 Bethania Mongi-Bragato
Bethania Mongi-Bragato María P. Avalos
María P. Avalos Andrea S. Guzmán
Andrea S. Guzmán Flavia A. Bollati
Flavia A. Bollati Liliana M. Cancela
Liliana M. Cancela
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 28 May 2018
Sec. Psychopharmacology
Volume 9 - 2018 | https://doi.org/10.3389/fpsyt.2018.00222
This article is part of the Research TopicCurrent Topics in Opioid ResearchView all 26 articles
Enkephalin expression is high in mesocorticolimbic areas associated with psychostimulant-induced behavioral and neurobiological effects, and may also modulate local neurotransmission in this circuit network. Psychostimulant drugs, like amphetamine and cocaine, significantly increase the content of enkephalin in these brain structures, but we do not yet understand the specific significance of this drug-induced adaptation. In this review, we summarize the neurochemical and molecular mechanism of psychostimulant-induced enkephalin activation in mesocorticolimbic brain areas, and the contribution of this opioid peptide in the pivotal neuroadaptations and long-term behavioral changes underlying psychostimulant addiction. There is evidence suggesting that adaptive changes in enkephalin content in the mesocorticolimbic circuit, induced by acute and chronic psychostimulant administration, may represent a key initial step in the long-term behavioral and neuronal plasticity induced by these drugs.
Psychostimulant addiction is a severe worldwide health problem. The most challenging aspects in its treatment are compulsive drug use and relapse. Currently, there are no effective pharmacotherapies for this disorder. New therapeutic approaches are required based on understanding the neurobiology of drug addiction. Numerous lines of research suggest that exposure to psychostimulant drugs causes neurochemical and molecular adaptations that explain the stability of the behavioral disorders characterizing the addictive state (1, 2). Attention has focused on how the mesocorticolimbic dopamine circuit is affected by drugs of abuse, and particularly on the role of glutamate and dopamine neurotransmission in determining the neuroplastic changes related to psychostimulant addiction (3–6). At molecular level, it has been shown that activation of glutamate and dopamine neurotransmission after repeated psychostimulant administrations, affects intracellular signaling cascades (7–9), alters the expression of membrane receptors (10, 11) and changes gene expression within the neural circuits (12, 13), which leads to sensitization of the drug's behavioral effects (14) and other behavioral alterations observed in addiction, like intense drug craving and relapse (15).
Enkephalin, an opioid peptide derived from proenkephalin (PENK), is widely expressed in the mesocorticolimbic circuit (16) and interacts with glutamate and dopamine in the brain reward structures related to psychostimulant-induced effects. Both delta-opioid (DOPr) and mu-opioid receptors (MOPr) can be activated by enkephalin, and each has its particular pattern of expression within the motivational circuit (17). Although several pharmacological and genetic approaches demonstrate a role of both MOPr and DOPr in psychostimulant-induced behavioral effects, the role of the endogenous opioid peptides in this process has not been fully examined. Previous studies from our lab have demonstrated a long-lasting increase in enkephalin levels within the mesocorticolimbic circuit after psychostimulant administration (18, 19). Enkephalin has also been shown to positively modulate dopamine and glutamate neurotransmission within this circuit (20–25). These data indicate that cocaine-induced enkephalin elevation may drive the neuronal plasticity induced by the drug and the long-term behavioral effects of psychostimulant exposure.
Mesolimbic dopamine activity is directly affected by psychostimulants (26). These drugs bind to monoamine transporters and block reuptake mechanisms (cocaine), or competitively inhibit dopamine uptake and disrupt vesicular storage (amphetamine). The activation of this system is a primary conditioner of their psychomotor stimulant and rewarding effects (27). Acute or chronic administration of psychostimulants also alters, among others, the levels of endogenous opioid peptides, including enkephalin, within areas of the mesocorticolimbic circuit.
Acute cocaine (28, 29) and amphetamine (30–33) elevate PENK mRNA levels in the striatum, and these levels are also increased after chronic cocaine exposure in different dopamine mesolimbic afferents (34, 35, 36). Elevated PENK levels were also observed within the caudate putamen on the second day of binge cocaine administration (37). Following chronic cocaine treatment, no changes were observed in the cortex in PENK mRNA levels (38), prefrontal cortex (39, 40), amygdala (41, 42), hypothalamus, pituitary, central gray and cerebellum, nucleus accumbens or caudate putamen (39). However, PENK mRNA levels were significantly elevated during long-term extinction (10 days) of a cocaine self-administration paradigm in the caudate putamen, nucleus accumbens, piriform cortex and olfactory tubercle regions, and decreased in the central amygdale of rats (43). Similarly, sensitized PENK mRNA expression was observed in the nucleus accumbens and/or caudate putamen in response to an amphetamine challenge following acute (44–47) or chronic (48) amphetamine pretreatment in animals after short-term abstinence from the drug.
Furthermore, data from our lab demonstrate an increase in the levels of met-enkephalin in the nucleus accumbens from rats after acute amphetamine (5 mg/kg i.p.) following 4, but not 7 or 21 days after the last drug injection (18, 49, 50). Met-enkephalin elevation was also observed after 4 days withdrawal period from chronic amphetamine (5 × 2 mg/kg i.p) (49). Interestingly, long-lasting sensitization to amphetamine-induced increases in met-enkephalin levels was evidenced in the same brain area following amphetamine challenge (1 mg/kg i.p.) 21 days after the last acute (5 mg/kg i.p.) administration of the drug (18). Similarly, persistent met-enkephalin immunoreactivity was evidenced in the nucleus accumbens from mice treated chronically with cocaine (9 × 15 mg/kg i.p.) after a long-term abstinence from the drug (12 days after last injection). Met-enkephalin immunoreactivity elevations induced by chronic cocaine is not dependent of cocaine challenge administration (7.5 mg/kg i.p., day 21), as this effect on met-enkephalin immunoreactivity was also observed after saline challenge injection (19).
Altogether these data demonstrate that PENK mRNA levels are increased in specific dopaminergic regions following psychostimulant administration, be the injection acute, chronic or remote.
Psychostimulant-induced PENK mRNA expression at striatal level may be the result of multiple neurotransmitter interactions (31, 51, 52). Cocaine and amphetamine stimulate the PENK mRNA expression in striatal neurons (19, 28, 31, 35, 49), which mostly express D2 receptors (53, 54), and also induce prodynorphin and substance P in striatal neurons (31), which mainly express D1 receptors (53, 55). Similarly, the full D1 receptor agonist SKF-82958 induced PENK, prodynorphin and substance P gene expression in both the dorsal and ventral striatum (33). Interestingly, the increase in met-enkephalin induced by amphetamine (50) or PENK mRNA levels stimulated by SKF-82958 in striatal neurons (33) was blocked by the D1 receptor antagonist SCH-23390 (50) and by scopolamine, the muscarinic receptor antagonist (32). Oppositely, the D2 receptor antagonist eticlopride did not affect SKF-82958-induced PENK mRNA expression (33). Similarly, amphetamine-induced met-enkephalin levels was not modified by raclopride, another D2 receptor antagonist (50). Thus, this evidence suggests that D1-mediated induction of PENK may involve trans-synaptic activation of cholinergic neurotransmission. That is, the psychostimulant-induced dopamine elevations stimulates acetylcholine release via a D1-dependent mechanism (56, 57). The acetylcholine released then activates muscarinic M1 receptors (32, 44) and associative signaling pathways in enkephalin-containing neurons thus facilitating PENK mRNA expression (Figure 1). Opioid receptors located at striatal level are also involved in psychostimulant-induced PENK mRNA expression. Selective kappa opioid receptor (KOPr) agonists appear to inhibit psychostimulant-induced alterations in PENK mRNA in the striatum (58), and DOPr antagonists significantly decreased amphetamine-induced mRNA PENK expression (45). In contrast to DOPr's inhibitory effects, MOPr antagonists, alone or combined with amphetamine, increase PENK mRNA levels in the dorsal striatum (45). Opioid receptors thus probably differentially regulate psychostimulant-induced PENK gene expression in the striatum, as a result of the predominantly MOPr expression at D1+ medium spiny neuron vs. D2+ medium spiny neuron and the selective pre-synaptic DOPr location in the local network. Similarly, pre-synaptic KOPr located at striatal dopamine and glutamate nerve terminals could regulates psychostimulant-evoked neurotransmitter release (59) indirectly affecting PENK expression within this brain area.
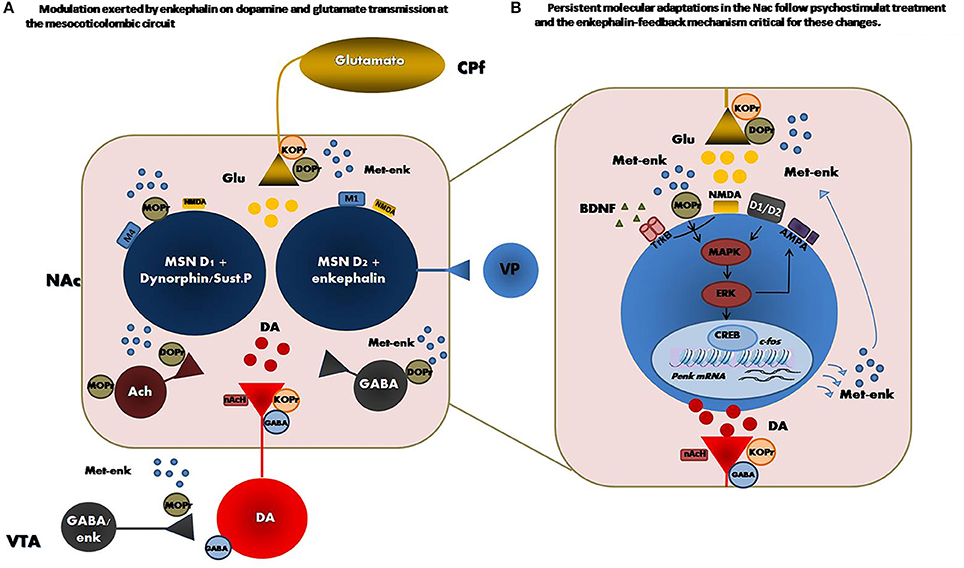
Figure 1. (Left) Principal met-enkephalin target nucleus in the mesocorticolimbic circuit. Distribution of opioid receptors within the circuit network is also shown and reveals the modulation exerted by enkephalin on dopamine and glutamate transmission at this level. (Right) Persistent adaptations in the enkephalin content followed by psychostimulant treatment and the subsequent activation of MOPr could result in a feedback mechanism critical for the neuronal plasticity induced by these drugs in the NAc. Enkephalin transmission activation promotes the development of psychostimulant-induced long-term neurochemical and molecular changes in the NAc, such as increases in BDNF/TrkB, phospho-ERK2/CREB signaling activation and GluR1 AMPA cell surface expression. VP, ventral pallidum; VTA, ventral tegmentalarea; PfC, Prefrontal cortex; DA, dopamine; Ach, acetylcholine; Glu, glutamate; met-enk, methionine encephalin; GABA, γ-aminobutyric, M1 and M4: muscarinic acetylcholine receptors type 1 and 4, respectively, acetylcholine nAch, nicotinic acetylcholine receptor.
Glutamate transmission actively regulates PENK gene expression under normal or stimulated conditions (51, 52). However, the precise mechanism by which glutamate participates in psychostimulant-stimulated PENK mRNA expression requires further study. Several reports indicate that cocaine (60, 61) and amphetamine (62–64) administration increases extracellular glutamate levels as well as dopamine levels in the striatum. Also, glutamate tone may be important for amphetamines to stimulate dopamine release from nerve terminals (65, 66, 67, 64). Thus, glutamate transmission could also play a role in regulating the stimulant effect of psychostimulants on PENK mRNA expression. There is evidence from our lab that pretreatment with NMDA receptor antagonists attenuates long-lasting amphetamine-induced PENK mRNA expression and met-enkephalin levels in the nucleus accumbens (18). In addition, there is evidence that glutamate transmission mediated by the AMPA receptor is involved in acute amphetamine-induced PENK levels in the striatum (52). Alternatively, elevated glutamate transmission seems to increase acetylcholine release (68, 69), and this induces acetylcholine-sensitive PENK gene expression, possibly through a NMDA receptor mechanism.
The regulation of PENK in the brain is usually preceded by the induction of AP-1, cAMP response element-binding protein (CREB) and c-Fos (70–74). Dopamine D1 receptor stimulation activates these transcription factors and, if dopamine D2 is also activated, there is a synergistic mechanism (75, 76). This initiates a sequence of molecular steps critically involved in psychostimulant-induced behavioral responses. CREB is the primary regulator of transcriptional activity in accumbal projection neurons and is phosphorylated by protein kinases, including the extracellular signaling-regulated kinase (ERK1/2) (77, 78). Glutamate-stimulated CREB phosphorylation in the striatum is attenuated by the ERK1/2 kinase inhibitor, PD98059 (77–79).
Psychostimulant drugs, which increase dopamine, glutamate and PENK content in mesocorticolimbic brain areas, also up-regulate ERK2/CREB phosphorylation (8, 9, 80). Consistent with this, the inhibition of the ERK2/CREB signaling pathway prevents the increase of psychostimulant-induced PENK mRNA expression (47). This strongly indicates that the long-term increase in met-enkephalin levels, induced by psychostimulants in mesocorticolimbic brain structures, is mediated by a dopamine- and glutamate-dependent mechanism, with the activation of dopamine D1 and glutamate NMDA receptors leading to ERK2/CREB signaling pathway activation in the same brain areas.
Although enkephalin seems to exert an influence on key areas involved in psychostimulant-induced behavioral effects, the mechanism underlying long-term effects has not yet been fully explained. Pharmacologically, PENK-derived opioid peptides seem to show high affinity for DOPr, but also good affinity for MOPr (17). Furthermore, dopamine release in the nuceleus accumbens appears to be promoted by enkephalin in the ventral tegmental area (20, 81), while MOPr antagonists administered intra-ventral tegmental area cause a decrease in dopamine neurotransmission (82). Pharmacological studies have shown that MOPr and DOPr contribute to increasing dopamine and glutamate release induced by psychostimulants in the nucleus accumbens (83–86). Consistently, pharmacological approaches using MOPr and DOPr antagonists, as well as MOPr knockout mice, demonstrate that the endogenous opioid system is involved in dopamine-related behaviors (87–89). This evidence, together with studies showing that PENK is one of the mediators of the positive reinforcing effects of nicotine, alcohol and marihuana (90–92), suggests that enkephalin may also have a role in psychostimulant-induced behaviors. However, further study is needed to explain the mechanism of its involvement.
Repeated intermittent exposure to cocaine steadily increases the locomotor response to the drug (behavioral sensitization) (14), which is mostly coupled to a greater drug-induced dopamine efflux in the nucleus accumbens (93–95). However, a reduction (96) or non-augmentation (97) in the levels of the neurotransmitter in the nucleus accumbens was found simultaneously with this phenomenon. Behavioral sensitization to psychostimulants may well be mediated by converging extracellular signals, which give rise to a number of specific molecular and cellular events, such as activating the ERK/CREB signaling pathway, and enhancing GluR1 AMPA receptor cell surface expression and brain-derived neurotrophic factor/tyrosine kinase B (BDNF/TrkB) receptor signaling within the nucleus accumbens (11, 98). As mentioned previously, the enkephalinergic system increases mesoaccumbal dopamine neurotransmission (25). Likewise, pharmacological studies have demonstrated that MOPr and DOPr receptors contribute to amphetamine (99, 100) and cocaine-induced enhancement of dopamine levels in the nucleus accumbens (84, 86), and there is data of the anatomical selectivity of MOPr receptors within the ventral tegmental area-nucleus accumbens pathway in cocaine-induced reward and locomotor-stimulating effects (101). It has also been proposed that cocaine may cause the release of endogenous opioid peptides. These then activate MOPr within the nucleus accumbens and ventral tegmental area and modulate the drug-induced behavioral effects (102).
The role of MOPr and DOPr in the development and expression of psychostimulant sensitization has been shown pharmacologically. It has been reported that naloxone and naltrexone, non-selective opioid receptor antagonists, attenuate the development of sensitization to cocaine in rats (103) and mice (19, 88, 104). Naltrindole, a DOPr antagonist (87) and CTAP (D-Phe-cyc(Cys-Tyr-D-Trp-Arg-Thr-Pen)-Thr-NH2), a selective MOPr antagonist(105), also reduce cocaine-induced sensitization in rats. Similarly, the development (106) and expression (107, 108) of amphetamine-induced behavioral sensitization were reduced following non-selective opioid receptor administration. Additionally, there is evidence of ERK1/2 signaling stimulation induced by MOPr/DOPr activation in the striatum (109). However, there is data showing that acute morphine caused a reduction in ERK 1/2 levels in the nucleus accumbens (110, 111). Interestingly, although chronic morphine, a MOPr agonist, caused a reduction (110) or tolerance to morphine-induced ERK1/2 activation (111), naloxone-precipitated withdrawal in morphine-dependent animals induced a robust stimulation of ERK1/2 in the striatum (109, 112). Together this evidence demonstrates a prominent role for MOPr in the regulation of molecular events, associated not only with psychostimulant induced-behavioral sensitization, but also with the underlying opiate dependence. However, studies using MOPr mice seem to be inconclusive (88) or did not show a significant influence of this receptor in cocaine sensitization (113–115). It is important to note that these behavioral evaluations were performed after short-term cocaine withdrawal [(88): 10 × 15 mg/kg i.p./7days withdrawal; (113): 5 × 20 mg/kg i.p./dose–response experiment; (114): 6 × 15 mg/kg i.p./6 days withdrawal; (115): 20 mg/kg i.p./3 days withdrawal], possibly masking the role of MOPr in long-term behavioral effects induced by cocaine (116). There is also evidence that, after long- but not short-term withdrawal, naloxone blockade is observed of the expression of behavioral sensitization to psychostimulants [(108): amphetamine 1.5 mg/kg i.p./14 days abstinence]. Despite all these studies, and reports demonstrating that met-enkephalin and MOPr have a prominent role in the ventral tegmental area at the initial step of sensitization (101, 117, 118), there is still no explanation in the literature of the influence of enkephalin on the psychostimulant-induced neuronal plasticity underpinning long-term sensitization. Data from our lab demonstrate an essential role of enkephalin in the development of neuroadaptations in the nucleus accumbens leading to cocaine-induced psychomotor sensitization (19). PENK knockout mice treated chronically with cocaine (9 days x 15 mg/kg) do not become sensitized to cocaine's properties stimulating locomotor activity and dopamine release in the nucleus accumbens 21 days after starting drug treatment. Additionally, the nucleus accumbens and dorsal striatum from PENK knockout mice showed no pivotal neuroadaptations such as the increase in phospho-TrkB receptor, phospho-ERK/CREB and GluR1 AMPA cell surface expression related to sensitized responses to cocaine. Consistent with these observations, full suppression of cocaine-induced behavioral and neuronal plasticity was observed in wild-type animals after naloxone pretreatment (1 mg/kg s.c. 15 min prior to cocaine injections). Reduced activity-dependent BDNF/TrkB signaling within the ventral tegmental area-nucleus accumbens circuit may attenuate the ability of cocaine to induce pathological changes in the nucleus accumbens that promote addiction (119, 120). Related with this, the lack of dopamine sensitization of a cocaine-induced increase in BDNF/TrkB signaling, identified in knockout- and naloxone-pretreated mice, strongly suggests that both enkephalin and BDNF have an important role in dopamine-sensitized behaviors. There is thus considerable evidence that the MOPr/endogenous enkephalin system has a prominent role in the establishment of long-term neuroadaptations within the nucleus accumbens underlying the expression of sensitization to cocaine.
Pharmacological evidence clearly demonstrates the role of MOPr and DOPr in the modulation of psychostimulant-induced rewarding properties by studying the development of conditioned place preference (CPP); i.e., acquisition of associative learning between a context and the rewarding effects of a drug. In this sense, the establishment of CPP induced by amphetamine was prevented by the non-selective opioid receptor antagonist naloxone (0.02, 0.2 or 2.0 mg/kg s.c.), administered during the conditioning sessions (121). Similarly, naltrexone implants can attenuate cocaine-induced CPP in rats (122), although high doses of the opioid antagonist were required. This effect could be due to the non-selective opioid receptor antagonism. Naltrindole, a highly selective DOPr antagonist, blocked the acquisition of cocaine and amphetamine-induced-CPP in rats (123, 124), indicating that a selective opioid receptor antagonism can fully attenuate the reinforcing properties of cocaine. Furthermore, several studies have demonstrated that selective MOPr receptor antagonists attenuate psychostimulant-induced CPP. Specifically, systemic pretreatment with the selective MOPr type-1 receptor antagonist naloxonazine (125) and intracerebroventricular administration (i.c.v.) of CTAP paired with peripheral injections of cocaine (105), prevented the development of cocaine-induced CPP. It has also been demonstrated that animals pre-treated with CTAP into the nucleus accumbens core or rostral ventral tegmental area, but not into the caudal ventral tegmental area, caudate putamen or medial nucleus accumbens shell, during cocaine conditioning, showed an attenuation of the establishment of cocaine-induced CPP, demonstrating the involvement of mesolimbic MOPr in cocaine-induced reward (101). Although all this evidence has focused on the role of MOPr and DOPr in the development of psychostimulant-induced CPP, their involvement in the expression of this behavior cannot be ruled out. In line, Gerrits et al. (126) assessed the effect of naloxone (0.01–0.1 mg/kg s.c.) administered prior the conditioning test, demonstrating the role of opioid receptors in the expression of cocaine's motivational effects.
Despite this pharmacological evidence, the data regarding cocaine-induced CPP in MOPr knockout mice seems to be inconsistent. For example, the development of cocaine induced-CPP has been reported to be attenuated (113), unchanged (127) or induced after higher doses of cocaine compared to that used in wild-type littermates (128). The mechanisms that underlie these discrepancies in behavioral effects induced by cocaine in MOPr knockout mice are unknown. One possible explanation involves the different protocols of conditioning and cocaine doses used [(128): 4 days conditioning/5 or 10 mg/kg; (127): 3 days–two conditioning sessions per day/10 mg/kg; (113): 2 days–two conditioning sessions per day/5 or 10 mg/kg]. Another explanation could be the genetic background of the mice [(128): 129/Ola × C57BL F2; (113): congenic C57B F10; (127): hybrid 129SV/C57BL/6 F1] that may influence the differences in the process of acquisition of cocaine-induced CPP.
Although the evidence indirectly indicates a potential role of enkephalin in psychostimulant-induced CPP, its role in this process has not been addressed yet. Moreover, the molecular mechanism that underlies the MOPr/DOPr contribution to psychostimulant-induced CPP and the potential role of enkephalin has not been fully studied. Interestingly, there is data suggesting that morphine (a MOPr agonist)-induced CPP is associated with neuroadaptations similar to that observed following chronic psychostimulant treatment in important brain areas associated with drug addiction and those related to memory consolidation. Augmented phosphorylation levels of the GluR1 AMPAR subunit and ERK/CREB were observed in the hippocampus (129–131) as well as in the nucleus accumbens (132) and ventral tegmental area (130, 133) following morphine-induced conditioned behavior. This, together with data from our lab demonstrating that the PENK gene regulates cocaine-induced long-lasting molecular changes, such as enhancement in dopamine transmission, GluR1 AMPA receptor cell surface expression, ERK/CREB signaling pathway activation and modulation of TrkB/BDNF levels in the nucleus accumbens (19), suggests that enkephalin and the MOPr system may favor neuronal plasticity within the mesolimbic circuit that underlies psychostimulant and opiate-induced CPP. Further genetic (PENK knockout mice) and pharmacological studies need to be carried out to confirm this hypothesis and demonstrate the role of enkephalin in psychostimulant-induced CPP.
There is now considerable pharmacological evidence of the important role that MOPr plays in mediating the reinforcing effects of cocaine in a self-administration paradigm. GSK1521498 (0.1, 1, and 3 mg/kg s.c.), a MOPr antagonist, and naltrexone administered at the same doses and route, reduced cocaine-seeking under a second-order schedule of reinforcement but did not affect cocaine self-administration under a simple fixed-ratio schedule (FR1) (134), indicating modulation of mechanisms regulating cocaine-seeking behavior rather than cocaine reinforcement (135). Additionally, GSK1521498 was more effective than naltrexone in reducing cocaine seeking, possibly because of different opioid receptor subtype selectivity. Similarly, low doses of naltrexone (0.1 mg/kg i.p.) showed no changes in cocaine self-administration (FR2 schedule), but attenuated cocaine- and cue-induced reinstatement of drug-seeking behavior administered 30 min prior to the reinstatement test (136). Consistently, the MOPr irreversible antagonist, beta-funaltrexamine, administered intra-ventral tegmental area or nucleus accumbens, had no effect on cocaine self-administration under a FR1 schedule of reinforcement. In contrast, MOPr blockade in both brain regions did attenuate the response to cocaine under a progressive ratio (PR) schedule, supporting the notion that MOPr within the mesolimbic system is involved in motivation to respond to cocaine (137). Regarding the role of MOPr, the selective MOPr antagonist CTAP (0.3 and 3 μg) administered in the ventral pallidum, but not in the nucleus accumbens or lateral hypothalamus, blocked the reinstatement of drug-seeking in rats that extinguished from cocaine self-administration (138). Given the GABA/enkephalin projection from the nucleus accumbens to the ventral pallidum, chronic cocaine may result in enkephalin release in this brain area, activating MOPr and eliciting cocaine relapse.
Data regarding the role of DOPr in mediating the rewarding effects of cocaine are conflicting. Naltrindole (0.03–3.0 mg/kg i.p. prior to self-administration session) did not alter the intake of cocaine (FR2 schedule of reinforcement) or the re-acquisition of cocaine self-administration (139). Similarly, a selective DOPr type-2 antagonist (administered i.c.v.) has been reported to have a slight effect on cocaine self-administration (FR1 schedule) (140). In contrast, there is data demonstrating that naltrindole (10 mg/kg i.p. 15 min prior FR1) reduced cocaine self-administration (141). These discrepancies regarding the role of DOPr in cocaine reinforcement may be due to the different types and doses of DOPr antagonist and cocaine-self-administration protocols. Importantly, none of these studies evaluated a possible role of DOPr within specific brain areas associated with cocaine reinforcement. DAMGO (1–3 ng) and DPDPE (300–3,000 ng), MOPr- and DOPr-selective ligands respectively, as well as β-endorphin (100–1,000 ng) and the enkephalinase inhibitor thiorphan (3–10 μg) microinjected into the nucleus accumbens, are sufficient to reinstate cocaine-seeking behavior in rats following extinction of cocaine self-administration (142). Thus, the stimulation of either accumbal MOPr or DOPr seems to be necessary to precipitate cocaine relapse.
Cocaine self-administration was reduced in MOPr knockout mice (143), suggesting a critical role of this receptor in cocaine reinforcement. In contrast, Gutiérrez-Cuesta et al. (144), found no changes in cocaine self-administration in this genotype. This discrepancy could be explained in the framework of the differences in experimental protocols used regarding cocaine dose and the time of the conditioning sessions, as in the study of Mathon et al. (143), which demonstrated significant differences in this genotype at high cocaine doses in shorter session times. Moreover, cocaine self-administration was reduced in both DOPr knockout and PENK knockout mice (144), mainly when animals were trained in FR3 and PR schedules. These findings suggest that DOPr and PENK are involved in the motivation to obtain cocaine, and the absence of these opioid components engenders an impaired response of cocaine self-administration, mainly when greater effort to obtain a reward is required. In addition, Gutiérrez-Cuesta et al. (144), demonstrated that cue-induced reinstatement of cocaine-seeking behavior was attenuated in both DOPr knockout and MOPr knockout. These data support previous pharmacological studies of Simmons and Self (142) addressing an important role of both receptors within the mesolimbic system in cocaine relapse. Consistent with these data, an enduring MOPr tone has been demonstrated within brain reward structures following extinction of cocaine self-administration (145), indicating that up regulating enkephalin levels may lead to long-lasting adaptations in response to repeated cocaine. Thus, all this evidence indicates that enkephalin, presumably acting on MOPr (although a role of DOPr cannot be ruled out) has a facilitatory influence on cocaine-induced behavioral and neuronal plasticity.
Importantly, the human literature shows encouraging evidence regarding the use of opioid antagonist in the treatment of psychostimulants relapse (146–148). Indeed, naltrexone (50 mg/day) administered in combination with relapse prevention therapy reduced cocaine use in a study of cocaine-addicted patients (n = 85). Thus, people receiving the combination of naltrexone (administered throughout 12 weeks) and relapse prevention therapy evidenced significantly reduced cocaine use than participants receiving other treatment combinations such us naltrexone alone or combined with drug counseling therapy (147). The same treatment protocol (naltrexone 50 mg/day during 12 weeks of medication and relapse prevention therapy) reduced amphetamine use as well as craving in amphetamine dependent patients (n = 55) (149). Additionally, naltrexone (50 mg/day) reduced the subjective effects of dexamphetamine (30 mg, oral) in amphetamine-dependent people (n = 20) (150). In constrast, patients who received oral naltrexone doses (0, 12.5, or 50 mg) before smoked cocaine (0, 12.5, 25, and 50 mg or placebo), or oral amphetamine (0, 10, and 20 mg or placebo) did not show alterations in positive subjective effects in cocaine users (n = 12) (146). This evidence suggests that this opioid antagonist did not alter positive subjective ratings after cocaine. Importantly, naltrexone did not alter physiological effects of psychostimulants in terms of cardiovascular function (146), cortisol levels and skin conductance (149, 150). Morever, naltrexone did significantly reduce craving for cocaine and tobacco during cocaine sessions (146) as well as amphetamine craving (149, 150). These data demonstrated that behavioral alterations observed in psychostimulants addiction, such us drug craving could be modulated by the endogenous opioid system.
It is important to address that in these studies, participants do not show evidence of any increase in the intake of other drugs of abuse during naltrexone protocol therapy to compensate for the reduction in the drug consumption that is being evaluated. On the other hand, these studies were restricted to short periods of naltrexone treatment and long-term effects in these patients are unknown. Thus, future longitudinal studies are required in order to follow patients over prolonged periods of time.
Similar effects on opioid antagonists were observed in patients with cocaine/alcohol comorbidity (148, 151–153) or cocaine/opiate dependence (154).
In summary, several studies show promising results for psychostimulants addiction treatment, suggesting a potential role of naltrexone as an anti-craving therapy for this psychiatric disorder.
This review emphasizes the important role of endogenous enkephalin during the development of the long-term neurobiological changes underlying psychostimulant addiction. It has been suggested that polymorphisms in genes encoding components of the endogenous opioid system are involved in predisposing to addiction to cocaine and opiates (155). Similarly, it is likely that genetic variations in the endogenous PENK gene (155–158) influence the development of behavioral and neurobiological adaptations in response to psychostimulant exposure, and thus modify vulnerability to psychostimulant addiction. This review also helps to understand how opioid antagonists can be effective in treating psychostimulant addiction (146, 147, 149), supporting their use as therapy for this disorder. Thus, the evidence presented in this review provides a basis for the development of new drug therapies for psychostimulant addiction based on specific modulation of the endogenous PENK system.
BMB wrote the article and performed the research related with the topic of this review; MPA, ASG and FAB contributed with the research related with the topic of this review and the writing of the review; LMC wrote the article, designed the research and provided the funds to perform papers related with the topic of this review.
This work has been supported by the grants from Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) PID 11420110100354, Secretaría de Ciencia y Técnica (SeCyT) 202/16, Fondo para la Investigación Científica y Tecnológica (FONCyT) and Ministerio de Ciencia y Técnica Argentina (MinCyT) PICT 2015-1622.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to Joss Heywood for his English technical assistance.
1. Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science (1997) 278 :58–63. doi: 10.1126/science.278.5335.58
2. White FJ, Kalivas PW. Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend. (1998) 51:141–53. doi: 10.1016/S0376-8716(98)00072-6
3. Kalivas PW. Cocaine and amphetamine-like psychostimulants: neurocircuitry and glutamate neuroplasticity. Dialogues Clin Neurosci. (2007) 9:389–397.
4. Pierce RC, Wolf ME. Psychostimulant-induced neuroadaptations in nucleus accumbens AMPA receptor transmission. Cold Spring Harbor Perspec Med. (2013) 3:a012021. doi: 10.1101/cshperspect.a012021
5. Steketee JD, Kalivas PW. Drug wanting: behavioral sensitization and relapse to drug-seeking behavior. Pharmacol Rev. (2011) 63:348–65. doi: 10.1124/pr.109.001933
6. Wolf ME, Sun X, Mangiavacchi S, Chao SZ. Psychomotor stimulants and neuronal plasticity. Neuropharmacol. (2004) 47:61–79. doi: 10.1016/j.neuropharm.2004.07.006
7. Girault JA, Valjent E, Caboche J, Hervé D. ERK2: a logical AND gate critical for drug-induced plasticity? Curr Opin Pharmacol. (2007) 7:77–85. doi: 10.1016/j.coph.2006.08.012
8. Valjent E, Corvol J-C, Pagès C, Besson M-J, Maldonado R, Caboche J. Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci. (2000) 20:8701–9. doi: 10.1523/JNEUROSCI.20-23-08701.2000
9. Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol J-C, et al. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci USA. (2005) 102:491–6. doi: 10.1073/pnas.0408305102
10. Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. (2005) 25:9144–51. doi: 10.1523/JNEUROSCI.2252-05.2005
11. Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci (2007) 27:10621–35. doi: 10.1523/JNEUROSCI.2163-07.2007
12. McGinty JF, Shi XD, Schwendt M, Saylor A. Regulation of psychostimulant-induced signaling and gene expression in the striatum. J Neurochem. (2008) 104:1440–9. doi: 10.1111/j.1471-4159.2008.05240.x
13. Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. (2011) 12:623–37. doi: 10.1038/nrn3111
14. Robinson TE, Berridge KC. Incentive-sensitization and addiction. Addiction (2001) 96:103–14. doi: 10.1046/j.1360-0443.2001.9611038.x
15. Scofield MD, Heinsbroek JA, Gipson CD, Kupchik YM, Spencer S, Smith ACW, et al. The nucleus accumbens: mechanisms of addiction across drug classes reflect the importance of glutamate homeostasis. Pharmacol Rev. (2016) 68:816–71. doi: 10.1124/pr.116.012484
16. Le Merrer J, Becker JAJ, Befort K, Kieffer BL. Reward processing by the opioid system in the brain. Physiol Rev. (2009) 89:1379–412. doi: 10.1152/physrev.00005.2009
17. Mansour A, Fox CA, Akil H, and Watson SJ. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. (1995) 18:22–9. doi: 10.1016/0166-2236(95)93946-U
18. Assis MA, Hansen C, Lux-Lantos V, Cancela LM. Sensitization to amphetamine occurs simultaneously at immune level and in met-enkephalin of the nucleus accumbens and spleen: an involved NMDA glutamatergic mechanism. Brain Behav Immun. (2009) 23:464–73. doi: 10.1016/j.bbi.2009.01.003
19. Mongi-Bragato B, Zamponi E, García-Keller C, Assis MA, Virgolini MB, Mascó DH, et al. Enkephalin is essential for the molecular and behavioral expression of cocaine sensitization. Addic Biol. (2016) 21:326–38. doi: 10.1111/adb.12200
20. Daugé V, Kalivas PW, Duffy T, Roques BP. Effect of inhibiting enkephalin catabolism in the VTA on motor activity and extracellular dopamine. Brain Res. (1992) 599:209–14. doi: 10.1016/0006-8993(92)90393-N
21. Dourmap N, Costentin J. Involvement of glutamate receptors in the striatal enkephalin-induced dopamine release. Eur J Pharmacol. (1994) 253:R9–11. doi: 10.1016/0014-2999(94)90210-0
22. Dourmap N, Michael-Titus A, Costentin J. Local enkephalins tonically modulate dopamine release in the striatum: a microdialysi study. Brain Res. (1990) 524:153–5. doi: 10.1016/0006-8993(90)90505-6
23. Hipólito L, Sánchez-Catalá M. J., Zanolini I, Polache A, Granero L. Shell/core differences in mu-and delta-opioid receptor modulation of dopamine efflux in nucleus accumbens. Neuropharmacol. (2008) 55:183–9. doi: 10.1016/j.neuropharm.2008.05.012
24. Kalivas PW, Duffy P. Effect of acute and daily neurotensin and enkephalin treatments on extracellular dopamine in the nucleus accumbens. J Neurosci. (1990b) 10:2940–9. doi: 10.1523/JNEUROSCI.10-09-02940.1990
25. Latimer LG, Duffy P, Kalivas PW. Mu opioid receptor involvement in enkephalin activation of dopamine neurons in the ventral tegmental area. J Pharmacol Exp Ther. (1987) 241:328–37.
26. Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Neurobiology (1988) 85:5274–8. doi: 10.1073/pnas.85.14.5274
27. Di Chiara G, Bassareo V, Fenu S, De Luca MA, Spina L, Cadoni C, et al. Dopamine and drug addiction: the nucleus accumbens shell connection. Neuropharmacology (2004) 47:227–41. doi: 10.1016/j.neuropharm.2004.06.032
28. Hurd YL, Herkenham M. Influence of a single injection of cocaine, amphetamine or Gbr-12909 on Messenger-Rna expression of striatal neuropeptides. Mol Brain Res. (1992) 16:97–104. doi: 10.1016/0169-328X(92)90198-K
29. Przewłocka B, Lason W. Adaptive changes in the proenkephalin and D2 dopamine receptor mRNA expression after chronic cocaine in the nucleus accumbens and striatum of the rat. Eur Neuropsychopharmacol. (1995) 5:465–9. doi: 10.1016/0924-977X(95)80005-M
30. Smith AJW, McGinty JF. Acute amphetamine or methamphetamine alters opioid peptide mRNA expression in rat striatum. Mol Brain Res. (1994) 21:359–62. doi: 10.1016/0169-328X(94)90268-2
31. Wang JQ, McGinty JF. D1 and D2 receptor regulation of preproenkephalin and preprodynorphin mRNA in rat striatum following acute injection of amphetamine or methamphetamine. Synapse (1996) 22:114–22. doi: 10.1002/(SICI)1098-2396(199602)22:2<114::AID-SYN4>3.0.CO;2-G
32. Wang JQ, McGinty JF. Intrastriatal injection of a muscarinic receptor agonist and antagonist regulates striatal neuropeptide mRNA expression in normal and amphetamine-treated rats. Brain Res. (1997a) 748:62–70. doi: 10.1016/S0006-8993(96)01244-9
33. Wang JQ, McGinty JF. The Full D1 dopamine receptor agonist SKF-82958 induces neuropeptide mRNA in the normosensitive striatum of rats: regulation of D1/D2 interactions by muscarinic receptors. J Pharmacol Exp Ther. (1997b) 281:972–82.
34. Mathieu-Kia A-M, Besson M-J. Repeated administration of cocaine, nicotine and ethanol: effects on preprodynorphin, preprotachykinin A and preproenkephalin mRNA expression in the dorsal and the ventral striatum of the rat. Mol Brain Res. (1998) 54:141–51. doi: 10.1016/S0169-328X(97)00338-0
35. Steiner H, Gerfen C. Cocaine-induced c-fos messenger RNA is inversely related to dynorphin expression in striatum. J Neurosci. (1993) 13:5066–81. doi: 10.1523/JNEUROSCI.13-12-05066.1993
36. Zhang Y, Schlussman SD, Butelman ER, Ho A, Kreek MJ. Effects of withdrawal from chronic escalating-dose binge cocaine on conditioned place preference to cocaine and striatal preproenkephalin mRNA in C57BL/6J mice. Neuropharmacology (2012) 63:322–9. doi: 10.1016/j.neuropharm.2012.03.021
37. Spangler R, Zhou Y, Maggos CE, Schlussman SD, Ho A, Kreek MJ. Prodynorphin, proenkephalin and κ opioid receptor mRNA responses to acute “binge” cocaine. Mol Brain Res. (1997) 44:139–42.
38. Daunais JB, McGinty JF. Acute and chronic cocaine administration differentially alters striatal opioid and nuclear transcription factor mRNAs. Synapse (1994) 18:35–45. doi: 10.1002/syn.890180106
39. Branch AD, Unterwald EM, Lee SE, Kreek MJ. Quantitation of preproenkephalin mRNA levels in brain regions from male Fischer rats following chronic cocaine treatment using a recently developed solution hybridization assay. Mol Brain Res. (1992) 14:231–8. doi: 10.1016/0169-328X(92)90178-E
40. Bailey A, Yuferov V, Bendor J, Schlussman SD, Zhou Y, Ho A, et al. Immediate withdrawal from chronic “binge” cocaine administration increases μ-opioid receptor mRNA levels in rat frontal cortex. Mol Brain Res. (2005) 137:258–62. doi: 10.1016/j.molbrainres.2005.02.017
41. Turchan J, Maj M, Przewocka B, Przewocki R. Effect of cocaine and amphetamine on biosynthesis of proenkephalin and prodynorphin in some regions of the rat limbic system. Pol J Pharmacol. (2002) 54:367–72.
42. Ziółkowska B, Stefanski R, Mierzejewski P, Zapart G, Kostowski W, Przewłocki R. Contingency does not contribute to the effects of cocaine self-administration on prodynorphin and proenkephalin gene expression in the rat forebrain. Brain Res. (2006) 1069:1–9. doi: 10.1016/j.brainres.2005.11.042
43. Crespo JA, Manzanares J, Oliva JM, Corchero J, Palomo T, Ambrosio E. Extinction of cocaine self-administration produces a differential time-related regulation of proenkephalin gene expression in rat brain. Neuropsychopharmacology (2001) 25:185–94. doi: 10.1016/S0893-133X(01)00221-4
44. Gonzalez-Nicolini V, McGinty JF. Gene expression profile from the striatum of amphetamine-treated rats: a cDNA array and in situ hybridization histochemical study. Gene Expr Patterns (2002) 1:193–8. doi: 10.1016/S1567-133X(02)00017-0
45. Gonzalez-Nicolini MV, Berglind W, Cole KS, Keogh CL, Mcginty JF. Local mu and deltaopioid receptors regulate amphetamine- induced behavior and neuropeptide mrna in the striatum. Neuroscience (2003) 121:387–98. doi: 10.1016/S0306-4522(03)00488-3
46. Morales-Mulia M, Panayi F, Lambás-Señas L, Scarna H, Méndez M. Changes in Proenkephalin mRNA expression in forebrain areas after amphetamine-induced behavioural sensitization. Pharmacol Biochem Behav. (2007) 87:232–40. doi: 10.1016/j.pbb.2007.04.019
47. Shi X, McGinty JF. Extracellular signal-regulated mitogen-activated protein kinase inhibitors decrease amphetamine-induced behavior and neuropeptide gene expression in the striatum. Neuroscience (2006) 138:1289–98. doi: 10.1016/j.neuroscience.2005.12.024
48. Wang JQ, McGinty JF. Alterations in striatal zif/268, preprodynorphin and preproenkephalin mRNA expression induced by repeated amphetamine administration in rats. Brain Res. (1995) 673:262–74. doi: 10.1016/0006-8993(94)01422-E
49. Assis MA, Collino C, Figuerola Mde L, Sotomayor C, Cancela LM. Amphetamine triggers an increase in met-enkephalin simultaneously in brain areas and immune cells. J Neuroimmunol. (2006) 178:62–75. doi: 10.1016/j.jneuroim.2006.05.009
50. Assis MA, Valdomero A, García-Keller C, Sotomayor C, Cancela LM. Decrease of lymphoproliferative response by amphetamine is mediated by dopamine from the nucleus accumbens: influence on splenic met-enkephalin levels. Brain Behav Immun. (2011) 25:647–57. doi: 10.1016/j.bbi.2011.01.001
51. Liste I, Rodriguez-Pallares J, Caruncho HJ, Labandeira-Garcia JL. Locomotor-activity-induced changes in striatal levels of preprotachykinin and preproenkephalin mRNA. Regulation by the dopaminergic and glutamatergic systems. Mol Brain Res. (1999) 70:74–83. doi: 10.1016/S0169-328X(99)00140-0
52. Mao L, Wang JQ. Contribution of ionotropic glutamate receptors to acute amphetamine- stimulated preproenkephalin mRNA expression in the rat striatum in vivo. Neurosci Lett. (2003) 346:17–20. doi: 10.1016/S0304-3940(03)00542-1
53. Gerfen C, Engber T, Mahan L, Susel Z, Chase T, Monsma F, et al. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science (1990) 250:1429–32. doi: 10.1126/science.2147780
54. Le Moine C, Normand E, Guitteny AF, Fouquet B, Teoulet R, Bloch B. Dopamine receptor gene expression by enkephalin neurons in rat forebrain (nigrostriatal pathway/D2 receptor/preproenkephalin A/in situ hybridization). Neurobiology (1990) 87:230–4.
55. Le Moine C, Bloch B. D1 and D2 dopamine receptor gene expression in the rat striatum: Sensitive cRNA probes demonstrate prominent segregation of D1 and D2 mRNAS in distinct neuronal populations of the dorsal and ventral striatum. J Com Neurol. (1995) 355:418–26. doi: 10.1002/cne.903550308
56. Damsma G, Robertson GS, Tham CS, Fibiger HC. Dopaminergic regulation of striatal acetylcholine release: importance of D1 and N-methyl-D-aspartate receptors. J Pharmacol Exp Ther. (1991) 259:1064–72.
57. Guix T, Hurd YL, Ungerstedt U. Amphetamine enhances extracellular concentrations of dopamine and acetylcholine in dorsolateral striatum and nucleus accumbens of freely moving rats. Neurosci Lett. (1992) 138:137–40. doi: 10.1016/0304-3940(92)90490-X
58. Tzaferis JA, McGinty JF. Kappa opioid receptor stimulation decreases amphetamine-induced behavior and neuropeptide mRNA expression in the striatum. Mol Brain Res. (2001) 93:27–35. doi: 10.1016/S0169-328X(01)00178-4
59. Gray AM, Rawls SM, Shippenberg TS, McGinty JF. The kappa-opioid agonist, U-69593, decreases acute amphetamine-evoked behaviors and calcium-dependent dialysate levels of dopamine and glutamate in the ventral striatum. J Neurochem. (1999) 73:1066–74. doi: 10.1046/j.1471-4159.1999.0731066.x
60. Pierce RC, Bell K, Duffy P, Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci. (1996) 16:1550–60. doi: 10.1523/JNEUROSCI.16-04-01550.1996
61. Vanderschuren LJ, Kalivas PW. Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology (2000) 151:99–120. doi: 10.1007/s002130000493
62. Del Arco A, González-Mora JL, Armas VR, Mora F. Amphetamine increases the extracellular concentration of glutamate in striatum of the awake rat: involvement of high affinity transporter mechanisms. Neuropharmacology (1999) 38:943–54. doi: 10.1016/S0028-3908(99)00043-X
63. Reid MS, Hsu K, Berger SP. Cocaine and amphetamine preferentially stimulate glutamate release in the limbic system: studies on the involvement of dopamine. Synapse (1997) 27:95–105. doi: 10.1002/(SICI)1098-2396(199710)27:2<95::AID-SYN1>3.0.CO;2-6
64. Wolf ME, Xue CJ. Amphetamine and D1 dopamine receptor agonists produce biphasic effects on glutamate efflux in rat ventral tegmental area: modification by repeated amphetamine administration. J Neurochem. (1998) 70:198–209. doi: 10.1046/j.1471-4159.1998.70010198.x
65. Darracq L, Drouin C, Blanc G, Glowinski J, Tassin J-P. Stimulation of metabotropic but not ionotropic glutamatergic receptors in the nucleus accumbens is required for the d-amphetamine-induced release of functional dopamine. Neuroscience (2001) 103:395–403. doi: 10.1016/S0306-4522(00)00578-9
66. Kalivas PW, Duffy P. Dopamine regulation of extracellular glutamate in the nucleus accumbens. Brain Res. (1997) 761:173–7. doi: 10.1016/S0006-8993(97)00464-2
67. Weihmuller FB, O'Dell SJ, Cole BN, Marshall JF. MK-801 attenuates the dopamine-releasing but not the behavioral effects of methamphetamine: an in vivo microdialysis study. Brain Res. (1991) 549:230–5.
68. Bickerdike MJ, Abercrombie ED. Enhanced acetylcholine release in striatum after chronic amphetamine is NMDA-dependent. Neuroreport (1999) 10:77–80. doi: 10.1097/00001756-199901180-00015
69. Knauber J, Kischka U, Roth M, Schmidt WJ, Hennerici M, Fassbender K. Modulation of striatal acetylcholine concentrations by NMDA and the competitive NMDA receptor-antagonist AP-5: an in vivo microdialysis study. J Neural Transm. (1999) 106:35–45. doi: 10.1007/s007020050139
70. Bacher B, Wang X, Schulz S, Höllt V. Induction of proenkephalin gene expression in cultured bovine chromaffin cells is dependent on protein synthesis of AP-1 proteins. J Neurochem. (1996) 66:2264–71. doi: 10.1046/j.1471-4159.1996.66062264.x
71. Cole RL, Konradi C, Douglass J, Hyman SE. Neuronal adaptation to amphetamine and dopamine: molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron (1995) 14:813–23. doi: 10.1016/0896-6273(95)90225-2
72. Kobierski LA, Wong AE, Srivastava S, Borsook D, Hyman SE. Cyclic AMP-dependent activation of the proenkephalin gene requires phosphorylation of CREB at serine-133 and a src-related kinase. J Neurochem. (1999) 73:129–38. doi: 10.1046/j.1471-4159.1999.0730129.x
73. Konradi C, Cole RL, Heckers S, Hymanls SE. Amphetamine regulates gene expression in rat striatum via transcription factor CREB. J Neurosci. (1994) 74:5623–6634. doi: 10.1523/JNEUROSCI.14-09-05623.1994
74. Monnier D, Loeffler JP. Pituitary adenylate cyclase-activating polypeptide stimulates proenkephalin gene transcription through AP1- and CREB-dependent mechanisms. DNA Cell Biol. (1998) 17:151–9. doi: 10.1089/dna.1998.17.151
75. Cho DI, Quan W, Oak MH, Choi HJ, Lee KY, Kim KM. Functional interaction between dopamine receptor subtypes for the regulation of c-fos expression. Biochem Biophys Res Commun. (2007) 357:1113–8. doi: 10.1016/j.bbrc.2007.04.066
76. Kashihara K, Ishihara T, Akiyama K, Abe K. D1/D2 receptor synergism on CREB DNA-binding activities in the caudate-putamen of rat. Neurol Res. (1999) 21:781–4. doi: 10.1080/01616412.1999.11741014
77. Sgambato V, Pagès C, Rogard M, Besson MJ, Caboche J. Extracellular Signal-Regulated Kinase (ERK) controls immediate early gene induction on corticostriatal stimulation. J Neurosci. (1998) 18:8814–25. doi: 10.1523/JNEUROSCI.18-21-08814.1998
78. Vanhoutte P, Barnier JV, Guibert B, Pagès C, Besson MJ, Hipskind RA, et al. Glutamate induces phosphorylation of Elk-1 and CREB, along with c -fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol. (1999) 19:136–46. doi: 10.1128/MCB.19.1.136
79. Choe ES, McGinty JF. Cyclic AMP, and mitogen-activated protein kinases are required for glutamate-dependent cyclic AMP response element binding protein and Elk-1 phosphorylation in the dorsal striatum in vivo. J Neurochem. (2001) 76:401–12. doi: 10.1046/j.1471-4159.2001.00051.x
80. Carlezon W Jr, Duman R, Nestler E. The many faces of CREB. Trends Neurosci. (2005) 28:436–45. doi: 10.1016/j.tins.2005.06.005
81. Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. (1992) 12:483–8. doi: 10.1523/JNEUROSCI.12-02-00483.1992
82. Spanagel R, Herz A, Shippenberg TS. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway (mirodialysis/nucleus accumbens/dopamine release and metabolism/opiate dependence). Pharmacology (1992) 89:2046–50.
83. Rawls SM, McGinty JF. Delta opioid receptors regulate calcium-dependent, amphetamine-evoked glutamate levels in the rat striatum: an in vivo microdialysis study. Brain Res. (2000) 861:296–304. doi: 10.1016/S0006-8993(00)02030-8
84. Shippenberg TS, Chefer VI. (2003) Opioid modulation of psychomotor stimulant effects. In: Maldonado R, editor. Molecular Biology of Drug Addiction. Totowa, NJ: Humana Press, 107–32.
85. Shippenberg TS, Elmer GI. The neurobiology of opiate reinforcement. Crit Rev Neurobiol (1988) 12:267–303.
86. Van Ree JM, Niesink RJ, Van Wolfswinkel L, Ramsey NF, Kornet MM, Vanderschuren LJ, et al. Endogenous opioids and reward. Eur J Pharmacol. (2000) 405:89–101. doi: 10.1016/S0014-2999(00)00544-6
87. Heidbreder C, Shoaib M, Shippenberg TS. Differential role of 6-opioid receptors in the development and expression of behavioral sensitization to cocaine. Eur J Pharmacol. (1996) 298:207–16. doi: 10.1016/0014-2999(95)00815-2
88. Hummel M, Ansonoff MA, Pintar JE, Unterwald EM. Genetic and pharmacological manipulation of mu opioid receptors in mice reveals a differential effect on behavioral sensitization to cocaine. Neuroscience (2004) 125:211–20. doi: 10.1016/j.neuroscience.2004.01.025
89. Racz I, Schürmann B, Karpushova A, Reuter M, Cichon S, Montag C, et al. The opioid peptides enkephalin and beta-endorphin in alcohol dependence. Biol Psychiatry (2008) 64:989–97. doi: 10.1016/j.biopsych.2008.05.008
90. Berrendero F. Nicotine-induced antinociception, rewarding effects, and physical dependence are decreased in mice lacking the preproenkephalin gene. J Neurosci. (2005) 25:1103–12. doi: 10.1523/JNEUROSCI.3008-04.2005
91. Gianoulakis C. Endogenous opioids and excessive alcohol consumption. J Psychiatr Neurosci. (1993) 18:148–56.
92. Marinelli PW, Bai L, Quirion R, Gianoulakis C. A microdialysis profile of Met-enkephalin release in the rat nucleus accumbens following alcohol administration. Alcohol Clin Exp Res. (2005) 29:1821–8. doi: 10.1097/01.alc.0000183008.62955.2e
93. Kalivas PW, Duffy P. Effect of acute and daily cocaine treatment on extracellular dopamine in the nucleus accumbens. Synapse (1990a) 5:48–58. doi: 10.1002/syn.890050104
94. Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Rev. (1991) 16:223–44. doi: 10.1016/0165-0173(91)90007-U
95. Kalivas PW, Duffy P. Time course of extracellular dopamine and behavioral sensitization to cocaine. I. Dopamine Axon Terminals. J Neurosci. (1993) 13:266–75. doi: 10.1523/JNEUROSCI.13-01-00266.1993
96. Segal DS, Kuczenski R. Repeated cocaine administration induces behavioral sensitization and corresponding decreased extracellular dopamine responses in caudate and accumbens. Brain Res. (1992) 577:351–5. doi: 10.1016/0006-8993(92)90297-M
97. Hurd YL, Weiss F, Koob GF, And N-E, Ungerstedt U. Cocaine reinforcement and extracellular dopamine overflow in rat nucleus accumbens: an in vivo microdialysis study. Brain Res. (1989) 498:199–203. doi: 10.1016/0006-8993(89)90422-8
98. Crooks KR, Kleven DT, Rodriguiz RM, Wetsel WC, McNamara JO. TrkB signaling is required for behavioral sensitization and conditioned place preference induced by a single injection of cocaine. Neuropharmacology (2010) 58:1067–77. doi: 10.1016/j.neuropharm.2010.01.014
99. Bosse KE, Jutkiewicz EM, Gnegy ME, Traynor JR. The selective delta opioid agonist SNC80 enhances amphetamine-mediated efflux of dopamine from rat striatum. Neuropharmacology (2008) 55:755–62. doi: 10.1016/j.neuropharm.2008.06.017
100. Schad CA, Justice JB, Holtzman SG. Differential effects of δ- and μ-Opioid receptor antagonists on the amphetamine-induced increase in extracellular dopamine in striatum and nucleus accumbens. J Neurochem. (1996) 67:2292–9. doi: 10.1046/j.1471-4159.1996.67062292.x
101. Soderman AR, Unterwald EM. Cocaine reward and hyperactivity in the rat: sites of mu opioid receptor modulation. Neuroscience (2008) 154:1506–16. doi: 10.1016/j.neuroscience.2008.04.063
102. Soderman AR, Unterwald EM. Cocaine-induced mu opioid receptor occupancy within the striatum is mediated by dopamine D2 receptors. Brain Res. (2009) 1296:63–71. doi: 10.1016/j.brainres.2009.08.035
103. Sala M, Braida D, Colombo M, Groppetti A, Sacco S, Gori E, et al. Behavioral and biochemical evidence of opioidergic involvement in cocaine sensitization. J Pharmacol Exp Ther. (1995) 274:450–7.
104. Kim HS, Park WK, Jang CG, Oh KW, Kong JY, Oh S, et al. Blockade by naloxone of cocaine-induced hyperactivity, reverse tolerance and conditioned place preference in mice. Behav Brain Res. (1997) 85:37–46. doi: 10.1016/S0166-4328(96)00162-3
105. Schroeder JA, Hummel M, Simpson AD, Sheikh R, Soderman AR, Unterwald EM. A role for mu opioid receptors in cocaine-induced activity, sensitization, and reward in the rat. Psychopharmacology (2007) 195:265–72. doi: 10.1007/s00213-007-0883-z
106. Balcells-Olivero M, Vezina P. Effects of naltrexone on amphetamine-induced locomotion and rearing: acute and repeated injections. Psychopharmacology (1997) 131:230–8. doi: 10.1007/s002130050288
107. Häggkvist J, Björkholm C, Steensland P, Lindholm S, Franck J, Schilström B. Naltrexone attenuates amphetamine-induced locomotor sensitization in the rat. Addict Biol. (2011) 16:20–9. doi: 10.1111/j.1369-1600.2009.00199.x
108. Magendzo K, Bustos G. Expression of amphetamine-induced behavioral sensitization after short-and long-term withdrawal periods: participation of m-and d-opioid receptors. Neuropsychopharmacology (2003) 28:468–77. doi: 10.1038/sj.npp.1300063
109. Asensio VJ, Miralles A, García-Sevilla JA. Stimulation of mitogen-activated protein kinase kinases (MEK1/2) by μ-, δ- and κ-opioid receptor agonists in the rat brain: Regulation by chronic morphine and opioid withdrawal. Eur J Pharmacol. (2006) 539:49–56. doi: 10.1016/j.ejphar.2006.04.001
110. Eitan S, Bryant CD, Saliminejad N, Yang YC, Vojdani E, Kaith D Jr, et al. Brain region-specific mechanisms for acute morphine- induced mitogen-activated protein kinase modulation and distinct patterns of activation during analgesic tolerance and locomotor sensitization. J Neurosci. (2003) 23:8360–9. doi: 10.1523/JNEUROSCI.23-23-08360.2003
111. Muller DL, Unterwald EM. In vivo regulation of extracellular signal-regulated protein kinase (ERK) and Protein Kinase B (Akt) phosphorylation by acute and chronic morphine. J Pharmacol Exp Ther. (2004) 10:774–82. doi: 10.1124/jpet.104.066548
112. Schulz S, Höllt V. Opioid withdrawal activates MAP kinase in locus coeruleus neurons in morphine-dependent rats in vivo. Eur J Neurosci. (1998) 10:1196–201. doi: 10.1046/j.1460-9568.1998.00103.x
113. Hall FS, Goeb M, Li X-F, Sora I, Uhl GR. A-Opioid receptor knockout mice display reduced cocaine conditioned place preference but enhanced sensitization of cocaine-induced locomotion. Mol Brain Res. (2004) 121:123–30. doi: 10.1016/j.molbrainres.2003.10.024
114. Yoo JH, Yang EM, Lee SY, Loh HH, Ho IK, Jang CG. Differential effects of morphine and cocaine on locomotor activity and sensitization in m-opioid receptor knockout mice. Neurosci Lett. (2003) 344:37–40. doi: 10.1016/S0304-3940(03)00410-5
115. Lesscher HM, Hordijk M, Bondar NP, Alekseyenko OV, Burbach JP, van Ree JM, et al. Mu-opioid receptors are not involved in acute cocaine-induced locomotor activity nor in development of cocaine-induced behavioral sensitization in mice. Neuropsychopharmacology (2005) 30:278–85. doi: 10.1038/sj.npp.1300529
116. Yoo JH, Kitchen I, Bailey A. The endogenous opioid system in cocaine addiction: what lessons have opioid peptide and receptor knockout mice taught us? Br J Pharmacol. (2012) 166:1993–2014. doi: 10.1111/j.1476-5381.2012.01952.x
117. DuMars LA, Rodger LD, Kalivas PW. Behavioral cross-sensitization between cocaine and enkephalin in the A 10 dopamine region. Behav Brain Res. (1988) 27:87–91. doi: 10.1016/0166-4328(88)90111-8
118. Kalivas PW, Widerlöv E, Stanley D, Breese G, Prange AJ. Enkephalin action on the mesolimbic system: a dopamine-dependent and a dopamine-independent increase in locomotor activity. J Pharmacol Exp Ther. (1983) 227:229–37.
119. Bahi A, Boyer F, Chandrasekar V, Dreyer J-L. Role of accumbens BDNF and TrkB in cocaine-induced psychomotor sensitization, conditioned-place preference, and reinstatement in rats. Psychopharmacology (2008) 199:169–82. doi: 10.1007/s00213-008-1164-1
120. Lobo MK, Iii HEC, Chaudhury D, Friedman AK, Sun H, Damez-werno D, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science (2011) 330:385–90. doi: 10.1126/science.1188472
121. Trujillo KA, Belluzzi JD, Stein L. Naloxone blockade of amphetamine place preference conditioning. Psychopharmacology (1991) 104:265–74. doi: 10.1007/BF02244190
122. Mitchem LD, Kruschel CK, Dallman E, Anders KA, Czapiga M, Panos JJ, et al. The effects of the naltrexone implant on rodent social interactions and cocaine-induced conditioned place preference. Pharmacol Biochem Behav. (1999) 62:97–102. doi: 10.1016/S0091-3057(98)00150-6
123. Menkens K, Bilsky EJ, Wild KD, Portoghese PS, Reid LD, Porreca F. Cocaine place preference is blocked by the 6-opioid receptor antagonist, naltrindole. Eur J Pharmacol. (1992) 219:345–6. doi: 10.1016/0014-2999(92)90319-Y
124. Suzuki T, Mori T, Tsuji M, Misawa M, Nagase H. The role of δ-opioid receptor subtypes in cocaine- and methamphetamine-induced place preferences. Life Sci. (1994) 55:PL339–4. doi: 10.1016/0024-3205(94)00774-8
125. Rademacher DJ, Steinpreis RE. Effects of the selective mu(1)-opioid receptor antagonist, naloxonazine, on cocaine-induced conditioned place preference and locomotor behavior in rats. Neurosci Lett. (2002) 332:159–62. doi: 10.1016/S0304-3940(02)00950-3
126. Gerrits MA, Patkina N, Zvartau EE, van Ree JM. Opioid blockade attenuates acquisition and expression of cocaine-induced place preference conditioning in rats.Psychopharmacology (1995) 119:92–8. doi: 10.1007/BF02246059
127. Contarino A, Picetti R, Matthes HW, Koob GF, Kieffer BL, Gold LH. Lack of reward and locomotor stimulation induced by heroin in A-opioid receptor-deficient mice. Eur J Pharmacol. (2002) 446:103–9. doi: 10.1016/S0014-2999(02)01812-5
128. Becker A, Grecksch G, Kraus J, Loh H, Schroeder H, Höllt V. Rewarding effects of ethanol and cocaine in μ opioid receptor-deficient mice. Naunyn Schmiedebergs Arch Pharmacol. (2002) 365:296–302. doi: 10.1007/s00210-002-0533-2
129. Billa SK, Sinha N, Rudrabhatla SR, Morón JA. Extinction of morphine-dependent conditioned behavior is associated with increased phosphorylation of the GluR1 subunit of AMPA receptors at hippocampal synapses. Eur J Neurosci. (2009) 29:55–64. doi: 10.1111/j.1460-9568.2008.06560.x
130. Morón JA, Gullapalli S, Taylor C, Gupta A, Gomes I, Devi LA. Modulation of opiate-related signaling molecules in morphine-dependent conditioned behavior: conditioned place preference to morphine induces CREB phosphorylation. Neuropsychopharmacology (2010) 35:955–66. doi: 10.1038/npp.2009.199
131. Pahlevani P, Fatahi Z, Moradi M, Haghparast A. Morphine-induced conditioned place preference and the alterations of p-ERK, p-CREB and c-fos levels in hypothalamus and hippocampus: the effects of physical stress. Cell Mol Biol. (2014) 60:48–55.
132. Haghparast A, Fatahi Z, Alamdary SZ, Reisi Z, Khodagholi F. Changes in the levels of p-ERK, p-CREB, and c-fos in rat mesocorticolimbic dopaminergic system after morphine-induced conditioned place preference: the role of acute and subchronic stress. Cell Mol Neurobiol. (2014) 34:277–88. doi: 10.1007/s10571-013-0011-z
133. Lin X, Wang Q, Ji A, Yu LC. Role of MEK-ERK pathway in morphine-induced conditioned place preference in ventral tegmental area of rats. J Neurosci Res. (2010) 88:1595–604. doi: 10.1002/jnr.22326
134. Giuliano C, Robbins TW, Wille DR, Bullmore ET, Everitt BJ. Attenuation of cocaine and heroin seeking by μ-opioid receptor antagonism. Psychopharmacology (2013) 227:137–47. doi: 10.1007/s00213-012-2949-9
135. Burattini C, Burbassi S, Aicardi G, Cervo L. Effects of naltrexone on cocaine- and sucrose-seeking behaviour in response to associated stimuli in rats. Int J Neuropsychopharmacol. (2008) 11:103–9. doi: 10.1017/S1461145707007705
136. Sushchyk S, Xi ZX, Wang JB. (2016). Combination of levo-tetrahydropalmatine and low dose naltrexone: a promising treatment for prevention of cocaine relapse. J Pharmacol Exp Ther. (2016) 357:248–57. doi: 10.1124/jpet.115.229542
137. Ward SJ, Martin TJ, Roberts DCS. Beta-funaltrexamine affects cocaine self-administration in rats responding on a progressive ratio schedule of reinforcement. Pharmacol Biochem Behav. (2003) 75:301–7. doi: 10.1016/S0091-3057(03)00087-X
138. Tang X-C, McFarland K, Cagle S, Kalivas PW. Cocaine-induced reinstatement requires endogenous stimulation of μ-opioid receptors in the ventral pallidum. J Neurosci. (2005) 25:4512–20. doi: 10.1523/JNEUROSCI.0685-05.2005
139. de Vries TJ, Babovic-Vuksanovic D, Elmer G, Shippenberg TS. Lack of involvement of delta-opioid receptors in mediating the rewarding effects of cocaine. Psychopharmacology (1995) 120:442–8. doi: 10.1007/BF02245816
140. Martin TJ, Kim SA, Cannon DG, Sizemore GM, Bian D, Porreca F, et al. Antagonism of delta(2)-Opioid Receptors by Naltrindole-5'- isothiocyanate attenuates heroin self-administration but not antinociception in rats. J Pharmacol Exp Ther. (2000) 294:975–82.
141. Reid LD, Glick SD, Menkens KA, French ED, Bilsky EJand Porreca F. Cocaine self-administration and naltrindole, a delta-selective opioid antagonist. Neuroreport (1995) 6:1409–12. doi: 10.1097/00001756-199507100-00012
142. Simmons D, Self DW. Role of Mu- and delta-opioid receptors in the nucleus accumbens in cocaine-seeking behavior. Neuropsychopharmacology (2009) 34:1946–57. doi: 10.1038/npp.2009.28
143. Mathon DS, Lesscher HM, Gerrits MA, Kamal A, Pintar JE, Schuller AG, et al. Increased gabaergic input to ventral tegmental area dopaminergic neurons associated with decreased cocaine reinforcement in mu-opioid receptor knockout mice. Neuroscience (2005) 130:359–67. doi: 10.1016/j.neuroscience.2004.10.002
144. Gutiérrez-Cuesta J, Burokas A, Mancino S, Kummer S, Martín-García E, Maldonado R. Effects of genetic deletion of endogenous opioid system components on the reinstatement of cocaine-seeking behavior in mice. Neuropsychopharmacology (2014) 39149:2974–88. doi: 10.1038/npp.2014.149
145. Kupchik YM, Scofield MD, Rice KC, Cheng K, Roques BP, Kalivas PW. (2014). Cocaine dysregulates opioid gating of GABA neurotransmission in the ventral pallidum. J Neurosci. (2014) 34:1057–66. doi: 10.1523/JNEUROSCI.4336-13.2014
146. Comer SD, Mogali S, Saccone PA, Askalsky P, Martinez D, Walker EA, et al. Effects of acute oral naltrexone on the subjective and physiological effects of oral D-amphetamine and smoked cocaine in cocaine abusers. Neuropsychopharmacology (2013) 38143:2427–38. doi: 10.1038/npp.2013.143
147. Schmitz JM, Stotts AL, Rhoades HM, Grabowski J. Naltrexone and relapse prevention treatment for cocaine-dependent patients. Addic Behav. (2001) 26:167–80. doi: 10.1016/S0306-4603(00)00098-8
148. Schmitz JM, Lindsay JA, Green CE, Herin DV, Stotts AL, Moeller FG. High-dose naltrexone therapy for cocaine-alcohol dependence. Am J Addic. (2009) 18:356–62. doi: 10.3109/10550490903077929
149. Jayaram-Lindström N, Hammarberg A, Beck O, Franck J. Naltrexone for the treatment of amphetamine dependence: a randomized, placebo-controlled trial. Am J Psychiatry (2008a) 165:1442–8. doi: 10.1176/appi.ajp.2008.08020304
150. Jayaram-Lindström N, Konstenius M, Eksborg S, Beck O, Hammarberg A, Franck J. Naltrexone attenuates the subjective effects of amphetamine in patients with amphetamine dependence. Neuropsychopharmacology (2008b) 33:1856–63. doi: 10.1038/sj.npp.1301572
151. Pettinati HM, Kampman KM, Lynch KG, Xie H, Dackis C, Rabinowitz AR, et al. A double blind, placebo-controlled trial that combines disulfiram and naltrexone for treating co-occurring cocaine and alcohol dependence. Addict Behav. (2008a) 33:651–67. doi: 10.1016/j.addbeh.2007.11.011
152. Pettinati HM, Kampman KM, Lynch KG, Suh JJ, Dackis C A, Oslin DW, et al. Gender differences with high-dose naltrexone in patients with co-occurring cocaine and alcoholdependence. J Subst Abuse Treat. (2008b) 34:378–90. doi: 10.1016/j.jsat.2007.05.011
153. Schmitz JM, Stotts AL, Sayre SL, DeLaune KA, Grabowski J. Treatment of cocaine-alcohol dependence with naltrexone and relapse prevention therapy. Am J Addict. (2004) 13:333–41. doi: 10.1080/10550490490480982
154. Dunn KE, Defulio A, Everly JJ, Donlin WD, Aklin WM, Nuzzo PA, et al. Employment-based reinforcement of adherence to oral naltrexone treatment in unemployed injection drug users. Exp Clin Psychopharmacol. (2013) 21:74–83. doi: 10.1037/a0030743
155. Kreek MJ. Pharmacogenetics and human molecular genetics of opiate and cocaine addictions and their treatments. Pharmacol Rev. (2005) 57:1–26. doi: 10.1124/pr.57.1.1
156. Moeller SJ, Beebe-Wang N, Schneider KE, Konova AB, Parvaz MA, Alia-Klein N, et al. Effects of an opioid (proenkephalin) polymorphism on neural response to errors in health and cocaine use disorder. Behav Brain Res. (2015) 293:18–26. doi: 10.1016/j.bbr.2015.07.004
157. Nikoshkov A, Drakenberg K, Wang X, Horvath MC, Keller E, Hurd YL. Opioid neuropeptide genotypes in relation to heroin abuse: dopamine tone contributes to reversed mesolimbic proenkephalin expression. Proc Natl Acad Sci USA. (2008) 105:786–91. doi: 10.1073/pnas.0710902105
Keywords: enkephalin, cocaine, amphetamine, neuroadaptations, addiction, opioid antagonists
Citation: Mongi-Bragato B, Avalos MP, Guzmán AS, Bollati FA and Cancela LM (2018) Enkephalin as a Pivotal Player in Neuroadaptations Related to Psychostimulant Addiction. Front. Psychiatry 9:222. doi: 10.3389/fpsyt.2018.00222
Received: 29 November 2017; Accepted: 08 May 2018;
Published: 28 May 2018.
Edited by:
Lawrence Toll, Florida Atlantic University, United StatesReviewed by:
Ellen M. Unterwald, Temple University, United StatesCopyright © 2018 Mongi-Bragato, Avalos, Guzmán, Bollati and Cancela. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liliana M. Cancela, bGNhbmNlbGFAZmNxLnVuYy5lZHUuYXI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.