 Dominik Langgartner
Dominik Langgartner Andrea M. Füchsl
Andrea M. Füchsl Nicole Uschold-Schmidt
Nicole Uschold-Schmidt David A. Slattery
David A. Slattery Stefan O. Reber
Stefan O. Reber- 1Laboratory for Molecular Psychosomatics, Clinic for Psychosomatic Medicine and Psychotherapy, University of Ulm, Ulm, Germany
- 2Laboratory of Molecular and Cellular Neurobiology, Department of Behavioural and Molecular Neurobiology, University of Regensburg, Regensburg, Germany
- 3Department of Behavioural and Molecular Neurobiology, University of Regensburg, Regensburg, Germany
Chronic, in particular chronic psychosocial, stress is a burden of modern societies and known to be a risk factor for numerous somatic and affective disorders (in detail referenced below). However, based on the limited existence of appropriate, and clinically relevant, animal models for studying the effects of chronic stress, the detailed behavioral, physiological, neuronal, and immunological mechanisms linking stress and such disorders are insufficiently understood. To date, most chronic stress studies in animals employ intermittent exposure to the same (homotypic) or to different (heterotypic) stressors of varying duration and intensity. Such models are only of limited value, since they do not adequately reflect the chronic and continuous situation that humans typically experience. Furthermore, application of different physical or psychological stimuli renders comparisons to the mainly psychosocial stressors faced by humans, as well as between the different stress studies almost impossible. In contrast, rodent models of chronic psychosocial stress represent situations more akin to those faced by humans and consequently seem to hold more clinical relevance. Our laboratory has developed a model in which mice are exposed to social stress for 19 continuous days, namely the chronic subordinate colony housing (CSC) paradigm, to help bridge this gap. The main aim of the current review article is to provide a detailed summary of the behavioral, physiological, neuronal, and immunological consequences of the CSC paradigm, and wherever possible relate the findings to other stress models and to the human situation.
Introduction
The Stress Concept
In the nineteenth century, the French physiologist Claude Bernard (1813–1878) noticed that relative constancy of the internal environment is critical for the functional integrity of an organism. Later, in his “emergency concept”, Walter Cannon (1871–1945) described the disruption of this internal equilibrium, thereafter referred to as homeostasis (1), by fear- or rage-induced “fight or flight” reactions. In 1936, it was Hans Selye (1907–1982), who first defined stress, and the stress response, as “the non-specific response of the body to any physical demand” (2), and made the distinction between “stress” and the “stressor” (3). According to him, “stressors” are defined as specific challenges that cause a physiological “stress” response (3). Until now, an overwhelming number of studies have focused on the physiological, in particular neuroendocrine, and behavioral consequences of an acute stress response, which are, in general, well understood.
Thus, it is commonly accepted that the physiological and behavioral responses to acute stressors are adaptive, and important to reinstate body homeostasis [(4–6); for review see (7, 8)]. While physical stressors are, thereby, defined as external challenges to homeostasis, psychological stressors are stated as the anticipation, justified or not, of a challenge to homeostasis (9). In contrast, repeated or chronic stressor exposure over several weeks or months, and the prolonged attempt of the body to reinstate homeostasis during this time – a process referred to as allostasis [for review see (5, 10)] – is thought to result in alterations of numerous body and brain systems, finally resulting in a disease state [(11, 12); for review see (5)]. However, although chronic stress-induced alterations in neuroendocrine, emotional, and immune parameters are likely to play a major role in the etiology of numerous diseases including anxiety and depressive disorders, chronic inflammatory disorders, or cancer [(13–21); for review see (22–27)], the detailed underlying mechanisms are less well understood due, at least in part, to the shortage of appropriate animal models.
Physiological responses to an acute stressor
In response to any acute stressor, two major stress systems become activated, namely the autonomic nervous system, especially its sympathetic (SNS) branch, and the hypothalamo–pituitary–adrenocortical (HPA) axis. Stimulation of these emergency systems, which differ in both their time course and processing, reflects the body’s attempt to deal with the immediate situation and to restore homeostasis [for review see (5, 10)]. Activation of the SNS occurs rapidly, within seconds, via exclusively neuronal pathways originating in the thoracolumbal regions of the spinal cord (splanchnic nerve), and results in the release of adrenaline from chromaffin cells of the adrenal medulla into the blood. Elevated adrenaline levels in the circulation act in synergy with an increased sympathetic noradrenergic innervation of essentially all organs in the body [referenced in (28, 29)]. As a result, cardiovascular and catabolic functions are promoted, and processes not vital in the immediate situation, such as anabolic processes and digestion, are inhibited.
In addition to the SNS, there is a slightly delayed activation of the HPA axis in response to acute stressors. The stimulation of the HPA axis is triggered by the secretion of corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) from parvocellular neurons of the paraventricular nucleus (PVN) of the hypothalamus into the portal blood stream of the pituitary stalk. CRH and AVP promote the synthesis and the secretion of adrenocorticotropic hormone (ACTH) from anterior pituitary corticotroph cells into the peripheral blood, which, in turn, stimulates cortical cells of the adrenal gland to produce and secrete glucocorticoids [GCs; cortisol in humans, corticosterone (CORT) in rats and mice] into the circulation. Within minutes of activation, termination of an acute HPA axis response is achieved by efficient negative feedback inhibition via GC acting at GC receptors and mineralocorticoid receptors at several brain levels (30). The degree and temporal dynamics of HPA axis activation are strongly dependent on the quality, intensity, and duration of the acute stressor (11, 31, 32). In addition, the acute neuroendocrine stress response was shown to be dependent on the time of day (33–35), age of an individual [(36); for review see (37)], reproductive status of an individual [e.g., in the peripartum period (38–40)], genetic background (41–45), and stressor exposure during life history (46, 47).
Taken together, stressor-induced activation of the SNS and HPA axis contribute to the restoration of the “internal equilibrium” by rapid mobilization of metabolic resources (glucose, oxygen); processes that are adaptive and essential for survival.
Behavioral responses to an acute stressor
In addition to, and facilitated by, the rapid activation of physiological systems (SNS, HPA axis) in response to an acute stressor, there is an instant behavioral response, such as arousal, anxiety/fear, or aggression. This behavioral flexibility is regulated via activation of a number of brain regions, including cortical areas, limbic regions, and the brainstem (48–50). A region of particular importance is the lateral septal area, which is thought to segregate the autonomic, neuroendocrine, and behavioral responses (51). In humans, the behavioral (emotional) response to acute stressor exposure is an important measure of mental health. It is mainly quantified retrospectively via questionnaires (52–54) or by analyzing behavioral patterns known to be linked with distress during, for instance, public speaking (55–57). In laboratory animal models, a variety of behavioral tests have been established in order to quantify signs of arousal (e.g., measurement of homecage activity/locomotion), fear and anxiety-/social anxiety-related behavior [e.g., novelty-supressed feeding, shock pole burying, elevated plus-maze (EPM) test, light–dark box (LDB) test, open arm exposure test, open field test, social preference/avoidance test (SPAT), elevated platform (EPF) exposure, resident-intruder test, Vogel test, 4-plate test, marble burying, stress-induced hyperthermia, contextual/cued fear conditioning, acoustic startle], learning deficits (e.g., Morris water maze, Y/T-maze, holeboard, Barnes maze), anhedonia (e.g., sucrose preference test, progressive ratio responding, psychostimulant-induced hyperactivity, female urine sniffing test), memory skills (contextual-/cued fear conditioning), aggression (resident-intruder test), and active versus passive stress coping strategies [e.g., forced swim test (FST), tail suspension test (TST), learned helplessness] [for reviews dealing with these, and additional tests to assess such behaviors see (58–65)]. Similar to the physiological stress response, the behavioral stress response is strongly dependent on the time of day of stressor exposure (33), quality, intensity, and duration of the stressor (66–70), as well as the genetic and environmental background of the organism (41, 46, 47, 71–74).
Chronic Stress in Humans
Mal-adaptive consequences of chronic stressor exposure
While the acute stressor-induced changes described in the sections above are adaptive, chronic activation of the two stress systems poses an acknowledged risk factor for numerous disorders, including somatic disorders, like cardiovascular diseases [(75–79); for review see (80)], chronic fatigue syndrome (81), fibromyalgia (82), bronchial asthma (83, 84), atopic dermatitis [for review see (85)], arthritis [(86); for review see (87)], inflammatory bowel disease (IBD) [(13, 14, 16, 18, 20, 21); for review see (22, 25, 26)], stomach ulcers (86), diarrhea and digestive problems (86, 88), chronic pelvic and abdominal pain (86, 88), infections (86, 88–90), headaches (86, 88), impaired wound healing (91–93), cancerogenesis [(17); for review see (27, 94)], as well as affective disorders, like anxiety disorders and depression [(95–98); for review see (24, 58, 60, 99, 100)]. While the underlying etiology of these diseases are not fully understood, due at least in part to a lack of animal models, chronic stress-induced dysregulation of almost all psycho-neuro-immunological systems including the HPA axis, the autonomic nervous system, the immune and cardiovascular systems, and emotional and cognitive brain circuits is highly likely to contribute to the complex, and multifactorial, etiology of such disorders. On closer inspection, one mechanism that appears to be common throughout all of these diseases and chronic stress models is altered GC signaling.
The link between chronic stress and impaired GC signaling
Raison and Miller defined decreased GC signaling as “any state in which the potential of GC is inadequate to restrain relevant stress-responsive systems”. Such inappropriate GC signaling can be the result of decreased hormone bioavailability (hypocorticism), attenuated GC sensitivity/enhanced GC resistance of target cells, or the combination of both [for review see (101)]. Although HPA axis hyperactivity (hypercorticism) has been generally linked to prolonged, or chronic, stressor exposure, there is accumulating evidence for additional, even opposite alterations [for review see (23)]. In this respect, chronic stress-induced hypocorticism regained consideration after being more or less ignored up until the beginning of the 2000s. For example, Friedman and colleagues in the early 1970s described decreased plasma and urinary cortisol levels in parents of children suffering from neoplastic disease, with a paradoxical decrease during periods of heightened stress (102). Lower basal GC levels were further reported in high work load employees (103) and patients suffering from post-traumatic stress disorder [for review see (104)]. While elevated basal GC levels have been repeatedly linked to stress-related disorders like major depression [(105); for review see (100, 106)], the overall GC signaling in these patients has been shown to be decreased both in vivo and in vitro as a consequence of GC insensitivity [for review see (106, 107)]. Taken together, this growing body of evidence has led to greater acceptance of the idea that chronic stress experiences in adulthood result in an insufficient GC signaling.
In addition, chronic stress experienced early in life, like loss of parents, emotional neglect, maltreatment, or abuse have also been linked to a reduced GC signaling capacity in humans. In this context, it has been shown that women maltreated during early life exhibited lower basal and ACTH-induced plasma cortisol levels, an effect that was probably mediated by adrenal dysregulation (108, 109). However, whether the reduction in the overall GC signaling poses a central and causal mechanism by which chronic stress causes the variety of somatic and affective disorders described above is still unknown, but likely.
Many stress-related disorders are linked to a decrease in GC signaling
Although a causal involvement still has to be proven, as stated above, several chronic stress-related pathologies have been shown to be concurrent with reduced GC signaling in a growing number of studies. For example, hypocorticism has been described in patients suffering from burnout and chronic fatigue syndrome, fibromyalgia, chronic pelvic pain, and geriatric depression [(105, 110–112; for review see (23)]. Low levels of plasma GC have been further reported when suffering from inflammatory disorders, including rheumatoid arthritis [for review see (113)] or asthma (114). In line with this, elevated levels of pro-inflammatory cytokines have been reported in patients suffering from acute GC deficiency after surgical removal of adrenal cortical tissue (115). Moreover, it has recently been shown that obese women have lower cortisol levels during pregnancy (116). Interestingly, based on human and animal studies, it has been hypothesized that the onset of IBD might be associated with hypo- rather than hypercorticism [for review see (117, 118)]. This is further supported by a recent finding showing an impaired HPA axis reactivity in 25% of Crohn’s patients during exposure to the ultra-low dose ACTH test (119). In addition, a positive correlation between plasma cortisol levels and the time patients were off steroid treatment has recently been described (120). Finally, Rodriguez and coworkers speculated that a down-regulated cortisol response to intero- and exteroceptive stressors might predispose patients suffering from irritable bowel syndrome to chronic inflammatory conditions, such as asthma, rheumatoid arthritis, or IBD (121).
Besides hypocorticism, GC resistance has been speculated to contribute to the reduced GC signaling and the pro-inflammatory immune shift in patients suffering from chronic stress-related pathologies (122). As mentioned above, the disorder that best fits this context is major depression [for review see (101)], as patients show a reduced response to GC both in vivo and in vitro [for review see (106, 107)], which is believed to be mediated, at least in part, by decreased GC receptor expression and/or functionality [(123–125); for review see (107)]. GC resistance has further been diagnosed in a subset of patients suffering from typically chronic inflammatory disorders like ulcerative colitis and Morbus Crohn [(126); for review see (127)], as well as rheumatoid arthritis (128).
To causally demonstrate that chronic psychosocial stress promotes the development of, at least some, somatic and affective disorders via a reduction in overall GC signaling, it is necessary to have appropriate animal stress models, which mimic the human situation in an adequate way. Thus, animal models are warranted that are of chronic psychosocial nature, to show face validity, and cause both somatic and affective disorders, as well as result in a reduced GC signaling (ideally both hypocorticism and decreased GC sensitivity), to provide predictive validity. Given that the vast majority of somatic and affective disorders are multifactorial diseases, for which the underlying etiological factors are only poorly understood, most of the animal models fail to satisfy construct validity. However, if insufficient GC signaling is indeed causally involved in the development of many such diseases, animal models resulting in either hypocorticism or a decreased GC sensitivity, or both, could be considered primarily as models displaying construct validity.
In the following paragraphs, we will in detail describe the chronic subordinate colony housing (CSC) paradigm, which fulfills all the criteria outlined above and, thus, represents an adequate and preclinically validated (face and predictive validity) animal model to investigate the underlying mechanisms related to chronic psychosocial stress-induced impaired GC signaling and its involvement in somatic and affective disorders.
Chronic Subordinate Colony Housing
General Description and Experimental Details
The CSC paradigm combines chronic, psychological, and social aspects of stress and, thus, represents a highly potent animal model to mimic the type of health compromising stressors faced by humans (high face validity). CSC further promotes development of both somatic and affective disorders, results in a reduced GC signaling (high predictive/construct validity) and, thus, provides a powerful experimental tool to study the mechanisms underlying several relevant stress-induced pathologies. Notably, given that the CSC paradigm is simply based on the fact that male mice instinctively establish a certain hierarchical structure within their colony, it additionally resembles the natural way of life of a male mouse in the wild [(129); for review see (9)]. However, before we detail the physiological, immunological, and behavioral consequences of the CSC paradigm unraveled to date, we will briefly introduce the experimental details of this chronic psychosocial stress model.
During CSC (for details see Figure 1), experimental mice (CSC mice) live in chronic subordination to a dominant resident mouse for 19 consecutive days (130). In detail, four CSC mice are put into the homecage of a larger male resident mouse on day 1 of the CSC paradigm, resulting in immediate subordination of the four intruder CSC mice. To avoid habituation, all four CSC mice are transferred into the homecage of a novel larger male resident mouse on days 8 and 15.
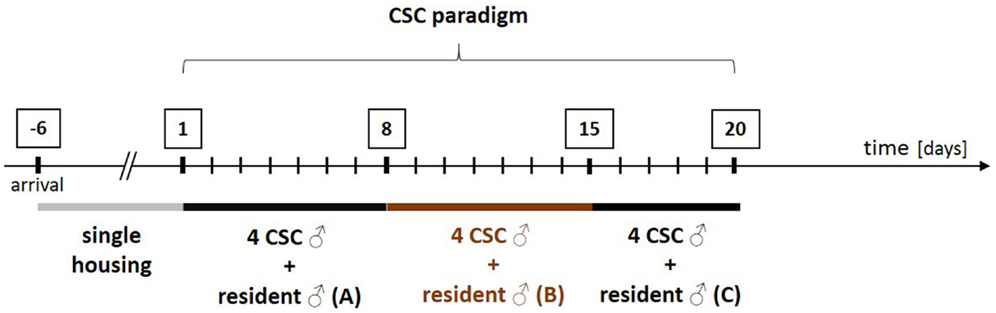
Figure 1. Schematic illustration of the experimental design of the chronic subordinate colony housing (CSC) paradigm. After arrival, all experimental male mice are housed singly for 1 week before they are assigned to the single-housed control (SHC) or the CSC group in a weight matched manner. In order to induce chronic psychosocial stress, CSC mice are housed together with a larger dominant male for 19 consecutive days. In detail, four experimental CSC mice are introduced into the homecage (polycarbonate observation cage; 38 cm × 22 cm × 35 cm) of resident A on day 1 of CSC, resulting in immediate subordination of the four intruder CSC mice. The latter are then housed together with this dominant resident (A) for eight consecutive days. On day 8, and again on day 15, of CSC, the four experimental CSC mice are transferred into the homecages of resident B (day 8) and resident C (day 15) to avoid habituation. Before the CSC procedure, the future dominant males are tested for their aggressive behavior. Males that start to injure their opponents by excessive and harmful bites during testing are not used.
All resident males are tested before CSC housing for their aggressive behavior and males that injure their opponents by excessive aggression (e.g., harmful bites) are not used. Notably, although this procedure strongly reduces the number of bite wounds delivered by the residents during CSC exposure, it does not 100% prevent them. As a matter of routine, the subordinate position of each CSC mouse is confirmed by behavioral analysis of the first 30 min after setting up the CSC colonies on days 1, 8, and 15 (131). Resident males reliably (>99% of CSC colonies) obtain the dominant position by displaying offensive behaviors toward the CSC mice, such as chasing, mounting, or attacking their four cage mates (131). In contrast, CSC mice can be considered as “subordinates” based on their defensive behaviors, including flight and submissive upright (131). So far, the CSC model has reproducibly been shown to work in different mouse strains, namely, C57BL/6 mice (130), BALB/c mice (132), and CD1 mice (41). Moreover, CSC effects are independent from the background of the residents, as the physiological, immunological, and behavioral consequences of the CSC paradigm are comparable using either C57BL/6 (130) or the male offspring of CD1 female mice [bred at the Max Planck Institute of Psychiatry in Munich (Germany) for high anxiety-related behavior (HAB mice)] and male C57BL/6 mice as dominant animals (133, 134). Recent own unpublished data reveal that using male CD1 mice as residents allows reliable reproduction of known CSC effects (see Figure 2).
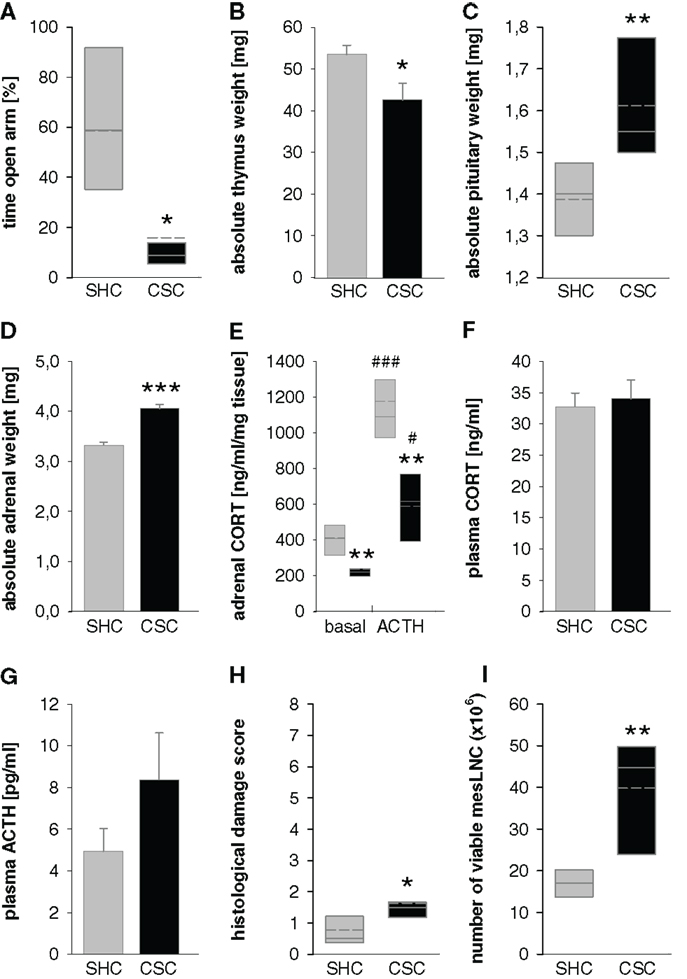
Figure 2. Effects of 19 days of CSC exposure on behavioral, physiological, and immunological parameters. Dominant resident male mice originated from the CD1 strain, CSC mice from the C57BL/6 strain. CSC exposure resulted in increased anxiety-related behavior measured on the elevated plus-maze [EPM; (A)] without affecting locomotor activity (data not shown), decreased thymus weight (B), increased pituitary weight (C), increased adrenal weight (D), reduced adrenal in vitro ACTH sensitivity (E), unaffected basal morning plasma corticosterone [CORT; (F)], a tendency toward increased basal morning plasma adrenocorticotropic hormone [ACTH; (G)], increased histological damage in colonic tissue (H), and increased number of viable mesenteric lymph node cells [mesLNC; (I)].  SHC (n = 7–8); ■ CSC (n = 7–8). Parametric data are represented as mean + SEM. Non-parametric data are represented in box-plot diagrams. Boxes signify the upper and lower quartiles, the median is represented as solid line, and the mean as dashed line within each box. *P < 0.05, **P < 0.01, ***P < 0.001 versus respective SHC; #P < 0.05, ###P < 0.001 versus respective basal values.
SHC (n = 7–8); ■ CSC (n = 7–8). Parametric data are represented as mean + SEM. Non-parametric data are represented in box-plot diagrams. Boxes signify the upper and lower quartiles, the median is represented as solid line, and the mean as dashed line within each box. *P < 0.05, **P < 0.01, ***P < 0.001 versus respective SHC; #P < 0.05, ###P < 0.001 versus respective basal values.
An important issue for the design of chronic psychosocial stress paradigms is the choice of adequate same-aged controls, with single housing [single-housed control (SHC) mice] or group housing [group-housed control (GHC) mice] being widely used options. For CSC experiments, we employ SHC mice based on own data indicating that group housing per se poses a stressful condition for male mice. Surprisingly, similar physiological and behavioral alterations after 3 weeks of GHC or CSC were observed, leading us to believe that the novel hierarchy formed by GHC mice is as almost as stressful as being subordinated by a dominant resident. For example, lower body weight gain and increased state anxiety were found in both CSC and GHC compared with SHC mice (135). In detail, the number and time of head dips and distance traveled on the open arm of the EPM were reduced in both CSC and GHC compared with SHC (135) mice; parameters related to risk assessment, anxiety, exploratory (136), and locomotor behavior (137), respectively. Given that isolation has been shown to lack effects on stress-related immune and/or endocrine functions in male mice by other stress laboratories (138, 139), single housing seems to be the most appropriate non-stressful control condition in non-sibling male mice. In line with this, Blanchard et al. [for review see (140)] and Palanza [for review see (141)] proposed that isolation is more stressful for female mice, while social grouping is more stressful for male mice. In males, any kind of group housing is likely to be accompanied by the establishment of subtle hierarchies with the result that in each cage dominant and more or less subordinate cage mates can be found (135).
CSC-Induced Consequences
Endocrine changes
Adrenal gland, pituitary, and acute stress reactivity. The CSC paradigm has been shown to result in profound and reproducible physiological changes, including a significant (41, 46, 130, 133, 142) and long-lasting (at least until day 8 after termination of CSC) (70) enlargement of the adrenal glands. This increase in absolute adrenal mass is mediated by cell hyperplasia (133), without alterations in adrenal cholesterol delivery pathways [cortical lipid droplets; protein expression of hormone-sensitive lipase, 3-hydroxy-3-methylglutaryl coenzyme A reductase, and low-density lipoprotein receptor, with the exception of the scavenger receptor class B type 1 protein, which was increased following CSC exposure (133)]. Notably, in an adrenocortical cell the CORT precursor molecule cholesterol can among others be derived from (i) hormone-sensitive lipase-mediated hydrolyzation of cholesteryl esters, stored in lipid droplets within the cytoplasm of mostly zona fasciculata cells [for review see (143)], (ii) hormone-sensitive lipase-mediated hydrolyzation of cholesteryl esters “selectively” taken up from high-density lipoproteins and low-density lipoprotein via the scavenger receptor class B type 1 [(144, 145); for review see (146–148)], (iii) lysosomal acid lipase-mediated hydrolyzation of cholesteryl esters from low-density lipoprotein taken up endocytotically via the low-density lipoprotein receptor (149), and (iv) endogenous de novo synthesis from acetyl coenzyme A via the 3-hydroxy-3-methylglutaryl coenzyme A reductase [for review see (150)]. Thus, given the elevated adrenal weight following 19 days of CSC, the molecular and cellular changes reported above in CSC versus SHC mice at the level of the adrenal glands strongly suggest an enhanced adrenal availability and/or mobilization capacity of the CORT precursor molecule cholesterol and, consequently, an increased adrenal functionality in CSC versus SHC mice. In line, analysis of plasma high-density lipoprotein cholesterol and low-density lipoprotein cholesterol revealed increased levels of the latter in CSC versus SHC mice. Similarly, a comparable or even increased relative expression of melanocortin 2 receptor protein and melanocortin 2 receptor accessory protein mRNA, as well as of steroidogenic acute regulatory protein mRNA, side-chain cleavage enzyme mRNA, 11β-hydroxylase and aldosterone-synthase mRNA – the latter enzymes are known to be essential in the progress of CORT synthesis from its precursor cholesterol [for review see (151, 152)] and to be controlled by ACTH signaling [for review see (153, 154)] – support the idea of an overall increased adrenal functionality following CSC exposure.
In confirmation of this hypothesis, mice exposed to 19 days of CSC indeed show exaggerated plasma CORT concentrations, as well as an increased adrenal CORT content, when killed 5 min following termination of a mild acute heterotypic stressor (EPF) at the beginning of the light phase, despite the increase in plasma ACTH concentrations not differing from EPF-exposed SHC mice (142). Notably, when exposed to a more severe acute heterotypic stressor, i.e., 6-min of forced swimming, CSC mice even show an exaggerated ACTH response compared with SHC mice (155), likely to further enhance HPA axis reactivity toward a novel and severe enough heterotypic challenge in CSC mice. A facilitated ACTH response to a novel heterotypic challenge is thereby in line with other chronic stress studies [(156); for review see (157)].
In line with what we found at the adrenal level, the increased capability of the CSC pituitary gland to produce and secret ACTH is mediated at least partly by corticotroph cell hyperplasia (155). Non-compromised functionality of those newly formed cells was suggested by comparable relative pituitary pro-opiomelanocortin protein expression between CSC and SHC mice (155). Interestingly, the idea that during conditions of prolonged/chronic stress AVP becomes the main pituitary ACTH secretagogue [for review see (157)] is supported by unaltered relative pituitary AVP1b receptor and decreased CRH receptor 1 protein expression in CSC versus SHC mice (155). Taken together, these data suggest that these newly formed corticotrophs shift their sensitivity from CRH to AVP. Increased AVP output at the level of the PVN – as suggested by other studies dealing with repeated/chronic stressor exposure [for review see (157)] – does not appear to enhance pituitary AVP stimulation and, thus, to contribute to the increased ACTH drive in CSC mice, as the number of AVP positive parvocellular PVN neurons is comparable between CSC and SHC mice (155). In line, mRNA expression of AVP is even lower in the PVN of CSC versus SHC mice (131), while CRH mRNA is not affected (130). Furthermore, neuronal activation in the parvocellular PVN (predominantly AVP and CRH neurons) is lower in CSC versus SHC mice following acute heterotypic stressor exposure (open arm; 5 min) (135). Similarly, the contributing role of changes in pituitary negative feedback inhibition to the increased ACTH secretion in CSC mice seems to be negligible, as the dexamethasone suppression test indicated a fully functional feedback system (155). Notably, the latter finding clearly indicates that a decrease in pituitary cytoplasmic GC receptor protein expression, as seen in CSC versus SHC mice (155), cannot generally be interpreted as an impairment of negative feedback function. This is of considerable importance for the stress field in general, as in many published studies this was common practice.
In addition to the above described data assessed on day 20 of CSC (after 19 days of CSC), a time course analysis revealed that relative adrenal mass was significantly increased in stressed mice at all time-points assessed (24, 48 h, days 7, 14, 20) during the 19 days of CSC exposure (130). A more recent study, confirming the increase in relative adrenal weight following 48 h of CSC, extended these findings to demonstrate that even 10 h of CSC are sufficient to cause such changes (134). However, in contrast to relative weight, absolute adrenal weight during this initial phase of CSC was increased after 10 h, but not 48 h, of CSC exposure. Considering the reduction in body weight at both these time-points (70, 132, 134), this clearly indicates that the increase in relative adrenal weight observed following 48 h of CSC is exclusively due to changes in body weight and not to changes at the adrenal level per se. Given the reliable increase of absolute adrenal mass following 19 days of CSC described earlier (41, 46, 130, 133, 142), these data for the first time indicate that the adrenal glands of an organism exposed to chronic psychosocial stress enlarge during the very initial phase of stressor exposure, normalize after about 48 h of continuous challenge and, given the stressor still persists, start to enlarge again.
Basal plasma CORT, ACTH and noradrenaline, and GC signaling. Interestingly, these changes in absolute adrenal weight during the initial phase of CSC seem to run in parallel with the fluctuations of basal morning plasma CORT levels. Following 10 h of CSC exposure, plasma morning CORT (132, 134, 158), as well as absolute adrenal mass (134), are significantly increased, whereas following 48 h both parameters return to baseline values. Thus, it seems that reversing the early increase in adrenal mass in CSC mice (10 h) poses some kind of adaptive mechanism, contributing, together with a reduction of stimulatory adrenal input (ACTH) from the pituitary and a possibly increased CORT metabolism (134), to prevent the organism from prolonged exposure to elevated plasma CORT concentrations. This is supported by studies showing a positive correlation between plasma CORT and adrenal weight under stress conditions in rats (159, 160). Chronically elevated CORT concentrations are known to have deleterious health consequences [(161); for review see (162–164)] and to cause increased anxiety- and depressive-like behavior in rats. Moreover, as greater insights can be obtained from rodent studies, chronically high CORT levels have also been shown to affect the brain serotonergic system (165), as well as to rapidly and dramatically increase body weight gain, adiposity, plasma leptin, insulin, and triglyceride levels, and also to decrease homecage locomotion (166) when delivered via the drinking water. However, future studies are needed to clarify whether these early adaptive changes in absolute adrenal weight during CSC exposure are mediated by hyper-/hypotrophy or by hyperplasia/apoptosis of adrenal cells.
Interestingly, although absolute adrenal mass increases again subsequent to the 48 h time point, plasma morning CORT concentrations on days 7, 14, 20 stay still comparable to those of SHC mice (41, 46, 130, 142). This suggests a mechanism different from the one involved during the initial phase of chronic stressor exposure to prevent the deleterious consequences of hypercorticism in the later phases of chronic stressor exposure. Given that isolated adrenal cells (130), as well as adrenal explants (142), from mice exposed to 19 days of CSC show a reduced in vitro CORT release when treated with different ACTH doses, it is likely that this is implemented via a reduced ACTH sensitivity of cortical adrenal cells. Notably, adrenal ACTH sensitivity seems to be not only diminished under in vitro conditions, as unaffected basal morning plasma CORT in 19-day CSC mice is paralleled by elevated plasma ACTH in comparison with SHC mice (41, 155). Support for this second mechanism to play a role only after prolonged stressor exposure and not to contribute to the initial normalization of plasma CORT is provided by our finding that adrenal in vitro ACTH sensitivity of CSC mice was not different from SHC at the 10 and 48 h time point (134).
While the reduction in adrenal ACTH sensitivity in the presence of increased absolute adrenal mass and plasma ACTH seems to ensure normal basal morning CORT concentrations, it is likely to promote the basal evening hypocorticism detected in 19-day CSC mice (130). SHC mice were able to show the expected (167, 168) diurnal rise in plasma GC concentrations at the beginning of their active period, whereas CSC mice were not and, thus, had lower plasma CORT concentrations than respective SHC mice in the evening of day 20 of CSC (130). The resulting decline in GC signaling was further amplified by a CSC-induced reduction in GC sensitivity. The latter was described in both lipopolysaccharide-stimulated splenocytes (130) and plate-bound anti-CD3-stimulated T helper (Th) 2 cells from peripheral lymph nodes (169) of 19-day CSC compared with SHC mice. Thus, given the accumulating evidence that a reduction in GC signaling might be involved in the development of somatic and affective disorders linked with an inflammatory component [for review see (101, 170–172)], it is likely that adrenal changes seen during CSC exposure, although preventing the negative consequences of prolonged hypercorticism, contribute to the development of spontaneous colitis (130, 158, 173), hepatic inflammation (174), increased anxiety-related behavior (41, 46, 70, 130, 131, 135, 142, 173, 175), hyperactivity (70), and the increased risk of inflammation-related colorectal cancer (CRC) (176). In support, we previously showed additive effects of early life stress (repeated maternal separation) and CSC exposure on both the development of hypocorticism and on the severity of a chemically induced colitis (46). To assess, adrenalectomy with CORT replacement needs to be performed to see what CSC-induced behavioral and physiological effects remain.
Notably, as already discussed above in detail, this decreased adrenal ACTH sensitivity is not preventing CSC mice from showing an exaggerated CORT response to subsequent EPF exposure, suggesting an additional, yet unknown, factor that is enhanced in CSC mice during acute heterotypic stressor exposure, unaffected by the diurnal rhythm, which rescues the attenuated adrenal ACTH responsiveness. For example, sympathetic innervation of the adrenal medulla via the splanchnic nerve is known to play a critical role in modulating adrenocortical sensitivity to ACTH (177–179). Following activation, adrenal medullary cells secrete neurotransmitters and neuropeptides such as adrenaline/noradrenaline, neuropeptide Y, vasoactive intestinal peptide, or substance P, which may, in a paracrine manner [for review see (180, 181)], influence adrenocortical CORT secretion. Moreover, neuropeptides such as prolactin and oxytocin (OXT), which are released during various types of acute stressor exposures [(39,182); for review see (183)], act as direct CORT secretagogues (184–186). Therefore, instead of rescuing ACTH signaling, it is also possible that this unknown factor is a CORT secretagogue itself, thereby simply replacing ACTH in the process of adrenal activation during heterotypic stressor exposure. However, future studies are required to elucidate the identity of this currently unknown determinant.
In contrast to the reduction of basal adrenal cortex function, increased basal plasma noradrenaline concentrations following CSC (130) indicate an over-activated adrenal medulla and, thus, uncoupling the activity of the HPA axis and the SNS during chronic psychosocial stressor exposure. As the concerted action of steroid hormones and neurotransmitters of the SNS is crucial for optimal immunosuppression, uncoupling of the HPA axis and the SNS is likely to further promote pro-inflammatory processes (187). Thus, future studies focusing on the role of the SNS in CSC-induced pathology are warranted.
Summary. In summary, exposure to CSC initially (10–24 h) triggers a pronounced HPA axis response, resulting in increased absolute adrenal mass and elevated basal morning plasma GC concentrations. Following 48 h of continuous CSC exposure, basal morning GC concentrations return to basal levels again, mediated most likely by a combination of decreased stimulatory input from the pituitary, enhanced CORT metabolism, and restoration of normal adrenal mass. Interestingly, during further stressor continuation, the recurrence of rising absolute adrenal mass is not paralleled by increased (morning), but rather decreased (evening) basal plasma GC concentrations, mediated at least partly via a pronounced reduction in adrenal ACTH responsiveness.
Body weight changes
While decreased body weight gain has been reported in many studies investigating the effects of repeated/chronic stressor exposure (67, 188–191), other studies have reported no alteration in body weight development (192–195). In line, the effects of CSC exposure on body weight development are not fully consistent, resulting in either decreased (130, 131, 135, 169, 173, 176) or unaffected body weight gain (41, 46, 70). Therefore, while CSC seems to reliably diminish body weight gain during the initial phase of CSC exposure (46, 70, 131, 132, 134), this in some sets of mice normalizes over the final days of stressor exposure. Notably, CSC mice further gained significantly more weight in the week after stressor termination than unstressed SHC controls leading to a normalization or even increased bodyweight of CSC versus SHC mice (70). Similar findings have previously been reported following subjection to the visible burrow system (196) and repeated social defeat (194) and may represent a general phenomenon following prolonged stressor exposure. This increase in body weight after termination of chronic stress may be an adaptive mechanism for ensuring sufficient resources in preparation for subsequent stressful events. Together, these data emphasize the necessity to further investigate the link between repeated/chronic stressor exposure and changes in body weight and, suggest that caution should be exhibited when interpreting a lack of reduced body weight gain as a sign of a non-effective chronic stress paradigm.
Somatic disorders
In addition to the consequences on endocrine parameters and body weight development, CSC represents an established model to study the immunological consequences of chronic psychosocial stress exposure. In agreement with other chronic social stress paradigms (188, 189, 197–201), CSC causes thymic involution, first detected after 24 h (130), which is in line with the thymus atrophy reported in rats following 24 h of resident-intruder confrontations (189, 201).
Inflammation. Interestingly, and again in line with others (198, 202, 203), CSC causes splenomegaly (41) and reduced in vitro GC sensitivity in isolated and lipopolysaccharide-stimulated splenocytes (130). Given that this is paralleled by pronounced immune activation in the social disruption (SDR) paradigm (203–205), it is very likely that the systemic immune status of CSC mice is enhanced as well. GC resistance of IL-4 producing Th2 cells, a reduced number of regulatory T cells, and an increased T cell effector function, all detected in peripheral lymph nodes following 19 days of CSC exposure (169) support this idea. Moreover, CSC mice show higher hepatic tumor necrosis factor alpha, monocyte chemotactic protein 1, and heme oxygenase mRNA expression, indicating noticeable oxidative stress and hepatic inflammation (174), and develop a more severe colitis when subsequently treated with dextran-sulfate sodium (DSS, 1%, 7 days) (46, 173). The latter was indicated by increased body weight loss, inflammatory reduction of colon length, and histological damage score in CSC versus SHC mice after 8 days of DSS treatment.
Interestingly, unlike SHC, CSC mice already on the second day of DSS treatment demonstrate an increased cytokine secretion from isolated and plate-bound anti-CD3-stimulated mesenteric lymph node cells (173), suggesting chronic subordination itself to trigger the development of a colonic inflammation. In support, stimulated cytokine secretion from isolated mesenteric lymph node cells is increased in non-DSS treated CSC mice 8 days following stressor termination (173). Finally, confirming chronic subordination-induced spontaneous colitis, CSC mice display an increased histological damage score in the colon – first detectable after 14 days of CSC (130), number of colonic macrophages, dendritic, and Th cells (158), and cytokine secretion from in vitro stimulated mesenteric lymph node (130) and lamina propria mononuclear cells (158). Notably, comparable to the CSC paradigm, a modified version of the SDR paradigm (202–204, 206) has recently been shown to also cause mild histological colonic damage in male mice (195).
Based on the absent CORT increase in the plasma of CSC mice on the second day of DSS treatment – despite increased in vitro stimulated cytokine secretion from mesenteric lymph node cells at this time – we recently hypothesized that CSC-induced adrenal insufficiency contributes to the increased severity of DSS-induced colitis [for review see (118)]. In contrast, increased cytokine secretion from in vitro stimulated mesenteric lymph node cells of SHC mice was first detected on the eighth day of DSS treatment, and immediately paralleled by high plasma CORT concentrations (173). However, the adrenal hyper-reactivity toward heterotypic stressors in CSC versus SHC mice clearly argues against a general break down of adrenal functioning in CSC mice. Thus, it is rather likely that cytokine levels secreted from mesenteric lymph node cells in vivo during DSS treatment were not high enough to spill over into the systemic circulation and, thereby, activate the HPA axis until day 4 of DSS treatment in both CSC and SHC mice. In turn, a more pronounced plasma CORT increase in CSC versus SHC mice on day 8 of DSS treatment suggests elevated systemic cytokine levels in both groups, and indicates HPA axis hyper-reactivity in CSC mice also to occur in response to heterotypic stressors of an inflammatory nature, given the assumption that inflammation is severe enough to activate the HPA axis.
Support for decreased basal GC signaling – caused by basal hypocorticism and/or GC resistance (130, 169) – to promote CSC-induced aggravation of DSS-induced colitis is provided by the finding that the combination of early-life stress (maternal separation, MS; 3 h/day, from postnatal day 1–14) and 19 days of CSC during adulthood has additive effects on DSS-induced colitis. In contrast to CSC mice, which are only unable to adequately increase plasma CORT at the beginning of the dark/active phase, mice exposed to both MS and CSC suffer from hypocorticism even during the morning hours (46).
With respect to the mechanisms underlying the development of CSC-induced spontaneous colitis, assessment of several functional levels of the colon following the initial stress phase (10 h of CSC) revealed a pronounced, adrenal hormone-mediated, local immune suppression in colonic tissue; probably allowing luminal- and translocated-bacteria to proliferate without constraint (158). Immune suppression was indicated by a reduced cytokine and immunoglobulin A secretion from isolated and anti-CD3/IL-2-stimulated lamina propria mononuclear cells, a decreased percentage of CD3+ cells within all isolated lamina propria mononuclear cells, a decreased pro-inflammatory colonic cytokine mRNA expression, and a lower number of F4/80+ macrophages, CD11c+ dendritic cells, CD3+ T cells, and CD4+ Th cells in colonic tissue of CSC compared with SHC mice. Whether or not this effect is mediated by cortical GC or medullary catecholamines still needs further investigation. The early decrease in colonic IgA secretion in combination with an obsolescent mucosa, indicated by reduced epithelial cell proliferation and apoptosis, additionally suggested the initiation of impaired epithelial barrier functions (158). In line, 10 h of CSC resulted in a reduced/deficient mucus production of colonic epithelial cells. Surprisingly, and in contrast to early CSC-induced immune suppression, our data clearly indicated that the reduction in epithelial barrier functions was not mediated by adrenal hormones. Given that intact local immune and epithelial barrier functions are essential for the control of commensal flora, it was not surprising to detect an increased bacterial load in colonic tissue and in stool samples from CSC mice following 10 h of stressor exposure. Furthermore, experiments employing prolonged antibiotic treatment have revealed a causal role of such bacterial translocation/proliferation during the initial phase of CSC in the initiation/induction of colonic inflammation (158). However, using adrenalectomized mice, we showed that the immunosuppressive effects of high levels of adrenal hormones during the initial CSC phase were required to develop a moderate colitis into a full-blown form (158). Direct evidence showing that the over-active immune system in the later stages of CSC, i.e., when hypocorticism (46, 130) and GC resistance (130, 169) have developed, targets this elevated presence of bacterial antigens in the colonic tissue of CSC mice, leading to the observed colitis (130, 158), still needs to be provided.
Inflammation-related colon carcinogenesis. Given that chronic stress is an acknowledged risk factor for numerous disorders, including IBD [(13, 14, 16, 18, 20, 21); for review see (22, 25, 26)] and cancer [(17); for review see (27)], and that CRC poses one of the most serious complications in IBD patients [(207); for review see (208, 209)], it is not surprising that CSC, besides causing spontaneous colitis (130, 158), also increases the risk for inflammation-related CRC. Combining a novel colitis-related CRC mouse model – in which CRC is initiated with azoxymethane and promoted by repeated cycles of DSS administration (210) – with CSC exposure, revealed that CSC mice show accelerated development of macroscopic suspect lesions, as well as a trend toward an increased incidence of low- and/or high-grade colonic dysplasia (176). Although only a small fraction of these polyps may finally become malignant, there is evidence indicating that a large majority of colorectal carcinomas develop from these adenomatous polyps (211). Similarly, humans who develop severe dysplasia in adenomas are considered to be at increased risk of developing cancer (211). CSC mice further showed an increased number of Ki-67+ and a decreased number of TUNEL+ colonic epithelial cells, indicating abnormal patterns of cell replication, as detected in several clinical conditions associated with an increased risk for colorectal malignancies [for review see (212)]. A reduction in epithelial cell apoptosis already following 10 h of CSC (158), thereby, indicates that this effect is fast in onset and, hence, likely to be causally involved in CSC-induced promotion of azoxymethane/DSS-induced CRC. The latter was further indicated by increased colonic mRNA and/or protein expression of liver receptor homolog-1, β-catenin, cyclooxygenase II, and tumor necrosis factor alpha in CSC compared with SHC mice. Both liver receptor homolog-1 (213) and β-catenin (214) are involved in the control of intestinal cell renewal, and known to be involved in gastrointestinal tumor development (215–217). The same is true for cyclooxygenase II, which modulates apoptosis, angiogenesis, and tumor invasiveness [for review see (218)] and is over-expressed in approximately 80% of CRC and 40% of colorectal adenomas relative to normal mucosa (219). Tumor necrosis factor has been shown to promote signaling via the β-catenin pathway, thereby contributing to tumor development in the gastric mucosa (220).
Interestingly, a shift from protective Th cells to regulatory T cells was recently hypothesized to mediate the increased susceptibility of mice to UV-induced skin cancer following repeated immobilization (6 h/day over 3 weeks) (12). Similarly, increased regulatory T cell infiltration into the tumor bed, predicted reduced survival in cancer-bearing patients [for review see (221)]. Therefore, development of GC resistance in Th2-, but not Th1-, cell subpopulations during 19 days of CSC (169), causing a potential down-regulation of tumor protective Th1 immunity during repeated post-CSC DSS cycles (heterotypic immune stressors; for details see before), might be involved in CSC-induced CRC progression. An increased number of colonic CD4+ Th cells and percentage of CD3+ mesenteric lymph node cells in CSC versus SHC mice, but a decreased colonic interferon-γ mRNA expression coupled with an unaltered interferon-γ secretion from stimulated mesenteric lymph node cells support this hypothesis. An increased colonic FoxP3 mRNA expression, as well as number of CD3+/Foxp3+ double-positive mesenteric lymph node cells, following CSC further suggests enhanced immune regulation. Together with the above described reduction in regulatory T cell counts in peripheral lymph node tissue immediately following termination of CSC (169), these data either suggest tissue specificity of CSC effects or that regulatory T cell numbers normalize and even increase gradually following CSC, ameliorating CSC effects on subsequent inflammatory episodes (repeated DSS treatment) but promoting those on CRC development. Notably, body weight development of CSC and SHC mice during second and third DSS cycles is comparable, indicative of an equally severe colitis, whereas three CSC but no SHC mice were dying off too severe colitis during the first DSS cycle.
Affective disorders
Hyperactivity. Humans exposed to severe stressors are at increased risk for developing affective disorders, including post-traumatic stress disorder, which is characterized by pronounced and long-lasting hyperarousal, among other symptoms [for review see (222)]. A link between stress and hyperactivity is also suggested by studies revealing that prenatal stress in humans is associated with attention deficits, hyperarousal, and hyperactivity during childhood [(223); for review see (224)]. Poor school and social functioning, behavioral problems, and parental conflicts, all representing chronic psychosocial stressors, are further well-known factors predicting persistence of childhood attention deficit hyperactivity disorder into adolescence and adulthood (for review see 224). Moreover, bipolar disorder, which affects between 1–3% of the population, is characterized by a cycling between depressive episodes and periods of overactivity, termed mania. Given this strong overactivity component, it is not surprising that the majority of animal models used to study mania have focused on manipulations leading to hyperactivity, e.g., psychostimulant-induced hyperlocomotion [for review see (60, 225, 226)]. In contrast, studies employing animal models of repeated/chronic stress more or less consistently report a stress-induced reduction in locomotor activity, both in the homecage (33, 227) and in a novel environment during behavioral testing (67, 188, 200, 228, 229). Notably, while locomotor activity during behavioral testing (e.g., EPM) should ideally be dissociated from the anxiety state of the animal, altered locomotion is a general confound in such tests. For instance, reduced locomotion during EPM testing of rats following a single cat exposure might also be interpreted as reduced exploration due to increased levels of predator-induced anxiety-related behavior (230). Moreover, highly-anxious rodents (rats bred for high-anxiety-related behavior, HAB rats) show a gender-independent decrease in the number of line crossings in the dark compartment during LDB testing compared with their respective low anxious counterparts (rats bred for low-anxiety-related behavior, LAB rats) (231). Given that genotype specific differences in anxiety-related behavior between these breeding lines have been convincingly shown in locomotion-independent (i.e., ultrasound vocalization) anxiety tests, this indicates that reduced locomotion may be one characteristic of anxious animals [for review see (232)]. This is further indicated by the fact that locomotion in the open field has been used not only as an index of general locomotor activity or exploratory behavior but also as index of anxiety [as referenced in (233)].
Assessment of homecage locomotion before, immediately after, and 1 week after CSC stressor exposure (70) confirmed the expected increase in locomotor activity at the beginning of the dark phase in both SHC and CSC mice prior to stress. While this increase is not seen immediately following CSC exposure, it is even more pronounced in CSC versus SHC mice 1 week later, indicating a long-lasting induction of dark phase hyperlocomotion/hyperactivity. Thus, the CSC paradigm poses one of the few animal models, which might help unraveling the mechanisms underlying stress-promoted hyperactivity.
State anxiety. Chronic psychosocial stressors have also been shown to reliably increase state anxiety in rodents (66, 67, 188, 200, 228, 229, 234) and to be a risk factor for anxiety disorders in humans [for review see (58)]. In keeping, CSC results in a profound and robust increase in state anxiety, which has been confirmed in at least five independent behavioral tests. In detail, exposure to 19 days of CSC reduces the time spent on the open arms of an EPM (130, 131, 173), specifically their distal parts during open arm exposure (135), as well as the time spent in the lit compartment of a LDB (131, 175). Moreover, CSC mice enter the central zone of an OF arena less often, explore novel objects less intensely during a novel object test (46, 70), and spend less time in the outer zone of a platform during EPF exposure (142). Importantly, in a recent study, we further described that CSC mice spent less time on the open arms of an EPM 4 and 8 days after stressor termination, indicating that the stressor-induced change in emotionality is a long-lasting phenomenon (70). With regard to the potential influence of the CSC-induced hyperlocomotion on the interpretation of these tests, they were performed in the early light phase, when home-cage locomotion is not affected. Moreover, no difference in locomotion parameters, such as closed arm entries or distance traveled, were observed between SHC and CSC mice. Therefore, the anxiogenic effect of CSC is robust and long lasting.
Given the anxiogenic effect of CSC exposure and that individuals vary in their response to chronic stressor exposure (234–237), in a recent study, we tested whether the genetic predisposition for high versus low anxiety-related behavior determines the vulnerability to CSC. Interestingly, and in line with our hypothesis, HAB CD1 mice and CD1 mice not selected for anxiety-related behavior (NAB) are equally vulnerable to the CSC-induced behavioral, physiological, neuroendocrine, and immunological effects, whereas CD1 mice bred for low-anxiety-related behavior (LAB) are found to be stress resilient (41). The latter is indicated by the fact that all stress-related parameters, including anxiety-related behavior, are comparable between CSC and SHC mice in the LAB group. In contrast, in both HAB and NAB genotypes, CSC results in an increased adrenal weight, a reduced adrenal in vitro ACTH responsiveness substantiated by a lower plasma CORT:ACTH ratio, and an enhanced pro-inflammatory cytokine secretion from isolated and stimulated mesenteric lymph node cells compared with respective SHC mice. Notably, the CSC-induced increase in anxiety described before in C57BL/6 mice (as referenced above) was only detectable in the NAB group, probably due to a ceiling effect in the anxious HAB line.
Social anxiety. Social anxiety disorder with a lifetime prevalence of 12.1% (238), is the “persistent fear of one or more situations in which an individual is exposed to unfamiliar people or possible scrutiny by others.” People suffering from social anxiety disorder attempt to avoid social situations that they fear, which only lead to a persistence of the disorder (239). CSC does not appear to result in social anxiety, despite the profound increase in state anxiety. In more detail, CSC mice spend a similar time investigating a novel object (empty cage) and a social contact (cage with an unknown conspecific) during the SPAT on day 20 of CSC, indicating if anything a lack of social preference (70). Interestingly, they show less investigation in both contexts, suggesting that anxiety of the novel environment may be, at least in part, involved. However, unlike following chronic social defeat, CSC mice do not show active social avoidance, i.e., less time investigating the social context than the non-social context (234, 240–242). Furthermore, when assessed 1 week after stressor termination, despite their still anxious-like phenotype (less time investigating the empty cage), CSC mice prefer to explore the novel conspecific (70). It is important to note that these findings were obtained with non-familiar conspecifics, and it is possible, if indeed not likely, that CSC mice exposed to one of the residents that they faced during the CSC paradigm, would show active social avoidance. For example, acute social defeat has repeatedly been shown to lead to social avoidance, but only to the aggressor [(243, 244); for review see (245)]. In agreement with Kalueff and coworkers (246), the initial lack of social preference following CSC is likely to reflect a temporary social deficit rather than depressive-like behavior (234, 240–242), particularly as CSC mice do not display depression-related behavior in the other tests (70).
Depressive-like behavior. In the literature, the majority of social stress paradigms lead to both increased depression- and anxiety-related behavior (67, 200, 228, 234, 240). This is perhaps not surprising, as there is high co-morbidity between the two disorders (247–250). However, in order to really dissect the mechanisms underlying anxiety or depression, animal models are sorely warranted, which specifically induce one phenotype. Importantly, the CSC paradigm seems to represent such a model, given that it does not lead to deficits in anhedonia (saccharine-preference) or depressive-like behaviour in the FST or TST for at least 1 week following stressor-termination. The use of more than one behavioral test is important, as, for example, GABAB receptor knockout mice were shown to display depression-related behavior in the FST, but not TST (251). Similarly, exposure to 10 days of social defeat did not alter FST or TST behavior, but lead to an anhedonic phenotype as assessed using the SPAT (234). Therefore, together with the SDR stress paradigm (252) and the social defeat/overcrowding stress paradigm (70, 199), CSC represents one of the few animal models that increase levels of anxiety without simultaneously increasing depression-related behavior. Of note, other depressive-like symptoms, such as cognitive dysfunction (63), have not yet been assessed following CSC.
Substance abuse disorders. Chronic psychosocial stress also represents a strong risk factor for the development of substance abuse disorders, such as alcoholism. Moreover, since CSC exposure reliably increases anxiety-related behavior (as referenced above), a known risk factor for developing ethanol- (EtOH) dependence in humans (253), we assessed whether CSC mice voluntarily consume more EtOH. Here, we could show that 14-day CSC exposure increases EtOH intake, as well as preference, without affecting taste preference or total fluid intake (175). This increased consumption is shown at all EtOH concentrations tested (2–8%), underlining the potency of CSC as a chronic stressor. This is in line with human studies, demonstrating a strong correlation between stressor exposure and the amount of EtOH consumed. It was, for instance, shown that individuals with increased numbers of stressful life events consume more EtOH and exhibit more indicators of EtOH dependence (254). In contrast, data gathered from rodent studies are less consistent. While, for instance, 5 min of daily social defeat over five consecutive days has the potential to increase EtOH consumption in male Long–Evans rats (255) and male C57BL/6 mice (256), there are also studies failing to detect a link between social stressor exposure and increased EtOH consumption [(257); for review see (258)]. These inconsistencies led us to consider our CSC model to be more relevant for the human situation, as it reliably induces an increase in EtOH consumption for a wide range of EtOH concentrations. At present, it is unclear whether CSC leads to abuse of other substances, such as cocaine or nicotine, remains to be determined in future studies.
Central mechanisms underlying the behavioral consequences of CSC. With respect to the central mechanisms underlying these CSC-induced behavioral consequences, we tested a possible involvement of the brain AVP, CRH, and OXT systems (130, 131, 155). These neuropeptides have all been linked with anxiety and substance abuse [(259–261); for review see (262)] and, thus, represent potential mediators of the CSC phenotype. While the expression patterns of hypothalamic OXT mRNA, generally known as an anxiolytic neuropeptide (38, 263–268), as well as the anxiogenic neuropeptide CRH (100) are not altered during CSC, the mRNA expression of the anxiogenic neuropeptide AVP (71, 269) is even reduced in the PVN following 20 days of CSC (131). Similarly, OXT mRNA expression in the PVN and supra optic nucleus is not affected on day 15 of CSC exposure (270). Immunohistochemistry further revealed unaffected numbers of AVP expressing parvo- and magnocellular PVN neurons in SHC and CSC mice (155), altogether making a substantial contribution of CRH, AVP, and OXT in CSC-induced anxiogenesis and EtOH preference, at least at the first glance, rather unlikely.
Recent findings strongly argue for a role of at least the oxytocinergic system in CSC-induced anxiety. Chronic central infusion of OXT (1 ng/h) via an osmotic minipump during 19-day CSC exposure – besides thymus atrophy, adrenal hypertrophy, and decreased adrenal in vitro ACTH sensitivity – further prevents CSC-induced anxiogenesis (270). The fact that chronic central OXT infusion additionally prevents the CSC-induced reduction in OXT receptor binding in the median raphe nucleus (270), a region in which OXT signaling has been recently implicated in serotonin release and subsequent anxiolytic effects (271), suggests a main role of the OXT system in this midbrain region in CSC-induced anxiety. Moreover, we have recent data showing that the same central OXT infusion procedure attenuates CSC-induced EtOH preference (Peters et al., unpublished observations). Since the raphe is hypothesized to be an important component of the circuitry involved in the reinforcing properties of drugs of abuse, including EtOH (272), and OXT can reduce drug intake and withdrawal symptoms (273), this region may be, at least in part, involved in CSC-induced heightened EtOH preference.
Besides these local changes in OXT-R binding, CSC and SHC mice show a different neuronal activity within various brain regions implicated in anxiety – under both basal and acute novel environment exposure conditions (135). For example, increased basal neuronal activation in the nucleus accumbens, as seen in CSC versus SHC, was shown in mice exposed to predator odor, which displayed increased anxiety-related behavior in the LDB (274). Furthermore, a decreased activation of the ventral and intermediate parts of the lateral septum, as seen in CSC mice following open arm exposure (135), has been described after acute stress in rats exposed to a learned-helplessness paradigm (275). Similarly, increasing the activity of lateral septum neurons was found to reduce feelings of fear and anxiety (276). Moreover, an increased activation of the dorsomedial part of the periaqueductal grey, as seen in open arm-exposed CSC versus SHC mice (135), has been reported in HAB rats after acute air jet exposure (277). Although acute stress-induced neuronal activation of the ventral hippocampus, a region well known to promote certain aspects of anxiety [for review see (278)], is not affected by CSC, given that cFOS activation in the hippocampal CA3 region is reduced in CSC versus SHC mice following open arm exposure (135). The latter may be explained by the well characterized effects of stress on retraction of dendritic spines (279), which is mainly restricted to this subfield (280). Of note, many of these changes occur in regions that form part of the reward circuitry, but whether acute administration of drugs of abuse (i.e., EtOH) would lead to differential activation between SHC versus CSC mice remains to be determined.
Together, these findings indicate that although the CSC-induced anxiety and substance abuse phenotype is pronounced, reliable, and long lasting, the detailed mechanisms behind are still poorly understood and await further investigation. Moreover, it remains to be seen whether traditional antidepressants or anxiolytics can reverse the CSC-induced behavioral and/or physiological phenotype. Furthermore, and more akin to the clinical situation, it will be interesting to assess whether post-CSC treatment of the mice can reverse the long-lasting behavioral and physiological consequences of stressor exposure.
Conclusion
In this review, we have highlighted the fact that numerous somatic and affective disorders, for which chronic psychosocial stress is an accepted risk factor, are characterized by insufficient GC signaling. Thus, hypocorticism and/or GC resistance are observed in many disorders and following chronic psychosocial stress. Consequently, animal stress models, utilizing a chronic psychosocial component, which result in a decreased GC signaling and concomitant somatic and affective pathologies are likely to hold more translational relevance than other stress models. Indeed, chronic psychosocial stress in mice induced by the CSC paradigm results in both an anxiogenic and substance abuse phenotype, resembling affective disorders, and an overall pro-inflammatory- and cancer-prone phenotype, akin to somatic disorders. CSC further causes basal evening hypocorticism and GC resistance, resembling decreased GC signaling (see Figure 3).
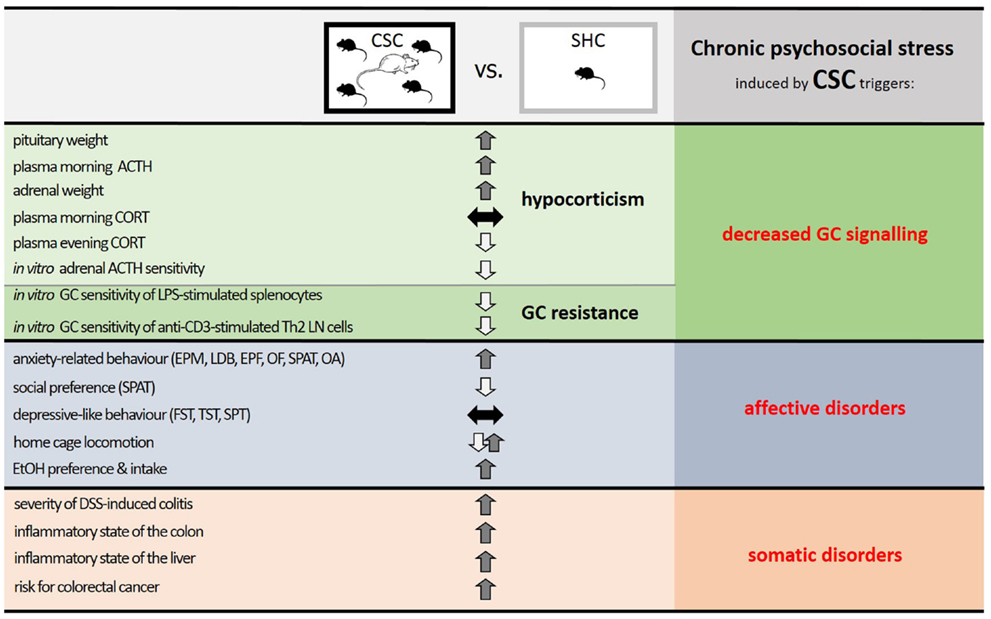
Figure 3. Summary of the effects of chronic psychosocial stress in male mice induced by 19 days of chronic subordinate colony housing (CSC) on physiological, immunological, and behavioral parameters. Compared with single-housed controls (SHC), CSC mice develop a decreased glucocorticoid (GC) signaling, induced by a combination of hypocorticism and GC resistance, and phenotypes characteristic of affective and somatic disorders. Given that stressors that can lead to somatic and affective disorders in humans are mainly chronic and psychosocial in nature, and result in a decreased GC signaling, the CSC paradigm represents a promising animal model to mimic stress-related pathologies in humans and to unravel the underlying mechanisms. Abbreviations: ACTH, adrenocorticotropic hormone; CORT, corticosterone; LPS, lipopolysaccharide; Th2, T helper 2; LN, lymph node; EPM, elevated plus-maze; LDB, light–dark box; EPF, elevated platform; OF, open field; SPAT, social preference/avoidance test; OA, open arm exposure; FST, forced swim test; TST, tail suspension test; SPT, saccharine preference test; EtOH, ethanol; DSS, dextran-sulfate sodium.
Therefore, we are convinced that the CSC paradigm represents an appropriate animal model for studying stress-related disorders in which altered GC signaling is a core feature. Such detailed knowledge will provide further insight into how such stress-related HPA axis changes ultimately lead to somatic and affective disorders, at both behavioral and mechanistic level. Such detailed knowledge, in turn, will allow us to identify novel targets for the treatment of a wide variety of somatic and affective disorders.
Conflict of Interest Statement
The Review Editor Karl Bechter declares that, despite being affiliated to the same institution as authors Dominik Langgartner, Andrea M. Füchsl and Stefan O. Reber, the review process was handled objectively and no conflict of interest exists. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Selye H. A syndrome produced by diverse nocuous agents. Nature (1936) 138:32. doi:10.1038/138032a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Selye H. Confusion and controversy in the stress field. J Human Stress (1975) 1(2):37–44. doi:10.1080/0097840X.1975.9940406
4. Dhabhar FS, McEwen BS. Stress-induced enhancement of antigen-specific cell-mediated immunity. J Immunol (1996) 156:2608–15.
5. McEwen BS. Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann N Y Acad Sci (2004) 1032:1–7. doi:10.1196/annals.1314.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Viswanathan K, Dhabhar FS. Stress-induced enhancement of leukocyte trafficking into sites of surgery or immune activation. Proc Natl Acad Sci U S A (2005) 102:5808–13. doi:10.1073/pnas.0501650102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Dhabhar FS. Stress-induced augmentation of immune function – the role of stress hormones, leukocyte trafficking, and cytokines. Brain Behav Immun (2002) 16:785–98. doi:10.1016/S0889-1591(02)00036-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. McEwen BS, Seeman T. Protective and damaging effects of mediators of stress. Elaborating and testing the concepts of allostasis and allostatic load. Ann N Y Acad Sci (1999) 896:30–47. doi:10.1111/j.1749-6632.1999.tb08103.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Sapolsky RM. The influence of social hierarchy on primate health. Science (2005) 308:648–52. doi:10.1126/science.1106477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Goldstein DS, Kopin IJ. Evolution of concepts of stress. Stress (2007) 10:109–20. doi:10.1080/10253890701288935
11. Dhabhar FS, McEwen BS. Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: a potential role for leukocyte trafficking. Brain Behav Immun (1997) 11:286–306. doi:10.1006/brbi.1997.0508
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Saul AN, Oberyszyn TM, Daugherty C, Kusewitt D, Jones S, Jewell S, et al. Chronic stress and susceptibility to skin cancer. J Natl Cancer Inst (2005) 97:1760–7. doi:10.1093/jnci/dji401
13. Bernstein CN, Singh S, Graff LA, Walker JR, Miller N, Cheang M. A prospective population-based study of triggers of symptomatic flares in IBD. Am J Gastroenterol (2010) 105:1994–2002. doi:10.1038/ajg.2010.140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Bitton A, Sewitch MJ, Peppercorn MA, Edwardes MD, Shah S, Ransil B, et al. Psychosocial determinants of relapse in ulcerative colitis: a longitudinal study. Am J Gastroenterol (2003) 98:2203–8. doi:10.1111/j.1572-0241.2003.07717.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Danese A, Pariante CM, Caspi A, Taylor A, Poulton R. Childhood maltreatment predicts adult inflammation in a life-course study. Proc Natl Acad Sci U S A (2007) 104:1319–24. doi:10.1073/pnas.0610362104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Duffy LC, Zielezny MA, Marshall JR, Byers TE, Weiser MM, Phillips JF, et al. Relevance of major stress events as an indicator of disease activity prevalence in inflammatory bowel disease. Behav Med (1991) 17:101–10. doi:10.1080/08964289.1991.9937553
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Levav I, Kohn R, Iscovich J, Abramson JH, Tsai WY, Vigdorovich D. Cancer incidence and survival following bereavement. Am J Public Health (2000) 90:1601–7. doi:10.2105/AJPH.90.10.1601
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Levenstein S, Prantera C, Varvo V, Scribano ML, Andreoli A, Luzi C, et al. Stress and exacerbation in ulcerative colitis: a prospective study of patients enrolled in remission. Am J Gastroenterol (2000) 95:1213–20. doi:10.1111/j.1572-0241.2000.02012.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH, et al. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry (2006) 163:1630–3. doi:10.1176/appi.ajp.163.9.1630
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Robertson DA, Ray J, Diamond I, Edwards JG. Personality profile and affective state of patients with inflammatory bowel disease. Gut (1989) 30:623–6. doi:10.1136/gut.30.9.1300
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Salem SN, Shubair KS. Non-specific ulcerative colitis in Bedouin Arabs. Lancet (1967) 1:473–5. doi:10.1016/S0140-6736(67)91094-X
22. Bernstein CN. New insights into IBD epidemiology: are there any lessons for treatment? Dig Dis (2010) 28:406–10. doi:10.1159/000320394
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Heim C, Ehlert U, Hellhammer DH. The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders. Psychoneuroendocrinology (2000) 25:1–35. doi:10.1016/S0306-4530(99)00035-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Heim C, Nemeroff CB. The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry (2001) 49:1023–39. doi:10.1016/S0006-3223(01)01157-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Mawdsley JE, Rampton DS. Psychological stress in IBD: new insights into pathogenic and therapeutic implications. Gut (2005) 54:1481–91. doi:10.1136/gut.2005.064261
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Mawdsley JE, Rampton DS. The role of psychological stress in inflammatory bowel disease. Neuroimmunomodulation (2006) 13:327–36. doi:10.1159/000104861
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Reiche EM, Nunes SO, Morimoto HK. Stress, depression, the immune system, and cancer. Lancet Oncol (2004) 5:617–25. doi:10.1016/S1470-2045(04)01597-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Bartolomucci A. Social stress, immune functions and disease in rodents. Front Neuroendocrinol (2007) 28:28–49. doi:10.1016/j.yfrne.2007.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Stratakis CA, Chrousos GP. Neuroendocrinology and pathophysiology of the stress system. Ann N Y Acad Sci (1995) 771:1–18. doi:10.1111/j.1749-6632.1995.tb44666.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology (1985) 117:2505–11. doi:10.1210/endo-117-6-2505
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Bowers SL, Bilbo SD, Dhabhar FS, Nelson RJ. Stressor-specific alterations in corticosterone and immune responses in mice. Brain Behav Immun (2008) 22:105–13. doi:10.1016/j.bbi.2007.07.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Koolhaas JM, Bartolomucci A, Buwalda B, de Boer SF, Flugge G, Korte SM, et al. Stress revisited: a critical evaluation of the stress concept. Neurosci Biobehav Rev (2011) 35(5):1291–301. doi:10.1016/j.neurobiorev.2011.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Bartlang MS, Neumann ID, Slattery DA, Uschold-Schmidt N, Kraus D, Helfrich-Förster C, et al. Time matters: pathological effects of repeated psychosocial stress during the active, but not inactive, phase of male mice. J Endocrinol (2012) 215:425–37. doi:10.1530/joe-12-0267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Kalsbeek A, Ruiter M, La Fleur SE, Van Heijningen C, Buijs RM. The diurnal modulation of hormonal responses in the rat varies with different stimuli. J Neuroendocrinol (2003) 15:1144–55. doi:10.1111/j.1365-2826.2003.01112.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Perez-Cruz C, Simon M, Flügge G, Fuchs E, Czéh B. Diurnal rhythm and stress regulate dendritic architecture and spine density of pyramidal neurons in the rat infralimbic cortex. Behav Brain Res (2009) 205:406–13. doi:10.1016/j.bbr.2009.07.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Meijer OC, Topic B, Steenbergen PJ, Jocham G, Huston JP, Oitzl MS. Correlations between hypothalamus-pituitary-adrenal axis parameters depend on age and learning capacity. Endocrinology (2005) 146(3):1372–81.
37. Sapolsky RM, Krey LC, McEwen BS. The adrenocortical axis in the aged rat: impaired sensitivity to both fast and delayed feedback inhibition. Neurobiol Aging (1986) 7:331–5. doi:10.1016/0197-4580(86)90159-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Neumann ID, Torner L, Wigger A. Brain oxytocin: differential inhibition of neuroendocrine stress responses and anxiety-related behaviour in virgin, pregnant and lactating rats. Neuroscience (2000) 95:567–75. doi:10.1016/S0306-4522(99)00433-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Torner L, Neumann ID. The brain prolactin system: involvement in stress response adaptations in lactation. Stress (2002) 5:249–57. doi:10.1080/1025389021000048638
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Torner L, Toschi N, Nava G, Clapp C, Neumann ID. Increased hypothalamic expression of prolactin in lactation: involvement in behavioural and neuroendocrine stress responses. Eur J Neurosci (2002) 15:1381–9. doi:10.1046/j.1460-9568.2002.01965.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Füchsl AM, Neumann ID, Reber SO. Stress resilience: a low-anxiety genotype protects male mice from the consequences of chronic psychosocial stress. Endocrinology (2014) 155:117–26. doi:10.1210/en.2013-1742
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Landgraf R, Wigger A, Holsboer F, Neumann ID. Hyper-reactive hypothalamo-pituitary-adrenocortical axis in rats bred for high anxiety-related behaviour. J Neuroendocrinol (1999) 11:405–7. doi:10.1046/j.1365-2826.1999.00342.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Ohta R, Shirota M, Adachi T, Tohei A, Taya K. Plasma ACTH levels during early, two-way avoidance acquisition in high- and low-avoidance rats (Hatano strains). Behav Genet (1999) 29:137–44. doi:10.1023/A:1021616723969
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Sternberg EM, Hill JM, Chrousos GP, Kamilaris T, Listwak SJ, Gold PW, et al. Inflammatory mediator-induced hypothalamic-pituitary-adrenal axis activation is defective in streptococcal cell wall arthritis-susceptible Lewis rats. Proc Natl Acad Sci U S A (1989) 86:2374–8. doi:10.1073/pnas.86.7.2374
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Touma C, Bunck M, Glasl L, Nussbaumer M, Palme R, Stein H, et al. Mice selected for high versus low stress reactivity: a new animal model for affective disorders. Psychoneuroendocrinology (2008) 33:839–62. doi:10.1016/j.psyneuen.2008.03.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Veenema AH, Reber SO, Selch S, Obermeier F, Neumann ID. Early life stress enhances the vulnerability to chronic psychosocial stress and experimental colitis in adult mice. Endocrinology (2008) 144:2727–36. doi:10.1210/en.2007-1469
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Wigger A, Neumann ID. Periodic maternal deprivation induces gender-dependent alterations in behavioral and neuroendocrine responses to emotional stress in adult rats. Physiol Behav (1999) 66:293–302. doi:10.1016/S0031-9384(98)00300-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Campeau S, Watson SJ. Neuroendocrine and behavioral responses and brain pattern of c-fos induction associated with audiogenic stress. J Neuroendocrinol (1997) 9:577–88.
49. Cullinan WE, Herman JP, Battaglia DF, Akil H, Watson SJ. Pattern and time course of immediate early gene expression in rat brain following acute stress. Neuroscience (1995) 64:477–505. doi:10.1016/0306-4522(94)00355-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Cullinan WE, Helmreich DL, Watson SJ. Fos expression in forebrain afferents to the hypothalamic paraventricular nucleus following swim stress. J Comp Neurol (1996) 368:88–99. doi:10.1002/(SICI)1096-9861(19960422)368:1<88::AID-CNE6>3.0.CO;2-G
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Reis DG, Scopinho AA, Guimaraes FS, Correa FM, Resstel LB. Behavioral and autonomic responses to acute restraint stress are segregated within the lateral septal area of rats. PLoS One (2011) 6:e23171. doi:10.1371/journal.pone.0023171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Balodis IM, Wynne-Edwards KE, Olmstead MC. The stress-response-dampening effects of placebo. Horm Behav (2011) 59:465–72. doi:10.1016/j.yhbeh.2011.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Elzinga BM, Spinhoven P, Berretty E, de Jong P, Roelofs K. The role of childhood abuse in HPA-axis reactivity in social anxiety disorder: a pilot study. Biol Psychol (2010) 83:1–6. doi:10.1016/j.biopsycho.2009.09.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Heinrichs M, Baumgartner T, Kirschbaum C, Ehlert U. Social support and oxytocin interact to suppress cortisol and subjective responses to psychosocial stress. Biol Psychiatry (2003) 54:1389–98. doi:10.1016/S0006-3223(03)00465-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Buske-Kirschbaum A, Geiben A, Hollig H, Morschhauser E, Hellhammer D. Altered responsiveness of the hypothalamus-pituitary-adrenal axis and the sympathetic adrenomedullary system to stress in patients with atopic dermatitis. J Clin Endocrinol Metab (2002) 87:4245–51. doi:10.1210/jc.2001-010872
56. Buske-Kirschbaum A, Gierens A, Hollig H, Hellhammer DH. Stress-induced immunomodulation is altered in patients with atopic dermatitis. J Neuroimmunol (2002) 129:161–7. doi:10.1016/S0165-5728(02)00168-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Buske-Kirschbaum A, Ebrecht M, Hellhammer DH. Blunted HPA axis responsiveness to stress in atopic patients is associated with the acuity and severeness of allergic inflammation. Brain Behav Immun (2010) 24:1347–53. doi:10.1016/j.bbi.2010.06.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Cryan JF, Holmes A. The ascent of mouse: advances in modelling human depression and anxiety. Nat Rev Drug Discov (2005) 4:775–90. doi:10.1038/nrd1825
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Cryan JF, Mombereau C, Vassout A. The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev (2005) 29:571–625. doi:10.1016/j.neubiorev.2005.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Cryan JF, Slattery DA. Animal models of mood disorders: recent developments. Curr Opin Psychiatry (2007) 20:1–7. doi:10.1097/YCO.0b013e3280117733
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Fuchs E. Social stress in tree shrews as an animal model of depression: an example of a behavioral model of a CNS disorder. CNS Spectr (2005) 10:182–90. doi:10.1017/S1092852900010038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Gould TD, Gottesman II. Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav (2006) 5:113–9. doi:10.1111/j.1601-183X.2005.00186.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Hales CA, Stuart SA, Anderson MH, Robinson ES. Modelling cognitive affective biases in major depressive disorder using rodents. Br J Pharmacol (2014) 171(20):4524–38. doi:10.1111/bph.12603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Harro J. Animal models of depression vulnerability. Curr Top Behav Neurosci (2013) 14:29–54. doi:10.1007/7854_2012_221
65. Henn FA, Vollmayr B. Stress models of depression: forming genetically vulnerable strains. Neurosci Biobehav Rev (2005) 29:799–804. doi:10.1016/j.neubiorev.2005.03.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Avgustinovich DF, Gorbach OV, Kudryavtseva NN. Comparative analysis of anxiety-like behavior in partition and plus-maze tests after agonistic interactions in mice. Physiol Behav (1997) 61:37–43. doi:10.1016/S0031-9384(96)00303-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Keeney AJ, Hogg S. Behavioural consequences of repeated social defeat in the mouse: preliminary evaluation of a potential animal model of depression. Behav Pharmacol (1999) 10:753–64. doi:10.1097/00008877-199912000-00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Keeney AJ, Jessop DS, Harbuz MS, Marsden CA, Hogg S, Blackburn-Munro RE. Differential effects of acute and chronic social defeat stress on hypothalamic-pituitary-adrenal axis function and hippocampal serotonin release in mice. J Neuroendocrinol (2006) 18:330–8. doi:10.1111/j.1365-2826.2006.01422.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Savignac HM, Finger BC, Pizzo RC, O’Leary OF, Dinan TG, Cryan JF. Increased sensitivity to the effects of chronic social defeat stress in an innately anxious mouse strain. Neuroscience (2011) 192:524–36. doi:10.1016/j.neuroscience.2011.04.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Slattery DA, Uschold N, Magoni M, Bar J, Popoli M, Neumann ID, et al. Behavioural consequences of two chronic psychosocial stress paradigms: anxiety without depression. Psychoneuroendocrinology (2012) 37(5):702–14. doi:10.1016/j.psyneuen.2011.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Bosch OJ, Krömer SA, Neumann ID. Prenatal stress: opposite effects on anxiety and hypothalamic expression of vasopressin and corticotropin-releasing hormone in rats selectively bred for high and low anxiety. Eur J Neurosci (2006) 23:541–51. doi:10.1111/j.1460-9568.2005.04576.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Neumann ID, Wigger A, Krömer S, Frank E, Landgraf R, Bosch OJ. Differential effects of periodic maternal separation on adult stress coping in a rat model of extremes in trait anxiety. Neuroscience (2005) 132:867–77. doi:10.1016/j.neuroscience.2005.01.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Neumann ID, Krömer SA, Bosch OJ. Effects of psycho-social stress during pregnancy on neuroendocrine and behavioural parameters in lactation depend on the genetically determined stress vulnerability. Psychoneuroendocrinology (2005) 30:791–806. doi:10.1016/j.psyneuen.2005.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Veenema AH, Meijer OC, de Kloet ER, Koolhaas JM. Genetic selection for coping style predicts stressor susceptibility. J Neuroendocrinol (2003) 15:256–67. doi:10.1046/j.1365-2826.2003.00986.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Buckley T, Stannard A, Bartrop R, McKinley S, Ward C, Mihailidou AS, et al. Effect of early bereavement on heart rate and heart rate variability. Am J Cardiol (2012) 110:1378–83. doi:10.1016/j.amjcard.2012.06.045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Dimsdale JE. Psychological stress and cardiovascular disease. J Am Coll Cardiol (2008) 51:1237–46. doi:10.1016/j.jacc.2007.12.024
77. Kivimaki M, Nyberg ST, Batty GD, Fransson EI, Heikkila K, Alfredsson L, et al. Job strain as a risk factor for coronary heart disease: a collaborative meta-analysis of individual participant data. Lancet (2012) 380:1491–7. doi:10.1016/S0140-6736(12)60994-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Mausbach BT, Chattillion E, Roepke SK, Ziegler MG, Milic M, von Kanel R, et al. A longitudinal analysis of the relations among stress, depressive symptoms, leisure satisfaction, and endothelial function in caregivers. Health Psychol (2012) 31:433–40. doi:10.1037/a0027783
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Unden AL, Orth-Gomer K, Elofsson S. Cardiovascular effects of social support in the work place: twenty-four-hour ECG monitoring of men and women. Psychosom Med (1991) 53:50–60. doi:10.1097/00006842-199101000-00005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Buckley T, Bartrop R, McKinley S, Ward C, Bramwell M, Roche D, et al. Prospective study of early bereavement on psychological and behavioural cardiac risk factors. Intern Med J (2009) 39:370–8. doi:10.1111/j.1445-5994.2009.01879.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Heim C, Nater UM, Maloney E, Boneva R, Jones JF, Reeves WC. Childhood trauma and risk for chronic fatigue syndrome: association with neuroendocrine dysfunction. Arch Gen Psychiatry (2009) 66:72–80. doi:10.1001/archgenpsychiatry.2008.508
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Almansa C, Rey E, Sanchez RG, Sanchez AA, Diaz-Rubio M. Prevalence of functional gastrointestinal disorders in patients with fibromyalgia and the role of psychologic distress. Clin Gastroenterol Hepatol (2009) 7:438–45. doi:10.1016/j.cgh.2008.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Wright RJ, Finn P, Contreras JP, Cohen S, Wright RO, Staudenmayer J, et al. Chronic caregiver stress and IgE expression, allergen-induced proliferation, and cytokine profiles in a birth cohort predisposed to atopy. J Allergy Clin Immunol (2004) 113:1051–7. doi:10.1016/j.jaci.2004.03.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Wright RJ, Mitchell H, Visness CM, Cohen S, Stout J, Evans R, et al. Community violence and asthma morbidity: the Inner-City Asthma Study. Am J Public Health (2004) 94:625–32. doi:10.2105/AJPH.94.4.625
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Buske-Kirschbaum A, Geiben A, Hellhammer D. Psychobiological aspects of atopic dermatitis: an overview. Psychother Psychosom (2001) 70:6–16. doi:10.1159/000056219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Coker AL, Smith PH, Bethea L, King MR, McKeown RE. Physical health consequences of physical and psychological intimate partner violence. Arch Fam Med (2000) 9:451–7. doi:10.1001/archfami.9.5.451
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Herrmann M, Scholmerich J, Straub RH. Stress and rheumatic diseases. Rheum Dis Clin North Am (2000) 26:737–763, viii. doi:10.1016/S0889-857X(05)70167-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Campbell J, Jones AS, Dienemann J, Kub J, Schollenberger J, O’Campo P, et al. Intimate partner violence and physical health consequences. Arch Intern Med (2002) 162:1157–63. doi:10.1001/archinte.162.10.1157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Cohen S, Tyrrell DA, Smith AP. Psychological stress and susceptibility to the common cold. N Engl J Med (1991) 325:606–12. doi:10.1056/NEJM199108293250903
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Kiecolt-Glaser JK, Glaser R, Gravenstein S, Malarkey WB, Sheridan J. Chronic stress alters the immune response to influenza virus vaccine in older adults. Proc Natl Acad Sci U S A (1996) 93:3043–7. doi:10.1073/pnas.93.7.3043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Kiecolt-Glaser JK, Marucha PT, Malarkey WB, Mercado AM, Glaser R. Slowing of wound healing by psychological stress. Lancet (1995) 346:1194–6. doi:10.1016/S0140-6736(95)92899-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Kiecolt-Glaser JK, Page GG, Marucha PT, MacCallum RC, Glaser R. Psychological influences on surgical recovery. Perspectives from psychoneuroimmunology. Am Psychol (1998) 53:1209–18. doi:10.1037/0003-066X.53.11.1209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Marucha PT, Kiecolt-Glaser JK, Favagehi M. Mucosal wound healing is impaired by examination stress. Psychosom Med (1998) 60:362–5. doi:10.1097/00006842-199805000-00025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Kiecolt-Glaser JK, Glaser R. Psychoneuroimmunology and health consequences: data and shared mechanisms. Psychosom Med (1995) 57:269–74. doi:10.1097/00006842-199505000-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Agid O, Shapira B, Zislin J, Ritsner M, Hanin B, Murad H, et al. Environment and vulnerability to major psychiatric illness: a case control study of early parental loss in major depression, bipolar disorder and schizophrenia. Mol Psychiatry (1999) 4:163–72. doi:10.1038/sj.mp.4000473
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci (2005) 6:463–75. doi:10.1038/nrn1683
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Virtanen M, Stansfeld SA, Fuhrer R, Ferrie JE, Kivimaki M. Overtime work as a predictor of major depressive episode: a 5-year follow-up of the Whitehall II study. PLoS One (2012) 7:e30719. doi:10.1371/journal.pone.0030719
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Virtanen M, Kivimaki M. Saved by the bell: does working too much increase the likelihood of depression? Expert Rev Neurother (2012) 12:497–9. doi:10.1586/ern.12.29
99. Hammen C. Stress and depression. Annu Rev Clin Psychol (2005) 1:293–319. doi:10.1146/annurev.clinpsy.1.102803.143938
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Holsboer F. Stress, hypercortisolism and corticosteroid receptors in depression: implications for therapy. J Affect Disord (2001) 62:77–91. doi:10.1016/S0165-0327(00)00352-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry (2003) 160:1554–65. doi:10.1176/appi.ajp.160.9.1554
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Friedman SB, Mason JW, Hamburg DA. Urinary 17-hydroxycorticosteroid levels in parents of children with neoplastic disease: a study of chronic psychological stress. Psychosom Med (1963) 25:364–76. doi:10.1097/00006842-196307000-00007
103. Caplan RD, Cobb S, French JR Jr. White collar work load and cortisol: disruption of a circadian rhythm by job stress? J Psychosom Res (1979) 23:181–92. doi:10.1016/0022-3999(79)90003-5
104. Yehuda R. Sensitization of the hypothalamic-pituitary-adrenal axis in posttraumatic stress disorder. Ann N Y Acad Sci (1997) 821:57–75. doi:10.1111/j.1749-6632.1997.tb48269.x
105. Penninx BW, Beekman AT, Bandinelli S, Corsi AM, Bremmer M, Hoogendijk WJ, et al. Late-life depressive symptoms are associated with both hyperactivity and hypoactivity of the hypothalamo-pituitary-adrenal axis. Am J Geriatr Psychiatry (2007) 15:522–9. doi:10.1097/JGP.0b013e318033ed80
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology (2000) 23:477–501. doi:10.1016/S0893-133X(00)00159-7
107. Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry (2001) 49:391–404. doi:10.1016/S0006-3223(00)01088-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Heim C, Newport DJ, Heit S, Graham YP, Wilcox M, Bonsall R, et al. Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA (2000) 284:592–7. doi:10.1001/jama.284.5.592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Heim C, Newport DJ, Bonsall R, Miller AH, Nemeroff CB. Altered pituitary-adrenal axis responses to provocative challenge tests in adult survivors of childhood abuse. Am J Psychiatry (2001) 158:575–81. doi:10.1176/appi.ajp.158.4.575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Crofford LJ, Pillemer SR, Kalogeras KT, Cash JM, Michelson D, Kling MA, et al. Hypothalamic-pituitary-adrenal axis perturbations in patients with fibromyalgia. Arthritis Rheum (1994) 37:1583–92. doi:10.1002/art.1780371105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Demitrack MA, Dale JK, Straus SE, Laue L, Listwak SJ, Kruesi MJ, et al. Evidence for impaired activation of the hypothalamic-pituitary-adrenal axis in patients with chronic fatigue syndrome. J Clin Endocrinol Metab (1991) 73:1224–34. doi:10.1210/jcem-73-6-1224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Heim C, Ehlert U, Hanker JP, Hellhammer DH. Abuse-related posttraumatic stress disorder and alterations of the hypothalamic-pituitary-adrenal axis in women with chronic pelvic pain. Psychosom Med (1998) 60:309–18. doi:10.1097/00006842-199805000-00017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Straub RH, Buttgereit F, Cutolo M. Alterations of the hypothalamic-pituitary-adrenal axis in systemic immune diseases – a role for misguided energy regulation. Clin Exp Rheumatol (2011) 29:S23–31.
114. Kruger U, Spiecker H. Diagnosis of adrenal cortex insufficiency in steroid-dependent bronchial asthma – the CRH test in comparison with diurnal cortisol profile in serum and cortisol in 24-hour urine. Pneumologie (1994) 48:793–8.
115. Papanicolaou DA, Tsigos C, Oldfield EH, Chrousos GP. Acute glucocorticoid deficiency is associated with plasma elevations of interleukin-6: does the latter participate in the symptomatology of the steroid withdrawal syndrome and adrenal insufficiency? J Clin Endocrinol Metab (1996) 81:2303–6. doi:10.1210/jc.81.6.2303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Stirrat L, O’Reilly J, Riley S, Howie A, Smith R, Walker B, et al. PPO.21 Altered maternal hypothalamic-pituitary-adrenal axis activity in obese pregnancy: a potential mechanism underlying macrosomia and prolonged pregnancy. Arch Dis Child Fetal Neonatal Ed (2014) 99:A157. doi:10.1136/archdischild-2014-306576.461
117. Mayer EA. The neurobiology of stress and gastrointestinal disease. Gut (2000) 47:861–9. doi:10.1136/gut.47.6.861
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Reber SO. Stress and animal models of inflammatory bowel disease – an update on the role of the hypothalamo-pituitary-adrenal axis. Psychoneuroendocrinology (2012) 37:1–19. doi:10.1016/j.psyneuen.2011.05.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Reinshagen M, Haefele T, Holicki S, Adler G, Klaus J, Dahlbender R, et al. The ultra low dose ACTH-test Identifies a subgroup of patients with Crohn’s disease and an impaired hypothalamo-pituitary-adrenal (HPA) axis. Gastroenterology (2002) 122:S1384.
120. Minderhoud IM, Oldenburg B, van Dam PS, van Berge Henegouwen GP. High prevalence of fatigue in quiescent inflammatory bowel disease is not related to adrenocortical insufficiency. Am J Gastroenterol (2003) 98:1088–93. doi:10.1111/j.1572-0241.2003.07414.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Garcia Rodriguez LA, Ruigomez A, Wallander MA, Johansson S, Olbe L. Detection of colorectal tumor and inflammatory bowel disease during follow-up of patients with initial diagnosis of irritable bowel syndrome. Scand J Gastroenterol (2000) 35:306–11. doi:10.1080/003655200750024191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Miller GE, Cohen S, Ritchey AK. Chronic psychological stress and the regulation of pro-inflammatory cytokines: a glucocorticoid-resistance model. Health Psychol (2002) 21:531–41. doi:10.1037/0278-6133.21.6.531
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Holsboer F. The dexamethasone suppression test in depressed patients: clinical and biochemical aspects. J Steroid Biochem (1983) 19:251–7. doi:10.1016/S0022-4731(83)80033-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Myers B, McKlveen JM, Herman JP. Glucocorticoid actions on synapses, circuits, and behavior: implications for the energetics of stress. Front Neuroendocrinol (2014) 35:180–96. doi:10.1016/j.yfrne.2013.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Uchoa ET, Aguilera G, Herman JP, Fiedler JL, Deak T, de Sousa MB. Novel aspects of glucocorticoid actions. J Neuroendocrinol (2014) 26:557–72. doi:10.1111/jne.12157
126. Faubion WA Jr, Loftus EV Jr, Harmsen WS, Zinsmeister AR, Sandborn WJ. The natural history of corticosteroid therapy for inflammatory bowel disease: a population-based study. Gastroenterology (2001) 121:255–60. doi:10.1053/gast.2001.26279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Creed TJ, Probert CS. Review article: steroid resistance in inflammatory bowel disease – mechanisms and therapeutic strategies. Aliment Pharmacol Ther (2007) 25:111–22. doi:10.1111/j.1365-2036.2006.03156.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Harbuz M, Korendowych E, Jessop D, Crown A, Li Pdfan S, Kirwan JR. Hypothalamo-pituitary-adrenal axis dysregulation in patients with rheumatoid arthritis after the dexamethasone/corticotrophin releasing factor test. J Endocrinol (2003) 178:55–60. doi:10.1677/joe.0.1780055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Reber SO, Birkeneder L, Veenema AH, Obermeier F, Falk W, Straub RH, et al. Adrenal insufficiency and colonic inflammation after a novel chronic psycho-social stress paradigm in mice: implications and mechanisms. Endocrinology (2007) 148:670–82. doi:10.1210/en.2006-0983
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Reber SO, Neumann ID. Defensive behavioral strategies and enhanced state anxiety during chronic subordinate colony housing are accompanied by reduced hypothalamic vasopressin, but not oxytocin, expression. Ann N Y Acad Sci (2008) 1148:184–95. doi:10.1196/annals.1410.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Forkwa TK, Neumann ID, Tamm ER, Ohlmann A, Reber SO. Short-term psychosocial stress protects photoreceptors from damage via corticosterone-mediated activation of the AKT pathway. Exp Neurol (2014) 252:28–36. doi:10.1016/j.expneurol.2013.11.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Füchsl AM, Uschold-Schmidt N, Reber SO. Chronic psychosocial stress in male mice causes an up-regulation of scavenger receptor class B type 1 protein in the adrenal glands. Stress (2013) 16:461–8. doi:10.3109/10253890.2013.793303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Uschold-Schmidt N, Peterlik D, Füchsl AM, Reber SO. HPA axis changes during the initial phase of psychosocial stressor exposure in male mice. J Endocrinol (2013) 218:193–203. doi:10.1530/joe-13-0027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Singewald GM, Nguyen NK, Neumann ID, Singewald N, Reber SO. Effect of chronic psychosocial stress-induced by subordinate colony (CSC) housing on brain neuronal activity patterns in mice. Stress (2009) 12:58–69. doi:10.1080/10253890802042082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Ohl F, Toschi N, Wigger A, Henniger MS, Landgraf R. Dimensions of emotionality in a rat model of innate anxiety. Behav Neurosci (2001) 115:429–36. doi:10.1037/0735-7044.115.2.429
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Salome N, Landgraf R, Viltart O. Confinement to the open arm of the elevated-plus maze as anxiety paradigm: behavioral validation. Behav Neurosci (2006) 120:719–23. doi:10.1037/0735-7044.120.3.719
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Bartolomucci A, Palanza P, Sacerdote P, Ceresini G, Chirieleison A, Panerai AE, et al. Individual housing induces altered immuno-endocrine responses to psychological stress in male mice. Psychoneuroendocrinology (2003) 28:540–58. doi:10.1016/S0306-4530(02)00039-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Gasparotto OC, Lopes DM, Carobrez SG. Pair housing affects anxiety-like behaviors induced by a social but not by a physiological stressor in male Swiss mice. Physiol Behav (2005) 85:603–12. doi:10.1016/j.physbeh.2005.06.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Blanchard RJ, McKittrick CR, Blanchard DC. Animal models of social stress: effects on behavior and brain neurochemical systems. Physiol Behav (2001) 73:261–71. doi:10.1016/S0031-9384(01)00449-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Palanza P. Animal models of anxiety and depression: how are females different? Neurosci Biobehav Rev (2001) 25:219–33. doi:10.1016/S0149-7634(01)00010-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Uschold-Schmidt N, Nyuyki KD, Füchsl AM, Neumann ID, Reber SO. Chronic psychosocial stress results in sensitization of the HPA axis to acute heterotypic stressors despite a reduction of adrenal in vitro ACTH responsiveness. Psychoneuroendocrinology (2012) 37:1676–87. doi:10.1016/j.psyneuen.2012.02.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Kraemer FB. Adrenal cholesterol utilization. Mol Cell Endocrinol (2007) 26(5–266):42–5. doi:10.1016/j.mce.2006.12.001
144. Rodrigueza WV, Thuahnai ST, Temel RE, Lund-Katz S, Phillips MC, Williams DL. Mechanism of scavenger receptor class B type I-mediated selective uptake of cholesteryl esters from high density lipoprotein to adrenal cells. J Biol Chem (1999) 274:20344–50. doi:10.1074/jbc.274.29.20344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Reaven E, Tsai L, Azhar S. Intracellular events in the “selective” transport of lipoprotein-derived cholesteryl esters. J Biol Chem (1996) 271:16208–17. doi:10.1074/jbc.271.27.16208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Gwynne JT, Strauss JF. The role of lipoproteins in steroidogenesis and cholesterol metabolism in steroidogenic glands. Endocr Rev (1982) 3:299–329. doi:10.1210/edrv-3-3-299
147. Krieger M. Charting the fate of the “good cholesterol”: identification and characterization of the high-density lipoprotein receptor SR-BI. Annu Rev Biochem (1999) 68:523–58. doi:10.1146/annurev.biochem.68.1.523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Williams DL, Connelly MA, Temel RE, Swarnakar S, Phillips MC, de la Llera-Moya M, et al. Scavenger receptor Bl and cholesterol trafficking. Curr Opin Lipidol (1999) 10:329–40. doi:10.1097/00041433-199908000-00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Kovanen PT, Faust JR, Brown MS, Goldstein JL. Low density lipoprotein receptors in bovine adrenal cortex. I. Receptor-mediated uptake of low density lipoprotein and utilization of its cholesterol for steroid synthesis in cultured adrenocortical cells. Endocrinology (1979) 104:599–609. doi:10.1210/endo-104-3-599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Rosol TJ, Yarrington JT, Latendresse J, Capen CC. Adrenal gland: structure, function, and mechanisms of toxicity. Toxicol Pathol. (2001) 29(1):41–8.
151. Biason-Lauber A. Molecular medicine of steroid hormone biosynthesis. Mol Aspects Med (1998) 19:155–220. doi:10.1016/S0098-2997(98)00004-1
152. Miller WL. Molecular biology of steroid hormone synthesis. Endocr Rev (1988) 9:295–318. doi:10.1210/edrv-9-3-295
153. Sewer MB, Waterman MR. ACTH modulation of transcription factors responsible for steroid hydroxylase gene expression in the adrenal cortex. Microsc Res Tech (2003) 61:300–7. doi:10.1002/jemt.10339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Sewer MB, Dammer EB, Jagarlapudi S. Transcriptional regulation of adrenocortical steroidogenic gene expression. Drug Metab Rev (2007) 39:371–88. doi:10.1080/03602530701498828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Füchsl AM, Langgartner D, Reber SO. Mechanisms underlying the increased plasma ACTH levels in chronic psychosocially stressed male mice. PLoS One (2013) 8:e84161. doi:10.1371/journal.pone.0084161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Bhatnagar S, Vining C. Facilitation of hypothalamic-pituitary-adrenal responses to novel stress following repeated social stress using the resident/intruder paradigm. Horm Behav (2003) 43:158–65. doi:10.1016/S0018-506X(02)00011-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Aguilera G. Regulation of pituitary ACTH secretion during chronic stress. Front Neuroendocrinol (1994) 15:321–50. doi:10.1006/frne.1994.1013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Reber SO, Peters S, Slattery DA, Hofmann C, Schölmerich J, Neumann ID, et al. Mucosal immunosuppression and epithelial barrier defects are key events in murine psychosocial stress-induced colitis. Brain Behav Immun (2011) 25:1153–61. doi:10.1016/j.bbi.2011.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Baranyi J, Bakos N, Haller J. Social instability in female rats: the relationship between stress-related and anxiety-like consequences. Physiol Behav (2005) 84:511–8. doi:10.1016/j.physbeh.2005.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Schwartz MW, Strack AM, Dallman MF. Evidence that elevated plasma corticosterone levels are the cause of reduced hypothalamic corticotrophin-releasing hormone gene expression in diabetes. Regul Pept (1997) 72:105–12. doi:10.1016/S0167-0115(97)01043-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Chiodini I, Scillitani A. Role of cortisol hypersecretion in the pathogenesis of osteoporosis. Recenti Prog Med (2008) 99:309–13.
162. Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol (2009) 5:374–81. doi:10.1038/nrendo.2009.106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med (1998) 338:171–9. doi:10.1056/NEJM199801153380307
164. McEwen BS. Allostasis, allostatic load, and the aging nervous system: role of excitatory amino acids and excitotoxicity. Neurochem Res (2000) 25:1219–31. doi:10.1023/A:1007687911139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Donner NC, Montoya CD, Lukkes JL, Lowry CA. Chronic non-invasive corticosterone administration abolishes the diurnal pattern of tph2 expression. Psychoneuroendocrinology (2011) 37:645–61. doi:10.1016/j.psyneuen.2011.08.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Karatsoreos IN, Bhagat SM, Bowles NP, Weil ZM, Pfaff DW, McEwen BS. Endocrine and physiological changes in response to chronic corticosterone: a potential model of the metabolic syndrome in mouse. Endocrinology (2010) 151(5):2117–27. doi:10.1210/en.2009-1436
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Lechner O, Dietrich H, Oliveira dos Santos A, Wiegers GJ, Schwarz S, Harbutz M, et al. Altered circadian rhythms of the stress hormone and melatonin response in lupus-prone MRL/MP-fasIpr mice. J Autoimmun (2000) 14:325–33. doi:10.1006/jaut.2000.0375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Miller AH, Spencer RL, Pearce BD, Pisell TL, Tanapat P, Leung JJ, et al. Effects of viral infection on corticosterone secretion and glucocorticoid receptor binding in immune tissues. Psychoneuroendocrinology (1997) 22:455–74. doi:10.1016/S0306-4530(97)00028-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Schmidt D, Reber SO, Botteron C, Barth T, Peterlik D, Uschold N, et al. Chronic psychosocial stress promotes systemic immune activation and the development of inflammatory Th cell responses. Brain Behav Immun (2010) 24:1097–104. doi:10.1016/j.bbi.2010.04.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Rook GA, Raison CL, Lowry CA. Can we vaccinate against depression? Drug Discov Today (2012) 17:451–8. doi:10.1016/j.drudis.2012.03.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Rook GA, Lowry CA, Raison CL. Microbial ‘old friends’, immunoregulation and stress resilience. Evol Med Public Health (2013) 2013:46–64. doi:10.1093/emph/eot004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Rook GA, Raison CL, Lowry CA. Childhood microbial experience, immunoregulation, inflammation and adult susceptibility to psychosocial stressors and depression in rich and poor countries. Evol Med Public Health (2013) 2013:14–7. doi:10.1093/emph/eos005
173. Reber SO, Obermeier F, Straub RH, Veenema AH, Neumann ID. Aggravation of DSS-induced colitis after chronic subordinate colony (CSC) housing is partially mediated by adrenal mechanisms. Stress (2008) 11:225–34. doi:10.1080/10253890701733351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Czech B, Neumann ID, Muller M, Reber SO, Hellerbrand C. Effect of chronic psychosocial stress on nonalcoholic steatohepatitis in mice. Int J Clin Exp Pathol (2013) 6:1585–93.
175. Peters S, Slattery DA, Flor PJ, Neumann ID, Reber SO. Differential effects of baclofen and oxytocin on the increased ethanol consumption following chronic psychosocial stress in mice. Addict Biol (2013) 18:66–77. doi:10.1111/adb.12001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Peters S, Grunwald N, Rummele P, Endlicher E, Lechner A, Neumann ID, et al. Chronic psychosocial stress increases the risk for inflammation-related colon carcinogenesis in male mice. Stress (2012) 15:403–15. doi:10.3109/10253890.2011.631232
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Edwards AV, Jones CT. The effect of splanchnic nerve section on the sensitivity of the adrenal cortex to adrenocorticotrophin in the calf. J Physiol (1987) 390:23–31. doi:10.1113/jphysiol.1987.sp016683
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Edwards AV, Jones CT. The effect of splanchnic nerve stimulation on adrenocortical activity in conscious calves. J Physiol (1987) 382:385–96. doi:10.1113/jphysiol.1987.sp016373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Ulrich-Lai YM, Arnhold MM, Engeland WC. Adrenal splanchnic innervation contributes to the diurnal rhythm of plasma corticosterone in rats by modulating adrenal sensitivity to ACTH. Am J Physiol Regul Integr Comp Physiol (2006) 290:R1128–35. doi:10.1152/ajpregu.00042.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Ehrhart-Bornstein M, Hinson JP, Bornstein SR, Scherbaum WA, Vinson GP. Intraadrenal interactions in the regulation of adrenocortical steroidogenesis. Endocr Rev (1998) 19(2):101–43. doi:10.1210/edrv.19.2.0326
181. Ehrhart-Bornstein M, Bornstein SR. Cross-talk between adrenal medulla and adrenal cortex in stress. Ann N Y Acad Sci (2008) 1148:112–7. doi:10.1196/annals.1410.053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Wotjak CT, Ganster J, Kohl G, Holsboer F, Landgraf R, Engelmann M. Dissociated central and peripheral release of vasopressin, but not oxytocin, in response to repeated swim stress: new insights into the secretory capacities of peptidergic neurons. Neuroscience (1998) 85:1209–22. doi:10.1016/S0306-4522(97)00683-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Landgraf R, Neumann I, Schwarzberg H. Central and peripheral release of vasopressin and oxytocin in the conscious rat after osmotic stimulation. Brain Res (1988) 457:219–25. doi:10.1016/0006-8993(88)90689-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Jaroenporn S, Nagaoka K, Ohta R, Watanabe G, Taya K. Direct effects of prolactin on adrenal steroid release in male hatano high-avoidance (HAA) rats may be mediated through Janus kinase 2 (Jak2) activity. J Reprod Dev (2007) 53:887–93. doi:10.1262/jrd.18174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Jaroenporn S, Nagaoka K, Ohta R, Shirota M, Watanabe G, Taya K. Differences in adrenocortical secretory and gene expression responses to stimulation in vitro by ACTH or prolactin between high- and low-avoidance Hatano rats. Informa Healthcare (2009) 12:22–9. doi:10.1080/10253890801976652
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Stachowiak A, Macchi C, Nussdorfer G, Malendowicz L. Effects of oxytocin on the function and morphology of the rat adrenal cortex: in vitro and in vivo investigations. Res Exp Med (1995) 195:265–74. doi:10.1007/BF02576797
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Straub RH, Herfarth H, Falk W, Andus T, Scholmerich J. Uncoupling of the sympathetic nervous system and the hypothalamic-pituitary-adrenal axis in inflammatory bowel disease? J Neuroimmunol (2002) 126:116–25. doi:10.1016/S0165-5728(02)00047-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Berton O, Aguerre S, Sarrieau A, Mormede P, Chaouloff F. Differential effects of social stress on central serotonergic activity and emotional reactivity in Lewis and spontaneously hypertensive rats. Neuroscience (1998) 82:147–59. doi:10.1016/S0306-4522(97)00282-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Engler H, Stefanski V. Social stress and T cell maturation in male rats: transient and persistent alterations in thymic function. Psychoneuroendocrinology (2003) 28:951–69. doi:10.1016/S0306-4530(02)00117-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Stefanski V. Social stress in loser rats: opposite immunological effects in submissive and subdominant males. Physiol Behav (1998) 63:605–13. doi:10.1016/S0031-9384(97)00492-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Zelena D, Haller J, Halasz J, Makara GB. Social stress of variable intensity: physiological and behavioral consequences. Brain Res Bull (1999) 48:297–302. doi:10.1016/S0361-9230(98)00176-2
192. Choi DC, Nguyen MM, Tamashiro KL, Ma LY, Sakai RR, Herman JP. Chronic social stress in the visible burrow system modulates stress-related gene expression in the bed nucleus of the stria terminalis. Physiol Behav (2006) 89:301–10. doi:10.1016/j.physbeh.2006.05.046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Lucas LR, Celen Z, Tamashiro KL, Blanchard RJ, Blanchard DC, Markham C, et al. Repeated exposure to social stress has long-term effects on indirect markers of dopaminergic activity in brain regions associated with motivated behavior. Neuroscience (2004) 124:449–57. doi:10.1016/j.neuroscience.2003.12.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Razzoli M, Carboni L, Andreoli M, Ballottari A, Arban R. Different susceptibility to social defeat stress of BalbC and C57BL6/J mice. Behav Brain Res (2011) 216:100–8. doi:10.1016/j.bbr.2010.07.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Savignac HM, Hyland NP, Dinan TG, Cryan JF. The effects of repeated social interaction stress on behavioural and physiological parameters in a stress-sensitive mouse strain. Behav Brain Res (2011) 216:576–84. doi:10.1016/j.bbr.2010.08.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Melhorn SJ, Krause EG, Scott KA, Mooney MR, Johnson JD, Woods SC, et al. Meal patterns and hypothalamic NPY expression during chronic social stress and recovery. Am J Physiol Regul Integr Comp Physiol (2010) 299:R813–22. doi:10.1152/ajpregu.00820.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
197. Blanchard DC, Spencer RL, Weiss SM, Blanchard RJ, McEwen B, Sakai RR. Visible burrow system as a model of chronic social stress: behavioral and neuroendocrine correlates. Psychoneuroendocrinology (1995) 20:117–34. doi:10.1016/0306-4530(94)E0045-B
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Engler H, Engler A, Bailey MT, Sheridan JF. Tissue-specific alterations in the glucocorticoid sensitivity of immune cells following repeated social defeat in mice. J Neuroimmunol (2005) 163:110–9. doi:10.1016/j.jneuroim.2005.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Reber SO, Obermeier F, Straub RH, Falk W, Neumann ID. Chronic intermittent psychosocial stress (social defeat/overcrowding) in mice increases the severity of an acute DSS-induced colitis and impairs regeneration. Endocrinology (2006) 147:4968–76. doi:10.1210/en.2006-0347
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. Schmidt MV, Sterlemann V, Ganea K, Liebl C, Alam S, Harbich D, et al. Persistent neuroendocrine and behavioral effects of a novel, etiologically relevant mouse paradigm for chronic social stress during adolescence. Psychoneuroendocrinology (2007) 32:417–29. doi:10.1016/j.psyneuen.2007.02.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
201. Stefanski V, Peschel A, Reber S. Social stress affects migration of blood T cells into lymphoid organs. J Neuroimmunol (2003) 138:17–24. doi:10.1016/S0165-5728(03)00076-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
202. Avitsur R, Stark JL, Sheridan JF. Social stress induces glucocorticoid resistance in subordinate animals. Horm Behav (2001) 39:247–57. doi:10.1006/hbeh.2001.1653
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
203. Engler H, Bailey MT, Engler A, Stiner-Jones LM, Quan N, Sheridan JF. Interleukin-1 receptor type 1-deficient mice fail to develop social stress-associated glucocorticoid resistance in the spleen. Psychoneuroendocrinology (2008) 33:108–17. doi:10.1016/j.psyneuen.2007.10.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
204. Bailey MT, Engler H, Powell ND, Padgett DA, Sheridan JF. Repeated social defeat increases the bactericidal activity of splenic macrophages through a toll-like receptor-dependent pathway. Am J Physiol Regul Integr Comp Physiol (2007) 293:R1180–90. doi:10.1152/ajpregu.00307.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
205. Quan N, Avitsur R, Stark JL, He L, Shah M, Caligiuri M, et al. Social stress increases the susceptibility to endotoxic shock. J Neuroimmunol (2001) 115:36–45. doi:10.1016/S0165-5728(01)00273-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
206. Bailey MT, Engler H, Sheridan JF. Stress induces the translocation of cutaneous and gastrointestinal microflora to secondary lymphoid organs of C57BL/6 mice. J Neuroimmunol (2006) 171:29–37. doi:10.1016/j.jneuroim.2005.09.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
207. van Hogezand RA, Eichhorn RF, Choudry A, Veenendaal RA, Lamers CB. Malignancies in inflammatory bowel disease: fact or fiction? Scand J Gastroenterol Suppl (2002) 236:48–53. doi:10.1080/003655202320621454
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
208. Eaden JA, Mayberry JF. Colorectal cancer complicating ulcerative colitis: a review. Am J Gastroenterol (2000) 95:2710–9. doi:10.1111/j.1572-0241.2000.02297.x
209. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut (2001) 48:526–35. doi:10.1136/gut.48.4.526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
210. Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxy- methane and dextran sodium sulfate. Cancer Sci (2003) 94:965–73. doi:10.1111/j.1349-7006.2003.tb01386.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
211. Konishi F, Morson BC. Pathology of colorectal adenomas: a colonoscopic survey. J Clin Pathol (1982) 35:830–41. doi:10.1136/jcp.35.8.830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
212. Tanaka T. Colorectal carcinogenesis: review of human and experimental animal studies. J Carcinog (2009) 8:5–24. doi:10.4103/1477-3163.49014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
213. Botrugno OA, Fayard E, Annicotte JS, Haby C, Brennan T, Wendling O, et al. Synergy between LRH-1 and beta-catenin induces G1 cyclin-mediated cell proliferation. Mol Cell (2004) 15:499–509. doi:10.1016/j.molcel.2004.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
214. Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through beta-catenin. Science (2002) 296:1644–6. doi:10.1126/science.1071549
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
215. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell (1996) 87:159–70. doi:10.1016/S0092-8674(00)81333-1
216. Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci U S A (1995) 92:4482–6. doi:10.1073/pnas.92.10.4482
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
217. Schoonjans K, Dubuquoy L, Mebis J, Fayard E, Wendling O, Haby CL, et al. Liver receptor homolog 1 contributes to intestinal tumor formation through effects on cell cycle and inflammation. Proc Natl Acad Sci U S A (2005) 102:2058–62. doi:10.1073/pnas.0409756102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
218. McConnell BB, Yang VW. The role of inflammation in the pathogenesis of colorectal cancer. Curr Colorectal Cancer Rep (2009) 5:69–74. doi:10.1007/s11888-009-0011-z
219. Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology (1994) 107:1183–8.
220. Oguma K, Oshima H, Aoki M, Uchio R, Naka K, Nakamura S, et al. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. EMBO J (2008) 27:1671–81. doi:10.1038/emboj.2008.105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
221. Martin F, Ladoire S, Mignot G, Apetoh L, Ghiringhelli F. Human FOXP3 and cancer. Oncogene (2010) 29:4121–9. doi:10.1038/onc.2010.174
222. Yehuda R. Post-traumatic stress disorder. N Engl J Med (2002) 346:108–14. doi:10.1056/NEJMra012941
223. Mulder EJH, Robles de Medina PG, Huizink AC, Van den Bergh BRH, Buitelaar JK, Visser GHA. Prenatal maternal stress: effects on pregnancy and the (unborn) child. Early Hum Dev (2002) 70:3–14. doi:10.1016/S0378-3782(02)00075-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
224. Cherkasova M, Sulla EM, Dalena KL, Ponde MP, Hechtman L. Developmental course of attention deficit hyperactivity disorder and its predictors. J Can Acad Child Adolesc Psychiatry (2013) 22:47–54.
225. Slattery DA, Cryan JF. The ups and downs of modelling mood disorders in rodents. ILAR J (2014) 55:297–309. doi:10.1093/ilar/ilu026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
226. Young JW, Henry BL, Geyer MA. Predictive animal models of mania: hits, misses and future directions. Br J Pharmacol (2011) 164:1263–84. doi:10.1111/j.1476-5381.2011.01318.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
227. Gorka Z, Moryl E, Papp M. Effect of chronic mild stress on circadian rhythms in the locomotor activity in rats. Pharmacol Biochem Behav (1996) 54:229–34. doi:10.1016/0091-3057(95)02173-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
228. Blanchard RJ, Yudko E, Dulloog L, Blanchard DC. Defense changes in stress nonresponsive subordinate males in a visible burrow system. Physiol Behav (2001) 72:635–42. doi:10.1016/S0031-9384(00)00449-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
229. Sterlemann V, Ganea K, Liebl C, Harbich D, Alam S, Holsboer F, et al. Long-term behavioral and neuroendocrine alterations following chronic social stress in mice: implications for stress-related disorders. Horm Behav (2008) 53:386–94. doi:10.1016/j.yhbeh.2007.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
230. Adamec RE, Shallow T. Lasting effects on rodent anxiety of a single exposure to a cat. Physiol Behav (1993) 54:101–9. doi:10.1016/0031-9384(93)90050-P
231. Henniger MSH, Ohl F, Holter SM, Weienbacher P, Toschi N, Lorscher P, et al. Unconditioned anxiety and social behaviour in two rat lines selectively bred for high and low anxiety-related behaviour. Behav Brain Res (2000) 111:153–63. doi:10.1016/S0166-4328(00)00151-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
232. Landgraf R, Wigger A. High vs low anxiety-related behavior rats: an animal model of extremes in trait anxiety. Behav Genet (2002) 32:301–14. doi:10.1023/A:1020258104318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
233. Tang X, Sanford LD. Home cage activity and activity-based measures of anxiety in 129P3/J, 129X1/SvJ and C57BL/6J mice. Physiol Behav (2005) 84:105–15. doi:10.1016/j.physbeh.2004.10.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
234. Krishnan V, Han M-H, Graham DL, Berton O, Renthal W, Russo SJ, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell (2007) 131:391–404. doi:10.1016/j.cell.2007.09.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
235. Castro JE, Diessler S, Varea E, Marquez C, Larsen MH, Cordero MI, et al. Personality traits in rats predict vulnerability and resilience to developing stress-induced depression-like behaviors, HPA axis hyper-reactivity and brain changes in pERK1/2 activity. Psychoneuroendocrinology (2012) 37:1209–23. doi:10.1016/j.psyneuen.2011.12.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
236. Krishnan V, Han MH, Mazei-Robison M, Iniguez SD, Ables JL, Vialou V, et al. AKT signaling within the ventral tegmental area regulates cellular and behavioral responses to stressful stimuli. Biol Psychiatry (2008) 64:691–700. doi:10.1016/j.biopsych.2008.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
237. Schmidt MV, Scharf SH, Sterlemann V, Ganea K, Liebl C, Holsboer F, et al. High susceptibility to chronic social stress is associated with a depression-like phenotype. Psychoneuroendocrinology (2010) 35:635–43. doi:10.1016/j.psyneuen.2009.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
238. Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the national comorbidity survey replication. Arch Gen Psychiatry (2005) 62:593–602. doi:10.1001/archpsyc.62.6.593
239. American-Psychiatric-Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: American Psychiatric Association Press (1994).
240. Berton O, McClung CA, DiLeone RJ, Krishnan V, Renthal W, Russo SJ, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science (2006) 311:864–8. doi:10.1126/science.1120972
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
241. Covington HE III, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, et al. Antidepressant actions of histone deacetylase inhibitors. J Neurosci (2009) 29:11451–60. doi:10.1523/jneurosci.1758-09.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
242. Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature (2008) 455:894–902. doi:10.1038/nature07455
243. Lukas M, Toth I, Reber SO, Slattery DA, Veenema AH, Neumann ID. The neuropeptide oxytocin facilitates pro-social behavior and prevents social avoidance in rats and mice. Neuropsychopharmacology (2011) 36:2159–68. doi:10.1038/npp.2011.95
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
244. Toth I, Neumann ID, Slattery DA. Social fear conditioning: a novel and specific animal model to study social anxiety disorder. Neuropsychopharmacology (2012) 37:1433–43. doi:10.1038/npp.2011.329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
245. Toth I, Neumann ID, Slattery DA. Social fear conditioning as an animal model of social anxiety disorder. Curr Protoc Neurosci (2013) Chapter 9:Unit 9.42. doi:10.1002/0471142301.ns0942s63
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
246. Kalueff AV, Avgustinovich DF, Kudryavtseva NN, Murphy DL. BDNF in anxiety and depression. Science (2006) 312:1598–9. doi:10.1126/science.312.5780.1598
247. Hirschfeld RM. The comorbidity of major depression and anxiety disorders: recognition and management in primary care. Prim Care Companion J Clin Psychiatry (2001) 3:244–54. doi:10.4088/PCC.v03n0609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
248. Kennedy SH. Core symptoms of major depressive disorder: relevance to diagnosis and treatment. Dialogues Clin Neurosci (2008) 10:271–7.
249. Kessler RC, Coccaro EF, Fava M, Jaeger S, Jin R, Walters E. The prevalence and correlates of DSM-IV intermittent explosive disorder in the national comorbidity survey replication. Arch Gen Psychiatry (2006) 63:669–78. doi:10.1001/archpsyc.63.6.669
250. Mackenzie CS, Reynolds K, Chou KL, Pagura J, Sareen J. Prevalence and correlates of generalized anxiety disorder in a national sample of older adults. Am J Geriatr Psychiatry (2011) 19:305–15. doi:10.1097/JGP.0b013e318202bc62
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
251. Cryan JF, Slattery DA. GABAB receptors and depression. Current status. Adv Pharmacol (2010) 58:427–51. doi:10.1016/S1054-3589(10)58016-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
252. Kinsey SG, Bailey MT, Sheridan JF, Padgett DA, Avitsur R. Repeated social defeat causes increased anxiety-like behavior and alters splenocyte function in C57BL/6 and CD-1 mice. Brain Behav Immun (2007) 21:458–66. doi:10.1016/j.bbi.2006.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
253. Cappell H, Herman CP. Alcohol and tension reduction. A review. Q J Stud Alcohol (1972) 33:33–64.
254. Linsky AS, Straus MA, Colby JP Jr. Stressful events, stressful conditions and alcohol problems in the United States: a partial test of Bales’s theory. J Stud Alcohol (1985) 46:72–80.
255. Caldwell EE, Riccio DC. Alcohol self-administration in rats: modulation by temporal parameters related to repeated mild social defeat stress. Alcohol (2010) 44:265–74. doi:10.1016/j.alcohol.2010.02.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
256. Croft AP, Brooks SP, Cole J, Little HJ. Social defeat increases alcohol preference of C57BL/10 strain mice; effect prevented by a CCKB antagonist. Psychopharmacology (Berl) (2005) 183:163–70. doi:10.1007/s00213-005-0165-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
257. van Erp AM, Miczek KA. Persistent suppression of ethanol self-administration by brief social stress in rats and increased startle response as index of withdrawal. Physiol Behav (2001) 73:301–11. doi:10.1016/S0031-9384(01)00458-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
258. Becker HC, Lopez MF, Doremus-Fitzwater TL. Effects of stress on alcohol drinking: a review of animal studies. Psychopharmacology (Berl) (2011) 218:131–56. doi:10.1007/s00213-011-2443-9
259. Cippitelli A, Damadzic R, Singley E, Thorsell A, Ciccocioppo R, Eskay RL, et al. Pharmacological blockade of corticotropin-releasing hormone receptor 1 (CRH1R) reduces voluntary consumption of high alcohol concentrations in non-dependent Wistar rats. Pharmacol Biochem Behav (2012) 100:522–9. doi:10.1016/j.pbb.2011.10.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
260. Molander A, Vengeliene V, Heilig M, Wurst W, Deussing JM, Spanagel R. Brain-specific inactivation of the Crhr1 gene inhibits post-dependent and stress-induced alcohol intake, but does not affect relapse-like drinking. Neuropsychopharmacology (2012) 37:1047–56. doi:10.1038/npp.2011.297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
261. Slattery DA, Neumann ID. Chronic icv oxytocin attenuates the pathological high anxiety state of selectively bred Wistar rats. Neuropharmacology (2010) 58:56–61. doi:10.1016/j.neuropharm.2009.06.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
262. Slattery DA, Neumann ID. Oxytocin and major depressive disorder: experimental and clinical evidence for links to aetiology and possible treatment. Pharmaceuticals (2010) 3:702–24. doi:10.3390/ph3030702
263. Blume A, Bosch OJ, Miklos S, Torner L, Wales L, Waldherr M, et al. Oxytocin reduces anxiety via ERK1/2 activation: local effect within the rat hypothalamic paraventricular nucleus. Eur J Neurosci (2008) 27:1947–56. doi:10.1111/j.1460-9568.2008.06184.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
264. Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science (2003) 301:386–9. doi:10.1126/science.1083968
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
265. McCarthy MM, McDonald CH, Brooks PJ, Goldman D. An anxiolytic action of oxytocin is enhanced by estrogen in the mouse. Physiol Behav (1997) 60:1209–15. doi:10.1016/S0031-9384(96)00212-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
266. Waldherr M, Neumann ID. Centrally released oxytocin mediates mating-induced anxiolysis in male rats. Proc Natl Acad Sci U S A (2007) 104:16681–4. doi:10.1073/pnas.0705860104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
267. Windle RJ, Shanks N, Lightman SL, Ingram CD. Central Oxytocin administration reduces stress-induced corticosterone release and anxiety behavior in rats. Endocrinology (1997) 138:2829–34. doi:10.1210/en.138.7.2829
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
268. Windle RJ, Kershaw YM, Shanks N, Wood SA, Lightman SL, Ingram CD. Oxytocin attenuates stress-induced c-fos mRNA expression in specific forebrain regions associated with modulation of hypothalamo-pituitary-adrenal activity. J Neurosci (2004) 24:2974–82. doi:10.1523/JNEUROSCI.3432-03.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
269. Landgraf R, Neumann ID. Vasopressin and oxytocin release within the brain: a dynamic concept of multiple and variable modes of neuropeptide communication. Front Neuroendocrinol (2004) 25:150–76. doi:10.1016/j.yfrne.2004.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
270. Peters S, Slattery DA, Uschold-Schmidt N, Reber SO, Neumann ID. Dose-dependent effects of chronic central infusion of oxytocin on anxiety, oxytocin receptor binding and stress-related parameters in mice. Psychoneuroendocrinology (2014) 42:225–36. doi:10.1016/j.psyneuen.2014.01.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
271. Yoshida M, Takayanagi Y, Inoue K, Kimura T, Young LJ, Onaka T, et al. Evidence that oxytocin exerts anxiolytic effects via oxytocin receptor expressed in serotonergic neurons in mice. J Neurosci (2009) 29:2259–71. doi:10.1523/jneurosci.5593-08.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
272. Kirby LG, Zeeb FD, Winstanley CA. Contributions of serotonin in addiction vulnerability. Neuropharmacology (2011) 61:421–32. doi:10.1016/j.neuropharm.2011.03.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
273. McGregor IS, Callaghan PD, Hunt GE. From ultrasocial to antisocial: a role for oxytocin in the acute reinforcing effects and long-term adverse consequences of drug use? Br J Pharmacol (2008) 154:358–68. doi:10.1038/bjp.2008.132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
274. Hebb ALO, Zacharko RM, Gauthier M, Trudel F, Laforest S, Drolet G. Brief exposure to predator odor and resultant anxiety enhances mesocorticolimbic activity and enkephalin expression in CD-1 mice. Eur J Neurosci (2004) 20:2415–29. doi:10.1111/j.1460-9568.2004.03704.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
275. Steciuk M, Kram M, Kramer GL, Petty F. Decrease in stress-induced c-Fos-like immunoreactivity in the lateral septal nucleus of learned helpless rats. Brain Res (1999) 822:256–9. doi:10.1016/S0006-8993(99)01134-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
276. Yadin E, Thomas E, Grishkat HL, Strickland CE. The role of the lateral septum in anxiolysis. Physiol Behav (1993) 53:1077–83. doi:10.1016/0031-9384(93)90362-J
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
277. Salchner P, Sartori SB, Sinner C, Wigger A, Frank E, Landgraf R, et al. Airjet and FG-7142-induced Fos expression differs in rats selectively bred for high and low anxiety-related behavior. Neuropharmacology (2006) 50:1048–58. doi:10.1016/j.neuropharm.2006.02.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
278. Bannerman DM, Rawlins JN, McHugh SB, Deacon RM, Yee BK, Bast T, et al. Regional dissociations within the hippocampus--memory and anxiety. Neurosci Biobehav Rev (2004) 28:273–83. doi:10.1016/j.neubiorev.2004.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
279. Yoon SH, Kim BH, Ye SK, Kim MH. Chronic non-social stress affects depressive behaviors but not anxiety in mice. Korean J Physiol Pharmacol (2014) 18:263–8. doi:10.4196/kjpp.2014.18.3.263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
280. Kulesskaya N, Karpova NN, Ma L, Tian L, Voikar V. Mixed housing with DBA/2 mice induces stress in C57BL/6 mice: implications for interventions based on social enrichment. Front Behav Neurosci (2014) 8:257. doi:10.3389/fnbeh.2014.00257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: chronic psychosocial stress, chronic subordinate colony housing, somatic and affective disorders, decreased glucocorticoid signaling, hypocorticism
Citation: Langgartner D, Füchsl AM, Uschold-Schmidt N, Slattery DA and Reber SO (2015) Chronic subordinate colony housing paradigm: a mouse model to characterize the consequences of insufficient glucocorticoid signaling. Front. Psychiatry 6:18. doi: 10.3389/fpsyt.2015.00018
Received: 20 October 2014; Paper pending published: 09 January 2015;
Accepted: 29 January 2015; Published online: 23 February 2015.
Edited by:
Ripu D. Jindal, University of Pittsburgh School of Medicine, USAReviewed by:
Osborne F. Almeida, Max Planck Institute of Psychiatry, GermanyKarl Bechter, University of Ulm, Germany
Copyright: © 2015 Langgartner, Füchsl, Uschold-Schmidt, Slattery and Reber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan O. Reber, Laboratory for Molecular Psychosomatics, Clinic for Psychosomatic Medicine and Psychotherapy, University Ulm, Albert-Einstein-Allee 23, Ulm 89081, Germany e-mail:c3RlZmFuLnJlYmVyQHVuaWtsaW5pay11bG0uZGU=