 Jeroen C. de Jonge1
Jeroen C. de Jonge1 Christiaan H. Vinkers
Christiaan H. Vinkers Hilleke E. Hulshoff Pol
Hilleke E. Hulshoff Pol Anouk Marsman
Anouk Marsman
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 11 August 2017
Sec. Neuroimaging
Volume 8 - 2017 | https://doi.org/10.3389/fpsyt.2017.00118
This article is part of the Research Topic MR Spectroscopy in Neuropsychiatry View all 10 articles
Schizophrenia is a psychiatric disorder characterized by hallucinations, delusions, disorganized thinking, and impairments in cognitive functioning. Evidence from postmortem studies suggests that alterations in cortical γ-aminobutyric acid (GABAergic) neurons contribute to the clinical features of schizophrenia. In vivo measurement of brain GABA levels using magnetic resonance spectroscopy (MRS) offers the possibility to provide more insight into the relationship between problems in GABAergic neurotransmission and clinical symptoms of schizophrenia patients. This study reviews and links alterations in the GABA system in postmortem studies, animal models, and human studies in schizophrenia. Converging evidence implicates alterations in both presynaptic and postsynaptic components of GABAergic neurotransmission in schizophrenia, and GABA may thus play an important role in the pathophysiology of schizophrenia. MRS studies can provide direct insight into the GABAergic mechanisms underlying the development of schizophrenia as well as changes during its course.
Schizophrenia is a severe chronic psychiatric disorder characterized by hallucinations, delusions, disorganized thinking, and impairments in cognitive functioning, affecting approximately 1% of the population. Several lines of evidence suggest that abnormalities of specific cortical inhibitory neurons and its neurotransmitter γ-aminobutyric acid (GABA) could play an important role in the pathophysiology of schizophrenia (1). The current evidence on GABAergic abnormalities in schizophrenia is mostly based on postmortem studies and has not yet provided a conclusive answer about GABAergic alterations and activity in schizophrenia. In vivo measurements of GABA in schizophrenia may reveal additional insights. The aim of this study is to review the findings of postmortem and animal studies on different components of GABAergic neurotransmission and in vivo magnetic resonance spectroscopy (MRS) findings on GABA levels in the brains of patients with schizophrenia. To collect relevant literature, a PubMed search was performed using the following terms: ((schizophrenia [tiab] OR schizophrenic* [tiab]) AND (glutamate decarboxylase [tiab] OR glutamic acid decarboxylase [tiab] OR GAD [tiab] OR GAD67 [tiab] OR GAD65 [tiab] OR GABA [tiab] OR gamma-aminobutyric acid [tiab] OR glutamate [tiab] OR glutamergic [tiab] OR gene expression [tiab])).
GABA is synthesized by decarboxylation of glutamate by glutamic acid decarboxylase (GAD) (Figure 1) (2). Based on its molecular weight, it is possible to distinguish two isotypes, the 65 kDa isotype GAD65 and the 67 kDa isotype GAD67, which are involved in different aspects of GABAergic neurotransmission (3). GAD65 is responsible for rapid synthesis of GABA during periods of high synaptic demand; it is predominantly located on axon terminals and synaptic vesicle membranes and is thus primarily associated with packaging and release of GABA (4–7). GAD67 is responsible for basal GABA levels (4, 5) and the majority (80–90%) of GABA synthesis (8); it is located in the cytosol and is thus primarily associated with GABA synthesis and non-vesicular release (6, 7).
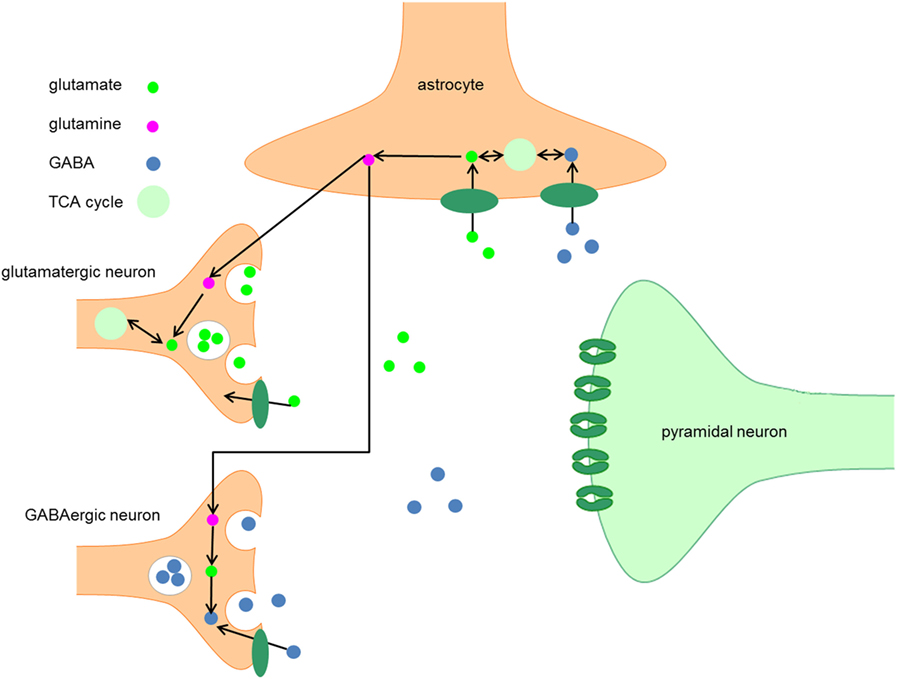
Figure 1. Metabolism of GABA. After synthesis in the presynaptic terminal of GABA neurons, GABA is packaged into vesicles by the vesicular GABA transporter, which is embedded in the vesicular membrane. The synaptic activity of GABA is terminated when GABA is taken up by GABA transporters embedded in the plasma membranes of neurons and astrocytes. When GABA is taken up by neurons, it can be either repacked in vesicles for neurotransmission or it can be degraded by the enzyme GABA transaminase to succinic semialdehyde (SSA). After conversion of SSA to succinate, it enters the TCA cycle and is subsequently converted into glutamate. The following conversion of glutamate to GABA by GAD65 and GAD67 completes the GABA cycle.
After synthesis in the presynaptic terminal, GABA is packaged into vesicles by the vesicular GABA transporter (VGAT), which is embedded in the vesicular membrane (9). A presynaptic action potential can induce a Ca2+-mediated fusion of the vesicle membrane and the presynaptic neuron membrane, which leads to release of GABA into the synaptic cleft. Alternatively, after strong depolarization or altered ion homeostasis, specific GABA transporters (GAT) may reverse their direction resulting in non-vesicular release of GABA (9, 10).
After release into the synaptic cleft, GABA exerts its inhibitory activity by binding to two types of receptors, such as GABAA and GABAB receptors. GABAA receptors are ligand-gated Cl− channels and produce most of the physiological actions of GABA (11). GABAA receptors have a pentameric subunit structure derived from different gene families and include α, β, γ, δ, ε, π, and θ subunits. Some of these subunits have several isoforms (α1–6, β1–3, and γ1–3) (12). In most cases, the pentamers of subunits include a pair of α subunits and a pair of β subunits in combination with a fifth subunit (γ or δ) (13).
The synaptic activity of GABA is terminated when GABA is taken up by GAT that are embedded in the membranes of neurons and astrocytes (10). In humans, four types of GAT can be distinguished, GAT-1 to 3 and the betaine GABA transporter (BGT-1). GAT-1 is widely expressed in the brain, predominantly in presynaptic GABA neurons, and is thus primarily responsible for GABA reuptake (9, 10). GAT-3 is primarily responsible for GABA uptake into local astrocytes (14). In contrast to GAT-1 and GAT-3, GAT-2 and BGT-1 play a very limited role in GABAergic neurotransmission (10).
When GABA is taken up by neurons, it can either be repacked into vesicles or it can be degraded to succinic semialdehyde (SSA) by the enzyme GABA transaminase. After conversion of SSA to succinate, the latter enters the TCA cycle and is subsequently converted into glutamate (10, 15). The following conversion of glutamate to GABA by GAD65 or GAD67 completes the GABA cycle (Figure 1).
One of the most consistent postmortem findings in schizophrenia is a reduction of mRNA encoding for GAD67 in the dorsolateral prefrontal cortex (DLPFC) in layers 1 through 5 (3–5, 16–29), which results in a reduction of GAD67 protein levels although this has been less extensively studied (Table 1) (4, 30, 31). Since the majority of studies reported unaltered or increased neuronal density, it is unlikely that the reduction of GAD67 mRNA can be attributed to a decrease in the number of neurons in schizophrenia (16, 27, 32, 33). Rather, the density of neurons expressing a detectable level of GAD67 mRNA is decreased (27); expression of GAD67 mRNA is decreased below a detectable level in 25–35% of GABAergic neurons, while the remaining neurons have GAD67 mRNA levels similar to controls (27, 29). It has therefore been suggested that impaired GAD67 gene expression is limited to a certain subset of GABAergic neurons (27, 31). This subset could concern the chandelier, double bouquet, or wide-arbor neurons, which can be distinguished by the presence of specific calcium-binding proteins (Box 1) (1).
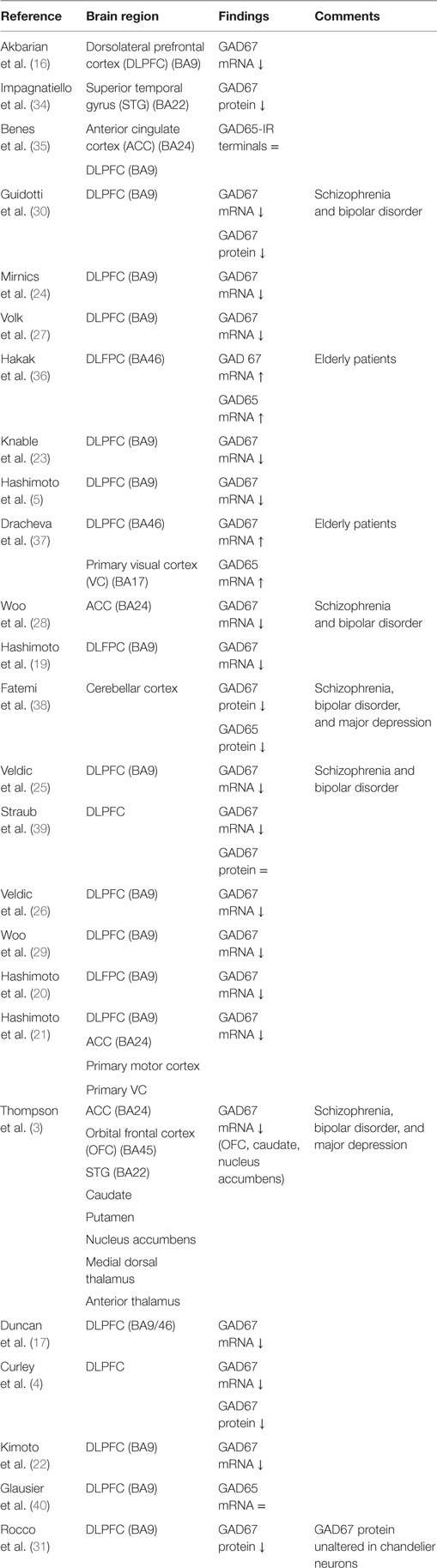
Table 1. Postmortem studies on glutamic acid decarboxylase (GAD) in schizophrenia.
Box 1. Subsets of GABAergic neurons.
Based on molecular, morphological, and physiological features, it is possible to distinguish different subsets of cortical GABA neurons, with the double bouquet, basket, and chandelier cells being the most abundant cortical GABAergic interneuron subsets (1, 18). The subpopulations have different influences on the regulation of information processing in the dorsolateral prefrontal cortex (DLPFC), partly because the axons of the GABAergic interneurons synapse at different locations on the pyramidal neuron (1, 41, 42). Furthermore, it is possible to identify certain morphological and functional subgroups of GABA neurons which contain different calcium-binding proteins (43–45).
Chandelier neurons synapse at axon initial segments (AIS) of pyramidal neurons and therefore provide inhibitory inputs to the AIS. These synaptic connections are formed in such a way that vertical arrays, so-called “cartridges,” are formed (1, 46). Furthermore, these neurons contain the calcium-binding protein parvalbumin (5, 47).
Basket or wide-arbor neurons synapse at cell bodies and proximal dendrites of pyramidal neurons. Similar to chandelier neurons, basket cells in the prefrontal cortex contain the protein parvalbumin (43).
Double bouquet neurons contain the calcium-binding protein calbindin and target the distal dendrites of pyramidal neurons (1, 48).
A third calcium-binding protein, calretinin, is expressed by approximately 50% GABAergic neurons, mainly double bouquet cells, in the DLPFC (43).
Since the parvalbumin-containing chandelier and basket neurons synapse at the AIS and soma, respectively, they provide a much stronger inhibitory regulation of the pyramidal neurons as compared to double bouquet cells, which synapse at the distal dendrites (49, 50). Given the heterogeneity in synaptic targets and specific features of the different subclasses of GABAergic neurons, altered interactions between different GABAergic neurons and pyramidal neurons may influence neuronal activity and hence functional output in different manners.
The subset that is affected in schizophrenia appears to include parvalbumin-containing GABAergic neurons. In schizophrenia, parvalbumin mRNA expression is reduced in prefrontal cortex (PFC) layers 3 and 4, but not layers 2, 5, or 6 (5, 27, 29, 51). The overall expression of parvalbumin mRNA is decreased whereas the density of neurons expressing detectable levels of parvalbumin is unaltered (5, 52, 53), implying that the reduction of parvalbumin mRNA is not accompanied by a loss of parvalbumin-containing neurons. The reduced parvalbumin mRNA expression is associated with the decreased density of GAD67 mRNA-positive GABAergic neurons. 50% of the parvalbumin-positive neurons lack detectable amounts of GAD67 mRNA (5), whereas calretinin mRNA (which is expressed by a different subset of neurons—see Box 1) expression and the density of calretinin-positive neurons remain unchanged in schizophrenia (5, 54). These findings imply that the reduced GAD67 mRNA expression may be selective for the parvalbumin-containing subgroup of GABA neurons in the PFC (5). Recent evidence suggests that GAD67 protein levels are unaltered in the chandelier neurons, suggesting that other parvalbumin-containing neurons, such as the basket cells, are involved (31).
The observed alterations regarding parvalbumin are not likely to be caused by exposure to antipsychotic medication. Long-term exposure to haloperidol and benzotropine did not lead to an altered expression of parvalbumin mRNA (5). Furthermore, transcript levels for parvalbumin were reduced to the same extent in the DLPFC of medication-naïve patients compared to patients receiving antipsychotic medication (20). Animal studies have shown that treatment with dopamine D2-receptor antagonists influences the expression of GAD67 mRNA in the basal ganglia (BG) (55–58) but not in the PFC; however, D2-receptor density in the PFC is much lower than in the BG (20, 27).
The transporter protein GAT-1 is present in the presynaptic neuron and is responsible for the synaptic reuptake of GABA (19, 59). It plays a role in both tonic and phasic GABA-mediated inhibition (60, 61). GAT-1 terminates the synaptic activity of GABA and regulates the duration and efficacy of synaptic GABAergic neurotransmission (62); therefore, reduced GAT-1 levels suggest increased availability of GABA in the synapse (63). Several studies found reduced mRNA levels encoding for the GAT-1 protein in schizophrenia. GAT-1 mRNA levels are decreased in GABAergic neurons in the DLPFC (Table 2) (20, 21, 59, 62, 64). Together with the diminished expression of GAD67 mRNA, it is unclear whether this results in a net increase or decrease of the inhibitory tone on pyramidal cells (63). Moreover, GAT-1 mRNA expression is reduced below detectable levels in a subset of GABAergic neurons and relatively unaltered in the majority of the GABAergic neurons (59). The affected subset appears to include parvalbumin-containing neurons (1, 59). The reduction of GAT-1 mRNA expression is limited to layers 2 through 5, the same layers in which parvalbumin-containing neurons are found (59, 65).
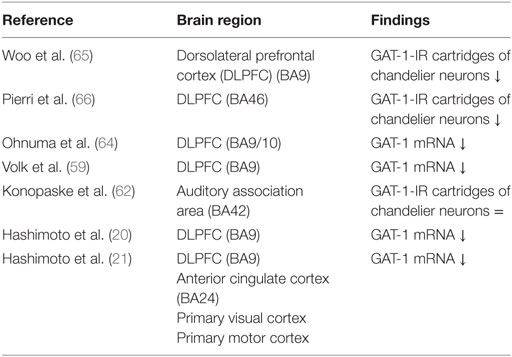
Table 2. Postmortem studies on GABA transporters (GAT)-1 in schizophrenia.
The subset of GABAergic neurons where reduced GAT-1 mRNA levels are detected is possibly the subset of chandelier neurons (see Box 1). A marker of chandelier neurons is their GAT-1 immunoreactivity; the density of GAT-1 immunoreactive cartridges is decreased in schizophrenia, while markers of other axon terminal populations remain unchanged (65, 66). The lower density of GAT-1 immunoreactive cartridges implies decreased GAT-1 protein, which is associated with decreased GAT-1 mRNA levels. Putting together these findings, reduced GAT-1 mRNA levels may therefore account for the decreased density of GAT-1 immunoreactive axon cartridges in chandelier neurons (59). The reduction of GAT-1 immunoreactive cartridges cannot be attributed to a reduction of chandelier neurons, since the density of GABAergic neurons [identified by parvalbumin (52, 53) and VGAT (67)] is unchanged. Thus concluding, the density of chandelier neurons containing GAT-1 protein in the DLPFC in patients with schizophrenia was reduced whereas the density of parvalbumin-containing neurons remains unaltered. This finding suggests that the reduced levels of GAT-1 mRNA are limited to the chandelier neurons (29, 65).
Long-term exposure to therapeutic blood levels of haloperidol in monkeys did not result in changes in the expression of GAT-1 mRNA or the expression of GAT-1 protein (65, 66, 68), nor did effects of alcohol abuse or benzodiazepine use explain the findings (20, 21).
GABAA receptors are ligand-gated chloride ion channels and produce most of the physiological actions of GABA (11). GABAA receptors have a pentameric subunit structure and the subunits are derived from different gene families encoding for different subunits including α1–6, β1–3, γ1–3, δ, ε, π, and θ (12). The pentamers of subunits include in most cases a pair of α subunits and a pair of β subunits in combination with a fifth subunit (γ or δ) (13). Early studies demonstrated increased binding of muscimol, a selective GABAA receptor agonist, in pyramidal neuronal cell bodies in patients with schizophrenia (69–71); however, muscimol can bind to all types of GABAA receptor subunits. Recent advancements in technology have enabled investigation of deficits of individual GABAA receptor subunits (72).
Subunits of the α-type can be characterized by their subcellular localization within the central nervous system. Over 95% of the GABAergic synapses on the axon initial segment (AIS) of pyramidal neurons contain the α2 subunit, while only 15% of cortical GABA receptors contain the α2 subunit (73, 74). It appears that this subunit is characterized by high affinity, fast activation, and slow deactivation (75). Given its anatomical position and functional features, the GABAA α2 subunit serves as a major source for inhibitory tone on pyramidal neurons (46). Parvalbumin-containing neurons, which appear to exhibit a reduced expression of GAT-1 and GAD67 mRNA in schizophrenia, target the AIS of pyramidal neurons. Indeed, it has been demonstrated that in schizophrenia, the GABAA α2-receptor subunit is upregulated in the AIS of pyramidal neurons (46, 69, 76). This increase in α2 subunit density may occur in response to reduced extracellular GABA concentrations due to diminished GABA synthesis (1, 46). Furthermore, GAT-1 immunoreactive cartridges and the density of α2 subunits at the postsynapse of pyramidal neurons demonstrate an inverse correlation, which implies that GABAA α2 subunits are upregulated at the AIS of pyramidal neurons and GAT-1 is downregulated to provide a synergetic compensation for the diminished GABAergic activity (46). In contrast to GAD67 mRNA and GAT-1 mRNA, mRNA expression levels of postsynaptic GABAA α2-receptor subunits seem to be unaltered (16, 17). Reductions of α2-receptor subunits are exclusively found at the AIS synapses; the lack of upregulation of α2 subunit mRNA might be explained by the fact that inhibitory synapses at the AIS of pyramidal neurons make up less than 10% of the total number of inhibitory synapses of the pyramidal neuron (16, 77).
mRNA levels of the GABAA α1, γ2, α4, α5, and δ receptor subunits are suggested to be downregulated in the DLPFC of patients with schizophrenia (Table 3) (20, 21, 47, 78–80). However, two studies reported an increase of α1 subunit mRNA expression (34, 64), one study revealed an increase of α5 subunit mRNA (34), one study observed an increase of the GABAA receptor α1 subunit protein (72), and one study demonstrated no change of the α4 receptor subunit (80). In contrast to the a2 subunit localized at the AIS of pyramidal neurons, GABAA receptors containing the α1, α5, γ2, and δ (often co-expressed by α4) subunits are predominantly localized in the dendrites of pyramidal neurons (73, 81, 82). The observed alterations in the postsynaptic GABAA receptors do not seem to be a consequence of an increased number of neurons, because the majority of studies have reported no change or an increase in neuron density (27, 32, 33, 51).
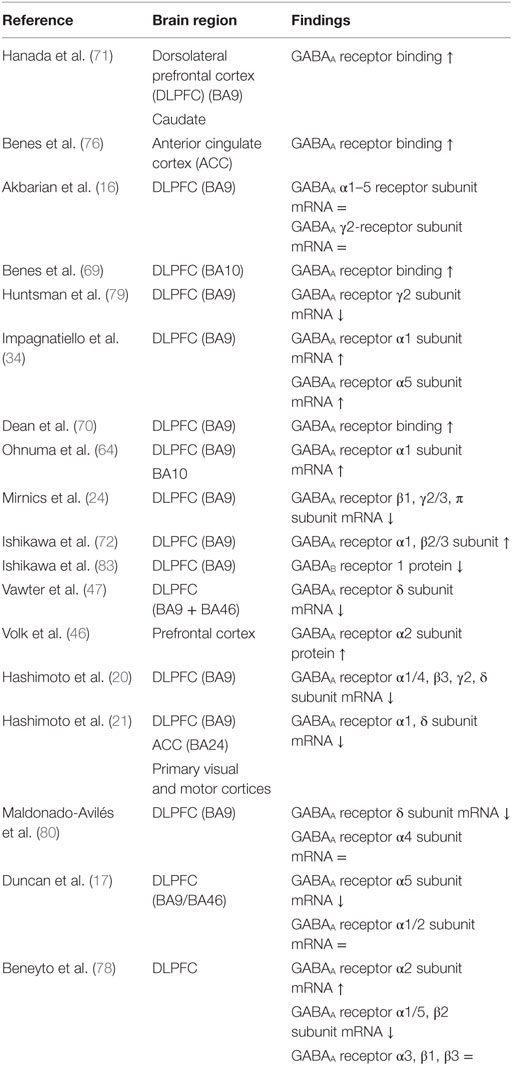
Table 3. Postmortem studies on postsynaptic GABA receptors in schizophrenia.
Animal studies in which rats were exposed to benzodiazepines did not reveal changes in the expression level of α2 subunit mRNA or protein levels and long-term exposure to haloperidol or olanzapine did not result in altered α1, α2, α5, β2, or δ subunit mRNA levels (20, 78, 84). Postmortem studies show that α1 and δ subunits are reduced to the same extent in the DLPFC of patients who were not taking antipsychotic medication at the time of death, which is unlikely to be driven by the effects of alcohol abuse or benzodiazepine use (20, 21). (For an overview of pre- and postsynaptic GABAergic alterations, see Figure 2.)
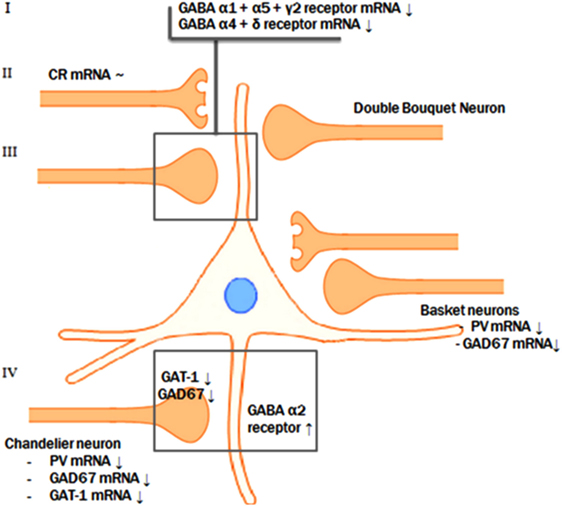
Figure 2. Pre- and postsynaptic GABAergic alterations. The reductions of GAD67 mRNA, parvalbumin mRNA, and GAT-1 mRNA levels in the parvalbumin-containing chandelier neurons seem to result in a compensatory postsynaptic upregulation of α2-receptor at the axon initial segment of the pyramidal neuron. Presynaptic alterations in neurons targeting the dendritic domain of the pyramid neuron might also be accompanied by abnormalities of the postsynaptic GABA α1, α5, and γ2 and the extrasynaptic α4 and δ receptor subunits.
There is sufficient histological–pathological evidence to link impairments in GABAergic neurotransmission in other cortical regions than the DLPFC to pathologies and cognitive dysfunctions observed in schizophrenia (63).
Similar to the DLPFC, the anterior cingulate cortex (ACC), primary visual cortex (VC), and primary motor cortex are characterized by the same deficits in GABAergic gene expression as seen in the DLPFC, including selective involvement of parvalbumin-containing subsets of GABA neurons. The largest declines were reported for the levels of mRNA encoding for parvalbumin (21). These brain areas also exhibit a decrease of GAD67 mRNA, GAD65 mRNA, GAT-1 mRNA, and GABAA receptor α1 and δ subunits (1, 21, 28). Calretinin levels remained unchanged (21). GABA-related transcript expression is suggested to be decreased to the same extent in all aforementioned brain regions, so there possibly is no preferential involvement of the DLPFC (21). The reduced expression of GABAA receptor α1 and δ subunits in these cortical areas also imply that reduced phasic and tonic inhibition, respectively, might be a feature shared by multiple cortical regions.
Furthermore, in addition to the ACC, primary VC, and primary motor cortex which demonstrated similar GABAergic expression deficits as the DLPFC, the orbital frontal cortex (OFC), superior temporal gyrus (STG), striatum, and thalamus show a diminished GAD67 mRNA expression as well (3). In addition, the STG and auditory gyri demonstrated reduced GAT-1 protein levels (34). Reduction in GABAergic activity in the OFC could lead to disturbances related to emotional and cognitive functioning and may therefore underlie symptoms regarding social withdrawal and apathetic behavior (85). In addition, abnormalities in the STG could contribute to deficit auditory processing and auditory hallucinations (3). These findings imply that the aberrations seen in the DLPFC may not be due to alterations in DLPFC circuitry only, but that the altered transcript levels appear to be the consequence of a common upstream mechanism that operates across multiple cortical areas.
A possible integrative model for the alterations in GABA neurotransmission is that a subset of prefrontal GABA neurons is affected in schizophrenia. In contrast to the reduced GAD67 and the consequent attenuation of inhibitory GABAergic neurotransmission, the reduction of GAT-1 mRNA expression tends to increase the synaptic activity of GABA (63). In addition, GABAA receptors are upregulated in postsynaptic pyramidal neurons, which suggests a compensatory increase in response to the decreased extracellular GABA concentrations (46, 70, 76). However, based on postmortem studies, it is not possible to identify the initial deficit in the pathological chain and, therefore, two scenarios are possible (see Figure 2).
The most likely scenario is an overall reduced GABAergic activity in schizophrenia. This implies that the initial step in this specific pathologic process is the presynaptic reduction of GABA synthesis, followed by a secondary, compensatory reduction of reuptake by means of GAT-1 and by compensatory upregulation of postsynaptic GABA receptors (1, 18, 86). This synergetic attempt, to improve the GABAergic neurotransmission at the synapse of the pyramidal neuron AIS, serves to compensate for the initial deficit in synthesis of GABA. Consistent with the theory that the reduction of synthesis is the first step in the pathological chain, mice lacking the GAT-1 gene do not develop diminished levels of GAD67 mRNA. This indicates that the reduction of GAD67 is the initial event (87). Furthermore, GABA hypofunction due to decreased synthesis reflected by the diminished levels of GAD67 mRNA was imitated in rats by means of pharmacological blockade of prefrontal GABAA receptors. This resulted in impaired working memory performance, a cognitive function characteristically disturbed in patients with schizophrenia (88, 89). However, it is still controversial whether the compensatory mechanisms are sufficient to overcome the decreased GABA synthesis. In other words, it is unknown if the net effect of the diminished presynaptic synthesis on the one hand and the decreased reuptake increased postsynaptic reception on the other hand result in an increase or decrease of the inhibitory tone on pyramidal cells by GABAergic neurons (63). In conclusion, the most likely scenario is that reduced presynaptic GABA production results in a reduced reuptake of GABA and in upregulated postsynaptic GABA receptors in schizophrenia.
Alternatively, an excessive increase of GABAergic activity due to both primary diminished reuptake and upregulated postsynaptic receptors may also be an initial step in the pathological process followed by secondary compensatory downregulation of GAD67 mRNA in chandelier neurons due to the excessive GABAergic activity. Furthermore, the effects of pharmaceuticals involved in GABAergic neurotransmission seem to be in line with the hypothesis of excess GABAergic activity. For example, lorazepam, a positive allosteric modulator of GABAergic neurotransmission, results in a deterioration of working memory aberrations while flumazenil, a partial inverse agonist, leads to improvement of the working memory deficits (63). Thus, according to this scenario, excessive GABAergic activity could be the result of an initial postsynaptic upregulation of the GABAA receptor and downregulation of the presynaptic GABA reuptake transporters as a first step in the pathological chain (63).
Finally, the aberrations seen in the DLPFC may not be due to alterations in DLPFC circuitry, but instead reflect transcript levels that are a consequence of a common upstream mechanism that operates across multiple cortical areas in schizophrenia.
In conclusion, the most likely scenario involves reduced GABA concentrations due to a compromised production of GABA reflected by the diminished concentration of GAD67 mRNA. However, due to the observation that presynaptic GAT-1 is reduced and postsynaptic receptors are upregulated, postmortem studies do not provide a conclusive answer about the net GABAergic concentrations and activity. Therefore, in vivo studies could provide additional insights into GABA levels in clinical states contributing to a more definitive formulation about the pathological cascade and GABAergic alterations in schizophrenia.
GABA can be measured in vivo using proton MRS (1H-MRS). MRS provides a means to non-invasively identify and quantify metabolites in tissue and can be carried out with an MR scanner. MRS makes use of the magnetic properties of nuclei, e.g., the proton (1H). Because the magnetic properties of a nucleus are influenced by its chemical environment, it is possible to identify signals from different molecules within the MR spectrum. However, measurement of GABA with 1H-MRS is challenging since its low concentration results in a relatively small signal which is overlapped by more intense signals from more abundant metabolites. It is possible to separate the GABA signal from other, more intense signals with spectral editing techniques. With spectral editing the magnetic properties of a specific molecule are used to improve detection of that molecule.
Based on presynaptic and postsynaptic GABAergic alterations in postmortem studies, it is possible to identify numerous brain areas such as the ACC, primary VC, primary motor cortex, OFC, BG, STG, thalamus, but especially the DLPFC in which it is expected to measure altered GABAergic concentrations by 1H-MRS. As mentioned before, postmortem studies do not provide a conclusive answer about the net GABAergic concentrations and activity. Therefore, 1H-MRS could provide additional insights, contributing to a more definitive formulation about the pathological cascade and GABAergic alterations in schizophrenia. However, up until now MRS studies on GABA in schizophrenia are rather scarce and only cross-sectional. Moreover, the current literature is inconsistent regarding the measured GABA levels in different brain regions of patients with schizophrenia. Currently, seven studies reported GABA reductions (90–96), six studies reported unchanged GABA levels (90, 92–94, 97, 98), and two studies reported increased levels (Table 4) (97, 99). Since GABA levels may differ in early (90, 91, 93, 94) and chronic schizophrenia (91, 93, 94, 98, 99), brain levels might also be dependent on the stage of the disease. Recent meta-analysis showed no changes in GABA levels in patients with schizophrenia in any given brain region, however, when averaging GABA levels across all measured brain regions per study, GABA appeared to be lower in patients compared to healthy controls (100).
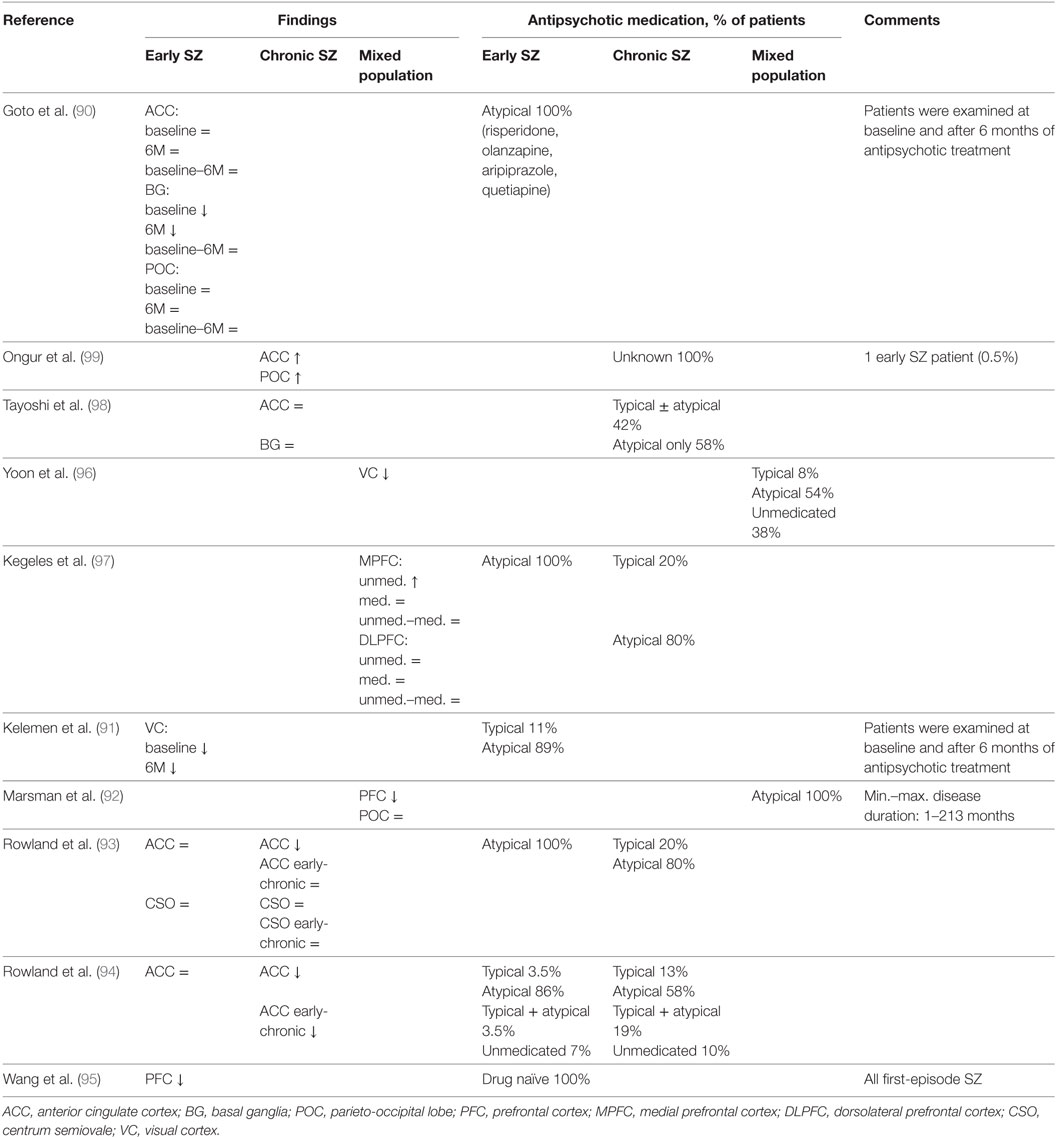
Table 4. In vivo magnetic resonance spectroscopy studies of GABA in schizophrenia.
The fluctuating and inconsistent findings of the few MRS studies that have been published so far in schizophrenia could be explained by several factors such as small and heterogeneous sample sizes, low magnetic field strengths resulting in a less robust measurement of GABA, methodological limitations leading to relatively large voxel volumes and marginal adjustments with regard to gray and white matter differences (Table 4) (15). Moreover, most studies measured GABA referenced to creatine and although this is a common approach, fluctuations in creatine concentrations could be, to a certain extent, responsible for the observed GABAergic findings. However, the most prominent limitation compromises the undetermined role of antipsychotic medication use with regard to GABA levels measured by 1H-MRS.
In 38 chronic schizophrenia patients, higher GABA concentrations were found in the left BG in patients using typical antipsychotics as compared to patients using atypical antipsychotics (82). Furthermore, a positive correlation was reported between GABA concentration in the left BG and anticholinergic medication (98). It is thus possible that antipsychotic medication influences GABA concentrations and different types of medications could have differing effects (98).
However, in patients diagnosed with schizophrenia and using antipsychotic medication at baseline, the use of atypical antipsychotics did not have any effects on GABA concentrations in the left BG, frontal lobe, and parieto-occipital lobe during a follow-up period of 6 months (90). At baseline, the concentration of GABA in the left BG in these first-episode patients was decreased (81), but this reduction was not reversed after 6 months of treatment with antipsychotic medication (84). Interestingly, clinical condition, assessed by PANSS scores, did improve during this time period. This suggests that medication use has no profound effect on GABA concentrations in patients with schizophrenia although there does occur a clinical improvement (90, 101). However, it is also possible that the medication regimen prevented further progressive reduction of GABA concentrations in these patients. Studying patients not taking antipsychotic medication may provide valuable additional insights regarding this matter. A recent study addressed this topic and evaluated GABA concentration in 16 unmedicated patients, consisting of 9 medication-naïve patients and 7 patients with no antipsychotic medication use 14 days prior to the investigation. This study observed higher GABA concentrations in never- and unmedicated patients compared to medicated patients (97). This implies that medication use might lead to a normalization of GABA concentrations (97). However, as mentioned before, medicated patients did not show any alterations regarding GABA concentrations after 6 months of antipsychotic therapy (90). Possibly, patients that were minimally treated at baseline differed from those that were medication naïve (90, 101), and the normalization of GABA concentrations due to antipsychotic treatment takes place at the beginning of the treatment. To formulate a conclusive answer, future studies are required, which assess both within-subject medication and medication-naïve study designs. In conclusion, many factors contribute to the inconsistency in literature and future studies need to take these factors into account to reconcile the fluctuating findings.
The observed changes in GABAergic neurotransmission may have functional significance (96). GABA measurement in the VC revealed reduced concentrations, and this decrease was positively correlated with orientation-specific surround suppression (OSSS) (96). OSSS is a behavioral measure of visual inhibition, and it is believed that this process relies on GABAergic neurotransmission in the VC (85). Furthermore, poorer performance on attention tests was correlated with decreased GABA concentrations in patients with schizophrenia (93). These observations are consistent with the GABA deficit hypothesis, which states that reduced GABAergic neurotransmission results in cognitive deficits, and imply that MRS is able to measure the pool of cortical GABA that has a direct relationship with GABA-mediated functions (15). Since the GABAergic expression deficits exhibit a widespread cortical involvement, it is likely that such aberrations generalize to other cortical areas (21, 96).
On the other hand, recent research showed a negative association between level of cognitive functioning and GABA level in the PFC in schizophrenia patients (92). Together with the finding that GABA levels are reduced in schizophrenia and albeit the finding that intelligence levels are lower in patients compared to matched healthy controls (102), this may imply that the GABA deficit hypothesis mainly applies to patients with lower intelligence (92). Alternatively, patients with higher intelligence may have better treatment compliance, possibly resulting in lower GABA levels (92).
The reported elevation of GABA levels in the MPFC by 1H-MRS in unmedicated patients seems to be inconsistent with the results of postmortem studies, which exhibit an impaired GABA synthesis of parvalbumin-containing subclasses of GABA neurons reflected by diminished GAD67 mRNA levels (97). This discrepancy could be explained by the extensive exposure of the postmortem brain samples to antipsychotic medication in predominantly chronically ill patients (18). Furthermore, the observed elevated GABA levels in the MPFC might also be an overcompensation of other subclasses of GABA neurons (97). The NMDA-receptor hypofunction hypothesis puts forward that an intrinsic deficit of GABA neurons, including impaired GABA synthesis, results in disinhibition of pyramidal neurons. The deficit regulation of pyramidal neurons by GABAergic neurotransmission leads to glutamate elevations (48, 103). Therefore, the remaining unimpaired subclasses (subclasses other than the parvalbumin-containing subclass) could be stimulated by the increased glutamergic activity, and this could serve as a compensation for the diminished synthesis in the parvalbumin-containing subclass (97).
Recent advantages in ultrahigh-field MR techniques allow for a more robust assessment of GABA levels, and future studies must point out whether in vivo measurement of GABA corresponds with the observed GABA deficiencies in postmortem tissues and whether the GABAergic deficits occur in a pan-cortical manner. Moreover, futures studies might point out if GABA concentrations predict functional outcome and if alterations in GABA concentrations relate to therapy response. It is clear that GABA measurement by in vivo MR spectroscopy could be of great value, but it is also evident that further work is needed to provide additional information on the validation of MR spectroscopy of GABA in schizophrenia.
Converging evidence implicates alterations in both presynaptic and postsynaptic components of GABA neurotransmission to fulfill an important role in the pathophysiology of schizophrenia. Multiple research sites using in situ hybridization, DNA microarray, or real-time quantitative PCR have consistently found reduced levels of GAD67 mRNA or a reduced density of neurons positive for GAD67 mRNA in the DLPFC as one of the most consistent findings with regard to pathological changes in schizophrenia. This decrease is the consequence of a reduction of GAD67 mRNA in a subset of GABA neurons. The affected neurons appear to include the parvalbumin-containing neurons. Parvalbumin-positive cells in the DLPFC include chandelier cells, targeting the upregulated α2-receptor subunit at the AIS of the pyramidal neuron. Furthermore, since GAD67 mRNA expression deficits were also observed in layers without parvalbumin expression, other subclasses may attribute to the observed GABAergic gene expression deficits as well. Furthermore, since other brain regions demonstrated similar GABAergic gene expression deficits as the DLPFC, disturbances in GABAergic neurotransmission could be the consequence of a common upstream effect. Therefore, identifying a common pathophysiology might give rise to new pharmacological opportunities in the treatment of schizophrenia. Measurement of GABA levels in vivo by means of MRS offers the possibility to approach the illness from a unique perspective and provides additional insights in the relationship between deficit components of GABA neurotransmission and GABA-mediated inhibitory activity. However, the current literature is inconsistent regarding the measured GABA levels in different brain regions of patients with schizophrenia. Future MRS studies using GABA editing are required to give us a better understanding of the pathophysiology of schizophrenia in different stages of the disease. Particularly GABA-editing at ultrahigh-field strengths will be beneficial for detection of the relatively small GABA signal, because of the increased sensitivity, resolution, and signal-to-noise ratio, allowing for an accurate and time-efficient assessment of GABA levels.
JJ contributed to the design of the study, performed literature research, and wrote and prepared the manuscript. CV contributed to the writing of the manuscript. HH contributed to the design of the study and the writing of the manuscript. AM contributed to the design of the study and supervised and contributed to the literature research, writing, and preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci (2005) 6(4):312–24. doi:10.1038/nrn1648
2. Petroff OA. GABA and glutamate in the human brain. Neuroscientist (2002) 8(6):562–73. doi:10.1177/1073858402238515
3. Thompson M, Weickert CS, Wyatt E, Webster MJ. Decreased glutamic acid decarboxylase(67) mRNA expression in multiple brain areas of patients with schizophrenia and mood disorders. J Psychiatr Res (2009) 43(11):970–7. doi:10.1016/j.jpsychires.2009.02.005
4. Curley AA, Arion D, Volk DW, Asafu-Adjei JK, Sampson AR, Fish KN, et al. Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: clinical, protein, and cell type-specific features. Am J Psychiatry (2011) 168(9):921–9. doi:10.1176/appi.ajp.2011.11010052
5. Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, et al. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci (2003) 23(15):6315–26.
6. Soghomonian JJ, Martin DL. Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci (1998) 19(12):500–5. doi:10.1016/S0165-6147(98)01270-X
7. Tian N, Petersen C, Kash S, Baekkeskov S, Copenhagen D, Nicoll R. The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc Natl Acad Sci U S A (1999) 96(22):12911–6. doi:10.1073/pnas.96.22.12911
8. Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, et al. Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem Biophys Res Commun (1996) 229(3):891–5. doi:10.1006/bbrc.1996.1898
9. Roth FC, Draguhn A. GABA metabolism and transport: effects on synaptic efficacy. Neural Plast (2012) 2012:805830. doi:10.1155/2012/805830
10. Rowley NM, Madsen KK, Schousboe A, Steve WH. Glutamate and GABA synthesis, release, transport and metabolism as targets for seizure control. Neurochem Int (2012) 61(4):546–58. doi:10.1016/j.neuint.2012.02.013
11. Sieghart W, Fuchs K, Tretter V, Ebert V, Jechlinger M, Höger H, et al. Structure and subunit composition of GABA(A) receptors. Neurochem Int (1999) 34(5):379–85. doi:10.1016/S0197-0186(99)00045-5
12. Mangan PS, Sun C, Carpenter M, Goodkin HP, Sieghart W, Kapur J. Cultured hippocampal pyramidal neurons express two kinds of GABAA receptors. Mol Pharmacol (2005) 67(3):775–88. doi:10.1124/mol.104.007385
13. Mohler H. GABA(A) receptor diversity and pharmacology. Cell Tissue Res (2006) 326(2):505–16. doi:10.1007/s00441-006-0284-3
14. Roettger VR, Amara SG. GABA and glutamate transporters: therapeutic and etiologic implications for epilepsy. Adv Neurol (1999) 79:551–60.
15. Maddock RJ, Buonocore MH. MR spectroscopic studies of the brain in psychiatric disorders. Curr Top Behav Neurosci (2012) 11:199–251. doi:10.1007/7854_2011_197
16. Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE Jr, et al. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry (1995) 52(4):258–66. doi:10.1001/archpsyc.1995.03950160008002
17. Duncan CE, Webster MJ, Rothmond DA, Bahn S, Elashoff M, Shannon WC. Prefrontal GABA(A) receptor alpha-subunit expression in normal postnatal human development and schizophrenia. J Psychiatr Res (2010) 44(10):673–81. doi:10.1016/j.jpsychires.2009.12.007
18. Guidotti A, Auta J, Davis JM, Dong E, Grayson DR, Veldic M, et al. GABAergic dysfunction in schizophrenia: new treatment strategies on the horizon. Psychopharmacology (Berl) (2005) 180(2):191–205. doi:10.1007/s00213-005-2212-8
19. Hashimoto T, Bergen SE, Nguyen QL, Xu B, Monteggia LM, Pierri JN, et al. Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J Neurosci (2005) 25(2):372–83. doi:10.1523/JNEUROSCI.4035-04.2005
20. Hashimoto T, Arion D, Unger T, Maldonado-Avilés JG, Morris HM, Volk DW, et al. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry (2008) 13(2):147–61. doi:10.1038/sj.mp.4002011
21. Hashimoto T, Bazmi HH, Mirnics K, Wu Q, Sampson AR, Lewis DA. Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am J Psychiatry (2008) 165(4):479–89. doi:10.1176/appi.ajp.2007.07081223
22. Kimoto S, Bazmi HH, Lewis DA. Lower expression of glutamic acid decarboxylase 67 in the prefrontal cortex in schizophrenia: contribution of altered regulation by Zif268. Am J Psychiatry (2014) 171(9):969–78. doi:10.1176/appi.ajp.2014.14010004
23. Knable MB, Barci BM, Bartko JJ, Webster MJ, Torrey EF. Molecular abnormalities in the major psychiatric illnesses: classification and regression tree (CRT) analysis of post-mortem prefrontal markers. Mol Psychiatry (2002) 7(4):392–404. doi:10.1038/sj.mp.4001034
24. Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron (2000) 28(1):53–67. doi:10.1016/S0896-6273(00)00085-4
25. Veldic M, Guidotti A, Maloku E, Davis JM, Costa E. In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proc Natl Acad Sci U S A (2005) 102(6):2152–7. doi:10.1073/pnas.0409665102
26. Veldic M, Kadriu B, Maloku E, Agis-Balboa RC, Guidotti A, Davis JM, et al. Epigenetic mechanisms expressed in basal ganglia GABAergic neurons differentiate schizophrenia from bipolar disorder. Schizophr Res (2007) 91(1–3):51–61. doi:10.1016/j.schres.2006.11.029
27. Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA. Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry (2000) 57(3):237–45. doi:10.1001/archpsyc.57.3.237
28. Woo TU, Walsh JP, Benes FM. Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-d-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry (2004) 61(7):649–57. doi:10.1001/archpsyc.61.7.649
29. Woo TU, Kim AM, Viscidi E. Disease-specific alterations in glutamatergic neurotransmission on inhibitory interneurons in the prefrontal cortex in schizophrenia. Brain Res (2008) 1218:267–77. doi:10.1016/j.brainres.2008.03.092
30. Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry (2000) 57(11):1061–9. doi:10.1001/archpsyc.57.11.1061
31. Rocco BR, Lewis DA, Fish KN. Markedly lower glutamic acid decarboxylase 67 protein levels in a subset of boutons in schizophrenia. Biol Psychiatry (2016) 79(12):1006–15. doi:10.1016/j.biopsych.2015.07.022
32. Benes FM, McSparren J, Bird ED, SanGiovanni JP, Vincent SL. Deficits in small interneurons in prefrontal and cingulate cortices of schizophrenic and schizoaffective patients. Arch Gen Psychiatry (1991) 48(11):996–1001. doi:10.1001/archpsyc.1991.01810350036005
33. Daviss SR, Lewis DA. Local circuit neurons of the prefrontal cortex in schizophrenia: selective increase in the density of calbindin-immunoreactive neurons. Psychiatry Res (1995) 59(1–2):81–96. doi:10.1016/0165-1781(95)02720-3
34. Impagnatiello F, Guidotti AR, Pesold C, Dwivedi Y, Caruncho H, Pisu MG, et al. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc Natl Acad Sci U S A (1998) 95(26):15718–23. doi:10.1073/pnas.95.26.15718
35. Benes FM, Todtenkopf MS, Logiotatos P, Williams M. Glutamate decarboxylase(65)-immunoreactive terminals in cingulate and prefrontal cortices of schizophrenic and bipolar brain. J Chem Neuroanat (2000) 20(3–4):259–69. doi:10.1016/S0891-0618(00)00105-8
36. Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A (2001) 98(8):4746–51. doi:10.1073/pnas.081071198
37. Dracheva S, Elhakem SL, McGurk SR, Davis KL, Haroutunian V. GAD67 and GAD65 mRNA and protein expression in cerebrocortical regions of elderly patients with schizophrenia. J Neurosci Res (2004) 76(4):581–92. doi:10.1002/jnr.20122
38. Fatemi SH, Stary JM, Earle JA, Araghi-Niknam M, Eagan E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res (2005) 72(2–3):109–22. doi:10.1016/j.schres.2004.02.017
39. Straub RE, Lipska BK, Egan MF, Goldberg TE, Callicott JH, Mayhew MB, et al. Allelic variation in GAD1 (GAD67) is associated with schizophrenia and influences cortical function and gene expression. Mol Psychiatry (2007) 12(9):854–69. doi:10.1038/sj.mp.4001988
40. Glausier JR, Fish KN, Lewis DA. Altered parvalbumin basket cell inputs in the dorsolateral prefrontal cortex of schizophrenia subjects. Mol Psychiatry (2014) 19(1):30–6. doi:10.1038/mp.2013.152
41. McBain CJ, Fisahn A. Interneurons unbound. Nat Rev Neurosci (2001) 2(1):11–23. doi:10.1038/35049047
42. Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci (2004) 5(10):793–807. doi:10.1038/nrn1519
43. Conde F, Lund JS, Jacobowitz DM, Baimbridge KG, Lewis DA. Local circuit neurons immunoreactive for calretinin, calbindin D-28k or parvalbumin in monkey prefrontal cortex: distribution and morphology. J Comp Neurol (1994) 341(1):95–116. doi:10.1002/cne.903410109
44. Gabbott PL, Bacon SJ. Local circuit neurons in the medial prefrontal cortex (areas 24a,b,c, 25 and 32) in the monkey: I. Cell morphology and morphometrics. J Comp Neurol (1996) 364(4):567–608. doi:10.1002/(SICI)1096-9861(19960122)364:4<567::AID-CNE1>3.3.CO;2-K
45. Kawaguchi Y, Kubota Y. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex (1997) 7(6):476–86. doi:10.1093/cercor/7.6.476
46. Volk DW, Pierri JN, Fritschy JM, Auh S, Sampson AR, Lewis DA. Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb Cortex (2002) 12(10):1063–70. doi:10.1093/cercor/12.10.1063
47. Vawter MP, Crook JM, Hyde TM, Kleinman JE, Weinberger DR, Becker KG, et al. Microarray analysis of gene expression in the prefrontal cortex in schizophrenia: a preliminary study. Schizophr Res (2002) 58(1):11–20. doi:10.1016/S0920-9964(01)00377-2
48. Lewis DA, Moghaddam B. Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch Neurol (2006) 63(10):1372–6. doi:10.1001/archneur.63.10.1372
49. Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature (1995) 378(6552):75–8. doi:10.1038/378075a0
50. Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science (2001) 293(5532):1159–63. doi:10.1126/science.1060342
51. Akbarian S, Huang HS. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res Rev (2006) 52(2):293–304. doi:10.1016/j.brainresrev.2006.04.001
52. Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry (2002) 52(7):708–15. doi:10.1016/S0006-3223(02)01360-4
53. Woo TU, Miller JL, Lewis DA. Schizophrenia and the parvalbumin-containing class of cortical local circuit neurons. Am J Psychiatry (1997) 154(7):1013–5. doi:10.1176/ajp.154.7.1013
54. Cotter D, Landau S, Beasley C, Stevenson R, Chana G, MacMillan L, et al. The density and spatial distribution of GABAergic neurons, labelled using calcium binding proteins, in the anterior cingulate cortex in major depressive disorder, bipolar disorder, and schizophrenia. Biol Psychiatry (2002) 51(5):377–86. doi:10.1016/S0006-3223(01)01243-4
55. Chen JF, Weiss B. Irreversible blockade of D2 dopamine receptors by fluphenazine-N-mustard increases glutamic acid decarboxylase mRNA in rat striatum. Neurosci Lett (1993) 150(2):215–8. doi:10.1016/0304-3940(93)90539-W
56. Delfs JM, Anegawa NJ, Chesselet MF. Glutamate decarboxylase messenger RNA in rat pallidum: comparison of the effects of haloperidol, clozapine and combined haloperidol-scopolamine treatments. Neuroscience (1995) 66(1):67–80. doi:10.1016/0306-4522(94)00572-M
57. Delfs JM, Ellison GD, Mercugliano M, Chesselet MF. Expression of glutamic acid decarboxylase mRNA in striatum and pallidum in an animal model of tardive dyskinesia. Exp Neurol (1995) 133(2):175–88. doi:10.1006/exnr.1995.1020
58. Jolkkonen J, Jenner P, Marsden CD. GABAergic modulation of striatal peptide expression in rats and the alterations induced by dopamine antagonist treatment. Neurosci Lett (1994) 180(2):273–6. doi:10.1016/0304-3940(94)90537-1
59. Volk D, Austin M, Pierri J, Sampson A, Lewis D. GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. Am J Psychiatry (2001) 158(2):256–65. doi:10.1176/appi.ajp.158.2.256
60. Bragina L, Marchionni I, Omrani A, Cozzi A, Pellegrini-Giampietro DE, Cherubini E, et al. GAT-1 regulates both tonic and phasic GABAA receptor-mediated inhibition in the cerebral cortex. J Neurochem (2008) 105:1781–93. doi:10.1111/j.1471-4159.2008.05273.x
61. Melone M, Ciappelloni S, Conti F. A quantitative analysis of cellular and synaptic localization of GAT-1 and GAT-3 in rat neocortex. Brain Struct Funct (2015) 220:885–97. doi:10.1007/s00429-013-0690-8
62. Konopaske GT, Sweet RA, Wu Q, Sampson A, Lewis DA. Regional specificity of chandelier neuron axon terminal alterations in schizophrenia. Neuroscience (2006) 138(1):189–96. doi:10.1016/j.neuroscience.2005.10.070
63. Menzies L, Ooi C, Kamath S, Suckling J, McKenna P, Fletcher P, et al. Effects of gamma-aminobutyric acid-modulating drugs on working memory and brain function in patients with schizophrenia. Arch Gen Psychiatry (2007) 64(2):156–67. doi:10.1001/archpsyc.64.2.156
64. Ohnuma T, Augood SJ, Arai H, McKenna PJ, Emson PC. Measurement of GABAergic parameters in the prefrontal cortex in schizophrenia: focus on GABA content, GABA(A) receptor alpha-1 subunit messenger RNA and human GABA transporter-1 (HGAT-1) messenger RNA expression. Neuroscience (1999) 93(2):441–8. doi:10.1016/S0306-4522(99)00189-X
65. Woo TU, Whitehead RE, Melchitzky DS, Lewis DA. A subclass of prefrontal gamma-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc Natl Acad Sci U S A (1998) 95(9):5341–6. doi:10.1073/pnas.95.9.5341
66. Pierri JN, Chaudry AS, Woo TU, Lewis DA. Alterations in chandelier neuron axon terminals in the prefrontal cortex of schizophrenic subjects. Am J Psychiatry (1999) 156(11):1709–19.
67. Rocco BR, DeDionisio AM, Lewis DA, Fish KN. Alterations in a unique class of cortical chandelier cell axon cartridges in schizophrenia. Biol Psychiatry (2017) 82(1):40–8. doi:10.1016/j.biopsych.2016.09.018
68. Volk DW, Lewis DA. Impaired prefrontal inhibition in schizophrenia: relevance for cognitive dysfunction. Physiol Behav (2002) 77(4–5):501–5. doi:10.1016/S0031-9384(02)00936-8
69. Benes FM, Vincent SL, Marie A, Khan Y. Up-regulation of GABAA receptor binding on neurons of the prefrontal cortex in schizophrenic subjects. Neuroscience (1996) 75(4):1021–31. doi:10.1016/0306-4522(96)00328-4
70. Dean B, Hussain T, Hayes W, Scarr E, Kitsoulis S, Hill C, et al. Changes in serotonin2A and GABA(A) receptors in schizophrenia: studies on the human dorsolateral prefrontal cortex. J Neurochem (1999) 72(4):1593–9. doi:10.1046/j.1471-4159.1999.721593.x
71. Hanada S, Mita T, Nishino N, Tanaka C. [3H]muscimol binding sites increased in autopsied brains of chronic schizophrenics. Life Sci (1987) 40(3):259–66. doi:10.1016/0024-3205(87)90341-9
72. Ishikawa M, Mizukami K, Iwakiri M, Hidaka S, Asada T. Immunohistochemical and immunoblot study of GABA(A) alpha1 and beta2/3 subunits in the prefrontal cortex of subjects with schizophrenia and bipolar disorder. Neurosci Res (2004) 50(1):77–84. doi:10.1016/j.neures.2004.06.006
73. Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol (1995) 359(1):154–94. doi:10.1002/cne.903590111
74. Nusser Z, Sieghart W, Benke D, Fritschy JM, Somogyi P. Differential synaptic localization of two major gamma-aminobutyric acid type A receptor alpha subunits on hippocampal pyramidal cells. Proc Natl Acad Sci U S A (1996) 93(21):11939–44. doi:10.1073/pnas.93.21.11939
75. Lavoie AM, Tingey JJ, Harrison NL, Pritchett DB, Twyman RE. Activation and deactivation rates of recombinant GABA(A) receptor channels are dependent on alpha-subunit isoform. Biophys J (1997) 73(5):2518–26. doi:10.1016/S0006-3495(97)78280-8
76. Benes FM, Sorensen I, Vincent SL, Bird ED, Sathi M. Increased density of glutamate-immunoreactive vertical processes in superficial laminae in cingulate cortex of schizophrenic brain. Cereb Cortex (1992) 2(6):503–12. doi:10.1093/cercor/2.6.503
77. Gonzalez-Burgos G, Fish KN, Lewis DA. GABA neuron alterations, cortical circuit dysfunction and cognitive deficits in schizophrenia. Neural Plast (2011) 2011:723184. doi:10.1155/2011/723184
78. Beneyto M, Abbott A, Hashimoto T, Lewis DA. Lamina-specific alterations in cortical GABA(A) receptor subunit expression in schizophrenia. Cereb Cortex (2011) 21(5):999–1011. doi:10.1093/cercor/bhq169
79. Huntsman MM, Tran BV, Potkin SG, Bunney WE Jr, Jones EG. Altered ratios of alternatively spliced long and short gamma2 subunit mRNAs of the gamma-amino butyrate type A receptor in prefrontal cortex of schizophrenics. Proc Natl Acad Sci U S A (1998) 95(25):15066–71. doi:10.1073/pnas.95.25.15066
80. Maldonado-Avilés JG, Curley AA, Hashimoto T, Morrow AL, Ramsey AJ, O’Donnell P, et al. Altered markers of tonic inhibition in the dorsolateral prefrontal cortex of subjects with schizophrenia. Am J Psychiatry (2009) 166(4):450–9. doi:10.1176/appi.ajp.2008.08101484
81. Ali AB, Thomson AM. Synaptic alpha 5 subunit-containing GABAA receptors mediate IPSPs elicited by dendrite-preferring cells in rat neocortex. Cereb Cortex (2008) 18(6):1260–71. doi:10.1093/cercor/bhm160
82. Hendry SH, Huntsman MM, Vinuela A, Mohler H, de Blas AL, Jones EG. GABAA receptor subunit immunoreactivity in primate visual cortex: distribution in macaques and humans and regulation by visual input in adulthood. J Neurosci (1994) 14(4):2383–401.
83. Ishikawa M, Mizukami K, Iwakiri M, Asada T. Immunohistochemical and immunoblot analysis of gamma-aminobutyric acid B receptor in the prefrontal cortex of subjects with schizophrenia and bipolar disorder. Neurosci Lett (2005) 383(3):272–7. doi:10.1016/j.neulet.2005.04.025
84. Chen S, Huang X, Zeng XJ, Sieghart W, Tietz EI. Benzodiazepine-mediated regulation of alpha1, alpha2, beta1-3 and gamma2 GABA(A) receptor subunit proteins in the rat brain hippocampus and cortex. Neuroscience (1999) 93(1):33–44. doi:10.1016/S0306-4522(99)00118-9
85. Gur RE, Cowell PE, Latshaw A, Turetsky BI, Grossman RI, Arnold SE, et al. Reduced dorsal and orbital prefrontal gray matter volumes in schizophrenia. Arch Gen Psychiatry (2000) 57(8):761–8. doi:10.1001/archpsyc.57.8.761
86. Lewis DA, Volk DW, Hashimoto T. Selective alterations in prefrontal cortical GABA neurotransmission in schizophrenia: a novel target for the treatment of working memory dysfunction. Psychopharmacology (Berl) (2004) 174(1):143–50. doi:10.1007/s00213-003-1673-x
87. Jensen K, Chiu CS, Sokolova I, Lester HA, Mody I. GABA transporter-1 (GAT1)-deficient mice: differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J Neurophysiol (2003) 90(4):2690–701. doi:10.1152/jn.00240.2003
88. Enomoto T, Tse MT, Floresco SB. Reducing prefrontal gamma-aminobutyric acid activity induces cognitive, behavioral, and dopaminergic abnormalities that resemble schizophrenia. Biol Psychiatry (2011) 69(5):432–41. doi:10.1016/j.biopsych.2010.09.038
89. Sawaguchi T, Matsumura M, Kubota K. Delayed response deficits produced by local injection of bicuculline into the dorsolateral prefrontal cortex in Japanese macaque monkeys. Exp Brain Res (1989) 75(3):457–69. doi:10.1007/BF00249897
90. Goto N, Yoshimura R, Kakeda S, Moriya J, Hori H, Hayashi K, et al. No alterations of brain GABA after 6 months of treatment with atypical antipsychotic drugs in early-stage first-episode schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2010) 34(8):1480–3. doi:10.1016/j.pnpbp.2010.08.007
91. Kelemen O, Kiss I, Benedek G, Keri S. Perceptual and cognitive effects of antipsychotics in first-episode schizophrenia: the potential impact of GABA concentration in the visual cortex. Prog Neuropsychopharmacol Biol Psychiatry (2013) 47:13–9. doi:10.1016/j.pnpbp.2013.07.024
92. Marsman A, Mandl RC, Klomp DW, Bohlken MM, Boer VO, Andreychenko A, et al. GABA and glutamate in schizophrenia: a 7 T (1)H-MRS study. Neuroimage Clin (2014) 6:398–407. doi:10.1016/j.nicl.2014.10.005
93. Rowland LM, Kontson K, West J, Edden RA, Zhu H, Wijtenburg SA, et al. In vivo measurements of glutamate, GABA, and NAAG in schizophrenia. Schizophr Bull (2012) 39(5):1096–104. doi:10.1093/schbul/sbs092
94. Rowland LM, Krause BW, Wijtenburg SA, McMahon RP, Chiappelli J, Nugent KL, et al. Medial frontal GABA is lower in older schizophrenia: a MEGA-PRESS with macromolecule suppression study. Mol Psychiatry (2016) 21(2):198–204. doi:10.1038/mp.2015.34
95. Wang J, Tang Y, Zhang T, Cui H, Xu L, Zeng B, et al. Reduced gamma-aminobutyric acid and glutamate+glutamine levels in drug-naive patients with first-episode schizophrenia but not in those at ultrahigh risk. Neural Plast (2016) 2016:3915703. doi:10.1155/2016/3915703
96. Yoon JH, Maddock RJ, Rokem A, Silver MA, Minzenberg MJ, Ragland JD, et al. GABA concentration is reduced in visual cortex in schizophrenia and correlates with orientation-specific surround suppression. J Neurosci (2010) 30(10):3777–81. doi:10.1523/JNEUROSCI.6158-09.2010
97. Kegeles LS, Mao X, Stanford AD, Girgis R, Ojeil N, Xu X, et al. Elevated prefrontal cortex gamma-aminobutyric acid and glutamate-glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Arch Gen Psychiatry (2012) 69(5):449–59. doi:10.1001/archgenpsychiatry.2011.1519
98. Tayoshi S, Nakataki M, Sumitani S, Taniguchi K, Shibuya-Tayoshi S, Numata S, et al. GABA concentration in schizophrenia patients and the effects of antipsychotic medication: a proton magnetic resonance spectroscopy study. Schizophr Res (2010) 117(1):83–91. doi:10.1016/j.schres.2009.11.011
99. Ongur D, Prescot AP, McCarthy J, Cohen BM, Renshaw PF. Elevated gamma-aminobutyric acid levels in chronic schizophrenia. Biol Psychiatry (2010) 68(7):667–70. doi:10.1016/j.biopsych.2010.05.016
100. Schür RR, Draisma LW, Wijnen JP, Boks MP, Koevoets MG, Joëls M, et al. Brain GABA levels across psychiatric disorders: a systematic literature review and meta-analysis of 1H-MRS studies. Hum Brain Mapp (2016) 37(9):3337–52. doi:10.1002/hbm.23244
101. Goto N, Yoshimura R, Moriya J, Kakeda S, Ueda N, Ikenouchi-Sugita A, et al. Reduction of brain gamma-aminobutyric acid (GABA) concentrations in early-stage schizophrenia patients: 3T proton MRS study. Schizophr Res (2009) 112(1–3):192–3. doi:10.1016/j.schres.2009.04.026
102. Hedman AM, Van Haren NE, Van Baal CG, Kahn RS, Hulshoff Pol HE. IQ change over time in schizophrenia and healthy individuals: a meta-analysis. Schizophr Res (2013) 146(1–3):201–8. doi:10.1016/j.schres.2013.01.027
Keywords: GABA, schizophrenia, magnetic resonance spectroscopy, postmortem studies, in vivo studies
Citation: De Jonge JC, Vinkers CH, Hulshoff Pol HE and Marsman A (2017) GABAergic Mechanisms in Schizophrenia: Linking Postmortem and In Vivo Studies. Front. Psychiatry 8:118. doi: 10.3389/fpsyt.2017.00118
Received: 31 March 2017; Accepted: 22 June 2017;
Published: 11 August 2017
Edited by:
André Schmidt, King’s College London, United KingdomReviewed by:
Fiorenzo Conti, Università Politecnica delle Marche, ItalyCopyright: © 2017 De Jonge, Vinkers, Hulshoff Pol and Marsman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anouk Marsman, YW5vdWttQGRyY21yLmRr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.