
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Psychiatry , 02 May 2019
Sec. Neurodegeneration
Volume 10 - 2019 | https://doi.org/10.3389/fpsyt.2019.00270
This article is part of the Research Topic Non-Motor Symptoms in Primary Motor Neurological Disorders: from Molecular Pathways to Clinical and Therapeutic Implications View all 11 articles
 Dominique Endres1,2
Dominique Endres1,2 Simon J. Maier1,2
Simon J. Maier1,2 Christiane Ziegler2
Christiane Ziegler2 Kathrin Nickel1,2
Kathrin Nickel1,2 Anne N. Riering2Benjamin Berger3
Anne N. Riering2Benjamin Berger3 Johann Lambeck3Miriam Fritz3Birgitta Gläser4
Johann Lambeck3Miriam Fritz3Birgitta Gläser4 Friedrich Stock5Michael Dacko6
Friedrich Stock5Michael Dacko6 Thomas Lange6
Thomas Lange6 Irina Mader7,8
Irina Mader7,8 Katharina Domschke2
Katharina Domschke2 Ludger Tebartz van Elst1,2*
Ludger Tebartz van Elst1,2*Background: Schizophrenic disorders are common and debilitating due to their symptoms, which can include delusions, hallucinations, and other negative symptoms. Organic forms can result from various cerebral disorders. In this paper, we discuss a potential association between schizophrenia and hereditary polyneuropathies (PNPs).
Case presentation: We present the case of a 55-year-old female patient with chronically paranoid–hallucinatory schizophrenia, severe cognitive deficits since the age of 30, and comorbid repeated focal pressure neuropathies beginning at age 20. At the age of 35, genetic testing revealed a deletion on chromosome 17p12 covering the peripheral myelin protein 22 gene (PMP22), which led to the diagnosis of hereditary neuropathy with liability to pressure palsy (HNPP). Cerebral magnetic resonance imaging showed internal atrophy, magnetic resonance spectroscopy found alteration of the glutamate and myo-inositol levels in the anterior cingulate cortex, neuropsychological testing showed deficits in working memory and psychomotor speed, and electrophysiological testing detected signs of sensorimotor demyelinating PNP (accentuated in the legs).
Conclusion: There may be an association between schizophrenia and HNPP. In observational studies, the deletion of interest (chromosome 17p12) was nearly 10 times more common in schizophreniform patients than in controls. This potential association could be pathophysiologically explained by the role of PMP22, which is mainly expressed in the peripheral nervous system. However, PMP22 mRNA and protein can also be found in the brain. PMP22 seems to play an important role in regulating cell growth and myelination, functions that are disturbed in schizophrenia. Such a connection obviously cannot be clarified on the basis of one case. Future studies should analyze whether patients with HNPP exhibit increased rates of psychotic disorders, and patients with schizophrenia and repeated focal pressure neuropathies should be examined for the PMP22 mutation. Alternatively, the co-occurrence of schizophrenia and HNPP could be coincidental.
Schizophrenia is a common disorder with a prevalence rate of about 1% (1). The clinical presentation is characterized by hallucinations, delusions, loss of self-boundaries, disorganized thinking and speech, cognitive deficits, lack of motivation, and social withdrawal (1). Secondary, organic forms can result from various cerebral disorders that are caused by genetic (22q11 deletion syndrome, cerebrotendinous xanthomatosis, Niemann–Pick type C, etc.), immunological (limbic encephalitis, anti-NMDA-R encephalitis, Hashimoto encephalopathy, etc.), infectious (neuroborreliosis, neurosyphilis, etc.), epileptic (paraepileptic psychosis, etc.), traumatic (traumatic brain injury), or neurodegenerative (frontotemporal dementia, etc). factors (2, 3).
The familial aggregation of schizophrenia is well established (1, 4). A variety of single-nucleotide polymorphisms (SNPs) in hundreds to thousands of genes have been implicated in the pathophysiology of the endogenous variants of schizophrenia (5). In these complex genetic forms, single genes only have a small effect size, and environmental factors are further important modulators. In contrast, secondary genetic forms are either monogenetic or oligogenetic: here, only one or a small number of genetic variants are important for the expression of clinical characteristics, such as copy number variants (CNVs), which are defined by deletions, duplications, or insertions of deoxyribonucleic acid (DNA) fragments, and chromosome aberrations (6, 7). In these forms, genetic variation in single genes has a high effect size, and environmental factors are considered to be less important (3). Secondary monogenic or oligogenic forms may potentially also occur in the context of hereditary polyneuropathies (hPNPs) (8). However, the association between schizophrenia and hPNPs is largely unknown. The hPNPs include isolated hPNPs (hereditary sensory neuropathy, hereditary sensory and autonomic neuropathy, and hereditary neuropathy with liability to pressure palsy or HNPP) and hPNPs in the context of systemic disorders (e.g., acute intermittent porphyria and Fabry’s disease). These systemic disorders are characterized by a monogenic or oligogenic background similar to that of secondary schizophrenia.
Searching PubMed for “hereditary polyneuropathy AND (schizophrenia OR psychosis)” yielded only eight results (as of 23 December 2018). One observational study discussed an association between hereditary spastic paraparesis and schizophrenia (8). In addition, an association between hereditary spastic paraplegia and psychosis in a female patient due to dysmorphic changes in her corpus callosum has been described independently (9). No results were returned when the same literature research was performed using “HNPP AND (schizophrenia OR psychosis)”. A nonsystematic literature search also showed that there might be a link between transthyretin-associated polyneuropathy (PNP) and schizophrenia or depression (10). Charcot–Marie–Tooth disorder, which is caused by a duplication of the PMP22 gene, and coincident psychosis were reported in monozygotic twins (11, 12).
We present the case of a 55-year-old female Caucasian patient trained as an occupational therapist who has suffered from chronic paranoid–hallucinatory schizophrenia since the age of 30. She continuously showed positive symptoms with superimposed exacerbations. At the age of 34, she was forced to retire early from her career due to her illness. Her delusions included the idea that she had sinned and needed to die, and she perceived diverse signs as confirmation of these delusions. She suffered from auditory hallucinations (voices from God, the devil, and her dead partner or mother), visual hallucinations (visions of angels), and a loss of self-boundaries (believing that other people could read her thoughts). Negative symptoms included a lack of motivation, flattened mood, and social withdrawal. Cognitive impairment has been observed since the onset of psychotic symptoms, with inattention and increasing deficits in working memory.
Intermittently, the patient abused alcohol (at least four beers per day) and benzodiazepines, but no illegal drugs. Her consumption of these substances increased during psychotic exacerbations with social withdrawal. The early death of her life partner reinforced this withdrawal. Since the onset of the disease, she had attempted suicide 10 times. Therefore, there were frequent inpatient stays in different psychiatric hospitals. Neither various neuroleptic treatments with average or high doses of aripiprazole, amisulpride, clozapine, haloperidol, perazine, pimozide, quetiapine, and risperidone, nor anticonvulsive treatment with valproate as an augmentation strategy led to full remission. Under different combination treatments, the described symptoms persisted at a reduced level.
At age 20, the patient developed clinical signs of HNPP. Initially, she was quickly fatigued and showed transient hypoesthesia of the left arm and foot. She developed transient foot dorsi-flexor paresis twice on the right side. The symptoms occurred after mechanical pressure on the corresponding body regions. In the further course of the disease, she developed transient left brachial plexus paresis at the age of 32 and again at the age of 35. When she was 35, genetic testing revealed a deletion on chromosome 17p12 involving the peripheral myelin protein 22 gene (PMP22), confirming the diagnosis of HNPP. The most recent neurological examination showed a discrete foot dorsi-flexor paresis on the right side (Medical Research Council score M4), absent Achilles tendon reflexes on both sides, slight pallhypesthesia of the lower extremity accentuated on the right side (5/8 on the right versus 6/8 on the left), and an ataxic and unsteady gait.
The patient’s developmental history was negative for in utero or birth complications, febrile convulsions, inflammatory brain diseases, and cerebral contusions. There was no evidence of any neurodevelopmental disorder. In the first two decades, the patient’s premorbid personality showed dependent traits. Her somatic medical history included only hypothyroidism, which was diagnosed at age 43. Her family history was positive for schizophrenia and HNPP. Her father’s half-brother suffered from schizophrenia. The patient’s older brother (transient hypesthesia of parts of one hand; side and explicit location unclear) and father (paresis of the shoulder abductors) potentially also suffered from HNPP (no genetic diagnostics performed, clinical reports not available). Her younger brother and her mother were healthy.
The serum, cerebrospinal fluid, and cerebral magnetic resonance imaging (cMRI) investigations did not show any evidence of an immunological cause for schizophrenia or PNPs. The electrophysiological tests revealed sensorimotor, demyelinating PNP (accentuated in the legs). Neurosonography showed bilateral enlargement of the median and ulnar nerve at typical sides of entrapment syndromes. Neuropsychological testing showed considerable deficits in working memory and a mild deficit in psychomotor speed. The electroencephalography (EEG) showed rare intermittent slowing. The cMRI showed generalized atrophy (Figure 1). Magnetic resonance spectroscopy (MRS) was performed in the dorsolateral prefrontal cortex (DLPFC), the dorsal anterior cingulate cortex (dACC), and the pregenual anterior cingulate cortex (pACC) using a MEGA PRESS sequence (repetition time = 1,500 ms, echo time = 68 ms, flip angle = 90°) and the acquired spectra were quantified with the software LCModel. Glutamate concentrations in the dACC and pACC were relatively high and outside the 90% reference intervals. The myo-inositol concentrations in the pACC were relatively low and within the 90% reference interval (Figure 2). The DLPFC measurement displayed poor spectral quality and was therefore not analyzed. The diagnostic findings are summarized in Table 1.
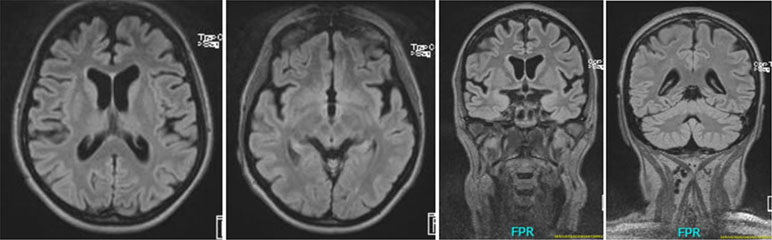
Figure 1 Cerebral magnetic resonance (cMRI) imaging showing generalized atrophy with perisylvian accentuation.
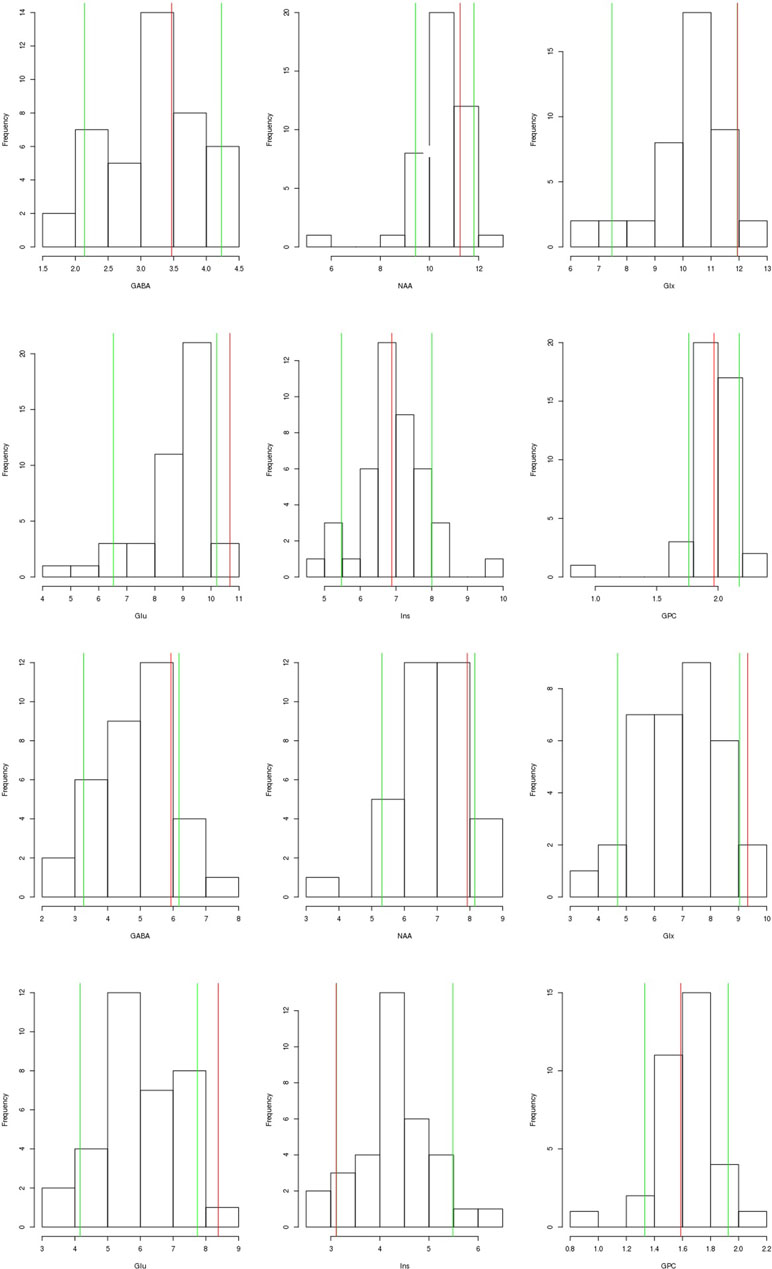
Figure 2 Magnetic resonance spectroscopy (MRS) findings: We present the findings of the dorsal anterior cingulate cortex (dACC; the two upper rows) and the pregenual anterior cingulate cortex (pACC; lower rows). The patient’s values (red line) are compared to the values of a healthy control group measured in another study (N = 43, mean age: 35, gender ratio: 14 females/29 males). The green line shows the 90% reference interval of the control group. For Glx in the dACC and Ins in the pACC the green (90% reference interval) and the red line (patient’s values) are superimposed, therefore only the red line is visible. The measured metabolite concentrations have been corrected for MRS voxel composition (content of gray matter, white matter, and cerebrospinal fluid), as well as for the influence of age and differences in the signal-to-noise ratio of the MRS measurements. Abbreviations: GABA, gamma-aminobutyric acid including coedited macromolecules; NAA, N-acetylaspartate; Glx, glutamate and glutamine; Glu, glutamate; Ins, myo-inositol; GPC, glycerophosphorylcholine.
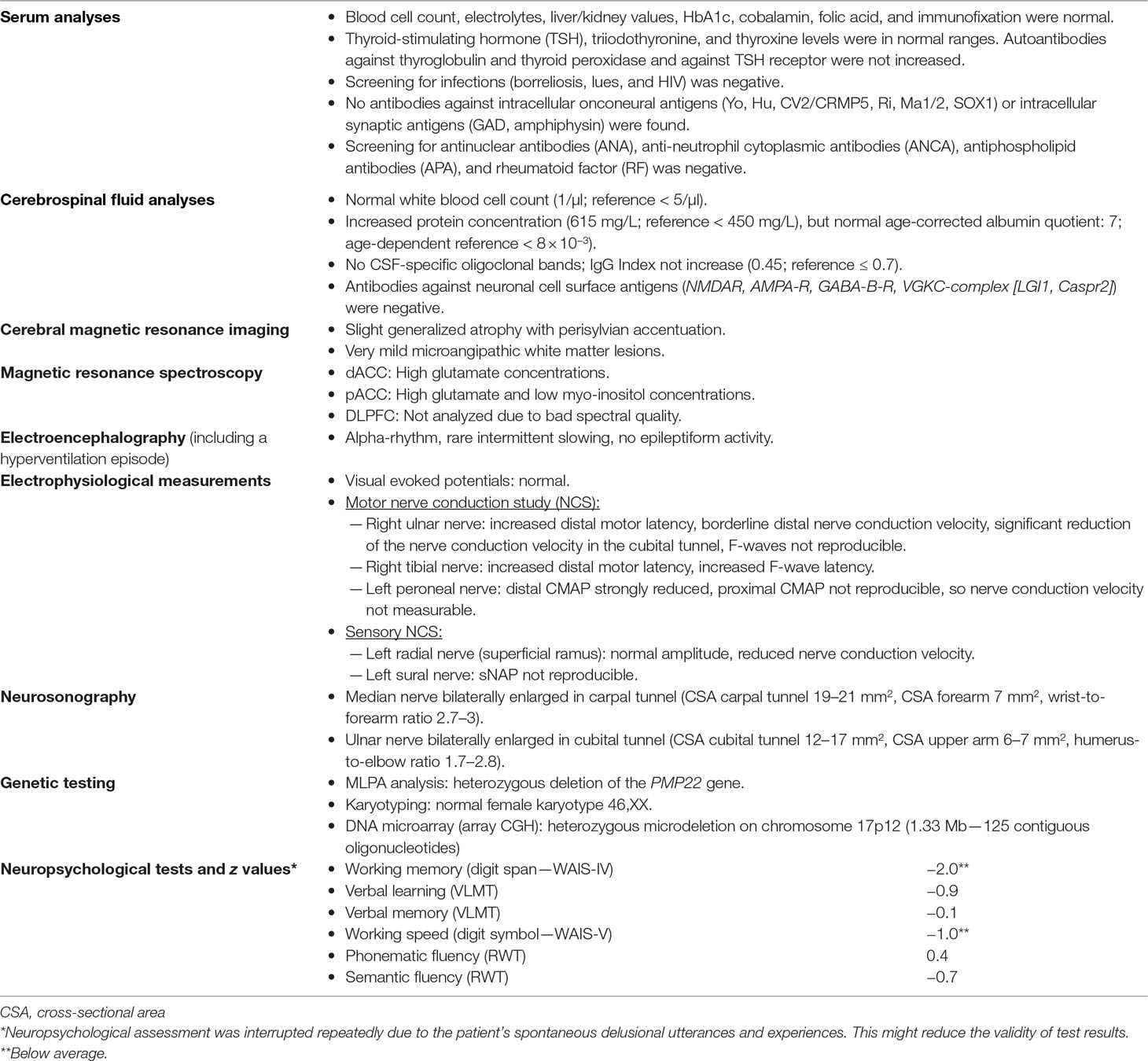
Table 1 Current diagnostic findings.
MLPA (multiplex ligation-dependent probe amplification) analysis revealed a heterozygous deletion of the PMP22 gene on chromosome 17p12. To delineate the extent of this deletion, we additionally performed conventional karyotyping and array CGH (comparative genomic hybridization).
Conventional R-banded karyotypes from the patient were analyzed according to standard protocols with a resolution of approximately 500 bphs and revealed a structurally and numerically normal female karyotype (46,XX) in all 28 metaphases examined. Furthermore, the genomic DNA of the patient was examined by microarray analysis (CytoSureTM constitutional v3 array 180k; Oxford Gene Technology) according to the manufacturer’s instructions. After hybridization, the array was scanned with the SureScan microarray scanner (Agilent); the results were analyzed using CytoSure interpret software v.4.9 (Oxford Gene Technology) and the Genome Reference Consortium human genome GRCh37 (hg19). Molecular karyotyping revealed a heterozygous deletion of approximately 1.33 Mb (125 contiguous oligonucleotides) out of the chromosomal region 17p12 (karyotype after the International System for Human Cytogenetic Nomenclature, 2016: arr[GRCh37] 17p12(14111972_15442257)x1). The deletion encompasses i.a. the genes COX10, HS3ST3B1, PMP22, TEKT2, and CDRT4. A deletion of that extent in 17p12 is found in nearly 80% of patients with HNPP (13). Further chromosomal deletions or duplications that might have etiologically contributed to our patient’s disease were not detected.
The patient’s psychiatric symptoms were compatible with paranoid–hallucinatory schizophrenia (ICD-10: F20.0). The PNPs in combination with the deletion of the PMP22 gene led to the diagnosis of HNPP (ICD-10: G60.0). Due to the potential secondary schizophrenia in the context of HNPP, a psychotic disorder with delusions due to a known physiological condition might ultimately be diagnosed (ICD-10: F06.2).
In this paper, we present the case of a female patient with chronic paranoid–hallucinatory schizophrenia with poor response to therapy and comorbid HNPP.
HNPP is an autosomal-dominant, peripheral neuropathy characterized by repeated and transient episodes of focal pressure neuropathies at compression-exposed sites (e.g., brachial plexus, sciatic nerve) (14). Our patient suffered from repeated foot flexor paresis and twice experienced brachial plexus paresis. In HNPP patients, nerve conduction study (NCS) often reveals demyelinating PNP with nerve suffering located predominantly in entrapment sites (15). In our patient, NCS showed findings compatible with sensorimotor, demyelinating PNPs with an entrapment syndrome in the right cubital tunnel. Histologically, sural nerve biopsies of HNPP patients typically display tomaculae, which are redundantly overfolded layers in the myelin coat (16). Genetically, a 1.5-Mb deletion on chromosome 17p12, including the PMP22 gene, is found in most cases (17). Molecular karyotyping in our patient showed a heterozygous deletion of 1.33 Mb.
The PMP22 gene is mainly expressed in the peripheral nervous system (PNS); however, mRNA and protein can also be found in the central nervous system (CNS) (18). Recent studies suggested the important involvement of the CNS in most patients with PMP22 deletion (18, 19). This is supported by prolonged latencies of visual evoked potentials, neurochemical alterations with decreased N-acetylaspartate (NAA) and creatine (Cre) concentrations, white matter (WM) volume reduction detected by cMRI, cognitive impairment (in 70% of patients), and fractional anisotropy alteration in several WM regions (e.g., in the columns of the fornix) (18–20). A large family study described CNS involvement and WM lesions predominantly in the subcortical frontal WM (21). Therefore, in line with the studies described, we hypothesizethat our patient’s schizophreniform symptoms may represent CNS involvement. This consideration is clinically supported by an insufficient therapy response to neuroleptics, severe cognitive deficits, EEG slowing, internal brain atrophy, and neurometabolic alterations (high glutamate in the dACC and pACC and low myo-inositol in the pACC). Compared with the patient group from Chanson and colleagues, we also found brain atrophy and neuropsychological deficits; however, in the presented case, NAA concentration and visual evoked potentials were normal, and DTI measurements were not performed (18). A causal relationship between the poor response to therapy and the PMP22 deletion remains speculative and is not proven by the case report. However, our hypothesis is also supported by several epidemiological and pathophysiological ideas, which we discuss in detail in the following paragraphs along with potential limitations.
Copy number variation in the chromosomal region 17p12 has repeatedly been implicated in schizophrenia (22–25). In a study analyzing the involvement of rare CNVs in 471 patients with schizophrenia, a deletion on 17p12 was found in two patients but not in controls. Therefore, the authors reanalyzed the data from two recent, large CNV studies of schizophrenia and found a PMP22 deletion in 6 out of 4,618 (0.13%) patients and 6 out of 36,092 (0.017%) controls (26–28). The data demonstrate that the described 17p12 deletion can be found nearly 10 times more often in schizophrenic patients compared to healthy controls (28). Moreover, there is at least one case report presenting a patient with schizophrenia in combination with mental retardation and PMP22 deletion without hPNPs (29).
PMP22 is a small, hydrophobic membrane glycoprotein that is mostly expressed by Schwann cells and comprises 2–5% of PNS myelin proteins in humans (16, 30). PMP22 mRNA and protein were also detected in the CNS, specifically in most parts of the brain (especially the corpus callosum) and the spinal cord. Changes in PMP22 mRNA may explain myelin abnormalities because it plays a role in the regulation of cell growth (even in the absence of protein), which was first described in fibroblasts (30, 31). Reductions in PMP22 mRNA have been observed in the hippocampus and anterior cingulate cortex in the postmortem brains of patients with schizophrenia (32). The PMP22 protein was restricted to a few areas [anterior horn and pia mater of the spinal cord, preganglionic sympathetic neurons; (33)]. In summary, PMP22 mRNA and protein are important for the regulation of cell growth and the maintenance of myelin integrity and therefore ensure the propagation of action potential (16). Heterozygous deletion of the PMP22 gene results in a loss-of-function phenotype (16) and altered myelination, as found in patients with schizophrenia (34).
It is important to note that the co-occurrence of schizophrenia and HNPP could be coincidental, which is supported by the absence of schizophreniform symptoms in our patient’s brother and father, who both very likely suffered from HNPP. However, genetic analyses of the patient’s brother and father were not performed to our knowledge. So, we can only speculate whether the discrepancy in psychiatric symptoms between our patient and her brother and father could be due to a variable expression and an incomplete penetrance, respectively, of the genetic effect of the PMP22 deletion. Besides environmental factors, further genetic variants might have contributed to our patient’s disease, particularly since the presently identified chromosomal deletion additionally encompasses COX10, HS3ST3B1, TEKT2, and CDRT4. However, the detected radiological and neuropsychological findings can also be found in schizophrenia; therefore, they are not clearly associated with the PMP22 deletion (34).
Case studies reporting such potential associations are essential to inspire further clinical trials (35). Retrospective studies analyzing psychiatric comorbidity in patients with HNPP could demonstrate whether there is a relevant association between the disorders and confirm or refute the pathophysiological role of PMP22 deletion in a subgroup of patients with schizophrenia.
There may be an association between schizophrenia and HNPP, which could be explained by the role of PMP22 in regulating cell growth and myelination. If such an association existed, which, of course, cannot be clarified from one case, then it might explain the poor control of psychiatric symptoms in our patient. But PMP22 deletion does not necessarily mean causality for the emergence of psychotic symptoms. For further clarification, patients with psychotic disorders and repeated transient focal neurological symptoms (which could be explained by pressure neuropathies) should be examined for PMP22 gene variation. Future studies should analyze whether patients with HNPP exhibit increased rates of schizophrenia. Alternatively, the co-occurrence of schizophrenia and HNPP could be coincidental.
The patient has given her signed, written informed consent for this case report, including the images presented, to be published.
DE and LT treated the patient. DE performed the data research and wrote the paper. SM, KN, MD, TL, and IM performed and interpreted the magnetic resonance spectroscopy. FS and BG performed the genetic testing. CZ and KD supported the interpretation of genetic findings. BB, JL, and MF performed and interpreted the electrophysiological measurements, neurosonography, neurological examination, and CSF measurements. IM performed and interpreted the cMRI. AR performed the neuropsychological testing. All authors were critically involved in the theoretical discussion and composition of the manuscript. All authors read and approved the final version of the manuscript.
The article processing charge was funded by the German Research Foundation (DFG) and the University of Freiburg in the funding program Open Access Publishing.
BB received travel grants and/or training expenses from Bayer Vital GmbH, Ipsen Pharma GmbH, Norvartis, Biogen GmbH, and Genzyme, as well as lecture fees from Ipsen Pharma GmbH, Alexion Pharma GmbH, Merck, Sanofi Genzyme, and Roche. IM received lecture fees from UCB Pharma GmbH, Germany. LTvE received advisory boards, lectures, or travel grants within the last 3 years: Eli Lilly, Janssen-Cilag, Novartis, Shire, UCB, GSK, Servier, Janssen, and Cyberonics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Owen MJ, Sawa A, Mortensen PB. Schizophrenia. Lancet (2016) 2388(10039):86–97. doi: 10.1016/S0140-6736(15)01121-6
2. Lishman A. Lishman’s organic psychiatry—a textbook of neuropsychiatry. Fourth Edition: Wiley-Blackwell (2009).
3. Tebartz van Elst L. Vom Anfang und Ende der Schizophrenie: Eine neuropsychiatrische Analyse des Schizophreniekonzepts. Stuttgart: Kohlhammer (2017).
4. Rees E, Kirov G, Walters JT, Richards AL, Howrigan D, Kavanagh DH, et al. Analysis of exome sequence in 604 trios for recessive genotypes in schizophrenia. Transl Psychiatry (2015) 215:e607. doi: 10.1038/tp.2015.99
5. Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kähler AK, Akterin S, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet (2013) 45(10):1150–9. doi: 10.1038/ng.2742
6. Grayton HM, Fernandes C, Rujescu D, Collier DA. Copy number variations in neurodevelopmental disorders. Prog Neurobiol (2012) 99(1):81–91. doi: 10.1016/j.pneurobio.2012.07.005
7. Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet (2017) 49(1):27–35. doi: 10.1038/ng.3725
8. McMonagle P, Hutchinson M, Lawlor B. Hereditary spastic paraparesis and psychosis. Eur J Neurol (2006) 13(8):874–9. doi: 10.1111/j.1468-1331.2006.01379.x
9. Osmolak AM, Wallenberg RB, Caplan JP. Hereditary spastic paraplegia and psychosis: connected by the corpus callosum? Psychosomatics (2012) 53(1):81–4. doi: 10.1016/j.psym.2011.05.004
10. Fleming CE, Nunes AF, Sousa MM. Transthyretin: more than meets the eye. Prog Neurobiol (2009) 89(3):266–76. doi: 10.1016/j.pneurobio.2009.07.007
11. Manyam NV, Cowell HR, Katz L. Charcot–Marie–Tooth disease and schizophrenia in identical twins. JAMA (1979) 241(1):54–5. doi: 10.1001/jama.1979.03290270044020
12. Birouk N, Gouider R, Le Guern E, Gugenheim M, Tardieu S, Maisonobe T, et al. Charcot-Marie-Tooth disease type 1A with 17p11.2 duplication. Brain (1997) 120(Pt 5):813–23. doi: 10.1093/brain/120.5.813
13. Chance PF, Abbas N, Lensch MW, Pentao L, Roa BB, Patel PI, et al. Two autosomal dominant neuropathies result from reciprocal DNA duplication/deletion of a region on chromosome 17. Hum Mol Genet (1994) 3(2):223–8. doi: 10.1093/hmg/3.2.223
14. Luigetti M, Del Grande A, Conte A, Lo Monaco M, Bisogni G, Romano A, et al. Clinical, neurophysiological and pathological findings of HNPP patients with 17p12 deletion: a single-centre experience. J Neurol Sci (2014) 341(1-2):46–50. doi: 10.1016/j.jns.2014.03.046
15. Hong YH, Kim M, Kim HJ, Sung JJ, Kim SH, Lee KW. Clinical and electrophysiologic features of HNPP patients with 17p11.2 deletion. Acta Neurol Scand (2003) 108(5):352–8. doi: 10.1034/j.1600-0404.2003.00132.x
16. Li J, Parker B, Martyn C, Natarajan C, Guo J. The PMP22 gene and its related diseases. Mol Neurobiol (2013) 47(2):673–98. doi: 10.1007/s12035-012-8370-x
17. Chance PF, Alderson MK, Leppig KA, Lensch MW, Matsunami N, Smith B, et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell (1993) 1572(1):143–51. doi: 10.1016/0092-8674(93)90058-X
18. Chanson JB, Echaniz-Laguna A, Blanc F, Lacour A, Ballonzoli L, Kremer S, et al. Central nervous system abnormalities in patients with PMP22 gene mutations: a prospective study. J Neurol Neurosurg Psychiatry (2013) 84(4):392–7. doi: 10.1136/jnnp-2012-303725
19. Brandt AU, Meinert-Bohn E, Rinnenthal JL, Zimmermann H, Mikolajczak J, Oberwahrenbrock T. Afferent visual pathway affection in patients with PMP22 deletion-related hereditary neuropathy with liability to pressure palsies. PLoS One (2016) 11(10):e0164617. doi: 10.1371/journal.pone.0164617
20. Wang WW, Song CL, Huang L, Song QW, Liang ZH, Wei Q, et al. DTI study of cerebral normal-appearing white matter in hereditary neuropathy with liability to pressure palsies (HNPP). Medicine (Baltimore) (2015) 94(43):e1909. doi: 10.1097/MD.0000000000001909
21. Sanahuja J, Franco E, Rojas-García R, Gallardo E, Combarros O, Begué R, et al. Central nervous system involvement in hereditary neuropathy with liability to pressure palsies: description of a large family with this association. Arch Neurol (2005) 62(12):1911–4. doi: 10.1001/archneur.62.12.1911
22. Magri C, Sacchetti E, Traversa M, Valsecchi P, Gardella R, Bonvicini C, et al. New copy number variations in schizophrenia. PLoS One (2010) 135(10):e13422. doi: 10.1371/journal.pone.0013422
23. Vassos E, Collier DA, Holden S, Patch C, Rujescu D, St Clair D, et al. Penetrance for copy number variants associated with schizophrenia. Hum Mol Genet (2010) 119(17):3477–81. doi: 10.1093/hmg/ddq259
24. Crespi BJ, Crofts HJ. Association testing of copy number variants in schizophrenia and autism spectrum disorders. J Neurodev Disord (2012) 304(1):15. doi: 10.1186/1866-1955-4-15
25. Grozeva D, Conrad DF, Barnes CP, Hurles M, Owen MJ, O’Donovan MC, et al. WTCCC. Schizophr Res (2012) 135(1-3):1–7. doi: 10.1016/j.schres.2011.11.004
26. Stefansson H, Rujescu D, Cichon S, Pietiläinen OP, Ingason A, Steinberg S, et al. Large recurrent microdeletions associated with schizophrenia. Nature (2008) 11455(7210):232–6. doi: 10.1038/nature07229
27. International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature (2008) 455(7210):237–41. doi: 10.1038/nature07239
28. Kirov G, Grozeva D, Norton N, Ivanov D, Mantripragada KK, Holmans P, International Schizophrenia Consortium; Wellcome Trust Case Control Consortium, Craddock N, Owen MJ, O’Donovan MC. Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum Mol Genet (2009) 1518(8):1497–503. doi: 10.1093/hmg/ddp043
29. Kunugi H, Ozeki Y, Mizuguchi T, Hirabayashi N, Ogawa M, Ohmura N, et al. A case of schizophrenia with chromosomal microdeletion of 17p11.2 containing a myelin-related gene PMP22. Open Psychiatry J (2008) 2:1–4. doi: 10.2174/1874354400802010001
30. Naef R, Suter U. Many facets of the peripheral myelin protein PMP22 in myelination and disease. Microsc Res Tech (1998) 41(5):359–71. doi: 10.1002/(SICI)1097-0029(19980601)41:5<359::AID-JEMT3>3.0.CO;2-L
31. Snipes GJ, Suter U, Welcher AA, Shooter EM. Characterization of a novel peripheral nervous system myelin protein (PMP-22/SR13). J Cell Biol (1992) 117(1):225–38. doi: 10.1083/jcb.117.1.225
32. Dracheva S, Davis KL, Chin B, Woo DA, Schmeidler J, Haroutunian V. Myelin-associated mRNA and protein expression deficits in the anterior cingulate cortex and hippocampus in elderly schizophrenia patients. Neurobiol Dis (2006) 21(3):531–40. doi: 10.1016/j.nbd.2005.08.012
33. Ohsawa Y, Murakami T, Miyazaki Y, Shirabe T, Sunada Y. Peripheral myelin protein 22 is expressed in human central nervous system. J Neurol Sci (2006) 15247(1):11–5. doi: 10.1016/j.jns.2006.03.004
34. Davis KL, Stewart DG, Friedman JI, Buchsbaum M, Harvey PD, Hof PR, et al. White matter changes in schizophrenia: evidence for myelin-related dysfunction. Arch Gen Psychiatry (2003) 60(5):443–56. doi: 10.1001/archpsyc.60.5.443
Keywords: hereditary polyneuropathy, schizophrenia, psychosis, PMP22, hereditary neuropathy with liability to pressure palsy
Citation: Endres D, Maier SJ, Ziegler C, Nickel K, Riering AN, Berger B, Lambeck J, Fritz M, Gläser B, Stock F, Dacko M, Lange T, Mader I, Domschke K and Tebartz van Elst L (2019) Schizophrenia and Hereditary Polyneuropathy: PMP22 Deletion as a Common Pathophysiological Link? Front. Psychiatry 10:270. doi: 10.3389/fpsyt.2019.00270
Received: 23 December 2018; Accepted: 09 April 2019;
Published: 02 May 2019.
Edited by:
Francesca Trojsi, Università degli Studi della Campania Luigi Vanvitelli Caserta, ItalyReviewed by:
Antonio Lucio Teixeira, University of Texas Health Science Center at Houston, United StatesCopyright © 2019 Endres, Maier, Ziegler, Nickel, Riering, Berger, Lambeck, Fritz, Gläser, Stock, Dacko, Lange, Mader, Domschke and Tebartz van Elst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ludger Tebartz van Elst dGViYXJ0enZhbmVsc3RAdW5pa2xpbmlrLWZyZWlidXJnLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.