 Madeleine Monique Uys
Madeleine Monique Uys Mohammed Shahid
Mohammed Shahid Brian Herbert Harvey
Brian Herbert Harvey
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 14 August 2017
Sec. Molecular Psychiatry
Volume 8 - 2017 | https://doi.org/10.3389/fpsyt.2017.00144
α2A- and α2C-adrenoceptors (ARs) are the primary α2-AR subtypes involved in central nervous system (CNS) function. These receptors are implicated in the pathophysiology of psychiatric illness, particularly those associated with affective, psychotic, and cognitive symptoms. Indeed, non-selective α2-AR blockade is proposed to contribute toward antidepressant (e.g., mirtazapine) and atypical antipsychotic (e.g., clozapine) drug action. Both α2C- and α2A-AR share autoreceptor functions to exert negative feedback control on noradrenaline (NA) release, with α2C-AR heteroreceptors regulating non-noradrenergic transmission (e.g., serotonin, dopamine). While the α2A-AR is widely distributed throughout the CNS, α2C-AR expression is more restricted, suggesting the possibility of significant differences in how these two receptor subtypes modulate regional neurotransmission. However, the α2C-AR plays a more prominent role during states of low endogenous NA activity, while the α2A-AR is relatively more engaged during states of high noradrenergic tone. Although augmentation of conventional antidepressant and antipsychotic therapy with non-selective α2-AR antagonists may improve therapeutic outcome, animal studies report distinct yet often opposing roles for the α2A- and α2C-ARs on behavioral markers of mood and cognition, implying that non-selective α2-AR antagonism may compromise therapeutic utility both in terms of efficacy and side-effect liability. Recently, several highly selective α2C-AR antagonists have been identified that have allowed deeper investigation into the function and utility of the α2C-AR. ORM-13070 is a useful positron emission tomography ligand, ORM-10921 has demonstrated antipsychotic, antidepressant, and pro-cognitive actions in animals, while ORM-12741 is in clinical development for the treatment of cognitive dysfunction and neuropsychiatric symptoms in Alzheimer’s disease. This review will emphasize the importance and relevance of the α2C-AR as a neuropsychiatric drug target in major depression, schizophrenia, and associated cognitive deficits. In addition, we will present new prospects and future directions of investigation.
The α2-adrenoceptor (AR) plays an important role in modulating the release of noradrenaline (NA) and various other important neurotransmitters in the central nervous system (CNS), providing a solid construct why drugs that target these receptors have clinical utility in several major neuropsychiatric disorders (1). The α2- (and α1-) AR plays a prominent role in the functioning of the prefrontal cortex (PFC) and as such mediates the effect of normal, aroused, and stressed NA levels on memory and other cognitive processes (2). To this end α2-AR antagonists mianserin and mirtazapine have seen widespread use in the therapy of major depressive disorder (MDD), while almost all atypical antipsychotics display moderate to potent levels of α2-AR antagonism, which has been suggested to underlie the atypical profile of antipsychotics such as clozapine, quetiapine, risperidone, and asenapine (3, 4). Importantly, both conventional antipsychotics (5–7) and antidepressants (8–10) show improved efficacy following augmentation with an α2-AR antagonist. Furthermore, cognitive parameters are also influenced by α2-AR modulation with α2-AR antagonism shown to improve attentional, verbal, and episodic memory deficits in patients with frontal dementia, although spatial working memory is unaffected (11). This is because stimulation of the cortical postsynaptic α2A-AR by NA is critical in the function of specific cognitive domains such as working memory (12), which is why α2-AR agonists are successfully used in the treatment of cognitive aspects of attention-deficit hyperactivity disorder (ADHD) (13). However, studies have indicated that α2-AR subtypes may not equally contribute to these beneficial effects on mood, psychotic, and cognitive disorders. In fact, findings from transgenic mouse studies have indicated distinct and sometimes opposing roles for the α2A-AR and α2C-AR (14–16), the two primary α2-AR subtypes involved in the regulation of CNS neurotransmission (refer to Table 1 for summary). Before the availability of sufficiently subtype-selective ligands, evidence from transgenic mouse studies have indicated a potential therapeutic role for selective antagonism of the α2C-AR in MDD, schizophrenia and associated cognitive impairment (16). More recently, the availability of highly selective α2C-AR antagonists for use in preclinical research has produced evidence confirming the antipsychotic-like, antidepressant-like, and pro-cognitive effects of this treatment strategy in animal models of schizophrenia and MDD (17–21). Genetic studies have also highlighted the potential involvement of the α2C-AR in these neuropsychiatric illnesses, with evidence that genetic polymorphism of the α2C-AR is associated with dysfunction in MDD (22), ADHD (23), and schizophrenia (24). With the first highly selective α2C-AR subtype antagonist, ORM-12741, showing improvement of cognitive parameters in Alzheimer’s Disease in Phase IIa clinical trials (25) and against a back-drop of evidence from transgenic mouse and other translational rodent models, the potential therapeutic benefit of selectively blocking α2C-ARs for the treatment of cognitive dysfunction in mood and psychotic disorders has attracted renewed interest. This review will summarize evidence from transgenic mouse models relating to the function of the α2C-AR in related neuropsychiatric function as well as present studies reporting on the therapeutic efficacy of selective α2C-AR antagonists in illness-specific models of MDD and schizophrenia in rats. Following a short overview of the functional roles for α2A and α2C-ARs, we will outline reasons for renewed interest in selective α2C-AR antagonism as a therapeutic target, its role in neurotransmitter regulation, and the evidence base for targeting this receptor for treating MDD and schizophrenia. We close with a brief discussion on the potential therapeutic benefits for α2C-AR modulation in other neuropsychiatric disorders and highlight progress in developing α2C-AR-related tools and technology to facilitate future basic and clinical research.
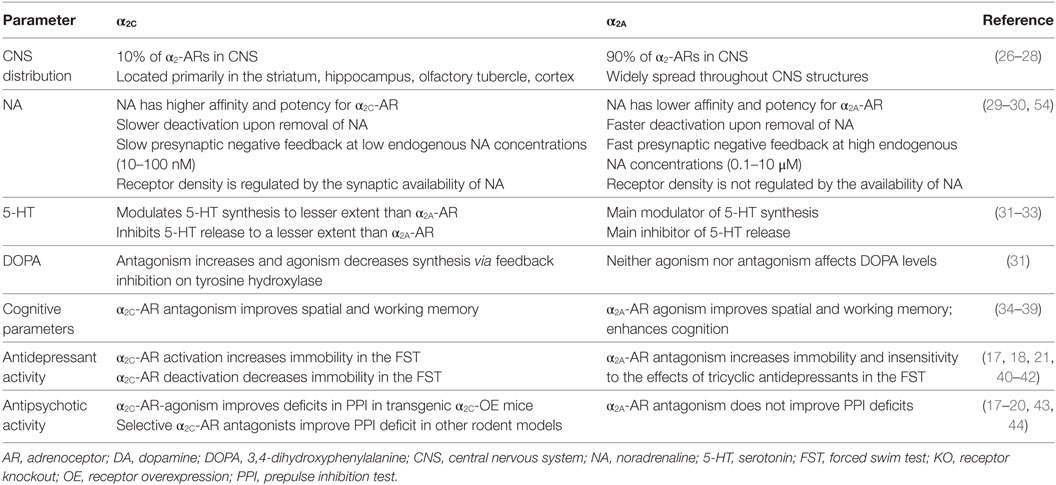
Table 1. Summary of opposing effects mediated through the α2C-AR and the α2A-AR.
The α2-AR is a member of the G-protein-coupled receptor (GPCR) superfamily, belonging to the rhodopsin-like or Class A GPCR receptors (45). α2-ARs couple to heterotrimeric Gi/o proteins when activated by their endogenous agonist, leading to inhibition of adenylyl cyclase and voltage-gated calcium channels, and activation of mitogen-activated protein kinase signaling cascades (15, 46). In the CNS, GPCRs and ion channels are targeted to the membrane of dendritic postsynaptic terminals in and around the postsynaptic density (PSD) via interaction with various scaffolding proteins (45). These proteins function as adaptors, regulators, and effectors of postsynaptic signaling to enable neural transmission and biological response. Spinophilin in particular is associated with the α2-AR (45), the relevance of which will be discussed later.
The presynaptic α2-AR autoreceptor inhibits NA synthesis and release and as such plays an important role in negative feedback, while presynaptic α2-AR heteroreceptors located on dopaminergic, serotoninergic, glutamatergic, and other terminals regulate the release of these latter transmitters (15, 46). Postsynaptic activation of α2-ARs in turn modulates neuronal excitability via regulation of ion channels, including the direct modulation of inwardly rectifying potassium channels and the indirect modulation of hyperpolarization-activated channels (46). While presynaptic action at α2-ARs affect neuropsychiatric processes through a cascade of effects on neurotransmitter feedback and regulation, postsynaptic activation of α2-ARs, specifically the α2A-AR, is associated with critical regulation and strengthening of working memory (12). Indeed, prefrontal cortical networks regulating various aspects of attention, cognition, and emotion require optimal catecholamine signaling, including stimulation of postsynaptic α2-ARs by NA to regulate “top-down” control of the PFC over subcortical regions (12, 47). This explains, for example, why α2-AR agonists favoring the α2A-AR have beneficial effects on memory and cognition in ADHD. However, α2-AR-mediated regulation of CNS function extends to the peripheral nervous system too. In this regard, the gut microbiome is increasingly being seen as a causal factor in psychiatric illness (48). Gut status is enabled to signal the CNS via a number of monoaminergic receptors located in the enteric nervous system (48), in particular dopamine (DA) (D2), serotonin (5-HT3; 5-HT4), and NA receptors, the latter via inhibition of vagal (parasympathetic) activity through presynaptic α2 receptors (49). Notwithstanding the neurophysiological importance of postsynaptic α2-AR activation, the literature increasingly points to selectively targeting specific α2-AR subtypes to exert control over presynaptic modulation of various neurotransmitter feedback systems associated with cognitive and affective functioning. While α2-ARs are collectively important in neural transmission, this review will delineate the therapeutic effects associated with modulation of the presynaptic α2C-AR.
The presynaptic α2-AR consists of three subtypes which are conserved across mammalian species, identified as the α2A/D, α2B, and α2C-AR-subtypes; the α2A/D designation refers to a small difference in amino acid sequence in rodents (α2D) as opposed to that in humans and rabbits (α2A) (50, 51). The rodent α2D-AR, however, is presumed to reflect the same physiological processes and pharmacological outcomes as the α2A-AR, and studies on this receptor in rodents is, therefore, reported as findings for the α2A-AR. The α2-AR subtypes have dissimilar tissue distribution patterns, along with distinct physiological and pharmacological profiles (51, 52). While all three receptors are present in the CNS, the α2B receptor is mainly expressed in the thalamus and does not seem to contribute to CNS auto- and heteroreceptor function (53). The α2A-ARs and α2C-ARs, on the other hand, are the primary α2-ARs modulating neurotransmission in the CNS (33, 53, 54), with the α2C-AR recognized to play a very distinct and specific role in memory, cognition, and mood disorders in a manner different to that of the α2A-AR. These separate effects will become evident in this review, and are summarized in Table 1.
Although 90% of α2-ARs in the CNS are contributed by the α2A-AR, the expression of the α2C-AR is more discrete, constituting approximately 10% of the total (26). Nevertheless, the α2C-AR seems to play a very important role in neurotransmission and potentially in the dysregulation observed in neuropsychiatric illness. Thus α2C-ARs densely populate the ventral and dorsal striatum and the hippocampus in humans (27, 51, 55), monkeys, and rodents (56). Dense population in the olfactory tubercle is also evident, while more subtle cortical expression is also evident (27, 28). The cerebellum is devoid of these receptors. Importantly, these same brain areas are populated by the α2A-AR, among others (27, 57, 58). The specific distribution pattern for the α2C-AR asserts its role in illnesses involving hippocampal and striatal dysfunction, such as schizophrenia and MDD, and in conditions characterized by cognitive deficits and cognitive decline involving these cortico-limbic structures (e.g., Alzheimer’s disease) (27, 59–61).
The distribution of α2C-ARs in human, monkey, and rodent brains are analogous (55, 56, 59, 62), implying that neuropharmacological data from transgenic mouse models and from rodent animal models may be relevant for humans also. Due to the paucity of sufficiently subtype-selective ligands, of which only a few have become available for preclinical investigation during the last decade (17–19, 63), transgenic mouse models have predominantly been used in earlier work to shed light on the physiology and pharmacology of the different α2-AR subtypes. Transgenic mouse models employ targeted genetic deletion or overexpression of the α2A-AR and/or α2C-AR to examine consequence of loss or gain of receptor function, respectively (16). Findings from these transgenic mouse models have suggested distinct and often seemingly opposing CNS roles for the α2A-AR and α2C-AR, with the implication that non-selective α2-AR modulation might potentially negate beneficial effects which could be attained by subtype-selective targeting.
Studies in genetically modified mouse models predicting antipsychotic-, antidepressant-, and pro-cognitive-like effects has brought to light an important role for the α2C-AR, as illustrated by a modulation of behavior and neurotransmission akin to that seen in neuropsychiatric disorders like MDD, schizophrenia, and their associated cognitive deficits (16, 40, 43, 64–67). However, transgenic mouse studies may suffer from the unknown contribution by physiological compensatory changes that take place as a consequence of lifelong absence or overexpression of α2-ARs (17). For example, Sallinen et al. (43) demonstrated deficient sensorimotor gating (see Cognitive Deficits Associated With MDD and Schizophrenia) in α2C-KO mice, suggesting that α2C-AR antagonism may induce effects likened to psychotomimetic agents such as phencyclidine (PCP). This contradicts recent findings described in the social isolation rearing (SIR) and N-methyl-d-aspartate (NMDA)-antagonist models of schizophrenia where selective α2C-AR antagonists, improved sensorimotor gating deficits (18, 20). This type of anomaly underscores the necessity to verify results obtained using transgenic mouse models with studies employing selective α2C-AR ligands in more naturalistic animal models with good validity for the chosen human disorder.
The next section discusses findings regarding the role of the α2C-AR as auto- and heteroreceptor in regulating neurotransmitters implicated in depressive and psychotic disorders. The findings from early studies in transgenic mouse models and studies using moderately selective α2-AR subtype ligands are reported and are aligned with new evidence using novel highly subtype-selective ligands, where available.
Despite a number of new theories that have been put forward to explain the underlying biology and development of mood and psychotic disorders, targeting monoaminergic transmission as a construct toward understanding and treating these disorders remains a relevant subject of investigation [reviewed in Ref. (68)]. The latter review emphasizes that while oxidative stress, neuroinflammation and neuroplastic/degenerative events are implicated in these disorders, selectively and appropriately targeting monoaminergic processes remains a core construct in novel antidepressant and antipsychotic drug development. The α2C-AR is associated with various effects on monoamine turnover. When treated with the subtype non-selective α2-AR agonist, dexmedetomidine, agonist-induced decreases in monoamine levels were absent in α2C-OE mice, while concentrations of DA, NA, and serotonin (5-HT) were shown to be increased in the brains of α2C-KO mice (67). Deactivation of α2C-ARs might thus facilitate increased CNS monoamine levels, which could be of benefit in disorders where monoamine dysfunction is apparent. However, α2C-heteroreceptors modulate other neurotransmitters implicated in the pathophysiology of these disorders, such as γ-aminobutyric acid (GABA), glutamate, and acetylcholine, as will be discussed.
The α2A-AR and α2C-AR are the main autoreceptors involved in presynaptic feedback inhibition of NA, with the α2B-AR making no significant contribution to NA feedback inhibition (14). However, the potency and affinity of NA at the α2C-AR is higher than that for the α2A-AR (14, 29, 69), and evidence from peripheral and CNS tissue demonstrates that the α2C-AR would inhibit NA release at low [10–100 nM, adapted from Ref. (14)] endogenous concentrations of NA as opposed to high [0.1 –10 μM, adapted from Ref. (14)] concentrations for the α2A-AR (14, 26). Deactivation kinetics also differs for the α2A-AR and α2C-AR, with the α2C-AR displaying much slower deactivation upon removal of NA than the α2A-AR (29). Despite their more modest presentation in the CNS, α2C-ARs will, therefore, have distinct effects on a number of important neurotransmitters (see below), while its effects on NA’ergic transmission cannot be underestimated. Along with the α2A-AR, α2C-ARs are involved in the presynaptic negative feedback loop on NA release in the cortex, although α2C-AR-mediated presynaptic inhibition occurs more slowly than that mediated by α2A-ARs (26). Figure 1 depicts this proposed differential regulation on NA feedback and receptor pharmacodynamics mediated by α2A-ARs and α2C-ARs. Furthermore, the α2C-AR produces a limited inhibition of NA release (maximum 20–30% in hippocampal tissue) in contrast to the α2A-AR (26), which would suggest that from a therapeutic perspective, α2C-AR modulation would provide a more subtle and targeted effect on NA release, while limited effects on NA release could potentially dampen the potential for cardiovascular side effects, which are a significant concern with α2A-AR antagonism (26). Ordway and co-workers demonstrated that the density of α2C-AR binding sites increases 3 weeks after the destruction of NA terminals in the rodent cerebral cortex, which suggests that α2C-AR density is regulated by the synaptic availability of NA. In contrast, altered α2A-AR density was not observed under the same conditions (30). This effect of synaptic availability on α2C-AR expression might imply a unique role for the α2C-AR in disorders characterized by noradrenergic dysregulation, such as MDD and schizophrenia.
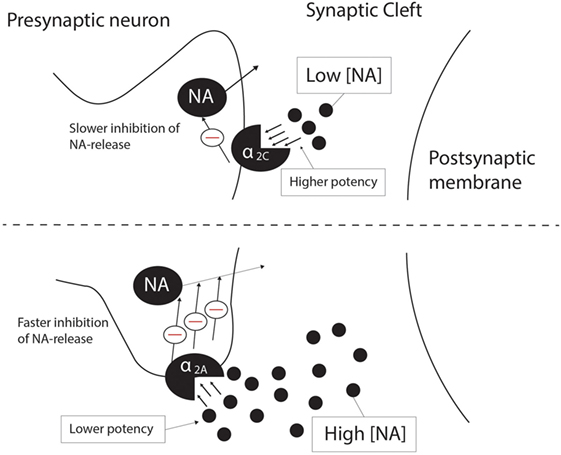
Figure 1. Differential presynaptic inhibition of NA release by the α2C-AR (top panel) and the α2A-AR (bottom panel). At low endogenous NA concentrations (10–100 nM), the α2C-AR is responsible for inhibition of NA release, while the α2A-AR inhibits NA release at high endogenous NA concentrations (0.1–10 μM). α2C-AR-mediated inhibition of NA release is a slower process than that of α2A-AR-mediated inhibition, although the potency and affinity of NA is higher at the α2C-AR than at the α2A-AR. See text for more detail. NA, noradrenaline; Θ, inhibition.
The α2C-AR has also been implicated in α2-autoreceptor-mediated modulation of hippocampal and cortical DA and NA synthesis via feedback inhibition on tyrosine hydroxylase, which converts tyrosine to the DA precursor 3,4-dihydroxyphenylalanine (DOPA) (31). These authors used early subtype-specific antagonists and agonists to measure levels of DOPA and NA in rodent hippocampus and cerebral cortex, with α2B/C-AR antagonists increasing synthesis of DOPA and α2B/C-AR agonists decreasing its synthesis. Although the ligands used in this study were α2B/C-AR specific ligands, the expression of α2B-ARs is limited to the hypothalamus and does not seem to contribute to auto- and heteroreceptor function in the CNS (53). This study also reported that α2A-AR specific antagonism and agonism were devoid of effects on DOPA. However, a limitation of this study is that the subtype-specific ligands used also present with some antagonist activity at 5-HT1A receptors (32). α2C-AR selective antagonism could, however, play a role in increasing DA and NA levels and thus be of benefit in the treatment of neuropsychiatric illness. Nevertheless, these findings need to be confirmed using novel, highly subtype-selective α2-AR ligands.
The high expression of α2C-ARs in the striatum allows it to modulate presynaptic DA release and DA-mediated behaviors (26). Of particular interest is that Zhang and co-workers (64) provided early evidence for the ability of DA to function as an activating ligand on striatal α2C-ARs, while Sallinen and co-workers (18) used a novel α2C-AR selective antagonist (ORM-10921) to show increased in vitro α2C-AR potency and selectivity ratios in the presence of DA as agonist (Figure 2). These authors also reported that ORM-10921 increases extracellular DA levels in the rodent PFC. In support of the correlation between DA activity and α2C-AR activity, early studies indicated changes in brain DA metabolism in α2C-KO and α2C-OE mice (67) (Figure 3). α2C-OE mice show higher levels of the DA metabolite homovanillic acid (HVA) in the frontal cortex but not in the striatum compared to wild-type controls, whereas α2C-KO animals showed lower HVA concentrations in the striatum (67), although not in the frontal cortex (Figure 3). These findings suggest decreased striatal but not frontal cortical DA turnover in response to α2C-AR deactivation and increased cortical DA turnover in response to α2C-AR stimulation. Therefore, an important relationship exists between DA and the α2C-AR. The therapeutic potential of this can be realized in the targeting of α2C-ARs in disorders characterized by mesolimbic-cortical DA imbalance, such as schizophrenia or as demonstrated in SIR rats (20).
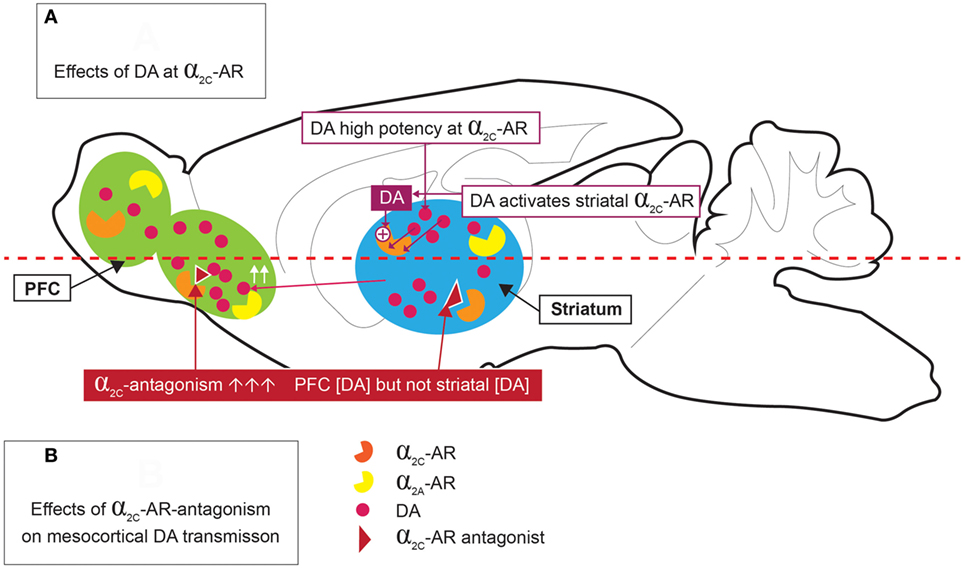
Figure 2. Dopamine (DA) stimulation of α2C-ARs [(A) top panel], and effects of α2C-AR-antagonism on mesocortical DA [(B) bottom panel]. DA is a high potency agonist at the α2C-AR, where it may have significant implications for DA release in the striatum and prefrontal cortex (PFC). α2C-AR antagonism increases PFC DA levels, but not striatal DA levels.
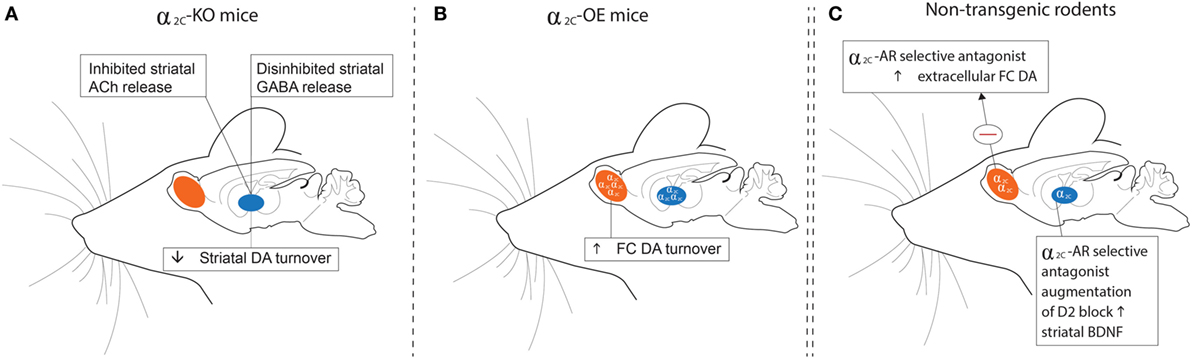
Figure 3. Schematic outline of findings relating to dopaminergic, GABAergic, and cholinergic transmission in the striata and frontal cortices of α2C-KO (left panel) and α2C-OE (center panel) mice and in non-transgenic rodents (right panel) treated with a selective α2C-AR antagonist. (A) HVA levels are decreased in the striata of α2C-KO mice, while α2-AR agonist-induced inhibition of striatal GABA release is disinhibited in α2C-KO mice. Striatal ACh release may be inhibited in α2C-KO mice, indicating a role for the α2C-AR in mediating striatal acetylcholine release. (B) HVA concentrations are increased in the FC of α2C-OE mice. (C) Microdialysis assays show that treatment with the α2C-AR selective antagonist, ORM-10921, increases extracellular DA levels in the frontal cortex of Han-Wistar rats, while augmentation of a D2 antagonist with ORM-10921 increases brain-derived neurotrophic factor (BDNF) in striatal brain tissue of SIR rats. Further support that extends the relevance of these findings to mood/psychosis, and referred to elsewhere in the text, include reduced plasma corticosterone and antidepressant behaviors (A); elevated plasma corticosterone and depressive behaviors (B); and increased sensorimotor gating, improved cognition, and antipsychotic-like behaviors (C). HVA, homovanillic acid; GABA, gamma-aminobutyric acid; KO, receptor knockout; OE, receptor overexpression; DA, dopamine; Ach, Acetylcholine; FC, frontal cortical; SIR, social isolation reared; Θ, inhibition.
α2C-ARs also modify d-amphetamine-induced hyperlocomotion. Here d-amphetamine administration is associated with increased DA and NA release in the caudate nucleus and nucleus accumbens of the dorsal and ventral striatum, respectively, as well as in the PFC, together with co-presenting hyperactive behavior (70–72). Hyperlocomotion was further increased in α2C-KO mice following d-amphetamine administration, while d-amphetamine-induced hyperlocomotion was attenuated in α2C-OE mice (66). Subsequent studies with methylphenidate, a drug which also increases DA release and blocks DA and NA reuptake, showed increased response rates in a cognitive task sensitive to alterations in striatal DA levels in α2C-KO mice (73). The effects of drugs that increase synaptic DA could, therefore, be enhanced by antagonism of the α2C-AR, further emphasizing the role of α2C-ARs in regulating DA release and metabolism.
Less evidence is available to delineate the role of the α2C-AR on serotonergic function. The hippocampal and cortical synthesis of the serotonin (5-HT) precursor, 5-hydroxytryptophan (5-HTP), via the rate-limiting enzyme tryptophan hydroxylase, seems to be dependent on both α2A-ARs and α2C-ARs in the rodent, with α2A-ARs emerging as the main α2-AR modulating 5-HT synthesis (31). Non-selective α2-AR agonism decreases 5-HTP levels in rodent hippocampus and cerebral cortex, while an increase in cortical 5-HTP levels seems to be largely induced by α2A-specific antagonism, with a α2B/C-AR antagonist producing an increase in 5-HTP levels (markedly less than that by a α2A-AR antagonist). These effects were not mirrored in the hippocampus, although α2B/C-AR specific antagonism decreased hippocampal 5-HTP levels in this brain region (31). Similarly, α2-AR-agonist-induced inhibition of 5-HT release is dependent on both α2A-ARs and α2C-ARs, although the α2C-AR exerts a more subtle effect on 5-HT release (33). These authors demonstrated that α2C-KO mice present with lower disinhibition of agonist-induced 5-HT release in hippocampal and occipito-parietal cortex slices compared to α2A-KO mice. The α2A-AR is, therefore, the main α2-AR regulating 5-HT release and possibly 5-HT synthesis. Nevertheless, selective antagonism of the α2C-AR could result in meaningful increases in 5-HT release and region-specific 5-HT synthesis (e.g., provoking serotonergic behaviours in Flinders Sensitive Line (FSL) rats (21)), which may be of importance in various neuropsychiatric illnesses characterized by altered serotonergic neurotransmission, such as obsessive compulsive disorder, MDD, and schizophrenia. Confirmation of these findings using highly selective α2C-AR subtype ligands and in appropriate animal models is, therefore, warranted (e.g., FSL rats; 21).
Apart from effects on the synthesis and release of monoamines, the α2C-AR is an important mediator of striatal, but not hippocampal GABA release (65) via heteroreceptor actions. While α2C-ARs and α2A-ARs are located on different striatal neurons, almost all GABAergic projection neurons in the striatum contains α2C-ARs (60), which project to the globus pallidus and substantia nigra (74). Inhibition of striatal GABA release by an α2-AR antagonist (RX821002) is completely blocked in α2C-KO mice, while enhancement of striatal GABA release by an α2-AR agonist is maintained in these mice, suggesting that inhibition of striatal GABA release is strongly mediated by the α2C-AR (65). Striatal GABA’ergic transmission and response to α2-AC modulation is depicted in Figure 3. This response was not found with respect to hippocampal GABA release (65). These findings could suggest that selective blockade of the α2C-AR may mediate disinhibited GABA release in brain regions with dense dopaminergic innervation and low noradrenergic innervation (3). Considering the presence of α2C-ARs in the striatum (particularly the reward centers), and the role of GABAergic transmission in mania and the action of mood stabilizers (75), selective α2C-AR antagonism could be of value in disorders like schizophrenia in which deficient GABAergic transmission may play a pathophysiological role (76).
Although it is known that α2A-AR modulate glutamate release via heteroreceptor-mediated cross-talk at glutamatergic neurons (77, 78), very little information is available on the specific role of the α2C-AR on central glutamatergic neurotransmission. Additional studies delineating the role of the α2C-AR on glutamatergic neurotransmission is warranted. Non-selective α2-AR antagonism per se does not seem to be beneficial in reversing NMDA-antagonist-induced cognitive impairment in rodent models (79), while non-selective α2-AR agonism may ameliorate these impairments (80–82). Contrasting the aforementioned findings, α2C-AR selective antagonists JP-1302, ORM-10921 and ORM-12741 reverse cognitive and social dysfunction in NMDA-antagonist-induced animal models of neuropsychiatric illness (17–19), indicating a beneficial role of selective α2C-AR antagonism (and not agonism) in attenuating symptoms induced by hypoglutamatergic states, although the mechanism is uncertain.
Disturbances in glutamate are well described in MDD and schizophrenia, while glutamatergic transmission represents an important target in pharmacological management of these disorders (68). Non-selective activation of α2 heteroreceptors on glutamatergic neurons by NA reduces glutamate release in various brain areas implicated in MDD and schizophrenia, including the frontal cortex, ventral tegmental area, hippocampus, and nucleus accumbens (77, 83). Moreover, the treatment arsenal for both MDD and schizophrenia include drugs that are α2-AR antagonists that would thus facilitate disinhibition of glutamate release. In support of this notion, the addition of a non-selective α2-AR-antagonist to a D2-blocker increases frontal cortical glutamatergic neurotransmission in rodents to a similar extent as the atypical antipsychotic clozapine, while at the same time improving cognitive and negative symptoms (3, 7, 84). Notably, clozapine has a threefold to fourfold higher α2C over α2A selectivity ratio and one of the highest α2C over D2 selectivity ratios compared to other antipsychotics. The novel antipsychotic asenapine also presents with increased affinity for the α2C-AR and has good efficacy in treating both positive and negative symptoms of schizophrenia (4). Like that observed with clozapine and following the combination of a α2-AR lytic with a DA antagonist (7, 84), asenapine enhances frontal cortical glutamate transmission via DA activation of D1 receptors (85). Considering the above described effects of α2-lytic activity on prefrontal cortical glutamatergic transmission (84), measuring frontal cortical NMDA currents in NMDA-antagonist model of schizophrenia might elucidate the effects whereby α2C-AR selective antagonists improve NMDA-induced behavioral deficits.
Thus, the above findings suggest that α2C-AR antagonism allows the regulation of cortical glutamatergic transmission, which may underscore a therapeutic option in schizophrenia and cognitive dysfunction in particular. The involvement of α2C-ARs in the inhibition of striatal GABA release as mentioned above (65), could also indicate an indirect role of the α2C-AR in glutamate release, since glutamate release is also tonically regulated by GABAergic interneurons (86).
Dysfunctional central cholinergic transmission has been implicated in the underlying pathophysiology of mood disorders, cognitive dysfunction, and schizophrenia [reviewed in Ref. (87)], while various drugs target the cholinergic system in an attempt at improving the above symptoms (88–90). Deficits in cholinergic transmission are also central to cognitive and memory dysfunction evident in Alzheimer’s disease (91). α2-adrenergic heteroreceptors, as well as D2 receptors, inhibit the release of acetylcholine (1). Similarly, the α2C-AR might be involved in the presynaptic regulation of cholinergic transmission. Since acetylcholine inhibits GABA release (92), Zhang and Ordway (65) have posited that α2C-AR effects on striatal GABA release (described above) might be attributed to the location of α2C-ARs on striatal cholinergic neurons. These authors have also reported that the α2C-AR mediates inhibition of striatal adenylyl cyclase and acetylcholine release, while these effects might be related to tonic activation of the α2C-AR by DA (64, 65). A selective α2C-AR antagonist might thus disinhibit striatal acetylcholine release that in turn may decrease extracellular striatal DA (87) (Figure 3). The findings of Zhang and Ordway (65) might thus be applicable to a neuropsychiatric disorder characterized by striatal dopaminergic over-activity, such as schizophrenia. A complex interplay of cortico-striatal cholinergic, GABAergic, and glutamatergic transmission has been described in the pathophysiology of schizophrenia (87), along with cholinergic regulation of dopaminergic and serotoninergic transmission and vice versa. However, more evidence in this regard using α2C-AR selective ligands is required to enable more definitive conclusions regarding the interplay of the α2C-AR, the cholinergic system and the effect of this interplay in neuropsychiatric disorders. Importantly though, the selective α2C-AR antagonist, ORM-12741, has demonstrated favorable effects on episodic memory in patients with Alzheimer’s disease (25), thus providing proof of concept regarding targeting of the α2C-AR in disorders of cognition, possibly via beneficial effects on cholinergic neurotransmission.
The α2C-AR thus seems to play a distinct role in monoaminergic, GABAergic, glutamatergic, and possibly cholinergic neurotransmission, making it a promising target in several neuropsychiatric illnesses characterized by dysregulation in the aforementioned pathways, in particular MDD, schizophrenia, and conditions associated with cognitive decline. The potential therapeutic role of the α2C-AR in these conditions, including an overview of evidence implicating its involvement in associated cognitive processes, will now be presented.
A genetic polymorphism of the α2C-AR has been associated with emotional dysfunction in MDD (22). The α2C-AR is densely expressed in the hippocampus, an area that is prominent in the pathophysiology of MDD (93). MDD is thought to be characterized, at least in some patients, by deficits in monoamine activity and diminished inhibitory neural control of the hippocampus and PFC over the hypothalamic–adrenal–pituitary axis (HPA-axis), resulting in HPA-axis over-activity with reduced negative feedback and hypercortisolaemia (94). Additionally sleep alterations, deficient neurotrophic signaling and the effects of chronic stress on neurotrophic factors and hippocampal atrophy has been hypothesized to underlie the complex pathophysiology of the disorder (95, 96). Aside from limbic function, the hippocampus plays an important role in learning and memory, and hippocampal atrophy could account for the cognitive deficits that accompany MDD (93).
Antidepressants generally increase the levels of NA, 5-HT and DA to varying degrees depending on the class of antidepressant (97). However, about 40% of patients do not respond to the most commonly used conventional antidepressants (98, 99). Considering that α2C-ARs are densely expressed in the hippocampus, this AR subtype might be a potential target to address hippocampal-related disturbances in MDD. α2-AR dysregulation in depressive disorders is widely described in the literature [Ref. (46) for review], with increased α2-AR density found in platelets and in post-mortem brain tissue of depressed suicide completers in the locus coeruleus, temporal and frontal cortex, hippocampus and hypothalamus (100–103). Moreover, receptor upregulation has been specifically associated with the α2A-AR subtype in depressed states (104–106). The role of the α2-AR in the action of antidepressants is also fairly well described, of particular relevance being the α2-AR antagonist antidepressants, mirtazapine and mianserin (107, 108). Indeed, α2-AR downregulation is induced by tricyclic antidepressants (TCAs) and mirtazapine in rodents and depressed humans (brain and platelets), although regional differences in α2-AR downregulation have been noted in the CNS [reviewed in Ref. (46)].
The rodent forced swim test (FST) is a well-described predictive model for antidepressant drug screening (109, 110). In this test, rodents are exposed to inescapable swim stress where the adoption of an immobile posture during re-exposure is thought to reflect failure in persistent escape-directed behavior, purported to model certain behavioral aspects of MDD such as the psychological feeling of “entrapment” and the replacement of active coping strategies with passivity (109, 111, 112), also resembling avolition and despair noted in MDD. Specifically, an increase in immobility time is considered to reflect the aforementioned depressive-like manifestations. The majority of antidepressants reduce immobility time in the FST (109).
The α2-AR has been implicated in mediating the antidepressant (or anti-immobility) effects of TCAs in the FST, while activation of the α2A-AR subtype seems to be essential in this regard (41, 46). Interestingly, the α2C-AR plays an opposite role in regulating antidepressant effects in the FST. Early studies in α2C-OE models in mice have suggested that α2C-AR activation increases depressive behaviour in the FST, with α2C-OE mice displaying increased immobility compared to wild-type-controls (40) (Figure 3B), an effect not attributed to altered locomotor activity (67). On the other hand, α2C-KO mice demonstrate an antidepressive phenotype (40) (Figure 3A). These findings might explain why relatively non-selective α2-AR agonists (113–115) and certain non-selective α2-AR antagonists have both shown antidepressant-like effects in the FST. Recently these findings have been confirmed in rodents using subtype-selective α2C-AR antagonists. Acute administration of highly subtype-selective α2C-AR antagonists, JP-1302 (17), ORM-10921 (18, 21) and ORM-12741 (19) to Sprague Dawley and Han-Wistar rats was found to decrease immobility in the FST (see Table 2), providing evidence that selective α2C-AR antagonism harbors therapeutic antidepressant effects. Although the aforementioned findings were predominantly from acute studies, we recently reported that chronic ORM-10921 reduced FST immobility time in the FSL rat, a genetic rodent model of MDD (21). Moreover, these effects were not seen with the non-selective α2-AR antagonist idazoxan (21). These findings constitute the first findings for an antidepressant-like effect of an α2C-AR antagonist within a translational and pathological construct-driven approach (16). The beneficial effect of α2A-AR agonism on immobility in the FST as mentioned earlier and the increased immobility of α2C-OE mice observed in this test emphasizes that both the absence/minimization of α2A-AR antagonism and the presence of α2C-AR antagonism might be required for antidepressant-like effects. Earlier, we discussed how α2A-AR antagonism bolsters 5-HT transmission (33). Various studies have supported a therapeutic advantage for augmenting conventional antidepressants with α2-AR antagonists. Dhir and Kulkarni demonstrated potentiated anti-immobility effects in the FST when fluoxetine and venlafaxine are augmented with yohimbine (9). This effect is mirrored in the clinic, where the addition of yohimbine to selective serotonin reuptake inhibitor (SSRI) treatment hastens antidepressant response and increases the number of responders compared to SSRI treatment alone (116). Enhanced clinical response to SSRI’s, venlafaxine, and bupropion is also evident following augmentation with the α2-AR antagonist antidepressant mirtazapine, showing an early-onset of action (107, 117) as well as an almost doubling of the remission rate (118, 119). Clearly there is strong argument for adding an α2-AR antagonist to conventional antidepressant therapy.
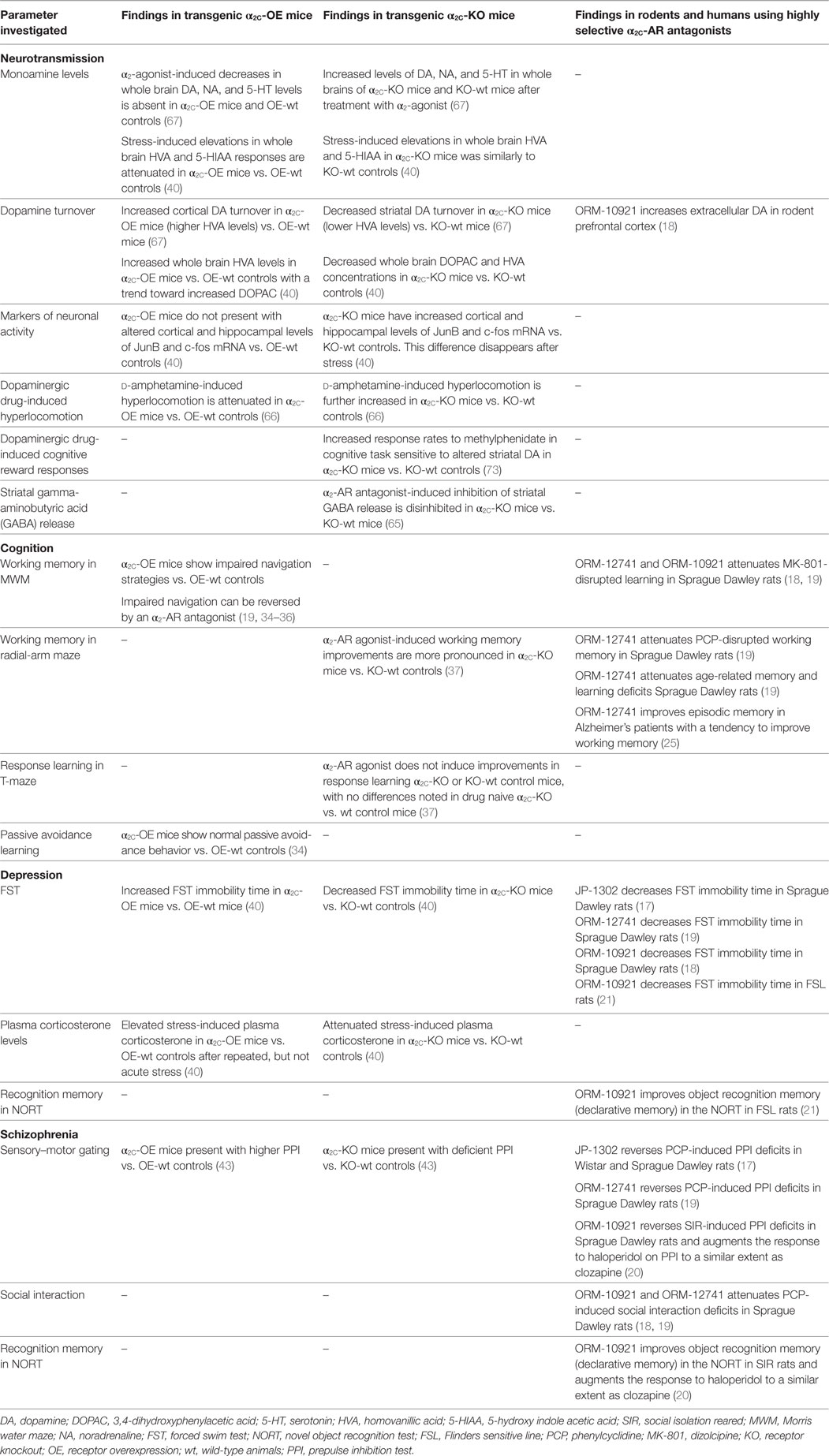
Table 2. Neurochemical and behavioral findings in transgenic α2C-OE or α2C-KO mice, and data from rodent and human studies employing highly selective α2C-AR antagonists.
The Novel Object Recognition Test (NORT) (see Cognitive Deficits Associated With MDD and Schizophrenia) measures recognition memory and is reliant on hippocampal function, while both this cognitive parameter (120, 121) and hippocampal function has been shown to be deficient in patients with MDD (93). Recently, an important role for the α2C-AR in this test has been demonstrated in the FSL rat, using the selective α2C-AR antagonist ORM-10921 in a chronic treatment paradigm (21). This study found that selective α2C-AR antagonism reversed deficits in novel object recognition memory in FSL rats, constituting the first findings for a pro-cognitive effect of a selective α2C-AR antagonist using an illness-specific construct-driven translational model of MDD.
Altered circadian rhythm is a well-recognized biomarker of MDD (68), with HPA-axis dysregulation and hypercortisolaemia underlying the pathophysiology of the disorder (94). Since stress and MDD are causally linked, stress-induced increases in glucocorticoids have been suggested to mediate hippocampal atrophy and neurodegeneration evident in depressed individuals (93, 122). This incapacitation of the hippocampus leads to impaired cognitive function as well as a perpetuation of the stress response, the latter due to an inability of the hippocampus to exert top-down control over the HPA-axis (122). Long-term exposure to elevated cortisol levels induces regional upregulation of α2-ARs (123), which in turn could result in further decreased NA levels. In this regard, the α2 antagonist and antidepressant mirtazapine has been associated with amelioration of HPA-axis hyperactivity in depressed patients (124, 125), albeit not necessarily related to clinical improvement. Interestingly, this amelioration of HPA-axis hyperactivity is not mirrored in rodents (126). In healthy volunteers, the acute administration of the α2-AR antagonist idazoxan has been associated with an attenuated normal diurnal fall in plasma cortisol, although dissipated following chronic treatment (127). Earlier studies on the other hand have shown that depressed patients exhibited much greater cortisol responses to yohimbine than controls (128). The α2C-KO mouse demonstrates attenuated plasma corticosterone elevations vs. wild-type controls following different stressors, while α2C-OE mice show more intense corticosterone responses compared to α2C-KO (40) (Figures 3A,B). Interestingly, non-selective α2-AR antagonism seems to elevate plasma corticosterone levels and to potentiate corticosterone responses to restraint stress in rodents (129). More selective α2C-AR antagonism might, therefore, elicit beneficial effects on HPA-axis functioning in depressive states. Previous studies have shown that both inhibition of corticosterone synthesis and injection of glucocorticoid receptor antisense oligonucleotides into the dentate gyrus of the hippocampus decreases immobility in the FST (130, 131). That the α2C-AR is the only α2-AR subtype expressed in this region in mice (67), together with the effects of α2C-AR modulation on corticosterone levels and FST immobility, consolidates a valuable role for α2C-AR antagonism in the treatment of MDD. Therefore, hypercortisolism in MDD may underscore a central dysfunctional adrenocortical feedback mechanism, with α2-ARs, and indeed the α2C-AR subtype specifically, being important in regulating glucocorticoid responses.
Associations between genetic polymorphism of the α2C-AR and certain aspects of psychotic disorders have been reported (24). Furthermore, α2C-ARs are most densely expressed in the striatum (132), where they are thought to play an inhibitory role (133). This dense expression has distinct importance when striatal dysfunction in schizophrenia is considered, especially its intricate connection to frontal cortical cognitive deficits (134). The α2C-AR, therefore, represents a potentially beneficial pharmacological approach to modulate striatal deficits in schizophrenia and possibly other psychotic disorders. The PFC, striatum and hippocampus are implicated in schizophrenia, where noradrenergic and dopaminergic terminals presenting with α2C auto and heteroreceptors are well-represented (27, 59). Despite the prominence of the DA hypothesis of schizophrenia, a hypothesis implicating noradrenergic dysfunction also has significant support in the literature (135).
The DA paradox is well described in schizophrenia (136), with mesolimbic hyperdopaminergic and mesocortical hypodopaminergic states being postulated. Excessive striatal DA is linked to positive symptoms, while cognitive dysfunction is linked to deficits in cortical dopaminergic function (137). In Section “Role of the α2C-AR in Regulating Key Neurotransmitters,” we discussed findings that suggest decreased striatal but not frontal cortical DA turnover in response to α2C-AR deletion (Figures 3A,B), while increased cortical DA turnover has been noted in response to α2C-AR overexpression (67). These early findings suggest a positive role for α2C-AR antagonism in regulating mesolimbic-cortical dopaminergic imbalances, which may have therapeutic value in schizophrenia. GABAergic and glutamatergic deficits are also implicated in schizophrenia disease pathology, where loss of GABAergic output onto secondary glutamatergic cortical neurons required for tonic control over subcortical dopaminergic neurons, results in increased mesolimbic dopaminergic firing (increased striatal DA release) and consequently the presentation of psychotic symptoms (86). As discussed earlier, the α2C-AR strongly mediates striatal GABA release, while α2C-AR deletion seems to disinhibit α2-AR antagonist-induced inhibition of GABA release (65). Here, α2C-AR subtype-selective antagonism might present with more beneficial effects on striatal GABA release than non-selective α2-AR antagonism when applied as pharmacological treatment of schizophrenia.
The atypicality of antipsychotic drugs primarily reflects their reduced risk of extra-pyramidal side effects and to some extent improved efficacy against negative and cognitive symptoms of schizophrenia (138), over and above their efficacy against positive symptoms. Atypicality has, apart from actions at serotonergic receptors, been proposed to revolve around α-AR modulation, with α1 and α2-AR antagonism suggested to contribute to stabilization of dysregulated dopaminergic activity (139). Indeed, in a thorough comparative study employing human receptor binding data, Shahid and colleagues (4) have shown that a number of atypical antipsychotics (clozapine, quetiapine, asenapine, risperidone, ziprasidone) possess significant α2-AR antagonist properties. Furthermore, quetiapine and in particular clozapine showed prominent α2C over D2 as well as α2C over α2A receptor selectivity. A pharmacological profile constituting a higher α2 vs. D2 receptor binding ratio (139), and specifically a higher α2C vs. D2 receptor selectivity ratio (3, 4), has been suggested to mediate the improved efficacy of drugs like clozapine that exhibit lower D2-receptor occupancy. The α2 over D2 receptor subtype selectivity ratios for various antipsychotics as well as the α2C-AR selective antagonist, ORM-10921 (which as described below has shown antipsychotic-like activity in animal models), are depicted in Figures 4A,B. Thus, reduced D2-receptor occupancy might be possible in therapy because of the beneficial effects of additional α2-AR antagonism on dysregulated dopaminergic activity, allowing for improved efficacy with less motor side effects. Support for this hypothesis has been demonstrated in studies employing non-selective α2-AR antagonists (e.g., idazoxan) as augmentation to D2-receptor antagonist antipsychotic treatment (7, 84, 140). While this combination of α2-AR and D2 receptor antagonism presents with improved antipsychotic-like effects in mouse models of schizophrenia, it also resulted in enhanced cortical glutamatergic transmission and increased dopaminergic output in the PFC, with subsequent improvement in cognitive parameters in rats (84). The effects of this augmentation strategy were comparable to that of clozapine. While clozapine requires approximately 45% D2 receptor occupancy compared to >70% required by other D2 receptor antagonists for antipsychotic efficacy (141, 142), the combination of idazoxan with a D2 receptor antagonist exhibited potent antipsychotic effects similar to that of clozapine at similar low D2 receptor occupancy rates (84).
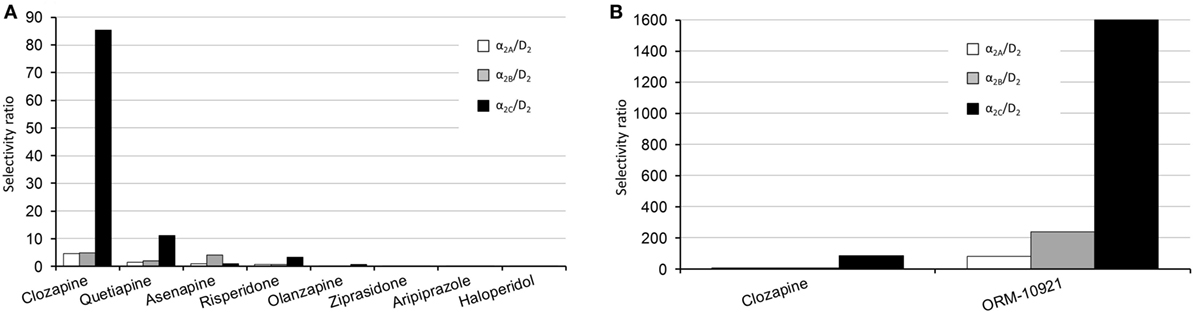
Figure 4. Human α2-AR subtype/D2 selectivity ratios of various antipsychotics, adapted from Ref. (4). (A) Left panel, comparative overview of the subtype selectivities of various antipsychotics. The α2C/D2 receptor selectivity ratios are as follows: clozapine, 85; quetiapine, 11; risperidone, 3.4; asenapine, 1; olanzapine, 0.53; ziprasidone, 0.02; haloperidol, 0.011. (B) Right panel, comparison between the subtype-selective ratios of clozapine and the α2C-AR antagonist ORM-10921, which has shown antipsychotic-like effects in preclinical studies (18, 20). The α2C/D2 receptor selectivity ratio for ORM-10921 is 1,600. Selectivity ratios were determined by dividing the D2 Ki value by the applicable α2 receptor Ki value.
Sensorimotor gating refers to the ability to integrate and process sensorimotor information, deficits of which are suggested to underlie the fragmentation of reality evident in schizophrenia (143). The prepulse inhibition (PPI) of startle test refers to the attenuation of a startle response produced by the presentation of a smaller prepulse, and is used to study the gating of sensorimotor information by the brain (143, 144). A typical example of the PPI test in humans employs the somatosensory eye blink reflex in response to acoustic, tactile (e.g., air puffs) or light stimuli (143, 145, 146). A PPI deficit can be induced in humans and animals by various psychotomimetic drugs, including dopaminergic and antiglutamatergic drugs. Animal models of schizophrenia, such as SIR (147–149) and various transgenic models including mice with altered DA, 5-HT, and glutamate receptor expression (150), present with deficits in PPI. Importantly, antipsychotic drugs normalize disrupted PPI in animals and humans (151–155). While the contribution of non-selective α2-blockade to modulation of PPI has been proposed, the literature is somewhat inconclusive in this regard. In fact, some papers have suggested that antagonism of the α2A-AR does not contribute to enhancement of the PPI (44, 156–158).
Considering the important role for α2-AR antagonism in managing schizophrenia (139), earlier studies in transgenic mouse models have demonstrated that antipsychotic-like effects are subtype dependent. In this regard, α2C-KO mice demonstrated clear PPI deficits compared to wild-type controls, while α2C-OE mice had markedly higher PPI scores than their wild-type controls (43), suggesting that α2C-receptor agonism may induce antipsychotic-like effects. However, this extrapolation from transgenic mouse studies has since been disproven following experiments with selective α2C-AR antagonists. JP-1302, ORM-10921, and ORM-12741 that consistently show improved PPI in Sprague Dawley and Wistar rats in NMDA-antagonist-induced models of schizophrenia (17–19). More recent findings in SIR rats, a putative neurodevelopmental model of schizophrenia (159, 160), corroborate these earlier findings, with ORM-10921 found to significantly improve SIR-associated PPI deficits in a manner comparable to clozapine (20) (Figure 3C). Moreover, ORM-10921 also enhanced the effects of haloperidol on the above-mentioned deficits in PPI (20). Earlier, in Section “Distinct Roles for α2-AR Subtypes,”, we highlighted this discrepancy, emphasizing the need to corroborate findings from transgenic mouse models with studies employing subtype-selective ligands in suitable animal models.
Cognitive deficits in schizophrenia make up some of the core elements of the disorder (161) and are often refractory to treatment (162). These impairments include deficits in working, recognition and spatial memory, cognitive flexibility, learning, and attention (163–165). However, antipsychotic treatments are not always reproducibly effective in reversing these cognitive deficits in animal models (166–170), which in fact reflects the relative lack of efficacy displayed by antipsychotics in treating cognitive impairment in the clinic (164, 165). Recently, the highly selective α2C-AR antagonist ORM-12741 showed improved effects on NMDA-antagonist-induced disruptions in working memory and spatial learning, navigation and memory in rodents (19). NMDA-antagonist models include the administration of the glutamate NMDA-receptor antagonists dizolcilpine (MK-801) or PCP which are known to induce behavioral, cognitive, and neurochemical disruptions in behavior akin to those seen in schizophrenia (171). ORM-12741 attenuates the disruption of learning in the Morris Water Maze (MWM) induced by MK-801, while also improving PCP-induced memory deficits in the 8-arm radial maze (19). Similar findings were reported for the selective α2C-AR antagonist ORM-10921 which attenuates MK-801-induced spatial navigation in the MWM (18), a finding consistent with effects described for atypical (167, 172) but not typical antipsychotics such as haloperidol (173). Additionally, ORM-10921 significantly improved object recognition memory in SIR rats, comparable to the atypical antipsychotic clozapine, while also significantly improving the efficacy of haloperidol in this regard (20) (Figure 3C). Evidence of improved cognition in NMDA-antagonist and neurodevelopmental models of schizophrenia with novel highly selective α2C-AR antagonists, therefore, demonstrates the therapeutic potential of targeting the α2C-AR in treating cognitive deficits associated with schizophrenia.
Another interesting observation concerns the neurotrophic hypothesis of schizophrenia, where reduced brain-derived neurotrophic factor (BDNF) is widely evident in the illness (174, 175), as well as being associated with the above-mentioned cognitive deficits (68). Although chronic treatment with the α2C-AR antagonist, ORM-10921, alone did not significantly reverse lowered BDNF levels in SIR rats on its own, combining haloperidol with ORM-10921 showed a significant increase in BDNF levels that exceeded that of either drug alone (20) (Figure 3C). These preliminary results further support a therapeutic role for α2C-AR antagonism in improving cognitive symptoms in schizophrenia.
Social isolation, decreased social cognition, and impaired social skills form part of the negative symptoms of schizophrenia and are refractory to most antipsychotic treatments (176). The social interaction test measures deficits in social motivation and self-directed behavior in rats and is used to measure predictive validity of antipsychotics in rodent models of schizophrenia (177). Although there are mixed results, generally atypical antipsychotics are more effective than typical antipsychotics at attenuating social deficits in these models (177, 178). In this regard, the α2C-AR antagonists ORM-10921 and ORM-12741 significantly attenuate PCP-induced deficits in social interaction in short-term single-housed and pair-housed rats (18, 19).
Since especially atypical antipsychotics have activity at the α2C-AR, it is important to consider data from functional assays on these compounds using cloned receptors in Chinese hamster ovary cell lines. Kalkman and Loetscher (3) found α2C over α2A receptor selectivity ratios for clozapine, chlorpromazine, risperidone, quetiapine, and iloperidone to be between 3 and 12, indicating that most atypical antipsychotics present with higher α2C-AR antagonist activity than α2A-AR antagonist activity. Additionally, the novel antipsychotics asenapine and lurasidone both present with potent α2C-AR binding affinity (4, 179). α2C over D2 selectivity ratio has, therefore, been suggested to be an important factor in antipsychotic efficacy (3). Clozapine, arguably the most efficacious antipsychotic in treatment refractory schizophrenia (180), presents with an α2C over D2 selectivity ratio of 85 compared to ratios of 0.01–11 for other tested antipsychotics (4) (see Figure 4A). Haloperidol, on the other hand, has the lowest potency at the α2C-AR as well as the lowest α2C over D2 ratio of tested compounds (3, 4), and is not regarded as an atypical antipsychotic. However, bolstering its antipsychotic-like and pro-cognitive effects with a selective α2C-AR-antagonist (20) supports the notion that an increased α2C over D2 ratio will translate to superior antipsychotic effects. Taken together, α2C-AR antagonism is involved in the mechanism of improved sensorimotor gating, cognitive, and social functioning in pharmacological and neurodevelopmental models of schizophrenia. These data are indicative of a therapeutic role for α2C-AR antagonism in the treatment of schizophrenia, and further study with more subtype-selective ligands is encouraged.
Many neuropsychiatric illnesses, including MDD and schizophrenia, present with cognitive deficits and memory impairments (122, 162, 163, 165). The α2C-AR has been shown to be involved in cognitive deficits evident in both non-pathological (34–37) and pathological (20, 21) animal models, with findings implicating a significant role in the treatment of cognitive deficits associated with these disorders. Although α2-AR agonists are associated with improved cognitive processing in humans and animals (38, 181–184) and in the treatment of cognitive decline associated with aging (185), these effects have been shown to be mediated via activation of the α2A-AR (37–39), which is also responsible for sedative and hypotensive effects (51, 186). In contrast, genetic deletion of the α2C-AR subtype, or by extrapolation selective α2C-AR antagonism, has been demonstrated to improve memory and cognition in the MWM, the 8-arm radial maze and the NORT, as discussed below. Furthermore, α2C-AR antagonism has been found to benefit neurotrophins and other biomarkers of neuronal resilience associated with cognition (20).
The MWM is a spatial water navigation task requiring the rodent to learn and remember the location of an escape platform in a water arena in order to locate a hidden (submerged) platform in subsequent trials by using various spatial cues. The escape latency is a measure of spatial working memory. The test is a reliable tool correlating with hippocampal synaptic plasticity as well as intact glutamate NMDA-receptor function (187). In early transgenic mouse studies, α2C-OE mice showed impaired spatial and non-spatial escape strategies and search patterns in the MWM. Administration of an α2-AR antagonist could reverse these impairments to a greater extent in α2C-OE than in wild-type mice, suggesting that α2C-AR antagonism might play a more prominent role than α2A-AR antagonism in brain areas involved in spatial navigation (34–36). Considering the dense expression of the α2C-AR in the hippocampus and striatum and that hippocampal (188) and striatal lesions (189) impair aspects of MWM navigation, α2C-AR selective antagonism may mediate information processing and behavioral adaptation to environmental change. α2C-OE mice display normal passive avoidance learning, suggesting that impaired water maze navigation in α2C-OE mice does not reflect defective stimulus-response learning and that α2C-AR deactivation is, therefore, associated with the processes underlying complex organization of escape behavior (34). This effect of α2C-AR antagonism might partially explain previous findings for pro-cognitive effects of the non-selective α2-AR antagonist, idazoxan, on planning, attention, episodic memory and verbal fluency in patients with frontal lobe dysfunction (11).
The radial-arm maze is a test used to measure reference and working memory in rodents and relies on intact functioning of the prefrontal cortical, hippocampal and striatal interconnections to locate food rewards hidden in various radial-arm target sites (190). Björklund and co-workers (37) demonstrated that the non-selective α2-AR agonist dexmedetomidine improves working memory in the radial-arm maze, and that this improvement is greater in α2C-KO mice, suggesting that the absence of α2C-AR agonism (or simultaneous α2C-AR antagonism) might result in enhanced performance with respect to working memory.
The NORT is a two-trial behavioral measure that relies on the rodent’s innate preference to explore novel objects over familiar objects, thereby enabling measurement of recognition memory (191, 192). The declarative memory processes underlying the NORT relies on the perirhinal cortex and the hippocampal complex (193–195). Uys and colleagues have demonstrated that selective α2C-AR antagonism with ORM-10921 markedly improves recognition memory in pathological animal models of schizophrenia (20) and MDD (21).
The above-mentioned benefits of selective α2C-AR antagonism on cognitive parameters have been corroborated with studies employing highly selective α2C-AR antagonists in animal models of schizophrenia, MDD, and age-related cognitive impairment (18–21), as described in Sections “Behavioural Deficits Associated With MDD” and “Behavioural Deficits Associated With Schizophrenia,” as well as in clinical trials investigating novel therapy for Alzheimer’s disease (25) (see Evidence for Targeting the α2C-AR in Other Neuropsychiatric Disorders).
Brain-derived neurotrophic factor is the most prevalent neurotrophic growth factor in the CNS where it is especially important in regulating synaptic plasticity and various aspects underlying cognitive performance, memory, and mood (196, 197). Acute and chronic stress purportedly have detrimental effects on rodent BDNF expression in the hippocampus, while altered BDNF levels are evident in depressive disorders (68, 198) and in schizophrenia (174, 175). While both antipsychotics and antidepressants alter BDNF levels to varying degrees (199–203), non-selective α2-AR antagonism has also been associated with neurogenesis and increased BDNF levels in the hippocampus (204, 205). Noradrenergic (202, 206), dopaminergic (207), serotonergic (208), and GABA-glutamate (209) interactions are involved in the expression of BDNF. With the α2C-AR acting as a heteroreceptor to modulate the release of many of the aforementioned neurotransmitters, this receptor might play an indirect role in regulating the expression of BDNF. Evidence for the involvement of the α2C-AR in the expression of BDNF has been demonstrated in the SIR animal model of schizophrenia, where SIR rats present with reduced striatal BDNF levels (20). While conventional antidopaminergic antipsychotics are not associated with correction of said reduced BDNF levels (201), a recent study reported that combining haloperidol with the selective α2C-AR antagonist ORM-10921 (but not α2C-AR antagonism per se) increases striatal BDNF levels in these animals, while at the same time improving deficits in cognition and sensorimotor gating (20). This study indicated that not only is augmentation with a α2C-AR antagonist associated with improved BDNF expression but also that this improvement is correlated with improved cognitive performance, thus supporting a role for α2C-AR antagonism in disorders associated with reduced cognitive flexibility and deficits in neurotrophin support.
Brain-derived neurotrophic factor is also important in regulating C-fos and JunB expression, biomarkers of neuronal activity that play an important role in synaptic function (210, 211). Upregulation of c-fos mRNA is induced by noxious stimuli, neurotransmitters, neurotrophins and other growth factors as well as learning and memory processes (212). Jun-B is also involved in the regulation of emotional memory (213). BDNF restores the expression of these transcription factors after neuronal insult (214), reinforcing BDNF’s role in neuroplasticity at gene-transcription level. Interestingly, cortical and hippocampal levels of c-fos and JunB mRNA are increased in α2C-KO mice compared to wild-type controls (40), while this is not the case in α2C-OE mice. Whether this increase is associated with altered BDNF levels in α2C-KO mice has not been investigated. Nevertheless, the increase in neuronal activity in α2C-KO mice is of interest considering the pro-cognitive behavioral characteristics of this transgenic strain and the above-mentioned beneficial effects of α2C-AR antagonists on BDNF expression and cognitive performance.
Thus, antagonism of the α2C-AR might benefit cognitive processes relevant to both MDD and schizophrenia. Since cognitive deficits are core symptoms of both disorders, the α2C-AR related effects on cognition and neuronal markers of plasticity support the therapeutic potential of targeting the α2C-AR in these disorders.
To summarize findings from transgenic mouse models and those gained from treatment with α2C-subtype-selective ligands, Table 2 presents neurochemical and behavioral findings reported in transgenic mice and in various rodent models predicting pro-cognitive-like, antidepressant-like and antipsychotic-like effects as described in Sections “Behavioural Deficits Associated With MDD,” “Behavioural Deficits Associated With Schizophrenia,” and “Cognitive Deficits Associated With MDD and Schizophrenia”. As a GPCR that functions within the PSD, the synaptic actions of the α2C-AR and indeed drugs that target this receptor, might involve regulatory PSD proteins to mediate the aforementioned effects.
The PSD is a specialized matrix located at excitatory postsynaptic terminals, described as a macromolecular complex of several hundreds of proteins that act as a molecular switch for multiple interacting neurotransmitter signaling pathways (215). Such proteins include those containing the PSD-95/disc large/zonula occludens-1 (PDZ) domain, and the membrane-associated guanylyl kinase, all of which comprise three PDZ peptide-binding domains (215). These proteins in turn promote binding to a variety of molecules within the PSD necessary for signal transduction (45). We have earlier noted the importance of the PSD in postsynaptic GPCR signaling. There is significant interest in the role of the PSD network in psychopharmacology and psychotropic drug action, although much of the extant evidence in support of this relates to DA and glutamate-dependent synaptic plasticity (45, 215). Nevertheless, this review has highlighted the importance of heteroreceptor-directed modulation of DA and glutamate signaling by the α2C-AR, while at least one prominent PDZ-domain binding protein, spinophilin, has been associated with the α2-AR (45). Spinophilin regulates α2-AR associated Gαi coupling, membrane localization, endocytosis, receptor desensitization and calcium signaling (216–218). Despite this evidence, however, spinophilin is not yet known to be involved in major neuropsychiatric disorders or to by modulated by main psychopharmacologic treatments (215). Nevertheless, it is perhaps worth discussing how and why a PSD protein such as spinophilin may mediate important pharmacological responses following ligand binding to the α2C-AR.
Although the specific role for PSD proteins in psychiatric illness remains speculative, clinical and preclinical studies have provided evidence for their involvement in aberrant synaptic plasticity [see Ref. (215) for review]. These processes are invariably associated with high-order cognitive alterations, which are essentially the core pathophysiology in a number of psychiatric diseases, including depression and schizophrenia (219–221).
When considering a therapeutic strategy in psychiatric diseases, psychotropic-mediated modulation of PSD molecules may occur either directly or indirectly, the latter as a consequence of drug interaction with their target non-PSD receptors. Currently, there is no known agent that directly targets a PSD protein for therapeutic effect. Since PSD molecules are modulated by antipsychotics and antidepressants (221–226), as well as play a key role in behavioral response (227, 228), they represent putative targets for pharmacological action. Moreover, concurrent administration of antipsychotics and antidepressants may induce synergistic modulation of specific PSD molecules (229–231), which provides at least conceptual support for targeting the PSD to address treatment resistance in mood and psychotic disorders. This is particularly relevant when discussing the α2-AR, since a number of studies have described enhanced efficacy of typical and atypical antipsychotic drugs by adjunctive α2-AR blockade (20, 84, 232). Concerning the α2C-AR, the α2C-AR antagonist, ORM-10921 has been found to bolster the response to haloperidol in social isolation reared rats at both the level of synaptic plasticity (i.e., BDNF) and cognition (i.e., object recognition memory) (20). That the combined response was similar to clozapine emphasizes the benefit of adjunctive α2C-AR antagonism with regard to treatment response. Such data holds promise for application in treatment resistance, and further studies in this regard, but combined with co-assessment of PSD proteins, are warranted.
This review has focused on the therapeutic potential of targeting the α2C-AR subtype in MDD and schizophrenia. However, cognitive dysfunction is common in patients with Alzheimer’s disease, MDD and schizophrenia, while symptoms of the latter two illnesses permeate through to patients suffering from Alzheimer’s disease. Indeed, recent preclinical and preliminary clinical evidence has revealed the promising therapeutic role for the α2C-AR in addressing cognitive decline in Alzheimer’s disease. Co-presentation of cognitive decline in this and other disorders, and the role of the α2C-AR, warrants brief discussion.
ORM-12741 is a novel highly selective α2C-AR antagonist with a 4000-fold selectivity for α2C-AR vs. the α2A/B-AR (19). Age-related memory and learning, as assessed in the rodent 8-arm radial maze (measuring spatial working memory and reference memory), was attenuated by sub-chronic administration of ORM-12741 (19), and more recently confirmed in a phase IIa randomized, double-blind, placebo-controlled clinical study in patients with moderate Alzheimer’s disease (25). Here, ORM-12741 was used as an add-on drug in patients already receiving donepezil, galantamine, rivastigmine or memantine. Significant improvements in episodic memory were observed, as well as a tendency to improve working memory. In addition, ORM-12741 produced significant improvement in perceived levels of distress with respect to symptoms of delusions, agitation and aggression, MDD, anxiety, disinhibition and other behavioral symptoms (25). These findings are not unlike the augmentation data described in preclinical studies with another α2C-AR antagonist, ORM-10921 (18). Moreover, there was a positive trend to lower caretaker distress scores which would also reflect reduced symptom severity and frequency (25). Thus improvements in cognitive performance in Alzheimer’s disease are supported by amelioration of co-presenting behavioral impairments, of which some are reminiscent of those presenting in MDD and schizophrenia.
Although the beneficial role of α2-AR agonism in strengthening prefrontal cortical function and enhancing working memory has been described in ADHD, these affects are associated with postsynaptic stimulation of the α2A-AR subtype (47). Early evidence has, however, also suggested a potential therapeutic role for selective targeting of the α2C-AR subtype in ADHD. A study in coloboma mice, a mouse model of ADHD (233), reported that the α2C-subtype preferring α2-AR antagonist MK912 (~10-fold selectivity over α2A-AR and α2B-AR) ameliorated NA-dependent hyperactivity (234), while α2A-AR and α2B-AR subtype-preferring drugs were ineffective. Considering the pronounced expression of the α2C-ARs in the basal ganglia, the authors suggest that α2C-AR antagonism might be a useful treatment for locomotor-related and hyperactivity functions in coloboma mice and by implication a potential therapeutic target for ADHD. It is conceivable that part of the mode of action of a selective α2C-AR antagonist may involve indirectly facilitating activation of postsynaptic α-AR including α2A-AR as a consequence of increase in synaptic NA. These effects need to be corroborated using subtype-selective ligands with higher selectivity ratios, and subsequent testing on cognition in models of ADHD.
A comment on the role of the α2C-AR in bipolar disorder is also warranted. Bipolar disorder is a mood disorder characterized by mixed symptoms of MDD and mania, with both antidepressants (235) and antipsychotics (236) in combination with mood stabilizers advocated as standard first-line treatment. Quetiapine is an atypical antipsychotic with a favorable α2C/α2A and a fairly high α2C/D2 ratio (3, 4) (see Figure 4A) that has shown marked clinical efficacy in treating mania and MDD in bipolar disorder (236, 237). In the light of evidence provided in the afore going sections, future studies investigating the therapeutic potential of targeting the α2C-AR in bipolar disorder using α2C-AR selective ligands could provide valuable insights.
Finally, given the prominent role of NA in the neurobiology and treatment of anxiety and fear-related manifestations (238), exploratory studies into the use of α2C-AR antagonists in anxiety disorders are also encouraged.
Recent developments and the current state of knowledge support the therapeutic potential of selectively targeting the α2C-AR in the treatment of MDD, schizophrenia and associated cognitive dysfunction. Antidepressant and antipsychotic treatment benefits are likely to include broader/enhanced efficacy (e.g., facilitation of postsynaptic cortical α2A-AR activity) as well as reduced side effects (e.g., liability for cardiovascular effects). There is, however, limited clinical data in this respect and further patient trials are urgently needed. In addition, despite recent progress there are still significant gaps in the knowledge base relating to the function, physiology, and pharmacology of α2C-ARs. Some areas requiring further research include:
• α2C-AR signal transduction pathways and trafficking in brain tissue from normal and disease model animals.
• Assessing treatment response following combined α2C-AR antagonism with a typical/atypical antipsychotic or antidepressant, using an animal model of treatment resistance, e.g., Ref. (239, 240), and combining with co-assessment of PSD proteins.
• Assess the effect of α2C-AR, alone or in combination with an antipsychotic/antidepressant, on the expression of PSD proteins, such as spinophilin, PSD-95, etc.
• α2C-AR receptor regulation; differences in human disease tissue or animal models, and whether existing treatments, e.g., for schizophrenia and MDD, alter α2C-AR density.
• Distribution and cellular localization of α2C-ARs at noradrenergic and non-adrenergic synapses and whether these receptors play an extra-synaptic role.
• Heteroreceptor function and mode of modulation of non-adrenergic neurotransmitter release, particularly in the hippocampus and frontal cortex.
• Insight on putative receptors (e.g., 5-HT1A, D1, AMPA receptors) that may be involved in mediating the in vivo central effects of selective α2C-AR receptor antagonism.
• Contribution toward modulation of stress and inflammation-linked pathways.
• Evaluation in animal models with strong disease construct (e.g., genetic, age, stress) and applying more translationally relevant approaches (e.g., chronic treatment, gender differences, altered circadian rhythms, augmentation strategies).
• Further experimental studies with new imaging tools [e.g., positron emission tomography (PET) ligand ORM-13070] to establish the role of the α2C-AR in the brain of healthy subjects and patients.
• Considering the high comorbidity of anxiety in these illnesses, and that it can significantly affect prognosis and treatment response (241, 242), to study the anxiolytic capabilities of α2C-AR modulators in appropriate models.
An array of tools is now available to facilitate further research. Highly selective α2C-AR subtype ligands, and specifically α2C-AR selective antagonists, have been developed over the past decade. Before that, drugs with marginal selectivity were used to delineate pharmacological effects of the α2-AR subtypes. For example, although BMY7378 is mainly an α1D-AR antagonist, it also presents with a 10-fold selectivity for α2C-ARs vs. α2A-ARs (243). Another example of an antagonist drug with marginal α2C-AR selectivity is MK912, which also displays approximate 10-fold greater selectivity for α2C-ARs vs. α2A-AR and α2B-ARs (57, 58) and has been used to delineate the role of the α2C-AR on hyperactive behavior in a mouse model of ADHD (234).
In 2008, N-{2-[4-(2,3-dihydro-benzo[1,4]dioxin-2-ylmethyl)-[1,4]diazepan-1-yl]-ethyl}-2-phenoxy-nicotinamide was synthesized, and found to display >100-fold selectivity for α2C-AR vs. α2A-AR, with excellent binding affinity and functional activity at the α2C-AR in rats. Although low selectivity vs. α2B-ARs was shown, the α2B-ARs have negligible distribution in the CNS. This compound displayed excellent binding affinity and functional activity for α2C-ARs in rats, with adequate CNS penetration (244). Further animal studies with this promising compound are eagerly awaited.
In 2007, Orion Pharma reported that their novel selective α2C-AR antagonist, JP-1302, presented with a minimum 50-fold selectivity for the α2C-AR with an α2C/2A ratio of 93 (17). However, this compound does not optimally enter the CNS. In 2013 another Orion Pharma compound with improved CNS penetration, ORM-10921, was characterized with an α2C/2A ratio of ~100 in rodents, although this ratio was found to be lower in human cells (~29) (18). Both JP-1302 and ORM-10921 have since been used safely in preclinical studies in rodent models of neuropsychiatric illness and highlighted in this review. On the other hand, the novel α2C-AR antagonist, ORM-12741, has been tested for safety and efficacy in both rodents and humans (19, 25) and presents with a 4000-fold selectivity for the α2C-AR vs. α2A-AR and α2B-AR. This highly selective α2C-AR antagonist is currently in clinical trials for the treatment of symptoms associated with Alzheimer’s disease (25).
An important recent development has been the development of ORM-13070, a selective α2C-AR which is amenable to labeling with 11C and has been successfully used as a α2C-AR PET tracer that readily enters the CNS (245). This compound has a binding affinity selectivity of over 200-fold vs. the α2A-AR, with weak or no activity at more than 100 other potential target sites and receptors, and will be highly valuable for facilitating forward and reverse translation between animal and human studies. An obvious application is determination of target engagement, through conducting receptor occupancy studies for novel drug candidate molecules for preclinical and clinical studies (62, 245, 246). However, it could also be used to gain more precise insight on the relative α2C-AR occupancy for antipsychotic (e.g., clozapine) and antidepressant (e.g., mirtazapine) agents at clinical doses thus enabling a better understanding on the mode of action of these drugs. The tracer could also be of potential value to investigate disease-related changes in receptor density and effects on neurotransmitter activity. The latter aspect has been investigated and in line with evidence that the α2C-AR is sensitive to low synaptic concentrations of NA, [11C]ORM-13070 shows increased CNS binding in response to decreased synaptic NA (132).
On the other side of the spectrum, novel α2C-AR agonists have also been characterized recently. [N-[3,4-dihydro-4-(1H-imidazol-4-ylmethyl)-2H-1,4-benzoxazin-6-yl]-N-ethyl-N′-methylurea] or “Compound A” and a chemically similar “Compound B” were found to be highly selective for the α2C-AR, albeit with poor brain penetration. These compounds are being investigated for effects on peripheral vasoconstriction (245, 247).
With the aim to stimulate further investigation into the value of the α2C-AR in neuropsychiatric disorders, genetic and molecular biology driven approaches will also be critical. Mice overexpressing or lacking the α2C-AR have been generated but have been phenotyped to a limited extent. Further behavioral and biological characterization, for example using -omics type molecular profiling, as well as regionally restricted genetic manipulation using genetic deletion technology in rats, would yield valuable data. The zebrafish is another platform of discovery that may provide a powerful model in which to study developmental and genetic factors that underlie human disease (248). Work in zebrafish has shown that the zebrafish α2-AR subtypes are markedly conserved compared to mammalian α2-AR subtypes with similar pharmacological profiles and functional effects compared to human α2-AR subtypes (249, 250). This model might also be beneficial in future studies when characterizing novel subtype-selective α2-AR ligands.
This review has provided an overview of recent developments and future direction in research investigating the role of the α2C-AR in neuropsychiatric illness and therapy, with specific focus on the effects of α2C-AR antagonism in cognition, MDD, and schizophrenia. Targeting this receptor could present with beneficial therapeutic effects and decreased noradrenergic side effects when used alone or as augmentation strategy in the treatment of these diseases, as well as disorders presenting more specifically with cognitive decline, such as Alzheimer’s disease. The recent advent of clinical grade subtype-selective α2C-AR antagonists has contributed toward delineating the neuropsychopharmacology of this receptor. Studies employing these novel highly selective α2C-AR ligands in putative translationally relevant animal models of psychiatric illness, to inform further experimental medicine evaluation in humans, will be vital in strengthening our understanding of the α2C-AR as a therapeutic target.
MU prepared the first draft of the manuscript, prepared all the figures and tables, as well as managed all subsequent changes and formatting. MS reviewed the manuscript and provided input on the manuscript design and content, as well as on the figures and tables. BH was the study leader and student supervisor to MU, developed the article concept and design, and finalized the manuscript for submission.
MS is an employee of Orion Pharma. No funding was received by Orion Pharma for this, or previous work by these authors. The authors declare that over the past 3 years, BH has participated in advisory boards and received honoraria from Servier®, and has received research funding from Servier® and Lundbeck®. ORM-10921, which was used in recent studies by the authors and cited in this paper was sponsored by Orion Pharma. BH declares that, except for income from the primary employer and research funding from the below-mentioned organizations and agencies, no financial support or compensation has been received from any individual or corporate entity over the past 3 years for research or professional services, and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest. The authors declare no other conflicts of interest.
The authors declare that projects leading up to this work has been funded by the South African Medical Research Council (BH) and the National Research Foundation (BH; grant number 77323). The grant-holder acknowledges that opinions, findings, and conclusions or recommendations expressed in any publication generated by NRF supported research are those of the authors, and that the NRF accepts no liability whatsoever in this regard. These funders have no other role in this study. ORM-10921 was sponsored by Orion Pharma in two studies cited in this manuscript and authored by MU, MS, and BH (20, 21).
ADHD, attention-deficit hyperactivity disorder; AR, adrenoceptor; BDNF, brain-derived neurotrophic factor; cAMP, cyclic adenosine monophosphate; CNS, central nervous system; DOPA, 3,4-dihydroxyphenylalanine; DA, dopamine; FST, forced swim test; FSL, Flinders Sensitive Line; FRL, Flinders resistant line; GABA, gamma-aminobutyric acid; HPA-axis, hypothalamic–pituitary–adrenal axis; HVA, homovanilic acid; KO, knockout; MAPK, mitogen-activated protein kinase; MK-801, dizolcipine; MWM, Morris Water Maze; NA, noradrenaline; NMDA, N-methyl-d-aspartate; NORT, novel object recognition test; OE, overexpressing; PCP, phenylcyclidine; PPI, prepulse inhibition; PSD, postsynaptic density; SIR, social isolation reared/social isolation rearing; SSRI, selective serotonin reuptake inhibitor; TCA, tricyclic antidepressant; 5-HIAA, 5-hydroxyindoleacetic acid; 5-HT, serotonin; 5-HTP, 5-hydroxytryptophan.
1. Langer SZ. α2-Adrenoceptors in the treatment of major neuropsychiatric disorders. Trends Pharmacol Sci (2015) 36:196–202. doi:10.1016/j.tips.2015.02.006
2. Berridge CW, Spencer RC. Differential cognitive actions of norepinephrine α2 and α1 receptor signaling in the prefrontal cortex. Brain Res (2016) 1641:189–96. doi:10.1016/j.brainres.2015.11.024
3. Kalkman HO, Loetscher E. α2C-Adrenoceptor blockade by clozapine and other antipsychotic drugs. Eur J Pharmacol (2003) 462:33–40. doi:10.1016/S0014-2999(03)01308-6
4. Shahid M, Walker GB, Zorn SH, Wong EHF. Asenapine: a novel psychopharmacologic agent with a unique human receptor signature. J Psychopharmacol (2009) 23:65–73. doi:10.1177/0269881107082944
5. Litman RE, Hong WW, Weissman EM, Su TP, Potter WZ, Pickar D. Idazoxan, an α2 antagonist, augments fluphenazine in schizophrenic patients: a pilot study. J Clin Psychopharmacol (1993) 13:264–7. doi:10.1097/00004714-199308000-00006
6. Litman RE, Su TP, Potter WZ, Hong WW, Pickar D. Idazoxan and response to typical neuroleptics in treatment-resistant schizophrenia: comparison with the atypical neuroleptic, clozapine. Br J Psychiatry (1996) 168:571–9. doi:10.1192/bjp.168.5.571
7. Marcus MM, Wiker C, Franberg O, Konradsson-Geuken A, Langlois X, Jardemark K, et al. Adjunctive alpha2-adrenoceptor blockade enhances the antipsychotic-like effect of risperidone and facilitates cortical dopaminergic and glutamatergic, NMDA receptor-mediated transmission. Int J Neuropsychopharmacol (2010) 13:891–903. doi:10.1017/S1461145709990794
8. Grossman F, Potter WZ, Brown EA, Maislin G. A double-blind study comparing idazoxan and bupropion in bipolar depressed patients. J Affect Disord (1999) 56:237–43. doi:10.1016/S0165-0327(99)00041-5
9. Dhir A, Kulkarni SK. Effect of addition of yohimbine (alpha-2-receptor antagonist) to the antidepressant activity of fluoxetine or venlafaxine in the mouse forced swim test. Pharmacology (2007) 80:239–43. doi:10.1159/000104877
10. Rénéric JP, Bouvard M, Stinus L. Idazoxan and 8-OH-DPAT modify the behavioral effects induced by either NA, or 5-HT, or dual NA/5-HT reuptake inhibition in the rat forced swimming test. Neuropsychopharmacology (2001) 24:379–90. doi:10.1016/S0893-133X(00)00214-1
11. Coull JT, Sahakian BJ, Hodges JR. The α2 antagonist idazoxan remediates certain attentional and executive dysfunction in patients with dementia of frontal type. Psychopharmacology (1996) 123:239–49. doi:10.1007/BF02246578
12. Arnsten AFT. Catecholamine influences on dorsolateral prefrontal cortical networks. Biol Psychiatry (2011) 69:e89–99. doi:10.1016/j.biopsych.2011.01.027
13. Sallee F, Connor DF, Newcorn JH. A review of the rationale and clinical utilization of alpha2-adrenoceptor agonists for the treatment of attention-deficit/hyperactivity and related disorders. J Child Adolesc Psychopharmacol (2013) 23:308–19. doi:10.1089/cap.2013.0028
14. Hein L, Altman JD, Kobilka BK. Two functionally distinct α2-adrenergic receptors regulate sympathetic neurotransmission. Nature (1999) 402:181–4. doi:10.1038/46040
15. Philipp M, Brede M, Hein L. Physiological significance of alpha(2)-adrenergic receptor subtype diversity: one receptor is not enough. Am J Physiol Regul Integr Comp Physiol (2002) 283:R287–95. doi:10.1152/ajpregu.00123.2002
16. Scheinin M, Sallinen J, Haapalinna A. Evaluation of the α2C-adrenoceptor as a neuropsychiatric drug target: studies in transgenic mouse models. Life Sci (2001) 68:2277–85. doi:10.1016/S0024-3205(01)01016-5
17. Sallinen J, Höglund I, Engström M, Lehtimäki J, Virtanen R, Sirviö J, et al. Pharmacological characterization and CNS effects of a novel highly selective α2C-adrenoceptor antagonist JP-1302. Br J Pharmacol (2007) 150:391–402. doi:10.1038/sj.bjp.0707005
18. Sallinen J, Holappa J, Koivisto A, Kuokkanen K, Chapman H, Lehtimäki J, et al. Pharmacological characterisation of a structurally novel α2C-adrenoceptor antagonist ORM-10921 and its effects in neuropsychiatric models. Basic Clin Pharmacol Toxicol (2013) 113:239–49. doi:10.1111/bcpt.12090
19. Sallinen J, Rouru J, Lehtimäki J, Marjamaeki P, Haaparanta-Solin M, Arponen E, et al. ORM-12741: receptor pharmacology of a novel alpha2c-adrenergic receptor subtype selective antagonist with multi-therapeutic potential. Neuropsychopharmacology (2013) 38:S558–558.
20. Uys M, Shahid M, Sallinen J, Dreyer W, Cockeran M, Harvey BH. The alpha2C-adrenoceptor antagonist, ORM-10921, has antipsychotic-like effects in social isolation reared rats and bolsters the response to haloperidol. Prog Neuropsychopharmacol Biol Psychiatry (2016) 71:108–16. doi:10.1016/j.pnpbp.2016.07.002
21. Uys MM, Shahid M, Sallinen J, Harvey BH. The alpha2C-adrenoceptor antagonist, ORM-10921, exerts antidepressant-like effects in the Flinders sensitive line rat. Behav Pharmacol (2017) 28:9–18. doi:10.1097/FBP.0000000000000261
22. Neumeister A, Drevets WC, Belfer I, Luckenbaugh DA, Henry S, Bonne O, et al. Effects of a α2C-adrenoreceptor gene polymorphism on neural responses to facial expressions in depression. Neuropsychopharmacology (2006) 31:1750–6. doi:10.1038/sj.npp.1301010
23. Cho S-C, Kim J-W, Kim B-N, Hwang J-W, Shin M-S, Park M, et al. Association between the alpha-2C-adrenergic receptor gene and attention deficit hyperactivity disorder in a Korean sample. Neurosci Lett (2008) 446:108–11. doi:10.1016/j.neulet.2008.09.058
24. Rivero G, Martin-Guerrero I, de Prado E, Gabilondo AM, Callado LF, Garcia-Sevilla JA, et al. Alpha2C-adrenoceptor Del322-325 polymorphism and risk of psychiatric disorders: significant association with opiate abuse and dependence. World J Biol Psychiatry (2016) 17:308–15. doi:10.3109/15622975.2016.1142608
25. Rinne JO, Wesnes K, Cummings JL, Hakulinen P, Hallikainen M, Hänninen J, et al. Tolerability of ORM-12741 and effects on episodic memory in patients with Alzheimer’s disease. Alzheimers Dement (N Y) (2017) 3:1–9. doi:10.1016/j.trci.2016.11.004
26. Bücheler MM, Hadamek K, Hein L. Two α2-adrenergic receptor subtypes, α2A and α2C, inhibit transmitter release in the brain of gene-targeted mice. Neuroscience (2002) 109:819–26. doi:10.1016/S0306-4522(01)00531-0
27. Scheinin M, Lomasney JW, Hayden-Hixson DM, Schambra UB, Caron MG, Lefkowitz RJ, et al. Distribution of α2-adrenergic receptor subtype gene expression in rat brain. Mol Brain Res (1994) 21:133–49. doi:10.1016/0169-328X(94)90386-7
28. Rosin DL, Talley EM, Lee A, Stornetta RL, Gaylinn BD, Guyenet PG, et al. Distribution of α(2C)-adrenergic receptor-like immunoreactivity in the rat central nervous system. JComp Neurol (1996) 372:135–65. doi:10.1002/(SICI)1096-9861(19960812)372:1<135::AID-CNE9>3.3.CO;2-B
29. Bunemann M, Bucheler MM, Philipp M, Lohse MJ, Hein L. Activation and deactivation kinetics of alpha 2A- and alpha 2C-adrenergic receptor-activated G protein-activated inwardly rectifying K+ channel currents. J Biol Chem (2001) 276:47512–7. doi:10.1074/jbc.M108652200
30. Ordway GA. Effect of noradrenergic lesions on subtypes of α2-adrenoceptors in rat brain. J Neurochem (1995) 64:1118–26. doi:10.1046/j.1471-4159.1995.64031118.x
31. Esteban S, Lladó J, García-Sevilla JA. α2-Autoreceptors and α2-heteroreceptors modulating tyrosine and tryptophan hydroxylase activity in the rat brain in vivo: an investigation into the α2-adrenoceptor subtypes. Naunyn Schmiedebergs Arch Pharmacol (1996) 353:391–9.
32. Meana JJ, Callado LF, Pazos A, Grijalba B, Garcia-Sevilla JA. The subtype-selective alpha 2-adrenoceptor antagonists BRL 44408 and ARC 239 also recognize 5-HT1A receptors in the rat brain. Eur J Pharmacol (1996) 312:385–8. doi:10.1016/0014-2999(96)00598-5
33. Scheibner J, Trendelenburg AU, Hein L, Starke K. α 2-Adrenoceptors modulating neuronal serotonin release: a study in α2-adrenoceptor subtype-deficient mice. Br J Pharmacol (2001) 132:925–33. doi:10.1038/sj.bjp.0703882
34. Björklund M, Sirviö J, Puoliväli J, Sallinen J, Jäkälä P, Scheinin M, et al. α(2C)-Adrenoceptor-overexpressing mice are impaired in executing nonspatial and spatial escape strategies. Mol Pharmacol (1998) 54:569–76.
35. Björklund M, Sirviö J, Riekkinen M, Sallinen J, Scheinin M, Riekkinen P Jr. Overexpression of alpha2c-adrenoceptors impairs water maze navigation. Neuroscience (1999) 95:481–7. doi:10.1016/S0306-4522(99)00428-5
36. Björklund M, Sirviö J, Sallinen J, Scheinin M, Kobilka BK, Riekkinen P Jr. Alpha2C-adrenoceptor overexpression disrupts execution of spatial and non-spatial search patterns. Neuroscience (1999) 88:1187–98. doi:10.1016/S0306-4522(98)00306-6
37. Björklund M, Siverina I, Heikkinen T, Tanila H, Sallinen J, Scheinin M, et al. Spatial working memory improvement by an α2-adrenoceptor agonist dexmedetomidine is not mediated through α2C-adrenoceptor. Prog Neuropsychopharmacology Biol Psychiatry (2001) 25:1539–54. doi:10.1016/S0278-5846(01)00209-3
38. Ramos BP, Arnsten AF. Adrenergic pharmacology and cognition: focus on the prefrontal cortex. Pharmacol Ther (2007) 113:523–36. doi:10.1016/j.pharmthera.2006.11.006
39. Franowicz JS, Kessler LE, Borja CM, Kobilka BK, Limbird LE, Arnsten AFT. Mutation of the α2A-adrenoceptor impairs working memory performance and annuls cognitive enhancement by guanfacine. J Neurosci (2002) 22:8771–7.
40. Sallinen J, Haapalinna A, MacDonald E, Viitamaa T, Lähdesmäki J, Rybnikova E, et al. Genetic alteration of the α2-adrenoceptor subtype c in mice affects the development of behavioral despair and stress-induced increases in plasma corticosterone levels. Mol Psychiatry (1999) 4:443–52. doi:10.1038/sj.mp.4000543
41. Schramm NL, McDonald MP, Limbird LE. The α2A-adrenergic receptor plays a protective role in mouse behavioral models of depression and anxiety. J Neurosci (2001) 21:4875–82.
42. Zhang HT, Whisler LR, Huang Y, Xiang Y, O’Donnell JM. Postsynaptic α-2 adrenergic receptors are critical for the antidepressant-like effects of desipramine on behavior. Neuropsychopharmacology (2009) 34:1067–77. doi:10.1038/npp.2008.184
43. Sallinen J, Haapalinna A, Viitamaa T, Kobilka BK, Scheinin M. Adrenergic α(2C)-receptors modulate the acoustic startle reflex, prepulse inhibition, and aggression in mice. J Neurosci (1998) 18:3035–42.
44. Lähdesmäki J, Sallinen J, MacDonald E, Scheinin M. Alpha2A-adrenoceptors are important modulators of the effects of d-amphetamine on startle reactivity and brain monoamines. Neuropsychopharmacology (2004) 29:1282–93. doi:10.1038/sj.npp.1300428
45. Dunn HA, Ferguson SS. PDZ protein regulation of G protein-coupled receptor trafficking and signaling pathways. Mol Pharmacol (2015) 88:624–39. doi:10.1124/mol.115.098509
46. Cottingham C, Wang Q. α2 Adrenergic receptor dysregulation in depressive disorders: implications for the neurobiology of depression and antidepressant therapy. Neurosci Biobehav Rev (2012) 36:2214–25. doi:10.1016/j.neubiorev.2012.07.011
47. Arnsten AFT. The use of α-2A adrenergic agonists for the treatment of attention-deficit/hyperactivity disorder. Expert Rev Neurother (2010) 10:1595–605. doi:10.1586/ern.10.133
48. Dinan TG, Cryan JF. The impact of gut microbiota on brain and behaviour: implications for psychiatry. Curr Opin Clin Nutr Metab Care (2015) 18:552–8. doi:10.1097/MCO.0000000000000221
49. Katzung BG. Introduction to autonomic pharmacology. In: Katzung BG, Trevor AJ, editors. Basic and Clinical Pharmacology. New York: McGraw-Hill Education (2015). p. 87–194.
50. Starke K. Presynaptic autoreceptors in the third decade: focus on α2-adrenoceptors. J Neurochem (2001) 78:685–93. doi:10.1046/j.1471-4159.2001.00484.x
51. MacDonald E, Kobilka BK, Scheinin M. Gene targeting – homing in on α2-adrenoceptor-subtype function. Trends Pharmacol Sci (1997) 18:211–9. doi:10.1016/S0165-6147(97)90625-8
52. Gilsbach R, Hein L. Are the pharmacology and physiology of α2adrenoceptors determined by α2-heteroreceptors and autoreceptors respectively? Br J Pharmacol (2012) 165:90–102. doi:10.1111/j.1476-5381.2011.01533.x
53. Trendelenburg AU, Klebroff W, Hein L, Starke K. A study of presynaptic α2-autoreceptors in α2A/D-, α2B- and α2C-adrenoceptor-deficient mice. Naunyn Schmiedebergs Arch Pharmacol (2001) 364:117–30. doi:10.1007/s002100100423
54. Scheibner J, Trendelenburg AU, Hein L, Starke K. Stimulation frequency-noradrenaline release relationships examined in α2A-,α2B- and α2C-adrenoceptor-deficient mice. Naunyn Schmiedebergs Arch Pharmacol (2001) 364:321–8. doi:10.1007/s002100100432
55. Fagerholm V, Rokka J, Nyman L, Sallinen J, Tiihonen J, Tupala E, et al. Autoradiographic characterization of α2C-adrenoceptors in the human striatum. Synapse (2008) 62:508–15. doi:10.1002/syn.20520
56. Finnema SJ, Hughes ZA, Haaparanta-Solin M, Stepanov V, Nakao R, Varnäs K, et al. Amphetamine decreases α(2C)-adrenoceptor binding of [(11)C]ORM-13070: a PET study in the primate brain. Int J Neuropsychopharmacol (2015) 18:yu081. doi:10.1093/ijnp/pyu081
57. Uhlen S, Muceniece R, Rangel N, Tiger G, Wikberg JE. Comparison of the binding activities of some drugs on alpha 2A, alpha 2B and alpha 2C-adrenoceptors and non-adrenergic imidazoline sites in the guinea pig. Pharmacol Toxicol (1995) 76:353–64. doi:10.1111/j.1600-0773.1995.tb00161.x
58. Uhlen S, Porter AC, Neubig RR. The novel alpha-2 adrenergic radioligand [3H]-MK912 is alpha-2C selective among human alpha-2A, alpha-2B and alpha-2C adrenoceptors. J Pharmacol Exp Ther (1994) 271:1558–65.
59. Holmberg M, Fagerholm V, Scheinin M. Regional distribution of α2C-adrenoceptors in brain and spinal cord of control mice and transgenic mice overexpressing the α2C-subtype: an autoradiographic study with [3H]RX821002 and [3H]rauwolscine. Neuroscience (2003) 117:875–98. doi:10.1016/S0306-4522(02)00966-1
60. Holmberg M, Scheinin M, Kurose H, Miettinen R. Adrenergic α(2C)-receptors reside in rat striatal GABAergic projection neurons: comparison of radioligand binding and immunohistochemistry. Neuroscience (1999) 93:1323–33. doi:10.1016/S0306-4522(99)00260-2
61. Winzer-Serhan UH, Raymon HK, Broide RS, Chen Y, Leslie FM. Expression of α2 adrenoceptors during rat brain development – II. α(2C) messenger RNA expression and [3H]rauwolscine binding. Neuroscience (1997) 76:261–72. doi:10.1016/S0306-4522(96)00369-7
62. Lehto J, Hirvonen MM, Johansson J, Kemppainen J, Luoto P, Naukkarinen T, et al. Validation of [11C]ORM-13070 as a PET tracer for alpha2c-adrenoceptors in the human brain. Synapse (2015) 69:172–81. doi:10.1002/syn.21798
63. Crassous PA, Cardinaletti C, Carrieri A, Bruni B, Di Vaira M, Gentili F, et al. α2-Adrenoreceptors profile modulation. 3.1 (R)-(+)-m-nitrobiphenyline, a new efficient and α2c-subtype selective agonist. J Med Chem (2007) 50:3964–8. doi:10.1021/jm061487a
64. Zhang W, Klimek V, Farley JT, Zhu MY, Ordway GA. α(2C) adrenoceptors inhibit adenylyl cyclase in mouse striatum: potential activation by dopamine. J Pharmacol Exp Ther (1999) 289:1286–92.
65. Zhang W, Ordway GA. The α2C-adrenoceptor modulates GABA release in mouse striatum. Mol Brain Res (2003) 112:24–32. doi:10.1016/S0169-328X(03)00026-3
66. Sallinen J, Haapalinna A, Viitamaa T, Kobilka BK, Scheinin M. d-Amphetamine and l-5-hydroxytryptophan-induced behaviours in mice with genetically-altered expression of the α(2C)-adrenergic receptor subtype. Neuroscience (1998) 86:959–65. doi:10.1016/S0306-4522(98)00100-6
67. Sallinen J, Link RE, Haapalinna A, Viitamaa T, Kulatunga M, Sjöholm B, et al. Genetic alteration of α(2C)-adrenoceptor expression in mice: influence on locomotor, hypothermic, and neurochemical effects of dexmedetomidine, a subtype-nonselective α2-adrenoceptor agonist. Mol Pharmacol (1997) 51:36–46.
68. Brand SJ, Moller M, Harvey BH. A review of biomarkers in mood and psychotic disorders: a dissection of clinical vs. preclinical correlates. Curr Neuropharmacol (2015) 13:324–68. doi:10.2174/1570159X13666150307004545
69. Link R, Daunt D, Barsh G, Chruscinski A, Kobilka B. Cloning of two mouse genes encoding α2-adrenergic receptor subtypes and identification of a single amino acid in the mouse α2-C10 homolog responsible for an interspecies variation in antagonist binding. Mol Pharmacol (1992) 42:16–27.
70. Zetterstrom T, Sharp T, Marsden CA, Ungerstedt U. In vivo measurement of dopamine and its metabolites by intracerebral dialysis: changes after d-amphetamine. J Neurochem (1983) 41:1769–73. doi:10.1111/j.1471-4159.1983.tb00893.x
71. Kuczenski R, Segal D. Concomitant characterization of behavioral and striatal neurotransmitter response to amphetamine using in vivo microdialysis. J Neurosci (1989) 9:2051–65.
72. Kuczenski R, Segal DS. Regional norepinephrine response to amphetamine using dialysis: comparison with caudate dopamine. Synapse (1992) 11:164–9. doi:10.1002/syn.890110210
73. Ihalainen JA, Tanila H, Scheinin M, Riekkinen P Jr. α2C-Adrenoceptors modulate the effect of methylphenidate on response rate and discrimination accuracy in an operant test. Brain Res Bull (2001) 54:553–7. doi:10.1016/S0361-9230(01)00449-X
74. Kawaguchi Y, Wilson CJ, Emson PC. Projection subtypes of rat neostriatal matrix cells revealed by intracellular injection of biocytin. J Neurosci (1990) 10:3421–38.
75. Monti B, Polazzi E, Contestabile A. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Curr Mol Pharmacol (2009) 2:95–109. doi:10.2174/1874467210902010095
76. Reynolds GP. The neurochemistry of schizophrenia. Psychiatry (2008) 7:425–9. doi:10.1016/j.mppsy.2008.07.014
77. Millan MJ. The neurobiology and control of anxious states. Prog Neurobiol (2003) 70:83–244. doi:10.1016/S0301-0082(03)00087-X
78. Harvey BH, Slabbert FN. New insights on the antidepressant discontinuation syndrome. Hum Psychopharmacol (2014) 29:503–16. doi:10.1002/hup.2429
79. Timofeeva OA, Levin ED. Idazoxan blocks the nicotine-induced reversal of the memory impairment caused by the NMDA glutamate receptor antagonist dizocilpine. Pharmacol Biochem Behav (2008) 90:372–81. doi:10.1016/j.pbb.2008.03.011
80. Bardgett ME, Points M, Ramsey-Faulkner C, Topmiller J, Roflow J, McDaniel T, et al. The effects of clonidine on discrete-trial delayed spatial alternation in two rat models of memory loss. Neuropsychopharmacology (2007) 33:1980–91. doi:10.1038/sj.npp.1301580
81. Jentsch JD, Anzivino LA. A low dose of the alpha2 agonist clonidine ameliorates the visual attention and spatial working memory deficits produced by phencyclidine administration to rats. Psychopharmacology (2004) 175:76–83. doi:10.1007/s00213-004-1772-3
82. Marrs W, Kuperman J, Avedian T, Roth RH, Jentsch JD. Alpha-2 adrenoceptor activation inhibits phencyclidine-induced deficits of spatial working memory in rats. Neuropsychopharmacology (2005) 30:1500–10. doi:10.1038/sj.npp.1300700
83. Jimenez-Rivera CA, Figueroa J, Vazquez-Torres R, Velez-Hernandez ME, Schwarz D, Velasquez-Martinez MC, et al. Presynaptic inhibition of glutamate transmission by alpha2 receptors in the VTA. Eur J Neurosci (2012) 35:1406–15. doi:10.1111/j.1460-9568.2012.08029.x
84. Marcus MM, Jardemark KE, Wadenberg ML, Langlois X, Hertel P, Svensson TH. Combined α2 and D2/3 receptor blockade enhances cortical glutamatergic transmission and reverses cognitive impairment in the rat. Int J Neuropsychopharmacol (2005) 8:315–27. doi:10.1017/S1461145705005328
85. Jardemark K, Marcus MM, Shahid M, Svensson TH. Effects of asenapine on prefrontal N-methyl-d-aspartate receptor-mediated transmission: involvement of dopamine D1 receptors. Synapse (2010) 64:870–4. doi:10.1002/syn.20803
86. Schwartz TL, Sachdeva S, Stahl SM. Glutamate neurocircuitry: theoretical underpinnings in schizophrenia. Front Pharmacol (2012) 3:195. doi:10.3389/fphar.2012.00195
87. Scarr E, Gibbons AS, Neo J, Udawela M, Dean B. Cholinergic connectivity: it’s implications for psychiatric disorders. Front Cell Neurosci (2013) 7:55. doi:10.3389/fncel.2013.00055
88. Friedman JI. Cholinergic targets for cognitive enhancement in schizophrenia: focus on cholinesterase inhibitors and muscarinic agonists. Psychopharmacology (Berl) (2004) 174:45–53. doi:10.1007/s00213-004-1794-x
89. Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry (2006) 63:1121–9. doi:10.1001/archpsyc.63.10.1121
90. Scarr E, Sundram S, Keriakous D, Dean B. Altered hippocampal muscarinic M4, but not M1, receptor expression from subjects with schizophrenia. Biol Psychiatry (2007) 61:1161–70. doi:10.1016/j.biopsych.2006.08.050
91. Mufson EJ, Counts SE, Perez SE, Ginsberg SD. Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert Rev Neurother (2008) 8:1703–18. doi:10.1586/14737175.8.11.1703
92. Raiteri M, Marchi M, Paudice P, Pittaluga A. Muscarinic receptors mediating inhibition of gamma-aminobutyric acid release in rat corpus striatum and their pharmacological characterization. J Pharmacol Exp Ther (1990) 254:496–501.
93. Sapolsky RM. Depression, antidepressants, and the shrinking hippocampus. Proc Natl Acad Sci U S A (2001) 98:12320–2. doi:10.1073/pnas.231475998
94. Kharade SM, Gumate DS, Naikwade NS. A review: hypothesis of depression and role of antidepressant drugs. Int J Pharm Pharm Sci (2010) 2:3–6.
95. Duman RS. Neurobiology of stress, depression, and rapid acting antidepressants: remodeling synaptic connections. Depress Anxiety (2014) 31:291–6. doi:10.1002/da.22227
96. Duman RS. Pathophysiology of depression: the concept of synaptic plasticity. Eur Psychiatry (2002) 17:306–10. doi:10.1016/S0924-9338(02)00654-5
97. Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature (2008) 455:894–902. doi:10.1038/nature07455
98. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J Psychiatry (2001) 178:234–41. doi:10.1192/bjp.178.3.234
99. Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR* D report. Am J Psychiatry (2006) 163:1905–17. doi:10.1176/ajp.2006.163.11.1905
100. De Paermentier F, Mauger JM, Lowther S, Crompton MR, Katona CLE, Horton RW. Brain α-adrenoceptors in depressed suicides. Brain Res (1997) 757:60–8. doi:10.1016/S0006-8993(97)00138-8
101. Ordway GA, Schenk J, Stockmeier CA, May W, Klimek V. Elevated agonist binding to α2-adrenoceptors in the locus coeruleus in major depression. Biol Psychiatry (2003) 53:315–23. doi:10.1016/S0006-3223(02)01728-6
102. Ordway GA, Widdowson PS, Smith KS, Halaris A. Agonist binding to α2-adrenoceptors is elevated in the locus coeruleus from victims of suicide. J Neurochem (1994) 63:617–24. doi:10.1046/j.1471-4159.1994.63020617.x
103. González AM, Pascual J, Meana JJ, Barturen F, Del Arco C, Pazos A, et al. Autoradiographic demonstration of increased α2-adrenoceptor agonist binding sites in the hippocampus and frontal cortex of depressed suicide victims. J Neurochem (1994) 63:256–65. doi:10.1046/j.1471-4159.1994.63010256.x
104. Callado LF, Meana JJ, Grijalba B, Pazos A, Sastre M, García-Sevilla JA. Selective increase of α(2A)-adrenoceptor agonist binding sites in brains of depressed suicide victims. J Neurochem (1998) 70:1114–23. doi:10.1046/j.1471-4159.1998.70031114.x
105. Javier Meana J, Barturen F, Garcia-Sevilla JA. α2-Adrenoceptors in the brain of suicide victims: increased receptor density associated with major depression. Biol Psychiatry (1992) 31:471–90. doi:10.1016/0006-3223(92)90259-3
106. Meana JJ, García-Sevilla JA. Increased α2;-adrenoceptor density in the frontal cortex of depressed suicide victims. J Neural Transm (1987) 70:377–81. doi:10.1007/BF01253612
107. Blier P. The pharmacology of putative early-onset antidepressant strategies. Eur Neuropsychopharmacol (2003) 13:57–66. doi:10.1016/S0924-977X(02)00173-6
108. Marshall RJ. The pharmacology of mianserin—an update. Br J Clin Pharmacol (1983) 15:263S–8S. doi:10.1111/j.1365-2125.1983.tb05874.x
109. Petit-Demouliere B, Chenu F, Bourin M. Forced swimming test in mice: a review of antidepressant activity. Psychopharmacology (2005) 177:245–55. doi:10.1007/s00213-004-2048-7
110. Castagné V, Moser P, Roux S, Porsolt RD. Rodent models of depression: forced swim and tail suspension behavioral despair tests in rats and mice. Curr Protoc Neurosci (2011) 55:1–14. doi:10.1002/0471142301.ns0810as55
111. Cryan JF, Mombereau C. In search of a depressed mouse: utility of models for studying depression-related behavior in genetically modified mice. Mol Psychiatry (2004) 9:326–57. doi:10.1038/sj.mp.4001457
112. Holmes PV. Rodent models of depression: reexamining validity without anthropomorphic inference. Crit Rev Neurobiol (2003) 15:143–74. doi:10.1615/CritRevNeurobiol.v15.i2.30
113. Cottingham C, Li X, Wang Q. Noradrenergic antidepressant responses to desipramine in vivo are reciprocally regulated by arrestin3 and spinophilin. Neuropharmacology (2012) 62:2354–62. doi:10.1016/j.neuropharm.2012.02.011
114. Cervo L, Samanin R. Clonidine causes antidepressant-like effects in rats by activating α2-adrenoceptors outside the locus coeruleus. Eur J Pharmacol (1991) 193:309–13. doi:10.1016/0014-2999(91)90144-F
115. Stone EA, Lin Y, Sarfraz Y, Quartermain D. Antidepressant-like action of intracerebral 6-fluoronorepinephrine, a selective full α-adrenoceptor agonist. Int J Neuropsychopharmacol (2011) 14:319–31. doi:10.1017/S1461145710000507
116. Sanacora G, Berman RM, Cappiello A, Oren DA, Kugaya A, Liu N, et al. Addition of the α2-antagonist yohimbine to fluoxetine: effects on rate of antidepressant response. Neuropsychopharmacology (2004) 29:1166–71. doi:10.1038/sj.npp.1300418
117. Davis R, Wilde MI. Mirtazapine: a review of its pharmacology and therapeutic potential in the management of major depression. CNS Drugs (1996) 5:389–402. doi:10.2165/00023210-199605050-00007
118. Blier P, Gobbi G, Turcotte JE, de Montigny C, Boucher N, Hébert C, et al. Mirtazapine and paroxetine in major depression: a comparison of monotherapy versus their combination from treatment initiation. Eur Neuropsychopharmacol (2009) 19:457–65. doi:10.1016/j.euroneuro.2009.01.015
119. Blier P, Ward HE, Tremblay P, Laberge L, Hébert C, Bergeron R. Combination of antidepressant medications from treatment initiation for major depressive disorder: a double-blind randomized study. Am J Psychiatry (2010) 167:281–8. doi:10.1176/appi.ajp.2009.09020186
120. Gotlib IH, Joormann J. Cognition and depression: current status and future directions. Annu Rev Clin Psychol (2010) 6:285–312. doi:10.1146/annurev.clinpsy.121208.131305
121. Hammar Å, Årdal G. Cognitive functioning in major depression – a summary. Front Hum Neurosci (2009) 3:26. doi:10.3389/neuro.09.026.2009
122. Campbell S, MacQueen G. The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci (2004) 29:417–26.
123. Flugge G. Effects of cortisol on brain alpha2-adrenoceptors: potential role in stress. Neurosci Biobehav Rev (1999) 23:949–56. doi:10.1016/S0149-7634(99)00028-7
124. Schule C, Baghai T, Zwanzger P, Rupprecht R. Attenuation of HPA axis hyperactivity and simultaneous clinical deterioration in a depressed patient treated with mirtazapine. World J Biol Psychiatry (2001) 2:103–5. doi:10.3109/15622970109027501
125. Schüle C, Baghai T, Zwanzger P, Ella R, Eser D, Padberg F, et al. Attenuation of hypothalamic-pituitary-adrenocortical hyperactivity in depressed patients by mirtazapine. Psychopharmacology (2003) 166:271–5. doi:10.1007/s00213-002-1356-z
126. Weber C-C, Eckert GP, Muller WE. Effects of antidepressants on the brain//plasma distribution of corticosterone. Neuropsychopharmacology (2006) 31:2443–8. doi:10.1038/sj.npp.1301076
127. Glue P, Wilson S, Campling GM, Knightly M, Franklin M, Cowen PJ, et al. Alpha-2-adrenoceptor control of cortisol and ACTH in normal volunteers: preliminary open trial of the effects of acute and chronic idazoxan. Psychoneuroendocrinology (1992) 17:261–6. doi:10.1016/0306-4530(92)90066-G
128. Price LH, Charney DS, Rubin AL, Heninger GR. Alpha 2-adrenergic receptor function in depression. The cortisol response to yohimbine. Arch Gen Psychiatry (1986) 43:849–58. doi:10.1001/archpsyc.1986.01800090035006
129. Finn DP, Hudson AL, Kinoshita H, Coventry TL, Jessop DS, Nutt DJ, et al. Imidazoline2 (I2) receptor- and alpha2-adrenoceptor-mediated modulation of hypothalamic-pituitary-adrenal axis activity in control and acute restraint stressed rats. J Psychopharmacol (2004) 18:47–53. doi:10.1177/0269881104040231
130. Báez M, Volosin M. Corticosterone influences forced swim-induced immobility. Pharmacol Biochem Behav (1994) 49:729–36. doi:10.1016/0091-3057(94)90093-0
131. Korte SM, De Kloet ER, Buwalda B, Bouman SD, Bohus B. Antisense to the glucocorticoid receptor in hippocampal dentate gyrus reduces immobility in forced swim test. Eur J Pharmacol (1996) 301:19–25. doi:10.1016/0014-2999(96)00064-7
132. Lehto J, Scheinin A, Johansson J, Marjamäki P, Arponen E, Scheinin H, et al. Detecting a dexmedetomidine-evoked reduction of noradrenaline release in the human brain with the alpha2C-adrenoceptor PET ligand [11C]ORM-13070. Synapse (2016) 70:57–65. doi:10.1002/syn.21872
133. Lu L, Ordway GA. Alpha2C-adrenoceptors mediate inhibition of forskolin-stimulated cAMP production in rat striatum. Brain Res Mol Brain Res (1997) 52:228–34. doi:10.1016/S0169-328X(97)00257-X
134. Simpson EH, Kellendonk C, Kandel E. A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron (2010) 65:585–96. doi:10.1016/j.neuron.2010.02.014
135. Yamamoto K, Hornykiewicz O. Proposal for a noradrenaline hypothesis of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2004) 28:913–22. doi:10.1016/j.pnpbp.2004.05.033
136. Moller M, Swanepoel T, Harvey BH. Neurodevelopmental animal models reveal the convergent role of neurotransmitter systems, inflammation, and oxidative stress as biomarkers of schizophrenia: implications for novel drug development. ACS Chem Neurosci (2015) 6:987–1016. doi:10.1021/cn5003368
137. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III – the final common pathway. Schizophr Bull (2009) 35:549–62. doi:10.1093/schbul/sbp006
138. Grunder G, Hippius H, Carlsson A. The ‘atypicality’ of antipsychotics: a concept re-examined and re-defined. Nat Rev Drug Discov (2009) 8:197–202. doi:10.1038/nrd2806
139. Svensson TH. α-Adrenoceptor modulation hypothesis of antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry (2003) 27:1145–58. doi:10.1016/j.pnpbp.2003.09.009
140. Hertel P, Fagerquist MV, Svensson TH. Enhanced cortical dopamine output and antipsychotic-like effects of raclopride by α2 adrenoceptor blockade. Science (1999) 286:105–7. doi:10.1126/science.286.5437.105
141. Farde L, Wiesel FA, Nordström AL, Sedvall G. D1- and D2-dopamine receptor occupancy during treatment with conventional and atypical neuroleptics. Psychopharmacology (1989) 99:S28–31. doi:10.1007/BF00442555
142. Farde L, Nordström AL. PET analysis indicates atypical central dopamine receptor occupancy in clozapine-treated patients. Br J Psychiatry Suppl (1992) 17:30–3.
143. Braff DL, Geyer MA. Sensorimotor gating and schizophrenia human and animal model studies. Arch Gen Psychiatry (1990) 47:181–8. doi:10.1001/archpsyc.1990.01810140081011
144. Geyer MA, Braff DL. Startle habituation and sensorimotor gating in schizophrenia and related animal models. Schizophr Bull (1987) 13:643–68. doi:10.1093/schbul/13.4.643
145. Flaten MA. Test-retest reliability of the somatosensory blink reflex and its inhibition. Int J Psychophysiol (2002) 45:261–5. doi:10.1016/S0167-8760(02)00034-X
146. Geyer MA, Swerdlow NR, Mansbach RS, Braff DL. Startle response models of sensorimotor gating and habituation deficits in schizophrenia. Brain Res Bull (1990) 25:485–98. doi:10.1016/0361-9230(90)90241-Q
147. Geyer MA, Wilkinson LS, Humby T, Robbins TW. Isolation rearing of rats produces a deficit in prepulse inhibition of acoustic startle similar to that in schizophrenia. Biol Psychiatry (1993) 34:361–72. doi:10.1016/0006-3223(93)90180-L
148. Bakshi VP, Swerdlow NR, Geyer MA. Clozapine antagonizes phencyclidine-induced deficits in sensorimotor gating of the startle response. J Pharmacol Exp Ther (1994) 271:787–94.
149. Varty GB, Higgins GA, Higgins GA. Examination of drug-induced and isolation-induced disruptions of prepulse inhibition as models to screen antipsychotic drugs. Psychopharmacology (1995) 122:15–26. doi:10.1007/BF02246437
150. Geyer MA, McIlwain KL, Paylor R. Mouse genetic models for prepulse inhibition: an early review. Mol Psychiatry (2002) 7:1039–53. doi:10.1038/sj.mp.4001159
151. Depoortere R, Perrault G, Sanger DJ. Potentiation of prepulse inhibition of the startle reflex in rats: pharmacological evaluation of the procedure as a model for detecting antipsychotic activity. Psychopharmacology (1997) 132:366–74. doi:10.1007/s002130050357
152. Swerdlow NR, Talledo J, Sutherland AN, Nagy D, Shoemaker JM. Antipsychotic effects on prepulse inhibition in normal ‘low gating’ humans and rats. Neuropsychopharmacology (2006) 31:2011–21. doi:10.1038/sj.npp.1301043
153. During S, Glenthoj BY, Andersen GS, Oranje B. Effects of dopamine D2/D3 blockade on human sensory and sensorimotor gating in initially antipsychotic-naive, first-episode schizophrenia patients. Neuropsychopharmacology (2014) 39:3000–8. doi:10.1038/npp.2014.152
154. Vollenweider FX, Barro M, Csomor PA, Feldon J. Clozapine enhances prepulse inhibition in healthy humans with low but not with high prepulse inhibition levels. Biol Psychiatry (2006) 60:597–603. doi:10.1016/j.biopsych.2006.03.058
155. Csomor PA, Preller KH, Geyer MA, Studerus E, Huber T, Vollenweider FX. Influence of aripiprazole, risperidone, and amisulpride on sensory and sensorimotor gating in healthy ‘low and high gating’ humans and relation to psychometry. Neuropsychopharmacology (2014) 39:2485–96. doi:10.1038/npp.2014.102
156. Larrauri JA, Levin ED. The alpha(2)-adrenergic antagonist idazoxan counteracts prepulse inhibition deficits caused by amphetamine or dizocilpine in rats. Psychopharmacology (Berl) (2012) 219:99–108. doi:10.1007/s00213-011-2377-2
157. Ozcetin A, Cevreli B, Uzbay T. Investigation of the role of alpha-2 adrenergic receptors on prepulse inhibition of acoustic startle reflex in rats. Synapse (2016) 70(12):501–7. doi:10.1002/syn.21923
158. Powell SB, Palomo J, Carasso BS, Bakshi VP, Geyer MA. Yohimbine disrupts prepulse inhibition in rats via action at 5-HT1A receptors, not alpha2-adrenoceptors. Psychopharmacology (Berl) (2005) 180:491–500. doi:10.1007/s00213-005-2193-7
159. Jones C, Watson D, Fone K. Animal models of schizophrenia. Br J Pharmacol (2011) 164:1162–94. doi:10.1111/j.1476-5381.2011.01386.x
160. Fone KC, Porkess MV. Behavioural and neurochemical effects of post-weaning social isolation in rodents-relevance to developmental neuropsychiatric disorders. Neurosci Biobehav Rev (2008) 32:1087–102. doi:10.1016/j.neubiorev.2008.03.003
161. Elvevag B, Goldberg TE. Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol (2000) 14:1–21. doi:10.1615/CritRevNeurobiol.v14.i1.10
162. Trivedi JK. Cognitive deficits in psychiatric disorders: current status. Indian J Psychiatry (2006) 48:10–20. doi:10.4103/0019-5545.31613
163. Bowie CR, Harvey PD. Cognitive deficits and functional outcome in schizophrenia. Neuropsychiatr Dis Treat (2006) 2:531–6. doi:10.2147/nedt.2006.2.4.531
164. Keefe RS, Harvey PD. Cognitive impairment in schizophrenia. Handb Exp Pharmacol (2012) 213:11–37. doi:10.1007/978-3-642-25758-2_2
165. Kahn RS, Keefe RSE. Schizophrenia is a cognitive illness: time for a change in focus. JAMA Psychiatry (2013) 70:1107–12. doi:10.1001/jamapsychiatry.2013.155
166. Skarsfeldt T. Differential effect of antipsychotics on place navigation of rats in the Morris water maze. A comparative study between novel and reference antipsychotics. Psychopharmacology (Berl) (1996) 124:126–33. doi:10.1007/BF02245612
167. Didriksen M, Skarsfeldt T, Arnt J. Reversal of PCP-induced learning and memory deficits in the Morris’ water maze by sertindole and other antipsychotics. Psychopharmacology (Berl) (2007) 193:225–33. doi:10.1007/s00213-007-0774-3
168. Grayson B, Idris NF, Neill JC. Atypical antipsychotics attenuate a sub-chronic PCP-induced cognitive deficit in the novel object recognition task in the rat. Behav Brain Res (2007) 184:31–8. doi:10.1016/j.bbr.2007.06.012
169. Wolff MC, Leander JD. Comparison of the effects of antipsychotics on a delayed radial maze task in the rat. Psychopharmacology (Berl) (2003) 168:410–6. doi:10.1007/s00213-003-1449-3
170. Amitai N, Markou A. Disruption of performance in the 5-choice serial reaction time task induced by administration of NMDA receptor antagonists: relevance to cognitive dysfunction in schizophrenia. Biol Psychiatry (2010) 68:5–16. doi:10.1016/j.biopsych.2010.03.004
171. Bubenikova-Valesova V, Horacek J, Vrajova M, Hoschl C. Models of schizophrenia in humans and animals based on inhibition of NMDA receptors. Neurosci Biobehav Rev (2008) 32:1014–23. doi:10.1016/j.neubiorev.2008.03.012
172. Enomoto T, Ishibashi T, Tokuda K, Ishiyama T, Toma S, Ito A. Lurasidone reverses MK-801-induced impairment of learning and memory in the Morris water maze and radial-arm maze tests in rats. Behav Brain Res (2008) 186:197–207. doi:10.1016/j.bbr.2007.08.012
173. Terry AV, Hill WD, Parikh V, Waller JL, Evans DR, Mahadik SP. Differential effects of haloperidol, risperidone, and clozapine exposure on cholinergic markers and spatial learning performance in rats. Neuropsychopharmacology (2003) 28:300–9. doi:10.1038/sj.npp.1300039
174. Favalli G, Li J, Belmonte-de-Abreu P, Wong AHC, Daskalakis ZJ. The role of BDNF in the pathophysiology and treatment of schizophrenia. J Psychiatr Res (2012) 46:1–11. doi:10.1016/j.jpsychires.2011.09.022
175. Nieto R, Kukuljan M, Silva H. BDNF and schizophrenia: from neurodevelopment to neuronal plasticity, learning, and memory. Front Psychiatry (2013) 4:45. doi:10.3389/fpsyt.2013.00045
176. Couture SM, Penn DL, Roberts DL. The functional significance of social cognition in schizophrenia: a review. Schizophr Bull (2006) 32:S44–63. doi:10.1093/schbul/sbl029
177. Wilson CA, Koenig JI. Social interaction and social withdrawal in rodents as readouts for investigating the negative symptoms of schizophrenia. Eur Neuropsychopharmacol (2014) 24:759–73. doi:10.1016/j.euroneuro.2013.11.008
178. Möller M, Du Preez JL, Emsley R, Harvey BH. Isolation rearing-induced deficits in sensorimotor gating and social interaction in rats are related to cortico-striatal oxidative stress, and reversed by sub-chronic clozapine administration. Eur Neuropsychopharmacol (2011) 21:471–83. doi:10.1016/j.euroneuro.2010.09.006
179. Ishibashi T, Horisawa T, Tokuda K, Ishiyama T, Ogasa M, Tagashira R, et al. Pharmacological profile of lurasidone, a novel antipsychotic agent with potent 5-hydroxytryptamine 7 (5-HT7) and 5-HT1A receptor activity. J Pharmacol Exp Ther (2010) 334:171–81. doi:10.1124/jpet.110.167346
180. Swartz MS, Wagner HR, Swanson JW, Stroup TS, McEvoy JP, Reimherr F, et al. The effectiveness of antipsychotic medications in patients who use or avoid illicit substances: results from the CATIE study. Schizophr Res (2008) 100:39–52. doi:10.1016/j.schres.2007.11.034
181. Arnsten A, Cai JX, Goldman-Rakic PS. The alpha-2 adrenergic agonist guanfacine improves memory in aged monkeys without sedative or hypotensive side effects: evidence for alpha-2 receptor subtypes. J Neurosci (1988) 8:4287–98.
182. Franowicz JS, Arnsten AF. Treatment with the noradrenergic alpha-2 agonist clonidine, but not diazepam, improves spatial working memory in normal young rhesus monkeys. Neuropsychopharmacology (1999) 21:611–21. doi:10.1016/S0893-133X(99)00060-3
183. Cai JX, Ma YY, Xu L, Hu XT. Reserpine impairs spatial working memory performance in monkeys: reversal by the alpha 2-adrenergic agonist clonidine. Brain Res (1993) 614:191–6. doi:10.1016/0006-8993(93)91034-P
184. Jäkälä P, Riekkinen M, Sirviö J, Koivisto E, Kejonen K, Vanhanen M, et al. Guanfacine, but not clonidine, improves planning and working memory performance in humans. Neuropsychopharmacology (1999) 20:460–70. doi:10.1016/S0893-133X(98)00127-4
185. Arnsten A, Goldman-Rakic PS. Alpha 2-adrenergic mechanisms in prefrontal cortex associated with cognitive decline in aged nonhuman primates. Science (1985) 230:1273–6. doi:10.1126/science.2999977
186. Lakhlani PP, MacMillan LB, Guo TZ, McCool BA, Lovinger DM, Maze M, et al. Substitution of a mutant alpha2a-adrenergic receptor via “hit and run” gene targeting reveals the role of this subtype in sedative, analgesic, and anesthetic-sparing responses in vivo. Proc Natl Acad Sci U S A (1997) 94:9950–5. doi:10.1073/pnas.94.18.9950
187. Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc (2006) 1:848–58. doi:10.1038/nprot.2006.116
188. Marston H, Everitt B, Robbins T. Comparative effects of excitotoxic lesions of the hippocampus and septum/diagonal band on conditional visual discrimination and spatial learning. Neuropsychologia (1993) 31:1099–118. doi:10.1016/0028-3932(93)90035-X
189. Annett L, McGregor A, Robbins T. The effects of ibotenic acid lesions of the nucleus accumbens on spatial learning and extinction in the rat. Behav Brain Res (1989) 31:231–42. doi:10.1016/0166-4328(89)90005-3
190. Floresco SB, Seamans JK, Phillips AG. Selective roles for hippocampal, prefrontal cortical, and ventral striatal circuits in radial-arm maze tasks with or without a delay. J Neurosci (1997) 17:1880–90.
191. Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: behavioral data. Behav Brain Res (1988) 31:47–59. doi:10.1016/0166-4328(88)90157-X
192. Antunes M, Biala G. The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn Process (2012) 13:93–110. doi:10.1007/s10339-011-0430-z
193. Broadbent NJ, Gaskin S, Squire LR, Clark RE. Object recognition memory and the rodent hippocampus. Learn Mem (2010) 17:794–800. doi:10.1101/lm.1650110
194. Cohen SJ, Stackman RW Jr. Assessing rodent hippocampal involvement in the novel object recognition task. a review. Behav Brain Res (2015) 285:105–17. doi:10.1016/j.bbr.2014.08.002
195. Reger ML, Hovda DA, Giza CC. Ontogeny of rat recognition memory measured by the novel object recognition task. Dev Psychobiol (2009) 51:672–8. doi:10.1002/dev.20402
196. Lu B, Nagappan G, Lu Y. BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol (2014) 220:223–50. doi:10.1007/978-3-642-45106-5_9
197. Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev (2012) 64:238–58. doi:10.1124/pr.111.005108
198. Neto FL, Borges G, Torres-Sanchez S, Mico JA, Berrocoso E. Neurotrophins role in depression neurobiology: a review of basic and clinical evidence. Curr Neuropharmacol (2011) 9:530–52. doi:10.2174/157015911798376262
199. Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci (1995) 15:7539–47.
200. Fumagalli F, Molteni R, Roceri M, Bedogni F, Santero R, Fossati C, et al. Effect of antipsychotic drugs on brain-derived neurotrophic factor expression under reduced N-methyl-d-aspartate receptor activity. J Neurosci Res (2003) 72:622–8. doi:10.1002/jnr.10609
201. Pillai A, Terry AV Jr, Mahadik SP. Differential effects of long-term treatment with typical and atypical antipsychotics on NGF and BDNF levels in rat striatum and hippocampus. Schizophr Res (2006) 82:95–106. doi:10.1016/j.schres.2005.11.021
202. Mannari C, Origlia N, Scatena A, Del Debbio A, Catena M, Dell’Agnello G, et al. BDNF level in the rat prefrontal cortex increases following chronic but not acute treatment with duloxetine, a dual acting inhibitor of noradrenaline and serotonin re-uptake. Cell Mol Neurobiol (2008) 28:457–68. doi:10.1007/s10571-007-9254-x
203. Pedrini M, Chendo I, Grande I, Lobato MI, Belmonte-de-Abreu PS, Lersch C, et al. Serum brain-derived neurotrophic factor and clozapine daily dose in patients with schizophrenia: a positive correlation. Neurosci Lett (2011) 491:207–10. doi:10.1016/j.neulet.2011.01.039
204. Rizk P, Salazar J, Raisman-Vozari R, Marien M, Ruberg M, Colpaert F, et al. The alpha2-adrenoceptor antagonist dexefaroxan enhances hippocampal neurogenesis by increasing the survival and differentiation of new granule cells. Neuropsychopharmacology (2005) 31:1146–57. doi:10.1038/sj.npp.1300954
205. Yanpallewar S, Fernandes K, Marathe S, Vadodaria K, Jhaveri D, Rommelfanger K, et al. Alpha2-adrenoceptor blockade accelerates the neurogenic, neurotrophic, and behavioral effects of chronic antidepressant treatment. J Neurosci (2010) 30:1096–109. doi:10.1523/JNEUROSCI.2309-09.2010
206. Francis BM, Yang J, Hajderi E, Brown ME, Michalski B, McLaurin J, et al. Reduced tissue levels of noradrenaline are associated with behavioral phenotypes of the TgCRND8 mouse model of Alzheimer’s disease. Neuropsychopharmacology (2012) 37:1934–44. doi:10.1038/npp.2012.40
207. Kuppers E, Beyer C. Dopamine regulates brain-derived neurotrophic factor (BDNF) expression in cultured embryonic mouse striatal cells. Neuroreport (2001) 12:1175–9. doi:10.1097/00001756-200105080-00025
208. Martinowich K, Lu B. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology (2007) 33:73–83. doi:10.1038/sj.npp.1301571
209. Marmigere F, Rage F, Tapia-Arancibia L. GABA-glutamate interaction in the control of BDNF expression in hypothalamic neurons. Neurochem Int (2003) 42:353–8. doi:10.1016/S0197-0186(02)00100-6
210. West AE, Griffith EC, Greenberg ME. Regulation of transcription factors by neuronal activity. Nat Rev Neurosci (2002) 3:921–31. doi:10.1038/nrn987
211. Tuvikene J, Pruunsild P, Orav E, Esvald EE, Timmusk T. AP-1 transcription factors mediate BDNF-positive feedback loop in cortical neurons. J Neurosci (2016) 36:1290–305. doi:10.1523/JNEUROSCI.3360-15.2016
212. Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev (2009) 89:121–45. doi:10.1152/physrev.00017.2008
213. Radwanska K, Schenatto-Pereira G, Ziółkowska M, Łukasiewicz K, Giese KP. Mapping fear memory consolidation and extinction-specific expression of JunB. Neurobiol Learn Mem (2015) 125:106–12. doi:10.1016/j.nlm.2015.08.007
214. Hsieh TF, Simler S, Vergnes M, Gass P, Marescaux C, Wiegand SJ, et al. BDNF restores the expression of Jun and Fos inducible transcription factors in the rat brain following repetitive electroconvulsive seizures. Exp Neurol (1998) 149:161–74. doi:10.1006/exnr.1997.6686
215. Tomasetti C, Iasevoli F, Buonaguro EF, De Berardis D, Fornaro M, Fiengo ALC, et al. Treating the synapse in major psychiatric disorders: the role of postsynaptic density network in dopamine-glutamate interplay and psychopharmacologic drugs molecular actions. Int J Mol Sci (2017) 18:135. doi:10.3390/ijms18010135
216. Brady AE, Wang Q, Colbran RJ, Allen PB, Greengard P, Limbird LE. Spinophilin stabilizes cell surface expression of alpha 2B-adrenergic receptors. J Biol Chem (2003) 278:32405–12. doi:10.1074/jbc.M304195200
217. Lu R, Chen Y, Cottingham C, Peng N, Jiao K, Limbird LE, et al. Enhanced hypotensive, bradycardic, and hypnotic responses to α(2)-adrenergic agonists in spinophilin-null mice are accompanied by increased G protein coupling to the α(2A)-adrenergic receptor. Mol Pharmacol (2010) 78:279–86. doi:10.1124/mol.110.065300
218. Wang X, Zeng W, Soyombo AA, Tang W, Ross EM, Barnes AP, et al. Spinophilin regulates Ca2+ signalling by binding the N-terminal domain of RGS2 and the third intracellular loop of G-protein-coupled receptors. Nat Cell Biol (2005) 7:405–11. doi:10.1038/ncb1237
219. Grabrucker S, Proepper C, Mangus K, Eckert M, Chhabra R, Schmeisser MJ, et al. The PSD protein ProSAP2/Shank3 displays synapto-nuclear shuttling which is deregulated in a schizophrenia-associated mutation. Exp Neurol (2014) 253:126–37. doi:10.1016/j.expneurol.2013.12.015
220. Qiao H, Li MX, Xu C, Chen HB, An SC, Ma XM. Dendritic spines in depression: what we learned from animal models. Neural Plast (2016) 2016:8056370. doi:10.1155/2016/8056370
221. Dean B, Gibbons AS, Boer S, Uezato A, Meador-Woodruff J, Scarr E, et al. Changes in cortical N-methyl-d-aspartate receptors and post-synaptic density protein 95 in schizophrenia, mood disorders and suicide. Aust N Z J Psychiatry (2016) 50:275–83. doi:10.1177/0004867415586601
222. de Bartolomeis A, Iasevoli F, Marmo F, Buonaguro EF, Eramo A, Rossi R, et al. Progressive recruitment of cortical and striatal regions by inducible postsynaptic density transcripts after increasing doses of antipsychotics with different receptor profiles: insights for psychosis treatment. Eur Neuropsychopharmacol (2015) 25:566–82. doi:10.1016/j.euroneuro.2015.01.003
223. de Bartolomeis A, Marmo F, Buonaguro EF, Latte G, Tomasetti C, Iasevoli F. Switching antipsychotics: imaging the differential effect on the topography of postsynaptic density transcripts in antipsychotic-naive vs. antipsychotic-exposed rats. Prog Neuropsychopharmacol Biol Psychiatry (2016) 70:24–38. doi:10.1016/j.pnpbp.2016.04.015
224. Iasevoli F, Tomasetti C, Marmo F, Bravi D, Arnt J, de Bartolomeis A. Divergent acute and chronic modulation of glutamatergic postsynaptic density genes expression by the antipsychotics haloperidol and sertindole. Psychopharmacology (Berl) (2010) 212:329–44. doi:10.1007/s00213-010-1954-0
225. Luoni A, Macchi F, Papp M, Molteni R, Riva MA. Lurasidone exerts antidepressant properties in the chronic mild stress model through the regulation of synaptic and neuroplastic mechanisms in the rat prefrontal cortex. Int J Neuropsychopharmacol (2015) 18(4):pyu061. doi:10.1093/ijnp/pyu061
226. O’Leary OF, Wu X, Castren E. Chronic fluoxetine treatment increases expression of synaptic proteins in the hippocampus of the ovariectomized rat: role of BDNF signalling. Psychoneuroendocrinology (2009) 34:367–81. doi:10.1016/j.psyneuen.2008.09.015
227. Peykov S, Berkel S, Schoen M, Weiss K, Degenhardt F, Strohmaier J, et al. Identification and functional characterization of rare SHANK2 variants in schizophrenia. Mol Psychiatry (2015) 20:1489–98. doi:10.1038/mp.2014.172
228. Zhang W, Wu J, Ward MD, Yang S, Chuang YA, Xiao M, et al. Structural basis of arc binding to synaptic proteins: implications for cognitive disease. Neuron (2015) 86:490–500. doi:10.1016/j.neuron.2015.03.030
229. Stan TL, Sousa VC, Zhang X, Ono M, Svenningsson P. Lurasidone and fluoxetine reduce novelty-induced hypophagia and NMDA receptor subunit and PSD-95 expression in mouse brain. Eur Neuropsychopharmacol (2015) 25:1714–22. doi:10.1016/j.euroneuro.2015.07.007
230. Dell’aversano C, Tomasetti C, Iasevoli F, de Bartolomeis A. Antipsychotic and antidepressant co-treatment: effects on transcripts of inducible postsynaptic density genes possibly implicated in behavioural disorders. Brain Res Bull (2009) 79:123–9. doi:10.1016/j.brainresbull.2009.01.006
231. de Bartolomeis A, Avvisati L, Iasevoli F, Tomasetti C. Intracellular pathways of antipsychotic combined therapies: implication for psychiatric disorders treatment. Eur J Pharmacol (2013) 718:502–23. doi:10.1016/j.ejphar.2013.06.034
232. Wadenberg ML, Wiker C, Svensson TH. Enhanced efficacy of both typical and atypical antipsychotic drugs by adjunctive alpha2 adrenoceptor blockade: experimental evidence. Int J Neuropsychopharmacol (2007) 10:191–202. doi:10.1017/S1461145706006638
233. Wilson MC. Coloboma mouse mutant as an animal model of hyperkinesis and attention deficit hyperactivity disorder. Neurosci Biobehav Rev (2000) 24:51–7. doi:10.1016/S0149-7634(99)00064-0
234. Bruno KJ, Hess EJ. The α2C-adrenergic receptor mediates hyperactivity of coloboma mice, a model of attention deficit hyperactivity disorder. Neurobiol Dis (2006) 23:679–88. doi:10.1016/j.nbd.2006.05.007
235. Gijsman HJ, Geddes JR, Rendell JM, Nolen WA, Goodwin GM. Antidepressants for bipolar depression: a systematic review of randomized, controlled trials. Am J Psychiatry (2004) 161:1537–47. doi:10.1176/appi.ajp.161.9.1537
236. Palazidou E. Quetiapine: a new option in bipolar depression. Future Prescr (2009) 10:9–14. doi:10.1002/fps.56
237. Sanford M, Keating GM. Quetiapine: a review of its use in the management of bipolar depression. CNS Drugs (2012) 26:435–60. doi:10.2165/11203840-000000000-00000
238. Garner M, Mohler H, Stein DJ, Mueggler T, Baldwin DS. Research in anxiety disorders: from the bench to the bedside. Eur Neuropsychopharmacol (2009) 19:381–90. doi:10.1016/j.euroneuro.2009.01.011
239. Brand SJ, Harvey BH. Exploring a post-traumatic stress disorder paradigm in Flinders sensitive line rats to model treatment-resistant depression I: bio-behavioural validation and response to imipramine. Acta Neuropsychiatr (2017) 29:193–206. doi:10.1017/neu.2016.44
240. Brand SJ, Harvey BH. Exploring a post-traumatic stress disorder paradigm in Flinders sensitive line rats to model treatment-resistant depression II: response to antidepressant augmentation strategies. Acta Neuropsychiatr (2017) 29:207–21. doi:10.1017/neu.2016.50
241. El-Hage W, Leman S, Camus V, Belzung C. Mechanisms of antidepressant resistance. Front Pharmacol (2013) 4:146. doi:10.3389/fphar.2013.00146
242. Millan MJ, Goodwin GM, Meyer-Lindenberg A, Ove Ogren S. Learning from the past and looking to the future: emerging perspectives for improving the treatment of psychiatric disorders. Eur Neuropsychopharmacol (2015) 25:599–656. doi:10.1016/j.euroneuro.2015.01.016
243. Cleary L, Murad K, Bexis S, Docherty JR. The α1D-adrenoceptor antagonist BMY 7378 is also an α2C-adrenoceptor antagonist. Auton Autacoid Pharmacol (2005) 25:135–41. doi:10.1111/j.1474-8673.2005.00342.x
244. Patel SD, Habeski WM, Min H, Zhang J, Roof R, Snyder B, et al. Identification and SAR around N-{2-[4-(2,3-dihydro-benzo[1,4]dioxin-2-ylmethyl)-[1,4]diazepan-1-yl]-ethyl}-2-phenoxy-nicotinamide, a selective α2C adrenergic receptor antagonist. Bioorg Med Chem Lett (2008) 18:5689–93. doi:10.1016/j.bmcl.2008.08.055
245. Corboz MR, Rivelli MA, McCormick KD, Wan Y, Shah H, Umland S, et al. Pharmacological characterization of a novel alpha2C-adrenoceptor agonist N-[3,4-dihydro-4-(1H-imidazol-4-ylmethyl)-2H-1, 4-benzoxazin-6-yl]-N-ethyl-N’-methylurea (compound A). J Pharmacol Exp Ther (2011) 337:256–66. doi:10.1124/jpet.110.175794
246. Luoto P, Suilamo S, Oikonen V, Arponen E, Helin S, Herttuainen J, et al. 11C-ORM-13070, a novel PET ligand for brain alpha2C-adrenoceptors: radiometabolism, plasma pharmacokinetics, whole-body distribution and radiation dosimetry in healthy men. Eur J Nucl Med Mol Imaging (2014) 41:1947–56. doi:10.1007/s00259-014-2782-y
247. Corboz MR, Rivelli MA, Shah H, Boyce CW, McCormick KD, Chapman RW, et al. Role of α2-adrenoceptors in electrical field stimulation-induced contraction of pig nasal mucosa and pharmacologic characterization of a novel α2C-adrenoceptor agonist. Am J Rhinol Allergy (2013) 27:84–90. doi:10.2500/ajra.2013.27.3842
248. Dooley K, Zon LI. Zebrafish: a model system for the study of human disease. Curr Opin Genet Dev (2000) 10:252–6. doi:10.1016/S0959-437X(00)00074-5
249. Ruuskanen JO, Peitsaro N, Kaslin JV, Panula P, Scheinin M. Expression and function of alpha-adrenoceptors in zebrafish: drug effects, mRNA and receptor distributions. J Neurochem (2005) 94:1559–69. doi:10.1111/j.1471-4159.2005.03305.x
Keywords: Alzheimer’s disease, α2C-antagonism, schizophrenia, depression, cognition, ORM-10921
Citation: Uys MM, Shahid M and Harvey BH (2017) Therapeutic Potential of Selectively Targeting the α2C-Adrenoceptor in Cognition, Depression, and Schizophrenia—New Developments and Future Perspective. Front. Psychiatry 8:144. doi: 10.3389/fpsyt.2017.00144
Received: 17 October 2016; Accepted: 24 July 2017;
Published: 14 August 2017
Edited by:
Ming D. Li, Zhejiang University, ChinaReviewed by:
Carlos M. Opazo, The University of Melbourne, AustraliaCopyright: © 2017 Uys, Shahid and Harvey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian Herbert Harvey, YnJpYW4uaGFydmV5QG53dS5hYy56YQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.