 Maria Shadrina
Maria Shadrina Elena A. Bondarenko
Elena A. Bondarenko Petr A. Slominsky
Petr A. Slominsky- Laboratory of Molecular Genetics of Hereditary Diseases, Institute of Molecular Genetics, Russian Academy of Sciences, Moscow, Russia
Depressive disorders (DDs) are one of the most widespread forms of psychiatric pathology. According to the World Health Organization, about 350 million people in the world are affected by this condition. Family and twin studies have demonstrated that the contribution of genetic factors to the risk of the onset of DDs is quite large. Various methodological approaches (analysis of candidate genes, genome-wide association analysis, genome-wide sequencing) have been used, and a large number of the associations between genes and different clinical DD variants and DD subphenotypes have been published. However, in most cases, these associations have not been confirmed in replication studies, and only a small number of genes have been proven to be associated with DD development risk. To ascertain the role of genetic factors in DD pathogenesis, further investigations of the relevant conditions are required. Special consideration should be given to the polygenic characteristics noted in whole-genome studies of the heritability of the disorder without a pronounced effect of the major gene. These observations accentuate the relevance of the analysis of gene-interaction roles in DD development and progression. It is important that association studies of the inherited variants of the genome should be supported by analysis of dynamic changes during DD progression. Epigenetic changes that cause modifications of a gene's functional state without changing its coding sequence are of primary interest. However, the opportunities for studying changes in the epigenome, transcriptome, and proteome during DD are limited by the nature of the disease and the need for brain tissue analysis, which is possible only postmortem. Therefore, any association studies between DD pathogenesis and epigenetic factors must be supplemented through the use of different animal models of depression. A threefold approach comprising the combination of gene association studies, assessment of the epigenetic state in DD patients, and analysis of different “omic” changes in animal depression models will make it possible to evaluate the contribution of genetic, epigenetic, and environmental factors to the development of different forms of depression and to help develop ways to decrease the risk of depression and improve the treatment of DD.
Introduction
Depressive disorders (DDs) comprise one of the most widespread forms of psychiatric pathology. According to the World Health Organization, about 350 million people are affected by a DD. The worldwide prevalence of DDs varies from 3% in Japan to 16.9% in the USA; in most countries, this prevalence ranges from 8 to 12% (1). It is predicted that, by 2020, DDs will be the second-leading cause of disability throughout the world after ischemic heart disease (2).
DD entails a number of unfavorable consequences with medical and sociological relevance, and affects significantly the quality of life and adaptive ability. Long-term and severe depression mixed with chronic somatic or neurological conditions might lead to attempted suicide.
Despite the great medical and social significance of DDs, there is no clear conceptualization to explain the causes and mechanisms of DD development. Several theories have been suggested to explain the onset of depression and have been confirmed by biochemical, immunological, and physiological studies. Parallel to well-known “monoamine,” “cytokine,” and “stress-induced” (hypothalamus–pituitary–adrenal (HPA) axis and stress theories) depression models, the phenomena of altered brain neural plasticity and neurogenesis and circadian rhythm desynchronosis (the chronobiological model) have been proposed to explain the onset of depression.
Family and twin studies have provided strong evidence for the contribution of genetic factors to the risk of depression. For instance, a meta-analysis of twin research data shows that the heritability rate for depression is 37% (95% CI: 31%−42%), and data from family studies show a two- to threefold increase in the risk of depression in first-degree offspring of patients with depression (3). Heritability has also been shown to be especially influential in severe forms of depression (4, 5). The illness severity depends on whether DDs are inherited maternally or paternally (6, 7).
Since 1978, when the first study devoted to identifying possible candidate genes related to DDs was published (8), many studies have searched for genes involved in the progression of depression worldwide. Based on the available data on the putative neurobiological mechanisms underlying DDs, more than 100 candidate genes have been analyzed to identify the possible associations between their alleles and the risk of depression onset or its symptoms. The studies of DD pathogenesis have yielded conflicting results. Development of DNA microchip technology has made it possible to conduct genome-wide associations studies (GWASs) to look for risk factors of depression onset, independent of the hypotheses to explain depression pathogenesis available at the time. However, GWASs using large sets of samples, including thousands of patients with different forms of DDs and tens of thousands of patients in meta-analyses, have failed to identify any specific loci responsible for predisposition to DDs. These studies have also not unambiguously defined the biological mechanisms underlying the pathogenesis of the pathology of DDs. This failure to identify clearly the genetic associations and underlying mechanisms indicate that depression is a complicated multifactor heterogeneous psychiatric disorder. It is likely that the predisposition to DDs is determined by the coordinated action of many genes and their interaction with each other and with diverse environmental factors. It is also likely that each gene by itself makes a relatively small contribution to the pathogenesis of the disease (9).
In this review, we discuss the principal theories to explain the development of depression and the genetic evidence in support of these theories. The final section discusses the results of GWASs and the possible contribution of epigenetic factors to the risk of onset of DDs.
Brief Characterization of Depressive Disorder
Depression
Depression (lat. depressio—gloominess, oppression) is a psychiatric disorder characterized by a pathologically low mood (hypothymia) and negative esteem about oneself, one's status in the real world, and one's future (10). In other words, the major depressive disorder is a complex and heterogeneous illness with an etiopathogenesis that is based upon multiple factors that may act at different levels, e.g., psychological, biological, genetic, and social (11).
Two current reference classifications provide a clinical description of depression: the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) and the International Classification of Diseases, 10th Edition (ICD-10).
According to the DSM-IV, one of the principal forms of depression is major depressive disorder (MDD). For an appropriate diagnosis, five or more of the following 10 DSM-IV symptoms must be exhibited:
• Depressive mood present continuously for a minimum 2-week period prevalent every day and a larger part of the day
• Pronounced elevated emotional psychomotor activity in children and teenagers
• Diminished ability to feel pleasure and rejoice
• Loss or gain of weight against a marked appetite alteration
• Sleep disturbances: insomnia at night and daytime sleepiness
• Objectively registered psychomotor agitation or motor retardation
• Feeling of weakness, loss of energy, marked fatigue even after minimal effort
• Lowered self-esteem and feeling of worthlessness, loss of self-confidence, ungrounded self-accusation to the extent of delirium
• Diminished ability to think or concentrate, mental slowness, lack of resolution
• Thoughts or actions leading to self-injurious or suicidal ideation.
The abovementioned symptoms must be present continuously for at least 2 weeks and cause disturbance of a person's normal vital activities.
In terms of the manifestation of depressive symptoms, the contemporary ICD-10 classification identifies a number of clinical variants of depression, which are classified according to their severity, the presence of psychiatric symptoms, and recurrence of DDs. Episodes of moderate depression (four or more symptoms exhibited; F32.1) and severe depression without psychotic symptoms (commonly with a number of symptoms, usually with lowered self-estimation and suicidal thoughts and attempts; F32.2), and a recurrent (repeated) DD of moderate or severe degree (F33.2–F33.3).
It is important to note that the main classifications of DDs have some differences and that this must be considered when analyzing the experimental data in studies of depression in humans. It is possible that some of the differences in results between studies will be related to the use of different samples of depressed patients who have been diagnosed according to different clinical criteria. Moreover, in our opinion, it is necessary to distinguish exo- and endogenous DD and analyze this type of DD separately. Exogenous, or reactive, depression is usually triggered by some situational stress, as losing a job, the loss of a member of the family, divorce, or relationship difficulties. As opposed to endogenous depression, exogenous is environmentally caused, and associated with anxiety and mood reactivity, and highly sensitive to psychosocial stressors. Endogenous or melancholic depression is a form of DD unrelated to any pronounced exogenous factors (primary severe somatic illness, illness in a close relative, perceived social problems. In exogenous DD can clearly determine the causes of this disease.
Major Hypotheses of Pathogenesis of Depression: From Clinical and Biochemical Data to Candidate Genes of Depression
The Monoamine Theory
The monoamine hypothesis of depression, the first theory historically, was proposed by Joseph Schildkraut in the 1960s. This theory was based on the successful use of iproniazid (monoamine oxidase inhibitor) (12–14) and imipramine (reuptake inhibitor of monoamine neuromediators) for depression (15, 16). As proposed by this theory, insufficiency of monoamine neuromediators (serotonin, norepinephrine, dopamine) in definite structures of the central nervous system (CNS) may lead to the development of depression. Detailed analysis of the mechanism of action of these preparations and of later designed tricyclic antidepressants and reuptake inhibitors of monoamine neuromediators confirmed the important role of the imbalance and insufficiency of neuromediators in DDs (17–20). According to the monoamine theory, the synthesis, vesicular transport, and receptors of monoamine neuromediators play an important role in the development of DDs. As a result, the first genetic studies focused on identifying and analyzing polymorphisms in genes associated with serotonin, noradrenalin, and dopamine neurotransmission.
Most studies have analyzed SLC6A4 (previously known as SERT), which encodes the serotonin transporter that is responsible for the reuptake of serotonin (5-HTT) from the synaptic cleft to the presynaptic neuron and thus plays a role in maintenance of the serotonin level in the presynaptic region. Interest in this transporter also arises from the observation that inhibitors of neural serotonin reuptake are used widely in psychiatry for the treatment of depression, anxiety, and other conditions.
The serotonin transporter is encoded by the solute carrier family 6 member 4 gene (SLC6A4) located on chromosome 17q11.1–17q12 (21). In the promotor region of SLC6A4, a 5-HTTLPR (5-hydroxytryptamine transporter-linked polymorphic region) polymorphism was shown to be associated with the availability (the long L allele) or absence (the short S allele) of the 44 bp fragment (22). The L allele bears 16 GC-rich repeated elements of 20–23 bp, whereas the S allele carries 14 similar repeated units that result from the deletion of the region from the 6th to 8th repeated elements (23). In vitro studies have shown that the S allele is associated with a lower expression level of SLC6A4 mRNA and lower serotonin transporter expression on membranes and, as a consequence, with a lower ability for serotonin reuptake compared with the L allele (23, 24). A number of other rare variants of 5-HTTLPR polymorphism, which contained 15, 19, and >20 repeats, were later reported (25).
In 2006, the single-nucleotide polymorphism (SNP) rs25531 (A → G) near the 5-HTTLPR polymorphism region was revealed by Hu and coauthors. This polymorphism appears to show linkage disequilibrium with 5-HTTLPR, and the G variant is only found in the L allele carriers. The A → G substitution evokes the appearance of the LG allele, the functional analog of the 5-HTTLPR S allele (14, 26). This is because the A → G substitution creates a strong AP2–DNA-binding site (TFBS) which, in turn, suppresses the transcription of SLC6A4 in LG allele carriers (13, 14). It appears that up to 15% of the individuals included in previous studies as L allele carriers should have been functionally classified as S allele carriers. This error may have distorted the results and created false-positive or false-negative evidence. Moreover, the situation appears to be more complicated and clearly demonstrates possible problems in interpreting the results of association analysis of individual polymorphic markers inside candidate genes.
In 2008, another SNP, rs25532 (C → T) also localized near 5-HTTLPR, was identified in the promotor region of SLC6A4. This SNP changes the activity of the 5-HTTLPR/rs2553 combination of polymorphisms. For instance, the LAC allele (the combination of the L allele at the 5-HTTLPR polymorphism with the A and C alleles at the rs25531 and rs25532 polymorphisms) is a variant that ensures a high level of SLC6A4 expression (27). Further studies revealed additional SNPs, with functionally significant changes, such as G56A in exon 2 and 1425V in exon 9. The 1425V mutation is located in the transmembrane area of 5-HTT, which is important for the formation of the secondary structure of this hydrophobic domain.
A polymorphic region comprising three alleles, Stin2.9, Stin2.10, and Stin2.12, was discovered in intron 2 of SLC6A4. This variable number tandem repeat polymorphism increases the expression in proportion to the number of repeated copies of the 16/17 bp element (12 > 10 > 9), as determined in the embryonic brain and in human JAR cells (28). Stin2 alleles respond differently to transcription factors YB-1 and CTCF which, in turn, can be regulated by lithium chloride, which is prescribed for the treatment of bipolar disorders (28, 29).
The structure of SLC6A4 may be far more complicated. Recent publications have reported that the expression of this gene is modulated by microRNA mir-16 binding sites in the 3′-nontranslating region of the gene (30). Therefore, polymorphisms localized within or near microRNA binding sites may be able to exert a strong effect on SLC6A4 expression and, consequently, on 5-HTT functions.
It is possible that the abovementioned complexity of SLC5A4 organization may be one reason for the conflicting results obtained by analyses of the association of this gene's polymorphic variants (primarily in the analysis of the L/S polymorphism of the 5-HTTLPR repeat) with the onset of depression. Meta-analyses of these studies allow no final conclusion to be drawn about the role of this polymorphism in the development of depression. For instance, the meta-analysis conducted by Lopez-Leon et al. (31) disclosed an elevated risk of DDs in S allele carriers, whereas no similar association was found in carriers of other alleles (32–34). The latest meta-analysis of the results of 23 original studies has shown that the S allele raises the risk of DDs; the risk of depression in S allele carriers is increased 1.14-fold (CI: 1.05–1.24). Nevertheless, the high level of heterogeneity of the data included in this meta-analysis should be noted. In all analyzed models, the association p-value does not reach 0.05. This may be due to the inclusion in the meta-analysis of studies of different size samples, including samples comprising fewer than 50 people (35).
In the context of the monoamine theory of DD development, analysis of a large number of candidate genes has been performed. They are, in particular, receptor genes for dopamine (DRD3, DRD4) and serotonin (HTR1A, HTR2A, HTR1B, HTR2C); genes for noradrenalin (SLC6A2) and dopamine (SLC6A3); genes for the enzymes monoamine oxidase A (MAOA), tyrosine hydroxylase (TH), tryptophan hydroxylase 1 (TPH1), catechol-o-methyl transferase (COMT); and the piccolo presynaptic cytomatrix protein (PCLO). For each of these genes, polymorphic variants were identified that were associated with point mutations or tandem repeat polymorphisms. These polymorphisms were analyzed in samples from patients of different ethnicity with DD. As for SLC6A4, different studies have produced conflicting results, and it seems reasonable not to analyze the results of individual studies but to consider only the meta-analyses that have shown the existence of associations between definite variants of the genes and DD development.
One of the first large-scale meta-analyses of genetic case–control research on DDs was conducted in 2008 by Lopez-Leon and coauthors. The final analysis focused on 20 polymorphisms in 18 genes. The pooled odds ratios (ORs) with 95% confidence intervals (CIs) were calculated. Among the genes of the monoaminergic system, statistically reliable associations were found for SLC6A4 and SLC6A3 (31).
In another meta-analysis, Gatt et al. (36) attempted to identify genes associated with DD and common genes shared by the five severe psychiatric disorders: MDD, anxiety (including panic disorders), schizophrenia, bipolar disorder, the attention deficit–hyperactivity syndrome (36). Table 1 displays the data for the genes whose products are involved in monoaminergic neurotransmission.

Table 1. Associations shown in meta-analyses between DDs and polymorphic variants of the genes linked to the exchange of monoamine neuromediators*.
It appears that polymorphic variants of the genes that are in some way associated with the monoamine theory of DD pathogenesis can influence the risk of developing depression. However, this influence is not large and cannot be currently regarded as unambiguously proven because of conflicting results of both individual studies and meta-analyses.
Stress as a Cause of Depressive Disorders
Chronic stress and stressful life events early in life are strong proximal predictors of the onset of depression. Although the response to stress implies stability or maintenance of homeostasis, long-time activation of the stress system can cause harmful or even fatal consequences by elevating the risk of obesity, heart diseases, depression, and other disorders (37). The Hypothalamic–pituitary–adrenal axis and its three main components—hypothalamic neurosecretory cells, pituitary gland, and adrenal cortex—are responsible for adaptation to changed environmental conditions and for mobilization of the organism's reserves during exposure to stress of different etiologies. The HPA system operates in the following way (Figure 1). In response to a stressor, neurons in the hypothalamic paraventricular nuclei secrete corticotropin-releasing hormone (CRH), which exerts its action on the hypophysis to initiate the release into the blood circulation of adrenocorticotropic hormone (ACTH), which stimulates the release of corticosteroids, particularly cortisol, from the adrenal cortex. The final hormonal product of the HPA axis, cortisol, binds to mineralocorticoid receptors (type 1) and glucocorticoid receptors (type 2) to form hormone–receptor complexes, which are then transported into the cell nucleus where they interact with specific DNA regions, the glucocorticoid-response elements, to activate the expression of hormone-dependent genes (38).
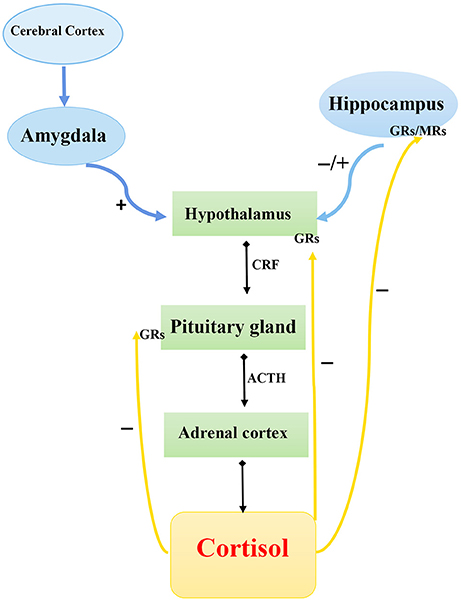
Figure 1. Schematic diagram of hypothalamic–pituitary–adrenal axis. CRF, corticotropin-releasing factor; ACTH, adrenocorticotropic hormone; GRs, glucocorticoid receptor; MRs, mineralocorticoid receptors.  , Secretion;
, Secretion;  , stimulation;
, stimulation;  , inhibition.
, inhibition.
The “stress-induced” theory of DD onset is based on the assumption that hyperactivity of the HPA system may be an important mechanism underlying the development of depression after exposure to stress. A number of examples of abnormal functioning of the HPA system during depression favor this hypothesis. First, stressful events during one's life are the strongest factors that can initiate depression onset (39, 40). Second, depressed patients frequently show elevated cortisol levels (the human endogenous glucocorticoid) in plasma, urine, and cerebrospinal fluid and corticotropin (ACTH) level in plasma (41). Depressed patients also exhibit increased size of the hypophysis and suprarenal glands (42) or decreased function of corticosteroid receptors (43). Excessive activation of the HPA axis is observed in 50% of depressed people, and continuous administration of antidepressants helps to attenuate this activation (44).
A large cohort of genes is likely to be involved in the normal functioning of the HPA axis, but only some of these genes have been actively investigated in the context of DDs. The genes most likely to be involved in DDs are those encoding the targets of cortisol and other glucocorticoid hormones secreted during stress. Polymorphic variants of these genes were analyzed in case-control studies and in some cases, associations between these variants and development of DD were shown. For instance, associations between DD onset and polymorphic sites of the genes coding for the GR (NR3C1) and mineralocorticoid receptor (MCR; NR3C2) have been reported (45–47). Moreover, postmortem studies by Klok et al. (46) have shown that MCR mRNA expression is reduced in the hippocampus in depressed patients (46).
Associations with DD were also shown for genes CRHR1 and CRHR2, which encode CRH receptors (48). Lui et al. (48) reported significant associations between depression and the SNV rs242939 in the CRHR1 gene. These authors also showed that the haplotype formed by G–G–T alleles of the rs1876828, rs242939, and rs242941 was most often represented in patients with MDD compared with controls (48). Szczepankiewicz et al. (49) found associations between DD and the SNVs rs4076452 and rs16940655 of CRHR1 gene (49). Xiao et al. (50) also reported associations between the rs242939 polymorphism of CRHR1 gene and recurrent depression (50).
We note that some of the associations described earlier were included in the meta-analyses by Lopez-Leon et al. (31) or Gatt et al. (36). These authors did not find any reliable associations between DDs and polymorphisms in the genes involved in the functioning and regulation of the HPA axis (31, 36).
Disturbance of Neurogenesis and Neuroplasticity
A large body of experimental data has recently provided evidence of a link between the development of depression with disturbance of normal neurogenesis during brain ontogenetic development and decreased neurogenesis of the adult brain. These effects are thought to be caused by metabolism disturbance of neurotrophic factors, primarily the brain-derived neurotrophic factor (BDNF) in nervous tissue (51–57).
BDNF is abundantly expressed in the adult brain's limbic structures. Some data have shown a connection between the BDNF-mediated signaling pathway and the functioning of serotoninergic neurons. For example, BDNF maintains the survivability and differentiation of serotoninergic neurons, and serotoninergic transmission exerts a strong influence on BDNF expression (58–60).
A functional missense polymorphism rs6265 (GI96A) was described in BDNF that is associated with the substitution of methionine (Met) with valine (Val) in codon 66 (Val66Met). The Met allele was shown to cause disturbed maturation of the protein and to be associated with decreased BDNF activity (61), which might be caused by two different mechanisms: the infective transport of the mutant protein over the regulatory secretory pathway and the defective transport of BDNF mRNA to dendrites (62). Val66Met is a frequent polymorphism whose frequency of alleles is determined by ethnicity. Met allele frequency is 25–32% in European populations and reaches 40–50% in Asian populations (63).
Some studies have reported an association of the Val66Met polymorphism in BDNF with DD onset (64–66), but different authors attribute the risk of DD to the effect of different allelic variants. For instance, Frielingsdorf et al. (66) showed that Met allele homozygotes are at significantly increased risk of MDD (66), whereas Ribeiro et al. (65) defined the Val allele as an allele for the risk for depression (65). This inconsistency motivated the publication of a meta-analysis that combined the results of 14 original studies, but this analysis did not confirm an association between the polymorphic variant in BDNF with DDs (63, 67). The effect of this polymorphism may have been observed only in interaction with other polymorphic systems, such as with 5-HTTLPR polymorphisms or after exposure to severe stress (68, 69).
The Cytokine Theory
The brain was earlier thought to be an “immune-privileged” organ that is protected from circulating immune cells by the blood–brain barrier. It is now well known that immune system cells can infiltrate into nervous tissue. Pro- and anti-inflammatory signals can be transmitted to nervous system from peripheral areas. Also, cytokines and their receptors can be produced in the CNS by astrocytes, microglia (70) and, in some cases, neurons. These molecules are believed to participate in the processes of neuronal development, plasticity, synaptogenesis, and tissue repair.
The hypothesis of bidirectional communication between the immune system and the CNS was suggested in the 1990s. According to this hypothesis, the immune system can interact with the CNS as well as being involved in neuropathological processes. In 1999, Maes M. proposed the inflammatory response system (IRS) model of depression, which claimed that the occurrence of depression depends on activation of the IRS. According to this model, depression can be regarded as a psychoneuroimmunological disease in which peripheral activation of the immune system through the release of anti-inflammatory cytokines can cause the various behavioral, neuroendocrine, and neurochemical changes observed in this disorder (71). This theory was later extended and is now referred to as the “cytokine theory.”
Cytokines constitute a heterogeneous class of mediator molecules produced as regulators of the immune response by immunocompetent cells such as lymphocytes and microphages. Cytokines can be classified into two groups: proinflammatory and anti-inflammatory cytokines. Proinflammatory cytokines are either immediately or indirectly involved in the inflammatory process: interleukin 1 (IL-1), IL-2, IL-6, and IL-12; interferon γ (IFNγ); and tumor necrosis factor α (TNFα). Anti-inflammatory cytokines include IL-4, IL-10, and IL-13, which suppress the immune response and thereby prevent both cell activation and the production of proinflammatory molecules. Some cytokines, such as IL-8, exert both the pro- and anti-inflammatory functions, according to their concentration.
Immunological changes during depression and psychiatric side effects caused by the use of cytokines in the treatments for hepatitis and cancer provide evidence in favor of this theory. For instance, stressors increase the expression of proinflammatory cytokines such as IL-1β, TNFα, and IL-6) in blood and brain (72, 73). DD patients display increased granulocyte and macrophage counts in peripheral blood (74).
In addition to associations with the depression, associations between inflammatory markers and some symptoms, such as apathy, cognitive dysfunction (75), and impaired sleep (76), have been reported. For instance, sleep impairment in depressed patients is associated with increased levels of IL-6 and the soluble forms of intercellular adhesion type 1 molecules in plasma (76) and with the activation in blood cells of nuclear factor-κB (NF-κB), the principal transcription factor involved in initiation of the inflammatory response (77). In other studies, about 50% of the patients chronically administered IFNα developed symptoms of depression, which further supports the role of inflammation in DD pathogenesis (78, 79).
We note, however, that the average cytokine levels in the plasma are elevated only slightly in DD patients compared with healthy people and that these levels are sometimes close to the physiological norm. In autoimmune or infectious diseases, the cytokine levels increase considerably more. Therefore, DD is not considered as a typical autoimmune disease (74).
IL1B is located at locus 2q14.1 on chromosome 2. Several SNPs have been reported for this gene, and two—rs1143627 (−31T/C) and rs16944 (−511C/T)—are associated with DD. Some studies have reported an association between rs16944 (−511C/T) and depression onset and with a positive response to treatment with some antidepressant medications (80–83). Borkowska et al. (81) reported a positive association with recurrent depression (P = 0.064) for the polymorphic haplotype comprising the rs1143627 SNP C allele and the rs16944 SNP T allele (81). By contrast, Yu et al. (82) did not confirm an association of the rs14944 SNP with major depression, although the severity of depression symptoms was elevated in allele C homozygotes (82). Similarly, Hwang et al. (84) failed to find any reliable associations of rs16944 SNP with senile depression or the severity of depression symptoms (84).
The SNPs rs1143627 (−31T/C) and rs16944 (−511C/T) are found in the promoter region of the gene and can affect the expression of this gene, which influences the IL-1β level. Chen et al. (85) showed that SNP rs1143627 localizes in the TATA box area in the IL1B promoter and that the T allele of this polymorphism is associated with increased production of IL-1β (85). However, other investigations have failed to find any reliable associations of SNP rs1143627 with IL-1β production in vitro, although one study found an association between elevated IL-1β expression in vivo and the C allele (86). Data concerning the influence of rs16944 (−511C/T) on IL1B expression are also conflicting. For instance, Hall et al. (87) showed that this SNP influences the expression directly (87). On the other hand, some data support the concept that the −511C/T polymorphism influences the expression of the gene only when acting in concert with the −31T/polymorphism (85).
Therefore, the current data concerning an association of polymorphic variants of the IL1B promoter area do not allow any unambiguous conclusions to be drawn about the role of this gene in the development of depression.
Another actively studied gene, IL6, encodes the proinflammatory cytokine IL-6 and is located at the 7p15.3 locus. The functional polymorphism rs1800795 (174G/C) is located in the gene's promoter region and influences gene expression both at the mRNA and protein levels. The IL-6 level is lower in C allele carriers under conditions of immune system activation (88). No association of this polymorphism with major depression, depression in children, or postbrain stroke depression has been found (89–91). However, this polymorphism has been reported to affect the progression of depressive symptoms in hepatitis C patients administered the immunomodulators IFNα and ribavirin (92). Another interesting observation in that study was the interaction between the 5-HTTLPR polymorphism in the serotonin transport gene (SLC6A4) and rs1800795. A “protective” effect of the 5-HTTLPR polymorphism was observed only in the presence of the low-expressing genotype for IL6 (CC). Consequently, in the genetics of depression, the transition from the analysis of individual polymorphic variants to the analysis of their combinations including two, three, or more loci seems to be very important.
The Circadian Rhythm Theory
Circadian rhythms oscillate with ~24-h periodicity and are responsible for regulating a wide variety of physiological and behavioral processes. Endogenous cyclic oscillations are regulated in humans and other mammals by the circadian pacemaker—the suprachiasmatic nucleus (SCN) neurons of the anterior hypothalamus (93). The circadian pacemaker can change its pattern so that the circadian rhythm may be advanced, delayed, or remain constant in various pathological states or when affected by different pharmacological preparations and hormones; for instance, melatonin regulates the function of the biological clock through melatoninergic receptors residing in the hypothalamic SCN (94). At the cellular level, the “molecular clock” refers to a network of “clock” genes, which are transcriptional regulators organized in a feedback-sustained transcription–translation network. This mechanism helps maintain the rhythmic expression of target genes during a 24-h cycle (95). The basis of the molecular clock is the negative feedback loop, in which the expression of PER and CRY proteins is inhibited by their interactions with the transcriptional factors CLOCK and BMAL1 and by blocking their binding to E-box regulatory elements in the promoters of the genes for PER and CRY protein family members. Posttranslational modifications of the molecular clock system components by signal molecules such as casein kinase δ/ε and glycogen synthase kinase 3-beta play important roles in the maintenance of circadian rhythms (96).
Sleep disorders (early awakening, insomnia, and inability to resume sleep after waking) are observed in 80–90% of patients with depression, and insomnia is regarded as a risk factor for the onset of depression (97). Therefore, disturbance of the normal functioning of circadian system proteins may play a role in DDs. Additional evidence for the role of circadian rhythm genes in DD onset was provided by studies of two familial syndromes of sleep disturbance: familial advanced sleep-phase syndrome and delayed sleep-phase syndrome (DSPS). Both syndromes are caused by mutations in the genes PER2, CKie, PER3, and CLOCK, which encode circadian system proteins (98). People with these mutations frequently experience depressive symptoms. In addition, people with a history of depression have elevated expression of the circadian system genes CLOCK, PER1, and BMAL1 compared with healthy volunteers (99). Case–control studies have shown associations between depression onset and polymorphic variants of the genes encoding circadian system proteins, including BMAL1, CLOCK, NPAS2, PER3, CRY1, and TIMELESS. However, none of these genes was confirmed in the meta-analysis (100).
Other Candidate Genes
Investigations of the candidate genes selected according to the principles of DD etiopathogenesis theories have identified only five genes whose polymorphic variants are reliably associated with DD onset according to the results of meta-analyses (Table 1). However, all of these genes have been discussed only in terms of the monoamine theory of depression.
The association research is not confined solely to the DD-related candidate genes studied in terms of DD etiopathogenesis theories. Other genes have also been actively studied, primarily those analyzed for other neurological and psychiatric disorders. Moreover, these studies have revealed a large amount of reliable associations that have passed through testing in large meta-analyzes (Table 2). These association studies have revealed genes encoding functionally diverse proteins, from chondroitin sulfate biosynthesis enzymes to the key enzyme of the renin–angiotensin system (RAS). On the one hand, this may reflect the limited understanding of DD etiopathogenesis, although the currently known associations suggest new hypotheses. On the other hand, many of the associations revealed are in some way linked to neurogenesis and neuroplasticity processes, and DDs may be regarded as disorders linked to disturbance of neural tissue ontogenesis.
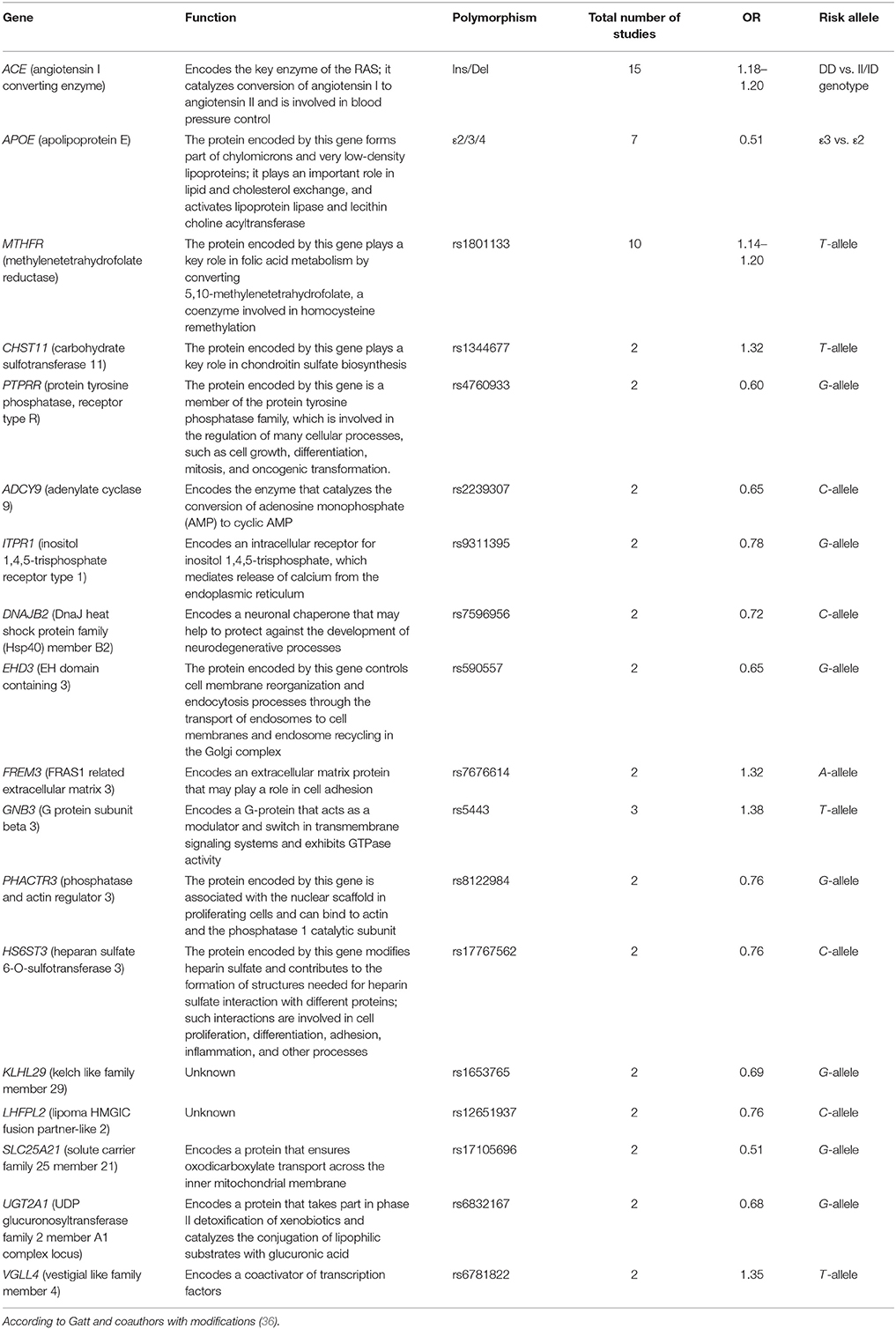
Table 2. Association shown by meta-analyses between DD and polymorphic variants of genes not linked with the general hypotheses of DD etiopathogenesis.
Associations between DD and polymorphic variants of the genes angiotensin-converting enzyme (ACE), apolipoprotein E (APOE), and methylenetetrahydrofolate reductase (MTHFR) are of special interest. These genes have been discussed in the context of another theory of DD etiopathogenesis—the vascular theory—which postulates that DD occurs because of disturbance in the blood supply to neural tissue. According to this theory, vascular disorders can cause DD as well as other mental illnesses such as schizophrenia and manic–depressive psychosis. It is possible that there is a continuum of these diseases, which are separated by only a thin line; for example, some clinical subtypes of depression are characterized by marked psychotic symptoms (F32.3 in ICD-10). Presumably, there may be a complex link between genetic variants and the occurrence of various pathologies along the continuum. For example, genetically determined vascular disorders provoke increased risk for different mental disorders. Progression of a particular disease (e.g., schizophrenia, depression, manic–depressive psychosis) may be determined by other genetic factors, such as those associated with neuromediator functions.
Genome-Wide Association Analysis of Depressive Disorder
As noted above, >20 genes have been associated with DD onset and confirmed by meta-analyses. In most cases, these associations involve genes that are not directly linked to the general theories of depression ethnopathogenesis. Association studies appeared to be connected with transition to genome-wide methods of association analysis without any suggestions about the genetic risk factors of depression.
In the first stage of GWASs, families with members that have experienced multiple depression events, severe course of the disorder, or an early age at its clinical onset were analyzed with special interest in patients with rare monogenic forms of depression. The results of these studies are summarized in Table 3. These studies have reported associations with extended genomic regions (even as long as full-length chromosomes), and the identification of the candidate genes seems to be provisional. This identification mainly based on the DD candidate genes mapped earlier in these genome regions.

Table 3. Mapping the loci associated with predisposition to different forms of depression in family studies using SNP panels of DNA markers.
GWASs have been used increasingly in the past decade to identify loci that control complex traits. In this analysis, as many as hundreds of thousands to several millions of SNPs distributed over the whole genome are identified in groups of persons having a particular trait of interest. Analysis of the genotype–phenotype associations makes it possible to establish a link between the allelic variant in some particular region of the genome with the trait studied. The principal difference between GWASs and candidate gene studies using the case–control method is that there is no preliminary hypothesis to explain the contribution of polymorphic variants of genes to the development of a pathology of interest. However, for a study to achieve statistically significant results, its algorithm requires very large samples of both patients and healthy persons. It can be extremely difficult to achieve clinical homogeneity in very large samples, especially when studying psychiatric diseases because there is always a subjectivity factor affecting the diagnostic accuracy in the relevant international classifications with almost no instrumental methods for assessing the patient's condition.
A number of studies have searched for loci associated with MDD or individual symptoms of depression. The results are summarized in Table 4. This table focuses mainly on those studies that analyzed primarily the risk of depression as a disease and not the endophenotypes (e.g., clinical onset age, severity of particular symptoms, patients' responses to therapy). As well, Table 4 includes the most statistically significant results from the analyzed articles.
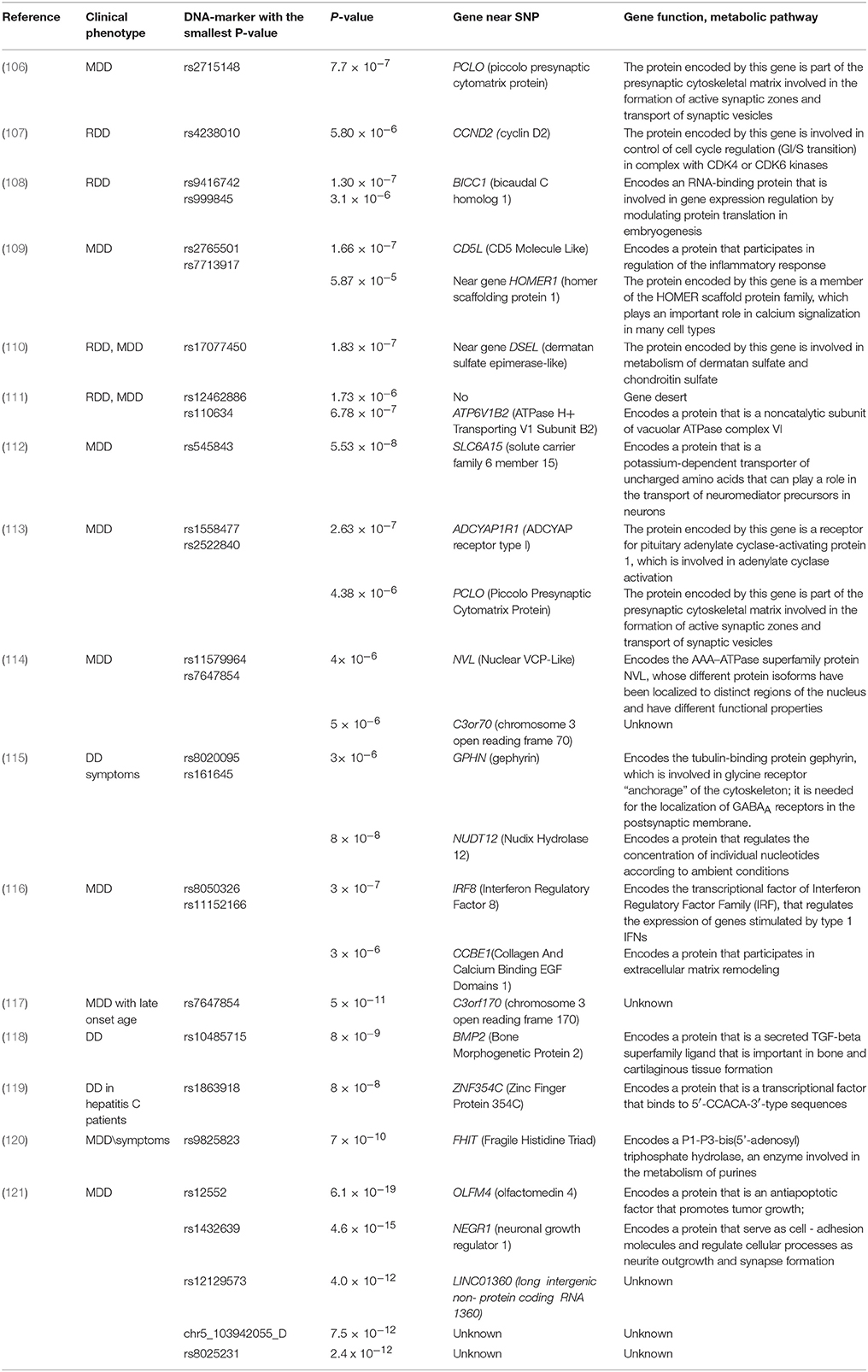
Table 4. Genome-wide association studies of major depressive disorders (MDDs) and recurrent depressive disorders (RDDs).
The first GWAS of a large representative sample (1738 DD patients, 1802 controls) was reported by Sullivan et al. (106). In this study, no association with any of SNPs achieved the value of genome wide significance. The maximum significance was found for the rs2715148 (p = 7.7 × 10−7). Also, in this genomic region near PCLO gene 10 more SNPs were associated with DD with relatively low significance (p = 10−5-10–6). They were mapped to a 167 kb region where PCLO was located (106). PCLO protein localizes in the cytoplasmic matrix of the presynaptic active zone and plays a significant role in brain monoaminergic neurotransmission. A possible role of this region in depression onset was confirmed by Hek et al. (115), who showed an association between the rs2522833 SNP in PCLO and DD in a population-based study from the Netherlands (115). Aragam et al. (113) found a close statistically significant association between DD development for the rs2715148 SNP (P = 5.64 × 10−7) in PCLO in women (113). This study found another SNP in LGSN that was associated with DD occurrence in men (rs9352774, P = 2.26 × 10−4). This gene is actively expressed in the human crystalline lens and encodes a protein related to GS-I and, to a lesser degree, to GS-II glutamine synthetases. This protein may play a role in glutamate exchange in both the retina and the nervous system.
A role of glutamate in DD was found in a GWAS conducted by Rietschel et al. (109). They found an association between DD and the rs7713917 SNP (P = 5.87 × 10−5) located in a putative regulatory region of HOMER1, which encodes proteins involved in glutaminergic processes via interaction with the metabotropic glutamate receptors mGluR1 and mGluR5.
We reiterate that the associations discovered in most GWASs did not attain a genome-wide significance level, primarily because of the genetic architecture of complex traits predisposing to depression. Adjustments of the genome-wide significance level are very rigorous, and we believe that SNP markers with a probability value close to the genome-wide threshold level should also be considered.
Some studies have achieved a genome-wide significance level. Kohli et al. (112) were the first to report an association between DDs and the rs1545843 SNP in SLC6A15 (solute carrier family 6, neutral amino acid transporter, member 15) in a recessive model of the effect of this polymorphism on the risk of DDs (112). This gene encodes the neutral amino acid transporter, and different rs1545843 alleles were shown to have different SLC6A15 expression levels in the hippocampus of epileptic patients. The authors presented additional evidence to support the involvement of this association and showed that the presence of the risk allele correlated with lower SLC6A15 expression in the hippocampus, smaller hippocampus volume, and neuronal integrity in vivo. Lower expression of Slc6a15 was also observed in the hippocampus of mice with elevated chronic stress susceptibility.
Kohli et al. (112) reported abundant data in support of the association between SLC6A15 and DD. However, subsequent GWAS disclosed no significant associations with this gene. The data obtained in GWASs are often not reproducible, and only one gene, PCLO, appeared to be associated with DD in two GWASs.
The Psychiatric Genomics Consortium (PGC) performed a meta-analysis of GWAS data. Unlike the conventional meta-analyses, which summarize the statistical data for each constituent analysis examined, the PGS study brought together and examined individual genotypic and phenotypic data from patients from different research centers. The PGS published the results of its genome-wide comparative analysis of 9240 samples collected from DD patients and 9519 samples from a control group of nine European populations (122). However, in the PGS analysis, none of SNPs identified in earlier studies achieved a genome-wide significance level. The SNPs with the most significant values were rs11579964 (P = 1.0 × 10−7), which mapped near CNIH4, NVL, and WDR26, and rs7647854 (P = 6.5 × 10−7), which mapped near C3orf70 and EHHADH. A subsequent replicative study conducted using an independent sample (6783 patients with MDD and 50,695 controls) did not confirm the associations mentioned.
Therefore, no locus has been shown to be consistently associated with a DD at a whole-genome significance level. Associations shown in independent samples have also not been reproduced. This lack of significance and reproducibility may reflect the particular features of the GWAS methodology, which has focused on polymorphic sites with a high minor allele frequency (>5%) in the associative analysis. These frequent polymorphic variants themselves are probably not pathogenically essential, but there may be disequilibrium linkages with rare variants of genes associated with DD pathogenesis. These rare variants may be specific for different populations. As a result, any association between the disease and a frequent polymorphic site may be found in one sample and may reflect the disequilibrium linkage of this polymorphic site with a rare, pathogenically significant variant in that sample. However, the pathogenically significant site may be missing in another sample and, as a consequence, no association of frequent polymorphism with DD occurrence will be found. In addition, the important role of rare genomic variants (a frequency <1%) has been reported in association with other mental disorders, such as schizophrenia and autism (123, 124).
To overcome these problems, transition from the analysis of polymorphic DNA markers using microarrays to low-coverage DNA sequencing may provide a new direction for research to identify DD-associated genetic variants. The first study of this kind was conducted within the CONVERGE Project (125) and included genome sequencing with an average coverage of 1.7 × in >9000 Chinese females; 5000 females out of this group were patients with melancholic depression, which is recognized as a more severe form of depression. This study found two loci bearing an association at a 10−8 significance level: one on the 5′-side of SIRT1 (SNP rs12415800) and the other in an LHPP intron (SNP rs35936514). This association was confirmed in an independent sample of melancholic Chinese women, and the significance values combined for the two samples were 2.53 × 10−10 for SIRT1 and 6.45 × 10−12 for LHPP. It is important to note that both associated SNPs occur frequently (e.g., the minimal allele frequencies were 45.3 and 26.2%), yet neither is included in the microarrays used widely for SNP marker typing and, therefore, may have been ignored in earlier GWASs.
Further analysis of the data in this project showed that frequent SNPs accounted for 20–30% of the DD risk dispersion, which suggested that the heritability of DD is evenly distributed over all chromosomes with preferential localization of DD-associated SNPs in both the coding and the 3′-untranslated areas of genes. DD patients showed an elevated frequency of unique mutations in gene coding regions, primarily in the genes actively expressed in nervous tissue (126).
Importantly, this study included a specific ethnic group (Han Chinese), which is sufficiently homogeneous, and only females, who show a higher heritability level as mentioned above, with a severe form of DD. This design included a more rigorous approach to inclusion of samples and consideration of factors such as the patients' sex, clinical DD variation, clinical onset age, and other factors that can affect the risk of disease and its progression. However, these factors may exert no influence on the risk of DD development; for example, the clinical onset age was recently shown to not affect the association analysis results in the Chinese CONVERGE sample (127).
Another study also found that ethnicity was important (128). That study included a combined analysis of the results obtained in the CONVERGE investigation of Chinese and of studies conducted by the PGC in different European populations. These studies found that some SNPs influence the risk of DD onset in both ethnic groups mentioned but, at the same time, detected a set of SNPs specific to each ethnic group. The highest contribution of genetic factors in both ethnic groups was observed in females and in recurrently depressed patients.
Powers et al. (116) attempted to include environmental factors into GWASs (116). They included as a factor stress-provoking events when including case–control pairs of patients in the study—a method referred to as propensity score matching. This analysis allowed them to reduce the heterogeneity of the samples with regard to the stress factor and to compare DD patients and healthy controls exposed to similar stressors.
The genetic structure of depression appears to be extremely complicated and involves a large number of loci, which cause various phenotypic effects and display complex interlocus interactions. Studies of the genetic structure suggest the need for a transition from the analysis of individual SNPs to that of sets of SNPs and, finally, to include a polygenic risk score, as used in genetics research of schizophrenia (129).
To address similar problem, a strategy for studying gene networks created by uniting signals from numerous SNPs and subsequent functional analysis of the signaling and metabolic pathways have been used with success. This approach provides for an increase in the power of comparative analysis of weak signals from numerous loci. The study by Song et al. (130) is an example of such an analysis. On the basis of a GWAS of samples from European cohorts, the authors conducted a search and analysis of DD-linked SNPs and genes with these SNPs to discover signal pathways linking these genes to each other (130). Five resulting signal paths were found to play a role in DD pathogenesis. Three of them were claimed to be connected in some way with the negative regulation of gene expression (GO:0016481, GO:0045934, GO:0010629) and were related to some DD-associated SNPs: rs3213764 in ATF7IP; rs2301721 in HOXA7; rs6720481 in LRRFIP1; rs2229742 in NRIP1.
Okbay et al. (118) and Hyde et al. (131) offered an alternative approach for sampling (118, 131). To diagnose DDs, they compiled a questionnaire to be completed by the respondents. Depression was diagnosed on the basis of the respondents' answers to the questionnaire with no clinical diagnosis by a psychiatrist. Although the accuracy of the diagnosis may be questioned, the questionnaire included questions on a wide range of phenotypic traits, and respondents could not associate them with any diagnoses. Data from biobanks or mass genotyping services such as 23 and Me allowed them to markedly increase the sample size. For example, the study by Hyde et al. (131) included >450,000 individuals, and analysis of their questionnaire data allowed them to diagnose depression in about 120,000 participants (131). Samples of this size are an order of magnitude greater than those included in the PGC studies or CONVERGE Project, and help to minimize the problems caused by DD diagnostic errors. The authors managed to identify 17 SNP markers in 15 loci whose significance level was >5 × 10−8, which reflects the size of the sample analyzed. The DNA markers detected differ from those associated with DD in the PGC studies, although they both analyzed samples of European origin. Therefore, the problem of the reproducibility of results obtained in the GWASs remains to be solved.
A possible way to solve this problem is to conduct a meta-analysis of GWA studies. This analysis was carried out by Wray et al. (121). This meta-analysis identified 44 independent loci that were statistically significant (P < 5 × 10−8). Of these loci, 30 are new and 14 were significant in a prior study of MDD or depressive symptoms, and 6 shared loci with schizophrenia. Thus, the increase in sample sizes in the meta-analysis, on the one hand, allows the confirmation of the results obtained earlier with GWAS for the previously described loci associated with MDD. On the other hand, it increases the power of the study, by increasing the sample size, making it possible to identify new loci associated with the MDD.
Several methods were proposed for calculating of genetic risk score (GRS): simple count genetic risk score (SC-GRS), odds ratio weighted genetic risk score (OR-GRS), direct logistic regression genetic risk score (DL-GRS), polygenic genetic risk score (PG-GRS) and explained variance weighted genetic risk score (EV-GRS). Currently, the most widely used method is polygenic risk score (PGRS) (132). This approach has been used to obtain evidence of a genetic effect even when no single markers are significant, to establish a common genetic basis for related disorders, and to construct risk prediction models (133). Currently, alternative approaches to statistical analysis of GWASs data are proposed, where the analysis is not of individual DNA markers, but their combinations. Recently, several papers have been published using PGRS for the MDD and other psychiatric disorders (134–136). The possibility of using the PGRS to evaluate the cumulative contribution of several polymorphic variants of genes to the formation of endophenotypes of MDD was demonstrated. Whalley et al. (135), using the PGRS, divided the MDD into two subtypes, one of which is close to schizophrenia (135).
Conclusions
Summing up the last quarter-century of investigation of the role of genetic factors in DD onset and progression, we note the polygenicity of inherited diseases with no pronounced effect of the principal gene. This has been made clear by recent studies undertaken in the CONVERGENCE and PGC studies and by analysis of candidate genes, which show that each of the genes implicated probably make a small contribution to DD progression. The important role of intergenic interactions has not been studied to date and will require new methods to analyze the data of association studies by including the contribution of combinations of two or more polymorphic DNA markers.
As noted above, DD typically has a high phenotypic heterogeneity, which may be manifested in differing severity of the main symptoms, and this heterogeneity limits the use of association studies. An approach to analyzing very large samples with minimally rigid assessment criteria for the DD phenotype (an extreme example is the variant identified by the 23andMe company associated with self-diagnostics of DD) is one possibility, but this must be supplemented by analysis of small and clinically highly homologous samples. This kind of analysis may not clear up the uncertainty about the genetics of DD on the whole, but it will make it possible to identify certain genetic variants underlying individual subphenotypes of DD.
A small but growing body of evidence suggests that mitochondrial dysfunction may play a role in the development of MDD. MDD was found to be associated with an increased production of mtROS, which could indicate a dysfunction of mitochondria (137, 138). Gardner et al. (139) found a decrease of mitochondrial ATP production rates and mitochondrial enzyme ratios in the muscle of patients with major depressive disorder and chronic physical conditions compared to controls (139). As well, decreased levels of an important part of electron transport chain, i.e., CoQ10, were found in the serum and peripheral blood mononuclear cells received from patients with MDD, which may also indicate a dysfunction of mitochondria (140).
Currently, however, there have been few studies on the role of genetic variants in mitochondrial DNA associated with MDD. A deletion in mtDNA in a child was associated with mitochondrial disease symptoms and with mild-moderate unipolar depression (141). Sequeira et al. (142) have analyzed post-mortem brain samples from human subjects and were failed to show associations of the mitochondrial haplogroups and major depression. However rare homoplasmic mutations with possible functional consequences were reveled in major depression cases, in the ATP synthase 8 (ATP8), ATP synthase 6 (ATP6), ND5 and cytochrome b (CYTB) genes, while another subject with depression demonstrated subthreshold heteroplasmy rate at a variant in the displacement loop (D-loop) part of mtDNA (142). Veronese et al. (143) found no significant associations between specific mitochondrial haplogroups and depressive symptoms either (143). Thus, changes in the functioning of mitochondria may be caused both by an abnormality of the mitochondrial DNA, and by variants of nuclear genes that encode mitochondrial proteins. The meta-analysis conducted by Huo et al. (141) identified SCL25A37 as a novel MDD risk gene, and Zhang et al. (144) have showed that a haplotype T-C consisting of rs12457810 and rs12964485 in the 5'-upstream region of NDUFV2 may be a protective factor for the development of MDD in Han Chinese (144, 145).
It is also important to supplement the association studies of inherited genome variants with analysis of dynamic modifications occurring during DD. There is much interest in the epigenetic changes that can modify a gene's functional status without changing its coding sequence. These epigenetic modifications can be caused by the action of different factors and can be stably inherited after disappearance of the factor causing the change. These epigenetic factors primarily involve DNA methylation and histone modification (methylation and acetylation). In recent years, several studies have analyzed changes in DNA methylation in DD. The first genome-wide analysis of methylation profiles in DD was by Sabuncian et al. (146), who assessed DNA methylation in postmortem frontal cortex material from DD patients and healthy people. In a number of regions, methylation differed reliably between healthy individuals and DD patients (146). A subsequent replicative investigation confirmed this modification of the methylation status in DD patients of the proline rich membrane anchor 1 gene, PRIMA1, which codes for the protein responsible for the assembly of acetylcholine esterase into tetramers and its “anchorage” in the neuron cellular membranes (147). This gene has not been mentioned in association with DD onset. Methylome changes were reported in peripheral blood of twins discordant with regard to DD occurrence. This study also reported on associative studies showing that, in some cases, the changes were related to genes linked to the development of the disorder (e.g., ZBTB20, AGTPB1, TBC1D8, and CLSTN1) (148). A number of studies have focused on associations between DD and histone methylation or acetylation (149, 150). Changes in lysine methylation of histone K27H3 were found postmortem in the BDNF promotor region in the prefrontal and frontal cortex of DD patients, and these changes correlated well with the expression level of BDNF. However, in this review, we do not analyze in detail the role of epigenetic factors in the development of the pathogenesis of DD.
Investigation of changes in the epigenome, transcriptome, and proteome in DD is probably limited by the nature of this disease and the need for brain tissue, which is possible only postmortem. Researchers must work with an extremely limited number of samples from patients, many of whom are on long-term treatment for both DD and somatic conditions. Therefore, to understand fully the entire process involved in DD onset and progression, studies of DD pathogenesis must be supplemented with experiments using different animal models of depression, which would permit an evaluation at different levels of the nervous system organization.
A threefold approach that combines gene-association studies with assessment of the epigenetic status of DD patients and analysis of the changes in animal models of depression, despite the limitations of such models (34), will enable researchers to identify the contributions of genetic, epigenetic, and environmental factors to different forms of DDs and to develop ways to reduce the risk of depression and to provide adequate treatment.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This study was supported by Russian Science Foundation (RSF) (project no. 16-15-10199).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Flint J, Kendler KS. The genetics of major depression. Neuron (2014) 81:484–503. doi: 10.1016/j.neuron.2014.01.027
2. Murray CJ, Lopez AD. Evidence-based health policy–lessons from the global burden of disease study. Science (1996) 274:740–3. doi: 10.1126/science.274.5288.740
3. Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry (2000) 157:1552–62. doi: 10.1176/appi.ajp.157.10.1552
4. McGuffin P, Cohen S, Knight J. Homing in on depression genes. Am J Psychiatry (2007) 164:195–7. doi: 10.1176/ajp.2007.164.2.195
5. Menke A, Klengel T, Binder EB. Epigenetics, depression and antidepressant treatment. Curr Pharm Des. (2012) 18:5879–89. doi: 10.2174/138161212803523590
6. Kendler KS, Gardner CO, Neale MC, Prescott CA. Genetic risk factors for major depression in men and women: similar or different heritabilities and same or partly distinct genes? Psychol Med. (2001) 31:605–16. doi: 10.1017/S0033291701003907
7. Kendler KS, Gatz M, Gardner CO, Pedersen NL. A Swedish national twin study of lifetime major depression. Am J Psychiatry (2006) 163:109–14. doi: 10.1176/appi.ajp.163.1.109
8. Beckman G, Beckman L, Cedergren B, Perris C, Strandman E. Serum protein and red cell enzyme polymorphisms in affective disorders. Hum Hered. (1978) 28:41–7. doi: 10.1159/000152929
9. Ebmeier KP, Donaghey C, Steele JD. Recent developments and current controversies in depression. Lancet (2006) 367:153–67. doi: 10.1016/S0140-6736(06)67964-6
11. Chirita AL, Gheorman V, Bondari D, Rogoveanu I. Current understanding of the neurobiology of major depressive disorder. Rom J Morphol Embryol. (2015) 56:651–8.
12. Deverteuil RL, Lehmann HE. Therapeutic trial of iproniazid (marsilid) in depressed and apathetic patients. Can Med Assoc J. (1958) 78:131–3.
13. Kraft JB, Slager SL, McGrath PJ, Hamilton SP. Sequence analysis of the serotonin transporter and associations with antidepressant response. Biol Psychiatry (2005) 58:374–81. doi: 10.1016/j.biopsych.2005.04.048
14. Hu XZ, Lipsky RH, Zhu G, Akhtar LA, Taubman J, Greenberg BD, et al. Serotonin transporter promoter gain-of-function genotypes are linked to obsessive-compulsive disorder. Am J Hum Genet. (2006) 78:815–26. doi: 10.1086/503850
15. Kuhn R. The treatment of depressive states with G 22355 (imipramine hydrochloride). Am J Psychiatry (1958) 115:459–64. doi: 10.1176/ajp.115.5.459
16. Schildkraut JJ, Kety SS. Biogenic amines and emotion. Science (1967) 156:21–37. doi: 10.1126/science.156.3771.21
17. Leonard BE. Evidence for a biochemical lesion in depression. J Clin Psychiatry (2000) 61 (Suppl. 6):12–17.
18. Ressler KJ, Nemeroff CB. Role of serotonergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depress Anxiety (2000) 12 (Suppl. 1):2–19. doi: 10.1002/1520-6394(2000)12:1+<2::AID-DA2>3.0.CO;2-4
19. Manji HK, Drevets WC, Charney DS. The cellular neurobiology of depression. Nat Med. (2001) 7:541–7. doi: 10.1038/87865
20. Nutt DJ. The neuropharmacology of serotonin and noradrenaline in depression. Int Clin Psychopharmacol. (2002) 17 (Suppl. 1):S1–12. doi: 10.1097/00004850-200206001-00002
21. Ramamoorthy S, Bauman AL, Moore KR, Han H, Yang-Feng T, Chang AS, et al. Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proc Natl Acad Sci USA. (1993) 90:2542–6. doi: 10.1073/pnas.90.6.2542
22. Heils A, Teufel A, Petri S, Seemann M, Bengel D, Balling U, et al. Functional promoter and polyadenylation site mapping of the human serotonin (5-HT) transporter gene. J Neural Transm Gen Sect. (1995) 102:247–54. doi: 10.1007/BF01281159
23. Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science (1996) 274:1527–31. doi: 10.1126/science.274.5292.1527
24. Heils A, Teufel A, Petri S, Stober G, Riederer P, Bengel D, et al. Allelic variation of human serotonin transporter gene expression. J Neurochem. (1996) 66:2621–4. doi: 10.1046/j.1471-4159.1996.66062621.x
25. Nakamura M, Ueno S, Sano A, Tanabe H. The human serotonin transporter gene linked polymorphism (5-HTTLPR) shows ten novel allelic variants. Mol Psychiatry (2000) 5:32–8. doi: 10.1038/sj.mp.4000698
26. Wendland JR, Martin BJ, Kruse MR, Lesch KP, Murphy DL. Simultaneous genotyping of four functional loci of human SLC6A4, with a reappraisal of 5-HTTLPR and rs25531. Mol Psychiatry (2006) 11:224–6. doi: 10.1038/sj.mp.4001789
27. Wendland JR, Moya PR, Kruse MR, Ren-Patterson RF, Jensen CL, Timpano KR, et al. A novel, putative gain-of-function haplotype at SLC6A4 associates with obsessive-compulsive disorder. Hum Mol Genet. (2008) 17:717–23. doi: 10.1093/hmg/ddm343
28. MacKenzie A, Quinn J. A serotonin transporter gene intron 2 polymorphic region, correlated with affective disorders, has allele-dependent differential enhancer-like properties in the mouse embryo. Proc Natl Acad Sci USA. (1999) 96:15251–5. doi: 10.1073/pnas.96.26.15251
29. Ali FR, Vasiliou Sa Haddley K, Haddley K Paredes UM, Roberts JC, Miyajima F, Klenova E, et al. Combinatorial interaction between two human serotonin transporter gene variable number tandem repeats and their regulation by CTCF. J Neurochem. (2010) 112:296–306. doi: 10.1111/j.1471-4159.2009.06453.x
30. Baudry A, Mouillet-Richard S, Schneider B, Launay JM, Kellermann O. miR-16 targets the serotonin transporter: a new facet for adaptive responses to antidepressants. Science (2010) 329:1537–41. doi: 10.1126/science.1193692
31. Lopez-Leon S, Janssens AC, Gonzalez-Zuloeta Ladd AM, Del-Favero J, Claes SJ, Oostra BA, et al. Meta-analyses of genetic studies on major depressive disorder. Mol Psychiatry (2008) 13:772–85. doi: 10.1038/sj.mp.4002088
32. Murphy DL, Maile MS, Vogt NM. 5HTTLPR: White Knight or Dark Blight? ACS Chem Neurosci. (2013) 4:13–5. doi: 10.1021/cn3002224
33. Chen D, Meng L, Pei F, Zheng Y, Leng J. A review of DNA methylation in depression. J Clin Neurosci. (2017) 20:30417–4. doi: 10.1016/j.jocn.2017.05.022
34. Wang Q, Timberlake MA II, Prall K, Dwivedi Y. The recent progress in animal models of depression. Prog Neuropsychopharmacol Biol Psychiatry (2017) 77:99–109. doi: 10.1016/j.pnpbp.2017.04.008
35. Oo KZ, Aung YK, Jenkins MA, Win AK. Associations of 5HTTLPR polymorphism with major depressive disorder and alcohol dependence: A systematic review and meta-analysis. Aust N Z J Psychiatry (2016) 50:842–57. doi: 10.1177/0004867416637920
36. Gatt JM, Burton KL, Williams LM, Schofield PR. Specific and common genes implicated across major mental disorders: a review of meta-analysis studies. J Psychiatr Res. (2015) 60:1–13. doi: 10.1016/j.jpsychires.2014.09.014
37. Bao AM, Meynen G, Swaab DF. The stress system in depression and neurodegeneration: focus on the human hypothalamus. Brain Res Rev. (2008) 57:531–53. doi: 10.1016/j.brainresrev.2007.04.005
38. Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology (2000) 23:477–501. doi: 10.1016/S0893-133X(00)00159-7
39. Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry (1999) 156:837–41. doi: 10.1176/ajp.156.6.837
40. Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev. (2005) 4:141–94. doi: 10.1016/j.arr.2005.03.003
41. Holsboer F, Barden N. Antidepressants and hypothalamic-pituitary-adrenocortical regulation. Endocr Rev. (1996) 17:187–205. doi: 10.1210/edrv-17-2-187
42. Nemeroff CB. The corticotropin-releasing factor (CRF) hypothesis of depression: new findings and new directions. Mol Psychiatry (1996) 1:336–42.
43. Modell S, Yassouridis A, Huber J, Holsboer F. Corticosteroid receptor function is decreased in depressed patients. Neuroendocrinology (1997) 65:216–22. doi: 10.1159/000127275
44. Maric NP, Adzic M. Pharmacological modulation of HPA axis in depression - new avenues for potential therapeutic benefits. Psychiatr Danub. (2013) 25:299–305.
45. DeRijk RH, Schaaf M, de Kloet ER. Glucocorticoid receptor variants: clinical implications. J Steroid Biochem Mol Biol. (2002) 81:103–22. doi: 10.1016/S0960-0760(02)00062-6
46. Klok MD, Alt SR, Irurzun Lafitte AJ, Turner JD, Lakke EA, Huitinga I, et al. Decreased expression of mineralocorticoid receptor mRNA and its splice variants in postmortem brain regions of patients with major depressive disorder. J Psychiatr Res. (2011) 45:871–8. doi: 10.1016/j.jpsychires.2010.12.002
47. Schatzberg AF, Keller J, Tennakoon L, Lembke A, Williams G, Kraemer FB, et al. HPA axis genetic variation, cortisol and psychosis in major depression. Mol Psychiatry (2014) 19:220–7. doi: 10.1038/mp.2013.129
48. Liu Z, Zhu F, Wang G, Xiao Z, Wang H, Tang J, et al. Association of corticotropin-releasing hormone receptor1 gene SNP and haplotype with major depression. Neurosci Lett. (2006) 404:358–62. doi: 10.1016/j.neulet.2006.06.016
49. Szczepankiewicz A, Leszczynska-Rodziewicz A, Pawlak J, Rajewska-Rager A, Wilkosc M, Zaremba D, et al. Epistatic interaction between CRHR1 and AVPR1b variants as a predictor of major depressive disorder. Psychiatr Genet. (2013) 23:239–46. doi: 10.1097/YPG.0000000000000007
50. Xiao Z, Liu W, Gao K, Wan Q, Yang C, Wang H, et al. Interaction between CRHR1 and BDNF genes increases the risk of recurrent major depressive disorder in Chinese population. PLoS ONE (2011) 6:e28733. doi: 10.1371/journal.pone.0028733
51. Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA. (1996) 93:3908–13. doi: 10.1073/pnas.93.9.3908
52. Jacobs BL, van Praag H, Gage FH. Adult brain neurogenesis and psychiatry: a novel theory of depression. Mol Psychiatry (2000) 5:262–9. doi: 10.1038/sj.mp.4000712
53. Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, et al. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci USA. (2004) 101:10827–32. doi: 10.1073/pnas.0402141101
54. Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry (2006) 59:1116–27. doi: 10.1016/j.biopsych.2006.02.013
55. Ressler KJ, Mayberg HS. Targeting abnormal neural circuits in mood and anxiety disorders: from the laboratory to the clinic. Nat Neurosci. (2007) 10:1116–24. doi: 10.1038/nn1944
56. McKinnon MC, Yucel K, Nazarov A, MacQueen GM. A meta-analysis examining clinical predictors of hippocampal volume in patients with major depressive disorder. J Psychiatry Neurosci. (2009) 34:41–54.
57. Cobb JA, Simpson J, Mahajan GJ, Overholser JC, Jurjus GJ, Dieter L, et al. Hippocampal volume and total cell numbers in major depressive disorder. J Psychiatr Res. (2013) 47:299–306. doi: 10.1016/j.jpsychires.2012.10.020
58. Gorski JA, Zeiler SR, Tamowski S, Jones KR. Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci. (2003) 23:6856–65. doi: 10.1523/JNEUROSCI.23-17-06856.2003
59. Martinowich K, Lu B. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology (2008) 33:73–83. doi: 10.1038/sj.npp.1301571
60. Schecterson LC, Sanchez JT, Rubel EW, Bothwell M. TrkB downregulation is required for dendrite retraction in developing neurons of chicken nucleus magnocellularis. J Neurosci. (2012) 32:14000–9. doi: 10.1523/JNEUROSCI.2274-12.2012
61. Chen ZY, Patel PD, Sant G, Meng CX, Teng KK, Hempstead BL, et al. Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci. (2004) 24:4401–11. doi: 10.1523/JNEUROSCI.0348-04.2004
62. Baj G, Carlino D, Gardossi L, Tongiorgi E. Toward a unified biological hypothesis for the BDNF Val66Met-associated memory deficits in humans: a model of impaired dendritic mRNA trafficking. Front Neurosci. (2013) 7:188. doi: 10.3389/fnins.2013.00188
63. Verhagen M, van der Meij A, van Deurzen PA, Janzing JG, Arias-Vasquez A, Buitelaar JK, et al. Meta-analysis of the BDNF Val66Met polymorphism in major depressive disorder: effects of gender and ethnicity. Mol Psychiatry (2010) 15:260–71. doi: 10.1038/mp.2008.109
64. Schumacher J, Jamra RA, Becker T, Ohlraun S, Klopp N, Binder EB, et al. Evidence for a relationship between genetic variants at the brain-derived neurotrophic factor (BDNF) locus and major depression. Biol Psychiatry (2005) 58:307–14. doi: 10.1016/j.biopsych.2005.04.006
65. Ribeiro L, Busnello JV, Cantor RM, Whelan F, Whittaker P, Deloukas P, et al. The brain-derived neurotrophic factor rs6265 (Val66Met) polymorphism and depression in Mexican-Americans. Neuroreport (2007) 18:1291–3. doi: 10.1097/WNR.0b013e328273bcb0
66. Frielingsdorf H, Bath KG, Soliman F, Difede J, Casey BJ, Lee FS. Variant brain-derived neurotrophic factor Val66Met endophenotypes: implications for posttraumatic stress disorder. Ann N Y Acad Sci. (2010) 1208:150–7. doi: 10.1111/j.1749-6632.2010.05722.x
67. Liu X, Xu Y, Jiang S, Cui D, Qian Y, Jiang K. Family-based association study between brain-derived neurotrophic factor gene and major depressive disorder of Chinese descent. Psychiatry Res. (2009) 169:169–72. doi: 10.1016/j.psychres.2008.06.014
68. Kaufman J, Yang BZ, Douglas-Palumberi H, Grasso D, Lipschitz D, Houshyar S, et al. Brain-derived neurotrophic factor-5-HTTLPR gene interactions and environmental modifiers of depression in children. Biol Psychiatry (2006) 59:673–80. doi: 10.1016/j.biopsych.2005.10.026
69. Pezawas L, Meyer-Lindenberg A, Goldman AL, Verchinski BA, Chen G, Kolachana BS, et al. Evidence of biologic epistasis between BDNF and SLC6A4 and implications for depression. Mol Psychiatry (2008) 13:709–16. doi: 10.1038/mp.2008.32
70. McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. (1995) 21:195–218. doi: 10.1016/0165-0173(95)00011-9
71. Maes M. Major depression and activation of the inflammatory response system. Adv Exp Med Biol. (1999) 461:25–46. doi: 10.1007/978-0-585-37970-8_2
72. Wohleb ES, McKim DB, Sheridan JF, Godbout JP. Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci. (2014) 8:447. doi: 10.3389/fnins.2014.00447
73. Hodes GE, Kana V, Menard C, Merad M, Russo SJ. Neuroimmune mechanisms of depression. Nat Neurosci. (2015) 18:1386–93. doi: 10.1038/nn.4113
74. Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry (2010) 67:446–57. doi: 10.1016/j.biopsych.2009.09.033
75. Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer (2005) 104:788–93. doi: 10.1002/cncr.21234
76. Motivala SJ, Sarfatti A, Olmos L, Irwin MR. Inflammatory markers and sleep disturbance in major depression. Psychosom Med. (2005) 67:187–94. doi: 10.1097/01.psy.0000149259.72488.09
77. Irwin MR, Wang M, Ribeiro D, Cho HJ, Olmstead R, Breen EC, et al. Sleep loss activates cellular inflammatory signaling. Biol Psychiatry (2008) 64:538–40. doi: 10.1016/j.biopsych.2008.05.004
78. Loftis JM, Hauser P. The phenomenology and treatment of interferon-induced depression. J Affect Disord. (2004) 82:175–90. doi: 10.1016/j.jad.2004.04.002
79. Asnis GM, De La Garza R. Interferon-induced depression in chronic hepatitis C: a review of its prevalence, risk factors, biology, and treatment approaches. J Clin Gastroenterol. (2006) 40:322–35. doi: 10.1097/01.mcg.0000210099.36500.fe
80. McCulley MC, Day INM, Holmes C. Association between interleukin 1-beta promoter (-511) polymorphism and depressive symptoms in Alzheimer's disease. Am J Med Genet Part B Neuropsyc Genet. (2004) 124B:50–3. doi: 10.1002/ajmg.b.20086
81. Borkowska P, Kucia K, Rzezniczek S, Paul-Samojedny M, Kowalczyk M, Owczarek A, et al. Interleukin-1beta promoter (-31T/C and−511C/T) polymorphisms in major recurrent depression. J Mol Neurosci. (2011) 44:12–6. doi: 10.1007/s12031-011-9507-5
82. Yu YW, Chen TJ, Hong CJ, Chen HM, Tsai SJ. Association study of the interleukin-1 beta (C-511T) genetic polymorphism with major depressive disorder, associated symptomatology, and antidepressant response. Neuropsychopharmacology (2003) 28:1182–5. doi: 10.1038/sj.npp.1300172
83. Tadic A, Rujescu D, Muller MJ, Kohnen R, Stassen HH, Szegedi A, Dahmen N, et al. Association analysis between variants of the interleukin-1beta and the interleukin-1 receptor antagonist gene and antidepressant treatment response in major depression. Neuropsychiatr Dis Treat. (2008) 4:269–76.
84. Hwang JP, Tsai SJ, Hong CJ, Yang CH, Hsu CD, Liou YJ. Interleukin-1 Beta-511C/T genetic polymorphism is associated with age of onset of geriatric depression. Neuromol Med. (2009) 11:322–7. doi: 10.1007/s12017-009-8078-x
85. Chen H, Wilkins LM, Aziz N, Cannings C, Wyllie DH, Bingle C, et al. Single nucleotide polymorphisms in the human interleukin-1B gene affect transcription according to haplotype context. Hum Mol Genet. (2006) 15:519–29. doi: 10.1093/hmg/ddi469
86. Hwang IR, Kodama T, Kikuchi S, Sakai K, Peterson LE, Graham DY, et al. Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1beta production in Helicobacter pylori infection. Gastroenterology (2002) 123:1793–803. doi: 10.1053/gast.2002.37043
87. Hall SK, Perregaux DG, Gabel CA, Woodworth T, Durham LK, Huizinga TW, et al. Correlation of polymorphic variation in the promoter region of the interleukin-1 beta gene with secretion of interleukin-1 beta protein. Arthritis Rheum. (2004) 50:1976–83. doi: 10.1002/art.20310
88. Fishman D, Faulds G, Jeffery R, Mohamed-Ali V, Yudkin JS, Humphries S, et al. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J Clin Invest. (1998) 102:1369–76. doi: 10.1172/JCI2629
89. Hong CJ, Yu YW, Chen TJ, Tsai SJ. Interleukin-6 genetic polymorphism and Chinese major depression. Neuropsychobiology (2005) 52:202–5. doi: 10.1159/000089003
90. Misener VL, Gomez L, Wigg KG, Luca P, King N, Kiss E, et al. Cytokine Genes TNF, IL1A, IL1B, IL6, IL1RN and IL10, and childhood-onset mood disorders. Neuropsychobiology (2008) 58:71–80. doi: 10.1159/000159775
91. Clerici M, Arosio B, Mundo E, Cattaneo E, Pozzoli S, Dell'osso B, et al. Cytokine polymorphisms in the pathophysiology of mood disorders. CNS Spectr. (2009) 14:419–25. doi: 10.1017/S1092852900020393
92. Bull SJ, Huezo-Diaz P, Binder EB, Cubells JF, Ranjith G, Maddock C, et al. Functional polymorphisms in the interleukin-6 and serotonin transporter genes, and depression and fatigue induced by interferon-alpha and ribavirin treatment. Mol Psychiatry (2009) 14:1095–104. doi: 10.1038/mp.2008.48
93. Weaver DR. The suprachiasmatic nucleus: a 25-year retrospective. J Biol Rhythms. (1998) 13:100–12. doi: 10.1177/074873098128999952
94. Albrecht U. Timing to perfection: the biology of central and peripheral circadian clocks. Neuron (2012) 74:246–60. doi: 10.1016/j.neuron.2012.04.006
95. Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. (2008) 9:764–75. doi: 10.1038/nrg2430
96. Reischl S, Kramer A. Kinases and phosphatases in the mammalian circadian clock. FEBS Lett. (2011) 585:1393–9. doi: 10.1016/j.febslet.2011.02.038
97. Armitage R. Sleep and circadian rhythms in mood disorders. Acta Psychiatr Scand Suppl. (2007) 433:104–15. doi: 10.1111/j.1600-0447.2007.00968.x
98. Gallego M, Virshup DM. Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Biol. (2007) 8:139–48. doi: 10.1038/nrm2106
99. Gouin JP, Connors J, Kiecolt-Glaser JK, Glaser R, Malarkey WB, Atkinson C, et al. Altered expression of circadian rhythm genes among individuals with a history of depression. J Affect Disord. (2010) 126:161–6. doi: 10.1016/j.jad.2010.04.002
100. Kishi T, Yoshimura R, Fukuo Y, Kitajima T, Okochi T, Matsunaga S, et al. The CLOCK gene and mood disorders: a case-control study and meta-analysis. Chronobiol Int. (2011) 28:825–33. doi: 10.3109/07420528.2011.609951
101. Holmans P, Zubenko GS, Crowe RR, DePaulo JRJr, Scheftner WA, Weissman MM, et al. Genomewide significant linkage to recurrent, early-onset major depressive disorder on chromosome 15q. Am J Hum Genet. (2004) 74:1154–67. doi: 10.1086/421333
102. Abkevich V, Camp NJ, Hensel CH, Neff CD, Russell DL, Hughes DC, et al. Predisposition locus for major depression at chromosome 12q22-12q23.2. Am J Hum Genet. (2003) 73:1271–81. doi: 10.1086/379978
103. Camp NJ, Lowry MR, Richards RL, Plenk AM, Carter C, Hensel CH, et al. Genome-wide linkage analyses of extended Utah pedigrees identifies loci that influence recurrent, early-onset major depression and anxiety disorders. Am J Med Genet B Neuropsychiatr Genet. (2005) 135B:85–93. doi: 10.1002/ajmg.b.30177
104. McGuffin P, Knight J, Breen G, Brewster S, Boyd PR, Craddock N, et al. Whole genome linkage scan of recurrent depressive disorder from the depression network study. Hum Mol Genet. (2005) 14:3337–45. doi: 10.1093/hmg/ddi363
105. Middeldorp CM, Sullivan PF, Wray NR, Hottenga JJ, de Geus EJ, van den Berg M, et al. Suggestive linkage on chromosome 2:8 and 17 for lifetime major depression. Am J Med Genet B Neuropsychiatr Genet. (2009) 150B:352–8. doi: 10.1002/ajmg.b.30817
106. Sullivan PF, de Geus EJ, Willemsen G, James MR, Smit JH, Zandbelt T, et al. Genome-wide association for major depressive disorder: a possible role for the presynaptic protein piccolo. Mol Psychiatry (2009) 14:359–75. doi: 10.1038/mp.2008.125
107. Muglia P, Tozzi F, Galwey NW, Francks C, Upmanyu R, Kong XQ, et al. Genome-wide association study of recurrent major depressive disorder in two European case-control cohorts. Mol Psychiatry (2010) 15:589–601. doi: 10.1038/mp.2008.131
108. Lewis CM, Ng MY, Butler AW, Cohen-Woods S, Uher R, Pirlo K, et al. Genome-wide association study of major recurrent depression in the U.K. population. Am J Psychiatry (2010) 167:949–57. doi: 10.1176/appi.ajp.2010.09091380
109. Rietschel M, Mattheisen M, Frank J, Treutlein J, Degenhardt F, Breuer R, et al. Genome-wide association-, replication-, and neuroimaging study implicates HOMER1 in the etiology of major depression. Biol Psychiatry (2010) 68:578–85. doi: 10.1016/j.biopsych.2010.05.038
110. Shi J, Potash JB, Knowles JA, Weissman MM, Coryell W, Scheftner WA, et al. Genome-wide association study of recurrent early-onset major depressive disorder. Mol Psychiatry (2011) 16:193–201. doi: 10.1038/mp.2009.124
111. Shyn SI, Shi J, Kraft JB, Potash JB, Knowles JA, Weissman MM, et al. Novel loci for major depression identified by genome-wide association study of Sequenced Treatment Alternatives to Relieve Depression and meta-analysis of three studies. Mol Psychiatry (2011) 16:202–15. doi: 10.1038/mp.2009.125
112. Kohli MA, Lucae S, Saemann PG, Schmidt MV, Demirkan A, Hek K, et al. The neuronal transporter gene SLC6A15 confers risk to major depression. Neuron (2011) 70:252–65. doi: 10.1016/j.neuron.2011.04.005
113. Aragam N, Wang KS, Pan Y. Genome-wide association analysis of gender differences in major depressive disorder in the Netherlands NESDA and NTR population-based samples. J Affect Disord. (2011) 133:516–21. doi: 10.1016/j.jad.2011.04.054
114. Wray NR, Pergadia ML, Blackwood DH, Penninx BW, Gordon SD, Nyholt DR, et al. Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol Psychiatry (2012) 17:36–48. doi: 10.1038/mp.2010.109
115. Hek K, Demirkan A, Lahti J, Terracciano A, Teumer A, Cornelis MC, et al. A genome-wide association study of depressive symptoms. Biol Psychiatry (2013) 73:667–78. doi: 10.1016/j.biopsych.2012.09.033
116. Power RA, Cohen-Woods S, Ng MY, Butler AW, Craddock N, Korszun A, et al. Genome-wide association analysis accounting for environmental factors through propensity-score matching: application to stressful live events in major depressive disorder. Am J Med Genet B Neuropsychiatr Genet. (2013) 162b:521–9. doi: 10.1002/ajmg.b.32180
117. Ikeda M, Shimasaki A, Takahashi A, Kondo K, Saito T, Kawase K, et al. Genome-wide environment interaction between depressive state and stressful life events. J Clin Psychiatry (2016) 77:e29–30. doi: 10.4088/JCP.15l10127
118. Okbay A, Baselmans BM, De Neve JE, Turley P, Nivard MG, Fontana MA, et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat Genet. (2016) 48:624–33. doi: 10.1038/ng.3552
119. Matsunami K, Nishida N, Kaneko N, Ikeo K, Toyo-Oka L, Takeuchi H, et al. Genome-wide association study identifies ZNF354C variants associated with depression from interferon-based therapy for chronic hepatitis, C. PLoS ONE (2016) 11:e0164418. doi: 10.1371/journal.pone.0164418
120. Direk N, Williams S, Smith JA, Ripke S, Air T, Amare AT, et al. An Analysis of Two Genome-wide Association Meta-analyses Identifies a New Locus for Broad Depression Phenotype. Biol Psychiatry. (2016) 82:322–9. doi: 10.1016/j.biopsych.2016.11.013
121. Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. (2018) 50:668–81. doi: 10.1038/s41588-018-0090-3
122. Major Depressive Disorder Working Group of the Psychiatric GC, Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry (2013) 18:497–511. doi: 10.1038/mp.2012.21
123. Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science (2007) 316:445–9. doi: 10.1126/science.1138659
124. Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature (2014) 506:185–90. doi: 10.1038/nature12975
125. Consortium C. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature (2015) 523:588–91. doi: 10.1038/nature14659
126. Peterson RE, Cai N, Bigdeli TB, Li Y, Reimers M, Nikulova A, et al. The genetic architecture of major depressive disorder in han chinese women. JAMA Psychiatry (2017) 74:162–8. doi: 10.1001/jamapsychiatry.2016.3578
127. Docherty AR, Edwards AC, Yang F, Peterson RE, Sawyers C, Adkins DE, et al. Age of onset and family history as indicators of polygenic risk for major depression. Depress Anxiety (2017) 34:446–52. doi: 10.1002/da.22607
128. Bigdeli TB, Ripke S, Peterson RE, Trzaskowski M, Bacanu SA, Abdellaoui A, et al. Genetic effects influencing risk for major depressive disorder in China and Europe. Transl Psychiatry (2017) 7:e1074. doi: 10.1038/tp.2016.292
129. Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. (2009) Nature 460:748–52. doi: 10.1038/nature08185
130. Song GG, Kim JH, Lee YH. Genome-wide pathway analysis in major depressive disorder. J Mol Neurosci. (2013) 51:428–36. doi: 10.1007/s12031-013-0047-z
131. Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet. (2016) 48:1031–6. doi: 10.1038/ng.3623
132. Wang C, Dai J, Sun Y, Xie L, Pan L, Hu Z, et al. Genetic risk score: principle, methods and application. Zhonghua Liu Xing Bing Xue Za Zhi (2015) 36:1062–4.
133. Dudbridge F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. (2013) 9:e1003348. doi: 10.1371/annotation/b91ba224-10be-409d-93f4-7423d502cba0
134. Peyrot WJ, Milaneschi Y, Abdellaoui A, Sullivan PF, Hottenga JJ, Boomsma DI, et al. Effect of polygenic risk scores on depression in childhood trauma. Br J Psychiatry (2014) 205:113–9. doi: 10.1192/bjp.bp.113.143081
135. Whalley HC, Adams MJ, Hall LS, Clarke TK, Fernandez-Pujals AM, Gibson J, et al. Dissection of major depressive disorder using polygenic risk scores for schizophrenia in two independent cohorts. Transl Psychiatry (2016) 6:e938. doi: 10.1038/tp.2016.207
136. Wang T, Zhang X, Li A, Zhu M, Liu S, Qin W, et al. Polygenic risk for five psychiatric disorders and cross-disorder and disorder-specific neural connectivity in two independent populations. Neuroimage Clin. (2017) 14:441–9. doi: 10.1016/j.nicl.2017.02.011
137. Alcocer-Gomez E, de Miguel M, Casas-Barquero N, Nunez-Vasco J, Sanchez-Alcazar JA, Fernandez-Rodriguez A, et al. NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain Behav Immun. (2014) 36:111–7. doi: 10.1016/j.bbi.2013.10.017
138. Klinedinst NJ, Regenold WT. A mitochondrial bioenergetic basis of depression. J Bioenerg Biomembr. (2015) 47:155–71. doi: 10.1007/s10863-014-9584-6
139. Gardner A, Johansson A, Wibom R, Nennesmo I, von Dobeln U, Hagenfeldt L, et al. Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J Affect Disord. (2003) 76:55–68. doi: 10.1016/S0165-0327(02)00067-8
140. Moreno-Fernandez AM, Cordero MD, Garrido-Maraver J, Alcocer-Gomez E, Casas-Barquero N, Carmona-Lopez MI, et al. Oral treatment with amitriptyline induces coenzyme Q deficiency and oxidative stress in psychiatric patients. J Psychiatr Res. (2012) 46:341–5. doi: 10.1016/j.jpsychires.2011.11.002
141. Koene S, Kozicz TL, Rodenburg RJ, Verhaak CM, de Vries MC, Wortmann S, et al. Major depression in adolescent children consecutively diagnosed with mitochondrial disorder. J Affect Disord. (2009) 114:327–32. doi: 10.1016/j.jad.2008.06.023
142. Sequeira A, Rollins B, Magnan C, van Oven M, Baldi P, Myers RM, et al. Mitochondrial mutations in subjects with psychiatric disorders. PLoS ONE (2015) 10:e0127280. doi: 10.1371/journal.pone.0127280
143. Veronese N, Stubbs B, Solmi M, Vaona A, Demurtas J, Carvalho AF, et al. Mitochondrial genetic haplogroups and depressive symptoms: A large study among people in North America. J Affect Disord. (2017) 217:55–9. doi: 10.1016/j.jad.2017.03.069
144. Zhang Z, Ni J, Zhang J, Tang W, Li X, Wu Z, et al. A haplotype in the 5'-upstream region of the NDUFV2 gene is associated with major depressive disorder in Han Chinese. J Affect Disord. (2016) 190:329–32. doi: 10.1016/j.jad.2015.10.034
145. Huo YX, Huang L, Zhang DF, Yao YG, Fang YR, Zhang C, et al. Identification of SLC25A37 as a major depressive disorder risk gene. J Psychiatr Res. (2016) 83:168–75. doi: 10.1016/j.jpsychires.2016.09.011
146. Sabunciyan S, Aryee MJ, Irizarry RA, Rongione M, Webster MJ, Kaufman WE, et al. Genome-wide DNA methylation scan in major depressive disorder. PLoS ONE (2012) 7:e34451. doi: 10.1371/journal.pone.0034451
147. Xie HQ, Choi RC, Leung KW, Chen VP, Chu GK, Tsim KW. Transcriptional regulation of proline-rich membrane anchor (PRiMA) of globular form acetylcholinesterase in neuron: an inductive effect of neuron differentiation. Brain Res. (2009) 1265:13–23. doi: 10.1016/j.brainres.2009.01.065
148. Davies MN, Krause L, Bell JT, Gao F, Ward KJ, Wu H, et al. Hypermethylation in the ZBTB20 gene is associated with major depressive disorder. Genome Biol. (2014) 15:R56. doi: 10.1186/gb-2014-15-4-r56
149. Chen ES, Ernst C, Turecki G. The epigenetic effects of antidepressant treatment on human prefrontal cortex BDNF expression. Int J Neuropsychopharmacol. (2011) 14:427–9. doi: 10.1017/S1461145710001422
Keywords: depressive disorder, candidate genes, brain-derived neurotrophic factor, hypothalamus-pituitary-adrenal axis, cytokines, GWAS, epigenome
Citation: Shadrina M, Bondarenko EA and Slominsky PA (2018) Genetics Factors in Major Depression Disease. Front. Psychiatry 9:334. doi: 10.3389/fpsyt.2018.00334
Received: 05 April 2018; Accepted: 02 July 2018;
Published: 23 July 2018.
Edited by:
Dimitrios Avramopoulos, Johns Hopkins University, United StatesReviewed by:
Camron D. Bryant, School of Medicine, Boston University, United StatesPippa Ann Thomson, Medical Research Council Institute of Genetics and Molecular Medicine (MRC), United Kingdom
Copyright © 2018 Shadrina, Bondarenko and Slominsky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena A. Bondarenko, ZWwuYm9uZGFyZW5rb0BnbWFpbC5jb20=