

 Jinxue Wei1
Jinxue Wei1
- 1 Department of Psychiatry and Neurobehavioral Sciences, University of Virginia, Charlottesville, VA, USA
- 2 Department of Biochemistry and Molecular Biology, Peking University, Beijing, China
Previous human and animal studies demonstrate that acute nicotine exposure has complicated influences on the function of the nervous system, which may lead to long-lasting effects on the behavior and physiology of the subject. To determine the genes and pathways that might account for long-term changes after acute nicotine exposure, a pathway-focused oligoarray specifically designed for drug addiction research was used to assess acute nicotine effect on gene expression in the neuron-like SH-SY5Y cells. Our results showed that 295 genes involved in various biological functions were differentially regulated by 1 h of nicotine treatment. Among these genes, the expression changes of 221 were blocked by mecamylamine, indicating that the majority of nicotine-modulated genes were altered through the nicotinic acetylcholine receptors (nAChRs)-mediated signaling process. We further identified 14 biochemical pathways enriched among the nicotine-modulated genes, among which were those involved in neural development/synaptic plasticity, neuronal survival/death, immune response, or cellular metabolism. In the genes significantly regulated by nicotine but blocked by mecamylamine, 13 enriched pathways were detected. Nine of these pathways were shared with those enriched in the genes regulated by nicotine, including neuronal function-related pathways such as glucocorticoid receptor signaling, p38 MAPK signaling, PI3K/AKT signaling, and PTEN signaling, implying that nAChRs play important roles in the regulation of these biological processes. Together, our results not only provide insights into the mechanism underlying the acute response of neuronal cells to nicotine but also provide clues to how acute nicotine exposure exerts long-term effects on the nervous system.
Introduction
Nicotine is the dependence-inducing constituent underlying tobacco smoking. Through interactions with nicotinic acetylcholine receptors (nAChRs) in the central nervous system (CNS), nicotine exposure not only stimulates the mesocorticolimbic dopamine system in the outer shell of the nucleus accumbens (NAc) and other brain regions (Benwell and Balfour, 1992, 1997; Gaddnas et al., 2001; Tammimaki et al., 2006) but also modulates the release of other neurotransmitters such as norepinephrine, serotonin, and GABA (Kenny et al., 2001; Barik and Wonnacott, 2006, 2009). Prolonged nicotine treatment can regulate the expression of genes/proteins involved in various functions such as ERK1/2 and CREB (Brunzell et al., 2003), as well as their downstream targets such as c-FOS and FOSB (Pagliusi et al., 1996; Nisell et al., 1997; Soderstrom et al., 2007). Further, biochemical pathways underlying various physiological processes; e.g., MAPK signaling, phosphatidylinositol phosphatase signaling, growth factor signaling, and ubiquitin– proteasome pathways, are modulated by nicotine (Tang et al., 1998; Konu et al., 2001; Li et al., 2004). Through its direct or indirect interaction with these genes and biological pathways, nicotine is involved in the regulation of various physiological processes, such as learning and memory, angiogenesis, energy metabolism, synaptic function, response to oxidative stress, and addiction (Harkness and Millar, 2002; Dajas-Bailador and Wonnacott, 2004; Kane et al., 2004; Robinson and Kolb, 2004; Dasgupta and Chellappan, 2006; Dasgupta et al., 2006; Hwang and Li, 2006).
Although nicotine dependence is attributed mainly to repeated and chronic nicotine exposure, acute administration also can evoke noticeable physiological effects. For example, in humans, acute nicotine exposure, while significantly increasing the plasma nicotine concentration (Argacha et al., 2008), modulates a series of physiological processes such as aortic wave reflection, arterial stiffness, and plasma asymmetrical dimethylarginine (ADMA) concentration (Adamopoulos et al., 2009). Experimentally, acute nicotine administration can significantly increase activation in multiple regions of mouse brain (Suarez et al., 2009) and induce spinal motor circuit output in embryonic zebrafish (Thomas et al., 2008). Previous studies have demonstrated the beneficial effect of acute nicotine exposure on cognitive performance (Foulds et al., 1996; Phillips and Fox, 1998; Ernst et al., 2001; Kumari et al., 2003), as well as its antidepressant effect (Tizabi et al., 2009).
Various genes and biochemical processes are involved in the biological response to acute nicotine exposure. These include immediate-early genes such as c-FOS, c-JUN, NURR77, and EGR1 (Ichino et al., 1999, 2002; Salminen et al., 1999) and the dendritically targeted early response genes activity-regulated cytoskeleton-associated protein (ARC) and dendrin in specific brain regions of rats (Schochet et al., 2005, 2008; Schmitt et al., 2008). Acute nicotine exposure not only increases the activity of dopaminergic neurons and promotes dopamine release (Levin and Rose, 1995), but also enhances dopamine turnover and metabolism (Grenhoff and Svensson, 1988). The administration of a single dose of nicotine enhances the synthesis and release of striatal dynorphin, a component of the circuit promoting negative motivational and affective states, possibly under the regulation of dopamine and glutamate (Isola et al., 2009). Acute nicotine exposure may impair nNOS-dependent dilation of cerebral arterioles via a mechanism related to the increase in oxidative stress (Arrick and Mayhan, 2007). Even a few minutes of nicotine exposure can significantly suppress arachidonic acid signaling in various brain regions in rats (Chang et al., 2009). In mice, acute administration of a low dose of nicotine increases phosphor-ERK1/2 immunoreactivity in the NAc, prefrontal cortex, and some other regions innervated by the mesolimbic dopamine pathway (Barik and Wonnacott, 2009).
Clearly, acute nicotine exposure can evoke multiple effects in the neuronal system, but its action mechanism is not completely understood. The neuron-like human neuroblastoma cell line SH-SY5Y endogenously expresses various nAChR subunits (e.g., α3, α5, α7, β2, and β4; Gould et al., 1992; Peng et al., 1994), which are assembled to form different types of nAChRs, such as α3*-nAChR or homomeric α7-nAChR receptors. The cells therefore provide a suitable in vitro model to investigate alterations in nAChRs in the presence of cholinergic ligands and have been widely used in nicotine-related study. In an earlier study, 17 genes with diverse biological functions were found to be significantly regulated in SH-SY5Y cells after exposed to 1 mM nicotine for 1 h (Dunckley and Lukas, 2003). The authors further showed that most of these genes were regulated via nAChRs-mediated mechanisms. However, because of the limitation of array technologies and bioinformatics tools at that time, a detailed analysis, especially at the biological pathway level, was impossible. With the rapid progress in the fields of genomics and bioinformatics, a detailed and comprehensive profiling of the genes and pathways responsive to acute nicotine treatment becomes feasible and necessary. In this study, by using an established and well-tested in vitro model in the field, we investigated the relation between acute nicotine exposure and neuronal function by examining the genes and biochemical pathways differentially regulated by nicotine in SH-SY5Y cells using a customized pathway-focused microarray (Cao et al., 2011; Wei et al., 2011).
Materials and Methods
Cell Culture and Drug Treatment
The human neuroblastoma SH-SY5Y cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured in a 1:1 mixture of ATCC-formulated Eagle’s Minimal Essential Medium and F12 medium supplemented with 10% fetal bovine serum (GIBCO Invitrogen, Grand Island, NY, USA) at 37°C in a humidified atmosphere of 5% CO2. At about 80% confluence, cells were treated with 1 mM nicotine (calculated as free base), 3 μM mecamylamine (nicotinic receptor antagonist), 1 mM nicotine in combination with 3 μM mecamylamine, or no drug (controls) for 1 h. For each experimental group, six independent cultures were prepared (N = 6).
RNA Isolation
Total RNA was isolated separately from each of the drug-treated and control cultures using Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. To eliminate potential residual DNA contamination, the RNA samples were treated with RNase-free DNase I at 37°C for 30 min followed by inactivation at 65°C for 10 min. The RNA concentration of each sample was measured at an optical density of 260 nm, and the integrity of the RNA was checked by examination of 28S and 18S ribosomal bands using agarose–formaldehyde gel electrophoresis.
Microarray Production
A pathway-focused oligoarray designed specifically for the study of drug addiction and related disorders was used. Briefly, 3,565 genes essential to CNS activities for the maintenance of neuronal homeostasis, as well as those associated with the neuron response to addictive substances such as nicotine, alcohol, and cocaine, were selected on the basis of an earlier version of a pathway-focused cDNA microarray (Konu et al., 2004a) and an extensive literature survey. These genes covered most of the major pathways related to cell metabolism, genetic information processing, cellular signaling transduction, neuron-related disease, and cell communication. OligoWiz1 was used to design the oligonucleotide for each gene. The average length of these oligonucleotides was 59.2 ± 3.8 bases (mean ± SD), with a GC content of 0.53 ± 0.05 and a Tm of 76.4 ± 1.7°C. The oligonucleotides and 10 control clones were synthesized by SIGMA Genosys (Sigma-Aldrich, St. Louis, MO, USA) and spotted at a concentration of 40 μM in 3× SSC and 1.5 M betaine buffer onto CMT-GAPS II slides (Corning Life Sciences, Lowell, MA, USA) using an OmniGrid MicroArrayer OGR-03 (GeneMachines, San Carlos, CA, USA).
cDNA Probe Labeling, Hybridization, and Scanning
To reduce the potential influence of any gene-specific dye effect, a universal reference design was used in the microarray analysis. For each experimental group, six independent cultures were prepared. Another 12 independent control cultures were used as “universal controls.” For each sample, 10 μg of total RNA was extracted for cDNA synthesis. Following purification with phenol:chloroform extraction and isopropanol precipitation, total RNA was dissolved in 28 μl of water, mixed with 4 μl of 10× buffer, 4 μl of 10 mM dTTP-free dNTP, 1 μl of 10 mM dTTP, 2 μl of 1 mM cyanine 3-dUTP (for either nicotine-treated or control sample) or cyanine 5-dUTP (for universal control sample; Enzo, Farmingdale, NY, USA), and 1 μl of Klenow fragment (50 units/μl), then incubated at 37°C for 3 h prior to purification with a QIAquick PCR kit (Qiagen, Valencia, CA, USA). The cyanine 3-labeled sample (either nicotine-treated or control) cDNA probes were mixed with the cyanine 5-labeled universal control cDNA probe and added to 7.5 μl of 20× SSC, 3 μg of CotI DNA, 3 μg of polyA, and 0.5 μl of 10% SDS adjusted to a final volume of 50 μl. The mixture was applied to the pathway-focused oligonucleotide microarray and hybridized overnight at 60°C. Slides were washed in 1× SSC and 0.2% SDS at 60°C for 5 min followed by washing in 0.1× SSC and 0.2% SDS and in 0.1× SSC at room temperature for 10 min each. Hybridized slides were scanned using the ScanArray Gx microarray scanner, and the intensities of each probe were quantified with the ScanArray Express microarray analysis system (PerkinElmer, Waltham, CA, USA).
Data Normalization and Statistical Analysis
After scanning the arrays, we obtained the raw hybridization intensity for each element printed on the array and then used the background-subtracted median intensity of each spot for further statistical analysis. The two replicates on each chip were initially processed separately. The weakest 5% of spots and the saturated spots in each replicate were discarded. An intensity-dependent method, locally weighed linear regress (Lowess), was used to normalize the data of each replicate (Yang et al., 2002). To minimize experimental error, genes with six or fewer valid measurements were removed from further analysis, and the two technical replicates per chip were averaged to obtain the measurement of each gene for a given sample. The normalized data from each drug treatment group were compared with the normalized data of controls, and significantly regulated genes were identified by unpaired two-tailed Student’s t-test. To control the multiple comparison error, the false discovery rate (FDR) was calculated by the method of Benjamini and Hochberg (1995) via MATLAB (The Mathworks Inc., Natick, MA, USA).
The genes significantly regulated by each drug treatment were analyzed by ingenuity pathway analysis (IPA)2 with the goal of revealing the enriched biochemical pathways. The core of IPA is the ingenuity pathways knowledge base (IPKB), which contains the biological function, interaction, and other related information of a curated gene set and more than 330 biochemical pathways. This pathway-based software is designed to identify global canonical pathways, dynamically generated biological networks, and global functions from a given list of genes. Basically, the genes with their symbols, corresponding GenBank Accession Numbers, or both were uploaded into the IPA and compared with the genes included in each canonical pathway using the whole gene set of IPKB as the background. All the pathways with one or more genes overlapping the candidate genes were extracted. In IPA, each of these pathways was assigned a p-value, which denoted the probability of overlap between the pathway and input genes, via Fisher’s exact test. Because a relatively large number of pathways were examined, multiple comparison correction for the individually calculated p-values was necessary to make reliable statistical inferences. The FDR was again calculated with the method of Benjamini and Hochberg (1995).
Quantitative real-time RT-PCR
The quantitative real-time RT-PCR (qRT-PCR) was carried out for eight representative genes selected from the aforementioned differentially expressed genes: cyclin d3 (CCND3), cell division cycle 42 (CDC42), death domain-associated protein 6 (DAXX), mitogen-activated protein kinase 14 (MAPK14), mads box transcription enhancer factor 2, polypeptide D (MEF2D), nerve growth factor receptor (NGFR), phospholipase A2, group IVA (PLA2G4A), and transformation-related protein 53 (TRP53). All primers and TaqMan probes for the eight genes were purchased from Applied Biosystems (ABI, Foster City, CA, USA), and qRT-PCR analysis was done as described previously (Gutala et al., 2004; Konu et al., 2004a). Briefly, PCR was carried out in a volume of 25 μl using the TaqMan assay on the ABI 7000 Sequence Detection System. The 18S ribosomal RNA was used as an internal control to normalize the expression patterns of target genes. Data analysis was performed using a comparative Ct method (Winer et al., 1999). Five independent cell cultures were prepared under the same conditions used for the microarray experiments for both the nicotine treatment and control groups (N = 5).
Western blotting analysis
The protein concentration of MAPK14 (p38) was measured in both nicotine-treated (1 mM for 1 h) and control samples. After appropriate treatment, cells were harvested using RIPA buffer followed by incubation on ice for 30 min. The cells were centrifuged at 12,000×g for 15 min to remove debris, and the total protein content of the lysates was determined using the Bio-Rad protein assay (Bio-Rad, Chicago, IL, USA). The samples were boiled at 90°C for 5 min and then cooled on ice. Twenty micrograms of protein was fractionated on 10% SDS-polyacrylamide gel (containing 30% acrylamide and 0.8% bisacrylamide) in Tris– glycine buffer containing SDS at 80 V for approximately 1.5 h. The separated proteins were then transferred to a PVDF membrane (Millipore, Bedford, MA, USA) overnight at 30 V at 4°C. The membrane was blocked for 1 h at room temperature with 5% non-fat dry milk diluted in TBST buffer and incubated overnight with primary antibody at 4°C. After being washed three times in TBST, the membranes were subjected to secondary antibody conjugated to horseradish peroxidase for 1 h at room temperature. Immunoreactivity was detected using an enhanced chemiluminescence kit (Bio-Rad), and the preparations were exposed to X-ray film. The MAPK14 antibody was from Calbiochem (San Diego, CA, USA), and anti-α-tubulin monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used to detect the housekeeping protein α-tubulin. The MAPK14 immunoreactivity bands were quantified using densitometry. After the films were developed, they were scanned on a Microtek ScanMaker i800 (Microtek Lab Inc., Cerritos, CA, USA) with ScanWizard 5.5 at a resolution of 600 dpi for quantitative analysis with ImageQuant 5.1 (Molecular Dynamics, Sunnyvale, CA, USA). The relative MAPK14 values were normalized to α-tubulin, and the significance of the difference between the nicotine-treated and control samples was determined by t-test via MATLAB (The Mathworks, Natick, MA, USA). Four independent cell cultures were prepared under the same conditions used for the microarray experiments for both nicotine treatment and control groups (N = 4).
Results
Gene Expression Changes in Response to Acute Nicotine Exposure
In the nicotine-treated group, 295 genes were significantly modulated (p < 0.05; Table A1 in Appendix), with FDR values ≤0.258. Among these genes, 98 had p-values <0.01, which corresponded to an FDR value of 0.155. Of the 295 differentially expressed genes, 221 showed no significant changes (p ≥ 0.05) in the cells treated with nicotine and mecamylamine, suggesting these genes were regulated through the mecamylamine-modulated nicotinic receptors.
Of the 295 genes, 128 were down-regulated by acute nicotine exposure. These included multiple transcription factors; e.g., activating transcription factor 2 (ATF2), general transcription factor IIH polypeptide 2 (GTF2H2), nuclear respiratory factor 1 (NRF1), and transcription factor EB (TRFEB). A few genes related to cell adhesion were also down-regulated by acute nicotine exposure; e.g., ninjurin 1 (NINJ1), kit ligand (KITLG), integrin beta 1 (ITGB1), neurexin 1 (NRXN1), and laminin gamma 1 (LAMC2). Some cell cycle-related genes were suppressed; e.g., cell division cycle 14 homolog B (CDC14B), cell division cycle 2 homolog A (CDC2A), cell division cycle 42 homolog (CDC42), G1 to S phase transition 1 (GSPT1), and meiotic recombination 11 homolog A (MRE11A).
The remaining 167 differentially expressed genes were up-regulated by nicotine treatment. Genes included in this category included those related to: (a) mitochondrial electron transport; e.g., cytochrome c oxidase, subunit VIIC (COX7C), polypeptide 1 of cytochrome p450 family 17 subfamily A (CYP17A1), rieske iron–sulfur protein (RISP), and NADH dehydrogenase (ubiquinone) flavoprotein 1 (NDUFV1); (b) ubiquitination; e.g., ubiquitin-activating enzyme E1 Chr X (UBE1X), ubiquilin 1 (UBQLN1), ubiquitin-specific protease 28 (USP28), and ubiquitin-conjugating enzyme e2a (UBE2A); and (c) transport; e.g., potassium channel TWIK (KCNK1), nucleotide-sensitive chloride channel 1A (CLNS1A), T type alpha 1G subunit of voltage-dependent calcium channel (CACNA1G), solute carrier family 8 (sodium/calcium exchanger) member 2 (SLC8A2), and solute carrier family 2 (facilitated glucose transporter) member 4 (SLC2A4).
Of the 295 genes significantly regulated by nicotine treatment, 74 were differentially expressed (p < 0.05) in the cells treated with nicotine + mecamylamine (Table A1 in Appendix). Most of these genes showed the same trend of regulation as those in the nicotine treatment group, suggesting that mecamylamine may have either no significant effect or a synergistic effect with nicotine on the expression changes in these genes.
Pathways Enriched in the Genes Regulated by Nicotine
As shown above, genes and biological processes involved in different functions were regulated by nicotine exposure. To further identify the biological themes underlying these genes, the biochemical pathways enriched in the nicotine-regulated genes were investigated. Two sets of genes were analyzed by IPA; i.e., those regulated significantly by 1 h of nicotine exposure (N = 295), and those with expression significantly changed by 1 h of nicotine with the effect being blocked by mecamylamine (N = 221). The second gene set included only those with p-values ≥0.05 during co-treatment with nicotine and mecamylamine. Although in this set, some genes may have considerable fold changes, the selection was based on p-value only.
Results from IPA analysis showed that 14 pathways were significantly enriched among the genes regulated by nicotine (p < 0.05; FDR < 0.05; Figure 1; Table 1), and 13 pathways were enriched in the second gene set. Among these pathways, nine were identified for both gene sets; i.e., glucocorticoid receptor signaling, p38 MAPK signaling, PI3K/AKT signaling, PTEN signaling, acute-phase response signaling, ERK/MAPK signaling, Toll-like receptor signaling, VEGF signaling, and T-cell receptor signaling. For the gene set regulated by nicotine treatment, five pathways were uniquely identified; i.e., mitochondrial dysfunction, PPAR signaling, IL-6 signaling, hepatic fibrosis/hepatic stellate cell activation, and death receptor signaling. The pathways uniquely identified in the second gene set were CD28 signaling in T-helper cells, CD40 signaling, role of NFAT in regulation of the immune response, and IL-8 signaling. In the pathways identified for the two gene sets, the genes affected were not identical (Table 1). For the nicotine + mecamylamine-treated samples, the pathways included only genes whose expression was modulated by nicotine exposure but blocked by simultaneous treatment with the two chemicals. For the nicotine-treated samples, the pathways also included genes whose expression was modulated in both nicotine- and nicotine + mecamylamine-treated samples.
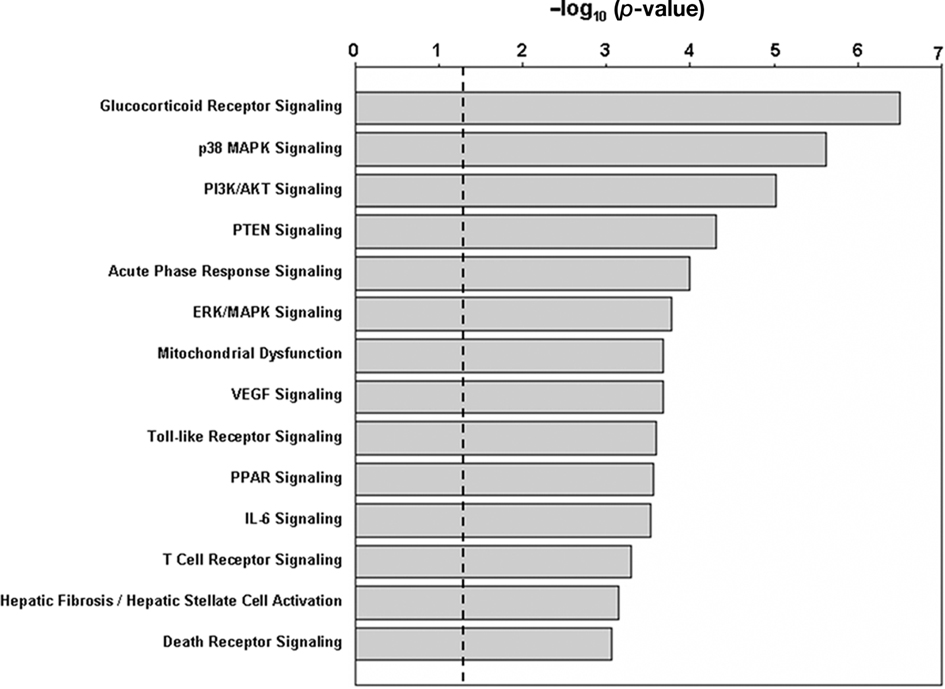
Figure 1. Pathways significantly enriched in the genes differentially regulated by nicotine exposure. The p-values are shown in negative logarithm form at base 10. The dashed line indicates the threshold corresponding to p = 0.05. All 14 pathways shown have an FDR < 0.05.
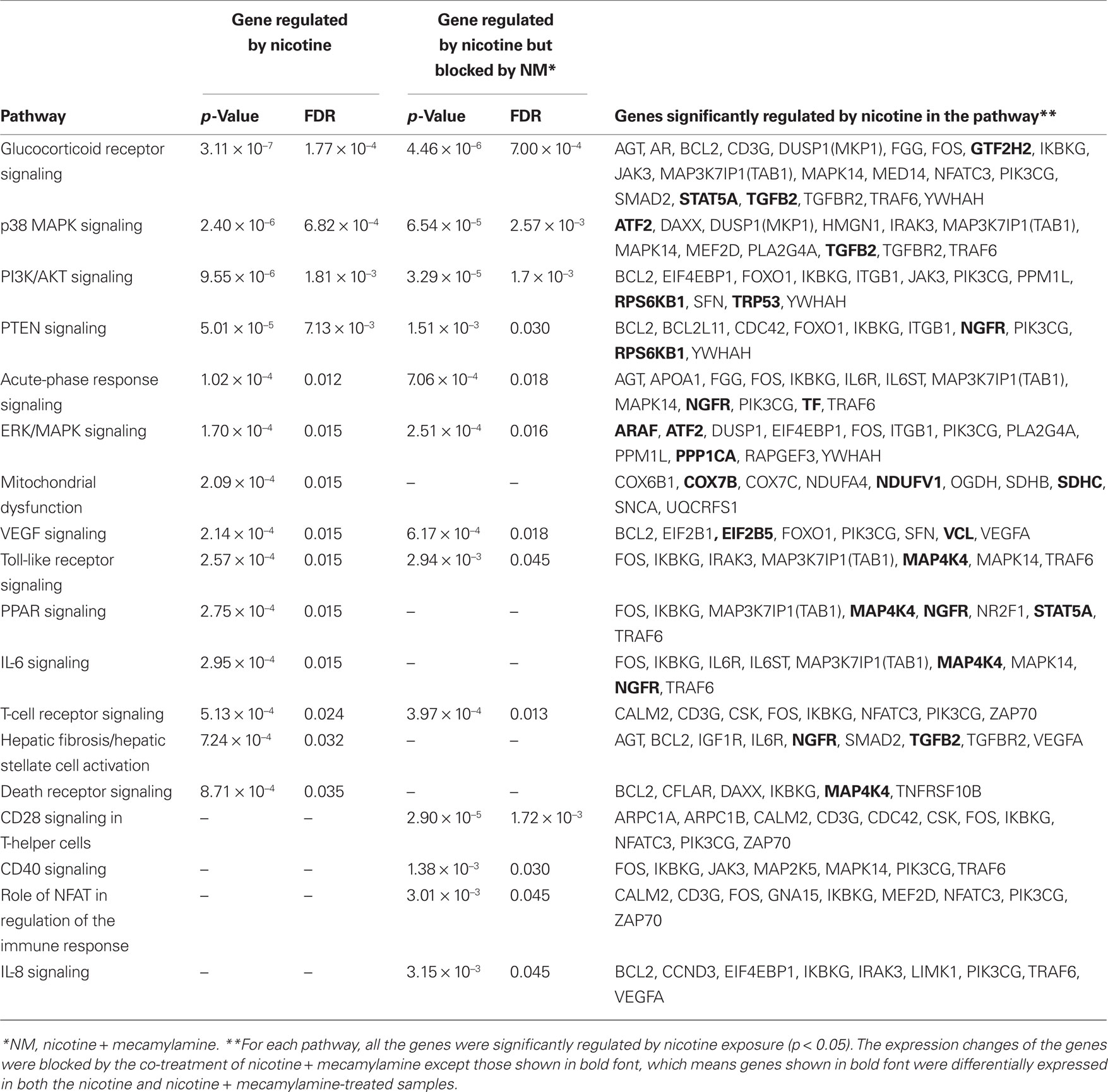
Table 1. Pathways enriched in genes significantly regulated by nicotine and genes significantly regulated by nicotine but not by nicotine + mecamylamine.
We also performed pathway analysis on the 74 genes significantly regulated in both nicotine-treated and the nicotine + mecamylamine-treated samples via IPA. Although a few pathways were assigned p-values <0.05, they are not reported here because of the relatively high FDR value after multiple comparison correction (with the minimum FDR being 0.262).
As shown in Table 1, multiple genes in each pathway were significantly regulated by nicotine exposure. Although the expression changes of some genes within each pathway were not statistically significant, they showed consistent patterns with those significantly modulated. One of the pathways, p38 MAPK signaling, is shown in Figure 2. Genes involved in this pathway were extensively regulated in response to nicotine exposure. The expression of a few genes, such as DAXX, MKP1/5, MAPK14, and MEF2, was significantly modulated by acute nicotine exposure (p < 0.05). Although the expression of other genes; e.g., ASK1, MKK4, and HPK1, was not significantly regulated under the experimental conditions, they showed patterns consistent with those of significantly regulated genes.
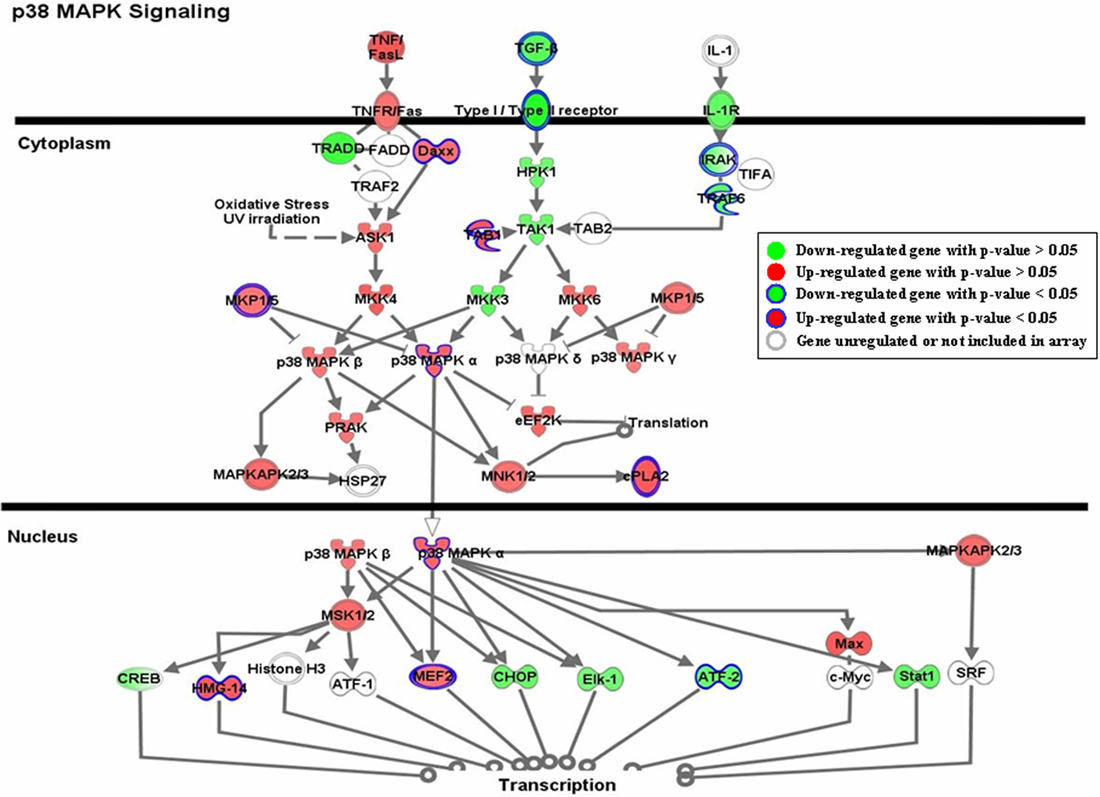
Figure 2. Gene expression pattern of the P38 MAPK signaling pathway. Genes shown in green are down-regulated, whereas those shown in red are induced by acute nicotine treatment. The genes significantly regulated by nicotine are those with closed blue edges. To demonstrate the expression pattern of the pathway, all the genes with expression ratios >1.1 or <0.90 relative to the controls are also shown. The genes in white with light gray edges are either not regulated by nicotine (expression ratios between 0.9 and 1.1) or were not measured in our analysis. The pathway was drawn by the IPA program.
Expression verification of representative genes
In an independent experiment involving the same treatments as in the microarray analysis, qPCR was performed on eight representative genes; i.e., CCND3, CDC42, DAXX, MAPK14, MEF2D, NGFR, PLA2G4A, and TRP53. All these genes were significantly regulated by nicotine exposure (Table A1 in Appendix), and each gene was part of one or more enriched pathways associated with nicotine treatment (Table 1). For example, CCND3 is part of the IL-8 signaling pathway; CDC42 of CD28 signaling and PTEN signaling pathways; DAXX of the p38 MAPK signaling pathway; MAPK14 of multiple pathways such as glucocorticoid receptor signaling, p38 MAPK signaling, acute-phase response signaling, and Toll-like receptor signaling; MEF2D of p38 MAPK signaling that plays a role of NFAT in regulation of the immune response; NGFR of PTEN signaling, acute-phase response signaling, PPAR signaling, and IL-6 signaling; PLA2G4A of p38 MAPK and ERK/MAPK signaling; and TRP53 of the PI3K/AKT signaling pathway.
Figure 3 shows the fold change in activity along with the standard deviation of the eight genes in response to nicotine treatment relative to the controls, as detected by microarray and quantitative RT-PCR. These analyses revealed significant up-regulation of seven genes and down-regulation of CDC42, which is consistent with the result from the microarray analysis. Furthermore, a comparison of the fold changes of each gene detected by the two molecular techniques revealed a correlation coefficient of 0.87 (p = 0.0068), indicating overall measurement of gene expression from the two approaches was consistent. Although only eight representative genes were selected for verification by qRT-PCR, a comparison of the two techniques demonstrates that the results from our microarray analyses were highly reproducible and reliable.
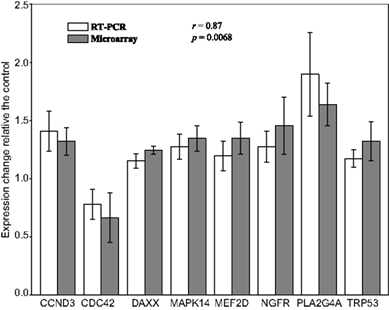
Figure 3. Comparative plot of results from microarray and real-time RT-PCR verification for eight representative genes. Y axis represents the fold change of expression in 1-h nicotine-treated SH-SY5Y cells compared with the average expression in the control sample for that particular gene. For the qRT-PCR results, the expression of each gene was normalized to the average of control samples. Abbreviations: CCND3, cyclin D3; CDC42, cell division cycle 42 homolog; DAXX, Fas death domain-associated protein; MAPK14, mitogen-activated protein kinase 14; MEF2D, myocyte enhancer factor 2d; NGFR, nerve growth factor receptor; PLA2G4A, phospholipase A2, group IVA; TRP53, transformation-related protein 53.
Furthermore, we used Western blotting to determine the total amount of MAPK14 (p38 MAPK) protein in lysates of cells treated with 1 mM nicotine and control samples. The results revealed significant induction (35.5%; p = 0.03) of MAPK14 expression in nicotine-treated samples (Figure 4).
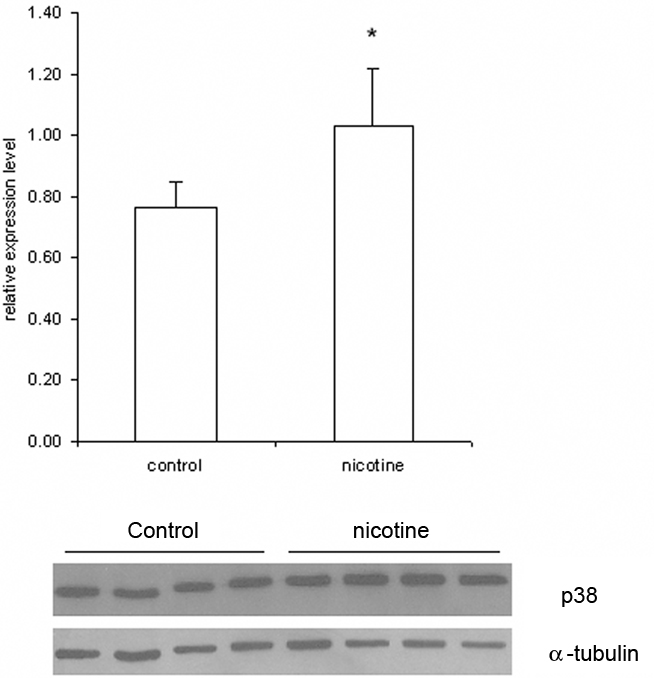
Figure 4. Expression of MAPK14 (p38 MAPK) protein in SH-SY5Y cells determined by Western blotting. The SH-SY5Y cells were treated for 1 h with or without (Controls) 1 mM nicotine. After treatment, protein was purified from the lysates and analyzed by Western blotting. In this figure, the MAPK14 concentration was determined using a specific monoclonal antibody. The histogram shows the relative expression of MAPK14 normalized to expression of α-tubulin, which demonstrated that the protein concentrations in nicotine-treated cells are significant higher than those in controls (p < 0.03). Data are presented as mean ± SEM (N = 4).
Discussion
In this study, we profiled gene expression in the SH-SY5Y cell line in response to 1 h of 1 mM nicotine exposure using a pathway-focused oligonucleotide microarray. Treatment with 1 mM nicotine maximally induces the up-regulation of numbers of nAChR radioligand-binding sites in SH-SY5Y cells, and this up-regulation has been implicated in the long-lasting effects of nicotine exposure, such as dependence and tolerance (Ke et al., 1998; Dunckley and Lukas, 2003, 2006). Given that the purpose of this study was to determine the pharmacologic effects of nicotine using an in vitro system, we adopted a 1-mM concentration for nicotine, as used in a previous study (Dunckley and Lukas, 2003). However, the current study is neither a simple modeling of the physiological conditions of smokers, nor repeat earlier studies in the same model. Instead, it focuses on exploring the molecular response at the pathway level to acute nicotine exposure that may maximally activate the function of molecules such as nAChRs and their downstream genes in SH-SY5Y cells.
Nicotine evokes its physiological effects mainly by binding with nAChRs. As a broad-spectrum antagonist of nAChRs, mecamylamine at a concentration of 3 μM can block most of the responses of α3*-nAChR to nicotine, as well as a fraction of the α7-nAChR responses (Chavez-Noriega et al., 1997; Papke et al., 2001; Dunckley and Lukas, 2003, 2006). Of the 295 genes significantly regulated by nicotine, 221 were not differentially expressed during co-treatment of nicotine + mecamylamine, providing evidence of direct involvement of nAChR in the process of modulation of SH-SY5Y cells by nicotine. Thus, our results indicate that the changes in 75% (221/295) of the differentially expressed genes in response to nicotine are attributable to direct interaction with nAChRs, especially α3*-nAChR receptors, which is consistent with the conclusions drawn from previous studies (Dunckley and Lukas, 2003, 2006). However, it is difficult to confirm a clear relation between nicotinic receptor subtype and gene expression from our findings because of the existence of different subtypes of nAChRs on the membrane of SH-SY5Y cells. These receptors have different properties, such as sensitivity to nicotine, permeability to calcium, and propensity to desensitize. It is possible that the regulation of some genes is related to multiple subtypes of nAChRs, including those α3- or α7-containing receptors. Partially blocking the function of some types of receptors (e.g., α3*- and α7-containing) may reduce the overall activation of nAChRs, which may lead to more subtle regulation of some genes compared with the response when mecamylamine is not applied. On the other hand, our results do not imply that nAChRs play little or no role in the regulation of genes significantly regulated in both nicotine and nicotine + mecamylamine treatment. The expression changes of these genes may have multiple explanations. A small fraction of α3*-nAChR receptors remain functionally active under the influence of mecamylamine, which may be sufficient to regulate the expression of some genes. At the same time, α7 and other types of nAChR receptors may be responsible for the expression change of some other genes as well. It is also possible that the expression changes of some genes in response to nicotine are caused by mechanisms not directly involving nAChRs, as nicotine can readily cross the plasma membrane and affect intracellular processes through an nAChR-independent mechanism.
Our analysis showed that acute nicotine exposure has effects on the expression of a large number of genes with diverse functions (Table A1 in Appendix). Among them are genes involved in the ubiquitin–proteasome pathway, such as proteasome subunit beta 4 (PSMB4), proteasome 26S subunit non-ATPase 2 (PSMD2), ubiquitin-activating enzyme E1 Chr X (UBE1X), ubiquitin-conjugating enzyme E2A (UBE2A), ubiquilin 1 (UBQLN1), and ubiquitin- specific protease 28 (USP28). Earlier studies showed that genes involved in the ubiquitin–proteasome pathway were regulated in SH-SY5Y cells by 1 mM nicotine after both 1- and 24-h treatment (Dunckley and Lukas, 2003, 2006). In the current study, more genes related to this pathway were found to be modulated by nicotine exposure. The ubiquitin–proteasome pathway is regulated by nicotine in cell lines (Konu et al., 2004b; Ficklin et al., 2005; Wang et al., 2007), animal models (Kane et al., 2004; Li et al., 2004; Rezvani et al., 2007; Vadasz et al., 2007; Wang et al., 2009), and human smokers (Mexal et al., 2005). Thus, the changes in ubiquitin–proteasome-related genes observed in this study may not be specific for the relatively high nicotine concentration; instead, they may be caused by mechanisms similar to those discovered in previous studies.
Multiple genes involved in mitochondrial functions also were found to be regulated by nicotine; these include acyl-coenzyme A thioesterase 3 (ACATE3), ATP synthase subunit g (ATP5L), mitochondrial ribosomal protein S7 (MRPS7), NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 4 (NDUFA4), and RISP. Previous studies have demonstrated that nicotine exposure can modulate the morphology (Onal et al., 2004) and function of mitochondria, such as protein turnover (Katyare and Shallom, 1988), enzyme activity (Xie et al., 2005), and generation of reactive oxygen species (Cormier et al., 2001; Soto-Otero et al., 2002). Recently, we showed that nicotine exposure regulates the electron transport system in multiple brain regions of rats (Wang et al., 2009). The results from the current study indicate that acute nicotine exposure also can regulate the expression of mitochondrial genes, potentially leading to the modulation of mitochondrial functions. In addition, several genes involved in synaptic transmission were identified, such as neuron-specific gene family member 1 (NSG1), synuclein alpha (SNCA), synapsin 2 (SYN2), synaptotagmin 2 (SYT2), synaptotagmin 7 (SYT7), and tachykinin 2 (TAC2). The expression of NSG1 in mouse cortical neurons is regulated by treatment with nicotine, alcohol, or both (Wang et al., 2007). SNCA is suggested as a candidate associated with alcohol, cocaine, and methamphetamine dependence (Kobayashi et al., 2004; Bonsch et al., 2005; Mash et al., 2008; Spence et al., 2009). The phosphorylation of synapsin is regulated by cocaine in a region-specific manner in rat brain (Edwards et al., 2007). The regulation of these synaptic transmission-related genes indicates that even a short nicotine exposure may have important effects on neuronal functions.
Among the 74 genes significantly regulated by both nicotine and nicotine + mecamylamine treatment, some have already been reported to be involved in the cellular response to nicotine exposure. Activating transcription factor 2 (ATF2), a DNA-binding protein that binds to cAMP response elements, was suppressed in both treatment conditions and is involved in mediating the activation of tyrosine hydroxylase transcription under nicotine treatment in PC12 cells (Gueorguiev et al., 2006). Nerve growth factor (NGF), a member of the neurotrophin family, is an essential mediator of neuronal activity and synaptic plasticity (Levi-Montalcini, 1987; Thoenen et al., 1987). Both NGF and its receptor, NGFR, were up-regulated in the two treatment groups. Nicotine has been reported to regulate the expression of NGF and NGFR in cell lines or tissues (Terry and Clarke, 1994; Jonnala et al., 2002; Garrido et al., 2003; Hernandez and Terry, 2005; French et al., 2006). Neurexin 1 (NRXN1) was down-regulated in both treatment groups. This and another member of the same family, NRXN3, were associated with smoking behaviors in several recent genetic studies (Bierut et al., 2007; Nussbaum et al., 2008; Novak et al., 2009). Although our data provide no further information on the mechanisms by which these genes are regulated, it is clear that acute nicotine exposure is responsible for their expression changes.
Consistently, the pathways enriched in nicotine-regulated genes were involved in many biological processes, such as neural development/synaptic plasticity, cell survival/death, immune response, and cellular metabolism. Of the identified pathways, nine were enriched in genes significantly regulated by nicotine, as well as those regulated by nicotine but blocked by mecamylamine, indicating these pathways were regulated mainly through nAChR-dependent mechanisms. Among these pathways, acute-phase response signaling, Toll-like receptor signaling, and T-cell receptor signaling play important roles in the immune response. Although the other pathways; i.e., glucocorticoid receptor signaling, p38 MAPK signaling, PI3K/AKT signaling, PTEN signaling, ERK/MAPK, and VEGF signaling, are involved in a large range of physiological processes, all of them represent key pathways for maintaining proper neuronal functions, such as cellular signaling and proliferation and differentiation of neurons. The modulation of the neuronal function-related pathways may have relatively long-lasting consequences. One example is that adolescent rats display different long-term neuroadaptive responses to acute nicotine than do adult animals, which is suggested to be related to the immature or still-developing plasticity mechanisms in the prefrontal cortex of young animals (Schochet et al., 2004). Among these pathways, particularly interesting are the glucocorticoid receptor and p38 MAPK signaling.
For the glucocorticoid receptor signaling pathway, 21 genes were significantly regulated by acute nicotine treatment (Table 1). The glucocorticoid receptor, GCCRα (also known as nuclear receptor subfamily 3, group c, member 1; NR3C1) was slightly suppressed by nicotine (fold change 0.83; p-value = 0.09). The regulation of these genes clearly indicates that the glucocorticoid receptor signaling pathway is modulated by acute nicotine exposure. Glucocorticoids, cortisol in humans, and corticosterone in the rodent, are secreted by the adrenal cortex and belong to the family of steroid hormones. Glucocorticoids and their receptors are essential for the development and survival of vertebrates (Cole et al., 1995). All the cellular responses to glucocorticoids are attributed to their binding to the intracellular glucocorticoid receptor (GCCR) and the translocation of the complexes to the nucleus, which then modulates gene expression through diverse mechanisms (Dittmar et al., 1997, 1998; Robinson-Rechavi et al., 2001). Although the activity of the GCCR is directly related to gene transactivation, considerable cross-talk occurs between the GCCR and a cohort of molecules to mediate their functions as transcriptional regulators. Studies on animals and humans have outlined the importance of glucocorticoids in mediating drug-seeking behavior and the rewarding properties of psychostimulant drugs (Piazza and Le Moal, 1997; Marinelli and Piazza, 2002). For example, in mice, activation of glucocorticoid receptors increases the propensity to self-administer cocaine by acting on the postsynaptic dopaminoceptive neurons of the dopaminergic system (Ambroggi et al., 2009). It was also found that learning and memory deficits in cocaine-dependent humans are associated with higher cortisol concentrations (Fox et al., 2009). The neuronal circuitry adaptations induced by heroin self-administration were correlated with elevated glucocorticoid production in certain brain regions of rats (Weber et al., 2009). Corticosteroid receptor modulation of the mesoaccumbens dopamine neurotransmission is believed to be a key neurobiological mechanism mediating the effects of stress in addiction (Tye et al., 2009). Nicotine can also interact with glucocorticoid receptor signaling cascades directly or indirectly. Furthermore, the relations between nicotine and glucocorticoids are bidirectional; i.e., not only can nicotine affect the hypothalamic–pituitary–adrenal (HPA) axis and circulating glucocorticoids, but adrenal steroids can modulate nicotine’s behavioral and physiological actions (Caggiula et al., 1998). Nicotine administration elevates plasma corticosterone in animals (Caggiula et al., 1998). Similarly, both cigaret smoking and nicotine infusion rapidly increase circulating cortisol concentrations in humans (Caggiula et al., 1998). In human smokers, the degree of nicotine addiction is reflected by changes in the sensitivity of central glucocorticoid receptors (Reuter et al., 2004). Nicotine administration to pregnant animals can lead to fetal overexposure to maternal glucocorticoid and result in fetal adrenocortical dysfunction and other diseases after birth (Chen et al., 2007). The significant regulation of the corticosteroid receptor signaling pathway by acute nicotine exposure provides additional evidence of the importance of this pathway in nicotine addiction, and the significantly regulated genes in this pathway are excellent candidates for further investigation. At the same time, the cross-talk between glucocorticoid receptor signaling and other pathways, such as MAPK signaling and PI3K/AKT signaling (Clark and Lasa, 2003; Gupta et al., 2007), implies that the regulation of this pathway indirectly induces modulations in other biological processes.
The MAPK signaling pathway is a major modulator of cell growth, division, and differentiation (Adams and Sweatt, 2002) and plays a key role in mediating neuronal activation induced by dopamine, glutamate, and drugs of abuse (Jiao et al., 2007). Nicotine activates the MAPK signaling pathway in a variety of tissues and cell types, including SH-SY5Y cells (Heusch and Maneckjee, 1998; Tang et al., 1998; Dajas-Nakayama et al., 2001; Wang et al., 2001; Bailador et al., 2002). Furthermore, many cellular processes are affected by both nicotine and MAPK signaling, such as cell survival and memory processing (Levin and Simon, 1998). Despite these known interactions between MAPK signaling and nicotine exposure, our work has shown a more detailed pattern of the effect of acute nicotine exposure on this pathway. A number of genes were significantly regulated by nicotine exposure (Table 1). There were also genes in this pathway that were regulated to a lesser extent (Figure 2). Whereas nicotine exposure decreases the amount of mRNA for TGF-beta 2 (TGFB2), TGFBR2, interleukin 1 receptor (IL1R), and interleukin 1 receptor-associated kinase 1 (IRAK), as well as a couple downstream genes, it also induces the expression of a number of genes, such as death-associated protein 6 (DAXX), MAPK14 (p38 MAPK), and mads box transcription enhancer factor 2 polypeptide D (MEF2D). The regulation pattern of this pathway clearly indicates the cross-talk and interaction between different cascades. Earlier studies showed that nicotine treatment decreases the amount of TGF-beta mRNA in different tissues. For example, nicotine treatment significantly suppresses the release of TGF-beta 1 in bovine aortic endothelial cells (Cucina et al., 1999) and several other cell lines (Lane et al., 2005). This protein has a bimodal dose-dependent effect on the proliferation of various cell types, being stimulatory at low concentration and inhibitory at high concentration (Pollman et al., 1999; Ghosh et al., 2005). The observed mRNA decrease of TGFB2 and its receptor, TGFBR2, may indicate an increase in cell proliferation, although this remains to be established. Our analysis further showed a moderate enhancement of MAPK14 as judged by both mRNA and protein expression. MEF2 is one of the many downstream targets regulated by MAPK14. In our analysis, the transcription of two members of the MEF2 family was induced in the nicotine-treated samples; i.e., MEF2D (fold change 1.35; p = 0.008) and MEF2C (fold change 1.27; p = 0.177). MEF2 genes are highly expressed in neurons of the CNS (Heidenreich and Linseman, 2004; Shalizi and Bonni, 2005) and play important roles in neuronal differentiation and maintenance. Blocking the function of MEF2 genes may result in the death of neurons, whereas their expression can promote neuronal survival (Mao et al., 1999; Li et al., 2001; Gaudilliere et al., 2002). Although the activity of MEF2 is regulated mainly via its phosphorylation by MAPK14 or other kinases, considering the expression induction of MAPK14 and MEF2 genes, it may be reasonable to assume the activity of MEF2 is enhanced by acute nicotine exposure in SH-SY5Y cells.
In an earlier study, Dunckley and Lukas (2003) analyzed the gene expression patterns in SH-SY5Y cells treated with nicotine under similar experimental conditions (i.e., 1 mM nicotine for 1 h) with different microarray platforms. Among the 17 genes identified, eight had valid expression measurements in our analysis, and their expression patterns were largely consistent in the two studies. While Dunckley and Lukas (2003) performed a more detailed investigation of the relation between the nicotine-regulated genes and nAChRs, we here provide a much more broad and comprehensive analysis and description of the biological processes/pathways underlying responses to acute nicotine exposure. Results from the two studies also indicate that more detailed and comprehensive information about the interaction between acute nicotine and SH-SY5Y cell line could be obtained.
Conclusion
By profiling the gene expression pattern in SH-SY5Y cell lines treated acutely with nicotine, we have identified a set of significantly modulated genes. Further, we showed that the majority of these differently expressed genes were regulated through the nAChR-mediated mechanism. From these significantly regulated genes, we have detected the enriched biochemical pathways, which included those potentially involved in the neuronal response to addictive drugs such as nicotine, including glucocorticoid receptor and p38 MAPK signaling. These findings can help us to understand the molecular mechanisms underlying the development of nicotine addiction. On the other hand, the identification of these genes and pathways further indicates that the physiological consequences of acute nicotine exposure are complicated. Thus, further investigation is necessary in order to obtain a more detailed and comprehensive understanding of the interaction between acute nicotine treatment and the nervous system.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was in part supported by NIH grants DA-13783 and DA-12844 to Ming D. Li. The authors thank Dr. David L. Bronson for his excellent editing of this manuscript.
Footnotes
References
Adamopoulos, D., Argacha, J. F., Gujic, M., Preumont, N., Degaute, J. P., and van de Borne, P. (2009). Acute effects of nicotine on arterial stiffness and wave reflection in healthy young non smokers. Clin. Exp. Pharmacol. Physiol. 36, 784–789.
Adams, J. P., and Sweatt, J. D. (2002). Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu. Rev. Pharmacol. Toxicol. 42, 135–163.
Ambroggi, F., Turiault, M., Milet, A., Deroche-Gamonet, V., Parnaudeau, S., Balado, E., Barik, J., van der Veen, R., Maroteaux, G., Lemberger, T., Schutz, G., Lazar, M., Marinelli, M., Piazza, P. V., and Tronche, F. (2009). Stress and addiction: glucocorticoid receptor in dopaminoceptive neurons facilitates cocaine seeking. Nat. Neurosci. 12, 247–249.
Argacha, J. F., Adamopoulos, D., Gujic, M., Fontaine, D., Amyai, N., Berkenboom, G., and van de Borne, P. (2008). Acute effects of passive smoking on peripheral vascular function. Hypertension 51, 1506–1511.
Arrick, D. M., and Mayhan, W. G. (2007). Acute infusion of nicotine impairs nNOS-dependent reactivity of cerebral arterioles via an increase in oxidative stress. J. Appl. Physiol. 103, 2062–2067.
Barik, J., and Wonnacott, S. (2006). Indirect modulation by alpha7 nicotinic acetylcholine receptors of noradrenaline release in rat hippocampal slices: interaction with glutamate and GABA systems and effect of nicotine withdrawal. Mol. Pharmacol. 69, 618–628.
Barik, J., and Wonnacott, S. (2009). Molecular and cellular mechanisms of action of nicotine in the CNS. Handb. Exp. Pharmacol. 173–207.
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B 57, 289–300.
Benwell, M. E., and Balfour, D. J. (1992). The effects of acute and repeated nicotine treatment on nucleus accumbens dopamine and locomotor activity. Br. J. Pharmacol. 105, 849–856.
Benwell, M. E., and Balfour, D. J. (1997). Regional variation in the effects of nicotine on catecholamine overflow in rat brain. Eur. J. Pharmacol. 325, 13–20.
Bierut, L. J., Madden, P. A., Breslau, N., Johnson, E. O., Hatsukami, D., Pomerleau, O. F., Swan, G. E., Rutter, J., Bertelsen, S., Fox, L., Fugman, D., Goate, A. M., Hinrichs, A. L., Konvicka, K., Martin, N. G., Montgomery, G. W., Saccone, N. L., Saccone, S. F., Wang, J. C., Chase, G. A., Rice, J. P., and Ballinger, D. G. (2007). Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum. Mol. Genet. 16, 24–35.
Bonsch, D., Lederer, T., Reulbach, U., Hothorn, T., Kornhuber, J., and Bleich, S. (2005). Joint analysis of the NACP-REP1 marker within the alpha synuclein gene concludes association with alcohol dependence. Hum. Mol. Genet. 14, 967–971.
Brunzell, D. H., Russell, D. S., and Picciotto, M. R. (2003) In vivo nicotine treatment regulates mesocorticolimbic CREB and ERK signaling in C57Bl/6J mice. J. Neurochem. 84, 1431–1441.
Caggiula, A. R., Donny, E. C., Epstein, L. H., Sved, A. F., Knopf, S., Rose, C., McAllister, C. G., Antelman, S. M., and Perkins, K. A. (1998). The role of corticosteroids in nicotine’s physiological and behavioral effects. Psychoneuroendocrinology 23, 143–159.
Cao, J., Dwyer, J. B., Mangold, J. E., Wang, J., Wei, J., Leslie, F. M., and Li, M. D. (2011). Modulation of cell adhesion systems by prenatal nicotine exposure in limbic brain regions of adolescent female rats. Int. J. Neuropsychopharmacol. 14, 157–174.
Chang, L., Rapoport, S. I., Nguyen, H. N., Greenstein, D., Chen, M., and Basselin, M. (2009). Acute nicotine reduces brain arachidonic acid signaling in unanesthetized rats. J. Cereb. Blood Flow Metab. 29, 648–658.
Chavez-Noriega, L. E., Crona, J. H., Washburn, M. S., Urrutia, A., Elliott, K. J., and Johnson, E. C. (1997). Pharmacological characterization of recombinant human neuronal nicotinic acetylcholine receptors h alpha 2 beta 2, h alpha 2 beta 4, h alpha 3 beta 2, h alpha 3 beta 4, h alpha 4 beta 2, h alpha 4 beta 4 and h alpha 7 expressed in Xenopus oocytes. J. Pharmacol. Exp. Ther. 280, 346–356.
Chen, M., Wang, T., Liao, Z. X., Pan, X. L., Feng, Y. H., and Wang, H. (2007). Nicotine-induced prenatal overexposure to maternal glucocorticoid and intrauterine growth retardation in rat. Exp. Toxicol. Pathol. 59, 245–251.
Clark, A. R., and Lasa, M. (2003). Crosstalk between glucocorticoids and mitogen-activated protein kinase signalling pathways. Curr. Opin. Pharmacol. 3, 404–411.
Cole, T. J., Blendy, J. A., Monaghan, A. P., Krieglstein, K., Schmid, W., Aguzzi, A., Fantuzzi, G., Hummler, E., Unsicker, K., and Schutz, G. (1995). Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 9, 1608–1621.
Cormier, A., Morin, C., Zini, R., Tillement, J. P., and Lagrue, G. (2001) In vitro effects of nicotine on mitochondrial respiration and superoxide anion generation. Brain Res. 900, 72–79.
Cucina, A., Corvino, V., Sapienza, P., Borrelli, V., Lucarelli, M., Scarpa, S., Strom, R., Santoro-D’Angelo, L., and Cavallaro, A. (1999). Nicotine regulates basic fibroblastic growth factor and transforming growth factor beta1 production in endothelial cells. Biochem. Biophys. Res. Commun. 257, 306–312.
Dajas-Bailador, F., and Wonnacott, S. (2004). Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol. Sci. 25, 317–324.
Dajas-Bailador, F. A., Soliakov, L., and Wonnacott, S. (2002). Nicotine activates the extracellular signal-regulated kinase 1/2 via the alpha7 nicotinic acetylcholine receptor and protein kinase A, in SH-SY5Y cells and hippocampal neurones. J. Neurochem. 80, 520–530.
Dasgupta, P., and Chellappan, S. P. (2006). Nicotine-mediated cell proliferation and angiogenesis: new twists to an old story. Cell Cycle 5, 2324–2328.
Dasgupta, P., Rastogi, S., Pillai, S., Ordonez-Ercan, D., Morris, M., Haura, E., and Chellappan, S. (2006). Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J. Clin. Invest. 116, 2208–2217.
Dittmar, K. D., Banach, M., Galigniana, M. D., and Pratt, W. B. (1998). The role of DnaJ-like proteins in glucocorticoid receptor.hsp90 heterocomplex assembly by the reconstituted hsp90.p60.hsp70 foldosome complex. J. Biol. Chem. 273, 7358–7366.
Dittmar, K. D., Demady, D. R., Stancato, L. F., Krishna, P., and Pratt, W. B. (1997). Folding of the glucocorticoid receptor by the heat shock protein (hsp) 90-based chaperone machinery. The role of p23 is to stabilize receptor.hsp90 heterocomplexes formed by hsp90.p60.hsp70. J. Biol. Chem. 272, 21213–21220.
Dunckley, T., and Lukas, R. J. (2003). Nicotine modulates the expression of a diverse set of genes in the neuronal SH-SY5Y cell line. J. Biol. Chem. 278, 15633–15640.
Dunckley, T., and Lukas, R. J. (2006). Nicotinic modulation of gene expression in SH-SY5Y neuroblastoma cells. Brain Res. 1116, 39–49.
Edwards, S., Graham, D. L., Bachtell, R. K., and Self, D. W. (2007). Region-specific tolerance to cocaine-regulated cAMP-dependent protein phosphorylation following chronic self-administration. Eur. J. Neurosci. 25, 2201–2213.
Ernst, M., Heishman, S. J., Spurgeon, L., and London, E. D. (2001). Smoking history and nicotine effects on cognitive performance. Neuropsychopharmacology 25, 313–319.
Ficklin, M. B., Zhao, S., and Feng, G. (2005). Ubiquilin-1 regulates nicotine-induced up-regulation of neuronal nicotinic acetylcholine receptors. J. Biol. Chem. 280, 34088–34095.
Foulds, J., Stapleton, J., Swettenham, J., Bell, N., McSorley, K., and Russell, M. A. (1996). Cognitive performance effects of subcutaneous nicotine in smokers and never-smokers. Psychopharmacology (Berl.) 127, 31–38.
Fox, H. C., Jackson, E. D., and Sinha, R. (2009). Elevated cortisol and learning and memory deficits in cocaine dependent individuals: relationship to relapse outcomes. Psychoneuroendocrinology 34, 1198–1207.
French, K. L., Granholm, A. C., Moore, A. B., Nelson, M. E., and Bimonte-Nelson, H. A. (2006). Chronic nicotine improves working and reference memory performance and reduces hippocampal NGF in aged female rats. Behav. Brain Res. 169, 256–262.
Gaddnas, H., Pietila, K., Piepponen, T. P., and Ahtee, L. (2001). Enhanced motor activity and brain dopamine turnover in mice during long-term nicotine administration in the drinking water. Pharmacol. Biochem. Behav. 70, 497–503.
Garrido, R., King-Pospisil, K., Son, K. W., Hennig, B., and Toborek, M. (2003). Nicotine upregulates nerve growth factor expression and prevents apoptosis of cultured spinal cord neurons. Neurosci. Res. 47, 349–355.
Gaudilliere, B., Shi, Y., and Bonni, A. (2002). RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J. Biol. Chem. 277, 46442–46446.
Ghosh, J., Murphy, M. O., Turner, N., Khwaja, N., Halka, A., Kielty, C. M., and Walker, M. G. (2005). The role of transforming growth factor beta1 in the vascular system. Cardiovasc. Pathol. 14, 28–36.
Gould, J., Reeve, H. L., Vaughan, P. F., and Peers, C. (1992). Nicotinic acetylcholine receptors in human neuroblastoma (SH-SY5Y) cells. Neurosci. Lett. 145, 201–204.
Grenhoff, J., and Svensson, T. H. (1988). Selective stimulation of limbic dopamine activity by nicotine. Acta Physiol. Scand. 133, 595–596.
Gueorguiev, V. D., Cheng, S. Y., and Sabban, E. L. (2006). Prolonged activation of cAMP-response element-binding protein and ATF-2 needed for nicotine-triggered elevation of tyrosine hydroxylase gene transcription in PC12 cells. J. Biol. Chem. 281, 10188–10195.
Gupta, V., Awasthi, N., and Wagner, B. J. (2007). Specific activation of the glucocorticoid receptor and modulation of signal transduction pathways in human lens epithelial cells. Invest. Ophthalmol. Vis. Sci. 48, 1724–1734.
Gutala, R., Wang, J., Kadapakkam, S., Hwang, Y., Ticku, M., and Li, M. D. (2004). Microarray analysis of ethanol-treated cortical neurons reveals disruption of genes related to the ubiquitin-proteasome pathway and protein synthesis. Alcohol. Clin. Exp. Res. 28, 1779–1788.
Harkness, P. C., and Millar, N. S. (2002). Changes in conformation and subcellular distribution of alpha4beta2 nicotinic acetylcholine receptors revealed by chronic nicotine treatment and expression of subunit chimeras. J. Neurosci. 22, 10172–10181.
Heidenreich, K. A., and Linseman, D. A. (2004). Myocyte enhancer factor-2 transcription factors in neuronal differentiation and survival. Mol. Neurobiol. 29, 155–166.
Hernandez, C. M., and Terry, A. V. Jr. (2005). Repeated nicotine exposure in rats: effects on memory function, cholinergic markers and nerve growth factor. Neuroscience 130, 997–1012.
Heusch, W. L., and Maneckjee, R. (1998). Signalling pathways involved in nicotine regulation of apoptosis of human lung cancer cells. Carcinogenesis 19, 551–556.
Hwang, Y. Y., and Li, M. D. (2006). Proteins differentially expressed in response to nicotine in five rat brain regions: identification using a 2-DE/MS-based proteomics approach. Proteomics 6, 3138–3153.
Ichino, N., Ishiguro, H., Yamada, K., Nishii, K., Sawada, H., and Nagatsu, T. (1999). Nicotine withdrawal up-regulates c-Fos transcription in pheochromocytoma cells. Neurosci. Res. 35, 63–69.
Ichino, N., Yamada, K., Nishii, K., Sawada, H., Nagatsu, T., and Ishiguro, H. (2002). Increase of transcriptional levels of egr-1 and nur77 genes due to both nicotine treatment and withdrawal in pheochromocytoma cells. J. Neural Transm. 109, 1015–1022.
Isola, R., Zhang, H., Tejwani, G. A., Neff, N. H., and Hadjiconstantinou, M. (2009). Acute nicotine changes dynorphin and prodynorphin mRNA in the striatum. Psychopharmacology (Berl.) 201, 507–516.
Jiao, H., Zhang, L., Gao, F., Lou, D., Zhang, J., and Xu, M. (2007). Dopamine D(1) and D(3) receptors oppositely regulate NMDA- and cocaine-induced MAPK signaling via NMDA receptor phosphorylation. J. Neurochem. 103, 840–848.
Jonnala, R. R., Terry, A. V. Jr., and Buccafusco, J. J. (2002). Nicotine increases the expression of high affinity nerve growth factor receptors in both in vitro and in vivo. Life Sci. 70, 1543–1554.
Kane, J. K., Konu, O., Ma, J. Z., and Li, M. D. (2004). Nicotine coregulates multiple pathways involved in protein modification/degradation in rat brain. Brain Res. Mol. Brain Res. 132, 181–191.
Katyare, S. S., and Shallom, J. M. (1988). Altered cerebral protein turnover in rats following prolonged in vivo treatment with nicotine. J. Neurochem. 50, 1356–1363.
Ke, L., Eisenhour, C. M., Bencherif, M., and Lukas, R. J. (1998). Effects of chronic nicotine treatment on expression of diverse nicotinic acetylcholine receptor subtypes. I. Dose- and time-dependent effects of nicotine treatment. J. Pharmacol. Exp. Ther. 286, 825–840.
Kenny, P. J., File, S. E., and Rattray, M. (2001). Nicotine regulates 5-HT(1A) receptor gene expression in the cerebral cortex and dorsal hippocampus. Eur. J. Neurosci. 13, 1267–1271.
Kobayashi, H., Ide, S., Hasegawa, J., Ujike, H., Sekine, Y., Ozaki, N., Inada, T., Harano, M., Komiyama, T., Yamada, M., Iyo, M., Shen, H. W., Ikeda, K., and Sora, I. (2004). Study of association between alpha-synuclein gene polymorphism and methamphetamine psychosis/dependence. Ann. N. Y. Acad. Sci. 1025, 325–334.
Konu, O., Kane, J. K., Barrett, T., Vawter, M. P., Chang, R., Ma, J. Z., Donovan, D. M., Sharp, B., Becker, K. G., and Li, M. D. (2001). Region-specific transcriptional response to chronic nicotine in rat brain. Brain Res. 909, 194–203.
Konu, O., Xu, X., Ma, J. Z., Kane, J., Wang, J., Shi, S. J., and Li, M. D. (2004a). Application of a customized pathway-focused microarray for gene expression profiling of cellular homeostasis upon exposure to nicotine in PC12 cells. Brain Res. Mol. Brain Res. 121, 102–113.
Konu, O., Xu, X., Ma, J. Z., Kane, J. K., Wang, J., Shi, S. J., and Li, M. D. (2004b). Application of a customized pathway-focused microarray for gene expression profiling of cellular homeostasis upon exposure to nicotine in PC12 cells. Brain Res. Mol. Brain Res. 110, 102–113.
Kumari, V., Gray, J. A., Ffytche, D. H., Mitterschiffthaler, M. T., Das, M., Zachariah, E., Vythelingum, G. N., Williams, S. C., Simmons, A., and Sharma, T. (2003). Cognitive effects of nicotine in humans: an fMRI study. Neuroimage 19, 1002–1013.
Lane, D., Gray, E. A., Mathur, R. S., and Mathur, S. P. (2005). Up-regulation of vascular endothelial growth factor-C by nicotine in cervical cancer cell lines. Am. J. Reprod. Immunol. 53, 153–158.
Levin, E. D., and Rose, J. E. (1995). Acute and chronic nicotinic interactions with dopamine systems and working memory performance. Ann. N. Y. Acad. Sci. 757, 245–252.
Levin, E. D., and Simon, B. B. (1998). Nicotinic acetylcholine involvement in cognitive function in animals. Psychopharmacology (Berl.) 138, 217–230.
Li, M., Linseman, D. A., Allen, M. P., Meintzer, M. K., Wang, X., Laessig, T., Wierman, M. E., and Heidenreich, K. A. (2001). Myocyte enhancer factor 2A and 2D undergo phosphorylation and caspase-mediated degradation during apoptosis of rat cerebellar granule neurons. J. Neurosci. 21, 6544–6552.
Li, M. D., Kane, J. K., Wang, J., and Ma, J. Z. (2004). Time-dependent changes in transcriptional profiles within five rat brain regions in response to nicotine treatment. Brain Res. Mol. Brain Res. 132, 168–180.
Mao, Z., Bonni, A., Xia, F., Nadal-Vicens, M., and Greenberg, M. E. (1999). Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 286, 785–790.
Marinelli, M., and Piazza, P. V. (2002). Interaction between glucocorticoid hormones, stress and psychostimulant drugs. Eur. J. Neurosci. 16, 387–394.
Mash, D. C., Adi, N., Duque, L., Pablo, J., Kumar, M., and Ervin, F. R. (2008). Alpha synuclein protein levels are increased in serum from recently abstinent cocaine abusers. Drug Alcohol Depend. 94, 246–250.
Mexal, S., Frank, M., Berger, R., Adams, C. E., Ross, R. G., Freedman, R., and Leonard, S. (2005). Differential modulation of gene expression in the NMDA postsynaptic density of schizophrenic and control smokers. Brain Res. Mol. Brain Res. 139, 317–332.
Nakayama, H., Numakawa, T., Ikeuchi, T., and Hatanaka, H. (2001). Nicotine-induced phosphorylation of extracellular signal-regulated protein kinase and CREB in PC12h cells. J. Neurochem. 79, 489–498.
Nisell, M., Nomikos, G. G., Chergui, K., Grillner, P., and Svensson, T. H. (1997). Chronic nicotine enhances basal and nicotine-induced Fos immunoreactivity preferentially in the medial prefrontal cortex of the rat. Neuropsychopharmacology 17, 151–161.
Novak, G., Boukhadra, J., Shaikh, S. A., Kennedy, J. L., and Le Foll, B. (2009). Association of a polymorphism in the NRXN3 gene with the degree of smoking in schizophrenia: a preliminary study. World J. Biol. Psychiatry 10, 929–935.
Nussbaum, J., Xu, Q., Payne, T. J., Ma, J. Z., Huang, W., Gelernter, J., and Li, M. D. (2008). Significant association of the neurexin-1 gene (NRXN1) with nicotine dependence in European- and African-American smokers. Hum. Mol. Genet. 17, 1569–1577.
Onal, A., Uysal, A., Ulker, S., Delen, Y., Yurtseven, M. E., and Evinc, A. (2004). Alterations of brain tissue in fetal rats exposed to nicotine in utero: possible involvement of nitric oxide and catecholamines. Neurotoxicol. Teratol. 26, 103–112.
Pagliusi, S. R., Tessari, M., DeVevey, S., Chiamulera, C., and Pich, E. M. (1996). The reinforcing properties of nicotine are associated with a specific patterning of c-fos expression in the rat brain. Eur. J. Neurosci. 8, 2247–2256.
Papke, R. L., Sanberg, P. R., and Shytle, R. D. (2001). Analysis of mecamylamine stereoisomers on human nicotinic receptor subtypes. J. Pharmacol. Exp. Ther. 297, 646–656.
Peng, X., Katz, M., Gerzanich, V., Anand, R., and Lindstrom, J. (1994). Human alpha 7 acetylcholine receptor: cloning of the alpha 7 subunit from the SH-SY5Y cell line and determination of pharmacological properties of native receptors and functional alpha 7 homomers expressed in Xenopus oocytes. Mol. Pharmacol. 45, 546–554.
Phillips, S., and Fox, P. (1998). An investigation into the effects of nicotine gum on short-term memory. Psychopharmacology (Berl.) 140, 429–433.
Piazza, P. V., and Le Moal, M. (1997). Glucocorticoids as a biological substrate of reward: physiological and pathophysiological implications. Brain Res. Brain Res. Rev. 25, 359–372.
Pollman, M. J., Naumovski, L., and Gibbons, G. H. (1999). Vascular cell apoptosis: cell type-specific modulation by transforming growth factor-beta1 in endothelial cells versus smooth muscle cells. Circulation 99, 2019–2026.
Reuter, M., Hennig, J., and Netter, P. (2004). Do smoking intensity-related differences in vigilance indicate altered glucocorticoid receptor sensitivity? Addict. Biol. 9, 35–41.
Rezvani, K., Teng, Y., Shim, D., and De Biasi, M. (2007). Nicotine regulates multiple synaptic proteins by inhibiting proteasomal activity. J. Neurosci. 27, 10508–10519.
Robinson, T. E., and Kolb, B. (2004). Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology 47(Suppl. 1), 33–46.
Robinson-Rechavi, M., Carpentier, A. S., Duffraisse, M., and Laudet, V. (2001). How many nuclear hormone receptors are there in the human genome? Trends Genet. 17, 554–556.
Salminen, O., Seppa, T., Gaddnas, H., and Ahtee, L. (1999). The effects of acute nicotine on the metabolism of dopamine and the expression of Fos protein in striatal and limbic brain areas of rats during chronic nicotine infusion and its withdrawal. J. Neurosci. 19, 8145–8151.
Schmitt, H. F., Huang, L. Z., Son, J. H., Pinzon-Guzman, C., Slaton, G. S., and Winzer-Serhan, U. H. (2008). Acute nicotine activates c-fos and activity-regulated cytoskeletal associated protein mRNA expression in limbic brain areas involved in the central stress-response in rat pups during a period of hypo-responsiveness to stress. Neuroscience 157, 349–359.
Schochet, T. L., Bremer, Q. Z., Brownfield, M. S., Kelley, A. E., and Landry, C. F. (2008). The dendritically targeted protein dendrin is induced by acute nicotine in cortical regions of adolescent rat brain. Eur. J. Neurosci. 28, 1967–1979.
Schochet, T. L., Kelley, A. E., and Landry, C. F. (2004). Differential behavioral effects of nicotine exposure in adolescent and adult rats. Psychopharmacology (Berl.) 175, 265–273.
Schochet, T. L., Kelley, A. E., and Landry, C. F. (2005). Differential expression of arc mRNA and other plasticity-related genes induced by nicotine in adolescent rat forebrain. Neuroscience 135, 285–297.
Shalizi, A. K., and Bonni, A. (2005). brawn for brains: the role of MEF2 proteins in the developing nervous system. Curr. Top. Dev. Biol. 69, 239–266.
Soderstrom, K., Qin, W., Williams, H., Taylor, D. A., and McMillen, B. A. (2007). Nicotine increases FosB expression within a subset of reward- and memory-related brain regions during both peri- and post- adolescence. Psychopharmacology (Berl.) 191, 891–897.
Soto-Otero, R., Mendez-Alvarez, E., Hermida-Ameijeiras, A., Lopez-Real, A. M., and Labandeira-Garcia, J. L. (2002). Effects of (−)-nicotine and (−)-cotinine on 6-hydroxydopamine-induced oxidative stress and neurotoxicity: relevance for Parkinson’s disease. Biochem. Pharmacol. 64, 125–135.
Spence, J. P., Liang, T., Liu, L., Johnson, P. L., Foroud, T., Carr, L. G., and Shekhar, A. (2009). From QTL to candidate gene: a genetic approach to alcoholism research. Curr. Drug Abuse Rev. 2, 127–134.
Suarez, S. V., Amadon, A., Giacomini, E., Wiklund, A., Changeux, J. P., Le Bihan, D., and Granon, S. (2009). Brain activation by short-term nicotine exposure in anesthetized wild-type and beta2-nicotinic receptors knockout mice: a BOLD fMRI study. Psychopharmacology (Berl.) 202, 599–610.
Tammimaki, A., Pietila, K., Raattamaa, H., and Ahtee, L. (2006). Effect of quinpirole on striatal dopamine release and locomotor activity in nicotine-treated mice. Eur. J. Pharmacol. 531, 118–125.
Tang, K., Wu, H., Mahata, S. K., and O’Connor, D. T. (1998). A crucial role for the mitogen-activated protein kinase pathway in nicotinic cholinergic signaling to secretory protein transcription in pheochromocytoma cells. Mol. Pharmacol. 54, 59–69.
Terry, A. V. Jr., and Clarke, M. S. (1994). Nicotine stimulation of nerve growth factor receptor expression. Life Sci. 55, PL91–PL98.
Thoenen, H., Bandtlow, C., and Heumann, R. (1987). The physiological function of nerve growth factor in the central nervous system: comparison with peiphery. Rev. Physiol. Biochem. Pharmacol. 109, 145–178.
Thomas, L., Welsh, L., Galvez, F., and Svoboda, K. (2008). Acute nicotine exposure and modulation of a spinal motor circuit in embryonic zebrafish. Toxicol. Appl. Pharmacol. 239, 1–12.
Tizabi, Y., Getachew, B., Rezvani, A. H., Hauser, S. R., and Overstreet, D. H. (2009). Antidepressant-like effects of nicotine and reduced nicotinic receptor binding in the Fawn-Hooded rat, an animal model of co-morbid depression and alcoholism. Prog. Neuropsychopharmacol. Biol. Psychiatry 33, 398–402.
Tye, S. J., Miller, A. D., and Blaha, C. D. (2009). Differential corticosteroid receptor regulation of mesoaccumbens dopamine efflux during the peak and nadir of the circadian rhythm: a molecular equilibrium in the midbrain? Synapse 63, 982–990.
Vadasz, C., Saito, M., O’Brien, D., Zavadil, J., Morahan, G., Chakraborty, G., and Wang, R. (2007). Ventral tegmental transcriptome response to intermittent nicotine treatment and withdrawal in BALB/cJ, C57BL/6ByJ, and quasi-congenic RQI mice. Neurochem. Res. 32, 457–480.
Wang, J., Chen, Y. B., Zhu, X. N., and Chen, R. Z. (2001). Activation of p42/44 mitogen-activated protein kinase pathway in long-term potentiation induced by nicotine in hippocampal CA1 region in rats. Acta Pharmacol. Sin. 22, 685–690.
Wang, J., Gutala, R., Sun, D., Ma, J. Z., Sheela, R. C., Ticku, M. K., and Li, M. D. (2007). Regulation of platelet-derived growth factor signaling pathway by ethanol, nicotine, or both in mouse cortical neurons. Alcohol. Clin. Exp. Res. 31, 357–375.
Wang, J., Kim, J. M., Donovan, D. M., Becker, K. G., and Li, M. D. (2009). Significant modulation of mitochondrial electron transport system by nicotine in various rat brain regions. Mitochondrion 9, 186–195.
Weber, R. J., Gomez-Flores, R., Smith, J. E., and Martin, T. J. (2009). Neuronal adaptations, neuroendocrine and immune correlates of heroin self-administration. Brain Behav. Immun. 23, 993–1002.
Wei, J., Wang, J., Dwyer, J. B., Mangold, J., Cao, J., Leslie, F. M., and Li, M. D. (2011). Gestational nicotine treatment modulates cell death/survival-related pathways in the brains of adolescent female rats. Int. J. Neuropsychopharmacol. 14, 91–106.
Winer, J., Jung, C. K., Shackel, I., and Williams, P. M. (1999). Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal. Biochem. 270, 41–49.
Xie, Y. X., Bezard, E., and Zhao, B. L. (2005). Investigating the receptor-independent neuroprotective mechanisms of nicotine in mitochondria. J. Biol. Chem. 280, 32405–32412.
Keywords: nicotine, neurons, mecamylamine, gene expression, p38
Citation: Wang J, Cui W, Wei J, Sun D, Gutala R, Gu J and Li MD (2011) Genome-wide expression analysis reveals diverse effects of acute nicotine exposure on neuronal function-related genes and pathways. Front. Psychiatry 2:5. doi: 10.3389/fpsyt.2011.00005
Received: 10 January 2011;
Paper pending published: 01 February 2011;
Accepted: 16 February 2011;
Published online: 08 March 2011.
Edited by:
Edythe D. London, University of California Los Angeles, USAReviewed by:
Herb Lachman, Albert Einstein College of Medicine of Yeshiva University, USAMikhail Pletnikov, Johns Hopkins University, USA
Copyright: © 2011 Wang, Cui, Wei, Sun, Gutala, Gu and Li. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Ming D. Li, Department of Psychiatry and Neurobehavioral Sciences, University of Virginia, 1670 Discovery Drive, Suite 110, Charlottesville, VA 22911, USA. e-mail:bWluZ19saUB2aXJnaW5pYS5lZHU=