 Adonis Sfera
Adonis Sfera Carolina Osorio
Carolina Osorio Luzmin Acosta Inderias
Luzmin Acosta Inderias Victoria Parker4
Victoria Parker4 Amy I. Price
Amy I. Price- 1Patton State Hospital, Psychiatry, Patton, CA, USA
- 2Loma Linda University, Psychiatry, Loma Linda, CA, USA
- 3Patton State Hospital, Medicine, Patton, CA, USA
- 4Patton State Hospital, Dietary, Patton, CA, USA
- 5Oxford University, Evidence Based Medicine, Oxford, UK
Pathological impulsivity is encountered in a broad range of psychiatric conditions and is thought to be a risk factor for aggression directed against oneself or others. Recently, a strong association was found between impulsivity and obesity which may explain the high prevalence of metabolic disorders in individuals with mental illness even in the absence of exposure to psychotropic drugs. As the overlapping neurobiology of impulsivity and obesity is being unraveled, the question asked louder and louder is whether they should be treated concomitantly. The treatment of obesity and metabolic dysregulations in chronic psychiatric patients is currently underutilized and often initiated late, making correction more difficult to achieve. Addressing obesity and metabolic dysfunction in a preventive manner may not only lower morbidity and mortality but also the excessive impulsivity, decreasing the risk for aggression. In this review, we take a look beyond psychopharmacological interventions and discuss dietary and physical therapy approaches.
Introduction
Although the prevalence of obesity and metabolic syndrome has been increasing worldwide over the past decades, it is significantly higher in mentally ill patients regardless of exposure to psychotropic drugs (PTDs) (1–9). In this regard, it was hypothesized by others that the parallel growth of psychiatric and metabolic disorders may indicate a shared pathoetiology (10–13).
Pathological impulsivity encompasses a heterogenous group of psychiatric disorders characterized by inability to resist impulses for engaging in behaviors harmful to self or others. It is encountered in numerous psychiatric conditions, ranging from intermittent explosive disorder, pathological gambling, kleptomania, trichotillomania, and pyromania on the one hand, to schizophrenia, mania, attention-deficit hyperactivity disorder (ADHD), antisocial personality disorder, and drug addictions on the other. The psychopharmacological treatment of these disorders is dependent on the primary pathology and may include almost all classes of PTDs: serotonin reuptake inhibitors, stimulants, antipsychotics, mood stabilizers, and in some instances opioid antagonists.
This observation is in line with the epidemiological studies, linking pathological impulsivity with weight gain and dysmetabolism (14–19). There is a growing body of evidence that includes studies in young individuals, indicating that obese/overweight adolescents are more likely to engage in risky behaviors (20). Along the same lines, nutritional studies reveal that perinatal exposure to high fat diets is more likely to bring about impulsive offspring (21, 22). Other research in nutrition connected a high trans-fat diet with aggression, while sociological studies pointed to a correlation between antisocial behavior and obesity (23, 24). Neuroimaging studies shed additional light on the impulsivity–obesity connection as functional magnetic resonance imaging and diffusion tensor imaging studies documented lower perfusion in the orbitofrontal, medial/ventrolateral prefrontal cortex, and middle/superior frontal gyri in impulsive and obese individuals (25–28). In addition, this body of evidence includes novel endocrinology studies demonstrating dysregulation of metabolic hormones ghrelin, leptin, and adiponectin in conditions associated with pathological impulsivity, including ADHD, aggression, and antisocial personality disorder (29–34). On the other hand, clozapine, an established anti-impulsivity drug, was found to directly alter the expression of leptin and adiponectin genes (35–37). Lithium, another anti-impulsivity drug, was found beneficial in correcting the glycemic parameters in diabetes mellitus type 2 (T2DM) by inhibiting glycogen synthase kinase-3 (GSK-3) a leptin-lowering enzyme (38–40). As mentioned earlier, lower leptin levels were associated with excessive impulsivity and dyslipidemias (41).
Multiple psychiatric studies over the past decades established that acetylcholine (ACh) and monoamines (MAs) including norepinephrine, dopamine, histamine, and serotonin are involved in a wide range of psychiatric disorders, including those marked by pathological impulsivity (14, 42–44). As these neurotransmitters were demonstrated to also modulate the hypothalamic feeding centers, it should not come as a surprise that impulsivity and obesity are intertwined (45, 46). Indeed, most antiobesity drugs present with agonistic action at the receptors in which many PTDs antagonize, suggesting the possibility of a psychometabolic continuum (46).
The development of PTDs with fewer metabolic adverse effects currently represents a major unmet need in psychiatry. As obesity and metabolic dysfunction in psychiatric patients trigger higher morbidity and mortality rates than in the general population, there is a heightened urgency for their prevention and early correction. Therefore, until this need is met, optimal utilization of available pharmacological and non-pharmacological tools is crucial for the overall well-being of these patients.
At present, the treatment of metabolic disorders in psychiatric patients is not only underutilized but also frequently delayed, rendering correction more difficult to achieve (6, 7). Indeed, after the diagnosis of obesity or metabolic syndrome is established, there are few effective interventions for reversing these conditions. In general, these interventions revolve around replacing an orexigenic with a less orexigenic drug, at the risk of clinical destabilization, or lifestyle and behavioral changes that have proved difficult to implement in this population (47, 48). Therefore, clinicians should adopt preemptive approaches, striving to avoid or delay the onset of obesity, and the metabolic syndrome instead of correcting them post hoc (7).
More studies are needed to assess the efficacy of preventive metabolic interventions, including the utilization of available antiobesity drugs in psychiatric disorders and PTD-induced obesity. There are even fewer studies on alternative modalities, including nutrition, physical therapy, and parasympathetic stimulation via cholinesterase inhibitors or transcutaneous auricular vagal nerve stimulation (taVNS).
In this article, we will review some of these modalities after a brief discussion on antiobesity drugs and their action in the hypothalamic feeding centers.
Arcuate (ARC) Nucleus: Where Psychopharmacology and Antiobesity Pharmacopeias Collide
Hunger, satiety, and energy homeostasis are controlled by the neuronal networks in the mediobasal hypothalamus and some brainstem areas. The ARC nucleus of the hypothalamus contains the first order neurons that balance hunger and satiety in order to maintain a stable body weight (49). This balancing act requires a constant cross-talk between the ARC nucleus and the peripheral organs, which takes place via metabolic hormones, neural input, and neurotransmitters, including MA and ACh (50).
The anorexigenic system that lowers appetite is composed of proopiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART) neurons. They produce the alpha-melanocyte stimulating hormone (alpha-MSH) that binds to the melanocortin 3 and 4 receptors (MC3Rs and MC4Rs) expressed by the second order neurons located in the periventricular nucleus and the lateral hypothalamic area (LHA) (51) (Figure 1).
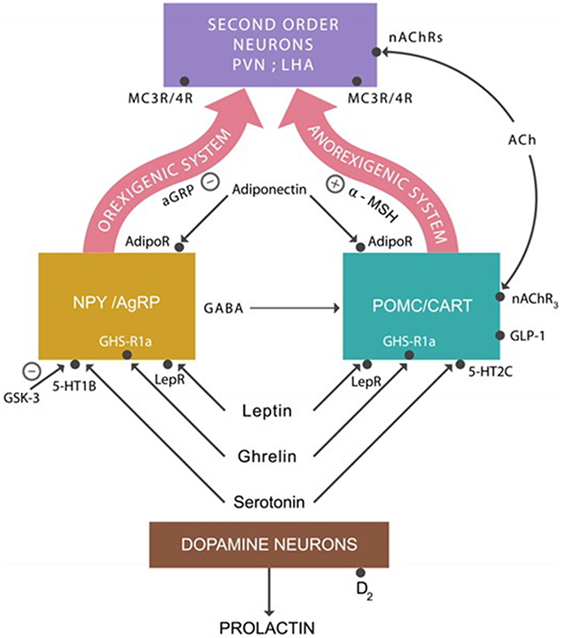
Figure 1. The anorexigenic system: alpha-melanocyte stimulating hormone (alpha-MSH) activates MC3/4 receptors expressed on second order neurons, inhibiting appetite. The orexigenic system: agouti-related protein (AgRP) is a competitor of alpha-MSH at MC3/4 receptors, augmenting appetite. Adiponectin, leptin, ghrelin, and serotonin activate the anorexigenic system. The action of these hormones on the orexigenic system results in the release of the inhibitory neurotransmitter GABA, which applies the brake on the anorexigenic system, increasing appetite. Abbreviations: AdipoR, adiponectin receptors; nAChRs, nicotinic cholinergic receptors; LepRs, leptin receptors; GHS-R1a, ghrelin receptors; serotonin 5-HT2C and 5-HT1A receptors.
The orexigenic system, which increases the appetite, consists of neuropeptide Y (NPY) and agouti-related protein (AgRP). They produce NPY, GABA, and the AgRP. AgRP is an antagonist of alpha-MSH at MC3Rs and MC4Rs prevents their activation, whereas GABA inhibits the POMC/CART neurons. Both of these actions result in appetite augmentation.
In addition to these neuronal groups, ARC also contains dopamine neurons which project to the anterior pituitary gland where they inhibit prolactin secretion (Figure 1). Blocking dopamine-2 (D-2) receptors by PTDs often results in hyperprolactinemia, a common adverse effect of high potency antipsychotic drugs (52).
Over the past few years, new drugs indicated for the long-term treatment of obesity obtained FDA approval. Interestingly, two of them are combinations of PTDs utilized in the treatment of psychiatric disorders. For example, phentermine–topiramate extended release (Qsymia) and naltrexone–bupropion extended release (Contrave) have been prescribed individually in affective disorders and drug addictions. These drugs may have a place in the preventive treatment of PTD-induced obesity as they may be prescribed “off label” in select patients needing more adequate mood stabilization. In this regard, four large randomized, double-blind, placebo controlled trials support the preventive use of topiramate alone for PTD-induced weight gain (53). Concomitant utilization of topiramate with orexigenic PTDs may constitute a preemptive intervention to avoid or delay the onset of obesity and metabolic dysregulation (Table 1). About 10% of patients may experience mild to moderate cognitive adverse effects early on topiramate treatment with verbal fluency being more affected compared to other antiepileptic drugs.
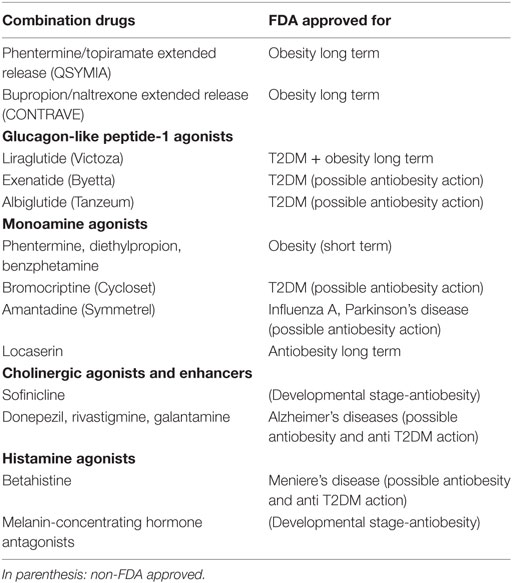
Table 1. Antiobesity and anti-diabetes mellitus type 2 (T2DM) drugs for potential use in psychiatry.
Metformin
Metformin is a drug widely prescribed in psychiatric patients diagnosed with T2DM, but rarely in a preventive manner, even though several studies found this drug to be efficient in preventing both obesity and T2DM (54–56). Since metformin is known for its relatively benign adverse effects, it could be prescribed from the treatment onset along with the orexigenic PTDs.
Metformin was demonstrated to result in 2.93–5 kg weight loss after 6 months of treatment even when prescribed after the onset of metabolic dysregulation (55). More studies are needed to assess the benefits of preventive metformin use alone or in combination with other antiobesity agents.
Glucagon-Like Peptide-1 (GLP-1)
Recently, there has been a strong interest in the GLP-1 agonists, including liraglutide (Victoza), exenatide (Byetta), and albiglutide (Tanzeum). These agents have been shown effective in halting weight gain in diabetic patients and their antiobesity action is most likely mediated via GLP-1 receptors expressed on POMC/CART neurons (57) (Figure 1).
In addition, novel studies demonstrate that GLP-1 inhibition may mediate clozapine-induced weight gain and metabolic dysregulation, suggesting that GLP-1 agonists could reverse both (58). To the best of our knowledge, currently, there is one ongoing study aiming at exploring liraglutide’s effects on glucose tolerance in patients on clozapine or olanzapine treatment (59). The once-a-week drug exenatide did not promote weight loss in this population (60). As liraglutide was demonstrated to be more effective than the GLP-1 mimetics, a clinical trial should be initiated to assess the efficacy of this drug for PTD-induced obesity either alone or in combination with another compound, such as locaserin (61). The once-a-week albiglutide was also not tested for weight loss in PTD-induced obesity. A large study found this drug effective, especially as an adjunct to metformin, suggesting that it could be helpful for psychiatric patients with inadequate glycemic control on metformin alone (62). In addition, since liraglutide has both T2DM and weight loss indication, it could be prescribed more often in overweight/obese psychiatric patients, especially when other agents cannot achieve adequate glycemic stabilization. Interestingly, novel studies demonstrate that GLP-1 agonists may have PTD-synergistic actions on impulsivity, suggesting that liraglutide-treated psychiatric patients may be managed with lower PTDs doses (63–67). Furthermore, since GLP-1 signaling was suggested as a common pathophysiological mechanism in both Alzheimer’s disease and T2DM, GLP-1 agonists may prevent cognitive decline (68).
Locaserin
The novel antiobesity drug, locaserin, is an agonist at serotonin 5-HT2C receptors that are antagonized by some PTDs, inducing the adverse effect of weight gain. Interestingly, preclinical studies demonstrated the efficacy 5-HT2C agonists for the treatment of excessive impulsivity (46, 69–71).
Different studies point to a connection between serotonin signaling and the metabolic hormones—ghrelin, leptin, and adiponectin—which were also linked to impulsivity (29–34, 41). For example, studies in rodents show that leptin modulates the biosynthesis and release of serotonin by the raphe nuclei (72–74). Other studies report that leptin receptors (LepRs) act synergistically with 5-HT2C expressed on POMC/CART neurons and with 5-HT1B receptors on NPY/AgRP neurons (75) (Figure 1). Clozapine, a drug with known anti-impulsivity actions and orexigenic adverse effects, is an antagonist at both 5-HT2C and 5-HT1B receptors. In addition, as mentioned earlier, clozapine alters the expression of leptin and adiponectin genes, suggesting direct effects on both impulsivity and weight gain.
To the best of our knowledge, locaserin has not been tested in PTD-induced weight gain, but its receptor profile suggests it may be beneficial as an adjunct in clozapine-, olanzapine-, or mirtazapine-treated patients.
A body of literature links the reduced serotonergic 5-HT1B receptor activity with impulsivity and drug addictions (76–79). In the hypothalamus, the 5-HT1B receptors are expressed by the NPY/AgRP neurons and their activation inhibits the release of GABA, thus disinhibiting the anorexigenic system, inducing appetite suppression (Figure 1). The 5-HT1B receptors were demonstrated to act synergistically in appetite suppression with the 5HT2C at POMC/CART receptors (75). At present, there are no concomitant agonists at 5-HT2C and 5-HT1B receptors, but such drugs may be expected to present with superior antiobesity and anti-impulsivity actions. As discussed above, lithium, a major anti-impulsivity drug, inhibits GSK-3, a 5-HT1B-blocking enzyme. Over-expressed GSK-3 was associated with excessive impulsivity in multiple psychiatric syndromes, including mania, suicide, and schizophrenia (80–84). In addition, GSK-3 upregulation was demonstrated in obesity and insulin resistance (38, 39, 85, 86). Since lithium was documented to possess anti-diabetic properties, GSK-3 inhibitors are currently T2DM targets (40). Indeed, three decades ago, it was noted that lithium acted synergistically with some antidiabetic drugs; however, the mechanism was unknown at that time (87–89) (Table 1).
Dopaminergic Drugs in Obesity–Impulsivity Axis
Some of the FDA approved antiobesity agents are agonists at dopamine D-2 receptors that are antagonized by all clinically utilized antipsychotic drugs (90). The antiobesity D-2 agonists include phentermine, diethylpropion, benzphetamine, and bupropion (as part of the combination drug Contrave). In addition, the D-2 stimulants bromocriptine and amantadine have been associated with weight loss (91, 92). For example, amantadine has demonstrated efficacy in overweight/obese patients treated with olanzapine (91). Bromocriptine under the market name Cycloset recently obtained FDA indication for the treatment of T2DM. This drug has been underutilized in PTD-associated glycemic dysfunction, although it may be beneficial for diabetic patients requiring treatment with risperidone, paliperidone, or some antidepressants that are often associated with hyperprolactinemia.
Recent studies revealed that elevated prolactin induces pancreatic beta cells to secrete insulin. Chronic hyperprolactinemia induces hyperinsulinemia, eventually resulting in beta cells exhaustion and T2DM (Figure 2). Aside from the anterior pituitary, prolactin is also released by the adipose tissue as a pro-inflammatory cytokine demonstrated to augment the metabolic inflammation associated with obesity (93, 94). This is one of the primary reasons clinicians should address hyperprolactinemia promptly in PTD-treated patients, after prolactinomas have been ruled out (94). Novel studies do not support the older concept that bromocriptine or other D-2 stimulants invariably precipitate psychosis. Persistently elevated prolactin was demonstrated by large studies to increase the all-cause mortality, emphasizing the need for treating this condition promptly (95, 96). In addition, other studies demonstrated the benefit of D-2 stimulation in patients with negative symptoms of schizophrenia (97, 98). Furthermore, some researchers hypothesized that hyperprolactinemia may exacerbate both schizophrenia and T2DM (99).
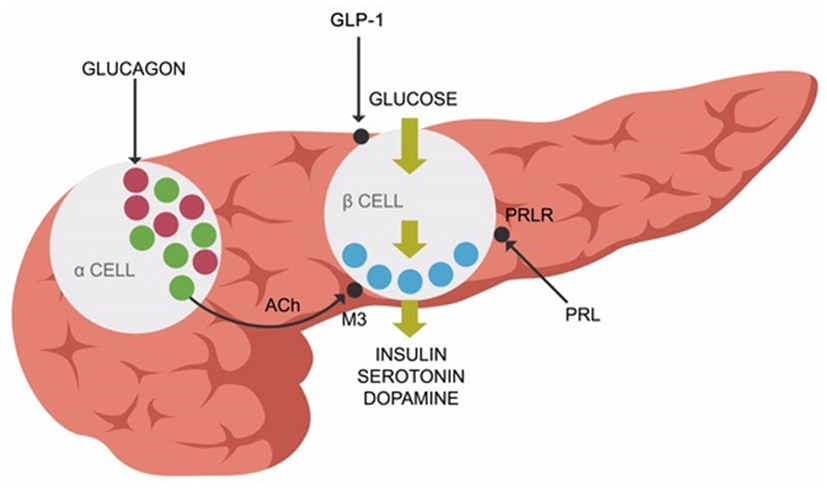
Figure 2. Prolactin receptors (PRLR), muscarinic-3 cholinergic, and glucagon-like peptide-1 (GLP-1) receptors are expressed on pancreatic beta-cells, facilitating insulin release along with serotonin and dopamine.
Appetite contains a large hedonic component mediated by the central reward system. A striatal hypodopaminergic state is believed to trigger overnutrition, which in turn augments dopamine transmission. According to this model, D-2 stimulants correct the hypodopaminergia, lowering the overnutrition reward (100–102). Indeed, reduced D-2 receptor density was demonstrated in obese individuals, implying impaired dopaminergic signaling in this metabolic disorder (103). Psychiatric patients treated with antipsychotic drugs (D-2 blockers) may present with an enduring iatrogenic hypodopaminergic state, which in turn triggers overnutrition to augment dopamine levels.
Amantadine, bromocriptine, or phentermine–topiramate extended release should be encouraged in psychiatric patients, especially when presenting with extrapyramidal symptoms or elevated prolactin.
Lisdexamfetamine, a dopaminergic pro-drug metabolized to dexamphetamine, was approved by the FDA for ADHD; however, its appetite suppressant activity and low abuse potential may render it useful in PTD-induced obesity. Recently, the effectiveness of this compound was demonstrated in binge eating (104). In addition, its stimulant action was associated with significant improvements in negative symptoms of schizophrenia without worsening the positive symptoms (105).
Acetylcholine
A novel line of research demonstrates that ACh signaling and metabolic pathways intersect, both in the pancreas and the hypothalamus (106, 107). Centrally, nicotine activates the nicotinic cholinergic receptors (nAChRs) expressed on POMC/CART neurons, turning the anorexigenic system “on” and lowering appetite (Figure 1). This was demonstrated to take place in smokers, while following smoking cessation, the lack of anorexigenic activation heightens the appetite, lading to weight gain. In addition, as nAChRs are also expressed on the second order neurons in the LHA and the perifornical organ. Smoking discontinuation affects these areas also, inducing weight gain (108).
Peripherally, ACh enables insulin release (in response to glucose) from the pancreatic beta cells via muscarinic-3 (M-3) receptors. Pancreatic alpha cells secrete ACh, facilitating the release of insulin from the beta cells (109, 110) (Figure 2).
As several PTDs are known to block the M-3 receptors, they may induce glycemic dysregulation (111). Interestingly, insulin is synthesized and released from the beta cells along with serotonin and dopamine molecules, demonstrating once more the interconnectedness between energy metabolism and ACh/MAs. This finding is in line with the epidemiological studies associating smoking cessation with increased risk for T2DM for up to 12 years (112).
In light of these data, it should not come as a surprise that obesity and T2DM have been associated with the over-expression of Ach-degrading enzymes—butyrylcholinesterase (BChE) and acetylcholinesterase (AChE)—which tone down cholinergic signaling. Indeed, cholinesterase inhibitors—donepezil, rivastigmine, and galantamine—were found beneficial in metabolic syndrome by some researchers (113–115). In addition, these drugs are currently utilized in patients with Alzheimer’s disease for the treatment of aggression, suggesting a psychometabolic role (116). Preventive use of these agents in psychiatric patients could preempt the development of metabolic dysfunction and obesity. Rivastigmine that inhibits both AChE and BChE was found to be a more effective anti-impulsivity agent in dementias (117). Interestingly, BChE was demonstrated to hydrolyze both ACh and ghrelin, thus comprising an antiobesity/impulsivity target (114, 118, 119). Moreover, LepRs were demonstrated to act synergistically with both serotonin and ACh receptors on POMC/CART neurons, indicating a close coordination between the metabolic hormones and these neurotransmitters (75). Furthermore, the existence of a leptin–dopamine axis was implied by preclinical studies which found that mesolimbic leptin signaling modulates the CNS reward system (120).
Melatonin and serotonin 5-HT2C receptors were demonstrated to be coexpressed in various areas of the CNS, possibly including POMC/CART neurons, explaining the frequently discussed antiobesity effect of melatonin (121).
The impaired expression of CHRNA gene, coding for the nicotinic acetylcholine receptors, has been associated with disorders marked by excessive impulsivity, including ADHD, drug addictions, and antisocial personality disorder (43, 122, 123). Sofinicline, a novel nicotinic agonist believed to augment CHRNA expression, is currently being tested for ADHD but appears promising in metabolic syndrome, T2DM, and eating disorders (124–126). The metabolic action of sofinicline is probably due to ghrelin modulation of POMC/CART neurons via nicotinic receptors coexpressed with 5-HT2C (33, 127). This recently revealed receptor–receptor interaction may lead to the development of novel compounds for metabolic syndrome and impulsivity (33, 128–130).
Clinicians should be encouraged to prescribe cholinesterase inhibitors in a preventive manner in psychiatric patients susceptible to PTD-induced obesity and dysmetabolism, especially in the presence of schizophrenia or dementia-related cognitive impairments. In this respect, rivastigmine patch represents a low risk intervention with several advantages over oral medications. Moreover, since impaired cholinergic signaling was involved in both metabolic and impulsivity disorders, clinicians should avoid the use of anticholinergic drugs even in younger patients and consider replacing them with amantadine for the extrapyramidal adverse effects of antipsychotic drugs and with melatonin or ramelteon for insomnia. Furthermore, cholinergic signaling may be enhanced via taVNS, which represents another low risk intervention for improving insulin sensitivity (131). Interestingly, taVNS has documented efficacy in depression, which may represent an additional benefit for psychiatric patients (132).
Histamine
The relationship between histamine and metabolism was first described in the 1950s, yet the development of histaminergic treatments for metabolic disorders and obesity is still in its infancy (133). The blockade of histamine-1 (H-1) receptors by PTDs was associated with both obesity and T2DM, suggesting the involvement of histaminergic pathways in energy homeostasis (131, 134–136). Briefly, H-1 and H-2 receptors’ blockade induces sedation, while antagonism at H-3 receptors results in wakefulness, as H-3 receptors are primarily autoreceptors.
Some clinical and preclinical studies demonstrated that betahistine, a centrally acting H-1 agonist and partial H-3 antagonist, was effective as an antiobesity drug, especially in younger women (137). Studies with olanzapine-induced obesity in rodents also demonstrated weight loss after exposure to betahistine (138). However, as other studies failed to establish a direct link between histamine and body weight, an indirect action is now suspected via other neurotransmitter systems or metabolic hormones, including thyrotropin-releasing hormone, growth hormone, and leptin (139–141). Indeed, novel human studies report that H-1 receptor antagonists inhibit alpha 7nAChRs which turn “off” the POMC/CART anorexigenic system (139). As alpha 7nAChRs are involved in cognition, histamine-blocking agents (including some PTDs) may represent both metabolic and cognitive risk factors.
Aside from these indirect actions on feeding, histamine was recently demonstrated to directly control the orexigenic melanin-concentrating hormone (MCH) (142). Indeed, the MCH receptor antagonists are currently thought of as promising antiobesity agents (143). As MCH neurons express H-3, but not H-1 or H-2 receptors, it is likely that histamine inhibits MCH secretion via H-3 receptors. Several studies indicate that histamine also exerts H-3-mediated anti-diabetic properties (144–146) (Table 1).
Furthermore, human microglial cells were documented to express H-3 receptors that are responsible for their activation (147). As numerous neuropsychiatric conditions were linked to microglial activation, H-3 receptors’ antagonists are currently evaluated for their efficacy in schizophrenia and Alzheimer’s disease (147, 148). Moreover, novel studies suggest that H-3 blockade may be associated with decreased impulsivity, especially in dementia, Parkinson’s disease, ADHD, and drug addiction (149–152). As histamine is known to be modulated by leptin and reduced leptin was associated with impulsivity, these finding are in line with the research connecting histamine with obesity–impulsivity axis (30–32).
The Brain–Gut Axis: May the Microbes be with You
Dietary interventions have been extremely underutilized in psychiatric patients despite accumulating evidence for their benefits.
The Alternate Healthy Eating Index 2010 (AHEI-2010) is a measure of diet quality. A high AHEI score was documented to lower the risk of chronic disease, including T2DM and cardiovascular disease and to reverse the metabolic syndrome (153–155). High AHEI score diets have demonstrated benefits in the affective and cognitive disorders, but they were not studied in schizophrenia or PTD-induced obesity (156, 157). These diets are thought to exert their beneficial effects by stabilizing the gut microbiome.
The human microbiome consists of over 100 trillion bacteria, fungi, and protozoa which inhabit the gastrointestinal (GI) tract, living in symbiosis with the host cells. Several lines of evidence point to the potential role of gut microbiota in modulating not only the host energy metabolism but also information processing and behavior (158, 159). Human and animal studies demonstrated that obesity was associated with GI “overpopulation” with phylum Firmicutes and concomitant decreases in phylum Bacteriodetes (160, 161). Interestingly, administration of an antibiotic along with olanzapine minimized the amount of the weight gain in rodents, a finding that may be in line with the antibiotic minocycline reducing psychotic symptoms in patients with schizophrenia. Minocycline is a tetracycline that was demonstrated to not only modulate the dopaminergic and glutamatergic CNS signaling but also to restore the physiologic Firmicutes/Bacteroidetes ratio in the gut with positive effect on hypertension (162).
These studies suggest that that gut microbiota may be involved not only in the etiology of obesity but also in schizophrenia, perhaps explaining the predisposition to metabolic disorders encountered in psychiatric patients (163, 164). In addition, preclinical studies show that transplantation of GI tract microorganisms from the obese into the lean mice was followed by weight gain in the later. Interestingly, fecal analysis of olanzapine treated rodents, demonstrated increases in Firmicutes and decreases in Bacteriodetes phyla, a pattern identical to the one found in obese humans (165, 166).
Other novel studies linked low dietary fiber with the dysregulation of Firmicutes/Bacteriodetes ratio. The colonic microbiota is known to induce fermentation of dietary fiber, resulting in the production of beneficial short-chain fatty acids (SCFAs), including butyrate and propionate. The SCFAs deficiency is believed to represent a risk factor in the pathogenesis of obesity (167). In this respect, butyrate was found protective against insulin resistance and inflammation, while propionate was demonstrated to lower cholesterol synthesis (168). Moreover, SCFAs were shown to augment the production of leptin which, as discussed above, was found to be decreased in disorders marked by impulsivity, thus linking SCFAs with psychiatric conditions (169).
Short-chain fatty acids were demonstrated not only to reach the bloodstream but also to cross the blood–brain barrier (BBB) and alter the hypothalamic leptin and adiponectin gene expression (170, 171). Activation of adiponectin receptors, expressed on both POMC/CART and NPY/AgRP are known to potentiate the function of leptin, lowering impulsivity (172, 173). Interestingly, many patients with schizophrenia treated with second-generation antipsychotic drugs were found to have lower plasma adiponectin and leptin levels (174, 175). Studies in metabolism indicate that hypoadiponectinemia lowers the skeletal muscle uptake of postprandial glucose (insulin resistance), eventually leading to T2DM (176, 177). Moreover, supplementation with dietary fiber was demonstrated to increase adiponectin levels by up to 115%, suggesting that SCFAs play a key role in adiponectin biosynthesis (178).
Western diets in general are known for lacking adequate amounts of dietary fiber. For example, typical Western adults were shown to consume 5–10 g of fiber daily, as opposed to the 35 or 50 g which is considered optimal (179). Patients with schizophrenia were demonstrated to consume an even lower amount of dietary fiber compared to the overall Western population, perhaps explaining the low adiponectin levels documented in this group (180). Mediterranean diets and variants aside from their demonstrated anti-inflammatory actions were found to facilitate SCFAs biosynthesis (181) (Table 2).

Table 2. Dietary interventions in metabolic syndrome.
Dietary polyphenols were documented to help maintain the adequate gut microbial balance (187). In addition, polyphenols were shown to be protective against T2DM and possibly psychiatric disorders manifested by impulsivity (182–186). To the best of our knowledge, at present, there are no studies on polyphenol/high fiber diets in psychiatric or PTD-treated patients. We suggest a trial with a modified Mediterranean diet, incorporating high dietary fiber, polyphenols, and SCFA. The efficacy of this approach can be monitored not only via BMI and the glycemic parameters but also by plasma adiponectin and leptin levels.
Skeletal Muscle and the “Exercise Factors”
In addition to enabling the human body mobility, the over 600 skeletal muscles comprise a complex endocrine organ, known for the secretion of a growing number of “myokines” which exert both local and distal effects. “Exercise factors” are a subgroup of myokines released into the circulation during the exercise (188, 189). Among them, the peroxisome proliferator-activated receptor-γ coactivator 1alpha (PGC-1alpha) is the most studied as it was shown to increase both the skeletal muscle glucose uptake and the oxidation of fatty acids (190). A sedentary lifestyle with lowered expression of PGC-1alpha is associated with weight gain, inflammation, and decreased insulin secretion, pathological changes that eventually culminate in T2DM and obesity (191) (Table 3).

Table 3. Specific therapeutic modalities described in metabolic syndrome.
Novel studies demonstrate the positive impact of physical exercise in disorders marked by excessive impulsivity, including ADHD, affective disorders, schizophrenia, drug addictions, and Alzheimer’s disease (196–199).
The sequence of events that may lead to T2DM is believed to be initiated by decreased uptake of glucose into the skeletal muscle or insulin resistance, followed by hyperinsulinemia which in time exhausts the beta cells, leading to insulin deficiency and T2DM. The skeletal muscle (assisted by insulin) uptakes about 80% circulating glucose immediately after meals, rapidly clearing the postprandial hyperglycemia (200). Impaired glucose uptake, elevated blood glucose levels, and hyperinsulinemia comprise act one of the T2DM drama. From this point on, it may take decades before the pancreatic beta cells become insufficient and unable to secrete insulin that triggers act two: insulin deficiency and frank T2DM. As insulin is cosecreted with serotonin and dopamine, these MAs also become deficient, contributing to circulatory and cardiovascular pathology which frequently accompany T2DM (Figure 2).
Tryptophan is an essential amino acid and the sole precursor of the human body serotonin. The CNS manufactures its own serotonin; however, exogenous tryptophan must be supplied to the brain, a process facilitated by insulin. Deficiency of this hormone manifested by lower brain tryptophan was documented in patients with T2DM, possibly explaining the higher prevalence of depression in this metabolic disorder (201). In addition, tryptophan depletion studies in humans and animals demonstrated that low plasma tryptophan levels were associated with aggression (202–204).
Aside from serotonin, tryptophan is catabolized into several other neuroactive compounds, including the neurotoxic kynurenine (KYN) and the neuroprotective kynurenic acid (KYNA) (Figure 3).
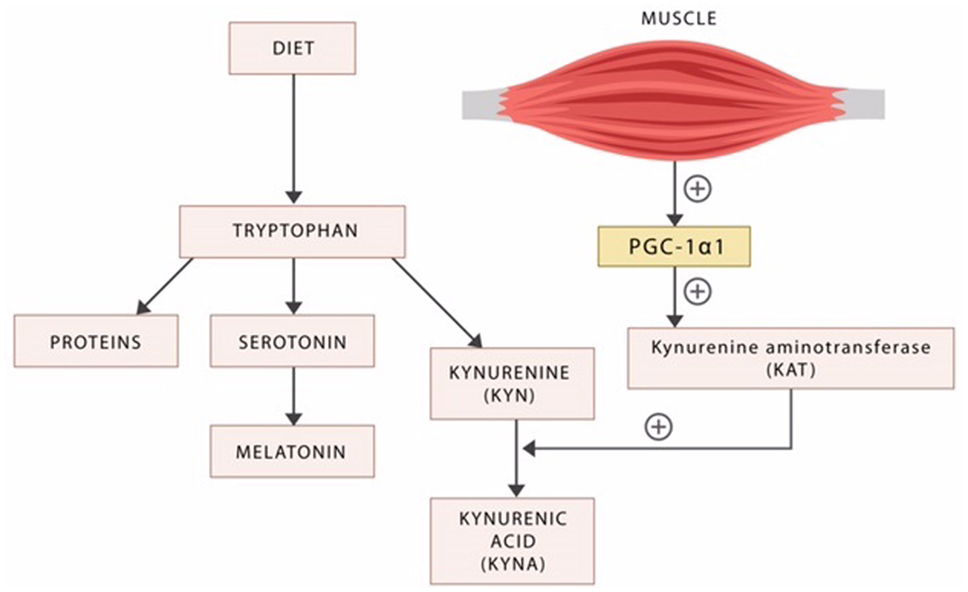
Figure 3. Dietary tryptophan is the precursor of proteins, serotonin, and the kynurenines. The exercise factor peroxisome proliferator-activated receptor-γ coactivator 1alpha (PGC-1alpha) released by the active skeletal muscle induces kynurenine aminotransferases (KAT), the enzyme responsible for the biosynthesis of the neuroprotective KYNA. In the absence of exercise, the neurotoxic KYN crosses the blood–brain barrier inducing psychopathology.
Kynurenine was demonstrated to cross the BBB and was implicated in the pathophysiology of disorders marked by impulsivity, including schizophrenia, ADHD, cognitive disorders, drug addictions, and mania (205). KYN is an antagonist at both the N-methyl-d-aspartate (NMDA) and alpha-7 nACh receptors, suggesting a possible pathoetiological mechanism of excessive impulsivity (206). This mechanism may be similar in nature to the antagonism at alpha-7 nACh receptors in POMC/CART neurons associated with weight gain and the T2DM risk (as referenced above in the discussion concerning smoking cessation).
It was recently demonstrated that the enzyme kynurenine aminotransferase (KAT) catalyzes the transformation of KYN into KYNA, in a reaction facilitated by the exercise factor PGC-1alpha (207) (Figure 3). As KYNA is neuroprotective, this molecular mechanism may provide an explanation for the known benefits of physical exercise in psychiatric disorders (208, 209) (Figure 3).
While the beneficial effect of exercise and physical activity are well established, specific recommendations for the metabolic dysregulation and regimen details are beginning to emerge (Table 2).
Conclusion: When the Seven Lean Cows Ate up the Seven Fat Cows
The energy–behavior link was intuited by the previous generations and currently we are merely rediscovering an old truth: the brain and energy are tightly interconnected. Indeed, the energy–hungry brain utilizes over 20% of the total body energy budget for the functioning of excitatory and inhibitory synapses believed to be dysregulated in impulsivity-connected psychopathology (210).
The metabolic hormones—leptin, adiponectin, and ghrelin—communicate the peripheral energy status to the hypothalamic feeding centers via receptors coexpressed with the major behavioral modulators, the MAs and ACh. As these receptors are blocked by the PTDs, they may become insensitive (for example, leptin resistance), inducing hypermetric hormonal responses with resultant obesity and metabolic dysregulation.
While PTDs may correct synaptic transmission, they alter the psychometabolic continuum, inducing weight gain in individuals already predisposed to it. Here, we suggest halting the development of metabolic dysregulation and obesity by initiating preventive treatment concurrently with the PTDs. Indeed, the management of impulsive aggression in psychiatric disorders may be incomplete without addressing the metabolic component and restoring the homeostasis of the psychometabolic continuum.
Addressing psychopathology and metabolic dysfunction simultaneously may result in superior outcomes compared to an intervention that addresses these conditions in a singular way. In addition, correcting energy homeostasis may lower the overall morbidity, mortality, and institutional cost in psychiatric population. A model can be inferred from clinical practice where it was found that correcting hypertension led to better outcomes in coronary artery disease, stroke, and renal failure compared to their post hoc treatment. After all, as prevention is preferable to treatment, what would be a valid reason for not addressing metabolic dysregulation in psychiatric patients in a preemptive manner?
Author Contributions
All the authors have contributed equally to this work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer (RG) declared a past co-authorship with the authors to the handling editor and the reviewer (NS) declared her shared affiliation with the handling editor, who ensured that the process met the standards of a fair and objective review.
References
1. Moeller FG, Barratt ES, Dougherty DM, Schmitz JM, Swann AC. Psychiatric aspects of impulsivity. Am J Psychiatry (2001) 158(11):1783–93. doi:10.1176/appi.ajp.158.11.1783
2. Fernandez-Egea E, Bernardo M, Donner T, Conget I, Parellada E, Justicia A, et al. Metabolic profile of antipsychotic-naive individuals with non-affective psychosis. Br J Psychiatry (2009) 194(5):434–8. doi:10.1192/bjp.bp.108.052605
3. Grover S, Nebhinani N, Chakrabarti S, Parakh P, Ghormode D. Metabolic syndrome in antipsychotic naïve patients diagnosed with schizophrenia. Early Interv Psychiatry (2012) 6(3):326–31. doi:10.1111/j.1751-7893.2011.00321.x
4. Darcin EA, Cavus YS, Dilbaz N, Kaya H, Dogan E. Metabolic syndrome in drug-naïve and drug-free patients with schizophrenia and in their siblings. Schizophr Res (2015) 166(1–3):201–6. doi:10.1016/j.schres.2015.05.004
5. Chadda RK, Ramshankar P, Deb KS, Sood M. Metabolic syndrome in schizophrenia: differences between antipsychotic-naïve and treated patients. J Pharmacol Pharmacother (2013) 4(3):176–86. doi:10.4103/0976-500X.114596
6. Nasrallah HA, Meyer JM, Goff DC, McEvoy JP, Davis SM, Stroup TS, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res (2006) 86(1–3):15–22. doi:10.1016/j.schres.2006.06.026
7. Mitchell AJ, Vancampfort D, De Herdt A, Yu W, De Hert M. Is the prevalence of metabolic syndrome and metabolic abnormalities increased in early schizophrenia? A comparative meta-analysis of first episode, untreated and treated patients. Schizophr Bull (2013) 39(2):295–305. doi:10.1093/schbul/sbs082
8. Guo X, Zhang Z, Wei Q, Lv H, Wu R, Zhao J. The relationship between obesity and neurocognitive function in Chinese patients with schizophrenia. BMC Psychiatry (2013) 13:109. doi:10.1186/1471-244X-13-109
9. Beltrán-Sánchez H, Harhay MO, Harhay MM, McElligott S. Prevalence and trends of metabolic syndrome in the adult US population, 1999–2010. J Am Coll Cardiol (2013) 62(8):697–703. doi:10.1016/j.jacc.2013.05.064
10. World Health Organization. Obesity: Preventing and Managing the Global Epidemic. Geneva: WHO (1998).
11. Merikangas KR, Nakamura EF, Kessler RC. Epidemiology of mental disorders in children and adolescents. Dialogues Clin Neurosci (2009) 11(1):7–20.
12. Sturm R, Hattori A. Morbid obesity rates continue to rise rapidly in the US. Int J Obes (2013) 37(6):889–91. doi:10.1038/ijo.2012.159
13. Kirkpatrick B, Miller B, Garcia-Rizo C, Fernandez-Egea E. Schizophrenia: a systemic disorder. Clin Schizophr Relat Psychoses (2014) 8:73–9. doi:10.3371/CSRP.KIMI.031513
14. Campbell BC, Eisenberg D. Obesity, attention deficit-hyperactivity disorder and the dopaminergic reward system. Coll Antropol (2007) 31(1):33–8.
15. Goldstein BI, Liu S-M, Zivkovic N, Schaffer A, Chien L-C, Blanco C. The burden of obesity among adults with bipolar disorder in the United States. Bipolar Disord (2011) 13(4):387–95. doi:10.1111/j.1399-5618.2011.00932.x
16. Chouinard VA, Pingali SM, Chouinard G, Henderson DC, Mallya SG, Cypess AM, et al. Factors associated with overweight and obesity in schizophrenia, schizoaffective and bipolar disorders. Psychiatry Res (2016) 237:304–10. doi:10.1016/j.psychres.2016.01.024
17. Wagner B, Klinitzke G, Brähler E, Kersting A. Extreme obesity is associated with suicidal behavior and suicide attempts in adults: results of a population-based representative sample. Depress Anxiety (2013) 30(10):975–81. doi:10.1002/da.22105
18. Kakoschke N, Kemps E, Tiggemann M. External eating mediates the relationship between impulsivity and unhealthy food intake. Physiol Behav (2015) 147(1):117–21. doi:10.1016/j.physbeh.2015.04.030
19. Sansone RA, Sansone LA. The relationship between borderline personality and obesity. Innov Clin Neurosci (2013) 10(4):36–40.
20. Farhat T, Iannotti RJ, Simons-Morton B. Overweight, obesity, youth, and health-risk behaviors. Am J Prev Med (2010) 38(3):258–67. doi:10.1016/j.amepre.2009.10.038
21. Sullivan EL, Nousen L, Chamlou K. Maternal high fat diet consumption during the perinatal period programs offspring behavior. Physiol Behav (2014) 123:236–42. doi:10.1016/j.physbeh.2012.07.014
22. Fields SA, Sabet M, Reynolds B. Dimensions of impulsive behavior in obese, overweight, and healthy-weight adolescents. Appetite (2013) 70:60–6. doi:10.1016/j.appet.2013.06.089
23. Golomb BA, Evans MA, White HL, Dimsdale JE. Trans fat consumption and aggression. PLoS One (2012) 7(3):e32175. doi:10.1371/journal.pone.0032175
24. Yu E, Lippert AM. Neighborhood crime rate, weight-related behaviors, and obesity: a systematic review of the literature. Sociol Comp (2016) 10(3):187–207. doi:10.1111/soc4.12356
25. Shott ME, Cornier MA, Mittal VA, Pryor TL, Orr JM, Brown MS, et al. Orbitofrontal cortex volume and brain reward response in obesity. Int J Obes (Lond) (2015) 39(2):214–21. doi:10.1038/ijo.2014.121
26. Horn NR, Dolan M, Elliott R, Deakin JF, Woodruff PW. Response inhibition and impulsivity: an fMRI study. Neuropsychologia (2003) 41(14):1959–66. doi:10.1016/S0028-3932(03)00077-0
27. Batterink L, Yokum S, Stice E. Body mass correlates inversely with inhibitory control in response to food among adolescent girls: an fMRI study. Neuroimage (2010) 52(4):1696–703. doi:10.1016/j.neuroimage.2010.05.059
28. Stoeckel LE, Murdaugh DL, Cox JE, Cook EW, Weller RE. Greater impulsivity is associated with decreased brain activation in obese women during a delay discounting task. Brain Imaging Behav (2013) 7(2):116–28. doi:10.1007/s11682-012-9201-4
29. Anderberg RH, Hansson C, Fenander M, Richard JE, Dickson SL, Nissbrandt H, et al. The stomach-derived hormone ghrelin increases impulsive behavior. Neuropsychopharmacology (2016) 41(5):1199–209. doi:10.1038/npp.2015.297
30. Sutin AR, Zonderman AB, Uda M, Deiana B, Taub DD, Longo DL, et al. Personality traits and leptin. Psychosom Med (2013) 75(5):505–9. doi:10.1097/PSY.0b013e3182919ff4
31. Atmaca M, Kuloglu M, Tezcan E, Gecici O, Ustundag B. Serum cholesterol and leptin levels in patients with borderline personality disorder. Neuropsychobiology (2002) 45(4):167–71. doi:10.1159/000063665
32. Tutuncu R, Gunay H, Balibey H, Abaslı D. Decreased leptin levels in antisocial personality disorder patients. Bull Clin Psychopharmacol (2016) 26(3):215–302. doi:10.5455/bcp.20130127022613
33. Ferrini F, Salio C, Lossi L, Merighi A. Ghrelin in central neurons. Curr Neuropharmacol (2009) 7(1):37–49. doi:10.2174/157015909787602779
34. Mavroconstanti T, Halmøy A, Haavik J. Decreased serum levels of adiponectin in adult attention deficit hyperactivity disorder. Psychiatry Res (2014) 216(1):123–30. doi:10.1016/j.psychres.2014.01.025
35. Bai YM, Chen JY, Yang WS, Chi YC, Liou YJ, Lin CC, et al. Adiponectin as a potential biomarker for the metabolic syndrome in Chinese patients taking clozapine for schizophrenia. J Clin Psychiatry (2007) 68(12):1834–9. doi:10.4088/JCP.v68n1202
36. Lu ML, Wang TN, Lin TY, Shao WC, Chang SH, Chou JY, et al. Differential effects of olanzapine and clozapine on plasma levels of adipocytokines and total ghrelin. Prog Neuropsychopharmacol Biol Psychiatry (2015) 58:47–50. doi:10.1016/j.pnpbp.2014.12.001
37. Lee AK, Bishop JR. Pharmacogenetics of leptin in antipsychotic-associated weight gain and obesity-related complications. Pharmacogenomics (2011) 12(7):999–1016. doi:10.2217/pgs.11.45
38. Freland L, Beaulieu J-M. Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front Mol Neurosci (2012) 5:14. doi:10.3389/fnmol.2012.00014
39. MacAulay K, Woodgett JR. Targeting glycogen synthase kinase-3 (GSK-3) in the treatment of Type 2 diabetes. Expert Opin Ther Targets (2008) 12(10):1265–74. doi:10.1517/14728222.12.10.1265
40. Gao C, Hölscher C, Liu Y, Li L. GSK3: a key target for the development of novel treatments for type 2 diabetes mellitus and Alzheimer disease. Rev Neurosci (2011) 23(1):1–11. doi:10.1515/rns.2011.061
41. Tutuncu R, Gunay H, Balibey H, Abaslı D. Decreased leptin levels in antisocial personality disorder patients. Bull Clin Psychopharmacol (2013) 23(1):72–6. doi:10.5455/bcp.20130127022613
42. Ohmura Y, Tsutsui-Kimura I, Yoshioka M. Impulsive behavior and nicotinic acetylcholine receptors. J Pharmacol Sci (2012) 118(4):413–22. doi:10.1254/jphs.11R06CR
43. Polina ER, Rovaris DL, de Azeredo LA, Mota NR, Vitola ES, Silva KL, et al. ADHD diagnosis may influence the association between polymorphisms in nicotinic acetylcholine receptor genes and tobacco smoking. Neuromolecular Med (2014) 16(2):389–97. doi:10.1007/s12017-013-8286-2
44. Dalley JW, Roiser JP. Dopamine, serotonin and impulsivity. Neuroscience (2012) 215:42–58. doi:10.1016/j.neuroscience.2012.03.065
45. Khorassani FE, Misher A, Garris S. Past and present of antiobesity agents: focus on monoamine modulators. Am J Health Syst Pharm (2015) 72(9):697–706. doi:10.2146/ajhp140034
46. Donovan MH, Tecott LH. Serotonin and the regulation of mammalian energy balance. Front Neurosci (2013) 7:36. doi:10.3389/fnins.2013.00036
47. Shrivastava A, Johnston ME. Weight-gain in psychiatric treatment: risks, implications, and strategies for prevention and management. Mens Sana Monogr (2010) 8(1):53–68. doi:10.4103/0973-1229.58819
48. Maayan L, Correll CU. Management of antipsychotic-related weight gain. Expert Rev Neurother (2010) 10(7):1175–200. doi:10.1586/ern.10.85
49. Minor RK, Chang JW, de Cabo R. Hungry for life: how the arcuate nucleus and neuropeptide Y may play a critical role in mediating the benefits of calorie restriction. Mol Cell Endocrinol (2009) 299(1):79–88. doi:10.1016/j.mce.2008.10.044
50. Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron (2012) 76(1):116–29. doi:10.1016/j.neuron.2012.08.036
51. Adan RAH, Tiesjema B, Hillebrand JJG, la Fleur SE, Kas MJH, de Krom M. The MC4 receptor and control of appetite. Br J Pharmacol (2006) 149(7):815–27. doi:10.1038/sj.bjp.0706929
52. Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev (2011) 63(1):182–217. doi:10.1124/pr.110.002642
53. Generali JA, Cada DJ. Topiramate: antipsychotic-induced weight gain. Hosp Pharm (2014) 49(4):345–7. doi:10.1310/hpj4904-354
54. Wu RR, Zhang FY, Gao KM, Ou JJ, Shao P, Jin H, et al. Metformin treatment of antipsychotic-induced dyslipidemia: an analysis of two randomized, placebo-controlled trials. Mol Psychiatry (2016) 21(11):1537–44. doi:10.1038/mp.2015.221
55. De Silva VA, Suraweera C, Ratnatunga SS, Dayabandara M, Wanniarachchi N, Hanwella R. Metformin in prevention and treatment of antipsychotic induced weight gain: a systematic review and meta-analysis. BMC Psychiatry (2016) 16:341. doi:10.1186/s12888-016-1049-5
56. Anagnostou E, Aman MG, Handen BL, Sanders KB, Shui A, Hollway JA, et al. Metformin for treatment of overweight induced by atypical antipsychotic medication in young people with autism spectrum disorder: a randomized clinical trial. JAMA Psychiatry (2016) 73(9):928–37. doi:10.1001/jamapsychiatry.2016.1232
57. Secher A, Jelsing J, Baquero AF, Hecksher-Sørensen J, Cowley MA, Dalbøge LS, et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Invest (2014) 124(10):4473–88. doi:10.1172/JCI75276
58. Mayfield K, Siskind D, Winckel K, Russell AW, Kisely S, Smith G, et al. Glucagon-like peptide-1 agonists combating clozapine-associated obesity and diabetes. J Psychopharmacol (2016) 30(3):227–36. doi:10.1177/0269881115625496
59. Larsen JR, Vedtofte L, Holst JJ, Oturai P, Kjaer A, Correll CU, et al. Does a GLP-1 receptor agonist change glucose tolerance in patients treated with antipsychotic medications? Design of a randomised, double-blinded, placebo-controlled clinical trial. BMJ Open (2014) 4(3):e004227. doi:10.1136/bmjopen-2013-004227
60. Ishøy PL, Knop FK, Broberg BV, Bak N, Andersen UB, Jørgensen NR, et al. Effect of GLP-1 receptor agonist treatment on body weight in obese antipsychotic-treated patients with schizophrenia: a randomized, placebo-controlled trial. Diabetes Obes Metab (2017) 19(2):162–71. doi:10.1111/dom.12795
61. Ebdrup BH, Knop FK, Madsen A, Mortensen HB, Søgaard B, Holst JJ, et al. Glucometabolic hormones and cardiovascular risk markers in antipsychotic-treated patients. J Clin Psychiatry (2014) 75(9):e899–905. doi:10.4088/JCP.13m08820
62. Kang JG, Park C-Y. Anti-obesity drugs: a review about their effects and safety. Diabetes Metab J (2012) 36(1):13–25. doi:10.4093/dmj.2012.36.1.13
63. Ahrén B, Johnson SL, Stewart M, Cirkel DT, Yang F, Perry C, et al. HARMONY 3: 104-week randomized, double-blind, placebo- and active-controlled trial assessing the efficacy and safety of albiglutide compared with placebo, sitagliptin, and glimepiride in patients with type 2 diabetes taking metformin. Diabetes Care (2014) 37(8):2141–8. doi:10.2337/dc14-0024
64. Fujioka K. Current and emerging medications for overweight or obesity in people with comorbidities. Diabetes Obes Metab (2015) 17(11):1021–32. doi:10.1111/dom.12502
65. Vilsbøll T, Christensen M, Junker AE, Knop FK, Gluud LL. Effects of glucagon-like peptide-1 receptor agonists on weight loss: systematic review and meta-analyses of randomised controlled trials. BMJ (2012) 344:d7771. doi:10.1136/bmj.d7771
66. Skibicka KP. The central GLP-1: implications for food and drug reward. Front Neurosci (2013) 7:181. doi:10.3389/fnins.2013.00181
67. McIntyre RS, Powell AM, Kaidanovich-Beilin O, Soczynska JK, Alsuwaidan M, Woldeyohannes HO, et al. The neuroprotective effects of GLP-1: possible treatments for cognitive deficits in individuals with mood disorders. Behav Brain Res (2013) 237:164–71. doi:10.1016/j.bbr.2012.09.021
68. Gejl M, Gjedde A, Egefjord L, Møller A, Hansen SB, Vang K, et al. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Front Aging Neurosci (2016) 8:108. doi:10.3389/fnagi.2016.00108
69. Schellekens H, De Francesco PN, Kandil D, Theeuwes WF, McCarthy T, van Oeffelen WE, et al. Ghrelin’s orexigenic effect is modulated via a serotonin 2C receptor interaction. ACS Chem Neurosci (2015) 6(7):1186–97. doi:10.1021/cn500318q
70. Palik E, Birkás KD, Faludi G, Karádi I, Cseh K. Correlation of serum ghrelin levels with body mass index and carbohydrate metabolism in patients treated with atypical antipsychotics. Diabetes Res Clin Pract (2005) 68(Suppl 1):S60–4. doi:10.1016/j.diabres.2005.03.008
71. Agnoli L, Carli M. Dorsal-striatal 5-HT2A and 5-HT2C receptors control impulsivity and perseverative responding in the 5-choice serial reaction time task. Psychopharmacology (Berl) (2012) 219(2):633–45. doi:10.1007/s00213-011-2581-0
72. Nonogaki K, Ohashi-Nozue K, Oka Y. A negative feedback system between brain serotonin systems and plasma active ghrelin levels in mice. Biochem Biophys Res Commun (2006) 341(3):703–7. doi:10.1016/j.bbrc.2006.01.021
73. Fletcher PJ, Soko AD, Higgins GA. Impulsive action in the 5-choice serial reaction time test in 5-HT2C receptor null mutant mice. Psychopharmacology (2013) 226(Issue 3):561–70. doi:10.1007/s00213-012-2929-0
74. Navarra R, Comery TA, Graf R, Rosenzweig-Lipson S, Day M. The 5-HT(2C) receptor agonist WAY-163909 decreases impulsivity in the 5-choice serial reaction time test. Behav Brain Res (2008) 188(2):412–5. doi:10.1016/j.bbr.2007.11.016
75. Doslikova B, Garfield AS, Shaw J, Evans ML, Burdakov D, Billups B, et al. 5-HT2C receptor agonist anorectic efficacy potentiated by 5-HT1B receptor agonist coapplication: an effect mediated via increased proportion of pro-opiomelanocortin neurons activated. J Neurosci (2013) 33(23):9800–4. doi:10.1523/JNEUROSCI.4326-12.2013
76. Clark MS, Neumaier JF. The 5-HT1B receptor: behavioral implications. Psychopharmacol Bull (2001) 35(4):170–85.
77. Anastasio NC, Stutz SJ, Fox RG, Sears RM, Emeson RB, DiLeone RJ, et al. Functional status of the serotonin 5-HT2C receptor (5-HT2CR) drives interlocked phenotypes that precipitate relapse-like behaviors in cocaine dependence. Neuropsychopharmacology (2014) 39(2):370–82. doi:10.1038/npp.2013.199
78. Nautiyal KM, Tanaka KF, Barr MM, Tritschler L, Le Dantec Y, David DJ, et al. Distinct circuits underlie the effects of 5-HT1B receptors on aggression and impulsivity. Neuron (2015) 86(3):813–26. doi:10.1016/j.neuron.2015.03.041
79. Olivier B, van Oorschot R. 5-HT1B receptors and aggression: a review. Eur J Pharmacol (2005) 526(1–3):207–17. doi:10.1016/j.ejphar.2005.09.066
80. Jiménez E, Arias B, Mitjans M, Goikolea JM, Roda E, Ruíz V, et al. Association between GSK3β gene and increased impulsivity in bipolar disorder. Eur Neuropsychopharmacol (2014) 24(4):510–8. doi:10.1016/j.euroneuro.2014.01.005
81. Koros E, Dorner-Ciossek C. The role of glycogen synthase kinase-3beta in schizophrenia. Drug News Perspect (2007) 20(7):437–45. doi:10.1358/dnp.2007.20.7.1149632
82. Ren X, Rizavi HS, Khan MA, Dwivedi Y, Pandey GN. Altered Wnt signalling in the teenage suicide brain: focus on glycogen synthase kinase-3β and β-catenin. Int J Neuropsychopharmacol (2013) 16(5):945–55. doi:10.1017/S1461145712001010
83. Polter AM, Li X. Glycogen synthase kinase-3 is an intermediate modulator of serotonin neurotransmission. Front Mol Neurosci (2011) 4:31. doi:10.3389/fnmol.2011.00031
84. Oreña SJ, Torchia AJ, Robert S, Garofalo RS. Inhibition of glycogen-synthase kinase 3 stimulates glycogen synthase and glucose transport by distinct mechanisms in 3T3-L1 adipocytes. J Biol Chem (2000) 275:15765–72. doi:10.1074/jbc.M910002199
85. Patel S, Doble B, Woodgett JR. Glycogen synthase kinase-3 in insulin and Wnt signalling: a double-edged sword? Biochem Soc Trans (2004) 32(5):803–8. doi:10.1042/BST0320803
86. Henriksen EJ, Dokken BB. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr Drug Targets (2006) 7(11):1435–41. doi:10.2174/1389450110607011435
87. Hu M, Wu H, Chao C. Assisting effects of lithium on hypoglycemic treatment in patients with diabetes. Biol Trace Elem Res (1997) 60(1–2):131–7. doi:10.1007/BF02783316
88. Hunt NT. Hypoglycemic effect of lithium. Biol Psychiatry (1987) 22:798–9. doi:10.1016/0006-3223(87)90219-8
90. Pijl H, Ohashi S, Matsuda M, Miyazaki Y, Mahankali A, Kumar V, et al. Bromocriptine: a novel approach to the treatment of type 2 diabetes. Diabetes Care (2000) 23(8):1154–61. doi:10.2337/diacare.23.8.1154
91. Graham KA, Gu H, Lieberman JA, Harp JB, Perkins DO. Double-blind, placebo-controlled investigation of amantadine for weight loss in subjects who gained weight with olanzapine. Am J Psychiatry (2005) 162(9):1744–6. doi:10.1176/appi.ajp.162.9.1744
92. Praharaj SK, Sharma PSVN. Amantadine for olanzapine-induced weight gain: a systematic review and meta-analysis of randomized placebo-controlled trials. Ther Adv Psychopharmacol (2012) 2(4):151–6. doi:10.1177/2045125312440441
93. Brandebourg T, Hugo E, Ben-Jonathan N. Adipocyte prolactin: regulation of release and putative functions. Diabetes Obes Metab (2007) 9(4):464–76. doi:10.1111/j.1463-1326.2006.00671.x
94. Lee MS, Song HC, An H, Yang J, Ko YH, Jung IK, et al. Effect of bromocriptine on antipsychotic drug-induced hyperprolactinemia: eight-week randomized, single-blind, placebo-controlled, multicenter study. Psychiatry Clin Neurosci (2010) 64(1):19–27. doi:10.1111/j.1440-1819.2009.02032.x
95. Friedrich N, Schneider HJ, Spielhagen C, Markus MR, Haring R, Grabe HJ, et al. The association of serum prolactin concentration with inflammatory biomarkers – cross-sectional findings from the population-based Study of Health in Pomerania. Clin Endocrinol (Oxf) (2011) 75(4):561–6. doi:10.1111/j.1365-2265.2011.04075.x
96. Haring R, Friedrich N, Völzke H, Vasan RS, Felix SB, Dörr M, et al. Positive association of serum prolactin concentrations with all-cause and cardiovascular mortality. Eur Heart J (2014) 35(18):1215–21. doi:10.1093/eurheartj/ehs23
97. Lindenmayer JP, Nasrallah H, Pucci M, James S, Citrome L. A systematic review of psychostimulant treatment of negative symptoms of schizophrenia: challenges and therapeutic opportunities. Schizophr Res (2013) 147(2–3):241–52. doi:10.1016/j.schres.2013.03.01
98. Levi-Minzi S, Bermanzohn PC, Siris SG. Bromocriptine for “negative” schizophrenia. Compr Psychiatry (1991) 32(3):210–6. doi:10.1016/0010-440X(91)90041-A
99. Gragnoli C, Reeves GM, Reazer J, Postolache TT. Dopamine–prolactin pathway potentially contributes to the schizophrenia and type 2 diabetes comorbidity. Transl Psychiatry (2016) 6(4):e785. doi:10.1038/tp.2016.50
100. Poulton AS, Hibbert EJ, Champion BL, Nanan RKH. Stimulants for the control of hedonic appetite. Front Pharmacol (2016) 7:105. doi:10.3389/fphar.2016.00105
101. Volkow ND, Wang G-J, Baler RD. Reward, dopamine and the control of food intake: implications for obesity. Trends Cogn Sci (2011) 15(1):37–46. doi:10.1016/j.tics.2010.11.001
102. Wang GJ, Volkow ND, Logan J, Pappas NR, Wong CT, Zhu W, et al. Brain dopamine and obesity. Lancet (2001) 357:354–7. doi:10.1016/S0140-6736(00)03643-6
103. Baik J-H. Dopamine signaling in food addiction: role of dopamine D2 receptors. BMB Rep (2013) 46(11):519–26. doi:10.5483/BMBRep.2013.46.11.207
104. Fornaro M, Solmi M, Perna G, De Berardis D, Veronese N, Orsolini L, et al. Lisdexamfetamine in the treatment of moderate-to-severe binge eating disorder in adults: systematic review and exploratory meta-analysis of publicly available placebo-controlled, randomized clinical trials. Neuropsychiatr Dis Treat (2016) 12:1827–36. doi:10.2147/NDT.S109637
105. Lasser RA, Dirks B, Nasrallah H, Kirsch C, Gao J, Pucci ML, et al. Adjunctive lisdexamfetamine dimesylate therapy in adult outpatients with predominant negative symptoms of schizophrenia: open-label and randomized-withdrawal phases. Neuropsychopharmacology (2013) 38(11):2140–9. doi:10.1038/npp.2013.111
106. Meister B, Gömüç B, Suarez E, Ishii Y, Dürr K, Gillberg L. Hypothalamic proopiomelanocortin (POMC) neurons have a cholinergic phenotype. Eur J Neurosci (2006) 24(10):2731–40. doi:10.1111/j.1460-9568.2006.05157.x
107. Jo Y-H, Talmage DA, Role LW. Nicotinic receptor-mediated effects on appetite and food intake. J Neurobiol (2002) 53(4):618–32. doi:10.1002/neu.10147
108. Zhou W-L, Gao X-B, Picciotto MR. Acetylcholine acts through nicotinic receptors to enhance the firing rate of a subset of hypocretin neurons in the mouse hypothalamus through distinct presynaptic and postsynaptic mechanisms. eNeuro (2015) 2(1). doi:10.1523/ENEURO.0052-14.2015
109. Rodriguez-Diaz R, Dando R, Jacques-Silva MC, Fachado A, Molina J, Abdulreda MH, et al. Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming human beta cell function. Nat Med (2011) 17(7):888–92. doi:10.1038/nm.2371
110. Yoshikawa H, Hellström-Lindahl E, Grill V. Evidence for functional nicotinic receptors on pancreatic beta cells. Metabolism (2005) 54(2):247–54. doi:10.1016/j.metabol.2004.08.020
111. Bymaster FP, Felder CC, Tzavara E, Nomikos GG, Calligaro DO, Mckinzie DL. Muscarinic mechanisms of antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry (2003) 27(7):1125–43. doi:10.1016/j.pnpbp.2003.09.008
112. Bajaj M. Nicotine and insulin resistance: when the smoke clears. Diabetes (2012) 61(12):3078–80. doi:10.2337/db12-1100
113. Rao AA, Sridhar GR, Das UN. Elevated butyrylcholinesterase and acetylcholinesterase may predict the development of type 2 diabetes mellitus and Alzheimer’s disease. Med Hypotheses (2007) 69(6):1272–6. doi:10.1016/j.mehy.2007.03.032
114. Chen VP, Gao Y, Geng L, Parks RJ, Pang Y-P, Brimijoin S. Plasma butyrylcholinesterase regulates ghrelin to control aggression. Proc Natl Acad Sci U S A (2015) 112(7):2251–6. doi:10.1073/pnas.142153611
115. Marrero MB, Lucas R, Salet C, Hauser TA, Mazurov A, Lippiello PM, et al. An alpha7 nicotinic acetylcholine receptor-selective agonist reduces weight gain and metabolic changes in a mouse model of diabetes. J Pharmacol Exp Ther (2010) 332(1):173–80. doi:10.1124/jpet.109.154633
116. Meyer JM, Cummings MA, Proctor G, Stahl SM. Psychopharmacology of persistent violence and aggression. Psychiatr Clin North Am (2016) 39(4):541–56. doi:10.1016/j.psc.2016.07.012
117. Figiel G, Sadowsky C. A systematic review of the effectiveness of rivastigmine for the treatment of behavioral disturbances in dementia and other neurological disorders. Curr Med Res Opin (2008) 24(1):157–66. doi:10.1185/030079908X260961
118. Brimijoin S, Chen VP, Pang YP, Geng L, Gao Y. Physiological roles for butyrylcholinesterase: a BChE-ghrelin axis. Chem Biol Interact (2016) 259(Pt B):271–5. doi:10.1016/j.cbi.2016.02.013
119. Maier C, Schaller G, Buranyi B, Luger A. The cholinergic system controls ghrelin release and ghrelin-induced growth hormone release in humans. J Clin Endocrinol Metab (2004) 89(9):4729–33. doi:10.1210/jc.2004-0656
120. Shen M, Jiang C, Liu P, Wang F, Ma L. Mesolimbic leptin signaling negatively regulates cocaine-conditioned reward. Transl Psychiatry (2016) 6:e972. doi:10.1038/tp.2016.223
121. Cipolla-Neto J, Amaral FG, Afeche SC, Tan DX, Reiter RJ. Melatonin, energy metabolism, and obesity: a review. J Pineal Res (2014) 56(4):371–81. doi:10.1111/jpi.12137
122. Haller G, Kapoor M, Budde J, Xuei X, Edenberg H, Nurnberger J, et al. Rare missense variants in CHRNB3 and CHRNA3 are associated with risk of alcohol and cocaine dependence. Hum Mol Genet (2014) 23(3):810–9. doi:10.1093/hmg/ddt463
123. Kamens HM, Corley RP, Richmond PA, Darlington TM, Dowell R, Hopfer CJ, et al. Evidence for association between low frequency variants in CHRNA6/CHRNB3 and antisocial drug dependence. Behav Genet (2016) 46(5):693–704. doi:10.1007/s10519-016-9792-4
124. Fleisher C, McGough J. Sofinicline: a novel nicotinic acetylcholine receptor agonist in the treatment of attention-deficit/hyperactivity disorder. Expert Opin Investig Drugs (2014) 23(8):1157–63. doi:10.1517/13543784.2014.934806
125. Liu H, Fu W, Wetter J, Xu H, Guan Z, Stuart P. Metabolism and disposition of ABT-894, a novel α4β2 neuronal acetylcholine receptor agonist, in mice and monkeys. Xenobiotica (2014) 44(6):531–40. doi:10.3109/00498254.2013.855836
126. Rowbotham MC, Arslanian A, Nothaft W, Duan WR, Best AE, Pritchett Y, et al. Efficacy and safety of the α4β2 neuronal nicotinic receptor agonist ABT-894 in patients with diabetic peripheral neuropathic pain. Pain (2012) 153(4):862–8. doi:10.1016/j.pain.2012.01.009
127. Martínez de Morentin PB, Whittle AJ, Fernø J, Nogueiras R, Diéguez C, Vidal-Puig A, et al. Nicotine induces negative energy balance through hypothalamic AMP-activated protein kinase. Diabetes (2012) 61(4):807–17. doi:10.2337/db11-1079
128. Kageyama H, Takenoya F, Shiba K, Shioda S. Neuronal circuits involving ghrelin in the hypothalamus-mediated regulation of feeding. Neuropeptides (2010) 44(2):133–8. doi:10.1016/j.npep.2009.11.010
129. Ghigo E, Broglio F, Arvat E, Maccario M, Papotti M, Muccioli G. Ghrelin: more than a natural GH secretagogue and/or an orexigenic factor. Clin Endocrinol (2005) 62:1–17. doi:10.1111/j.1365-2265.2004.02160.x
130. Horvath TL, Diano S, Sotonyi P, Heiman M, Tschop M. Minireview: ghrelin and the regulation of energy balance – a hypothalamic perspective. Endocrinology (2001) 142:4163–9. doi:10.1210/en.142.10.4163
131. Montastruc F, Palmaro A, Bagheri H, Schmitt L, Montastruc JL, Lapeyre-Mestre M. Role of serotonin 5-HT2C and histamine H1 receptors in antipsychotic-induced diabetes: a pharmacoepidemiological-pharmacodynamic study in VigiBase. Eur Neuropsychopharmacol (2015) 25(10):1556–65. doi:10.1016/j.euroneuro.2015.07.010
132. Huang F, Dong J, Kong J, Wang H, Meng H, Spaeth RB, et al. Effect of transcutaneous auricular vagus nerve stimulation on impaired glucose tolerance: a pilot randomized study. BMC Complement Altern Med (2014) 14:203. doi:10.1186/1472-6882-14-203
133. Pini A, Obara I, Battell E, Chazot PL, Rosa AC. Histamine in diabetes: is it time to reconsider? Pharmacol Res (2016) 111:316–24. doi:10.1016/j.phrs.2016.06.021
134. Deng C, Weston-Green K, Huang XF. The role of histaminergic H1 and H3 receptors in food intake: a mechanism for atypical antipsychotic-induced weight gain? Prog Neuropsychopharmacol Biol Psychiatry (2010) 34:1–4. doi:10.1016/j.pnpbp.2009.11.009
135. Masaki T, Chiba S, Yasuda T, Noguchi H, Kakuma T, Watanabe T, et al. Involvement of hypothalamic histamine H1 receptor in the regulation of feeding rhythm and obesity. Diabetes (2004) 53(9):2250–60. doi:10.2337/diabetes.53.9.2250
136. Barak N, Greenway FL, Fujioka K, Aronne LJ, Kushner RF. Effect of histaminergic manipulation on weight in obese adults: a randomized placebo controlled trial. Int J Obes (Lond) (2008) 32(10):1559–65. doi:10.1038/ijo.2008.135
137. Ali AH, Yanoff LB, Stern EA, Akomeah A, Courville A, Kozlosky M, et al. Acute effects of betahistine hydrochloride on food intake and appetite in obese women: a randomized, placebo-controlled trial. Am J Clin Nutr (2010) 92(6):1290–7. doi:10.3945/ajcn.110.001586
138. Lian J, Huang X-F, Pai N, Deng C. Preventing olanzapine-induced weight gain using betahistine: a study in a rat model with chronic olanzapine treatment. PLoS One (2014) 9(8):e104160. doi:10.1371/journal.pone.0104160
139. Sadek B, Khanian SS, Ashoor A, Prytkova T, Ghattas MA, Atatreh N, et al. Effects of antihistamines on the function of human α7-nicotinic acetylcholine receptors. Eur J Pharmacol (2015) 746:308–16. doi:10.1016/j.ejphar.2014.10.046
140. Gotoh K, Fukagawa K, Fukagawa T, Noguchi H, Kakuma T, Sakata T, et al. Hypothalamic neuronal histamine mediates the thyrotropin-releasing hormone-induced suppression of food intake. J Neurochem (2007) 103(3):1102–10. doi:10.1111/j.1471-4159.2007.04802.x
141. Yoshimatsu H, Itateyama E, Kondou S, Tajima D, Himeno K, Hidaka S, et al. Hypothalamic neuronal histamine as a target of leptin in feeding behavior. Diabetes (1999) 48(12):2286–91. doi:10.2337/diabetes.48.12.2286
142. Parks GS, Olivas ND, Ikrar T, Sanathara NM, Wang L, Wang Z, et al. Histamine inhibits the melanin-concentrating hormone system: implications for sleep and arousal. J Physiol (2014) 592(10):2183–96. doi:10.1113/jphysiol.2013.268771
143. Igawa H, Takahashi M, Kakegawa K, Kina A, Ikoma M, Aida J, et al. Melanin-concentrating hormone receptor 1 antagonists lacking an aliphatic amine: synthesis and structure-activity relationships of novel 1-(Imidazo[1,2-a]pyridin-6-yl)pyridin-2(1H)-one derivatives. J Med Chem (2016) 59(3):1116–39. doi:10.1021/acs.jmedchem.5b01704
144. Henry MB, Zheng S, Duan C, Patel B, Vassileva G, Sondey C, et al. Antidiabetic properties of the histamine H3 receptor protean agonist proxyfan. Endocrinology (2011) 152(3):828–35. doi:10.1210/en.2010-0757
145. Rao AU, Shao N, Aslanian RG, Chan TY, Degrado SJ, Wang L, et al. Discovery of a potent thiadiazole class of histamine H3 receptor antagonist for the treatment of diabetes. ACS Med Chem Lett (2012) 3(3):198–202. doi:10.1021/ml200250t
146. Cacabelos R. Histamine: the missing link in the pathogenesis of some brain disorders. J Clin Exp Neuroimmunol (2016) 1:103. doi:10.4172/jceni.1000103
147. Mahmood D. Histamine H3 receptors and its antagonism as a novel mechanism for antipsychotic effect: a current preclinical & clinical perspective. Int J Health Sci (2016) 10(4):564–75.
148. Vohora D, Bhowmik M. Histamine H3 receptor antagonists/inverse agonists on cognitive and motor processes: relevance to Alzheimer’s disease, ADHD, schizophrenia, and drug abuse. Front Syst Neurosci (2012) 6:72. doi:10.3389/fnsys.2012.00072
149. Ellenbroek BA. Histamine H3 receptors, the complex interaction with dopamine and its implications for addiction. Br J Pharmacol (2013) 170(1):46–57. doi:10.1111/bph.12221
150. Lethbridge NL, Chazot PL. Ligand autoradiographical quantification of histamine H3 receptor in human dementia with Lewy bodies. Pharmacol Res (2016) 113(Pt A):245–56. doi:10.1016/j.phrs.2016.08.034
151. Esbenshade TA, Browman KE, Bitner RS, Strakhova M, Cowart MD, Brioni JD. The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br J Pharmacol (2008) 154(6):1166–81. doi:10.1038/bjp.2008.147
152. Weisler RH, Pandina GJ, Daly EJ, Cooper K, Gassmann-Mayer C; 31001074-ATT2001 Study Investigators. Randomized clinical study of a histamine H3 receptor antagonist for the treatment of adults with attention-deficit hyperactivity disorder. CNS Drugs (2012) 26(5):421–34. doi:10.2165/11631990-000000000-00000
153. Guenther PM, Casavale KO, Reedy J, Kirkpatrick SI, Hiza HA, Kuczynski KJ, et al. Update of the Healthy Eating Index: HEI-2010. J Acad Nutr Diet (2013) 113(4):569–80. doi:10.1016/j.jand.2012.12.016
154. Akbaraly TN, Singh-Manoux A, Tabak AG, Jokela M, Virtanen M, Ferrie JE, et al. Overall diet history and reversibility of the metabolic syndrome over 5 years: the Whitehall II prospective cohort study. Diabetes Care (2010) 33(11):2339–41. doi:10.2337/dc09-2200
155. Wu PY, Huang CL, Lei WS, Yang SH. Alternative health eating index and the dietary guidelines from American Diabetes Association both may reduce the risk of cardiovascular disease in type 2 diabetes patients. J Hum Nutr Diet (2016) 29(3):363–73. doi:10.1111/jhn.12317
156. Sánchez-Villegas A, Henríquez-Sánchez P, Ruiz-Canela M, Lahortiga F, Molero P, Toledo E, et al. A longitudinal analysis of diet quality scores and the risk of incident depression in the SUN Project. BMC Med (2015) 13:197. doi:10.1186/s12916-015-0428-y
157. Ozawa M, Shipley M, Kivimaki M, Singh-Manoux A, Brunner EJ. Dietary pattern, inflammation and cognitive decline: the Whitehall II prospective cohort study. Clin Nutr (2016). doi:10.1016/j.clnu.2016.01.013
158. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature (2006) 444:1027–31. doi:10.1038/nature05414
159. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature (2009) 457(7228):480–4. doi:10.1038/nature07540
160. Clarke SF, Murphy EF, Nilaweera K, Ross PR, Shanahan F, O’Toole PW, et al. The gut microbiota and its relationship to diet and obesity: new insights. Gut Microbes (2012) 3(3):186–202. doi:10.4161/gmic.20168
161. Moreno-Indias I, Cardona F, Tinahones FJ, Queipo-Ortuño MI. Impact of the gut microbiota on the development of obesity and type 2 diabetes mellitus. Front Microbiol (2014) 5:190. doi:10.3389/fmicb.2014.00190
162. Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, et al. Gut dysbiosis is linked to hypertension. Hypertension (2015) 65(6):1331–40. doi:10.1161/HYPERTENSIONAHA.115.05315
163. Davey KJ, Cotter PD, O’Sullivan O, Crispie F, Dinan TG, Cryan JF, et al. Antipsychotics and the gut microbiome: olanzapine-induced metabolic dysfunction is attenuated by antibiotic administration in the rat. Transl Psychiatry (2013) 3(10):e309. doi:10.1038/tp.2013.83
164. Xiang Y-Q, Zheng W, Wang S-B, Yang X-H, Cai D-B, Ng CH, et al. Adjunctive minocycline for schizophrenia: a meta-analysis of randomized controlled trials. Eur Neuropsychopharmacol (2017) 27(1):8–18. doi:10.1016/j.euroneuro.2016.11.012
165. Morgan AP, Crowley JJ, Nonneman RJ, Quackenbush CR, Miller CN, Ryan AK, et al. The antipsychotic olanzapine interacts with the gut microbiome to cause weight gain in mouse. PLoS One (2014) 9(12):e115225. doi:10.1371/journal.pone.0115225
166. Bahra SM, Weidemann BJ, Castro AN, Walsh JW, deLeon O, Burnett CM, et al. Risperidone-induced weight gain is mediated through shifts in the gut microbiome and suppression of energy expenditure. EBioMedicine (2015) 2(11):1725–34. doi:10.1016/j.ebiom.2015.10.018
167. Chakraborti CK. New-found link between microbiota and obesity. World J Gastrointest Pathophysiol (2015) 6(4):110–9. doi:10.4291/wjgp.v6.i4.110
168. Simpson HL, Campbell BJ. Review article: dietary fibre-microbiota interactions. Aliment Pharmacol Ther (2015) 42(2):158–79. doi:10.1111/apt.13248
169. Den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud D-J, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res (2013) 54(9):2325–40. doi:10.1194/jlr.R036012
170. Frost G, Sleeth ML, Sahuri-Arisoylu M, Lizarbe B, Cerdan S, Brody L, et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat Commun (2014) 5:3611. doi:10.1038/ncomms4611
171. MacFabe DF. Enteric short-chain fatty acids: microbial messengers of metabolism, mitochondria, and mind: implications in autism spectrum disorders. Microb Ecol Health Dis (2015) 26:28177. doi:10.3402/mehd.v26.28177
172. Suyama S, Maekawa F, Maejima Y, Kubota N, Kadowaki T, Yada T. Glucose level determines excitatory or inhibitory effects of adiponectin on arcuate POMC neuron activity and feeding. Sci Rep (2016) 6:30796. doi:10.1038/srep30796
173. Sun J, Gao Y, Yao T, Huang Y, He Z, Kong X, et al. Adiponectin potentiates the acute effects of leptin in arcuate Pomc neurons. Mol Metab (2016) 5(10):882–91. doi:10.1016/j.molmet.2016.08.007
174. Bartoli F, Crocamo C, Clerici M, Carrà G. Second-generation antipsychotics and adiponectin levels in schizophrenia: a comparative meta-analysis. Eur Neuropsychopharmacol (2015) 25(10):1767–74. doi:10.1016/j.euroneuro.2015.06.011
175. Bai YM, Chen TT, Yang WS, Chi YC, Lin CC, Liou YJ, et al. Association of adiponectin and metabolic syndrome among patients taking atypical antipsychotics for schizophrenia: a cohort study. Schizophr Res (2009) 111(1–3):1–8. doi:10.1016/j.schres.2009.03.014
176. Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest (2006) 116(7):1784–92. doi:10.1172/JCI29126
177. Bruce CR, Mertz VA, Heigenhauser GJ, Dyck DJ. The stimulatory effect of globular adiponectin on insulin-stimulated glucose uptake and fatty acid oxidation is impaired in skeletal muscle from obese subjects. Diabetes (2005) 54(11):3154–60. doi:10.2337/diabetes.54.11.3154
178. Silva FM, de Almeida JC, Feoli AM. Effect of diet on adiponectin levels in blood. Nutr Rev (2011) 69(10):599–612. doi:10.1111/j.1753-4887.2011.00414.x
179. Slavin J. Fiber and prebiotics: mechanisms and health benefits. Nutrients (2013) 5(4):1417–35. doi:10.3390/nu5041417
180. Henderson DC, Borba CP, Daley TB, Boxill R, Nguyen DD, Culhane MA, et al. Dietary intake profile of patients with schizophrenia. Ann Clin Psychiatry (2006) 18(2):99–105. doi:10.1080/10401230600614538
181. Gutiérrez-Díaz I, Fernández-Navarro T, Sánchez B, Margolles A, González S. Mediterranean diet and faecal microbiota: a transversal study. Food Funct (2016) 7(5):2347–56. doi:10.1039/c6fo00105j
182. Chang LC, Huang KC, Wu YW, Kao HL, Chen CL, Lai LP, et al. The clinical implications of blood adiponectin in cardiometabolic disorders. J Formos Med Assoc (2009) 108(5):353–66. doi:10.1016/S0929-6646(09)60079-6
183. Papathanasopoulos A, Camilleri M. Dietary fiber supplements: effects in obesity and metabolic syndrome and relationship to gastrointestinal functions. Gastroenterology (2010) 138(1):.e1–2. doi:10.1053/j.gastro.2009.11.045
184. Lyon MR, Kacinik V. Is there a place for dietary fiber supplements in weight management? Curr Obes Rep (2012) 1(2):59–67. doi:10.1007/s13679-012-0016-9
185. Xiao JB, Högger P. Dietary polyphenols and type 2 diabetes: current insights and future perspectives. Curr Med Chem (2015) 22(1):23–38. doi:10.2174/0929867321666140706130807
186. Trebatická J, Ďuračková Z. Psychiatric disorders and polyphenols: can they be helpful in therapy? Oxid Med Cell Longev (2015) 2015:248529. doi:10.1155/2015/248529
187. Dueñas M, Muñoz-González I, Cueva C, Jiménez-Girón A, Sánchez-Patán F, Santos-Buelga C, et al. A survey of modulation of gut microbiota by dietary polyphenols. Biomed Res Int (2015) 2015:850902. doi:10.1155/2015/850902
188. Catoire M, Kersten S. The search for exercise factors in humans. FASEB J (2015) 29(5):1615–28. doi:10.1096/fj.14-263699
189. Schnyder S, Handschin C. Skeletal muscle as an endocrine organ: PGC-1α, myokines and exercise. Bone (2015) 80:115–25. doi:10.1016/j.bone.2015.02.008
190. Wueest S, Item F, Boyle CN, Jirkof P, Cesarovic N, Ellingsgaard H, et al. Interleukin-6 contributes to early fasting-induced free fatty acid mobilization in mice. Am J Physiol Regul Integr Comp Physiol (2014) 306(11):R861–7. doi:10.1152/ajpregu.00533.2013
191. Oropeza D, Jouvet N, Bouyakdan K, Perron G, Ringuette LJ, Philipson LH, et al. PGC-1 coactivators in β-cells regulate lipid metabolism and are essential for insulin secretion coupled to fatty acids. Mol Metab (2015) 4(11):811–22. doi:10.1016/j.molmet.2015.08.001
192. Van Leusden JWR, Sellaro R, Colzato LS. Transcutaneous vagal nerve stimulation (tVNS): a new neuromodulation tool in healthy humans? Front Psychol (2015) 6:102. doi:10.3389/fpsyg.2015.00102
193. Kanaley JA, Goulopoulou S, Franklin R, Baynard T, Carhart RL Jr, Weinstock RS, et al. Exercise training improves hemodynamic recovery to isometric exercise in obese men with type 2 diabetes but not in obese women. Metabolism (2012) 61(12):1739–46. doi:10.1016/j.metabol.2012.07.014
194. Rojhani-Shirazi Z, Barzintaj F, Salimifard MR. Comparison the effects of two types of therapeutic exercises Frenkele vs. Swiss ball on the clinical balance measures in patients with type II diabetic neuropathy. Diabetes Metab Syndr (2016). doi:10.1016/j.dsx.2016.08.020
195. Sá-Caputo Dda C, Ronikeili-Costa P, Carvalho-Lima RP, Bernardo LC, Bravo-Monteiro MO, Costa R, et al. Whole body vibration exercises and the improvement of the flexibility in patient with metabolic syndrome. Rehabil Res Pract (2014) 2014:628518. doi:10.1155/2014/628518
196. Cerrillo-Urbina AJ, García-Hermoso A, Sánchez-López M, Pardo-Guijarro MJ, Santos Gómez JL, Martínez-Vizcaíno V. The effects of physical exercise in children with attention deficit hyperactivity disorder: a systematic review and meta-analysis of randomized control trials. Child Care Health Dev (2015) 41(6):779–88. doi:10.1111/cch.12255
197. Malchow B, Reich-Erkelenz D, Oertel-Knöchel V, Keller K, Hasan A, Schmitt A, et al. The effects of physical exercise in schizophrenia and affective disorders. Eur Arch Psychiatry Clin Neurosci (2013) 263(6):451–67. doi:10.1007/s00406-013-0423-2
198. Smith MA, Lynch WJ. Exercise as a potential treatment for drug abuse: evidence from preclinical studies. Front Psychiatry (2011) 2:82. doi:10.3389/fpsyt.2011.00082
199. Vidoni ED, Van Sciver A, Johnson DK, He J, Honea R, Haines B, et al. A community-based approach to trials of aerobic exercise in aging and Alzheimer’s disease. Contemp Clin Trials (2012) 33(6):1105–16. doi:10.1016/j.cct.2012.08.002
200. DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care (2009) 32(Suppl 2):S157–63. doi:10.2337/dc09-S302
201. Banks WA, Owen JB, Erickson MA. Insulin in the brain: there and back again. Pharmacol Ther (2012) 136(1):82–93. doi:10.1016/j.pharmthera.2012.07.006
202. Young SN. The effect of raising and lowering tryptophan levels on human mood and social behaviour. Philos Trans R Soc Lond B Biol Sci (2013) 368(1615):20110375. doi:10.1098/rstb.2011.0375
203. Mosienko V, Bert B, Beis D, Matthes S, Fink H, Bader M, et al. Exaggerated aggression and decreased anxiety in mice deficient in brain serotonin. Transl Psychiatry (2012) 2(5):e122. doi:10.1038/tp.2012.44
204. Marsh DM, Dougherty DM, Moeller FG, Swann AC, Spiga R. Laboratory-measured aggressive behavior of women: acute tryptophan depletion and augmentation. Neuropsychopharmacology (2002) 26(5):660–71. doi:10.1016/S0893-133X(01)00369-4
205. Myint AM. Kynurenines: from the perspective of major psychiatric disorders. FEBS J (2012) 279(8):1375–85. doi:10.1111/j.1742-4658.2012.08551.x
206. Beggiato S, Antonelli T, Tomasini MC, Tanganelli S, Fuxe K, Schwarcz R, et al. Kynurenic acid, by targeting α7 nicotinic acetylcholine receptors, modulates extracellular GABA levels in the rat striatum in vivo. Eur J Neurosci (2013) 37(9):1470–7. doi:10.1111/ejn.12160
207. Mudry JM, Alm PS, Erhardt S, Goiny M, Fritz T, Caidahl K, et al. Direct effects of exercise on kynurenine metabolism in people with normal glucose tolerance or type 2 diabetes. Diabetes Metab Res Rev (2016) 32(7):754–61. doi:10.1002/dmrr.2798
208. Olsson SK, Samuelsson M, Saetre P, Lindström L, Jönsson EG, Nordin C, et al. Elevated levels of kynurenic acid in the cerebrospinal fluid of patients with bipolar disorder. J Psychiatry Neurosci (2010) 35(3):195–9. doi:10.1503/jpn.090180
209. Cho HS, Baek DJ, Baek SS. Effect of exercise on hyperactivity, impulsivity and dopamine D2 receptor expression in the substantia nigra and striatum of spontaneous hypertensive rats. J Exerc Nutrition Biochem (2014) 18(4):379–84. doi:10.5717/jenb.2014.18.4.379
Keywords: monoamines, acetylcholine, short-chain fatty acids, impulsivity, microbiome
Citation: Sfera A, Osorio C, Inderias LA, Parker V, Price AI and Cummings M (2017) The Obesity–Impulsivity Axis: Potential Metabolic Interventions in Chronic Psychiatric Patients. Front. Psychiatry 8:20. doi: 10.3389/fpsyt.2017.00020
Received: 24 December 2016; Accepted: 25 January 2017;
Published: 13 February 2017
Edited by:
Stefan Borgwardt, University of Basel, SwitzerlandReviewed by:
Stefania Schiavone, University of Foggia, ItalyRoberto Gradini, Sapienza University of Rome, Italy
Nina Schweinfurth, University of Basel, Switzerland
Copyright: © 2017 Sfera, Osorio, Inderias, Parker, Price and Cummings. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adonis Sfera, ZHIuc2ZlcmFAZ21haWwuY29t