br J Pharmacol (2004) 143(8):960–7. doi: 10.1371/journal.pone.0020402
173. Yang SB, Major F, Tietze LF, Rupnik M. Block of delayed-rectifier potassium channels by reduced haloperidol and related compounds in mouse cortical neurons. J Pharmacol Exp Ther (2005) 315(1):352–62. doi: 10.1038/sj.bjp.0706017
174. Lee HJ, Sung KW, Hahn SJ. Effects of haloperidol on Kv4.3 potassium channels. Eur J Pharmacol (2014) 740:1–8. doi: 10.1124/jpet.105.086561
175. Hahn J, Tse TE, Levitan ES. Long-term K+ channel-mediated dampening of dopamine neuron excitability by the antipsychotic drug haloperidol. J Neurosci (2003) 23(34):10859–66. doi: 10.1016/j.ejphar.2014.06.043
176. Martel P, Leo D, Fulton S, Berard M, Trudeau LE. Role of Kv1 potassium channels in regulating dopamine release and presynaptic D2 receptor function. PLoS One (2011) 6(5):e20402. doi: 10.1016/j.neuron.2012.04.019
177. van Horne C, Hoffer BJ, Stromberg I, Gerhardt GA. Clearance and diffusion of locally applied dopamine in normal and 6-hydroxydopamine-lesioned rat striatum. J Pharmacol Exp Ther (1992) 263(3):1285–92. doi: 10.1523/JNEUROSCI.23-34-10859.2003
178. Nishino H, Kumazaki M, Fukuda A, Fujimoto I, Shimano Y, Hida H, et al. Acute 3-nitropropionic acid intoxication induces striatal astrocytic cell death and dysfunction of the blood–brain barrier: involvement of dopamine toxicity. Neurosci Res (1997) 27(4):343–55.
179. Heinz A, Saunders RC, Kolachana BS, Jones DW, Gorey JG, Bachevalier J, et al. Striatal dopamine receptors and transporters in monkeys with neonatal temporal limbic damage. Synapse (1999) 32(2):71–9. doi: 10.1016/S0168-0102(97)01170-X
180. Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci (2003) 4(1):13–25. doi: 10.1002/(SICI)1098-2396(199905)32:2<71::AID-SYN1>3.3.CO;2-H
181. Cragg SJ, Rice ME. DAncing past the DAT at a DA synapse. Trends Neurosci (2004) 27(5):270–7. doi: 10.1038/nrn1008
182. Sulzer D, Cragg SJ, Rice ME. Striatal dopamine neurotransmission: regulation of release and uptake. Basal Ganglia (2016) 6(3):123–48.
183. Flatmark T, Almas B, Ziegler MG. Catecholamine metabolism: an update on key biosynthetic enzymes and vesicular monoamine transporters. Ann N Y Acad Sci (2002) 971:69–75. doi: 10.1016/j.tins.2004.03.011
184. Wightman RM, Amatore C, Engstrom RC, Hale PD, Kristensen EW, Kuhr WG, et al. Real-time characterization of dopamine overflow and uptake in the rat striatum. Neuroscience (1988) 25(2):513–23. doi: 10.1111/j.1749-6632.2002.tb04436.x
185. Wightman RM, Zimmerman JB. Control of dopamine extracellular concentration in rat striatum by impulse flow and uptake. Brain Res Brain Res Rev (1990) 15(2):135–44. doi: 10.1016/0306-4522(88)90255-2
186. Fusar-Poli P, Meyer-Lindenberg A. Striatal presynaptic dopamine in schizophrenia, Part I: meta-analysis of dopamine active transporter (DAT) density. Schizophr Bull (2013) 39(1):22–32. doi: 10.1016/0165-0173(90)90015-G
187. Rothblat DS, Schneider JS. Regionally specific effects of haloperidol and clozapine on dopamine reuptake in the striatum. Neurosci Lett (1997) 228(2):119–22. doi: 10.1093/schbul/sbr111
188. Reader TA, Ase AR, Huang N, Hebert C, van Gelder NM. Neuroleptics and dopamine transporters. Neurochem Res (1998) 23(1):73–80. doi: 10.1016/S0304-3940(97)00377-7
189. Meiergerd SM, Patterson TA, Schenk JO. D2 receptors may modulate the function of the striatal transporter for dopamine: kinetic evidence from studies in vitro and in vivo. J Neurochem (1993) 61(2):764–67. doi: 10.1023/A:1022405621365
190. Parsons LH, Schad CA, Justice JB Jr. Co-administration of the D2 antagonist pimozide inhibits up-regulation of dopamine release and uptake induced by repeated cocaine. J Neurochem (1993) 60(1):376–9. doi: 10.1111/j.1471-4159.1993.tb02185.x
191. Cass WA, Gerhardt GA. Direct in vivo evidence that D2 dopamine receptors can modulate dopamine uptake. Neurosci Lett (1994) 176(2):259–63. doi: 10.1111/j.1471-4159.1993.tb05864.x
192. Benoit-Marand M, Ballion B, Borrelli E, Boraud T, Gonon F. Inhibition of dopamine uptake by D2 antagonists: an in vivo study. J Neurochem (2011) 116(3):449–58. doi: 10.1016/0304-3940(94)90096-5
193. Benoit-Marand M, Suaud-Chagny MF, Gonon F. Presynaptic regulation of extracellular dopamine as studied by continuous amperometry in anesthetized animals. In: Michael AC, Borland LM, editors. Electrochemical methods for neuroscience. Boca Raton (FL): CRC Press/Taylor & Francis (2007). doi: 10.1111/j.1471-4159.2010.07125.x
194. Suaud-Chagny MF, Dugast C, Chergui K, Msghina M, Gonon F. Uptake of dopamine released by impulse flow in the rat mesolimbic and striatal systems in vivo. J Neurochem (1995) 65(6):2603–11. doi: 10.1201/9781420005868.ch3
195. Lee SH, Oh DY, Jung SC, Kim YM, Cho HK, Koh JK, et al. Neuroleptic drugs alter the dopamine transporter-mediated uptake and release of dopamine: a possible mechanism for drug-induced tardive dyskinesia. Pharmacol Res (1997) 35(5):447–50. doi: 10.1046/j.1471-4159.1995.65062603.x
196. Siebert GA, Pond SM, Bryan-Lluka LJ. Further characterisation of the interaction of haloperidol metabolites with neurotransmitter transporters in rat neuronal cultures and in transfected COS-7 cells. Naunyn Schmiedebergs Arch Pharmacol (2000) 361(3):255–64. doi: 10.1006/phrs.1997.0159
197. Ewing AG, Wightman RM. Monitoring the stimulated release of dopamine with in vivo voltammetry. J Neurochem (1984) 43(2):570–7. doi: 10.1007/s002109900202
198. Zetterstrom T, Sharp T, Ungerstedt U. Effect of neuroleptic drugs on striatal dopamine release and metabolism in the awake rat studied by intracerebral dialysis. Eur J Pharmacol (1984) 106(1):27–37. doi: 10.1111/j.1471-4159.1984.tb00936.x
199. Imperato A, Di Chiara G. Dopamine release and metabolism in awake rats after systemic neuroleptics as studied by trans-striatal dialysis. J Neurosci (1985) 5(2):297–306. doi: 10.1016/0014-2999(84)90674-5
200. Hernandez L, Hoebel BG. Haloperidol given chronically decreases basal dopamine in the prefrontal cortex more than the striatum or nucleus accumbens as simultaneously measured by microdialysis. Brain Res Bull (1989) 22(4):763–9. doi: 10.1523/JNEUROSCI.05-02-00297.1985
201. Invernizzi R, Morali F, Pozzi L, Samanin R. Effects of acute and chronic clozapine on dopamine release and metabolism in the striatum and nucleus accumbens of conscious rats. Br J Pharmacol (1990) 100(4):774–8. doi: 10.1016/0361-9230(89)90097-X
202. Meltzer HY, Chai BL, Thompson PA, Yamamoto BK. Effect of scopolamine on the efflux of dopamine and its metabolites after clozapine, haloperidol or thioridazine. J Pharmacol Exp Ther (1994) 268(3):1452–61. doi: 10.1111/j.1476-5381.1990.tb14091.x
203. Zahniser NR, Sorkin A. Rapid regulation of the dopamine transporter: role in stimulant addiction? Neuropharmacology (2004) 47(Suppl 1):80–91.
204. Furman CA, Chen R, Guptaroy B, Zhang M, Holz RW, Gnegy M. Dopamine and amphetamine rapidly increase dopamine transporter trafficking to the surface: live-cell imaging using total internal reflection fluorescence microscopy. J Neurosci (2009) 29(10):3328–36. doi: 10.1016/j.neuropharm.2004.07.010
205. Nikolaus S, Antke C, Kley K, Beu M, Wirrwar A, Muller HW. Pretreatment with haloperidol reduces (123)I-FP-CIT binding to the dopamine transporter in the rat striatum: an in vivo imaging study with a dedicated small-animal SPECT camera. J Nucl Med (2009) 50(7):1147–52. doi: 10.1523/JNEUROSCI.5386-08.2009
206. Cumming P, Maschauer S, Riss PJ, Tschammer N, Fehler SK, Heinrich MR, et al. Radiosynthesis and validation of (1)(8)F-FP-CMT, a phenyltropane with superior properties for imaging the dopamine transporter in living brain. J Cereb Blood Flow Metab (2014) 34(7):1148–56. doi: 10.2967/jnumed.109.061952
207. Xu M, Xing Q, Li S, Zheng Y, Wu S, Gao R, et al. Pharmacogenetic effects of dopamine transporter gene polymorphisms on response to chlorpromazine and clozapine and on extrapyramidal syndrome in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2010) 34(6):1026–32. doi: 10.1038/jcbfm.2014.63
208. Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci (1984) 4(11):2866–76. doi: 10.1016/j.pnpbp.2010.05.017
209. Marcott PF, Mamaligas AA, Ford CP. Phasic dopamine release drives rapid activation of striatal D2-receptors. Neuron (2014) 84(1):164–76. doi: 10.1523/JNEUROSCI.04-11-02866.1984
210. Bueschbell B, Barreto CAV, Preto AJ, Schiedel AC, Moreira IS. A complete assessment of dopamine receptor–ligand interactions through computational methods. Molecules (2019) 24(7):1–26. doi: 10.1016/j.neuron.2014.08.058
211. Bertolino A, Fazio L, Caforio G, Blasi G, Rampino A, Romano R, et al. Functional variants of the dopamine receptor D2 gene modulate prefronto-striatal phenotypes in schizophrenia. Brain (2009) 132(Pt 2):417–25. doi: 10.3390/molecules24071196
212. Roberts DA, Balderson D, Pickering-Brown SM, Deakin JF, Owen F. The relative abundance of dopamine D4 receptor mRNA in post mortem brains of schizophrenics and controls. Schizophr Res (1996) 20(1–2):171–4. doi: 10.1093/brain/awn248
213. Benoit-Marand M, Borrelli E, Gonon F. Inhibition of dopamine release via presynaptic D2 receptors: time course and functional characteristics in vivo. J Neurosci (2001) 21(23):9134–41. doi: 10.1016/0920-9964(96)88526-4
214. Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience (1991) 41(1):1–24. doi: 10.1523/JNEUROSCI.21-23-09134.2001
215. Laruelle M, Abi-Dargham A, van Dyck CH, Gil R, D’Souza CD, Erdos J, et al. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci U S A (1996) 93(17):9235–40. doi: 10.1016/0306-4522(91)90196-U
216. Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, et al. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry (1998) 155(6):761–7. doi: 10.1073/pnas.93.17.9235
217. Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry (1999) 46(1):56–72. doi: 10.1176/ajp.155.6.761
218. Amato D, Canneva F, Nguyen HP, Bauer P, Riess O, von Horsten S, et al. Capturing schizophrenia-like prodromal symptoms in a spinocerebellar ataxia-17 transgenic rat. J Psychopharmacol (2016b) 31(4):461–73. doi: 10.1016/S0006-3223(99)00067-0
219. Creese I, Stewart K, Snyder SH. Species variation in dopamine receptor binding. Eur J Pharmacol (1979) 60(1):55–66. doi: 10.1177/0269881116675510
220. Bannon MJ, Whitty CJ. Age-related and regional differences in dopamine transporter mRNA expression in human midbrain. Neurology (1997) 48(4):969–77. doi: 10.1016/0014-2999(79)90052-9
221. Heinz A, Goldman D, Jones DW, Palmour R, Hommer D, Gorey JG, et al. Genotype influences in vivo dopamine transporter availability in human striatum. Neuropsychopharmacology (2000) 22(2):133–9. doi: 10.1212/WNL.48.4.969
222. Faraone SV, Spencer TJ, Madras BK, Zhang-James Y, Biederman J. Functional effects of dopamine transporter gene genotypes on in vivo dopamine transporter functioning: a meta-analysis. Mol Psychiatry (2014) 19(8):880–9. doi: 10.1016/S0893-133X(99)00099-8
223. Volkow ND, Ding YS, Fowler JS, Wang GJ, Logan J, Gatley SJ, et al. Dopamine transporters decrease with age. J Nucl Med (1996) 37(4):554–9. doi: 10.1038/mp.2013.126
224. Antonini A, Leenders KL. Dopamine D2 receptors in normal human brain: effect of age measured by positron emission tomography (PET) and [11C]-raclopride. Ann N Y Acad Sci (1993) 695:81–5.
225. Volkow ND, Fowler JS, Wang GJ, Logan J, Schlyer D, MacGregor R, et al. Decreased dopamine transporters with age in health human subjects. Ann Neurol (1994) 36(2):237–9. doi: 10.1111/j.1749-6632.1993.tb23033.x
226. Volkow ND, Gur RC, Wang GJ, Fowler JS, Moberg PJ, Ding YS, et al. Association between decline in brain dopamine activity with age and cognitive and motor impairment in healthy individuals. Am J Psychiatry (1998a) 155(3):344–9. doi: 10.1002/ana.410360218
227. Volkow ND, Wang GJ, Fowler JS, Ding YS, Gur RC, Gatley J, et al. Parallel loss of presynaptic and postsynaptic dopamine markers in normal aging. Ann Neurol (1998b) 44(1):143–7. doi: 10.1176/ajp.155.3.344
228. Harada N, Nishiyama S, Satoh K, Fukumoto D, Kakiuchi T, Tsukada H. Age-related changes in the striatal dopaminergic system in the living brain: a multiparametric PET study in conscious monkeys. Synapse (2002) 45(1):38–45. doi: 10.1002/ana.410440125
229. Troiano AR, Schulzer M, de la Fuente-Fernandez R, Mak E, McKenzie J, Sossi V, et al. Dopamine transporter PET in normal aging: dopamine transporter decline and its possible role in preservation of motor function. Synapse (2010) 64(2):146–51. doi: 10.1002/syn.10082
230. Uchida H, Suzuki T, Graff-Guerrero A, Mulsant BH, Pollock BG, Arenovich T, et al. Therapeutic window for striatal dopamine D(2/3) receptor occupancy in older patients with schizophrenia: a pilot PET study. Am J Geriatr Psychiatry (2014) 22(10):1007–16. doi: 10.1002/syn.20708
231. Li SC, Papenberg G, Nagel IE, Preuschhof C, Schroder J, Nietfeld W, et al. Aging magnifies the effects of dopamine transporter and D2 receptor genes on backward serial memory. Neurobiol Aging (2013) 34358(1):e351–310. doi: 10.1016/j.jagp.2013.01.045
232. Kanaan NM, Kordower JH, Collier TJ. Age-related changes in dopamine transporters and accumulation of 3-nitrotyrosine in rhesus monkey midbrain dopamine neurons: relevance in selective neuronal vulnerability to degeneration. Eur J Neurosci (2008) 27(12):3205–15. doi: 10.1016/j.neurobiolaging.2012.08.001
233. Rieckmann A, Hedden T, Younger AP, Sperling RA, Johnson KA, Buckner RL. Dopamine transporter availability in clinically normal aging is associated with individual differences in white matter integrity. Hum Brain Mapp (2016) 37(2):621–31. doi: 10.1111/j.1460-9568.2008.06307.x
234. Reis Marques T, Taylor H, Chaddock C, Dell’acqua F, Handley R, Reinders AA, et al. White matter integrity as a predictor of response to treatment in first episode psychosis. Brain (2014) 137(Pt 1):172–82. doi: 10.1002/hbm.23054
235. Szeszko PR, Robinson DG, Ikuta T, Peters BD, Gallego JA, Kane J, et al. White matter changes associated with antipsychotic treatment in first-episode psychosis. Neuropsychopharmacology (2014) 39(6):1324–31. doi: 10.1093/brain/awt310
236. Blanchard JJ, Brown SA, Horan WP, Sherwood AR. Substance use disorders in schizophrenia: review, integration, and a proposed model. Clin Psychol Rev (2000) 20(2):207–34. doi: 10.1038/npp.2013.288
237. Chambers RA, Krystal JH, Self DW. A neurobiological basis for substance abuse comorbidity in schizophrenia. Biol Psychiatry (2001) 50(2):71–83. doi: 10.1016/S0272-7358(99)00033-1
238. Volkow ND. Substance use disorders in schizophrenia–clinical implications of comorbidity. Schizophr Bull (2009) 35(3):469–72. doi: 10.1016/S0006-3223(01)01134-9
239. Muller CP, Schumann G. Drugs as instruments: a new framework for non-addictive psychoactive drug use. Behav Brain Sci (2011) 34(6):293–310. doi: 10.1093/schbul/sbp016
240. Lopez-Quintero C, Anthony JC. Drug use disorders in the polydrug context: new epidemiological evidence from a foodborne outbreak approach. Ann N Y Acad Sci (2015) 1349:119–26. doi: 10.1017/S0140525X11000057
241. Khantzian EJ. The self-medication hypothesis of addictive disorders: focus on heroin and cocaine dependence. Am J Psychiatry (1985) 142(11):1259–64. doi: 10.1111/nyas.12868
242. Schneier FR, Siris SG. A review of psychoactive substance use and abuse in schizophrenia. J Nerv Ment Dis (1987) 175(11):641–52. doi: 10.1176/ajp.142.11.1259
243. Swendsen J, Conway KP, Degenhardt L, Glantz M, Jin R, Merikangas KR, et al. Mental disorders as risk factors for substance use, abuse and dependence: results from the 10-year follow-up of the National Comorbidity Survey. Addiction (2010) 105(6):1117–28. doi: 10.1097/00005053-198711000-00001
244. Nestler EJ. Is there a common molecular pathway for addiction? Nat Neurosci (2005) 8(11):1445–9. doi: 10.1111/j.1360-0443.2010.02902.x
245. Volkow ND, Koob GF, McLellan AT. Neurobiologic advances from the brain disease model of addiction. N Engl J Med (2016) 374(4):363–71. doi: 10.1038/nn1578
246. Haber SN. Corticostriatal circuitry. Dialogues Clin Neurosci (2016) 18(1):7–21. doi: 10.1056/NEJMra1511480
247. Zhu J, Reith ME. Role of the dopamine transporter in the action of psychostimulants, nicotine, and other drugs of abuse. CNS Neurol Disord Drug Targets (2008) 7(5):393–409. doi: 10.1007/978-1-4614-6434-1_135-1
248. Volkow ND, Chang L, Wang GJ, Fowler JS, Franceschi D, Sedler M, et al. Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J Neurosci (2001) 21(23):9414–8. doi: 10.2174/187152708786927877
249. Leroy C, Karila L, Martinot JL, Lukasiewicz M, Duchesnay E, Comtat C, et al. Striatal and extrastriatal dopamine transporter in cannabis and tobacco addiction: a high-resolution PET study. Addict Biol (2012) 17(6):981–90. doi: 10.1523/JNEUROSCI.21-23-09414.2001
250. Liu Y, Han M, Liu X, Deng Y, Li Y, Yuan J, et al. Dopamine transporter availability in heroin-dependent subjects and controls: longitudinal changes during abstinence and the effects of Jitai tablets treatment. Psychopharmacology (Berl) (2013) 230(2):235–44. doi: 10.1111/j.1369-1600.2011.00356.x
251. Yen CH, Yeh YW, Liang CS, Ho PS, Kuo SC, Huang CC, et al. Reduced dopamine transporter availability and neurocognitive deficits in male patients with alcohol dependence. PLoS One (2015) 10(6):e0131017. doi: 10.1007/s00213-013-3148-z
252. Xu S, Liu Y, Li Y, Deng Y, Yuan J, Lv R, et al. Availability of dopamine transporters in heroin-dependent subjects: a (18)F-FECNT PET imaging study. Psychiatry Res Neuroimaging (2017) 263:121–6. doi: 10.1371/journal.pone.0131017
253. Yuan J, Liu XD, Han M, Lv RB, Wang YK, Zhang GM, et al. Comparison of striatal dopamine transporter levels in chronic heroin-dependent and methamphetamine-dependent subjects. Addict Biol (2017) 22(1):229–34. doi: 10.1016/j.pscychresns.2017.03.011
254. Volkow ND, Wang GJ, Fischman MW, Foltin RW, Fowler JS, Abumrad NN, et al. Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature (1997) 386(6627):827–30. doi: 10.1111/adb.12271
255. Thompson JL, Urban N, Slifstein M, Xu X, Kegeles LS, Girgis RR, et al. Striatal dopamine release in schizophrenia comorbid with substance dependence. Mol Psychiatry (2013) 18(8):909–15. doi: 10.1038/386827a0
256. Peraile I, Torres E, Mayado A, Izco M, Lopez-Jimenez A, Lopez-Moreno JA, et al. Dopamine transporter down-regulation following repeated cocaine: implications for 3,4-methylenedioxymethamphetamine-induced acute effects and long-term neurotoxicity in mice. Br J Pharmacol (2010) 159(1):201–11. doi: 10.1038/mp.2012.109
257. Ferris MJ, Calipari ES, Mateo Y, Melchior JR, Roberts DC, Jones SR. Cocaine self-administration produces pharmacodynamic tolerance: differential effects on the potency of dopamine transporter blockers, releasers, and methylphenidate. Neuropsychopharmacology (2012) 37(7):1708–16. doi: 10.1111/j.1476-5381.2009.00522.x
258. Calipari ES, Ferris MJ, Zimmer BA, Roberts DC, Jones SR. Temporal pattern of cocaine intake determines tolerance vs sensitization of cocaine effects at the dopamine transporter. Neuropsychopharmacology (2013) 38(12):2385–92. doi: 10.1038/npp.2012.17
259. Kawa AB, Allain F, Robinson TE, Samaha AN. The transition to cocaine addiction: the importance of pharmacokinetics for preclinical models. Psychopharmacology (Berl) (2019). doi: 10.1038/npp.2013.136
260. Kapur S, Remington G. Serotonin-dopamine interaction and its relevance to schizophrenia. Am J Psychiatry (1996) 153(4):466–76. doi: 10.1007/s00213-019-5164-0
261. Guiard BP, El Mansari M, Merali Z, Blier P. Functional interactions between dopamine, serotonin and norepinephrine neurons: an in-vivo electrophysiological study in rats with monoaminergic lesions. Int J Neuropsychopharmacol (2008) 11(5):625–39. doi: 10.1176/ajp.153.4.466
Keywords: schizophrenia, drug addiction, antipsychotic efficacy, antipsychotic-resistant schizophrenia, dopamine transporter, dopamine synthesis, dopamine release
Citation: Amato D, Kruyer A, Samaha A-N and Heinz A (2019) Hypofunctional Dopamine Uptake and Antipsychotic Treatment-Resistant Schizophrenia. Front. Psychiatry 10:314. doi: 10.3389/fpsyt.2019.00314
Received: 30 January 2019; Accepted: 23 April 2019;
Published: 28 May 2019.
Edited by:
Felice Iasevoli, University of Naples Federico II, ItalyReviewed by:
Mette Ødegaard Nielsen, Center for Neuropsychiatric Schizophrenia Research (CNSR), DenmarkMark Slifstein, Stony Brook University, United States
Hiroyoshi Takeuchi, Keio University, Japan
Copyright © 2019 Amato, Kruyer, Samaha and Heinz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Davide Amato, YW1hdG9kQG11c2MuZWR1; YW1hdG9kYXZpZGVAZ21haWwuY29t