

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 18 January 2013
Sec. Molecular Psychiatry
Volume 3 - 2012 | https://doi.org/10.3389/fpsyt.2012.00118
Autism is a neurodevelopmental disorders characterized by impairments in communication and social behavior, and by repetitive behaviors. Although genetic factors might be largely responsible for the occurrence of autism they cannot fully account for all cases and it is likely that in addition to a certain combination of autism-related genes, specific environmental factors might act as risk factors triggering the development of autism. Thus, the role of environmental factors in autism is an important area of research and recent data will be discussed in this review. Interestingly, the results show that many environmental risk factors are interrelated and their identification and comparison might unveil a common scheme of alterations on a contextual as well as molecular level. For example, both, disruption in the immune system and in zinc homeostasis may affect synaptic transmission in autism. Thus, here, a model is proposed that interconnects the most important and scientifically recognized environmental factors. Moreover, similarities in how these risk factors impact synapse function are discussed and a possible influence on an already well described genetic pathway leading to the development of autism via zinc homeostasis is proposed.
In the last decade, multiple genes have been implicated in autism, collectively accounting for approximately 15% of cases including autism spectrum disorders (ASDs; Abrahams and Geschwind, 2010). In this context, an increasing number of ASDs can be attributed to rare genetic changes that are either inherited or appear de novo and further mutations and candidate genes are increasingly identified. Besides a handful of single specific genes that can be associated with autism, the current theory supports the idea of a polygenic inheritance, meaning that multiple genes are likely to be involved that may predispose an individual to develop autism. Although inherited factors might be largely responsible for the occurrence of autism, it is equally clear that genetic aberrations cannot fully account for all cases of autism. The California Autism Twins Study (CATS) for example, with 192 identical and fraternal twin pairs shows a concordance rate of 77% for male monozygotic twins and 50% for female identical twins. The rates among fraternal twins were 31% (male) and 36% (female; Hallmayer et al., 2011). The concordance rate of 31/36% of fraternal twins is higher than the observed rate of 3–14% between siblings of different ages. Thus, in addition to a genetic heritability, common factors such as the shared prenatal environment might play a role in the formation of ASD. Moreover, the increasing prevalence of autism has drawn attention to the potential involvement of toxins in our environment. Extending the theory of pure genetic causes, it seems likely that in addition to a certain combination of autism-related genes, exposure to specific environmental factors might be necessary to trigger the development of autism in some individuals. However, environmental risk factors do not solely cover the exposure to toxins but include all changes other than those on a DNA – level such as maternal nutrition, infection during pregnancy, and prematurity as well as parental age at conception.
This review highlights the role of environmental risk factors in autism in the context of emerging genetic research which suggests that the synapse is an organelle particularly vulnerable to genetic disruption and possibly disruption by related environmental influences. In particular, recent data suggest that immune system abnormalities and altered zinc homeostasis may affect synaptic transmission. Understanding how genetic and environmental risk factors in autism converge at synapses might provide a valuable starting point for future work towards uncovering the patho-mechanisms of autism.
Autism is a developmental disorder and most cases are diagnosed by the age of three and as early as 14 months (Landa, 2008). Nonetheless, autism might be present from birth on rather than develop within this developmental time window. Thus, prenatal environmental factors are of considerable interest for the development of autism (Table 1).

Table 1. Summary of the presented environmental risk factors for autism.
Failures in early fetal brain development have been linked to a higher risk for autism and attention has been drawn to offspring exposed to viral or bacterial infections in utero (Arndt et al., 2005; Libbey et al., 2005; Miller et al., 2005; Patterson, 2009). For instance, infections that have been associated with autism include prenatal influenza, rubella, and cytomegalovirus infections (Pardo et al., 2005). However, the outcome of exposure to a prenatal viral infection depends on many factors such as maternal immune status, susceptibility of the maternal and fetal host, the developmental stage of the fetus, the amount of virus reaching the fetus, the route of access and the infecting virus, and strain of virus (Blattner, 1974). Nevertheless, given the variety of viruses and their pathogenic effects that can be associated with autism, immunological imbalance in general might be an underlying risk factor for autism (Pardo et al., 2005). This hypothesis is underlined by many reports of abnormalities in peripheral immune cells, as well as associations between variants of genes for cytokines, their receptors, and human leukocyte antigens (HLA) in autism (Patterson, 2009). It was shown that immune changes early in development persist throughout development and aging and are still detectable in the adult brain (Patterson, 2009). Despite these long lasting changes, specific gestational windows may be associated with a higher susceptibility to infection-mediated deregulations in brain development (Meyer et al., 2007).
The risk for autism associated with prenatal infections is most likely dependent on the individual immune status of the mother and fetus. This is substantiated by the finding of a possible association of autism with autoimmune disease and allergy, particularly in the mother (Fox et al., 2012). Consequentially, maternal infection could alter the immune status of the fetal brain and the fetal immune system in general, since the placenta serves as the source of hematopoietic stem cells for the fetus (Gekas et al., 2005). A pathology of the placenta has been linked to the development of autism (Anderson et al., 2007).
Metal ion homeostasis is essential for proper brain function and its disruption is implicated in severe neurological symptoms and cognitive diseases. Zinc (Zn2+) -deficiency has many different effects on the brain. Zn2+ is the second most abundant trace element in the body and thus not surprisingly, plays a role in many different processes, such as cell division and differentiation. For instance, hundreds of enzymes require Zn2+ for their functions (Takeda, 2001). Many studies have shown that Zn2+-deficiency affects several brain functions and interferes with neuronal maturation via impairment of Zn2+ dependent systems during early development, resulting in severe brain dysfunctions such as irrecoverable impairment of learning and memory. Approximately 90% of the total brain Zn2+ is bound to proteins with free Zn2+ mostly located in presynaptic vesicles (Frederickson, 1989). However, transgenic animals without presynaptic Zn2+ were not shown to develop symptoms of autism. Recent studies have shown that Zn2+ ions are able to modulate the PSD scaffold of synapses via the autism-associated proteins Shank2 and Shank3 (Jan et al., 2002; Baron et al., 2006).
Within the body, copper (Cu2+) and Zn2+ have competing roles in a way that Cu2+ overload leads to Zn2+-deficiency (Hall et al., 1979; Huster, 2010). Former studies describe a significant elevation of Cu2+ in the hair and nail samples of subjects with autism (Lakshmi Priya and Geetha, 2011). Moreover, further studies indicate that the Cu2+/Zn2+ ratio is increased in the serum of subjects with autism (Faber et al., 2009) and was suggested as biomarker for children with autism. However, Zn2+ deficiency might not only be a biomarker for autism, but indeed a risk factor. Many autistic children have Zn2+-deficiency (Walsh et al., 2001, 2002; Jen and Yan, 2010; Yasuda et al., 2011). In a recent study, hair Zn2+ concentrations from 1,967 children with autism were investigated and an incidence rate of Zn2+-deficiency in the infant-group aged 0–3-year-old was estimated 43.5% in male and 52.5% in female. These findings suggest that infantile Zn2+-deficiency may contribute to the pathogenesis of autism (Yasuda et al., 2011).
Zn2+-deficiency causes neuropsychological symptoms, learning, and memory impairments (Golub et al., 1995), behavioral problems and an enhancement of glutamate excitotoxicity. Therefore, Zn2+-deficiency is also associated with the occurrence of seizures (Grabrucker et al., 2011a) and many patients with autism suffer from epilepsy.
Some candidate genes that have been identified so far to be associated with the development of autism, such as COMMD1 (COMM domain-containing protein 1), MTF1 (metal regulatory transcription factor 1), metallothioneins (MTs), ZnT5 (zinc transporter 5), ERK1 (extracellular signal-regulated kinase 1), TrkB (tyrosine-related kinase B), SHANK2, and SHANK3 (SH3 and multiple ankyrin repeat domains 2/3, also known as proline-rich synapse-associated protein (ProSAP1 and ProSAP2; Serajee et al., 2004; Huang et al., 2008; Grabrucker et al., 2011b; Levy et al., 2011; O’Roak et al., 2011; Nuttall and Oteiza, 2012; Sanders et al., 2012), are influenced by Zn2+ or themselves involved in Zn2+-signaling and metal ion homeostasis. Thus, Zn2+ might provide a mechanism of gene/environment interaction and maternal Zn2+-deficiency might act as risk factor for autism. Zn2+-deficiency in pregnant rhesus monkeys has effects on the behavior of infants (Sandstead et al., 1978). Intriguingly Zn2+-deficiency is known to compromise the immune system, providing a possible link to prenatal infections.
It was hypothesized that abnormalities in melatonin secretion may play a role in the development of autism. Melatonin is an endogenous neurohormone produced in the dark predominantly in the pineal gland and levels of melatonin or melatonin derivatives were found to be often below average in individuals with ASD (Rossignol and Frye, 2011). Along with the decreased levels of melatonin, children with ASD have a higher prevalence of sleep abnormalities such as longer “sleep onset latency,” frequent night-time awakenings and reduced sleep duration (Rossignol and Frye, 2011). Sleep problems in ASD might occur as a result of complex interactions between genetic and social/environmental factors (Cortesi et al., 2010). Circadian abnormalities in autism might be the result of genetic abnormalities related to melatonin synthesis. Deletions of the ASMT (acetylserotonin O-methyltransferase) gene, encoding the last enzyme of melatonin synthesis, have been found in several individuals with ASD (Melke et al., 2008). Additionally, environmental factors can influence melatonin synthesis. For instance, maternal stress was shown to result in reduced melatonin levels (Feng et al., 2012) as well as light pollution (Reiter et al., 2006; Falchi et al., 2011). Interestingly, Zn2+-deficiency has an effect on various hormones such as melatonin. A reduction in Zn2+ levels in pinealectomized rats suggests a reciprocal affect between Zn2+ and melatonin and a significant decrease in melatonin levels was observed in Zn2+-deficient animals (Bediz et al., 2003).
Although melatonin is best known for its role as a key regulator of the circadian rhythm, it is also a potent antioxidant, has anti-inflammatory properties, is involved in the immune response, and helps regulate synaptic plasticity (Rossignol and Frye, 2011). Given that a low melatonin level can also be found in healthy individuals, it cannot be considered as a direct cause of ASD, but as a susceptibility factor. The effects of maternal melatonin deficiency on the offspring have not been investigated thoroughly in terms of an association with ASDs.
Meta-analysis revealed diabetes in the mother during pregnancy as a risk factor for autism (Gardener et al., 2009) with a twofold increased risk. Still, the pathological mechanisms remain unknown. In line with these data, a study (Krakowiak et al., 2012) found that metabolic conditions such as obesity, hypertension, or diabetes could occur more frequently in mothers of ASD cases. However, obesity alone was significantly associated with this risk in this study, while only a trend was observed for hypertension and diabetes.
Several lines of evidence show significant effects of prenatal stress activating the hypothalamic-pituitary-adrenal (HPA) axis on postnatal behavior in human and animal studies. If autism can result from prenatal stress is currently investigated and the duration, severity, and type of stress are under debate. So far, autism has been reported to be associated with prenatal stress both with retrospective studies (Ward, 1990; Beversdorf et al., 2005) and by comparison to historical events such as storm catastrophes. Moreover, animal studies have reported that prenatal stress produces behaviors resembling symptoms of autism (Kinney et al., 2008). Along with behavioral symptoms significant changes have been found in the regulation of the HPA axis of these animals, regardless of the specific prenatal stressor used. Additionally, prenatal exposure to either stress hormones or psychological stress of the mother in rhesus monkeys resulted in abnormalities in postnatal immune function in infancy that were persistent throughout late childhood (Coe et al., 1999). These immune functions such as proliferation of lymphocytes, natural killer cell activity, and production of cytokines, might decrease the ability to resist viral and bacterial infections and thus the pathological mechanisms of prenatal stress might be related to the increased risk of autism seen with prenatal infections. Stress can also be produced by complications of labor and delivery. Both are associated with greater risk of hypoxia and cerebral hemorrhage in the newborn and a study shows that those who survived cerebellar hemorrhagic injury had a significantly increased risk to develop autism (Limperopoulos et al., 2007).
Independent from maternal health, environmental agents are threatening the development of a fetus, causing birth defects. These so called teratogens have been suggested as potential risk factor for autism. For example, the incidence of autism is significantly higher in children prenatally exposed to the anticonvulsant agent valproic acid (Moore et al., 2000) or the immunomodulatory agent thalidomide (Stromland et al., 1994) early in the first trimester of gestation. Valproic acid for instance, an anticonvulsant used in the treatment of epilepsy and also bipolar disorder due to its mood-modifying ability, is a histone deacetylase inhibitor and might change the methylation pattern of autism – associated genes, thereby affecting their expression rate. Intriguingly, administration of valproic acid in utero leads to developmental delays and lifelong deficits in motor performance, social behavior, and anxiety – like behavior in the offspring (Kolozsi et al., 2009). Along with this, Neuroligin 3 (NLGN3) mRNA expression was significantly decreased in hippocampal CA1 region, dentate gyrus, and somatosensory cortex of these animals (Kolozsi et al., 2009). Nlgn3, a member of neuroligin family, is expressed in the postsynaptic compartment of neurons and mediates transsynaptic signaling by interacting with neurexin (NRXN; Südhof, 2008). Mutations in NLGN3 and NRXN have been reported as genetic cause for ASD (Jamain et al., 2003).
Thalidomide, which was infamous for causing birth defects when used as an antiemetic in pregnancy in 1950/1960 still is of pharmacological interest given its immunomodulatory effects (Kumar and Chhibber, 2011). The observed immunomodulation by thalidomide might be related to the increased risk observed through prenatal infection. Thalidomide, for instance, modulates cytokine levels and macrophage pro-inflammatory function (Kumar et al., 2010). It was also found to have anti-angiogenic and immunomodulatory properties including T cell co-stimulation, and activation of NK (natural killer) cells (Quach et al., 2010).
Prenatal exposure to organophosphate pesticides such as diazinon and chlorpyrifos, agents that have been shown to be neurotoxic (Karr et al., 2007), may contribute to autism. A genetic predisposition seems to enhance vulnerability for these substances (Dufault et al., 2012). Additionally, a study found that women who are in the first 8 weeks of pregnancy in closer contact with the organochlorine pesticides dicofol and endosulfan due to their residential proximity to sprayed fields, are several times more likely to give birth to children with autism (Roberts et al., 2007). However, a limited number of cases and studies makes these findings hard to interpret and the concordance rate is not 100%, which suggests that a genetic predisposition might be necessary for a toxin to act as trigger (Szpir, 2006).
A more solid association for a factor for the development of autism can be found in the use of psychiatric drugs in the mother during pregnancy (Gardener et al., 2009). The association of psychoactive medications could be achieved through various ways. Since most of these drugs are able to cross the placenta, the effect might indeed be caused by direct exposure to the psychoactive substance. Nevertheless, the actually treated condition of the mother might itself have an influence on fetal development or reflect certain genetic traits that are possibly shared between autism and the mother’s condition. Moreover, the need of psychoactive medication in the mother may hint towards a prenatal stress situation, a condition that has been linked to autism before.
The risk to develop autism is associated with advanced age in either parent (Gardener et al., 2009; Shelton et al., 2010; Parner et al., 2012). A meta-analysis of epidemiological studies including 25,687 ASD cases and 8,655,576 control subjects investigating the association between maternal age and autism supports advanced maternal age as a risk factor for autism. The association still persisted after considering effects of paternal age (Sandin et al., 2012). Further studies indicate that the father’s age is of similar importance given that men transmit a much higher number of mutations to their children than women (Kong et al., 2012) and that the age of the father is the dominant factor in determining the number of de novo mutations in the child (Kong et al., 2012). Thus, advanced paternal age was similarly associated with increased risk of ASDs in offspring (Croen et al., 2007; Grether et al., 2009; Reichenberg et al., 2010; Shelton et al., 2010; Buizer-Voskamp et al., 2011; van Balkom et al., 2012). However, advanced paternal and maternal age may act as risk factors through different mechanisms. While most small-scale de novo mutations accumulate in the paternal germline over time related to parental age (Iossifov et al., 2012), eggs are fully developed at the time of a mother’s birth. Thus, advanced maternal age might contribute as risk factor for autism through mechanisms such as increased pregnancy complications (Gardener et al., 2009) and maternal autoimmunity (Enstrom et al., 2009). The risk factor of maternal autoimmunity again hints towards the involvement of an immune response during pregnancy as a risk factor for autism. Pro-inflammatory cytokines possibly arising from maternal autoimmunity might cause aberrant neuronal growth and function within the developing fetal brain (Buehler, 2011).
Although it is assumed that children are born with autism, a wide variety of postnatal factors contributing to autism have been proposed (Table 1). However, the evidence for these risk factors, such as gastrointestinal or immune system abnormalities, allergies, and exposure of children to drugs, infection, certain foods, or heavy metals needs further substantiation. Vaccination can no longer be regarded as risk factor for autism (Doja and Roberts, 2006). Evidence from several rigorous scientific studies examining an association between vaccine use and autism have not identified such a link (Miller and Reynolds, 2009) and the data of former studies claiming an association were shown to be scientifically fraudulent (Flaherty, 2011).
Gastrointestinal abnormalities, in terms of the “Leaky gut syndrome” as risk factor for autism are based on parents reporting gastrointestinal disturbances in autistic children, and several studies have investigated possible associations between autism and the gut (Liu et al., 2005; Iebba et al., 2011). Additionally, a connection between autism and opiates was proposed (Sahley and Panksepp, 1987). Caused by a digestive disorder, gluten, and casein might be converted to the opioid peptides gliadorphin and casomorphin, thus influencing the development of autism. However, more evidence is needed to verify that autistic children are more likely to have gastrointestinal pathologies than control children. Nevertheless, it is tempting to speculate that a possible link between malnutrition, gastrointestinal abnormalities, and Zn2+-deficiency reported as risk factors for autism may exist. Similarly, Zn2+-deficiency might influence the immune system and it was hypothesized that autoantibodies that compromise brain development or important metabolic pathways may be a risk factor for autism. Similar to maternal antibodies, via an autoimmune mechanism, own antibodies may cause an aberrant immune activity (Wills et al., 2007).
Heavy metal poisoning by mercury and lead have been associated with autism. Given that elevated lead levels in the blood of autistic children have been found, lead poisoning has been suggested as a possible risk factor (Cohen et al., 1982). Lead and stress share biological substrates and produce common adverse effects (Rossi-George et al., 2011). However, the evidence so far is indirect for the association between autism and mercury or lead exposure after birth. Moreover, atypical eating behaviors of autistic children, such as seen in “pica” (the persistent eating of non-nutritive substances for a period of at least 1 month; Kinnell, 1985) may influence lead levels more than being a consequence of autism (Cohen et al., 1976).
Taken together, there is overwhelming evidence that autism is a genetic disorder. Twin studies indicate a genetic basis for autism and genome wide association studies have revealed some interesting candidate genes (Miles, 2011; Li et al., 2012). Nevertheless, environmental factors can have an obvious influence on the development of autism. It is tempting to speculate that most of the identified risk factors so far, such as prenatal stress, prenatal infection, maternal Zn2+-deficiency, and maternal exposure to toxins as well as potential novel factors that will be identified in the future might indeed all be different sides of the same coin. Thus, the search for similarities in all the identified genetic as well as environmental factors might lead to a time window in brain development, where certain brain regions and specific pathways are prone to disruption caused by various agents but all leading to the phenotype of autism. Figure 1 shows an attempt to unite the several environmental risk factors identified in autism so far and tries to bridge the gap between these and genetic factors (Figure 1). From this analysis, two central motifs, immune system abnormalities, and Zn2+-deficiency, emerge.
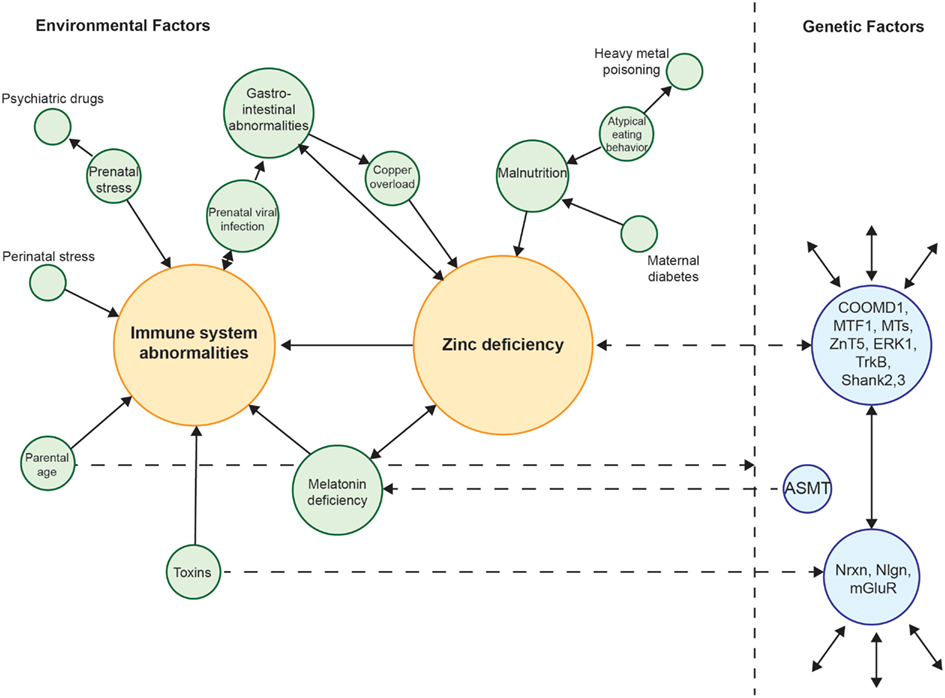
Figure 1. Interconnection of various environmental risk factors for autism. Many risk factors for autism can be related to each other. All factors are shown weighted for their interconnectedness with other factors. Zn2+-deficiency and immune system aberrations receive most connections and thus can be placed in the center of the network. Zn2+-deficiency, toxins, and the parental age may act on genetic factors providing a crosstalk between genes and environment. Zn2+-deficiency and melatonin deficiency in turn can also be caused by genetic alterations.
A close connection exists between a functional immune system and the development of the central nervous system and a successful neurodevelopment requires a normal balance of immune responses (Ashwood et al., 2006). Immune system abnormalities might result from toxins that compromise important players of the system, stress, parental age, Zn2+-deficiency, and infections.
Prenatal stress in turn was associated with an abnormally developing HPA system, an affected placenta as well as hippocampal volume reduction. Smaller hippocampal volumes in turn are associated with an increased secretion of cortisol (Frodl and O’Keane, 2012). Intriguingly, patients with ASD were characterized by a significant reduction of gray matter volume in the amygdala-hippocampus complex (Via et al., 2011). Cortisol as the primary end product of the HPA axis is an important component of the stress system in humans and essential for fetal growth and the induction of specific enzymes (Charil et al., 2010). Therefore, women have naturally elevated levels of cortisol during pregnancy. However, it is possible that maternal cortisol concentrations can reach abnormally high levels under particular stressful conditions. Cortisol that reaches the fetus may potentially alter fetal development and growth. Animal studies have shown that fetal exposure to high levels of cortisol alters neuronal development and results in a smaller hippocampal size (Charil et al., 2010).
The placenta is similarly affected by prenatal stress and releases Corticotropin releasing factor (CRF) from the placenta during sub-acute and chronic prenatal stress that may cross the blood-brain barrier of the fetus. Consequently, the function and integrity of the hippocampus is affected, for instance by an increase in release of acetylcholine, inhibiting the influence of cholinergic receptors on the activity of the HPA axis (Charil et al., 2010).
Parental age is correlated with an increased risk of maternal autoimmunity as the majority of autoimmune disorders are manifest between 30 years of age and upward (Ashwood et al., 2006). Autoimmune disorders arise when the immune system is inappropriately directed to recognize and response to self-components and maternal autoantibodies may affect fetal brain development during critical periods of neurodevelopment (Chonchaiya et al., 2010). Components of the immune system such as cytokines and autoantibodies, may act to increase fetal cytokine signaling and expression, disrupt trophic factor, and apoptotic signaling, as well as modulate cytoarchitecture in the CNS (Murphy et al., 2010). Prevalence rates suggest that autoimmune diseases, such as rheumatoid arthritis, autoimmune thyroid diseases, myasthenia gravis, multiple sclerosis, systemic lupus erythematosus (SLE), and primary biliary cirrhosis are not uncommon, affecting approximately 5% of women and 3% of men in the USA (Murphy et al., 2010).
Prenatal infections could persistently alter the immune status of the fetus. An inflammatory-like state has been detected in postmortem brains of autism patients, indicated by elevated cytokines and activated microglia and astrocytes. Moreover, cytokine elevation was also found in the cerebrospinal fluid (CSF) of living autistic children (Pardo et al., 2005). Cytokine IL-6 thereby attracted special attention. IL-6 leads to a reduction in insulin-like growth factor (IGFs)-binding protein 3 and IGF1 (IGFs 1; Patterson, 2009). Intriguingly, IL-6 also regulates Zn2+ binding MTs (Manso et al., 2011). Animal studies suggest that these inflammatory changes might ultimately alter NMDA receptor mediated excitatory synaptic transmission and plasticity in the hippocampus (Escobar et al., 2011). The inflammatory-like state was also detected in the peripheral immune system. Immune cell may infiltrate into the colon, ileum, and duodenum and an increased T cell activation can be present in the intestinal mucosa. Thus, these inflammatory changes may also lead to gastrointestinal abnormalities. However, in terms of prenatal infections it is unclear, if the infection itself causes an imbalance of the immune system or if a compromised immune system in the first place leads to an increased susceptibility for infections.
The second central environmental factor is Zn2+-deficiency. Zinc is an essential trace element and plays various roles in biological processes. For instance, Zn2+ is a structural component of proteins and involved in enzymatic processes and a transcriptional regulator (Tuerk and Fazel, 2009). Zn2+-deficiency itself influences the immune system and low Zn2+ status has been associated with an increased susceptibility for infections (Wong and Ho, 2012). Zn2+-deficiency significantly impairs both adaptive and innate immune responses, and promotes systemic inflammation. Zn2+ is essential for normal development and function of cell-mediating innate immunity, neutrophils, and natural killer cells. For instance, macrophages, phagocytosis, intracellular killing, and the growth and function of T and B cells are all affected by Zn2+-deficiency. Zn2+-deficiency also affects the production, secretion, and functions of cytokines (see thalidomide), the basic messengers of the immune system (Prasad, 2009). Cytokines have widespread effects on neuronal pathways and are able to show behavioral effects such as alteration of mood and sleep (Ashwood et al., 2006). Imbalances in the immune system were seen in humans with induced mild Zn2+-deficiency within 12 weeks of a Zn2+-restricted diet (Prasad, 2009).
Zn2+ homeostasis is also highly regulated via the gastrointestinal tract (Tuerk and Fazel, 2009) and gastrointestinal abnormalities have been mentioned as risk factor for autism. Moreover, viral infections might affect the gastrointestinal system providing another link between immune system abnormalities and Zn2+-deficiency. However, Zn2+-deficiency can be caused by many factors that have been associated with autism in the past, such as maternal diabetes, malnutrition, copper overload, and atypical eating behavior. Maternal diabetes and obesity have been associated with this risk to develop autism and may influence the developing child similar to malnutrition. Vegetarian diet usually provides a low intake of Zn2+ (Craig, 2010) and also fiber rich diet, suggested for diabetes (Wolfram and Ismail-Beigi, 2011), increases the risk of Zn2+-deficiency (Foster et al., 2012). Intriguingly the consumption of fiber rich diet within the USA increased in the last decades (Hiza and Bente, 2007).
Within the body Cu2+ and Zn2+ have competing roles in a way, where Cu2+ overload leads to Zn2+-deficiency (Hall et al., 1979; Huster, 2010). Cu2+ overload in an extreme form is seen in patients suffering from Wilson’s disease, where homozygous mutation of the ATP7B gene results in Cu2+ accumulation in tissues manifesting in neurological or psychiatric symptoms among others. Although the disease is rather rare, the heterozygous form with only one faulty copy of the affected gene is not so rare. It was speculated that in this case, the developing fetus of a pregnant woman may experience major difficulties in the early development of the brain due to a Cu2+ overload – induced Zn2+-deficiency, which may later manifest as autism (Johnson, 2001). Atypical eating behaviors in autism, such as pica, in turn may influence again the nutritional status but also cause heavy metal poisoning. In this case, both scenarios would be postnatally affecting the development of autism. Nevertheless, a heavy metal intoxication during pregnancy might again disturb Zn2+ homeostasis.
Melatonin deficiency may be the result and cause of a Zn2+-deficiency. Melatonin also has an impact on the immune system, thus possibly influencing both major identified environmental factors for the development of autism.
Severe Zn2+-deficiency is rare and mostly occurs in developing countries. Marginal Zn2+-deficiency in contrast, is a potentially widespread problem. Based on a study of 14,770 individuals aged 3–74 years, the prevalence of Zn2+-deficiency in the USA was estimated with 1–3 and 10% of the US population consumes less than half the recommended level for Zn2+ and thus is at risk for Zn2+-deficiency (Tuerk and Fazel, 2009; Wong and Ho, 2012). Moreover, women may not ingest adequate amounts of Zn2+ during pregnancy (Halas and Sandstead, 1975) and suffer from decreased levels of Zn2+ during the latter part of pregnancy (Halsted and Smith, 1970; Sandstead, 1973). However, since Zn2+-deficiencies can be found in many brain disorders such as schizophrenia and depression (Grabrucker et al., 2011a), more research is needed to investigate the cause of these Zn2+-deficiencies.
Intriguingly, environmental factors such as Zn2+-deficiency might have a crosstalk with genetic factors. Some autism candidate genes that have been identified so far, such as COMMD1, MTF1, MTs, ZnT5, ERK1, TrkB, Shank2, and Shank3 (Serajee et al., 2004; Huang et al., 2008; Grabrucker et al., 2011b; Levy et al., 2011; O’Roak et al., 2011; Nuttall and Oteiza, 2012; Sanders et al., 2012), are influenced by Zn2+ or themselves involved in Zn2+ signaling and metal ion homeostasis. The idea that many genes implicated in autism might converge on a single pathway present at excitatory glutamatergic synapses has recently been raised by multiple studies and a Nrxn-Nlgn-Shank pathway has been proposed (Bourgeron, 2009). Thus, Shank proteins, via their dependency on Zn2+ ions (Grabrucker et al., 2011c), might influence further autism-associated proteins. For instance, mGluR5, an important target in fragile-X-syndrome is an interaction partner of Shank proteins (Lim et al., 1999). However, it is possible that other identified autism-associated genes have different crossing points with the environmental risk factors for autism discussed here and future research will picture the model of environmental factors influencing genetic predispositions for autism in more detail.
Given this interconnectedness of many environmental and genetic factors associated with the development of autism, one can speculate how these risk factors might converge an a single hypothetical pathway by looking at possible similarities in the mode of action. Autism is caused by abnormalities in the structure or functions of the brain, but the underlying biological patho-mechanisms are currently not well understood. However, various recent studies have identified copy number variations (CNVs) as well as single nucleotide polymorphisms (SNPs) of neuronal genes in the genomes of ASD patients. Many of these genes encode proteins that are crucial components of excitatory glutamatergic synapses, including scaffold proteins such as Shank2, Shank3, SAPAP2 from the GKAP/SAPAP family of Shank interacting proteins, neural adhesion molecules, such as Neuroligins, Neurexins as well as Cadherins, Contactin, Contactin-associated protein, and other cell-adhesion molecules (CAMs), calcium channels, and neurotransmitter receptors (Betancur et al., 2009; Pinto et al., 2010; Toro et al., 2010). Moreover, protein kinases and proteins that regulate protein syntheses have been identified as candidate genes associated with ASD. Mouse models for these genes underline their role in autism and hint towards a deregulation of excitatory synapses since all these proteins play a role at the synapse of glutamatergic neurons. Thus, these data imply a link between a synaptic pathology and the characteristics of ASD and shift the synaptic compartment in the focus of interest.
To study a crossing point of environmental factors with genetic predispositions and the development of autism, excitatory synapses may provide a useful starting point. Given that a synaptopathy is the underlying mechanism for autism and focusing on the two central environmental factors “immune system abnormalities” and “zinc deficiency,” similarities can be found influencing synaptic function and plasticity (Figure 2).
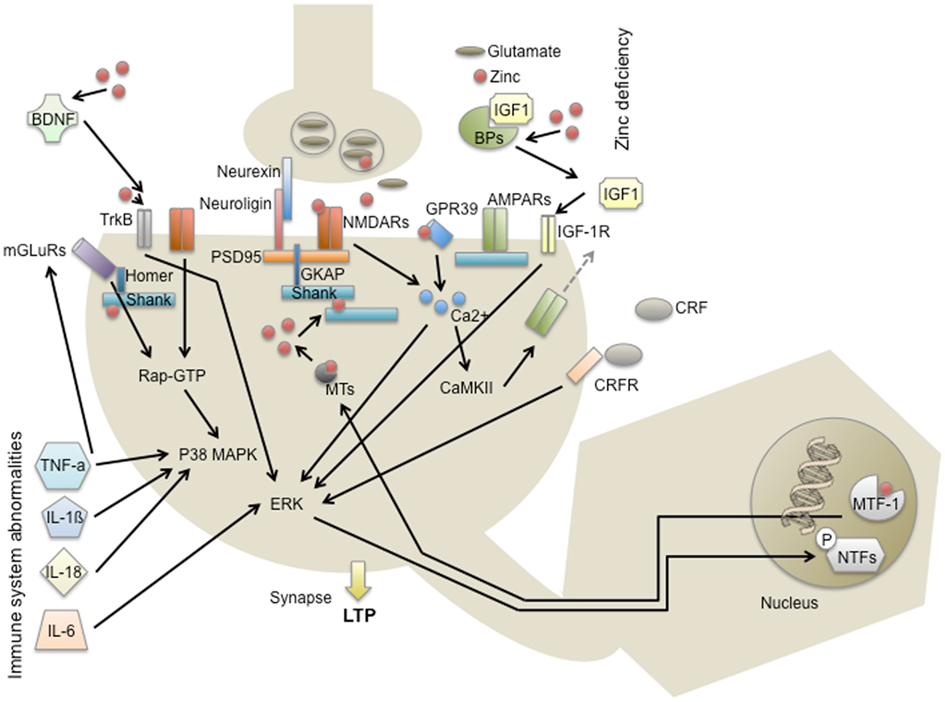
Figure 2. Modes of action of environmental risk factors for autism at excitatory synapses in the CNS. Scaffold proteins within the PSD such as proteins of the Shank family build a dense meshwork of interacting protein complexes including Homer, mGluR, AMPAR, and GKAP further connecting to PSD95 and NMDAR. These synaptic components are regulated via signaling cascades such as the p38 MAPK and ERK pathway and altogether ultimately determine the immediate and long-term response to a synaptic stimulus. Environmental factors that have been discussed in autism may influence this signaling outcome on various levels. Zn2+-deficiency for instance might be able to alter the PSD scaffold via Shank2 and Shank3 and additionally influences NMDAR, TrkB receptor, and GPR39 receptor signaling. Moreover, Zn2+ that might be released from presynaptic vesicles or postsynaptic metallothioneins can affect IGF1 signaling and gene expression via MTF1. Immune system abnormalities, including the expression of cytokines such as TNF-α, Il-1β, IL-18, and IL-6 will affect mGluR, p38 MPAK, and ERK signaling, pathways that are also activated by the receptors mentioned above.
Abnormalities in the immune system are known to result in altered cytokine and major histocompatibility complex (MHC) expression and release of nitric oxide (NO) and cyclo-oxygenase products on a molecular level. Thus, inflammatory responses can have several effects on the brain. Circulating immune cells may infiltrate the CNS, resident cells are activated and pro-inflammatory mediators such as cytokines are produced and released. These immune diffusible mediators may influence synaptic processes such as long-term potentiation (LTP; Di Filippo et al., 2008), a molecular mechanism thought to underlie synaptic plasticity in learning and memory formation. LTP manifests as a persistent increase of the evoked response following repeated synaptic activation. The generation of LTP requires activation of postsynaptic NMDA receptors, subsequent postsynaptic Ca2+ entry together with the induction of several signaling systems and finally gene transcription and new protein synthesis (Malenka and Nicoll, 1999). Cytokines and other molecules overexpressed during the neuroinflammatory process act as neuromodulators with pro-inflammatory cytokines showing a detrimental effect on neuronal function, viability, and plasticity (Allan and Rothwell, 2001) even when expressed at low levels. In particular, the release of neurotransmitter is influenced as well as signal transduction mechanisms within the postsynaptic density (PSD) and, finally, the ability to induce LTP.
For instance, IL-1 crucially influences several key molecular components of LTP – induction, such as NMDA and AMPA receptor signaling (Coogan and O’Connor, 1997; Lai et al., 2006) and glutamate release (Murray et al., 1997). Similarly, IL-18, a member of the IL-1 family, is able to impair the induction of LTP, probably by negatively modulating NMDA receptor mediated transmission in the hippocampus (Curran and O’Connor, 2001). However, the effects of IL-1 on NMDA and AMPA receptors have not been completely elucidated to date and might be brain region specific. IL-6 is thought to modulate the ERK pathway, a signaling pathway involved in the generation of LTP. Additionally, an increase in IL-6 leads to a reduction in IGFs-binding protein 3 and IGF1 (Patterson, 2009). Intriguingly, IL-6 also influences one of the major Zn2+ stores in postsynapses, the Zn2+ binding MT proteins (Manso et al., 2011).
IL-18, IL-1β, and TNF-α (Tumor necrosis factor alpha) act on AMPA and NMDA as well as metabotropic receptor signaling, thereby modulating the activity of the stress activated p38 MAPK. TNF-α, a pro-inflammatory cytokine binds to receptors, which are expressed on neurons and glial cells throughout the CNS. Interestingly, the inhibition of mGluR1 and mGluR5 can block the TNF-α – dependent inhibition of early and late LTP. Thus, a combined activation of TNF receptors and mGluRs may impair the formation of LTP, although the exact mechanism of TNF and mGlu receptor activation is still not well understood (Pickering et al., 2005).
Besides their role as modulators of brain growth, IGFs also influence brain plasticity. Specifically, IGFs regulate synapse formation, neurotransmitter release, and neuronal excitability via posttranslational modification of NMDA and AMPA receptors (Torres-Aleman, 1999). IGF1 acts primarily on the IGF1 receptor, which is neuronally expressed and coupled to the PI3K-AKT signal transduction pathway that positively influences cellular growth and thus affects the maturation of outgrowing neurites and synapse like structures. In the case of IGF1, this feature has already been used therapeutically in preclinical settings, where the application of recombinant IGF1 was demonstrated to improve synaptic maturation in a mouse model for Rett syndrome, a type of ASD (Tropea et al., 2009). Moreover, a crosstalk between IGFR activation and the ERK pathway can be found.
Thus, immune system abnormalities might ultimately alter NMDA receptor mediated excitatory synaptic transmission and plasticity (Escobar et al., 2011). Similar results have already been shown in animal studies where changes in NMDAR responses and NMDAR dependent LTP as well as an imbalance in NMDAR levels and subunit composition were reported in recent mouse models for autism (Bangash et al., 2011; Wang et al., 2011; Schmeisser et al., 2012; Won et al., 2012).
Prenatal stress might additionally contribute to the above – mentioned effects via CRF secretion. Stress in known to influence synaptic transmission in brain regions, such as hippocampus, ultimately affecting learning, and memory. Whereas physiological levels of CRF augment memory processes, exposure of high levels of the peptide results in retraction of dendritic spines of excitatory synapses in the hippocampus and amygdala and thus to synapse loss. Although only a minority of dendritic spines is lost due to long lasting CRF exposure, a profound memory impairment and loss of LTP can be observed (Chen et al., 2012). However, the molecular mechanisms of this synapse loss are not fully understood. Intriguingly, CRF activates, through CRF1, the ERK pathway in the area CA3 of the hippocampus.
The idea of Zn2+ being a regulatory core component within PSDs results in novel insights and understanding concerning synapse formation and might hint towards a mechanism of Zn2+-deficiency in autistic patients. Indeed several autism-associated genes that can be found at excitatory postsynapses, such as Shank2 and Shank3, MTs and ERK are Zn2+ binding, and Zn2+ regulated proteins. Shank proteins further connect to mGluR, GKAP, Neuroligins, and Neurexins. To date, the source of postsynaptic Zn2+ involved in synaptic plasticity is still unclear and might include presynaptic vesicular Zn2+ and/or postsynaptic activity dependent release from Zn2+ stores such as MTs. Focusing on excitatory synapses, it has been shown that Zn2+ influences the activity of glutamate receptors. NMDA receptors have a very high sensitivity to extracellular Zn2+ and the activity of GluN2A subunit containing NMDA receptors is inhibited allosterically at low nanomolar concentrations, while at micromolar Zn2+ concentrations that may occur during synaptic activity, Zn2+ binds to the GluN2B subunit, inhibiting GluN2B-containing receptors (Sensi et al., 2011).
Zn2+ also activates a specific metabotropic Zn2+-sensing receptor GPR39, which triggers IP3 production, followed by intracellular Ca2+ and subsequent activation of ERK and CAMKII-dependent pathways (Besser et al., 2009). Moreover, Zn2+ directly promotes the transactivation of the BDNF (brain-derived neurotrophic factor)-related TrkB pathway (Huang et al., 2008) and maternal Zn2+-deficiency decreases BDNF expression in the offspring (Chowanadisai et al., 2005). In a recent study, NTFs (neurotrophic factors) were measured in 414 ASD cases and 820 controls. ASD cases were more likely to have significantly decreased BDNF levels and significantly lower TGF-β (transforming growth factor beta) levels were detected in females with ASD. These data hint towards decreased NTFs levels during neonatal period as risk factor for ASD contributing to the pathophysiology of ASD through impairments of neuroplasticity (Abdallah et al., 2012).
Given that Zn2+ increases IGF1 receptor sensitivity, Zn2+ also affects IGF1 signaling in a way that Zn2+-deficiency may decrease IGF1 receptor binding (McCusker and Novakofski, 2004). In general, neurons control cytosolic Zn2+ levels tightly using several ZnT transporters, ZIP transporters, and Zn2+ buffering MTs and Zn2+-deficiency might influence all of the above – mentioned pathways and proteins. Thus, the many faces of Zn2+ taking part in such a high number of biological and synaptic processes makes it hard to pinpoint the mechanism involved in the development of autism and the presented model might have to be expanded in the future.
Taken together, two major targets of environmental factors contributing to the development of autism can be found on a synaptic level. The first is the structural composition of synapses that might be altered through deletions or mutations in key scaffold or CAMs such as Neurexin, Neuroligins, GKAP, and proteins of the ProSAP/Shank family. This might ultimately result in changes in receptor number, or composition influencing NMDAR, AMPAR, and mGluR and affect signaling components within the PSD. For instance, Zn2+ depletion was shown to cause a decrease in Shank3 and NMDAR levels of forming synapses in vitro (Grabrucker et al., 2011c). Loss of Shank3 in turn affects transsynaptic signaling via Neuroligin-Neurexin complexes (Arons et al., 2012) and mGluR5 signaling (Verpelli et al., 2011) and Shank3 knock-out mice display a reduction of AMPAR and NMDAR (Bozdagi et al., 2010; Bangash et al., 2011). Intriguingly, prenatal exposure to valproic acid leads to reduced expression of the synaptic adhesion molecule Neuroligin 3 in mice (Kolozsi et al., 2009).
The second major target is the p38 MAPK (mitogen-activated protein kinase) and ERK pathway. ERK regulates two cellular processes closely associated with plasticity: synaptic delivery of AMPAR (Zhu et al., 2002), and activity dependent dendritic spine modifications (Goldin and Segal, 2003). ERK kinases are themselves heavily influenced by the local Zn2+ concentration (Nuttall and Oteiza, 2012). Additionally, ERK2 knock-out mice exhibit marked anomalies in multiple aspects of social behaviors related to facets of ASDs and impaired long-term memory (Satoh et al., 2011). Moreover, Zn2+ signaling via GPR39 further alters the receptor composition of excitatory synapses. Thus, both, alterations in synapse morphology induced by structural changes and disturbed signaling by kinases might converge leading to disrupted LTP formation and plasticity at synapses. Therefore, a specific decrement in excitatory signaling caused by various factors, such as genetic mutations or environmental factors like immune system abnormalities or Zn2+-deficiency, will ultimately lead to an imbalance of inhibition and excitation. An imbalance of excitation and inhibition was already proposed as underlying cause of autism (Rubenstein and Merzenich, 2003).
Of course, the proposed model is an oversimplification and should be considered a starting point rather than a finalized concept. Many studies have not been considered here and might add, revise, or refine the interactions described above. For instance, the incorporation of novel risk factors or brain region specific effects might deepen the understanding of environmental factors in autism and of autism itself.
Experimental research often is designed in a way that one component of a system is manipulated and the various outcomes documented. For example, a transgenic mouse model with the knock-out of a specific gene might display a pleiotropy of symptoms depending on the specific role of the encoded protein. However, the cause for these various symptoms is known, since only a single gene has been manipulated.
In the case of autism, current research faces the inverse situation. Many symptoms are observed and many significant associations of environmental as well as genetic factors were found. What is missing so far is a hypothesis, unifying all those different factors chasing autism back to a single cause, for instance the disturbance of a specific synaptic pathway that is influenced by genetic and environmental factors. The fact that many autism-associated genes are interconnected and that also many environmental factors seem to be related and impact genetic factors makes this goal look achievable in the future. Like a necklace of pearls, where a cut between two single parts at a random position in the chain will lead to the drop of every pearl, the deregulation of this environmentally influenced genetic pathway at any level may results in autism. However, it is likely that the cut in the signaling chain might influence the occurrence and severity of specific features of autism.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Andreas M. Grabrucker is supported by Baustein 3.2 (L.SBN.0083) and the DAAD.
Abdallah, M. W., Mortensen, E. L., Greaves-Lord, K., Larsen, N., Bonefeld-Jørgensen, E. C., Nørgaard-Pedersen, B., et al. (2012). Neonatal levels of neurotrophic factors and risk of autism spectrum disorders. Acta Psychiatr. Scand. 1–9.
Abrahams, B. S., and Geschwind, D. H. (2010). Connecting genes to brain in the autism spectrum disorders. Arch. Neurol. 67, 395–299.
Allan, S. M., and Rothwell, N. J. (2001). Cytokines and acute neurodegeneration. Nat. Rev. Neurosci. 2, 734–744.
Anderson, G. M., Jacobs-Stannard, A., Chawarska, K., Volkmar, F. R., and Kliman, H. J. (2007). Placental trophoblast inclusions in autism spectrum disorder. Biol. Psychiatry 61, 487–491.
Arndt, T. L., Stodgell, C. J., and Rodier, P. M. (2005). The teratology of autism. Int. J. Dev. Neurosci. 23, 189–199.
Arons, M., Thynne, C. J., Grabrucker, A. M., Li, D., Schoen, M., Cheyne, J. E., et al. (2012). Autism associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin mediated transsynaptic signaling. J. Neurosci. 32, 14966–14978.
Ashwood, P., Wills, S., and Van de Water, J. (2006). The immune response in autism: a new frontier for autism research. J. Leukoc. Biol. 80, 1–15.
Bangash, M. A., Park, J. M., Melnikova, T., Wang, D., Jeon, S. K., Lee, D., et al. (2011). Enhanced polyubiquitination of Shank3 and NMDA receptor in a mouse model of autism. Cell 145, 758–772.
Baron, M. K., Boeckers, T. M., Vaida, B., Faham, S., Gingery, M., Sawaya, M. R., et al. (2006). An architectural framework that may lie at the core of the postsynaptic density. Science 311, 531–535.
Bediz, C. S., Baltaci, A. K., and Mogulkoc, R. (2003). Both zinc deficiency and supplementation affect plasma melatonin levels in rats. Acta Physiol. Hung. 90, 335–339.
Besser, L., Chorin, E., Sekler, I., Silverman, W. F., Atkin, S., Russell, J. T., et al. (2009). Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J. Neurosci. 29, 2890–2901.
Betancur, C., Sakurai, T., and Buxbaum, J. D. (2009). The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 32, 402–412.
Beversdorf, D. Q., Manning, S. E., Hillier, A., Anderson, S. L., Nordgren, R. E., Walters, S. E., et al. (2005). Timing of prenatal stressors and autism. J. Autism Dev. Disord. 35, 471–478.
Bozdagi, O., Sakurai, T., Papapetrou, D., Wang, X., Dickstein, D. L., Takahashi, N., et al. (2010). Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 1, 15.
Buehler, M. R. (2011). A proposed mechanism for autism: an aberrant neuroimmune response manifested as a psychiatric disorder. Med. Hypotheses 76, 863–870.
Buizer-Voskamp, J. E., Laan, W., Staal, W. G., Hennekam, E. A., Aukes, M. F., Termorshuizen, F., et al. (2011). Paternal age and psychiatric disorders: findings from a Dutch population registry. Schizophr. Res. 129, 128–132.
Charil, A., Laplante, D. P., Vaillancourt, C., and King, S. (2010). Prenatal stress and brain development. Brain Res. Rev. 65, 56–79.
Chen, Y., Andres, A. L., Frotscher, M., and Baram, T. Z. (2012). Tuning synaptic transmission in the hippocampus by stress: the CRH system. Front. Cell Neurosci. 6:13. doi:10.3389/fncel.2012.00013
Chonchaiya, W., Tassone, F., Ashwood, P., Hessl, D., Schneider, A., Campos, L., et al. (2010). Autoimmune disease in mothers with the FMR1 premutation is associated with seizures in their children with fragile X syndrome. Hum. Genet. 128, 539–548.
Chowanadisai, W., Kelleher, S. L., and Lönnerdal, B. (2005). Maternal zinc deficiency reduces NMDA receptor expression in neonatal rat brain, which persists into early adulthood. J. Neurochem. 94, 510–519.
Coe, C. L., Lubach, G. R., and Karaszewski, J. W. (1999). Prenatal stress and immune recognition of self and nonself in the primate neonate. Biol. Neonate 76, 301–310.
Cohen, D. J., Johnson, W. T., and Caparulo, B. K. (1976). Pica and elevated blood lead level in autistic and atypical children. Am. J. Dis. Child. 130, 47–48.
Cohen, D. J., Paul, R., Anderson, G. M., and Harcherik, D. F. (1982). Blood lead in autistic children. Lancet 2, 94–95.
Coogan, A., and O’Connor, J. J. (1997). Inhibition of NMDA receptor-mediated synaptic transmission in the rat dentate gyrus in vitro by IL-1 beta. Neuroreport 8, 2107–2110.
Cortesi, F., Giannotti, F., Ivanenko, A., and Johnson, K. (2010). Sleep in children with autistic spectrum disorder. Sleep Med. 11, 659–664.
Craig, W. J. (2010). Nutrition concerns and health effects of vegetarian diets. Nutr. Clin. Pract. 25, 613–620.
Croen, L. A., Najjar, D. V., Fireman, B., and Grether, J. K. (2007). Maternal and paternal age and risk of autism spectrum disorders. Arch. Pediatr. Adolesc. Med. 161, 334–340.
Curran, B., and O’Connor, J. J. (2001). The pro-inflammatory cytokine interleukin-18 impairs long-term potentiation and NMDA receptor-mediated transmission in the rat hippocampus in vitro. Neuroscience 108, 83–90.
Di Filippo, M., Sarchielli, P., Picconi, B., and Calabresi, P. (2008). Neuroinflammation and synaptic plasticity: theoretical basis for a novel, immune-centred, therapeutic approach to neurological disorders. Trends Pharmacol. Sci. 29, 402–412.
Doja, A., and Roberts, W. (2006). Immunizations and autism: a review of the literature. Can. J. Neurol. Sci. 33, 341–346.
Dufault, R., Lukiw, W. J., Crider, R., Schnoll, R., Wallinga, D., and Deth, R. (2012). A macroepigenetic approach to identify factors responsible for the autism epidemic in the United States. Clin. Epigenetics 4, 6.
Enstrom, A. M., Van de Water, J. A., and Ashwood, P. (2009). Autoimmunity in autism. Curr. Opin. Investig. Drugs 10, 463–473.
Escobar, M., Crouzin, N., Cavalier, M., Quentin, J., Roussel, J., Lanté, F., et al. (2011). Early, time-dependent disturbances of hippocampal synaptic transmission and plasticity after in utero immune challenge. Biol. Psychiatry 70, 992–999.
Faber, S., Zinn, G. M., Kern, J. C., and Kingston, H. M. (2009). The plasma zinc/serum Cu2+ ratio as a biomarker in children with autism spectrum disorders. Biomarkers 14, 171–180.
Falchi, F., Cinzano, P., Elvidge, C. D., Keith, D. M., and Haim, A. (2011). Limiting the impact of light pollution on human health, environment and stellar visibility. J. Environ. Manage. 92, 2714–2722.
Feng, P., Hu, Y., Vurbic, D., and Guo, Y. (2012). Maternal stress induces adult reduced REM sleep and melatonin level. Dev. Neurobiol. 72, 677–687.
Flaherty, D. K. (2011). The vaccine-autism connection: a public health crisis caused by unethical medical practices and fraudulent science. Ann. Pharmacother. 45, 1302–1304.
Foster, M., Karra, M., Picone, T., Chu, A., Hancock, D. P., Petocz, P., et al. (2012). Dietary fiber intake increases the risk of zinc deficiency in healthy and diabetic women. Biol. Trace Elem. Res. 149, 135–142.
Fox, E., Amaral, D., and Van de Water, J. (2012). Maternal and fetal antibrain antibodies in development and disease. Dev. Neurobiol. 72, 1327–1334.
Frederickson, C. J. (1989). Neurobiology of zinc and zinc-containing neurons. Int. Rev. Neurobiol. 31, 145–238.
Frodl, T., and O’Keane, V. (2012). How does the brain deal with cumulative stress? A review with focus on developmental stress, HPA axis function and hippocampal structure in humans. Neurobiol. Dis. [in press].
Gardener, H., Spiegelman, D., and Buka, S. L. (2009). Prenatal risk factors for autism: comprehensive meta-analysis. Br. J. Psychiatry 195, 7–14.
Gekas, C., Dieterlen-Lièvre, F., Orkin, S. H., and Mikkola, H. K. (2005). The placenta is a niche for hematopoietic stem cells. Dev. Cell 8, 365–375.
Goldin, M., and Segal, M. (2003). Protein kinase C and ERK involvement in dendritic spine plasticity in cultured rodent hippocampal neurons. Eur. J. Neurosci. 17, 2529–2539.
Golub, M. S., Keen, C. L., Gershwin, M. E., and Hendrickx, A. G. (1995). Developmental zinc deficiency and behavior. J. Nutr. 125, 2263–2271.
Grabrucker, A. M., Rowan, M., and Garner, C. C. (2011a). Brain-delivery of Zinc-ions as potential treatment for neurological diseases. Drug Deliv. Lett. 1, 13–23.
Grabrucker, A. M., Schmeisser, M. J., Schoen, M., and Boeckers, T. M. (2011b). Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol. 21, 594–603.
Grabrucker, A. M., Knight, M. J., Proepper, C., Bockmann, J., Joubert, M., Rowan, M., et al. (2011c). Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 30, 569–581.
Grether, J. K., Anderson, M. C., Croen, L. A., Smith, D., and Windham, G. C. (2009). Risk of autism and increasing maternal and paternal age in a large north American population. Am. J. Epidemiol. 170, 1118–1126.
Halas, E. S., and Sandstead, H. H. (1975). Some effects of prenatal zinc deficiency on behavior of the adult rat. Pediatr. Res. 9, 94–97.
Hall, A. C., Young, B. W., and Bremner, I. (1979). Intestinal metallothionein and the mutual antagonism between Cu2+ and zinc in the rat. J. Inorg. Biochem. 11, 57–66.
Hallmayer, J., Cleveland, S., Torres, A., Phillips, J., Cohen, B., Torigoe, T., et al. (2011). Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 68, 1095–1102.
Hiza, H. A. B., and Bente, L. (2007). Nutrient Content of the U.S. Food Supply, 1909–2004: A Summary Report. Home Economics Research Report, No. 57. U.S. Department of Agriculture, Center for Nutrition and Promotion.
Huang, Y. Z., Pan, E., Xiong, Z. Q., and McNamara, J. O. (2008). Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron 57, 546–558.
Iebba, V., Aloi, M., Civitelli, F., and Cucchiara, S. (2011). Gut microbiota and pediatric disease. Dig. Dis. 29, 531–539.
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299.
Jamain, S., Quach, H., Betancur, C., Råstam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29.
Jan, H. H., Chen, I. T., Tsai, Y. Y., and Chang, Y. C. (2002). Structural role of zinc ions bound to postsynaptic densities. J. Neurochem. 83, 525–534.
Jen, M., and Yan, A. C. (2010). Syndromes associated with nutritional deficiency and excess. Clin. Dermatol. 28, 669–685.
Johnson, S. (2001). Micronutrient accumulation and depletion in schizophrenia, epilepsy, autism and Parkinson’s disease? Med. Hypotheses 56, 641–645.
Karr, C. J., Solomon, G. M., and Brock-Utne, A. C. (2007). Health effects of common home, lawn, and garden pesticides. Pediatr. Clin. North Am. 54, 63–80.
Kinney, D. K., Munir, K. M., Crowley, D. J., and Miller, A. M. (2008). Prenatal stress and risk for autism. Neurosci. Biobehav. Rev. 32, 1519–1532.
Kolozsi, E., MacKenzie, R. N., Roullet, F. I., deCatanzaro, D., and Foster, J. A. (2009). Prenatal exposure to valproic acid leads to reduced expression of synaptic adhesion molecule neuroligin 3 in mice. Neuroscience 163, 1201–1210.
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475.
Krakowiak, P., Walker, C. K., Bremer, A. A., Baker, A. S., Ozonoff, S., Hansen, R. L., et al. (2012). Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics 129, e1121–e1128.
Kumar, V., and Chhibber, S. (2011). Thalidomide: an old drug with new action. J. Chemother. 23, 326–334.
Kumar, V., Harjai, K., and Chhibber, S. (2010). Thalidomide treatment modulates macrophage pro-inflammatory function and cytokine levels in Klebsiella pneumoniae B5055 induced pneumonia in BALB/c mice. Int. Immunopharmacol. 10, 777–783.
Lai, A. Y., Swayze, R. D., El-Husseini, A., and Song, C. (2006). Interleukin-1 beta modulates AMPA receptor expression and phosphorylation in hippocampal neurons. J. Neuroimmunol. 175, 97–106.
Lakshmi Priya, M. D., and Geetha, A. (2011). Level of trace elements (Cu2+, zinc, magnesium and selenium) and toxic elements (lead and mercury) in the hair and nail of children with autism. Biol. Trace Elem. Res. 142, 148–158.
Landa, R. J. (2008). Diagnosis of autism spectrum disorders in the first 3 years of life. Nat. Clin. Pract. Neurol. 4, 138–147.
Levy, D., Ronemus, M., Yamrom, B., Lee, Y. H., Leotta, A., Kendall, J., et al. (2011). Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70, 886–897.
Li, X., Zou, H., and Brown, W. T. (2012). Genes associated with autism spectrum disorder. Brain Res. Bull. 88, 543–552.
Libbey, J. E., Sweeten, T. L., McMahon, W. M., and Fujinami, R. S. (2005). Autistic disorder and viral infections. J. Neurovirol. 11, 1–10.
Lim, S., Naisbitt, S., Yoon, J., Hwang, J. I., Suh, P. G., Sheng, M., et al. (1999). Characterization of the Shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J. Biol. Chem. 274, 29510–29518.
Limperopoulos, C., Bassan, H., Gauvreau, K., Robertson, R. L., Sullivan, N. R., Benson, C. B., et al. (2007). Does cerebellar injury in premature infants contribute to the high prevalence of long-term cognitive, learning, and behavioral disability in survivors? Pediatrics 120, 584–593.
Liu, Z., Li, N., and Neu, J. (2005). Tight junctions, leaky intestines, and pediatric diseases. Acta Paediatr. 94, 386–393.
Malenka, R. C., and Nicoll, R. A. (1999). Long-term potentiation – a decade of progress? Science 285, 1870–1874.
Manso, Y., Adlard, P. A., Carrasco, J., Vašák, M., and Hidalgo, J. (2011). Metallothionein and brain inflammation. J. Biol. Inorg. Chem. 16, 1103–1113.
McCusker, R. H., and Novakofski, J. (2004). Zinc partitions IGFs from soluble IGF binding proteins (IGFBP)-5, but not soluble IGFBP-4, to myoblast IGF type 1 receptors. J. Endocrinol. 180, 227–246.
Melke, J., Goubran Botros, H., Chaste, P., Betancur, C., Nygren, G., Anckarsäter, H., et al. (2008). Abnormal melatonin synthesis in autism spectrum disorders. Mol. Psychiatry 13, 90–98.
Meyer, U., Yee, B. K., and Feldon, J. (2007). The neurodevelopmental impact of prenatal infections at different times of pregnancy: the earlier the worse? Neuroscientist 13, 241–256.
Miller, L., and Reynolds, J. (2009). Autism and vaccination-the current evidence. J. Spec. Pediatr. Nurs. 14, 166–172.
Miller, M. T., Stromland, K., Ventura, L., Johansson, M., Bandim, J. M., and Gillberg, C. (2005). Autism associated with conditions characterized by developmental errors in early embryogenesis: a mini review. Int. J. Dev. Neurosci. 23, 201–219.
Moore, S. J., Turnpenny, P., Quinn, A., Glover, S., Lloyd, D. J., Montgomery, T., et al. (2000). A clinical study of 57 children with fetal anti-convulsant syndromes. J. Med. Genet. 37, 489–497.
Murphy, T. K., Storch, E. A., Turner, A., Reid, J. M., Tan, J., and Lewin, A. B. (2010). Maternal history of autoimmune disease in children presenting with tics and/or obsessive-compulsive disorder. J. Neuroimmunol. 229, 243–247.
Murray, C. A., McGahon, B., McBennett, S., and Lynch, M. A. (1997). Interleukin-1 beta inhibits glutamate release in hippocampus of young, but not aged, rats. Neurobiol. Aging 18, 343–348.
Nuttall, J. R., and Oteiza, P. I. (2012). Zinc and the ERK kinases in the developing brain. Neurotox. Res. 21, 128–141.
O’Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S., et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589.
Pardo, C. A., Vargas, D. L., and Zimmerman, A. W. (2005). Immunity, neuroglia and neuroinflammation in autism. Int. Rev. Psychiatry 17, 485–495.
Parner, E. T., Baron-Cohen, S., Lauritsen, M. B., Jørgensen, M., Schieve, L. A., Yeargin-Allsopp, M., et al. (2012). Parental age and autism spectrum disorders. Ann. Epidemiol. 22, 143–150.
Patterson, P. H. (2009). Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav. Brain Res. 204, 313–321.
Pickering, M., Cumiskey, D., and O’Connor, J. J. (2005). Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp. Physiol. 90, 663–670.
Pinto, D., Pagnamenta, A. T., Klei, L., Anney, R., Merico, D., Regan, R., et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372.
Prasad, A. S. (2009). Zinc: role in immunity, oxidative stress and chronic inflammation. Curr. Opin. Clin. Nutr. Metab. Care 12, 646–652.
Quach, H., Ritchie, D., Stewart, A. K., Neeson, P., Harrison, S., Smyth, M. J., et al. (2010). Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 24, 22–32.
Reichenberg, A., Gross, R., Sandin, S., and Susser, E. S. (2010). Advancing paternal and maternal age are both important for autism risk. Am. J. Public Health 100, 772–773.
Reiter, R. J., Gultekin, F., Manchester, L. C., and Tan, D. X. (2006). Light pollution, melatonin suppression and cancer growth. J. Pineal Res. 40, 357–358.
Roberts, E. M., English, P. B., Grether, J. K., Windham, G. C., Somberg, L., and Wolff, C. (2007). Maternal residence near agricultural pesticide applications and autism spectrum disorders among children in the California Central Valley. Environ. Health Perspect. 115, 1482–1489.
Rossi-George, A., Virgolini, M. B., Weston, D., Thiruchelvam, M., and Cory-Slechta, D. A. (2011). Interactions of lifetime lead exposure and stress: behavioral, neurochemical and HPA axis effects. Neurotoxicology 32, 83–99.
Rossignol, D. A., and Frye, R. E. (2011). Melatonin in autism spectrum disorders: a systematic review and meta-analysis. Dev. Med. Child Neurol. 53, 783–792.
Rubenstein, J. L., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267.
Sahley, T. L., and Panksepp, J. (1987). Brain opioids and autism: an updated analysis of possible linkages. J. Autism Dev. Disord. 17, 201–216.
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241.
Sandin, S., Hultman, C. M., Kolevzon, A., Gross, R., MacCabe, J. H., and Reichenberg, A. (2012). Advancing maternal age is associated with increasing risk for autism: a review and meta-analysis. J. Am. Acad. Child Adolesc. Psychiatry 51, 477.e1–486.e1.
Sandstead, H. H., Strobel, D. A., Logan, G. M., Marks, E. O., and Jacob, R. A. (1978). Zinc deficiency in pregnant rhesus monkeys: effects on behavior of infants. Am. J. Clin. Nutr. 31, 844–849.
Satoh, Y., Endo, S., Nakata, T., Kobayashi, Y., Yamada, K., Ikeda, T., et al. (2011). ERK2 contributes to the control of social behaviors in mice. J. Neurosci. 31, 11953–11967.
Schmeisser, M. J., Ey, E., Wegener, S., Bockmann, J., Stempel, A. V., Kuebler, A., et al. (2012). Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260.
Sensi, S. L., Paoletti, P., Koh, J. Y., Aizenman, E., Bush, A. I., and Hershfinkel, M. (2011). The neurophysiology and pathology of brain zinc. J. Neurosci. 31, 16076–16085.
Serajee, F. J., Nabi, R., Zhong, H., and Huq, M. (2004). Polymorphisms in xenobiotic metabolism genes and autism. J. Child Neurol. 19, 413–417.
Shelton, J. F., Tancredi, D. J., and Hertz-Picciotto, I. (2010). Independent and dependent contributions of advanced maternal and paternal ages to autism risk. Autism Res. 3, 30–39.
Stromland, K., Nordin, V., Miller, M., Akerstrom, B., and Gillberg, C. (1994). Autism in thalidomide embryopathy: a population study. Dev. Med. Child Neurol. 36, 351–356.
Südhof, T. C. (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911.
Szpir, M. (2006). Tracing the origins of autism: a spectrum of new studies. Environ. Health Perspect. 114, A412–A418.
Toro, R., Konyukh, M., Delorme, R., Leblond, C., Chaste, P., Fauchereau, F., et al. (2010). Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 26, 363–372.
Torres-Aleman, I. (1999). Insulin-like growth factors as mediators of functional plasticity in the adult brain. Horm. Metab. Res. 31, 114–119.
Tropea, D., Giacometti, E., Wilson, N. R., Beard, C., McCurry, C., Fu, D. D., et al. (2009). Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 106, 2029–2034.
van Balkom, I. D., Bresnahan, M., Vuijk, P. J., Hubert, J., Susser, E., and Hoek, H. W. (2012). Paternal age and risk of autism in an ethnically diverse, non-industrialized setting: aruba. PLoS ONE 7:e45090. doi:10.1371/journal.pone.0045090
Verpelli, C., Dvoretskova, E., Vicidomini, C., Rossi, F., Chiappalone, M., Schoen, M., et al. (2011). Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. J. Biol. Chem. 286, 34839–34850.
Via, E., Radua, J., Cardoner, N., Happé, F., and Mataix-Cols, D. (2011). Meta-analysis of gray matter abnormalities in autism spectrum disorder: should Asperger disorder be subsumed under a broader umbrella of autistic spectrum disorder? Arch. Gen. Psychiatry 68, 409–418.
Walsh, W. J., Usman, A., and Tarpey, J. (2001). “Disordered metal metabolism in a large autism population,” in Proceedings of the American Psychological Association; New Research: Abstract NR109, New Orleans.
Walsh, W. J., Usman, A., Tarpey, J., and Kelly, T. (2002). Metallothionein and Autism, 2nd Edn. Naperville, IL: Pfeiffer Treatment Center.
Wang, X., McCoy, P. A., Rodriguiz, R. M., Pan, Y., Je, H. S., Roberts, A. C., et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108.
Ward, A. J. (1990). A comparison and analysis of the presence of family problems during pregnancy of mothers of “autistic” children and mothers of normal children. Child Psychiatry Hum. Dev. 20, 279–288.
Wills, S., Cabanlit, M., Bennett, J., Ashwood, P., Amaral, D., and Van de Water, J. (2007). Autoantibodies in autism spectrum disorders (ASD). Ann. N. Y. Acad. Sci. 1107, 79–91.
Wolfram, T., and Ismail-Beigi, F. (2011). Efficacy of high-fiber diets in the management of type 2 diabetes mellitus. Endocr. Pract. 17, 132–142.
Won, H., Lee, H. R., Gee, H. Y., Mah, W., Kim, J. I., Lee, J., et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265.
Wong, C. P., and Ho, E. (2012). Zinc and its role in age-related inflammation and immune dysfunction. Mol Nutr Food Res 56, 77–87.
Yasuda, H., Yoshida, K., Yasuda, Y., and Tsutsui, T. (2011). Infantile zinc deficiency: association with autism spectrum disorders. Sci. Rep. 1, 129.
Keywords: zinc deficiency, immune system, cytokines, ASD, Shank3, melatonin, risk factor
Citation: Grabrucker AM (2013) Environmental factors in autism. Front. Psychiatry 3:118. doi: 10.3389/fpsyt.2012.00118
Received: 18 September 2012; Accepted: 28 December 2012;
Published online: 18 January 2013.
Edited by:
Andreas Reif, University of Wuerzburg, GermanyReviewed by:
Konrad Prasad, University of Pittsburgh School of Medicine, USACopyright: © 2013 Grabrucker. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Andreas M. Grabrucker, WG Molecular Analysis of Synaptopathies, Anatomy and Cell Biology, Ulm University, Albert-Einstein Allee 11, Ulm 89081, Germany. e-mail:YW5kcmVhcy5ncmFicnVja2VyQHVuaS11bG0uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.