 Nicole Mons
Nicole Mons Daniel Beracochea
Daniel Beracochea
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 06 October 2016
Sec. Addictive Disorders
Volume 7 - 2016 | https://doi.org/10.3389/fpsyt.2016.00165
This article is part of the Research Topic Memory systems of the addicted brain: the underestimated role of drug-induced cognitive biases in addiction and its treatment View all 14 articles
A prime mechanism that contributes to the development and maintenance of alcoholism is the dysregulation of the hypothalamic–pituitary–adrenal axis activity and the release of glucocorticoids (cortisol in humans and primates, corticosterone in rodents) from the adrenal glands. In the brain, sustained, local elevation of glucocorticoid concentration even long after cessation of chronic alcohol consumption compromises functional integrity of a circuit, including the prefrontal cortex (PFC), the hippocampus (HPC), and the amygdala (AMG). These structures are implicated in learning and memory processes as well as in orchestrating neuroadaptive responses to stress and anxiety responses. Thus, potentiation of anxiety-related neuroadaptation by alcohol is characterized by an abnormally AMG hyperactivity coupled with a hypofunction of the PFC and the HPC. This review describes research on molecular and epigenetic mechanisms by which alcohol causes distinct region-specific adaptive changes in gene expression patterns and ultimately leads to a variety of cognitive and behavioral impairments on prefrontal- and hippocampal-based tasks. Alcohol-induced neuroadaptations involve the dysregulation of numerous signaling cascades, leading to long-term changes in transcriptional profiles of genes, through the actions of transcription factors such as [cAMP response element-binding protein (CREB)] and chromatin remodeling due to posttranslational modifications of histone proteins. We describe the role of prefrontal–HPC–AMG circuit in mediating the effects of acute and chronic alcohol on learning and memory, and region-specific molecular and epigenetic mechanisms involved in this process. This review first discusses the importance of brain region-specific dysregulation of glucocorticoid concentration in the development of alcohol dependence and describes how persistently increased glucocorticoid levels in PFC may be involved in mediating working memory impairments and neuroadaptive changes during withdrawal from chronic alcohol intake. It then highlights the role of cAMP–PKA–CREB signaling cascade and histone acetylation within the PFC and limbic structures in alcohol-induced anxiety and behavioral impairments, and how an understanding of functional alterations of these pathways might lead to better treatments for neuropsychiatric disorders.
Alcoholism is a chronic, often relapsing brain disorder characterized by periods of sustained, compulsive alcohol intake, relying in part on allostatic changes within the prefrontal cortex (PFC) and limbic structures [i.e., the hippocampus (HPC) and the amygdala (AMG)] [for review, see Ref. (1)]. This circuit plays key roles in behavior and cognitive function as well as in orchestrating neuroadaptive responses to stress and anxiety. The transition from recreational to alcohol dependence and compulsive alcohol drinking takes place via neuroadaptive changes in the stress-related neural circuits, caused partly by repeated cycles of alcohol intoxication and withdrawal (2, 3). A prime mechanism that contributes to the development and maintenance of alcoholism is the dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis activity (4) and the release of glucocorticoids (cortisol in humans and primates, corticosterone in rodents) from the adrenal glands. Clinical and preclinical evidence in both humans (5–7) and rodents (4, 8, 9) have shown that acute and chronic alcohol consumption, as well as withdrawal, markedly affects plasma glucocorticoid levels. The release of glucocorticoids can influence brain function by readily crossing the blood–brain barrier and exert effects through a dual glucocorticoid binding receptor system, i.e., the type I high affinity mineralocorticoid receptors (MRs) or the type II low affinity glucocorticoid receptors (GRs) (10), which act as ligand-dependent transcription factors to modulate target gene transcription. The MRs display a restricted expression in the brain, with highest densities in the HPC (11–13). The GRs are widely distributed throughout the brain (10, 14, 15) with a predominant expression in the three areas involved in learning and memory and particularly sensitive to the effects of stress, namely, the PFC, the dorsal HPC, and the AMG (16–18). Indeed, human studies of Cushing’s syndrome have shown that sustained cortisol elevation over the years compromises the integrity of the HPC–PFC circuitry and thus influences the onset and/or the severity of cognitive decline in various tasks, including spatial, decision-making and working memory processes (19–23). Further, sustained, high local concentration of glucocorticoids is responsible for long-lasting cognitive impairments occurring several weeks after the cessation of alcohol in rodents (24, 25) and abstinent patients (26, 27). As to how elevation of glucocorticoids might be implicated in the enduring cellular, molecular, and behavioral changes, it has been suggested that neuroadaptation induced by alcohol exposure involves the dysregulation of numerous signaling cascades, leading to long-term changes in transcriptional profiles of genes, through the actions of transcription factors such as [cAMP response element-binding protein (CREB)] and chromatin remodeling due to modifications of the posttranslational properties of histone proteins [for review, see Ref. (28)]. In the following, we provide an overview of how transcriptional and histone acetylation changes in the PFC, the HPC, and the AMG play a central role in the glucocorticoid-dependent neuroadaptation and behavioral deficits that occur during acute and chronic alcohol exposure. While this review focuses on aspects of how spatial and temporal changes in histone acetylation drive alcohol-induced alterations in neural plasticity and behavior, it should be emphasized that other histone modifications marks, such as histone phosphorylation and histone lysine methylation, occur in parallel and are also involved in the long-term adaptations in neural function and behavioral responses to alcohol exposure.
Surprisingly, little is known about the long-lasting neuroadaptive changes of glucocorticoids caused by prolonged alcohol consumption and withdrawal within neural circuits involved in learning and memory and emotional events and about their behavioral consequences. Studies, including our own, have shown that the initial phase of alcohol withdrawal period produces elevation in both circulating and brain glucocorticoids levels (29–31). Importantly, Little and colleagues (30) were first to show that during the initial phase of withdrawal from chronic (8 months in rats) alcohol consumption, rats and mice display an abnormal, exaggerated corticosterone level selectively in the medial PFC and the dorsal HPC. Strikingly, the authors found that withdrawal-associated excessive corticosterone response in the PFC persists for up to 2 months; therefore, long after, plasma corticosterone levels returned to baseline levels. In the PFC, the sustained elevation of corticosterone concentration was associated with enhanced GRs activation in mice undergoing a 2-week withdrawal period from chronic alcohol consumption (30). Further, administration of the GRs antagonist mifepristone or the dihydropyridine calcium channel nimodipine, given just prior to withdrawal from chronic alcohol exposure, not only reduced the rises in brain corticosterone but also prevented persistent memory deficits seen several weeks later in mice (24) or rats (32), suggesting that withdrawn-associated rise in glucocorticoid levels specifically within medial PFC may be an early index of maladaptive persistent behaviors in alcohol-dependent subjects. Indeed, chronic treatment with the GR antagonist mifepristone attenuated escalation of ethanol intake following intermittent ethanol vapor exposure (33) as well as the development of alcohol dependence and ultimately withdrawal-associated behavioral deficits (34). Endogenous glucocorticoids have been suggested to play an essential role in maintaining PFC-dependent cognitive functions, mainly via complex interaction with dopaminergic and glutamatergic receptors (35–37). Both human and animal studies have demonstrated that alcohol withdrawal impairs a variety of the cognitive functions during tests that require cortical prefrontal processing (38–40). As regards, pharmacological (hydrocortisone administration) or pathological (Cushing’s disease) increase of cortisol was found to predict frontal cortex-based cognitive impairments including alterations in executive processes and working memory dysfunction (19, 23, 41–43). Long-lasting deficits on tasks that rely on the PFC are also observed in rodent models in which chronic alcohol dependence is induced by chronic alcohol exposure or chronic intermittent ethanol that involves repeated cycles of exposure to alcohol vapors (44, 45). However, in addition to PFC dysfunction, there is evidence that a functional disconnection of brain network connectivity between the (dorsomedial) PFC and the central nucleus of the AMG may also contribute to the alcohol-induced working memory impairments in rats (46).
Recent work in our laboratory has employed in vivo microdialysis in freely moving mice to investigate effects of chronic alcohol treatment and withdrawal (early and prolonged) periods on brain corticosterone concentrations by simultaneously measuring time-course evolution of corticosterone concentration in the medial PFC and dorsal HPC seen before, during, and after completion of a working memory task in a T-maze (31, 47). This task is based on spontaneous alternation behavior, known to require intact connections between the two structures for successful performance (48, 49). Specifically, alternation behavior is the innate tendency of rodents to alternate at each successive trial the choice of the goal arm over a series of trials run in a T-maze (except for the first trial). From trial to trial, accurate performance at a given trial (N) requires for subjects to be able to discriminate the specific target trial N − 1 from the interfering trial N − 2. Thus, the target information required for successful performance varies from trial to trial, so that the subject is not only required to temporarily keep specific information in short-term storage but also reset it over successive runs. The resetting mechanisms and cognitive flexibility required to alternate over successive runs are major components of working memory processes. Working memory is a component of the sequential alternation task, since spontaneous alternation rates are dependent on the length of the inter-trial delay interval and/or the place of the trial in the series. Indeed, repetitive testing constitutes a potent source of proactive interference. Thus, the sequential alternation procedure is relevant to assess delay-dependent working memory in mice (50–52). Using in vivo microdialysis in freely moving mice, we observed that early (1 week) and protracted (6 weeks) withdrawal periods from prolonged (6 months) alcohol exposure causes an exaggerated corticosterone rise in the medial PFC. In addition, withdrawn mice having abnormal corticosterone concentration in the PFC displayed impaired working memory performance, effects that were not observed in animals still submitted to chronic alcohol consumption. Moreover, early and protracted withdrawal periods had no effect on the dynamic pattern of corticosterone response in the dorsal HPC, indicating that alcohol impacts glucocorticoid regulation in a brain region-specific fashion. During the 6-week withdrawal period, the degree of working memory impairment correlated with the magnitude of prefrontal corticosterone concentration, which is in accordance with the notion that there is a functional link between excessive corticosteroid signaling and PFC dysfunction (53–55). Many neuroimaging studies have indicated consistently that structural and functional deficits in PFC regulatory regions are associated with chronic alcoholism [for review, see Ref. (56)]. Another study using SPECT imaging showed that detoxified alcoholic patients who relapsed 2 months later displayed working memory deficits associated with low blood flow in the medial frontal lobe (57). Given the importance of frontal cortical regions in the modulation of AMG reactivity and the mediation of effective emotion regulation, weakened PFC function associated with a specific functional disconnection between the PFC and the AMG has been proposed as an early index of neuroadaptation in alcohol dependence that predicts PFC-dependent cognitive impairments observed during abstinence (38, 39, 46).
Subsequently, we have studied whether local glucocorticoid blockade in the medial PFC would prevent the long-term deficits in working memory induced by protracted withdrawal from chronic alcohol consumption (31). Intraperitoneal administration of the corticosterone synthesis inhibitor metyrapone prior to testing prevented the withdrawal-associated working memory impairments, confirming the essential role of persistently increased glucocorticoid levels in behavioral impairments during withdrawal from chronic alcohol intake. Similarly, a single bilateral infusion of spironolactone into the medial PFC that diminished MR activation and to a lesser extent of mifepristone that diminished GRs activation fully restored working memory function in withdrawn mice. In contrast, neither spironolactone nor mifepristone had any effect when infused into the dorsal HPC, thus highlighting the importance of glucocorticoids specific to the PFC in neural substrates mediating the prolonged, detrimental effects of alcohol on behavioral performance. These findings are reminiscent of data showing that elevated glucocorticoid levels, via either systemic injection of corticosterone or local infusion of the GRs agonist RU 28362 into the medial PFC shortly before testing, similarly impair working memory (55), while the GRs antagonist RU 38486 infused into the PFC can restore stress-induced deficits in executive function (58). Collectively, these data support the view that long-term adaptive behavioral effects of chronic alcohol exposure are mediated in large part through long-lasting glucocorticoid dysregulation within the PFC circuitry.
The transcription factor CREB is a key downstream target of a variety of kinases, including cAMP–protein kinase A (PKA), Ca2+/calmodulin-dependent kinase, and extracellular-regulated kinase/mitogen-associated protein kinase (ERK/MAPK) (59, 60). The resulting activation/phosphorylation of CREB and recruitment of CREB-binding protein (CBP) along with other transcriptional components enables transcription of specific CREB target genes, including those implicated in long-term memory and plasticity as well as in the development of anxiety-like and alcohol-drinking behaviors, such as the neuropeptide Y (NPY) and the brain-derived neurotrophic factor (BDNF) (61–64). There is mounting evidence to support a role for phosphorylated CREB (pCREB) through a PKA-dependent mechanism and downstream CREB target genes, in the adaptive changes and behavioral effects associated with acute and chronic alcohol exposure [for review, see Ref. (65–68)]. Acute and chronic ethanol exposures have long been known to modulate the various steps of the cAMP-dependent pathways in the rodent brain and in other cell systems (69–71). Exposure to ethanol affects a cascade of events allowing for sustained translocation of PKA catalytic subunit into the nucleus (72), ultimately resulting in long-lasting increased CREB activation/phosphorylation (73) and downstream expression of many target genes (74). In this context, abnormal PKA-dependent CREB functioning has been implicated in the molecular mechanisms of neuroplasticity that underlie alcoholism and alcohol drinking. There is evidence for a biphasic temporal effect of ethanol on cAMP–PKA-dependent signaling cascade with acute and prolonged exposure to ethanol potentiating (75) and decreasing (76, 77), respectively, adenylyl cyclase–cAMP–PKA activity in the cortex and HPC (78) in mice. Using a combination of genetic or pharmacological approaches, Drosophila and rodents studies have shown that maintaining integrity of the cAMP–PKA activity is central to establishing sensitivity to the sedative effect of ethanol as well as in modulating ethanol consumption (79–81). Acute withdrawal (24 h) from chronic ethanol treatment produced a decrease in Ser133–pCREB within specific neurocircuitry of the frontal, parietal, and piriform cortex in rats (82), suggesting the possibility that CREB-dependent events in these cortical structures may be involved in the development of alcohol dependence. Among the mechanisms responsible for reduced pCREB and downregulation of cAMP-dependent genes, chronic intermittent alcohol exposure has been shown to increase expression of the protein kinase inhibitor-α (PKI-α) in the PFC, nucleus accumbens, and AMG in Wistar rats (83). Given the wealth of data for the recruitment of the cAMP–PKA signaling pathways upon acute ethanol exposure, it has been proposed that the increased PKI-α expression may be part of the adaptation of the cAMP–PKA pathway induced by intermittent alcohol exposure.
Investigations into the role of CREB in amygdaloid brain structures with regard to anxiety-like and alcohol-drinking behaviors have shown that CREB activity fluctuates depending on brain structures and alcohol “condition” (acute, chronic, or withdrawal). For instance, a series of studies by Pandey’s group conducted in the rat AMG clearly indicate a strong relationship between decreased CREB phosphorylation and high anxiety-like responses associated with acute withdrawal from 2-week ethanol treatment (62, 82). Decreases in CREB phosphorylation and downstream cAMP-inducible genes, including NPY in the central and medial, but not the basolateral, nuclei of the AMG, have been associated with a predisposition to both anxiety-like and excessive alcohol-drinking behaviors in alcohol-preferring rats (60, 84–86). Restoring CREB function to optimal level or enhancing NPY signaling in the central AMG prevented the onset of anxiety-like behaviors (84, 87, 88), while alcohol-associated anxiety disorders can be mimicked by pharmacological blockade of PKA in ethanol-naïve-preferring rats or non-preferring rats (60, 84). Thus, anxiety-induced downregulation of CREB function in the AMG may constitute a critical neuroadaptation central to the development and maintenance of alcohol dependence. As regards, dysregulation of the PFC associated with a functional disconnection between the PFC and AMG central nucleus during abstinence and renewed access to alcohol has been implicated in long-lasting cognitive impairment and excessive alcohol drinking in rats (46).
Clinical evidence from alcohol-dependent patients also indicates that acute and protracted withdrawal/abstinence is strongly associated with depressive-like behaviors, such as anhedonia.
The catecholamines dopamine and noradrenaline via the cAMP–PKA–CREB signaling cascade provide an essential modulatory influence on PFC-dependent behaviors producing an inverted “U-shaped” dose–response influence, whereby moderate levels improve PFC function while either too little or too much catecholamines lead to cognitive impairments [for review, see Ref. (89)]. A number of studies including work in our laboratory (51) have shown that blocking the cAMP–PKA–CREB signaling cascade via local infusion of Rp-cAMPS (a compound known to inhibit CREB phosphorylation) into the PFC prevents the impairing effect of stress or aging on working memory performance, while drugs that increase cAMP–PKA signaling either by direct intra-PFC infusion of the cAMP analog Sp-cAMPS or dopamine D1 receptor agonist or i.p. administration of the phosphodiesterase (PDE) inhibitor Rolipram impair cognitive functions [for reviews, see Ref. (89–91)]. As mentioned above, we recently reported that consumption of an alcohol-containing liquid diet for 6 months followed by a 1-week withdrawal period produces working memory impairment in a T-maze spontaneous alternation task in mice, which persists for at least 6 weeks after the cessation of alcohol intake (31, 47). Moreover, withdrawn mice displaying impaired working memory performance were those that had the lowest pCREB level in the PFC along with a persistent rise of prefrontal corticosterone concentration. Because glucocorticoids in the PFC interact with β-adrenoceptor–cAMP/PKA activity to influence working memory function (92), one route by which elevated glucocorticoid levels may impair PFC-mediated cognitive function long after the cessation of alcohol exposure is by inhibiting the cAMP–PKA cascade. In this context, growing evidence supports a central role for PDE, which is responsible for the breakdown of cAMP, in the regulation of alcohol drinking in rodents [for review, see Ref. (93)]. For example, treatment with various PDE4 inhibitors, including rolipram, produces long-lasting reduction of alcohol intake and preference in C57BL/6J mice (94). Chronic rolipram treatment also results in sustained reduction of alcohol seeking and consumption in alcohol-preferring rats (95, 96). As mentioned earlier, mice subjected to 1- or 6-week alcohol withdrawal from chronic alcohol consumption exhibited working memory impairments accompanied by enhanced anxiety level (at 1 week only) as well as persistently elevated corticosterone and sustained decreased pCREB levels in the PFC. Intraperitoneal administration of the PDE4 inhibitor, rolipram, before working memory testing abolished these withdrawal-associated behavioral, endocrine, and neuronal alterations (31) – a finding consistent with other observation, which demonstrated that in rats, heightened anxiety during acute alcohol withdrawal was accompanied by elevated expression of Pde10a isoform mRNA levels in interconnected medial PFC–AMG circuit, which persisted in the AMG after protracted (6 weeks) alcohol withdrawal (97). Together, these observations strongly support further research with regard to isoform-specific PDE-selective inhibitors that are promising pharmacotherapy targets for alcohol use disorders.
As discussed above, long-term adaptive behavioral effects of chronic alcohol exposure are mediated in large part through long-lasting glucocorticoid dysregulation within the PFC but not the dorsal HPC. Confirming differential sensitivity of the PFC and dorsal HPC to chronic alcohol-induced damage, recent work in our laboratory has shown that, unlike the PFC in which withdrawal from prolonged alcohol intake caused persistent working memory impairments along with sustained inhibition of the cAMP–PKA–CREB signaling cascade, both alcohol (unimpaired) and alcohol withdrawal (impaired) mice display reduced levels of pCREB in the dorsal HPC (namely, the CA1 region), compared with water-drinking mice (31, 47). Furthermore, intraperitoneal administration of rolipram was able to correct the deficit in pCREB in the dorsal HPC but did not reverse working memory impairments in withdrawn animals (47). Together, these observations support the notion that disruption of the cAMP–PKA–CREB signaling cascade specifically in the PFC (but not in the dorsal HPC) has an essential role in promoting long-term neuroadaptive changes accompanying persistent behavioral changes during withdrawal from chronic alcohol intake. Interestingly, early pioneering work in our laboratory emphasized a key role for PKA–CREB signaling as a sustained “molecular switch” that gradually converts acute “drug” responses into relatively stable adaptations that contribute to drug and alcohol addiction-mediated long-lasting neural and behavioral plasticity. Under conditions of drug- and food-reinforced behavior, drug-induced reward impaired spatial discrimination learning in a Y-maze task and caused drastic decreases in pCREB and downstream target c-Fos expression in the dorsal HPC and the PFC while sparing the cued version of the task and pCREB in the dorsal striatum in mice (98). Further, pharmacological blockade of cAMP–PKA cascade into the striatum before training normalized CREB activity within the HPC–PFC circuit and, as subsequently, prevented the drug-induced modulation of multiple memory systems.
Emerging evidence indicates that brain region-specific alteration of CREB signaling is also an important regulator involved in depression-like behavior that emerges during abstinence following alcohol drinking. As a key symptom of clinical depression, anhedonia reflects reduced interest in enjoying pleasure-seeking behavior and plays a key role in relapse (99, 100) and in the perpetuation of excessive alcohol consumption in dependent individuals (101). Important clinical evidence clearly demonstrated that the persistence and intensity of some behavioral withdrawal symptoms positively correlated with anhedonia scales in detoxified alcohol-dependent subjects (102), extending previous findings of strong correlation between anhedonia and substance-related symptoms particularly in detoxified opiate-dependent subjects (103). The presence of depression-related behavioral phenotypes during protracted abstinence was also reported in rodent models (104–106). In mice undergoing 2 weeks of abstinence from chronic alcohol consumption, the persistent increase in plasma corticosterone response and upregulation of GR expression correlated with the development of depressive-like phenotypes, including anhedonia and helplessness (105), and reduced hippocampal neurogenesis (104). Further, there are several lines of evidence that suggest that downregulation of BDNF–TrKB–CREB signaling pathway may serve as a common link between the development of alcohol-induced depression-like symptoms and reduced hippocampal neurogenesis (104, 105, 107, 108). Finally, since enhancing the BDNF–CREB activity through pharmacological treatments with various classes of antidepressant drugs or environmental enrichment abolished the alcohol-induced anhedonia and depressive behaviors seen during protracted abstinence (104, 107, 108), supporting the hypothesis that BDNF–CREB signaling pathway may be a potential therapeutic target for interventions in alcoholism–depression coincidence.
Equally important for providing precise, long-lasting changes in brain function associated with alcohol intake are histone modifications, which exert lasting control over transcriptional activity of target genes through modifications of the chromatin structure and function that make the DNA less or more accessible to transcription factors and enzymes. The basic unit of chromatin, the nucleosome, is a histone octamer wrapped by approximately 147 base pairs of DNA. Each core histone (H2A, H2B, H3, and H4) has a highly conserved amino (N)-terminal tail, which is subject through a range of posttranslational modification (PTM) marks at distinct residues/sites including acetylation and methylation of lysine residues and phosphorylation of serine residues (109). Histones acetylation and phosphorylation are associated with transcriptional activation, whereas histone methylation reflects both transcriptional activation and repression depending on the specific site and context of the modification. An important feature of histone PTMs is that they can influence each other in a synergistic or antagonistic manner, leading to a complex “histone code” (110). Of these histone PTMs, histone acetylation is the most widely investigated in terms of epigenetic mechanisms underlying region-specific changes in brain gene networks required for long-term memory processes. Many rodent studies have detailed how different learning paradigms trigger distinct histone acetylation patterns in the brain, which are accompanied by region-, task-, and age-specific changes in memory-associated genes [for reviews, see Ref. (111–115)]. For instance, increased acetylation of histones, H3 and H4, occurred in the dorsal HPC or the dorsal striatum, depending on whether mice were subjected to a spatial or cued training in the water maze task, respectively (116, 117).
The degree of histone acetylation/deacetylation is finely orchestrated by dynamic balance of antagonistic enzymes that “write” (HATs) and “erase” [histone deacetylases (HDACs)] acetylation sites (113, 118–121). Systemic administration of HDAC inhibitors (HDACi), such as sodium butyrate (NaB) or trichostatin A (TSA), can improve memory formation and also prevent or reverse cognitive impairments associated with normal and pathological aging. However, this enhancing effect of HDACi on HPC-dependent memory required accurate CREB activity (116, 117, 122). Furthermore, infusing HDACi directly to the HPC was not only effective in promoting HPC-dependent learning and memory processes but can also influence relative use of multiple memory processes by affecting transcriptional events within subcortical and PFC cortical structures (116, 123).
A growing set of studies in both humans and animals have indicated that alcohol exposure causes widespread, dynamic changes of histone acetylation patterns, and thereby dysregulation in gene expression profiles across multiple brain regions (28, 124–126). Most of the studies have focused on the two histones H3 and H4 acetylation and chromatin-related events within the PFC, the HPC, and the AMG. In mouse and rat brain, studies reported that alcohol’s effects on histone acetylation patterns depend on the alcohol treatment paradigm, the timing of alcohol exposure or withdrawal, and brain structures examined, and even within a structure, alcohol can affect differently subregions. For example, work from Pandey’s lab has shown that anxiolytic-like responses caused by acute ethanol i.p. injection were accompanied by increased HAT CBP activity and associated increased acetylation of histone H3 at lysine 9 and histone H4 at lysine 8 (H3K9 and H4K8, respectively) leading to rapid elevation of NPY (mRNA and protein level) specifically in the central and medial, but not the basolateral amygdaloid, nuclei (125). The same group observed that a 2-week ethanol exposure followed by acute ethanol withdrawal (24 h) switches alcohol’s effect to anxiogenic-like responses, effects that involve a shift from HDAC hypoactivity to HDAC hyperactivity and subsequently decreased histone acetylation and transcriptional repression of NPY function in the two AMG nuclei (125, 127, 128). Correcting histone acetylation deficits in the AMG via administration of the pan HDACi TSA can reverse the rapid tolerance to the anxiolytic effects of ethanol (128) and prevent the development of alcohol withdrawal-related anxiety in rat (125). Alcohol-induced neuroadaptation in the AMG also implicated deficits of BDNF activity and its target [activity-regulated cytoskeleton-associated protein (Arc)], two key signaling factors involved in synaptic transmission and plasticity. While acute ethanol exposure caused an upregulation of BDNF–Arc signaling pathway and subsequently increased dendritic spine densities in the central and medial AMG nuclei, withdrawal from prolonged ethanol exposure or binge ethanol consumption potently inhibited BDNF and Arc expression and reduced dendritic arborization in these nuclei and other regions, leading to increased anxiety-like and drinking behaviors (66, 129, 130). Importantly, these long-lasting adaptive changes associated with alcohol dependence were reversed upon treatment with the HDACi TSA (61, 128–130). In another study by Moonat and colleagues (61) examining the role of HDAC2 in the development of alcohol dependence, investigators found lower baseline BDNF protein levels in the AMG (and also the bed nucleus of stria terminalis) of alcohol-preferring rats, a well-established model used to study the genetic predisposition to alcoholism (131), relative to the low-drinking NP rats. In addition, innate HDAC2 overexpression and decreased H3K9 acetylation in the central nucleus of alcohol-preferring rats correlated with low levels of BDNF, Arc, and NPY and was accompanied with high levels of anxiety-like and alcohol-drinking behaviors. These HDAC2-associated molecular and behavioral deficits were rescued via specific knockdown of HDAC2 expression either by direct infusion of small interfering RNA (siRNA) against HDAC2 into the central AMG nucleus (61, 66) or by TSA treatment (127, 130, 132). Collectively, these observations raised the possibility that adaptive epigenetic changes involving HDACs, and in particular HDAC2, in the AMG may be important regulatory mechanisms that underlie expression of genes implicated in the development and pathogenesis of alcohol dependence.
Using a chronic intermittent ethanol exposure model, a robust H3K9 hyperacetylation was seen in the AMG and cortical areas of rats, which displayed motivation to self-administer ethanol after a 6-h withdrawal period, compared with non-dependent rats (133). Treatment with the HDACi NaB or MS-275 (i.p. or i.c.v.) was able to counteract the effects of alcohol in dependent rats but not in non-dependent rats. Treatment with NaB, when administrated prior to ethanol self-administration, was also able to reverse H3K9 hyperacetylation and counteract excessive alcohol intake and relapse in alcohol-dependent rats. In order to identify brain region-specific regulatory molecular (epigenetic) signatures potentially involved in adaptive processes that lead to alcohol tolerance and dependence, Smith and colleagues (134) recently examined brain regional expression network responses to acute (0–8 h) and late (72 h to 7 days) withdrawal from chronic intermittent ethanol exposure in mice. Remarkably, the authors showed that neuroinflammatory responsive genes can be seen across all brain regions at 0–8 h after the beginning of alcohol withdrawal, while sustained over-representation for subset groups of genes related to neurodevelopment and synaptic plasticity (such as Bdnf) and to histone acetylation (such as HDAC4 and HDAC6) and histone/DNA methylation are found at 3- to 7-day-withdrawal periods specifically in the PFC and the HPC. These results illustrate how transient and persistent histone acetylation changes could serve as a key mechanism for tight regulation of the expression of large sets of genes within specific brain regions of animals predisposed to excessive ethanol drinking or exposed to protracted abstinence. A functional disconnection of the CeA–PFC circuit during abstinence (72 h) and renewed access to alcohol has been recently implicated in long-lasting PFC-dependent cognitive dysfunction and the development of anxiety-like behavior, and more specifically, the resulting PFC hypofunction was shown to facilitate the transition from moderate to excessive and uncontrolled alcohol intake in rats (46).
Persistent changes of the HAT CBP activity and H4 acetylation were observed in the frontal cortex of C57BL/6 mice given 5-month chronic alcohol consumption followed by a 15-day withdrawal period (135). In that study, withdrawal-associated H4 hypoacetylation correlated with neuroinflammatory damage and the persistently altered memory and anxiety-related behaviors. Nonetheless, these changes were absent in mice lacking the Toll-like receptor 4 (TLR4) that have undergone the same treatment, suggesting a critical role for TLR4-mediated epigenetic modifications in mediating long-lasting deleterious effects of chronic alcohol on PFC-dependent behaviors (135). This is in line with findings in our laboratory showing a robust decrease in histone H4 acetylation in the medial PFC of C57/BL mice at 1 week after withdrawal from chronic alcohol consumption; this decrease was maintained for at least 6 weeks after alcohol withdrawal and correlated with the persistently impairment of working memory noted during abstinence (31, 47). Alcohol’s effects on H4 acetylation closely paralleled effects on CREB activation in the PFC. Further, systemic delivery of corticosterone inhibitor metyrapone or local intra-PFC blockade of MRs (via spironolactone) or GRs (via mifepristone) similarly reversed long-lasting deficits in pCREB and H4 acetylation levels in the PFC and alleviated working memory deficits associated with alcohol withdrawal (31). Thus, these findings suggest that long-lasting glucocorticoid-induced neuroadaptive changes in CREB and H4 acetylation in the PFC may be involved in the enduring working memory impairments caused by prolonged alcohol consumption and withdrawal. Cumulative evidence indicates that structural and functional integrity of the HPC was also compromised in rats after prolonged alcohol exposure and even greatest alterations were found after cessation of alcohol exposure (136–138). Prolonged ethanol intake caused enduring deficits in HPC-dependent spatial reference memory in the water maze (138–140). Chronic ethanol treatment also caused long-lasting decrease of histone acetylation in the dorsal HPC. However, contrary to the PFC where there was strong relationship between alcohol-induced decrease of H4 acetylation and long-lasting working memory impairments, H4 acetylation in the HPC (the CA1 region) was decreased in behaviorally “unimpaired” alcohol-treated mice and even continued to decrease in “impaired” withdrawal-treated mice, compared with water-treated mice (31, 47). However, the drugs that prevented alcohol’s effects in the PFC did not rescue alcohol’s effects on HPC function, underscoring a region-specific influence of regulatory epigenetic signature on adaptive processes that lead to alcohol tolerance and dependence.
Ethanol’s effects on histone H3 phosphorylation at serine 10 (H3ser10phos) and concurrent H3 phosphoacetylation are of particular interest as their rapid elevation is critical for leaning/memory-associated induction of immediate-early genes (e.g., c-fos and egr-1) (141–143), an effect shown to mediate adaptive responses to psychology stressful events such as forced swimming or novelty stress paradigm exposure (142–145). In rats, acute ethanol dose dependently alters the number of H3ser10phos in the dentate granular cells of the HPC, and these changes are paralleled by changes in c-fos protein expression (146). The same group has shown that, in ethanol-dependent rats, both H3ser10phos and c-fos levels are reduced in dentate granule cells during excessive alcohol intake, while opposite effects are evident at withdrawal peak in the HPC. Elevation of H3ser10phos and histone H3 phosphoacetylation is achieved through a direct interaction of the GR with [mitogen- and stress-activated protein kinase 1 (MSK1)] and ETS-domain protein Elk-1 that are downstream of the ERK/MAPK signaling cascade (143, 145, 147, 148). Conversely, nuclear type 1 protein phosphatase (PP1), a nuclear protein Ser/Thr phosphatase that acts as a universal negative regulator of memory and synaptic plasticity, interfered with H3Ser10phos in several brain areas such as the HPC and the AMG (149–152).
Combinatorial modifications of acetylated H3 and histone H3 lysine 4 trimethylation (H3K4me3) have been implicated in long-term adaptive changes in the HPC resulting from prolonged alcohol intake (126, 153). Using a 3-week mouse model of chronic ethanol consumption, Stragier and colleagues (154) recently reported that ethanol-induced BDNF-mediated neuroplastic changes in the HPC are controlled by combinatorial modifications of acetylated H3 and H3K4me3 around individual Bdnf gene promoters in dorsal CA3 region and the dentate gyrus and by decreased Bdnf DNA methylation in CA1–CA3 regions of the HPC. These ethanol-induced changes were associated with a deficit in HPC-dependent (contextual fear and novel recognition object) memory while sparing AMG-based cued fear memory. Chronic intermittent ethanol vapor exposure followed by 2–5 days of abstinence robustly and selectively increased histone H3K9 acetylation and DNA demethylation in PFC neurons with a parallel decrease of H3K9 methylation repressive mark as well as a downregulation of a set of histone methyltransferases (HMT) (155, 156). These changes mostly occurred after ethanol removal and contributed to the development of physical dependence on alcohol through an adaptive long-lasting upregulation of the NMDA receptor 2B (NR2B) gene expression (155). Moreover, systemic treatment with TSA during ethanol exposure increased H3K9 acetylation at the NR2B promoter in PFC neurons and potentiated voluntary ethanol consumption (157). Together, these data suggest that persistent upregulation of the NR2B-containing NMDA receptors through deregulation of the balance between histone H3K9 acetylation and methylation states in the PFC may act as a potentially important contributor to the development of alcohol dependence.
This review summarizes recent advances in our comprehension of endocrine, epigenetic, and transcriptional changes that serve as determining factors in controlling alcohol-associated changes in the expression of gene networks and behavior and play a central role in the regulation of alcohol dependence, withdrawal, and relapse (Figure 1). Most of the studies conducted thus far focused mainly on epigenetic and transcriptional regulation of adaptive responses to acute and chronic alcohol that occur within a single brain region (mostly the AMG). This review highlights new evidence from clinical and preclinical studies on how long-term adaptations arising from disruption of the fine coordination of highly interconnected brain structures within a circuit, including, but not limited to, the PFC, the HPC, and the AMG, may contribute to excessive alcohol consumption and alcohol dependence as well as behavior impairments. The findings reviewed in this article support the view that brain region- and cell type-specific histone acetylation modification (both in terms of global/genome-wide changes as well as promoter-specific changes) is a key mechanism underlying anxiety-like and alcohol-drinking behaviors. Thus, treatments designed to counteract alcohol-associated epigenetic changes may be promising targets for novel medications in the treatment of alcoholism.
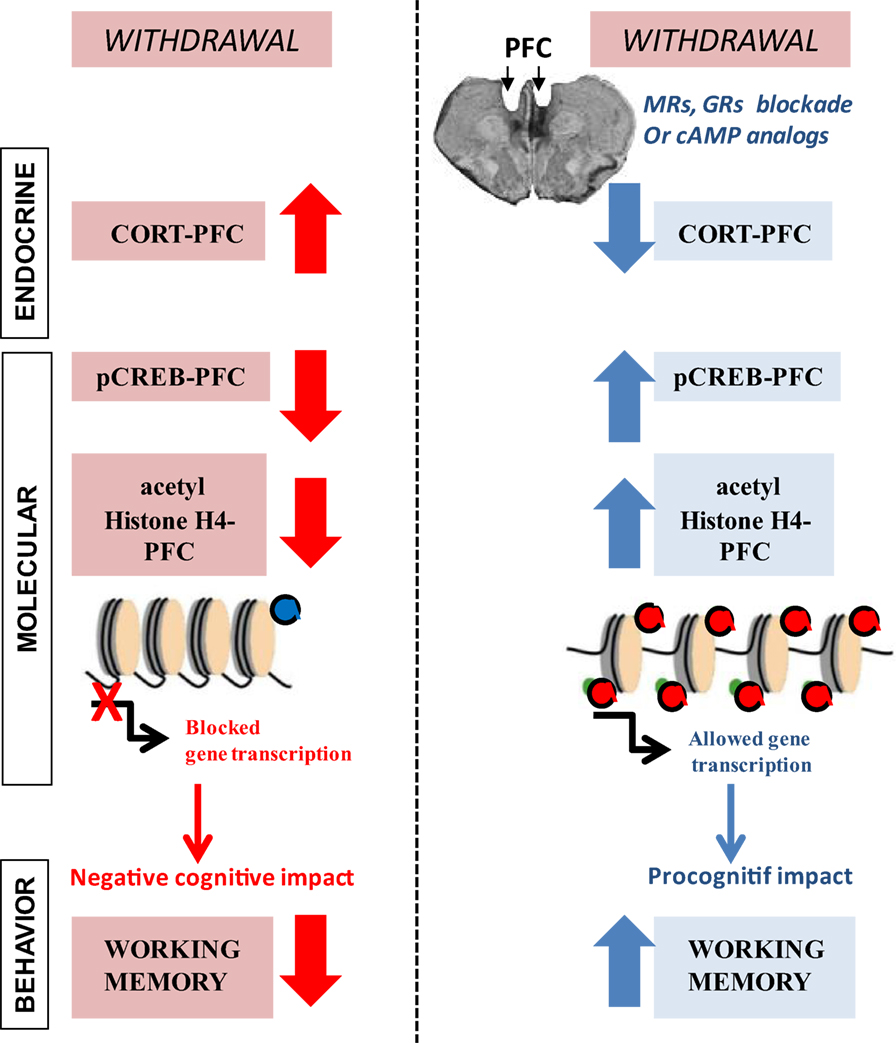
Figure 1. Endocrine and molecular events that are associated with long-lasting prefrontal cortex (PFC)-based working memory impairment during withdrawal from prolonged ethanol consumption. (Left) in C57BL/6 mice, chronic ethanol consumption (6 months) followed by a 6-week withdrawal period causes excessive peak corticosterone (CORT) response, specifically in the PFC that lasts for several weeks. Protracted withdrawal also produces long-lasting deficits in pCREB and histone H4 acetylation levels in the PFC along with enduring working memory impairments. (Red) pharmacological glucocorticoid blockade in the PFC through bilateral infusion of drugs that diminish corticosteroid receptors [mineralocorticoid (spironolactone; MR) or glucocorticoid (mifepristone; GR)] activity as well as pharmacological elevation of cAMP–PKA-mediated signaling cascade through bilateral infusion of the cAMP analog Sp-cAMPS into the PFC fully prevent long-lasting alcohol-related endocrine, molecular, and behavioral changes. Adapted from Ref. (31, 47).
NM and DB contributed to the writing of this review article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by a grant from FRA (Fondation pour la Recherche en Alcoologie, Paris, France).
1. McEwen BS. The brain on stress: toward an integrative approach to brain, body, and behavior. Perspect Psychol Sci (2013) 8(6):673–5. doi:10.1177/1745691613506907
2. Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci (2007) 30(8):399–406. doi:10.1016/j.tins.2007.06.006
3. Breese GR, Sinha R, Heilig M. Chronic alcohol neuroadaptation and stress contribute to susceptibility for alcohol craving and relapse. Pharmacol Ther (2011) 129(2):149–71. doi:10.1016/j.pharmthera.2010.09.007
4. Richardson HN, Lee SY, O’Dell LE, Koob GF, Rivier CL. Alcohol self-administration acutely stimulates the hypothalamic-pituitary-adrenal axis, but alcohol dependence leads to a dampened neuroendocrine state. Eur J Neurosci (2008) 28(8):1641–53. doi:10.1111/j.1460-9568.2008.06455.x
5. Lovallo WR, Dickensheets SL, Myers DA, Thomas TL, Nixon SJ. Blunted stress cortisol response in abstinent alcoholic and polysubstance-abusing men. Alcohol Clin Exp Res (2000) 24(5):651–8. doi:10.1111/j.1530-0277.2000.tb02036.x
6. Adinoff B, Ruether K, Krebaum S, Iranmanesh A, Williams MJ. Increased salivary cortisol concentrations during chronic alcohol intoxication in a naturalistic clinical sample of men. Alcohol Clin Exp Res (2003) 27(9):1420–7. doi:10.1097/01.ALC.0000087581.13912.64
7. Sinha R, Fox HC, Hong KI, Hansen J, Tuit K, Kreek MJ. Effects of adrenal sensitivity, stress- and cue-induced craving, and anxiety on subsequent alcohol relapse and treatment outcomes. Arch Gen Psychiatry (2011) 68(9):942–52. doi:10.1001/archgenpsychiatry.2011.49
8. Rasmussen DD, Boldt BM, Bryant CA, Mitton DR, Larsen SA, Wilkinson CW. Chronic daily ethanol and withdrawal: 1. Long-term changes in the hypothalamo-pituitary-adrenal axis. Alcohol Clin Exp Res (2000) 24(12):1836–49. doi:10.1111/j.1530-0277.2000.tb01988.x
9. Zorrilla EP, Valdez GR, Weiss F. Changes in levels of regional CRF-like-immunoreactivity and plasma corticosterone during protracted drug withdrawal in dependent rats. Psychopharmacology (Berl) (2001) 158(4):374–81. doi:10.1007/s002130100773
10. Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology (1985) 117(6):2505–11. doi:10.1210/endo-117-6-2505
11. Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science (1987) 237(4812):268–75. doi:10.1126/science.3037703
12. Arriza JL, Simerly RB, Swanson LW, Evans RM. The neuronal mineralocorticoid receptor as a mediator of glucocorticoid response. Neuron (1988) 1(9):887–900. doi:10.1016/0896-6273(88)90136-5
13. Reul JM, van den Bosch FR, de Kloet ER. Relative occupation of type-I and type-II corticosteroid receptors in rat brain following stress and dexamethasone treatment: functional implications. J Endocrinol (1987) 115(3):459–67. doi:10.1677/joe.0.1150459
14. Seckl JR, Dickson KL, Yates C, Fink G. Distribution of glucocorticoid and mineralocorticoid receptor messenger RNA expression in human postmortem hippocampus. Brain Res (1991) 561(2):332–7. doi:10.1016/0006-8993(91)91612-5
15. Joels M, De Kloet ER. Coordinative mineralocorticoid and glucocorticoid receptor-mediated control of responses to serotonin in rat hippocampus. Neuroendocrinology (1992) 55(3):344–50. doi:10.1159/000126135
16. Fuxe K, Cintra A, Harfstrand A, Agnati LF, Kalia M, Zoli M, et al. Central glucocorticoid receptor immunoreactive neurons: new insights into the endocrine regulation of the brain. Ann N Y Acad Sci (1987) 512:362–93. doi:10.1111/j.1749-6632.1987.tb24974.x
17. Cintra A, Bhatnagar M, Chadi G, Tinner B, Lindberg J, Gustafsson JA, et al. Glial and neuronal glucocorticoid receptor immunoreactive cell populations in developing, adult, and aging brain. Ann N Y Acad Sci (1994) 746:42–61; discussion 61–3. doi:10.1111/j.1749-6632.1994.tb39210.x
18. Patel PD, Lopez JF, Lyons DM, Burke S, Wallace M, Schatzberg AF. Glucocorticoid and mineralocorticoid receptor mRNA expression in squirrel monkey brain. J Psychiatr Res (2000) 34(6):383–92. doi:10.1016/S0022-3956(00)00035-2
19. Starkman MN, Giordani B, Berent S, Schork MA, Schteingart DE. Elevated cortisol levels in Cushing’s disease are associated with cognitive decrements. Psychosom Med (2001) 63(6):985–93. doi:10.1097/00006842-200111000-00018
20. Starkman MN. Neuropsychiatric findings in Cushing syndrome and exogenous glucocorticoid administration. Endocrinol Metab Clin North Am (2013) 42(3):477–88. doi:10.1016/j.ecl.2013.05.010
21. Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NP, et al. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci (1998) 1(1):69–73. doi:10.1038/1149
22. Patil CG, Prevedello DM, Lad SP, Vance ML, Thorner MO, Katznelson L, et al. Late recurrences of Cushing’s disease after initial successful transsphenoidal surgery. J Clin Endocrinol Metab (2008) 93(2):358–62. doi:10.1210/jc.2007-2013
23. Terfehr K, Wolf OT, Schlosser N, Fernando SC, Otte C, Muhtz C, et al. Hydrocortisone impairs working memory in healthy humans, but not in patients with major depressive disorder. Psychopharmacology (Berl) (2011) 215(1):71–9. doi:10.1007/s00213-010-2117-z
24. Jacquot C, Croft AP, Prendergast MA, Mulholland P, Shaw SG, Little HJ. Effects of the glucocorticoid antagonist, mifepristone, on the consequences of withdrawal from long term alcohol consumption. Alcohol Clin Exp Res (2008) 32(12):2107–16. doi:10.1111/j.1530-0277.2008.00799.x
25. Kesner RP, Churchwell JC. An analysis of rat prefrontal cortex in mediating executive function. Neurobiol Learn Mem (2011) 96(3):417–31. doi:10.1016/j.nlm.2011.07.002
26. Keedwell PA, Kumari V, Poon L, Marshall EJ, Checkley SA. Information processing deficits in withdrawing alcoholics. Addict Biol (2001) 6(3):239–45. doi:10.1080/13556210120056571
27. Errico AL, King AC, Lovallo WR, Parsons OA. Cortisol dysregulation and cognitive impairment in abstinent male alcoholics. Alcohol Clin Exp Res (2002) 26(8):1198–204. doi:10.1111/j.1530-0277.2002.tb02656.x
29. Rose AK, Shaw SG, Prendergast MA, Little HJ. The importance of glucocorticoids in alcohol dependence and neurotoxicity. Alcohol Clin Exp Res (2010) 34(12):2011–8. doi:10.1111/j.1530-0277.2010.01298.x
30. Little HJ, Croft AP, O’Callaghan MJ, Brooks SP, Wang G, Shaw SG. Selective increases in regional brain glucocorticoid: a novel effect of chronic alcohol. Neuroscience (2008) 156(4):1017–27. doi:10.1016/j.neuroscience.2008.08.029
31. Dominguez G, Belzung C, Pierard C, David V, Henkous N, Decorte L, et al. Alcohol withdrawal induces long-lasting spatial working memory impairments: relationship with changes in corticosterone response in the prefrontal cortex. Addict Biol (2016). doi:10.1111/adb.12371
32. Brooks SP, Croft AP, Norman G, Shaw SG, Little HJ. Nimodipine prior to alcohol withdrawal prevents memory deficits during the abstinence phase. Neuroscience (2008) 157(2):376–84. doi:10.1016/j.neuroscience.2008.09.010
33. Vendruscolo LF, Barbier E, Schlosburg JE, Misra KK, Whitfield TW Jr, Logrip ML, et al. Corticosteroid-dependent plasticity mediates compulsive alcohol drinking in rats. J Neurosci (2012) 32(22):7563–71. doi:10.1523/JNEUROSCI.0069-12.2012
34. Sharrett-Field L, Butler TR, Berry JN, Reynolds AR, Prendergast MA. Mifepristone pretreatment reduces ethanol withdrawal severity in vivo. Alcohol Clin Exp Res (2013) 37(8):1417–23. doi:10.1111/acer.12093
35. Mizoguchi K, Ishige A, Aburada M, Tabira T. Chronic stress attenuates glucocorticoid negative feedback: involvement of the prefrontal cortex and hippocampus. Neuroscience (2003) 119(3):887–97. doi:10.1016/S0306-4522(03)00105-2
36. Mizoguchi K, Ishige A, Takeda S, Aburada M, Tabira T. Endogenous glucocorticoids are essential for maintaining prefrontal cortical cognitive function. J Neurosci (2004) 24(24):5492–9. doi:10.1523/JNEUROSCI.0086-04.2004
37. Yuen EY, Liu W, Karatsoreos IN, Feng J, McEwen BS, Yan Z. Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proc Natl Acad Sci U S A (2009) 106(33):14075–9. doi:10.1073/pnas.0906791106
38. O’Daly OG, Trick L, Scaife J, Marshall J, Ball D, Phillips ML, et al. Withdrawal-associated increases and decreases in functional neural connectivity associated with altered emotional regulation in alcoholism. Neuropsychopharmacology (2012) 37(10):2267–76. doi:10.1038/npp.2012.77
39. Pfefferbaum A, Desmond JE, Galloway C, Menon V, Glover GH, Sullivan EV. Reorganization of frontal systems used by alcoholics for spatial working memory: an fMRI study. Neuroimage (2001) 14(1 Pt 1):7–20. doi:10.1006/nimg.2001.0785
40. Moselhy HF, Georgiou G, Kahn A. Frontal lobe changes in alcoholism: a review of the literature. Alcohol Alcohol (2001) 36(5):357–68. doi:10.1093/alcalc/36.5.357
41. Cerqueira JJ, Pego JM, Taipa R, Bessa JM, Almeida OF, Sousa N. Morphological correlates of corticosteroid-induced changes in prefrontal cortex-dependent behaviors. J Neurosci (2005) 25(34):7792–800. doi:10.1523/JNEUROSCI.1598-05.2005
42. Leon-Carrion J, Atutxa AM, Mangas MA, Soto-Moreno A, Pumar A, Leon-Justel A, et al. A clinical profile of memory impairment in humans due to endogenous glucocorticoid excess. Clin Endocrinol (Oxf) (2009) 70(2):192–200. doi:10.1111/j.1365-2265.2008.03355.x
43. Vaz LJ, Pradella-Hallinan M, Bueno OF, Pompeia S. Acute glucocorticoid effects on the multicomponent model of working memory. Hum Psychopharmacol (2011) 26(7):477–87. doi:10.1002/hup.1230
44. Abernathy K, Chandler LJ, Woodward JJ. Alcohol and the prefrontal cortex. Int Rev Neurobiol (2010) 91:289–320. doi:10.1016/S0074-7742(10)91009-X
45. Vetreno RP, Hall JM, Savage LM. Alcohol-related amnesia and dementia: animal models have revealed the contributions of different etiological factors on neuropathology, neurochemical dysfunction and cognitive impairment. Neurobiol Learn Mem (2011) 96(4):596–608. doi:10.1016/j.nlm.2011.01.003
46. George O, Sanders C, Freiling J, Grigoryan E, Vu S, Allen CD, et al. Recruitment of medial prefrontal cortex neurons during alcohol withdrawal predicts cognitive impairment and excessive alcohol drinking. Proc Natl Acad Sci U S A (2012) 109(44):18156–61. doi:10.1073/pnas.1116523109
47. Dominguez G, Dagnas M, Decorte L, Vandesquille M, Belzung C, Beracochea D, et al. Rescuing prefrontal cAMP-CREB pathway reverses working memory deficits during withdrawal from prolonged alcohol exposure. Brain Struct Funct (2016) 221(2):865–77. doi:10.1007/s00429-014-0941-3
48. Lee I, Kesner RP. Differential roles of dorsal hippocampal subregions in spatial working memory with short versus intermediate delay. Behav Neurosci (2003) 117(5):1044–53. doi:10.1037/0735-7044.117.5.1044
49. Spellman T, Rigotti M, Ahmari SE, Fusi S, Gogos JA, Gordon JA. Hippocampal-prefrontal input supports spatial encoding in working memory. Nature (2015) 522(7556):309–14. doi:10.1038/nature14445
50. Chauveau F, Celerier A, Ognard R, Pierard C, Beracochea D. Effects of ibotenic acid lesions of the mediodorsal thalamus on memory: relationship with emotional processes in mice. Behav Brain Res (2005) 156(2):215–23. doi:10.1016/j.bbr.2004.05.026
51. Vandesquille M, Baudonnat M, Decorte L, Louis C, Lestage P, Beracochea D. Working memory deficits and related disinhibition of the cAMP/PKA/CREB are alleviated by prefrontal alpha4beta2*-nAChRs stimulation in aged mice. Neurobiol Aging (2013) 34(6):1599–609. doi:10.1016/j.neurobiolaging.2012.10.006
52. Beracochea D, Jaffard R. Memory deficits subsequent to chronic consumption of alcohol in mice: an analysis based on spontaneous alternation behavior. Behav Brain Res (1985) 15(1):15–25. doi:10.1016/0166-4328(85)90014-2
53. Dalley JW, Cardinal RN, Robbins TW. Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neurosci Biobehav Rev (2004) 28(7):771–84. doi:10.1016/j.neubiorev.2004.09.006
54. Runyan JD, Dash PK. Distinct prefrontal molecular mechanisms for information storage lasting seconds versus minutes. Learn Mem (2005) 12(3):232–8. doi:10.1101/lm.92405
55. Roozendaal B, McReynolds JR, McGaugh JL. The basolateral amygdala interacts with the medial prefrontal cortex in regulating glucocorticoid effects on working memory impairment. J Neurosci (2004) 24(6):1385–92. doi:10.1523/JNEUROSCI.4664-03.2004
56. Buhler M, Mann K. Alcohol and the human brain: a systematic review of different neuroimaging methods. Alcohol Clin Exp Res (2011) 35(10):1771–93. doi:10.1111/j.1530-0277.2011.01540.x
57. Noel X, Sferrazza R, Van Der Linden M, Paternot J, Verhas M, Hanak C, et al. Contribution of frontal cerebral blood flow measured by (99m)Tc-Bicisate spect and executive function deficits to predicting treatment outcome in alcohol-dependent patients. Alcohol Alcohol (2002) 37(4):347–54. doi:10.1093/alcalc/37.4.347
58. Butts KA, Weinberg J, Young AH, Phillips AG. Glucocorticoid receptors in the prefrontal cortex regulate stress-evoked dopamine efflux and aspects of executive function. Proc Natl Acad Sci U S A (2011) 108(45):18459–64. doi:10.1073/pnas.1111746108
59. Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron (2002) 35(4):605–23. doi:10.1016/S0896-6273(02)00828-0
60. Pandey SC, Roy A, Zhang H, Xu T. Partial deletion of the cAMP response element-binding protein gene promotes alcohol-drinking behaviors. J Neurosci (2004) 24(21):5022–30. doi:10.1523/JNEUROSCI.5557-03.2004
61. Moonat S, Sakharkar AJ, Zhang H, Tang L, Pandey SC. Aberrant histone deacetylase2-mediated histone modifications and synaptic plasticity in the amygdala predisposes to anxiety and alcoholism. Biol Psychiatry (2013) 73(8):763–73. doi:10.1016/j.biopsych.2013.01.012
62. Pandey SC. Anxiety and alcohol abuse disorders: a common role for CREB and its target, the neuropeptide Y gene. Trends Pharmacol Sci (2003) 24(9):456–60. doi:10.1016/S0165-6147(03)00226-8
63. Valdez GR, Koob GF. Allostasis and dysregulation of corticotropin-releasing factor and neuropeptide Y systems: implications for the development of alcoholism. Pharmacol Biochem Behav (2004) 79(4):671–89. doi:10.1016/j.pbb.2004.09.020
64. Janak PH, Wolf FW, Heberlein U, Pandey SC, Logrip ML, Ron D. BIG news in alcohol addiction: new findings on growth factor pathways BDNF, insulin, and GDNF. Alcohol Clin Exp Res (2006) 30(2):214–21. doi:10.1111/j.1530-0277.2006.00026.x
65. Moonat S, Starkman BG, Sakharkar A, Pandey SC. Neuroscience of alcoholism: molecular and cellular mechanisms. Cell Mol Life Sci (2010) 67(1):73–88. doi:10.1007/s00018-009-0135-y
66. Moonat S, Sakharkar AJ, Zhang H, Pandey SC. The role of amygdaloid brain-derived neurotrophic factor, activity-regulated cytoskeleton-associated protein and dendritic spines in anxiety and alcoholism. Addict Biol (2011) 16(2):238–50. doi:10.1111/j.1369-1600.2010.00275.x
67. Crews FT, Nixon K. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol (2009) 44(2):115–27. doi:10.1093/alcalc/agn079
68. Pandey SC, Chartoff EH, Carlezon WA Jr, Zou J, Zhang H, Kreibich AS, et al. CREB gene transcription factors: role in molecular mechanisms of alcohol and drug addiction. Alcohol Clin Exp Res (2005) 29(2):176–84. doi:10.1097/01.ALC.0000153550.31168.1D
69. Hoffman PL, Tabakoff B. Ethanol and guanine nucleotide binding proteins: a selective interaction. FASEB J (1990) 4(9):2612–22.
70. Wand GS, Levine MA. Hormonal tolerance to ethanol is associated with decreased expression of the GTP-binding protein, Gs alpha, and adenylyl cyclase activity in ethanol-treated LS mice. Alcohol Clin Exp Res (1991) 15(4):705–10. doi:10.1111/j.1530-0277.1991.tb00583.x
71. Coe IR, Dohrman DP, Constantinescu A, Diamond I, Gordon AS. Activation of cyclic AMP-dependent protein kinase reverses tolerance of a nucleoside transporter to ethanol. J Pharmacol Exp Ther (1996) 276(2):365–9.
72. Dohrman DP, Chen HM, Gordon AS, Diamond I. Ethanol-induced translocation of protein kinase A occurs in two phases: control by different molecular mechanisms. Alcohol Clin Exp Res (2002) 26(3):407–15. doi:10.1111/j.1530-0277.2002.tb02553.x
73. Constantinescu A, Diamond I, Gordon AS. Ethanol-induced translocation of cAMP-dependent protein kinase to the nucleus. Mechanism and functional consequences. J Biol Chem (1999) 274(38):26985–91. doi:10.1074/jbc.274.38.26985
74. Asher O, Cunningham TD, Yao L, Gordon AS, Diamond I. Ethanol stimulates cAMP-responsive element (CRE)-mediated transcription via CRE-binding protein and cAMP-dependent protein kinase. J Pharmacol Exp Ther (2002) 301(1):66–70. doi:10.1124/jpet.301.1.66
75. Nagy LE, DeSilva SE. Ethanol increases receptor-dependent cyclic AMP production in cultured hepatocytes by decreasing G(i)-mediated inhibition. Biochem J (1992) 286(Pt 3):681–6. doi:10.1042/bj2860681
76. Saito T, Luthin GR, Lee JM, Hoffman PL, Tabakoff B. Differential effects of ethanol on the striatal and cortical adenylate cyclase system. Jpn J Pharmacol (1987) 43(2):133–41. doi:10.1254/jjp.43.133
77. Saito T, Lee JM, Hoffman PL, Tabakoff B. Effects of chronic ethanol treatment on the beta-adrenergic receptor-coupled adenylate cyclase system of mouse cerebral cortex. J Neurochem (1987) 48(6):1817–22. doi:10.1111/j.1471-4159.1987.tb05741.x
78. Valverius P, Hoffman PL, Tabakoff B. Hippocampal and cerebellar beta-adrenergic receptors and adenylate cyclase are differentially altered by chronic ethanol ingestion. J Neurochem (1989) 52(2):492–7. doi:10.1111/j.1471-4159.1989.tb09147.x
79. Rodan AR, Kiger JA Jr, Heberlein U. Functional dissection of neuroanatomical loci regulating ethanol sensitivity in Drosophila. J Neurosci (2002) 22(21):9490–501.
80. Thiele TE, Willis B, Stadler J, Reynolds JG, Bernstein IL, McKnight GS. High ethanol consumption and low sensitivity to ethanol-induced sedation in protein kinase A-mutant mice. J Neurosci (2000) 20(10):RC75.
81. Wand G, Levine M, Zweifel L, Schwindinger W, Abel T. The cAMP-protein kinase A signal transduction pathway modulates ethanol consumption and sedative effects of ethanol. J Neurosci (2001) 21(14):5297–303.
82. Pandey SC, Roy A, Mittal N. Effects of chronic ethanol intake and its withdrawal on the expression and phosphorylation of the creb gene transcription factor in rat cortex. J Pharmacol Exp Ther (2001) 296(3):857–68.
83. Repunte-Canonigo V, Lutjens R, van der Stap LD, Sanna PP. Increased expression of protein kinase A inhibitor alpha (PKI-alpha) and decreased PKA-regulated genes in chronic intermittent alcohol exposure. Brain Res (2007) 1138:48–56. doi:10.1016/j.brainres.2006.09.115
84. Pandey SC, Zhang H, Roy A, Xu T. Deficits in amygdaloid cAMP-responsive element-binding protein signaling play a role in genetic predisposition to anxiety and alcoholism. J Clin Invest (2005) 115(10):2762–73. doi:10.1172/JCI24381
85. Wand G. The anxious amygdala: CREB signaling and predisposition to anxiety and alcoholism. J Clin Invest (2005) 115(10):2697–9. doi:10.1172/JCI26436
86. Zhang H, Pandey SC. Effects of PKA modulation on the expression of neuropeptide Y in rat amygdaloid structures during ethanol withdrawal. Peptides (2003) 24(9):1397–402. doi:10.1016/j.peptides.2003.08.008
87. Badia-Elder NE, Stewart RB, Powrozek TA, Roy KF, Murphy JM, Li TK. Effect of neuropeptide Y (NPY) on oral ethanol intake in Wistar, alcohol-preferring (P), and -nonpreferring (NP) rats. Alcohol Clin Exp Res (2001) 25(3):386–90. doi:10.1111/j.1530-0277.2001.tb02225.x
88. Primeaux SD, Wilson SP, Bray GA, York DA, Wilson MA. Overexpression of neuropeptide Y in the central nucleus of the amygdala decreases ethanol self-administration in “anxious” rats. Alcohol Clin Exp Res (2006) 30(5):791–801. doi:10.1111/j.1530-0277.2006.00092.x
89. Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci (2009) 10(6):410–22. doi:10.1038/nrn2648
90. Shansky RM, Lipps J. Stress-induced cognitive dysfunction: hormone-neurotransmitter interactions in the prefrontal cortex. Front Hum Neurosci (2013) 7:123. doi:10.3389/fnhum.2013.00123
91. Gamo NJ, Lur G, Higley MJ, Wang M, Paspalas CD, Vijayraghavan S, et al. Stress impairs prefrontal cortical function via D1 dopamine receptor interactions with hyperpolarization-activated cyclic nucleotide-gated channels. Biol Psychiatry (2015) 78(12):860–70. doi:10.1016/j.biopsych.2015.01.009
92. Barsegyan A, Mackenzie SM, Kurose BD, McGaugh JL, Roozendaal B. Glucocorticoids in the prefrontal cortex enhance memory consolidation and impair working memory by a common neural mechanism. Proc Natl Acad Sci U S A (2010) 107(38):16655–60. doi:10.1073/pnas.1011975107
93. Logrip ML. Phosphodiesterase regulation of alcohol drinking in rodents. Alcohol (2015) 49(8):795–802. doi:10.1016/j.alcohol.2015.03.007
94. Hu W, Lu T, Chen A, Huang Y, Hansen R, Chandler LJ, et al. Inhibition of phosphodiesterase-4 decreases ethanol intake in mice. Psychopharmacology (Berl) (2011) 218(2):331–9. doi:10.1007/s00213-011-2290-8
95. Wen RT, Zhang M, Qin WJ, Liu Q, Wang WP, Lawrence AJ, et al. The phosphodiesterase-4 (PDE4) inhibitor rolipram decreases ethanol seeking and consumption in alcohol-preferring Fawn-Hooded rats. Alcohol Clin Exp Res (2012) 36(12):2157–67. doi:10.1111/j.1530-0277.2012.01845.x
96. Franklin KM, Hauser SR, Lasek AW, McClintick J, Ding ZM, McBride WJ, et al. Reduction of alcohol drinking of alcohol-preferring (P) and high-alcohol drinking (HAD1) rats by targeting phosphodiesterase-4 (PDE4). Psychopharmacology (Berl) (2015) 232(13):2251–62. doi:10.1007/s00213-014-3852-3
97. Logrip ML, Zorrilla EP. Differential changes in amygdala and frontal cortex Pde10a expression during acute and protracted withdrawal. Front Integr Neurosci (2014) 8:30. doi:10.3389/fnint.2014.00030
98. Baudonnat M, Guillou JL, Husson M, Vandesquille M, Corio M, Decorte L, et al. Disrupting effect of drug-induced reward on spatial but not cue-guided learning: implication of the striatal protein kinase A/cAMP response element-binding protein pathway. J Neurosci (2011) 31(46):16517–28. doi:10.1523/JNEUROSCI.1787-11.2011
99. Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology (2001) 24(2):97–129. doi:10.1016/S0893-133X(00)00195-0
100. Volkow ND, Fowler JS, Wang GJ, Goldstein RZ. Role of dopamine, the frontal cortex and memory circuits in drug addiction: insight from imaging studies. Neurobiol Learn Mem (2002) 78(3):610–24. doi:10.1006/nlme.2002.4099
101. Ahmed SH, Koob GF. Transition from moderate to excessive drug intake: change in hedonic set point. Science (1998) 282(5387):298–300. doi:10.1126/science.282.5387.298
102. Martinotti G, Nicola MD, Reina D, Andreoli S, Foca F, Cunniff A, et al. Alcohol protracted withdrawal syndrome: the role of anhedonia. Subst Use Misuse (2008) 43(3–4):271–84. doi:10.1080/10826080701202429
103. Janiri L, Martinotti G, Dario T, Reina D, Paparello F, Pozzi G, et al. Anhedonia and substance-related symptoms in detoxified substance-dependent subjects: a correlation study. Neuropsychobiology (2005) 52(1):37–44. doi:10.1159/000086176
104. Stevenson JR, Schroeder JP, Nixon K, Besheer J, Crews FT, Hodge CW. Abstinence following alcohol drinking produces depression-like behavior and reduced hippocampal neurogenesis in mice. Neuropsychopharmacology (2009) 34(5):1209–22. doi:10.1038/npp.2008.90
105. Pang TY, Renoir T, Du X, Lawrence AJ, Hannan AJ. Depression-related behaviours displayed by female C57BL/6J mice during abstinence from chronic ethanol consumption are rescued by wheel-running. Eur J Neurosci (2013) 37(11):1803–10. doi:10.1111/ejn.12195
106. Lee KM, Coehlo M, McGregor HA, Waltermire RS, Szumlinski KK. Binge alcohol drinking elicits persistent negative affect in mice. Behav Brain Res (2015) 291:385–98. doi:10.1016/j.bbr.2015.05.055
107. Briones TL, Rogozinska M, Woods J. Environmental experience modulates ischemia-induced amyloidogenesis and enhances functional recovery. J Neurotrauma (2009) 26(4):613–25. doi:10.1089/neu.2008.0707
108. Yan T, Xu M, Wan S, Wang M, Wu B, Xiao F, et al. Schisandra chinensis produces the antidepressant-like effects in repeated corticosterone-induced mice via the BDNF/TrkB/CREB signaling pathway. Psychiatry Res (2016) 243:135–42. doi:10.1016/j.psychres.2016.06.037
109. Kouzarides T. Chromatin modifications and their function. Cell (2007) 128(4):693–705. doi:10.1016/j.cell.2007.02.005
110. Latham JA, Dent SY. Cross-regulation of histone modifications. Nat Struct Mol Biol (2007) 14(11):1017–24. doi:10.1038/nsmb1307
111. Jarome TJ, Thomas JS, Lubin FD. The epigenetic basis of memory formation and storage. Prog Mol Biol Transl Sci (2014) 128:1–27. doi:10.1016/B978-0-12-800977-2.00001-2
112. Graff J, Mansuy IM. Epigenetic codes in cognition and behaviour. Behav Brain Res (2008) 192(1):70–87. doi:10.1016/j.bbr.2008.01.021
113. Graff J, Tsai LH. Histone acetylation: molecular mnemonics on the chromatin. Nat Rev Neurosci (2013) 14(2):97–111. doi:10.1038/nrn3427
114. Day JJ, Sweatt JD. Epigenetic mechanisms in cognition. Neuron (2011) 70(5):813–29. doi:10.1016/j.neuron.2011.05.019
115. Zovkic IB, Guzman-Karlsson MC, Sweatt JD. Epigenetic regulation of memory formation and maintenance. Learn Mem (2013) 20(2):61–74. doi:10.1101/lm.026575.112
116. Dagnas M, Guillou JL, Prevot T, Mons N. HDAC inhibition facilitates the switch between memory systems in young but not aged mice. J Neurosci (2013) 33(5):1954–63. doi:10.1523/JNEUROSCI.3453-12.2013
117. Dagnas M, Micheau J, Decorte L, Beracochea D, Mons N. Post-training, intrahippocampal HDAC inhibition differentially impacts neural circuits underlying spatial memory in adult and aged mice. Hippocampus (2015) 25(7):827–37. doi:10.1002/hipo.22406
118. Graff J, Tsai LH. Cognitive enhancement: a molecular memory booster. Nature (2011) 469(7331):474–5. doi:10.1038/469474a
119. Graff J, Tsai LH. The potential of HDAC inhibitors as cognitive enhancers. Annu Rev Pharmacol Toxicol (2013) 53:311–30. doi:10.1146/annurev-pharmtox-011112-140216
120. Haggarty SJ, Tsai LH. Probing the role of HDACs and mechanisms of chromatin-mediated neuroplasticity. Neurobiol Learn Mem (2011) 96(1):41–52. doi:10.1016/j.nlm.2011.04.009
121. Schneider A, Chatterjee S, Bousiges O, Selvi BR, Swaminathan A, Cassel R, et al. Acetyltransferases (HATs) as targets for neurological therapeutics. Neurotherapeutics (2013) 10(4):568–88. doi:10.1007/s13311-013-0204-7
122. Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci (2007) 27(23):6128–40. doi:10.1523/JNEUROSCI.0296-07.2007
123. Stafford JM, Raybuck JD, Ryabinin AE, Lattal KM. Increasing histone acetylation in the hippocampus-infralimbic network enhances fear extinction. Biol Psychiatry (2012) 72(1):25–33. doi:10.1016/j.biopsych.2011.12.012
124. Kyzar EJ, Pandey SC. Molecular mechanisms of synaptic remodeling in alcoholism. Neurosci Lett (2015) 601:11–9. doi:10.1016/j.neulet.2015.01.051
125. Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci (2008) 28(14):3729–37. doi:10.1523/JNEUROSCI.5731-07.2008
126. Ponomarev I. Epigenetic control of gene expression in the alcoholic brain. Alcohol Res (2013) 35(1):69–76.
127. Sakharkar AJ, Zhang H, Tang L, Baxstrom K, Shi G, Moonat S, et al. Effects of histone deacetylase inhibitors on amygdaloid histone acetylation and neuropeptide Y expression: a role in anxiety-like and alcohol-drinking behaviours. Int J Neuropsychopharmacol (2014) 17(8):1207–20. doi:10.1017/S1461145714000054
128. Sakharkar AJ, Zhang H, Tang L, Shi G, Pandey SC. Histone deacetylases (HDAC)-induced histone modifications in the amygdala: a role in rapid tolerance to the anxiolytic effects of ethanol. Alcohol Clin Exp Res (2012) 36(1):61–71. doi:10.1111/j.1530-0277.2011.01581.x
129. Pandey SC, Zhang H, Ugale R, Prakash A, Xu T, Misra K. Effector immediate-early gene arc in the amygdala plays a critical role in alcoholism. J Neurosci (2008) 28(10):2589–600. doi:10.1523/JNEUROSCI.4752-07.2008
130. You C, Zhang H, Sakharkar AJ, Teppen T, Pandey SC. Reversal of deficits in dendritic spines, BDNF and Arc expression in the amygdala during alcohol dependence by HDAC inhibitor treatment. Int J Neuropsychopharmacol (2014) 17(2):313–22. doi:10.1017/S1461145713001144
131. Prakash A, Zhang H, Pandey SC. Innate differences in the expression of brain-derived neurotrophic factor in the regions within the extended amygdala between alcohol preferring and nonpreferring rats. Alcohol Clin Exp Res (2008) 32(6):909–20. doi:10.1111/j.1530-0277.2008.00650.x
132. Agudelo M, Gandhi N, Saiyed Z, Pichili V, Thangavel S, Khatavkar P, et al. Effects of alcohol on histone deacetylase 2 (HDAC2) and the neuroprotective role of trichostatin A (TSA). Alcohol Clin Exp Res (2011) 35(8):1550–6. doi:10.1111/j.1530-0277.2011.01492.x
133. Simon-O’Brien E, Alaux-Cantin S, Warnault V, Buttolo R, Naassila M, Vilpoux C. The histone deacetylase inhibitor sodium butyrate decreases excessive ethanol intake in dependent animals. Addict Biol (2015) 20(4):676–89. doi:10.1111/adb.12161
134. Smith ML, Lopez MF, Archer KJ, Wolen AR, Becker HC, Miles MF. Time-course analysis of brain regional expression network responses to chronic intermittent ethanol and withdrawal: implications for mechanisms underlying excessive ethanol consumption. PLoS One (2016) 11(1):e0146257. doi:10.1371/journal.pone.0146257
135. Pascual M, Balino P, Alfonso-Loeches S, Aragon CM, Guerri C. Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain Behav Immun (2011) 25(Suppl 1):S80–91. doi:10.1016/j.bbi.2011.02.012
136. Paula-Barbosa MM, Pereira PA, Cadete-Leite A, Dulce Madeira M. NGF and NT-3 exert differential effects on the expression of neuropeptides in the suprachiasmatic nucleus of rats withdrawn from ethanol treatment. Brain Res (2003) 983(1–2):64–73. doi:10.1016/S0006-8993(03)03030-0
137. Phillips SC, Cragg BG. Blood-brain barrier dysfunction in thiamine-deficient, alcohol-treated rats. Acta Neuropathol (1984) 62(3):235–41. doi:10.1007/BF00691858
138. Farr SA, Scherrer JF, Banks WA, Flood JF, Morley JE. Chronic ethanol consumption impairs learning and memory after cessation of ethanol. Alcohol Clin Exp Res (2005) 29(6):971–82. doi:10.1097/01.ALC.0000171038.03371.56
139. Lukoyanov NV, Pereira PA, Paula-Barbosa MM, Cadete-Leite A. Nerve growth factor improves spatial learning and restores hippocampal cholinergic fibers in rats withdrawn from chronic treatment with ethanol. Exp Brain Res (2003) 148(1):88–94. doi:10.1007/s00221-002-1290-7
140. Lukoyanov NV, Madeira MD, Paula-Barbosa MM. Behavioral and neuroanatomical consequences of chronic ethanol intake and withdrawal. Physiol Behav (1999) 66(2):337–46. doi:10.1016/S0031-9384(98)00301-1
141. Collins A, Hill LE, Chandramohan Y, Whitcomb D, Droste SK, Reul JM. Exercise improves cognitive responses to psychological stress through enhancement of epigenetic mechanisms and gene expression in the dentate gyrus. PLoS One (2009) 4(1):e4330. doi:10.1371/journal.pone.0004330
142. Bilang-Bleuel A, Ulbricht S, Chandramohan Y, De Carli S, Droste SK, Reul JM. Psychological stress increases histone H3 phosphorylation in adult dentate gyrus granule neurons: involvement in a glucocorticoid receptor-dependent behavioural response. Eur J Neurosci (2005) 22(7):1691–700. doi:10.1111/j.1460-9568.2005.04358.x
143. Chandramohan Y, Droste SK, Arthur JS, Reul JM. The forced swimming-induced behavioural immobility response involves histone H3 phospho-acetylation and c-Fos induction in dentate gyrus granule neurons via activation of the N-methyl-d-aspartate/extracellular signal-regulated kinase/mitogen- and stress-activated kinase signalling pathway. Eur J Neurosci (2008) 27(10):2701–13. doi:10.1111/j.1460-9568.2008.06230.x
144. Reul JM, Hesketh SA, Collins A, Mecinas MG. Epigenetic mechanisms in the dentate gyrus act as a molecular switch in hippocampus-associated memory formation. Epigenetics (2009) 4(7):434–9. doi:10.4161/epi.4.7.9806
145. Gutierrez-Mecinas M, Trollope AF, Collins A, Morfett H, Hesketh SA, Kersante F, et al. Long-lasting behavioral responses to stress involve a direct interaction of glucocorticoid receptors with ERK1/2-MSK1-Elk-1 signaling. Proc Natl Acad Sci U S A (2011) 108(33):13806–11. doi:10.1073/pnas.1104383108
146. McClain JA, Nixon K. Alcohol induces parallel changes in hippocampal histone H3 phosphorylation and c-fos protein expression in male rats. Alcohol Clin Exp Res (2016) 40(1):102–12. doi:10.1111/acer.12933
147. Chwang WB, O’Riordan KJ, Levenson JM, Sweatt JD. ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn Mem (2006) 13(3):322–8. doi:10.1101/lm.152906
148. Chwang WB, Arthur JS, Schumacher A, Sweatt JD. The nuclear kinase mitogen- and stress-activated protein kinase 1 regulates hippocampal chromatin remodeling in memory formation. J Neurosci (2007) 27(46):12732–42. doi:10.1523/JNEUROSCI.2522-07.2007
149. Genoux D, Haditsch U, Knobloch M, Michalon A, Storm D, Mansuy IM. Protein phosphatase 1 is a molecular constraint on learning and memory. Nature (2002) 418(6901):970–5. doi:10.1038/nature00928
150. Koshibu K, Graff J, Mansuy IM. Nuclear protein phosphatase-1: an epigenetic regulator of fear memory and amygdala long-term potentiation. Neuroscience (2011) 173:30–6. doi:10.1016/j.neuroscience.2010.11.023
151. Koshibu K, Graff J, Beullens M, Heitz FD, Berchtold D, Russig H, et al. Protein phosphatase 1 regulates the histone code for long-term memory. J Neurosci (2009) 29(41):13079–89. doi:10.1523/JNEUROSCI.3610-09.2009
152. Graff J, Kim D, Dobbin MM, Tsai LH. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol Rev (2011) 91(2):603–49. doi:10.1152/physrev.00012.2010
153. Zhou Z, Yuan Q, Mash DC, Goldman D. Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc Natl Acad Sci U S A (2011) 108(16):6626–31. doi:10.1073/pnas.1018514108
154. Stragier E, Massart R, Salery M, Hamon M, Geny D, Martin V, et al. Ethanol-induced epigenetic regulations at the Bdnf gene in C57BL/6J mice. Mol Psychiatry (2015) 20(3):405–12. doi:10.1038/mp.2014.38
155. Qiang M, Denny A, Lieu M, Carreon S, Li J. Histone H3K9 modifications are a local chromatin event involved in ethanol-induced neuroadaptation of the NR2B gene. Epigenetics (2011) 6(9):1095–104. doi:10.4161/epi.6.9.16924
156. Qiang M, Denny A, Chen J, Ticku MK, Yan B, Henderson G. The site specific demethylation in the 5’-regulatory area of NMDA receptor 2B subunit gene associated with CIE-induced up-regulation of transcription. PLoS One (2010) 5(1):e8798. doi:10.1371/journal.pone.0008798
Keywords: alcoholism, epigenetic, learning and memory, glucocorticoid, anxiety, signaling, CREB, brain
Citation: Mons N and Beracochea D (2016) Behavioral Neuroadaptation to Alcohol: From Glucocorticoids to Histone Acetylation. Front. Psychiatry 7:165. doi: 10.3389/fpsyt.2016.00165
Received: 30 May 2016; Accepted: 21 September 2016;
Published: 06 October 2016
Edited by:
Stefan Borgwardt, University of Basel, SwitzerlandReviewed by:
Giovanni Martinotti, Università degli Studi “G. d’Annunzio” Chieti – Pescara, ItalyCopyright: © 2016 Mons and Beracochea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicole Mons, bmljb2xlLm1vbnNAdS1ib3JkZWF1eC5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.