 Mu-N Liu
Mu-N Liu Chi-Ieong Lau4,5,6
Chi-Ieong Lau4,5,6 Ching-Po Lin
Ching-Po Lin- 1Institute of Brain Science, National Yang-Ming University, Taipei, Taiwan
- 2Department of Psychiatry, Taipei Veterans General Hospital, Taipei, Taiwan
- 3Department of Neurology, Memory and Aging Centre, University of California, San Francisco, San Francisco, CA, United States
- 4Department of Neurology, Shin Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan
- 5Applied Cognitive Neuroscience Group, Institute of Cognitive Neuroscience, University College London, London, United Kingdom
- 6College of Medicine, Fu-Jen Catholic University, Taipei, Taiwan
- 7Institute of Neuroscience, National Yang-Ming University, Taipei, Taiwan
- 8Aging and Health Research Center, National Yang Ming University, Taipei, Taiwan
Frontotemporal dementia (FTD) is a common young-onset dementia presenting with heterogeneous and distinct syndromes. It is characterized by progressive deficits in behavior, language, and executive function. The disease may exhibit similar characteristics to many psychiatric disorders owing to its prominent behavioral features. The concept of precision medicine has recently emerged, and it involves neurodegenerative disease treatment that is personalized to match an individual's specific pattern of neuroimaging, neuropathology, and genetic variability. In this paper, the pathophysiology underlying FTD, which is characterized by the selective degeneration of the frontal and temporal cortices, is reviewed. We also discuss recent advancements in FTD research from the perspectives of clinical, imaging, molecular characterizations, and treatment. This review focuses on the approach of precision medicine to manage the clinical and biological complexities of FTD.
Introduction
Frontotemporal dementia (FTD) is an insidious neurodegenerative clinical syndrome that is characterized by progressive disturbances in behavior as well as deficits in executive function and language. FTD is a common early-onset dementia (occurring in patients aged < 65 years), has a prevalence rate of 3–26%, and is one of the most common forms of dementia across all age groups (1). Arnold Pick, a Czech psychiatrist, first identified the clinical syndrome of FTD in 1892 (2). He described a patient with aphasia, focal frontal and temporal lobar atrophy, and presenile dementia. Alois Alzheimer, a German psychiatrist and neuropathologist, later characterized Pick bodies as being associated with FTD and named the disorder Pick's disease in 1911 (3). Although, the term Pick's disease initially referred to both the clinical syndrome and the pathological diagnosis, modern nomenclature designates Pick's disease as only the pathological diagnosis, whereas a clinical diagnosis for prominent behavioral changes is known as behavioral-variant FTD (bvFTD). Mesulam described primary progressive aphasia (PPA), the language subtype of FTD, in 1982 (4). Revised diagnostic criteria were issued in 2011 (5, 6).
Precision medicine, also called “personalized medicine” or “individualized medicine,” is a rapidly advancing field in medical, clinical, and research settings. It aims to optimize the effectiveness of disease prevention and treatment and simultaneously minimize side effects in individuals who are less likely to respond to a particular therapy, by considering an individual's specific makeup with regard to genetics, biomarkers, phenotype, and psychosocial characteristics. In this review, we discuss the precision medicine of FTD, from clinical phenotypes, epidemiology, genetics, neuroimaging to neuropathological biomarkers. We further review recent advancements in therapeutic strategies and potential personalized treatment for FTD (7–13). This review improves the understanding of accurate diagnosis and personalized effective disease treatment strategies.
Cognitive and Behavioral Markers
FTD is an umbrella term for three recognizable clinical syndromes, namely bvFTD, semantic-variant PPA (svPPA), and non-fluent-variant PPA (nfvPPA) (Table 1). FTD also frequently overlaps clinically with three neurodegenerative diseases that exhibit motor deficits, namely corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), and amyotrophic lateral sclerosis (14).
Behavioral-Variant Frontotemporal Dementia
The symptoms of bvFTD include progressive personality and behavioral changes, apathy, and disinhibition in interpersonal interactions. Patients may experience early changes in disinhibition, stereotypic behavior, alterations in food preferences and eating behavior, alterations in empathy, apathy, and dysexecutive symptoms (5, 15). Some of these early symptoms, such as decreased empathy, may have diagnostic value for bvFTD, but they have not been ascertained in clinical practice. Apathy may manifest as reduced interest in work, hobbies, social interaction, and hygiene; however, apathy can be misdiagnosed as depression.
Symptoms similar to those detected in psychiatric disorders are frequently observed in patients with bvFTD. Thus, discriminating the behavioral features of bvFTD from those of primary psychiatric disorders such as depression, schizophrenia, bipolar disorder, and borderline personality disorder may be challenging (16, 17). Although psychotic symptoms such as hallucinations and delusions are rare in bvFTD, cases of these symptoms have been reported (17), particularly in patients carrying the chromosome 9 open reading frame 72 (C9orf72) repeat expansion (18).
Primary Progressive Aphasia
Patients with PPA exhibit a progressive decline in linguistic skills during the early phase of the disease. Language dysfunction is the main symptom during the first 2 years of PPA. Deficits in object naming, syntax, or word comprehension may become apparent during conversation or may be identified using speech and language assessment. The subtypes of PPA are differentiated by specific types of speech or language deficits. The three PPA subtypes are the semantic, non-fluent, and logopenic variants (19). Each subtype has a distinct pattern of language deficits. Naming difficulty is common to all three subtypes; therefore, it is not a distinguishing feature. The non-fluent (or agrammatic) variant and the semantic variant are classified as FTD, whereas the logopenic variant, most often associated with temporoparietal atrophy, is typically due to underlying Alzheimer's pathology; hence, it is not discussed in this review.
Semantic-Variant Primary Progressive Aphasia
In svPPA, a syndrome characterized by semantic aphasia and associative agnosia, anterior temporal lobe degeneration disrupts semantic memory (Table 1) (6). Anomia and single-word comprehension deficits, starting with low-frequency items, are essential for diagnosis (20). In contrast to patients with nfvPPA, those with svPPA maintain fluent speech and correct grammar during the early stages of this disease. Early symptoms of semantic PPA include anomia, word-finding difficulties, and repetitive speech, whereas early behavioral syndrome presents with irritability and emotional distance or coldness.
Non-fluent/Agrammatic-Variant Primary Progressive Aphasia
Articulation deficits resulting in slow, labored, and halting speech production as well as incorrect grammar or syntax (agrammatism) characterize nfvPPA. The core criteria of nfvPPA are agrammatism and effortful speech, and at least one of the criteria should be present (Table 1) (6). Patients tend to exhibit motor speech disorders characterized by a slow speech rate, abnormal prosody, and distorted sound substitutions, additions, repetitions, and prolongations, which are occasionally accompanied by groping, trial-and-error articulatory movements (21), or agrammatic errors. Repetition is less impaired than is spontaneous speech, and semantic knowledge for words typically remains well-preserved throughout the disease process.
Motor Symptoms
The three FTD-spectrum motor syndromes are FTD with motor neuron disease (FTD-MND) and two variants with parkinsonism, namely corticobasal syndrome (CBS) and progressive supranuclear palsy syndrome (PSP-S). Up to 15% of patients with FTD have concomitant MND, and nearly 30% of patients present with mild features of MND (9, 22). MND may include upper motor neuron signs (hyperreflexia, extensor plantar response, and spasticity), lower motor neuron signs (weakness, muscle atrophy, and fasciculations), dysarthria, dysphagia, and pseudobulbar affect (22). Up to 20% of patients with FTD present with parkinsonism, which is most often observed in patients with bvFTD, followed by those with nfvPPA (23). Patients with FTD may exhibit features of CBS or PSP-S. CBS is a heterogeneous syndrome featuring behavioral, cognitive, and motor changes. The clinical criteria for probable CBS include asymmetric presentation with any two symptoms among (A) limb rigidity or akinesia, (B) limb dystonia, and (C) limb myoclonus, as well as any two symptoms among (D) orobuccal or limb apraxia, (E) cortical sensory deficit, and (F) alien limb phenomena (more than simple levitation) (24). Finally, PSP-S is characterized by atypical parkinsonism with axial and symmetrical rigidity, supranuclear gaze palsy (most prominent in the vertical plane), decreased saccadic velocity, early postural instability with falls, and prominent frontal lobe dysfunction (25, 26).
Taken together, the vast heterogeneity and overlap of clinical phenotypes in FTD often poses diagnostic challenges for clinicians, in particular the presenting psychiatric symptoms that may easily be mistaken for psychiatric disorders. The accurate diagnosis of each subtype of FTD, therefore, requires a precision medicine approach.
Imaging Biomarkers
Neuroimaging has the potential to aid the differential diagnosis of FTD. For example, FTD is characterized by predominant frontal or temporal atrophy, particularly in the frontoinsular region, as revealed by structural brain imaging (Figure 1; Table 2) (8). Using voxel-based morphometry, Rosen et al. demonstrated that core neuropsychiatric symptoms of bvFTD, including apathy, disinhibition, and aberrant motor behavior, are localized to the right frontal structures. Moreover, atrophy in the right-hemispheric anterior cingulate cortex and adjacent ventromedial superior frontal gyrus, posterior ventromedial prefrontal cortex, lateral middle frontal gyrus, caudate head, orbitofrontal cortex, and anterior insula was correlated with symptom severity (27). Very mild bvFTD targets paralimbic networks, including the anterior cingulate, insular, medial frontal, and orbitofrontal cortices (28). Specifically, atrophy of the right ventromedial superior frontal gyrus was associated with apathy; atrophy of the right ventromedial prefrontal cortex was associated with disinhibition (27); and atrophy of the dorsolateral prefrontal was associated with executive deficit (7). A widespread alteration in white matter connectivity between the frontal and temporal lobes was noted using diffusion tensor imaging. The uncinate fasciculus, anterior parts of the superior and inferior longitudinal fasciculi, genu of the corpus callosum, cingulum, and inferior fronto-occipital fasciculus were affected (Table 2) (10, 29).
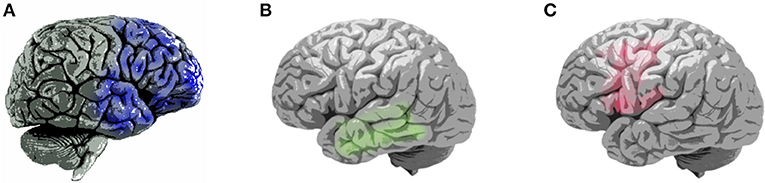
Figure 1. Patterns of brain atrophy in clinical subtypes of frontotemporal dementia (7–9). (A) Areas of brain atrophy in behavioral-variant frontotemporal dementia (blue), right hemisphere lateral view; (B) Areas of brain atrophy in semantic-variant primary progressive aphasia(green), left hemisphere lateral view; (C) Areas of brain atrophy in non-fluent variant primary progressive aphasia, left hemisphere lateral view.
By contrast, language symptoms are usually localized to the left hemisphere and are associated with a deficit of the language circuit. Degeneration of the left anterior temporal lobe is associated with linguistic semantic loss, whereas that of the right anterior temporal lobe is associated with prominent behavioral and personality changes, including lack of empathy and increased rigidity (30). Patients with PPA present with focal and asymmetric changes in specific networks fundamental to language processing.
Patients with svPPA present with semantic memory deficit that is localized to the anterior temporal lobes. Abnormalities in white matter connectivity are predominantly distributed over the left fronto-temporal areas, including the uncinate fasciculus, inferior longitudinal fasciculus, corpus callosum, and cingulum (Figure 1; Table 2) (10, 31, 32). The occipital lobe, cerebellum, and brainstem are spared (10, 31). The left temporal lobe variant is approximately three times more common than the right temporal lobe variant (33). Eventually, degeneration spreads to the other side; accordingly, patients with svPPA develop right temporal pole atrophy (and vice versa). The following observations are relatively likely to be associated with major right temporal atrophy and difficulty with person identification (34). Patients with the right temporal variant may have difficulty recognizing famous faces because of semantic loss. As the disease spreads from the temporal lobes into the orbitofrontal cortex, the patients start exhibiting behavioral changes, such as irritability, emotional withdrawal, insomnia, strict or selective eating (often focusing on one particular type of food), and occasionally depression (30). Despite the loss of semantic knowledge in the left temporal lobe variant, functions such as visual attention on the right side are sometimes enhanced. Patients with the left temporal lobe variant are more likely to develop visual compulsions such as repetitions in activities, collecting brightly colored objects, jewelry beading, gardening, or painting (30, 33). By contrast, individuals with the right temporal lobe variant develop verbal compulsions on words and symbols (such as compulsively writing letters, playing solitaire, writing telephone numbers, and making puns). In summary, left temporal atrophy has been associated with loss of verbal semantic knowledge, whereas behavioral symptoms dominate the right temporal variant.
In nfvPPA, the deficits target the frontoinsular cortex. Atrophy is most frequently noted in the left inferior frontal and insular cortices, which disrupts language fluency and grammar (7, 35). White matter abnormalities are predominantly distributed over the left frontal-temporal-parietal regions, including the left superior longitudinal fasciculus, corpus callosum, cingulum, inferior and orbital frontal, anterior temporal, and inferior parietal areas (Figure 1; Table 2) (10, 36). Except for structural MRI, functional MRI, fluorodeoxyglucose (FDG)- PET, and single-photon-emission computed tomography all show disturbances in perfusion and metabolism over these areas (8, 10). In summary, due to cerebral hemispheric differences, patients with right-predominant patterns of atrophy tend to present with behavioral disturbance, such as bvFTD and right-sided svPPA, whereas patients with left-predominant atrophy may lead to language-related impairment, such as left-sided svPPA and nfvPPA.
Neuropathological Biomarkers: Tau, TDP-43, and Fused in Sarcoma
FTD is caused by FTLD, a pathological process of cortical and subcortical degeneration over the frontal and temporal areas. Abnormal intracellular aggregates of tau and transactive response (TAR) DNA-binding protein of 43 kDa (TDP-43) are the leading causes of FTD (accounting for ~90% of cases). Fused in sarcoma (FUS), characterized by abnormal intracellular FUS inclusions, is associated with most of the remaining cases (Figure 2) (7).
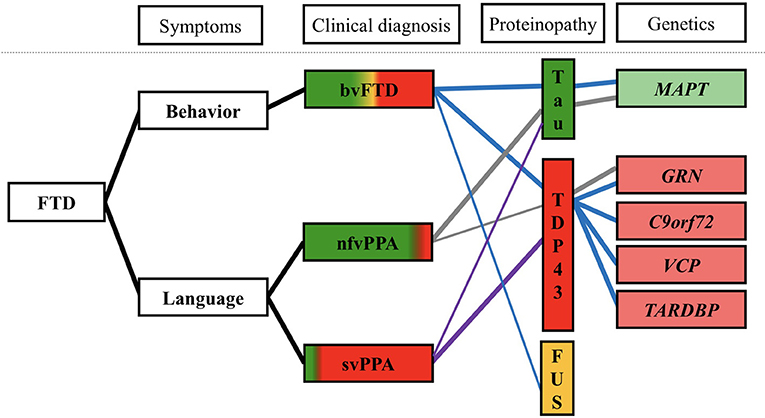
Figure 2. Clinical, pathological, and genetic associations in FTD (7–9, 37). Clinical symptoms are shown at left, colored regions of clinical diagnosis represent relative percentages of patients found to have each underlying neuropathological diagnosis. Blue line, genetic and pathological aspects of bvFTD; gray line, genetic, and pathological aspects of nfvPPA; purple line, genetic, and pathological aspects of svPPA. bvFTD, behavioral-variant frontotemporal dementia; svPPA, semantic-variant primary progressive aphasia; nfvPPA, non-fluent variant primary progressive aphasia; FUS, fused in sarcoma; TDP-43, transactive response (TAR) DNA-binding protein of 43 kDa; C9orf72, chromosome 9 open reading frame 72; GRN, progranulin mutations; MAPT, microtubule-associated protein tau; and VCP, valosin-containing protein.
Frontotemporal Lobar Degeneration -Tau Pathology
FTLD-tau accounts for one-third to one-half of all cases of FTLD, characterized by neuronal and glial tau aggregation (8, 38). Tau is critical for cellular morphology and function by binding to and stabilizing microtubules. In neurodegenerative disorders, tau becomes excessively hyperphosphorylated, dissociates from microtubules, and aberrantly aggregates within neurons and glia. Tau is encoded by microtubule-associated protein tau (MAPT). MAPT mutations mainly result in FTLD-tau pathology (Figure 2). Alternative splicing of MAPT mRNA leads to the production of three or four microtubule-binding domain repeats (3R or 4R). FTLD-tau is further subdivided into 3R, 4R, and 3R/4R tauopathies. Pick's disease, a 3R tauopathy, accounts for up to 30% of FTLD-tau cases (8, 38). A striking atrophy over the frontal, cingulate, and temporal gyri was noted (8). CBD, a 4R tauopathy, has been observed in approximately 35% of patients with FTLD-tau and involves the dorsal prefrontal cortex, supplemental motor area, perirolandic cortex, and subcortical nuclei (8, 38). PSP, also a 4R tauopathy, accounts for approximately 30% of patients with FTLD-tau (38). PSP is associated with frontal atrophy and with subcortical atrophy of the globus pallidus, subthalamic nucleus, and brainstem nuclei (8).
Approximately half of all bvFTD patients and the majority of nfvPPA patients have FTLD-tau pathology (Figure 2) (11). Furthermore, in most bvFTD or nfvPPA cases, the presence of extrapyramidal symptoms suggestive of CBS/PSP likely reflect an underlying tauopathy (12). Behavioral changes seen in bvFTD, or non-fluent motor speech difficulties in nfvPPA, may be presented in patients with CBS/PSP clinical syndrome before or after development of movement disorder. PSP neuropathology is strongly associated with patients with PSP clinical syndrome, particularly presence of the supranuclear vertical gaze palsy and early postural instability (11). Thus, PSP patients have become popular for the emergence of tau-targeting therapies in precision medicine. On the other hand, CBS is a highly variable clinical syndrome. In contrast, only 23% of the clinical CBS patients possessed Alzheimer's disease (AD) neuropathology, 13% with PSP neuropathology and 35% with CBD neuropathology in a large series of autopsy (39).
Frontotemporal Lobar Degeneration-TDP Pathology
FTLD-TDP accounts for approximately half of all patients with FTLD (38). TDP-43, a nuclear protein, is crucial for exon skipping and transcription regulation (40). In FTLD-TDP, TDP-43 becomes aberrantly localized from the nucleus to the cytoplasm, where it forms cytoplasmic inclusions (41). The cytoplasmic inclusions cause neurodegeneration though the potential toxicity of pathological TDP-43 aggregates and loss of normal TDP-43 function (i.e., nuclear clearance, RNA regulation) (11, 42). Four subtypes of FTLD-TDP (types A, B, C, and D) are recognized on the basis of the shape and distribution of TDP-43-positive lesions within the associative cortex (8). Type A accounts for approximately half of nfvPPA patients, one-quarter of suspected CBD patients, and one-quarter of bvFTD patients. Type C accounts for the majority of svPPA patients (Figure 2) (8).
Frontotemporal Lobar Degeneration -FUS Pathology
FUS is an RNA-binding protein involved in splicing and nuclear export of mRNA. FTLD-FUS has the following three subtypes: atypical FTLD with ubiquitin-positive inclusions, neuronal intermediate filament inclusion disease, and basophilic inclusion body disease (11, 43). A majority of patients with FTLD-FUS pathology are diagnosed as having bvFTD, characterized by sporadic, early-onset FTD with behavioral disturbance, disinhibition, and psychotic symptoms (Figure 2) (44).
Collectively, the diversity of pathology FTLD gives rise to a vast complexity of clinical phenotypes, with often overlapping neuropsychiatric features. Understanding the underlying neuropathological biomarkers, through personalized medicine, may in the future, offer more targeted and precise therapeutic options (Figure 2).
Genetics Biomarkers
Up to 40% of FTLD patients have a family history of dementia, thus suggesting a familial transmission; however, a clear autosomal-dominant history accounts for only 10% of all patients (45). To date, mutations in MAPT, chromosome 9 open reading frame 72 (C9orf72), and progranulin (GRN) have accounted for more than half of patients in FTLD families with a strong autosomal-dominant history (8). Mutations in MAPT and GRN account for 5–20% of patients with familial FTLD; C9orf72 mutations account for approximately 13–50% of familial FTLD patients and is a common genetic cause of FTD (8, 46, 47).
MAPT
MAPT mutations cause impaired microtubule assembly, impaired axonal transport, and increased pathological tau aggregation (48). As mentioned above, pathological tau formation in the cortical and subcortical brain areas results in neurodegeneration and is neuroanatomically correlated with the development of clinical symptoms (13). Patients with MAPT mutations are relatively young at onset (<50 years) and have a relatively short duration of illness (compared with those with other mutations), characterized by psychiatric and behavioral features (i.e., disinhibition, stereotyped repetitive behavior, and obsessions), parkinsonism, and oculomotor dysfunction (13, 49, 50). Neuroimaging studies revealed brain atrophy in the anterior temporal, orbitofrontal, caudate, insula, and anterior cingulate cortices (51), and different MAPT mutations may target different brain areas. For example, the medial temporal lobe indicates mutations in the splicing of exon 10, whereas mutations affecting the coding region target the lateral temporal lobe (52).
C9orf72
The expansion of a noncoding GGGGCC hexanucleotide repeat in C9orf72 is the most common cause of inherited FTD worldwide, and it accounts for a relatively small proportion of sporadic cases (47, 53). Normally, the number of GGGGCC hexanucleotide repeats is < 20; however, the presence of 65 or more repeats is considered pathogenic. Typically, the number of pathogenic repeats is in the hundreds (54). These expansion mutations are highly associated with TDP-43 pathology (13). FTD patients with the C9orf72 expansion mutation commonly present with bvFTD, MND, or a combination of the two, characterized by behavioral features (i.e., apathy, loss of empathy, and disinhibition), complex stereotyped behaviors, executive dysfunction, and Parkinson-like symptoms (i.e., gait disturbance, tremor, and rigidity) (13). Approximate 38% of patients present with psychotic symptoms, characterized by delusions, hallucinations, somatic symptoms, agitation, and anxiety (13, 55). Imaging-genetic studies in FTD patients with C9orf72 expansions have revealed a distributed symmetric pattern of brain atrophy in the frontal lobe (medial, dorsolateral, and orbitofrontal FTD patients with the C9orf72 expansion may have a more rapid cognitive decline related to cortical atrophy compared with other forms of FTLD-TDP (56, 57), and most C9orf72 expansion carriers with Parkinson-like symptoms respond poorly to levodopa usage (13). Therefore, C9orf72 genotyping could potentially be useful for precision medicine approach, not only to classify patients with FTLD but also offer prognostic values.
GRN
Progranulin is a secreted protein involved in cell-cycle regulation, wound repair, axonal growth, and inflammation modulation (58). GRN mutations are associated with haploinsufficiency and the reduction of progranulin production and secretion (59). They are clinically associated with bvFTD, nfvPPA, and CBS, thus conferring relatively low penetrance until 70 years (60, 61). Psychiatric symptoms, including delusions, hallucinations, ritualistic behaviors, apathy, and social withdrawal, are common. Language function is involved early in FTD patients with GRN mutations compared with patients with C9orf72 or MAPT mutations (13). Imaging study results showed asymmetric atrophy in the inferior frontal, temporal, and inferior parietal lobes, whereas C9orf72 or MAPT mutations are associated with symmetrical brain atrophy (62).
Other Genetic Biomarkers
Mutations in TAR DNA-binding protein (TARDBP), valosin-containing protein (VCP), TIA1, TBK1, and CCNF genes (associated with TDP-43 pathology); and FUS and CHMP2B (associated with tau-negative, TDP-negative, ubiquitin-positive inclusions) account for a minority of familial FTD (8). Mutations of TARDBP are commonly associated with ALS and FTD-ALS, and may be associated with PSP-like symptoms and chorea. Patients with “ALS-plus” symptom (clinical features extending beyond pyramidal and neuromuscular systems) have an increased likelihood of carrying a pathogenic TARDBP, C9orf72, or VCP mutation in contrast with sporadic cases (63).
Accordingly, genetic assessment for the above known genetic variants may improve diagnosis of FTD amid an overly complicated clinical picture. In addition, it may offer biological information to predict personal disease risk, understand underlying pathophysiology, identify presymptomatic individuals at risk for FTD and even provide future options for personalized therapeutics.
Applications of FTD Biomarkers for Precision Medicine
Although abnormal tau and TDP protein deposits may not be the only cause of FTD pathogenesis, they can define FTD as a unique neurodegenerative disease in the differential diagnosis of dementia. In addition, the initial differentiation of FTD from atypical AD using FTD precision medicine is crucial because FTD symptoms may become more severe following the application of approved AD therapies (64). A research framework focusing on biomarker diagnosis of FTD is urgently required because of the complex clinicopathological relationships of the disease. With the advent of biomarkers diagnostic techniques such as neuroimaging, biofluid dynamics, and genetics, FTD pathology can be identified; this can greatly improve diagnostic precision regarding the underlying pathophysiology of clinical syndromes. The National Institute on Aging and Alzheimer's Association recently introduced a new research framework for AD diagnosis based on biomarkers (65). A future diagnostic framework for FTD could adopt a similar approach for precision medicine. The best neuroimaging approach to separate AD from FTD is by FDG-PET, as evident by the diffuse posterior temporoparietal hypometabolism in AD vs. the focal frontotemporal hypometabolism in FTD (8, 10). Although a high proportion of FTLD patients have low levels of comorbid AD neuropathology and amyloid-beta (Aβ1−42) imaging is non-specific, amyloid PET scanning may still be useful for such differentiation (11). However, total tau (t-tau) and Aβ1−42 are extensively studied cerebrospinal fluid (CSF) biomarkers that may be used to accurately distinguish autopsy-confirmed FTD from AD (66); FTD patients have lower t-tau–Aβ1−42 ratios (37). The CSF t-tau–Aβ1−42 ratio may lead to substantial improvement in clinical diagnosis for differentiating FTD from atypical AD (67). Levels of CSF biomarkers of axonal injury and neuronal loss such as neurofilament light chains are reportedly elevated in clinical FTD cohorts compared with cohorts of other neurodegenerative diseases (68, 69) and have been associated with FTD disease severity (69).
A key task is to distinguish FTD from transmissible spongiform encephalopathies [i.e., Creutzfeldt-Jakob disease (CJD)], especially when early symptoms are subtle. CJD is a typical human prion disease caused by the aggregation and propagation of scrapie prion protein (PrPSC)—a misfolded form of normal prion protein (PrPC). CJD has many forms, including familial, variant, iatrogenic, and sporadic. Sporadic is the most common form (appropriately 85%) (70). Symptoms include myoclonus, rapidly progressive dementia, pyramidal/extrapyramidal signs, visual/cerebellar symptoms, and akinetic mutism. Clinical diagnosis of sporadic CJD (sCJD) is supported by the identification of 14-3-3 protein in CSF, periodic sharp electroencephalographic spikes, or MRI T2 FLAIR or diffusion-weighted imaging hyperintensities in the basal ganglia and cerebral or cerebellar cortex (71, 72). Compared with FTD, in sCJD, the CSF t-tau level is higher and the p-tau-t-tau ratio is lower (73). The aforementioned clinical, imaging, and biofluid markers are helpful in discriminating patients with FTD from those with CJD when clinical images are early and subtle.
Based on protein-targeting therapies such as those targeting tau (74), differentiation of FTLD-tau from FTLD-TDP after the exclusion of other neurodegenerative diseases (i.e., atypical AD and CJD) is the third step. Compared with FTLD-TDP and ALS, FTLD-tau has a higher p-tau level and higher p-tau-t-tau ratio (11). FTLD-TDP does not have significant p-tau pathology, and thus less p-tau may be released into the CSF compared with FTLD-tau (68, 75). The quantitative immunoprecipitation approach has been developed to detect specific forms of tau [extended (55 kDa) and truncated (33 kDa)] (76) to differentiate FTLD-tau from its subtypes. Through the exploratory proteomics-based approach, many novel CSF biomarkers have been identified for discriminating FTLD-tau from the main FTLD pathological subtypes as well as from non-demented controls and other forms of dementia with maximal accuracy (77). Studies to demonstrate the use of non-invasive methods such as the application of tau-specific radio ligands to identify FTLD-tau cases are ongoing (78, 79). Imaging and CSF biomarkers may assist in the development of successful therapies by facilitating the appropriate selection of cases for clinical trials targeting specific proteinopathies.
The longitudinal progression of biomarkers at early disease stages may be understood through the investigation of presymptomatic individuals within families that possess pathogenic mutations such as an earlier age of FTLD-tau onset with MAPT mutations (64). Brain network dysfunction has been observed in presymptomatic FTD with GRN (80) and C9orf72 (81) mutations. C9orf72 disease exhibits additional protein inclusion, additional clinical symptoms, and worse prognosis compared with its sporadic forms (11).
In line with the popular use of the APOE genotype in AD clinical trials, hereditary FTLD may be a popular choice for clinical trial development of therapies specific to this mutation. An autopsy-confirmed sporadic FTLD genetic study revealed several single-nucleotide polymorphisms (SNPs) that were overexpressed in patients with FTLD-tau and those with FTLD-TDP (82). The risk allele in the FTLD-tau-related SNP was associated with a shorter disease duration and white matter loss in the midbrain and long association fibers in sporadic bvFTD (83). This indicates the usefulness of SNP genotyping as a diagnostic and prognostic tool. A syndrome constitutes a clinical outcome of one or multiple diseases as opposed to an etiology. Similar to AD, a biological definition of FTD as opposed to a syndromal one is a superior means of enhancing the understanding of the underlying mechanisms of the clinical expression of FTD (65). Future precision-medicine approaches for FTD treatment must include biologically defined targets alongside the establishment of biomarker profiles and categories.
Treatments
Nonpharmacological interventions are considered for the management of dementia before the use of pharmacological treatments that may exacerbate medical comorbidities affecting elderly patients. Healthy lifestyle changes, social connections, physical activity, and environmental intervention may mitigate the effects of dementia (42, 43). The main purpose of nonpharmacological interventions is to prevent disruptive behaviors, provide symptom remission, and reduce caregiver distress. For example, environmental approaches (i.e., reduction of noise, limitation of stimuli, and simplification of daily activities and social parameters) intend to reduce irritability, aggression, and anxiety caused by daily external stimuli.
Currently, no disease-modifying drugs approved by the U.S. Food and Drug Administration are available for the treatment of FTD. Most treatments are focused on the management of behavioral symptoms. The use of selective serotonin reuptake inhibitors can reduce the severity of agitation, aggressiveness, impulsivity, aberrant eating behaviors, and compulsions (8). Herrmann et al. reported that behavioral and psychiatric symptoms (including irritability, disinhibition, and depression) were alleviated after citalopram treatment at a target dose of 40 mg once daily; furthermore, they observed a decrease in the overall Neuropsychiatric Inventory and Frontal Behavioral Inventory scores (84). The findings suggest that antidepressants may be beneficial in the treatment of neuropsychiatric and behavioral disturbances in FTD.
Behavioral disturbance (agitation or impulsivity) may also be controlled using atypical antipsychotics such as risperidone, olanzapine, and quetiapine. However, these medications could have side effects and increase the risk of mortality in patients with FTD (13, 85). In FTD patients with C9orf72 expansion, antipsychotic drugs cause noticeable adverse effects, and the adverse effects could not be reversed in some patients even after drug withdrawal (86, 87). In summary, low doses of atypical antipsychotic drugs may be useful for managing behavioral disturbance (8), but such drugs should be used with caution in patients with FTD because of the risk of mortality associated with cardiac events and falls secondary to the side effects (88).
Cholinesterase inhibitors, such as donepezil, do not alleviate but can even exacerbate behavioral disturbance in patients with FTD (89). This is likely because cholinergic deficit may not contribute to the pathophysiology of FTD (90). Memantine, an N-methyl-d-aspartate (NMDA) antagonist, has been reported to have no benefits in alleviating or delaying the progression of FTD symptoms, but it is generally well-tolerated by patients (91, 92). In summary, cholinesterase inhibitors and NMDA antagonist have no clinical efficacy in the treatment of FTD, and they potentially have detrimental effects on cognitive performance and behavioral symptoms (91, 93). Therefore, preferred management options should be based on risk assessment, and nonpharmacological interventions and caregiver support are preferred for first-line interventions (93).
Tau-Targeting Therapeutics
Due to knowledge advancements in molecular biology, pathophysiology, and neuropathology, precision medicine could be applied in FTD treatment by targeting the underlying pathogenesis (85). Based on the knowledge of the pathological tau protein spreading through prion-like propagation, the prevention of transneuronal spreading of pathological abnormalities is potentially an effective therapeutic strategy (94, 95). Administering tau aggregation inhibitors (i.e., methylthioninium chloride), microtubule-stabilizing drugs, tau-targeted immunotherapy, and tau vaccines may be useful therapeutic approaches in patients with tau pathology (96, 97).
Multiple approaches are available for tau-targeting therapeutics. First, tau aggregation and the various tau species formed (monomers, oligomers, prefilaments, granules, fibrils, and insoluble aggregates) during aggregation are of interest for potential therapeutic intervention. Hence, tau aggregation inhibitors have been proven effective in various in vitro studies (98). A proprietary formulation of non-neuroleptic phenothiazine methylene blue (methylthioninium chloride), which is used to treat malaria (99), has risen in the ranks in clinical development in recent years. This compound readily crosses the blood-brain barrier and prevents tau aggregation in vitro as well as in cell and animal models (100, 101). Safety and efficacy in a randomized, double-blind, placebo-controlled, multinational, and parallel-group clinical trial was demonstrated in 220 patients with bvFTD after 12 months of oral treatment; the results are yet to be published (74). Clarifying the efficacy of tau aggregation inhibitors in vivo is critical for preventing cognitive decline.
Second, microtubule stabilizers may be used as FTLD-tau therapeutic agents. Detachment of tau from microtubules leads to the loss of normal microtubule-stabilizing function, resulting in axonal transport defects and synaptic dysfunction. Davunetide—an eight-amino-acid peptide that arises from an activity-dependent neuroprotective protein-exerted substantial effects on behavior and cognition in tau-transgenic mice (102). In addition, intranasal or intravenous administration of davunetide established the safety and tolerability profile of davunetide in patients with mild cognitive impairment (103). However, whether microtubule destabilization is directly related to tau toxicity in tauopathies remains unclear. No therapeutic effect of davunetide for PSP treatment was detected in a double-blind, placebo-controlled, randomized phase II/III clinical trial (104).
Third, various anti-tau immunotherapy strategies have been successfully tested, suggesting that such strategies could be feasible options for clearing toxic protein species in tauopathies (105). Targeting abnormally phosphorylated tau epitopes (or pathologically relevant conformational epitopes) may be favorable for inducing antibody responses that promote tau clearance, as suggested by evidence found in animal models (106). The humanized anti-tau monoclonal antibody named ABBV-8E12 is also available for PSP treatment. A satisfactory safety and tolerability profile for ABBV-8E12 was demonstrated in a placebo-controlled, double-blind, phase I single-ascending-dose trial of 30 patients with PSP (107). Finally, modulating tau phosphorylation and targeting other posttranslational tau modifications (i.e., tau acetylation inhibitors) are also potential therapeutic strategies for FTLD-tau and other tauopathies (74). However, the current consensus is that tau-centric targeted treatment has no marked effect on long-term clinical outcomes (108, 109); further research is required to elucidate the potential roles of such therapies in treating FTD.
Other targets for therapy include disrupting the downstream effects of C9orf72 and GRN mutations. An approach that entails developing antisense oligonucleotides to reduce the concentrations of potentially toxic C9orf72 mRNAs has been applied to FTD patients (110, 111). This approach has also been implicated in the reduction of the total amount of pathological tau species (112). Because GRN mutation is related to progranulin haploinsufficiency and reduced progranulin concentrations, studies are attempting to adopt molecular approaches to prevent the reduction of progranulin concentrations and instead increase progranulin concentrations; such approaches include applying the histone deacetylase inhibitor suberoylanilide hydroxamic acid, which enhances progranulin transcription and alkalizing compounds that stimulate progranulin production (113, 114). Collectively, advances in the understanding of genetic mutations causing FTD have created new potential therapeutic targets for the development of effective disease-modifying drugs (85). These findings may enable physicians to develop precision medicine for the treatment of FTD patients with C9orf72, MAPT, or GRN mutations at a single-patient levels (10, 115).
Future Direction- Treatment Through Personalized Medicine
The implication of precision medicine is to enable physicians to identify highly selective and effective treatments with relatively few side effects for patients with specific illness. Currently, precision medicine is applied to FTD diagnosis and treatment. Based on advances in neuroimaging and genomic research that has explored underlying genetic risk variants and cerebral structural and functional change in order to determine specific molecular pathways and pathophysiological processes, precision medicine is currently applied in clinical trials; such trials focus on subgroups of individuals and the development of therapeutic targets with known genetic risk for FTD (116). For example, in patients with clinical diagnosis of bvFTD, tau-targeting therapeutics (e.g., tau aggregation inhibitors) may be administered in those with suspected FTLD-tau pathology. On the other hand, in bvFTD patients with suspected FTLD-TDP pathology, progranulin -related therapies may be a viable treatment for those with GRN mutation. Likewise, for those with C9orf72 repeat expansions, candidate antisense therapeutics could be used to reduce C9ORF72 expression. However, this is only the beginning of the precision medicine approach targeting the clinical process and treatment response of FTD. Collaboration among parents, family caregivers, and professionals (e.g., clinicians, scientists, and medical technologists) is crucial for identifying the pathological processes underlying FTD and for developing new interventions for successful application of precision medicine.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This research was supported by V108B-009, MOST 107-2634-F-010-001, MOST 107-2420-H-010 -001, MOST 104-2218-E-010-007-MY3, and NHRI-EX106-10611EI.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the support received from the MRI Core Laboratory of National Yang-Ming University, Taiwan. M-NL enormously grateful to the UCSF Memory and Aging Center to offer training for the clinical assessment of frontotemporal dementia.
References
1. Vieira RT, Caixeta L, Machado S, Silva AC, Nardi AE, Arias-Carrion O, et al. Epidemiology of early-onset dementia: a review of the literature. Clin Pract Epidemiol Ment Health (2013) 9:88–95. doi: 10.2174/1745017901309010088
2. Pick A. Über die Beziehungen der senilen Hirnatrophie zur Aphasie. Prager Med Wochenschr. (1892) 17:165–67.
3. Alzheimer A. Über eigenartige Krankheitsfälle der späteren Alters. Z Gesamte Neurol Psychiatr. (1911) 4:356–85.
5. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain (2011) 134(Pt 9):2456–77. doi: 10.1093/brain/awr179
6. Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology (2011) 76:1006–14. doi: 10.1212/WNL.0b013e31821103e6
7. Elahi FM, Miller BL. A clinicopathological approach to the diagnosis of dementia. Nat Rev Neurol. (2017) 13:457–76. doi: 10.1038/nrneurol.2017.96
8. Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet (2015) 386:1672–82. doi: 10.1016/S0140-6736(15)00461-4
9. Olney NT, Spina S, Miller BL. Frontotemporal dementia. Neurol Clin. (2017) 35:339–74. doi: 10.1016/j.ncl.2017.01.008
10. Gordon E, Rohrer JD, Fox NC. Advances in neuroimaging in frontotemporal dementia. J Neurochem. (2016) 138(Suppl. 1):193–210. doi: 10.1111/jnc.13656
11. Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM, et al. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol. (2015) 129:469–91. doi: 10.1007/s00401-014-1380-1
12. Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB, et al. Frontotemporal dementia: clinicopathological correlations. Ann Neurol. (2006) 59:952–62. doi: 10.1002/ana.20873
13. Young JJ, Lavakumar M, Tampi D, Balachandran S, Tampi RR. Frontotemporal dementia: latest evidence and clinical implications. Ther Adv Psychopharmacol. (2018) 8:33–48. doi: 10.1177/2045125317739818
14. Rogalski E, Cobia D, Martersteck A, Rademaker A, Wieneke C, Weintraub S, et al. Asymmetry of cortical decline in subtypes of primary progressive aphasia. Neurology (2014) 83:1184–91. doi: 10.1212/WNL.0000000000000824
15. Garcin B, Lillo P, Hornberger M, Piguet O, Dawson K, Nestor PJ, et al. Determinants of survival in behavioral variant frontotemporal dementia. Neurology (2009) 73:1656–61. doi: 10.1212/WNL.0b013e3181c1dee7
16. Rankin KP, Santos-Modesitt W, Kramer JH, Pavlic D, Beckman V, Miller BL. Spontaneous social behaviors discriminate behavioral dementias from psychiatric disorders and other dementias. J Clin Psychiatry (2008) 69:60–73. doi: 10.4088/JCP.v69n0109
17. Velakoulis D, Walterfang M, Mocellin R, Pantelis C, McLean C. Frontotemporal dementia presenting as schizophrenia-like psychosis in young people: clinicopathological series and review of cases. Br J Psychiatry (2009) 194:298–305. doi: 10.1192/bjp.bp.108.057034
18. Sha SJ, Takada LT, Rankin KP, Yokoyama JS, Rutherford NJ, Fong JC, et al. Frontotemporal dementia due to C9ORF72 mutations: clinical and imaging features. Neurology (2012) 79:1002–11. doi: 10.1212/WNL.0b013e318268452e
19. Gorno-Tempini ML, Brambati SM, Ginex V, Ogar J, Dronkers NF, Marcone A, et al. The logopenic/phonological variant of primary progressive aphasia. Neurology (2008) 71:1227–34. doi: 10.1212/01.wnl.0000320506.79811.da
20. Kramer JH, Jurik J, Sha SJ, Rankin KP, Rosen HJ, Johnson JK, et al. Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol. (2003) 16:211–8. doi: 10.1097/00146965-200312000-00002
21. Josephs KA, Duffy JR, Strand EA, Whitwell JL, Layton KF, Parisi JE, et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain (2006) 129(Pt 6):1385–98. doi: 10.1093/brain/awl078
22. Burrell JR, Kiernan MC, Vucic S, Hodges JR. Motor neuron dysfunction in frontotemporal dementia. Brain (2011) 134(Pt 9):2582–94. doi: 10.1093/brain/awr195
23. Le Ber I, Guedj E, Gabelle A, Verpillat P, Volteau M, Thomas-Anterion C, et al. Demographic, neurological and behavioural characteristics and brain perfusion SPECT in frontal variant of frontotemporal dementia. Brain (2006) 129(Pt 11):3051–65. doi: 10.1093/brain/awl288
24. Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology (2013) 80:496–503. doi: 10.1212/WNL.0b013e31827f0fd1
25. Hoglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord. (2017) 32:853–64. doi: 10.1002/mds.26987
26. Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology (1996) 47:1–9. doi: 10.1212/WNL.47.1.1
27. Rosen HJ, Allison SC, Schauer GF, Gorno-Tempini ML, Weiner MW, Miller BL. Neuroanatomical correlates of behavioural disorders in dementia. Brain (2005) 128(Pt 11):2612–25. doi: 10.1093/brain/awh628
28. Seeley WW, Crawford R, Rascovsky K, Kramer JH, Weiner M, Miller BL, et al. Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Arch Neurol. (2008) 65:249–55. doi: 10.1001/archneurol.2007.38
29. Diehl-Schmid J, Onur OA, Kuhn J, Gruppe T, Drzezga A. Imaging frontotemporal lobar degeneration. Curr Neurol Neurosci Rep. (2014) 14:489. doi: 10.1007/s11910-014-0489-x
30. Seeley WW, Bauer AM, Miller BL, Gorno-Tempini ML, Kramer JH, Weiner M, et al. The natural history of temporal variant frontotemporal dementia. Neurology (2005) 64:1384–90. doi: 10.1212/01.WNL.0000158425.46019.5C
31. Lam BY, Halliday GM, Irish M, Hodges JR, Piguet O. Longitudinal white matter changes in frontotemporal dementia subtypes. Hum Brain Mapp. (2014) 35:3547–57. doi: 10.1002/hbm.22420
32. Tu S, Leyton CE, Hodges JR, Piguet O, Hornberger M. Divergent longitudinal propagation of white matter degradation in logopenic and semantic variants of primary progressive aphasia. J Alzheimers Dis. (2016) 49:853–61. doi: 10.3233/JAD-150626
33. Thompson SA, Patterson K, Hodges JR. Left/right asymmetry of atrophy in semantic dementia: behavioral-cognitive implications. Neurology (2003) 61:1196–203. doi: 10.1212/01.WNL.0000091868.28557.B8
34. Chan D, Anderson V, Pijnenburg Y, Whitwell J, Barnes J, Scahill R, et al. The clinical profile of right temporal lobe atrophy. Brain (2009) 132(Pt 5):1287–98. doi: 10.1093/brain/awp037
35. Gorno-Tempini ML, Dronkers NF, Rankin KP, Ogar JM, Phengrasamy L, Rosen HJ, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. (2004) 55:335–46. doi: 10.1002/ana.10825
36. Galantucci S, Tartaglia MC, Wilson SM, Henry ML, Filippi M, Agosta F, et al. White matter damage in primary progressive aphasias: a diffusion tensor tractography study. Brain (2011) 134(Pt 10):3011–29. doi: 10.1093/brain/awr099
37. Struyfs H, Niemantsverdriet E, Goossens J, Fransen E, Martin JJ, De Deyn PP, et al. Cerebrospinal fluid P-Tau181P: biomarker for improved differential dementia diagnosis. Front Neurol. (2015) 6:138. doi: 10.3389/fneur.2015.00138
38. Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. (2011) 122:137–53. doi: 10.1007/s00401-011-0839-6
39. Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, DeArmond SJ, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol. (2011) 70:327–40. doi: 10.1002/ana.22424
40. Bhardwaj A, Myers MP, Buratti E, Baralle FE. Characterizing TDP-43 interaction with its RNA targets. Nucleic Acids Res. (2013) 41:5062–74. doi: 10.1093/nar/gkt189
41. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (2006) 314:130–3. doi: 10.1126/science.1134108
42. Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. (2011) 13:38–50. doi: 10.1038/nrn3121
43. Mackenzie IR, Munoz DG, Kusaka H, Yokota O, Ishihara K, Roeber S, et al. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol. (2011) 121:207–18. doi: 10.1007/s00401-010-0764-0
44. Riedl L, Mackenzie IR, Forstl H, Kurz A, Diehl-Schmid J. Frontotemporal lobar degeneration: current perspectives. Neuropsychiatr Dis Treat. (2014) 10:297–310. doi: 10.2147/NDT.S38706
45. Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology (2009) 73:1451–6. doi: 10.1212/WNL.0b013e3181bf997a
46. Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. (2012) 8:423–34. doi: 10.1038/nrneurol.2012.117
47. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
48. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature (1998) 393:702–5. doi: 10.1038/31508
49. Ng AS. Genetics of frontotemporal dementia in Asia: advancing knowledge through collaboration. Neurology (2015) 85:2060–2. doi: 10.1212/WNL.0000000000002045
50. Boeve BF, Hutton M. Refining frontotemporal dementia with parkinsonism linked to chromosome 17: introducing FTDP-17 (MAPT) and FTDP-17 (PGRN). Arch Neurol. (2008) 65:460–4. doi: 10.1001/archneur.65.4.460
51. Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. (2015) 41:24–46. doi: 10.1111/nan.12213
52. Whitwell JL, Jack CR Jr, Boeve BF, Senjem ML, Baker M, Ivnik RJ, et al. Atrophy patterns in IVS10+16, IVS10+3, N279K, S305N, P301L, and V337M MAPT mutations. Neurology (2009) 73:1058–65. doi: 10.1212/WNL.0b013e3181b9c8b9
53. Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
54. Loy CT, Schofield PR, Turner AM, Kwok JB. Genetics of dementia. Lancet (2014) 383:828–40. doi: 10.1016/S0140-6736(13)60630-3
55. Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain (2012) 135(Pt 3):693–708. doi: 10.1093/brain/awr355
56. Floeter MK, Gendron TF. Biomarkers for amyotrophic lateral sclerosis and frontotemporal dementia associated with hexanucleotide expansion mutations in C9orf72. Front Neurol. (2018) 9:1063. doi: 10.3389/fneur.2018.01063
57. Irwin DJ, McMillan CT, Brettschneider J, Libon DJ, Powers J, Rascovsky K, et al. Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry (2013) 84:163–9. doi: 10.1136/jnnp-2012-303507
58. Toh H, Chitramuthu BP, Bennett HP, Bateman A. Structure, function, and mechanism of progranulin; the brain and beyond. J Mol Neurosci. (2011) 45:538–48. doi: 10.1007/s12031-011-9569-4
59. Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, et al. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem. (2008) 283:1744–53. doi: 10.1074/jbc.M705115200
60. Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. (2006) 15:2988–3001. doi: 10.1093/hmg/ddl241
61. Mackenzie IR, Neumann M. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochem. (2016) 138 (Suppl 1):54–70. doi: 10.1111/jnc.13588
62. Rohrer JD, Ridgway GR, Modat M, Ourselin S, Mead S, Fox NC, et al. Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage (2010) 53:1070–6. doi: 10.1016/j.neuroimage.2009.12.088
63. McCluskey L, Vandriel S, Elman L, Van Deerlin VM, Powers J, Boller A, et al. ALS-Plus syndrome: non-pyramidal features in a large ALS cohort. J Neurol Sci. (2014) 345:118–24. doi: 10.1016/j.jns.2014.07.022
64. Boxer AL, Gold M, Huey E, Hu WT, Rosen H, Kramer J, et al. The advantages of frontotemporal degeneration drug development (part 2 of frontotemporal degeneration: the next therapeutic frontier). Alzheimers Dement. (2013) 9:189–98. doi: 10.1016/j.jalz.2012.03.003
65. Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. (2018) 14:535–62. doi: 10.1016/j.jalz.2018.02.018
66. Irwin DJ, Trojanowski JQ, Grossman M. Cerebrospinal fluid biomarkers for differentiation of frontotemporal lobar degeneration from Alzheimer's disease. Front Aging Neurosci. (2013) 5:6. doi: 10.3389/fnagi.2013.00006
67. Irwin DJ, McMillan CT, Toledo JB, Arnold SE, Shaw LM, Wang LS, et al. Comparison of cerebrospinal fluid levels of tau and Abeta 1-42 in Alzheimer disease and frontotemporal degeneration using 2 analytical platforms. Arch Neurol. (2012) 69:1018–25. doi: 10.1001/archneurol.2012.26
68. Kortvelyessy P, Heinze HJ, Prudlo J, Bittner D. CSF biomarkers of neurodegeneration in progressive non-fluent aphasia and other forms of frontotemporal dementia: clues for pathomechanisms? Front Neurol. (2018) 9:504. doi: 10.3389/fneur.2018.00504
69. Scherling CS, Hall T, Berisha F, Klepac K, Karydas A, Coppola G, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol. (2014) 75:116–26. doi: 10.1002/ana.24052
70. Danev SI, St Stoyanov D. Early noninvasive diagnosis of neurodegenerative diseases. Folia Med. (2010) 52:5–13. doi: 10.2478/v10153-010-0041-y
71. Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain (2009) 132(Pt 10):2659–68. doi: 10.1093/brain/awp191
72. Vitali P, Maccagnano E, Caverzasi E, Henry RG, Haman A, Torres-Chae C, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology (2011) 76:1711–9. doi: 10.1212/WNL.0b013e31821a4439
73. Riemenschneider M, Wagenpfeil S, Vanderstichele H, Otto M, Wiltfang J, Kretzschmar H, et al. Phospho-tau/total tau ratio in cerebrospinal fluid discriminates Creutzfeldt-Jakob disease from other dementias. Mol Psychiatry (2003) 8:343–7. doi: 10.1038/sj.mp.4001220
74. Medina M. An Overview on the clinical development of tau-based therapeutics. Int J Mol Sci. (2018) 19:E1160. doi: 10.3390/ijms19041160
75. Hu WT, Watts K, Grossman M, Glass J, Lah JJ, Hales C, et al. Reduced CSF p-Tau181 to Tau ratio is a biomarker for FTLD-TDP. Neurology (2013) 81:1945–52. doi: 10.1212/01.wnl.0000436625.63650.27
76. Borroni B, Gardoni F, Parnetti L, Magno L, Malinverno M, Saggese E, et al. Pattern of Tau forms in CSF is altered in progressive supranuclear palsy. Neurobiol Aging (2009) 30:34–40. doi: 10.1016/j.neurobiolaging.2007.05.009
77. Teunissen CE, Elias N, Koel-Simmelink MJ, Durieux-Lu S, Malekzadeh A, Pham TV, et al. Novel diagnostic cerebrospinal fluid biomarkers for pathologic subtypes of frontotemporal dementia identified by proteomics. Alzheimers Dement. (2016) 2:86–94. doi: 10.1016/j.dadm.2015.12.004
78. Brendel M, Yousefi BH, Blume T, Herz M, Focke C, Deussing M, et al. Comparison of F-T807 and F-THK5117 PET in a mouse model of tau pathology. Front Aging Neurosci. (2018) 10:174. doi: 10.3389/fnagi.2018.00174
79. Goedert M, Yamaguchi Y, Mishra SK, Higuchi M, Sahara N. Tau Filaments and the development of positron emission tomography Tracers. Front Neurol. (2018) 9:70. doi: 10.3389/fneur.2018.00070
80. Dopper EG, Rombouts SA, Jiskoot LC, den Heijer T, de Graaf JR, de Koning I, et al. Structural and functional brain connectivity in presymptomatic familial frontotemporal dementia. Neurology (2014) 83:e19–26. doi: 10.1212/WNL.0000000000000583
81. Lee SE, Khazenzon AM, Trujillo AJ, Guo CC, Yokoyama JS, Sha SJ, et al. Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain (2014) 137(Pt 11):3047–60. doi: 10.1093/brain/awu248
82. McMillan CT, Toledo JB, Avants BB, Cook PA, Wood EM, Suh E, et al. Genetic and neuroanatomic associations in sporadic frontotemporal lobar degeneration. Neurobiol Aging (2014) 35:1473–82. doi: 10.1016/j.neurobiolaging.2013.11.029
83. Irwin DJ, McMillan CT, Suh E, Powers J, Rascovsky K, Wood EM, et al. Myelin oligodendrocyte basic protein and prognosis in behavioral-variant frontotemporal dementia. Neurology (2014) 83:502–9. doi: 10.1212/WNL.0000000000000668
84. Herrmann N, Black SE, Chow T, Cappell J, Tang-Wai DF, Lanctot KL. Serotonergic function and treatment of behavioral and psychological symptoms of frontotemporal dementia. Am J Geriatr Psychiatry (2012) 20:789–97. doi: 10.1097/JGP.0b013e31823033f3
85. Tsai RM, Boxer AL. Therapy and clinical trials in frontotemporal dementia: past, present, and future. J Neurochem. (2016) 138 (Suppl. 1):211–21. doi: 10.1111/jnc.13640
86. Landqvist Waldo M, Gustafson L, Nilsson K, Traynor BJ, Renton AE, Englund E, et al. Frontotemporal dementia with a C9ORF72 expansion in a Swedish family: clinical and neuropathological characteristics. Am J Neurodegener Dis. (2013) 2:276–86.
87. Ducharme S, Bajestan S, Dickerson BC, Voon V. Psychiatric presentations of C9orf72 mutation: what are the diagnostic implications for clinicians? J Neuropsychiatry Clin Neurosci. (2017) 29:195–205. doi: 10.1176/appi.neuropsych.16090168
88. Steinberg M, Lyketsos CG. Atypical antipsychotic use in patients with dementia: managing safety concerns. Am J Psychiatry (2012) 169:900–6. doi: 10.1176/appi.ajp.2012.12030342
89. Mendez MF, Shapira JS, McMurtray A, Licht E. Preliminary findings: behavioral worsening on donepezil in patients with frontotemporal dementia. Am J Geriatr Psychiatry (2007) 15:84–7. doi: 10.1097/01.JGP.0000231744.69631.33
90. Chow TW. Treatment approaches to symptoms associated with frontotemporal degeneration. Curr Psychiatry Rep. (2005) 7:376–80. doi: 10.1007/s11920-005-0040-5
91. Vercelletto M, Boutoleau-Bretonniere C, Volteau C, Puel M, Auriacombe S, Sarazin M, et al. Memantine in behavioral variant frontotemporal dementia: negative results. J Alzheimers Dis. (2011) 23:749–59. doi: 10.3233/JAD-2010-101632
92. Boxer AL, Knopman DS, Kaufer DI, Grossman M, Onyike C, Graf-Radford N, et al. Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. (2013) 12:149–56. doi: 10.1016/S1474-4422(12)70320-4
93. Mocellin R, Scholes A, Walterfang M, Looi JC, Velakoulis D. Clinical update on frontotemporal dementia: diagnosis and treatment. Australas Psychiatry (2015) 23:481–7. doi: 10.1177/1039856215582276
94. Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron (2014) 82:1271–88. doi: 10.1016/j.neuron.2014.04.047
95. Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron (2013) 80:402–14. doi: 10.1016/j.neuron.2013.07.046
96. Wischik CM, Harrington CR, Storey JM. Tau-aggregation inhibitor therapy for Alzheimer's disease. Biochem Pharmacol. (2014) 88:529–39. doi: 10.1016/j.bcp.2013.12.008
97. Coughlin D, Irwin DJ. Emerging diagnostic and therapeutic strategies for tauopathies. Curr Neurol Neurosci Rep. (2017) 17:72. doi: 10.1007/s11910-017-0779-1
98. Mancini RS, Wang Y, Weaver DF. Phenylindanes in brewed coffee inhibit amyloid-beta and tau aggregation. Front Neurosci. (2018) 12:735. doi: 10.3389/fnins.2018.00735
99. Buchholz K, Schirmer RH, Eubel JK, Akoachere MB, Dandekar T, Becker K, et al. Interactions of methylene blue with human disulfide reductases and their orthologues from Plasmodium falciparum. Antimicrob Agents Chemother. (2008) 52:183–91. doi: 10.1128/AAC.00773-07
100. Heard DS, Tuttle CSL, Lautenschlager NT, Maier AB. Repurposing proteostasis-modifying drugs to prevent or treat age-related dementia: a systematic review. Front Physiol. (2018) 9:1520. doi: 10.3389/fphys.2018.01520
101. Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci USA. (1996) 93:11213–8. doi: 10.1073/pnas.93.20.11213
102. Matsuoka Y, Jouroukhin Y, Gray AJ, Ma L, Hirata-Fukae C, Li HF, et al. A neuronal microtubule-interacting agent, NAPVSIPQ, reduces tau pathology and enhances cognitive function in a mouse model of Alzheimer's disease. J Pharmacol Exp Ther. (2008) 325:146–53. doi: 10.1124/jpet.107.130526
103. Morimoto BH, Schmechel D, Hirman J, Blackwell A, Keith J, Gold M. A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement Geriatr Cogn Disord. (2013) 35:325–36. doi: 10.1159/000348347
104. Boxer AL, Lang AE, Grossman M, Knopman DS, Miller BL, Schneider LS, et al. Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol. (2014) 13:676–85. doi: 10.1016/S1474-4422(14)70088-2
105. Pedersen JT, Sigurdsson EM. Tau immunotherapy for Alzheimer's disease. Trends Mol Med. (2015) 21:394–402. doi: 10.1016/j.molmed.2015.03.003
106. Medina M, Avila J. New insights into the role of glycogen synthase kinase-3 in Alzheimer's disease. Expert Opin Ther Targets (2014) 18:69–77. doi: 10.1517/14728222.2013.843670
107. West T, Hu Y, Verghese PB, Bateman RJ, Braunstein JB, Fogelman I, et al. Preclinical and clinical development of ABBV-8E12, a humanized anti-tau antibody, for treatment of Alzheimer's disease and other tauopathies. J Prev Alzheimers Dis. (2017) 4:236–41. doi: 10.14283/jpad.2017.36
108. Panza F, Solfrizzi V, Seripa D, Imbimbo BP, Lozupone M, Santamato A, et al. Tau-centric targets and drugs in clinical development for the treatment of Alzheimer's disease. Biomed Res Int. (2016) 2016:3245935. doi: 10.1155/2016/3245935
109. O'Brien JT, Holmes C, Jones M, Jones R, Livingston G, McKeith I, et al. Clinical practice with anti-dementia drugs: a revised (third) consensus statement from the British Association for Psychopharmacology. J Psychopharmacol. (2017) 31:147–68. doi: 10.1177/0269881116680924
110. Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA. (2013) 110:E4530–9. doi: 10.1073/pnas.1318835110
111. Kramer NJ, Carlomagno Y, Zhang YJ, Almeida S, Cook CN, Gendron TF, et al. Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science (2016) 353:708–12. doi: 10.1126/science.aaf7791
112. DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. (2013) 33:12887–97. doi: 10.1523/JNEUROSCI.2107-13.2013
113. Cenik B, Sephton CF, Dewey CM, Xian X, Wei S, Yu K, et al. Suberoylanilide hydroxamic acid (vorinostat) up-regulates progranulin transcription: rational therapeutic approach to frontotemporal dementia. J Biol Chem. (2011) 286:16101–8. doi: 10.1074/jbc.M110.193433
114. Capell A, Liebscher S, Fellerer K, Brouwers N, Willem M, Lammich S, et al. Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J Neurosci. (2011) 31:1885–94. doi: 10.1523/JNEUROSCI.5757-10.2011
115. Whitwell JL, Weigand SD, Boeve BF, Senjem ML, Gunter JL, DeJesus-Hernandez M, et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain (2012) 135(Pt 3):794–806. doi: 10.1093/brain/aws001
Keywords: frontotemporal dementia, frontotemporal lobar degeneration, genetics, precision medicine, neuroimaging, primary progressive aphasia
Citation: Liu M-N, Lau C-I and Lin C-P (2019) Precision Medicine for Frontotemporal Dementia. Front. Psychiatry 10:75. doi: 10.3389/fpsyt.2019.00075
Received: 07 August 2018; Accepted: 01 February 2019;
Published: 21 February 2019.
Edited by:
Stefan Borgwardt, Universität Basel, SwitzerlandReviewed by:
Stefan Klöppel, Universität Bern, SwitzerlandDrozdstoy Stoyanov Stoyanov, Plovdiv Medical University, Bulgaria
Copyright © 2019 Liu, Lau and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ching-Po Lin, Y2hpbmdwb2xpbkBnbWFpbC5jb20=