 Yuyan Zhang
Yuyan Zhang Xin Guo2
Xin Guo2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 27 March 2025
Sec. Plant Bioinformatics
Volume 16 - 2025 | https://doi.org/10.3389/fpls.2025.1558619
This article is part of the Research TopicEvolutionary Dynamics, Functional Variation and Application of Plant Organellar GenomeView all 17 articles
Prunus davidiana is an excellent woody species with high resistance to various abiotic and biotic stresses. The species is also a potential gene donor due to its important developmental prospects. However, the characterisation of the mitochondrial genome (mitogenome) remains unexplored. In this study, we sequenced and assembled the mitogenome of P. davidiana using reads from Illumina sequencing and the Oxford Nanopore platform. According to the results, the P. davidiana mitogenome is 407,608 bp in length, with a GC content of 45.5%. It contains 64 genes, including 39 protein-coding genes, 22 tRNA genes and 3 rRNA genes. Codon usage, repetitive sequences, nonsynonymous to synonymous substitution ratios, RNA editing, synteny, and phylogenetic relationships among Rosaceae species and gene migration events were investigated. In addition, two novel indel markers were developed to distinguish several Prunus species (i.e. P. pseudocerasus, P. armeniaca, P. salicina and P. davidiana). The results of our analyses provide valuable information and a theoretical basis for future research on P. davidiana.
Prunus davidiana is a small shrub belonging to the subfamily Prunoideae of the family Rosaceae (Liu et al., 2017). The species is native to China and is reported to be resistant to cold, saline alkalinity and drought stress, as well as some pests and diseases (Hesse, 1975). Therefore, it is widely used as a grafting rootstock for peach and plum in northern China due to its adaptability to different environmental conditions (Xu et al., 2020). Accordingly, P. davidiana is considered as a potential gene donor in future peach breeding programmes.
As the ‘energy factories’ of cells, mitochondria are important for energy synthesis and conversion in cell physiological activities (Ye et al., 2017). They play a critical role in plant growth and development (Ogihara et al., 2005). Plant organelle genomes have genetic systems that are independent of the nuclear genome and share stable regulatory mechanisms with the nuclear genome (Li et al., 2022). The chloroplast (cp) genome is usually structurally conserved with double-stranded stable circular genome (Ma et al., 2019). However, other mitogenome structures have been observed, including circular, linear, branched and numerous small circular molecules (Sloan, 2013). Furthermore, plant mitochondrial genomes (mitogenomes) vary in size even among related species. Most genomes are 200-800 kb in size, and the largest land plant mitogenome is about 11.3 Mb (Silene conica) (Sloan et al., 2012). The smallest is only about 66 kb (Viscum scurruloideum) (Skippington et al., 2015). Mitogenomes contain abundant repetitive sequences and mediate intergenomic gene transfer, which can lead to variation in genome size and rapid genome rearrangement. Reconstructing the structure of plant mitogenomes is challenging due to their redundant sequences and genomic recombination (Christensen, 2013). Plant mitochondrial markers provide an important complement to plastid DNA markers, indel and minisatellite regions are relatively easy to detect, and are often used to develop markers for breeding and species identification (Sebbenn et al., 2019; Ma et al., 2019). Furthermore, gene sequence transfer between nuclear and organellar genomes is a common phenomenon, as reported in recent studies (Bergthorsson et al., 2003; Li et al., 2022; Ma et al., 2022). As a consequence, the gene content also varies considerably (Wynn and Christensen, 2019).
P. davidiana is an excellent germplasm material for the peach breeding research due to its high resistance to many biotic and abiotic stresses. In previous studies, several mitogenomes of Prunus species have been sequenced and assembled, such as P. avium (Yan et al., 2019) and P. salicina (Fang et al., 2021) Although the cp genome of P. davidiana has been reported, the mitogenome has not been reported. In the present study, we report the sequencing, assembly and annotation of the P. davidiana mitogenome using a combination of the Illumina and PacBio sequencing platforms. In addition, we also analysed repetitive sequences, selective pressures, RNA editing sites and phylogenetic relationships among Rosaceae species in the present study. Gene transfer between nuclear, cp and mitochondrial genomes was also investigated. The results of the present study could improve our understanding of organelle genome evolution in P. davidiana and facilitate genomic breeding of P. davidiana.
P. davidiana plants were grown under natural conditions at the germplasm base of the Jiangsu Academy of Agricultural Sciences (Xuanwu District, Nanjing, China, lat.32°02’N, long.118°46’E). Total DNA was isolated from fresh leaves using the CTAB method (Varré et al., 2019), and quality was assessed by 1% agarose gel electrophoresis. Qualified samples were sent to Nanjing Jisi Huiyuan Biotechnology Co. (http://www.genepioneer.com/) for Illumina sequencing and Oxford sequencing. Experimental procedures were performed according to a standard protocol (Bi et al., 2020). To obtain the full-length mitochondrial genome with high accuracy. Paired end sequencing (PE) reads (150 bp) were obtained using an Illumina Novaseq 6000 (Illumina, San Diego, CA, USA) and we filtered the original data using fastp (v 0.20.0, https://github.com/OpenGene/fastp) software to obtain high quality clear reads. Long read sequences were obtained from Nanopore PromethION (Nanopore, Oxford, UK) and filtered using filtlong (v0.2.1, https://mirrors.sjtug.sjtu.edu.cn/kali/pool/main/f/filtlong/) software.
Plant mitochondrial genes (CDS and rRNA) are highly conserved. Taking advantage of this feature, reads were mapped to the reference gene sequences (plant 2mitochondrial core genes (https://github.com/xul962464/plant_mt_ref_gene)) using Minimap2 (v2.1) (Ma et al., 2017). Candidate sequences containing multiple core genes and higher alignment quality (covering more complete core genes) were selected as seed sequences. The seed sequences were then compared to the long-read sequencing data with a minimum overlap of 1 kb and at least 70% similarity. Canu (Ye et al., 2017) was used to correct the long-read sequencing data, and bowtie2 (v2.3.5.1) (Cheng et al., 2021) was used to align the short-read sequencing data to the corrected sequence. The preliminary assembly drawing was obtained in Unicycler (Wick et al., 2017), then the final assembly of mtDNA was performed using the long-read sequencing data. Finally, the longest path was selected as the mitochondrial genome of P. davidiana on based on the relationship between the linkage and sequence depth.
The structure annotation of the mitochondrial genome was divided into the following steps: (1) The encoding protein and rRNA were aligned with published plant mitochondrial sequences using BLAST and further manual alignment with related species. (2) The tRNA was annotated using tRNAs-canSE (http://lowelab.ucsc.edu/tRNAscan-SE/) with default settings (Goremykin et al., 2012). To obtain more accurate annotation results, the above results were checked and manually corrected using Apollo (Gualberto et al., 2014). The mitochondrial genome was then mapped using OGDRAW (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html).
We performed codon preference analysis on the PCGs of the mitogenome and calculated RSCU values using MEGA 7.0 (Xiong et al., 2021). SSRs were detected using the Perl script MISA (http://pgrc.ipkgatersleben.de/misa/) (Liu et al., 2023), with sizes ranging from one to six nucleotides and thresholds of 10, 5, 4, 3, 3 and 3. In addition, REPuter (Clifton et al., 2004) was used to identify the forward, palindromic and tandem repeats.
We calculated the non-synonymous (Ka) and synonymous (Ks) substitution rates with PCGs in P. davidiana, M. domestica, S. aucuparia, R. bibas, P. betulifolia and F. orientalis. DnaSP was used to calculate Ka, Ks and Ka/Ks values (Rice et al., 2013).
The P. davidiana cp genome (MH460864) was downloaded from the NCBI Organelle Genome Resources Database. The P. davidiana mitogenome was searched against the cp genome using BLASTN 2.9.0+ (Zhao et al., 2019). In addition, a BLASTN search (E-value=10-50) was performed between the mitogenome and the P. davidiana nuclear genome sequence (PRJNA655343). PREP suit (http://prep.unl.edu/) was used to predict the RNA editing sites in the PCGs of the P. davidiana mitogenome (the cut-off value was set to 0.2).
A pairwise comparison dot plot was generated and the conserved co-linear blocks were plotted. A multiple synteny plot of the P. davidiana mitogenome with closely related species was then visualised using MCscanX (Arseneau et al., 2017). A total of 29 complete mitogenomes were downloaded (https://www.ncbi.nlm.nih.gov/) to determine the phylogenetic position of P. davidiana. We aligned the 32 common genes of the analysed species in Muscle (Bolger et al., 2014) and constructed the maximum likelihood (ML) phylogenetic tree using MEGA X (1000 bootstrap replicates) (Xiong et al., 2021).
The P. davidiana mitogenome was sequenced, assembled and annotated. The Oxford Nanopore (9.09 Gb, 22, 312 × coverage, with a read length N50 of ~15.82 kb) and Illumina data (13, 275× coverage) have been submitted in the Sequence Read Archive (SRA) under study accession number SRR25468552 and SRR25468515, respectively (Supplementary Table S1). The graph-based mitochondrial genome of P. davidiana was assembled, and a specific linear structure was used to represent the entire mitogenome sequence. The total length of the P. davidiana mitogenome was 407, 608 bp and the GC content was 45.5% (Figure 1A). The genome sequence of P. davidiana was submitted to the GenBank database (PQ825745).
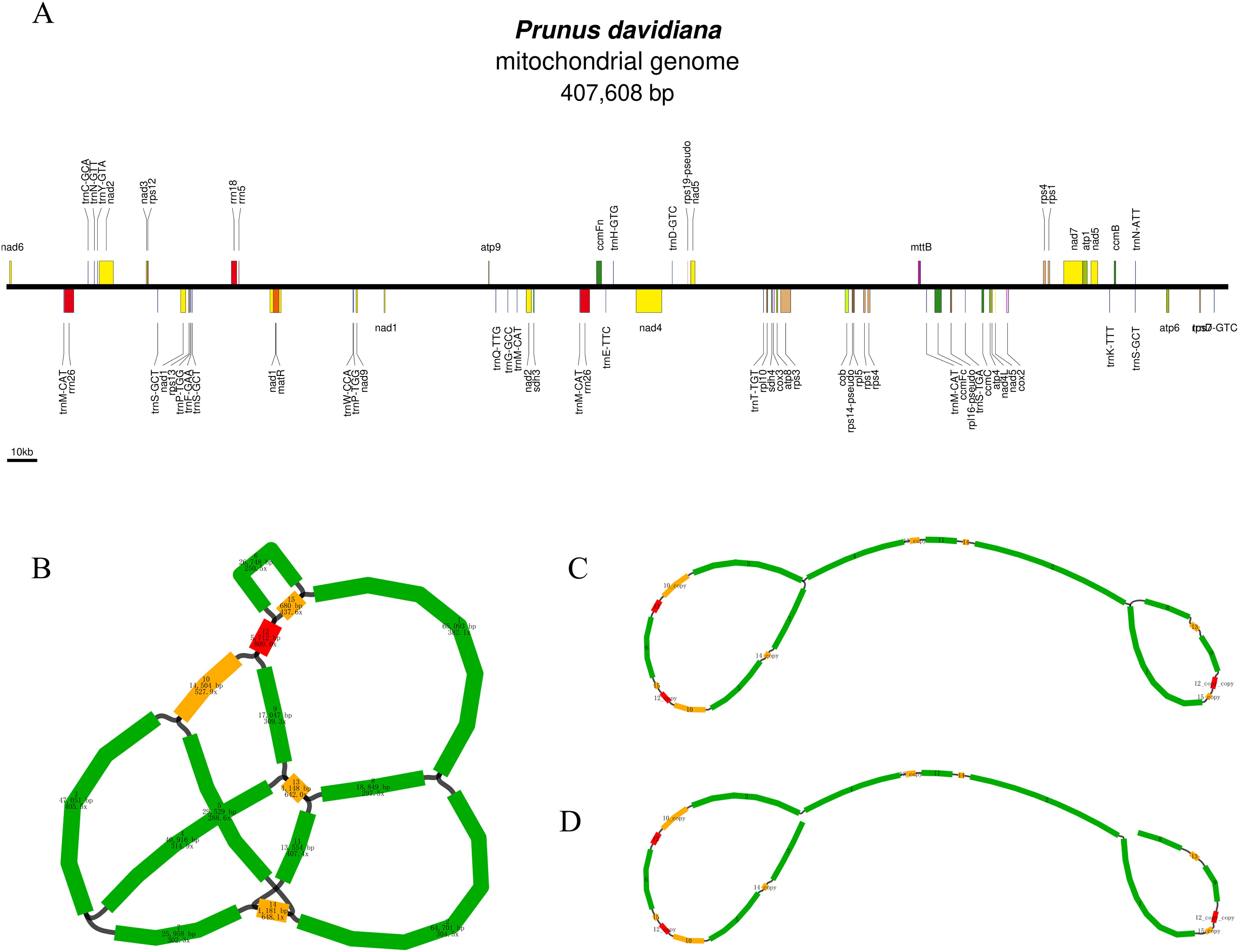
Figure 1. (A) The order, orientation and size of gene within the P. davidiana mitogenome. Each box is proportional to size of the gene including introns. (B) Schematic of mtDNA of P. davidianas. (C) Unicycler hybrid assembly-based mtDNA of P. davidianas. (D) The specific solution path is 8-13-9-12-copy-copy-15-copy-1-2-14-11-13-copy-4-3-10-copy-12-6-15-12-copy-10-5-14-copy-7.
A total of 64 genes were annotated in the P. davidiana mitogenome, including 39 protein-coding genes (PCGs), 22 tRNA genes and 3 rRNA genes (Table 1). Among the 39 PCGs, six contained introns (nad1, nad2, nad4, nad7, ccmFc and rps3). Interestingly, two copy genes (nad1, nad2 and cox1) and one three copy gene (nad5) were found. We also observed five tRNA genes and one rRNA gene located in repeat sequences (trnN-GTT, trnM-CAT, trnP-TGG, trnH-GTG, trnW-CCA and rrn5) (Figure 1A).
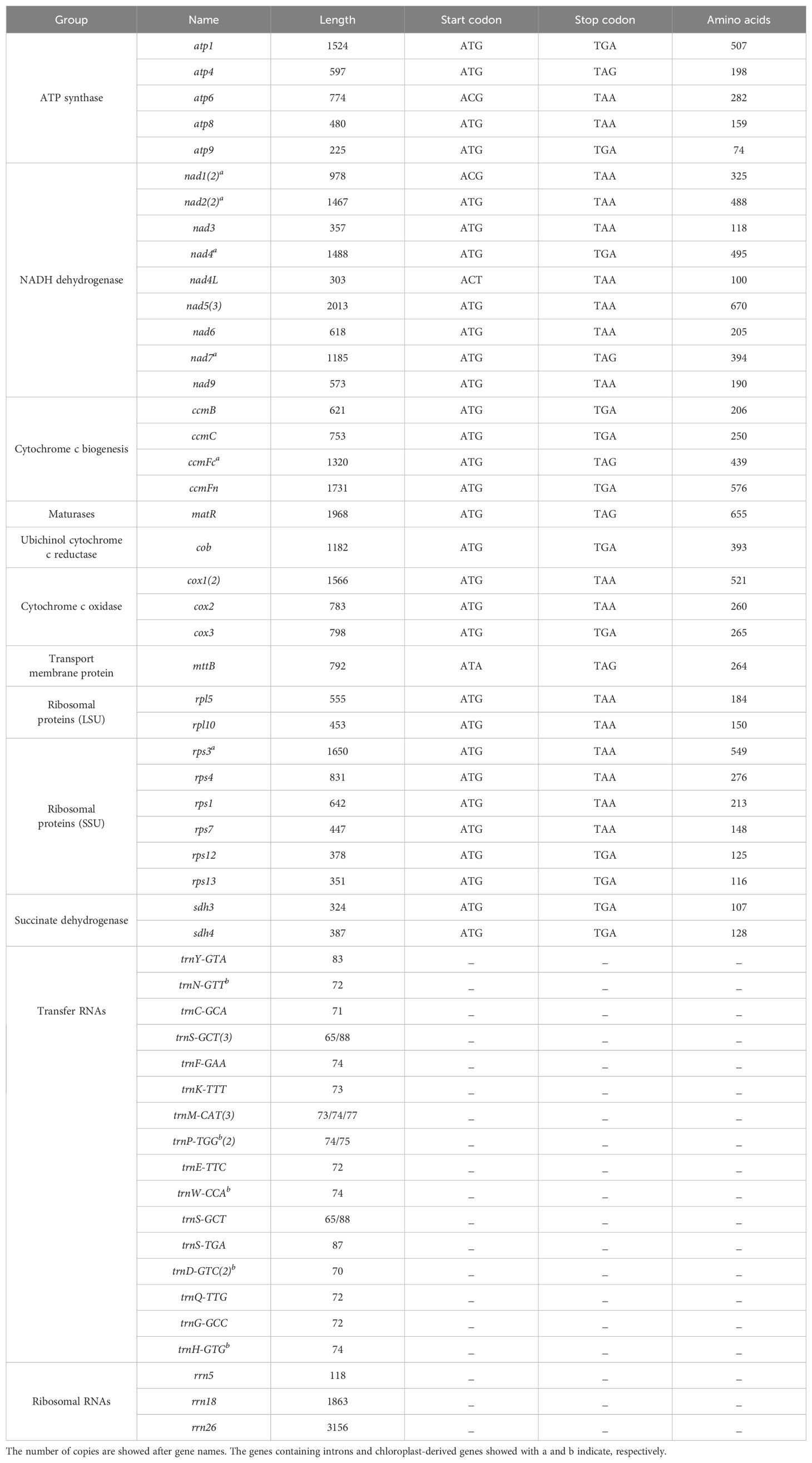
Table 1. The characteristics of genes in P. davidiana mitogenome.
We visualised the schematic of the mitogenome assembled from the long read data using Bandage software. The schematic contained 14 contigs, the sequence length and sequencing depth of the contigs are shown in Figure 1B. The black line indicates the overlapping region between two contigs. These contigs were used to construct a complex multibranched structure. The multiple major branch nodes were resolved using Unicycler software with long reads (Wick et al., 2017). In short, for branch nodes with multiple connections, those connections supported by more long reads are preferred. The genome sequence of a line obtained after solving the branch nodes using Unicycler is shown in Figure 1B. The specific solution path is 8-13-9-12-copy-copy-15-copy-1-2-14-11-13-copy-4-3-10-copy-12-6-15-12-copy-10-5-14-copy-7 (Figures 1C, D). The plant mitogenome is not unique, and the assembly is not the only form, but is in the midst of dynamic changes, which has facilitated the subsequent analysis.
Codon usage analysis was performed on 39 PCGs. The codon usage of each amino acid is shown in Figure 2. The PCGs in P. davidiana had a total length of 30, 234 nucleotides. The typical ATG start codon was found in most PCGs, whereas nad4L and nad1 were ACG, which can be altered by C-to-U RNA editing (Table 1). As in other mitogenomes, TAA, TGA and TAG served as stop codons.
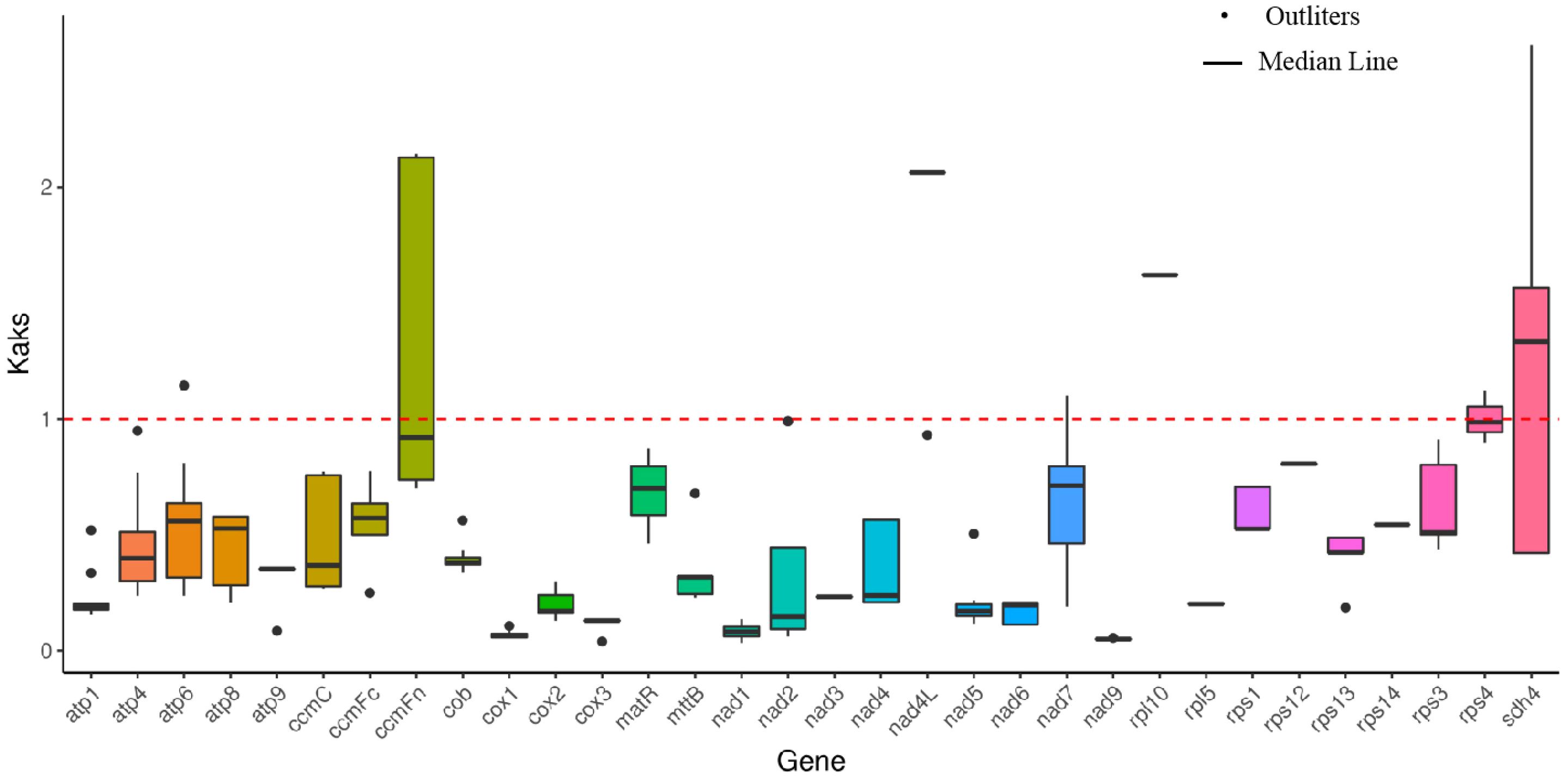
Figure 2. Ka/Ks calculated based on 32 shared genes among the six Rosaceae mitogenome.
The relative synonymous codon usage (RSCU) of 39 PCGs was also analysed. The 39 PCGs comprised 30, 234 bp encoding 10, 078 codons, excluding termination codons. Codons with RSCU >1 were considered to be preferentially used by amino acids. All RSCU values of NNT and NNA codons, except Ile (ATA), Leu (CTA) and Thr (ACA), were higher than 1.0 (Figure 3). The result indicates the existence of a strong As or Ts bias at the third codon position in P. davidiana mitogenomes. This phenomenon is also present in other mitogenomes of plant species (Ma et al., 2022).

Figure 3. The RSCU analysis in the P. davidiana mitogenome. The x-axis was Codon families. RSCU values displayed on bar.
The non-synonymous to synonymous substitution ratios (Ka/Ks) between any two species among six species (Malus domestica, Sorbus aucuparia, Rhaphiolepis bibas, Pyrus betulifolia, Fragaria orientalis, and P. davidiana) were calculated based on the 32 shared genes in Rosaceae. According to Figure 2, the mean values of pairwise Ka/Ks of ccmFn, rps4 and adh4 were higher than those of other genes. The result suggests that these genes were under positive selection during evolution in the six plant species.
RNA editing events, which are post-transcriptional processes, are enriched in metagenomes (Li, 2018). A total of 502 potential RNA editing sites were identified on 32 mitochondrial PCGs according to predictions from the online PREP suit website (http://prep.unl.edu/) (cut-off value = 0.2). All were found to be C to U base editing. As shown in Figure 4, the nad4 gene encoded the most RNA editing sites (40 sites), whereas nad4L and rps7 encoded only one site. Of the 502 predicted sites, the results were hydrophilic to hydrophobic (13.15%; 66 sites), hydrophobic to hydrophilic (47.01%; 236 sites), hydrophilic to hydrophilic (7.97%; 40 sites), hydrophobic to hydrophobic (31.27%; 157 sites), and hydrophilic to stop (0.60%, 3 sites).
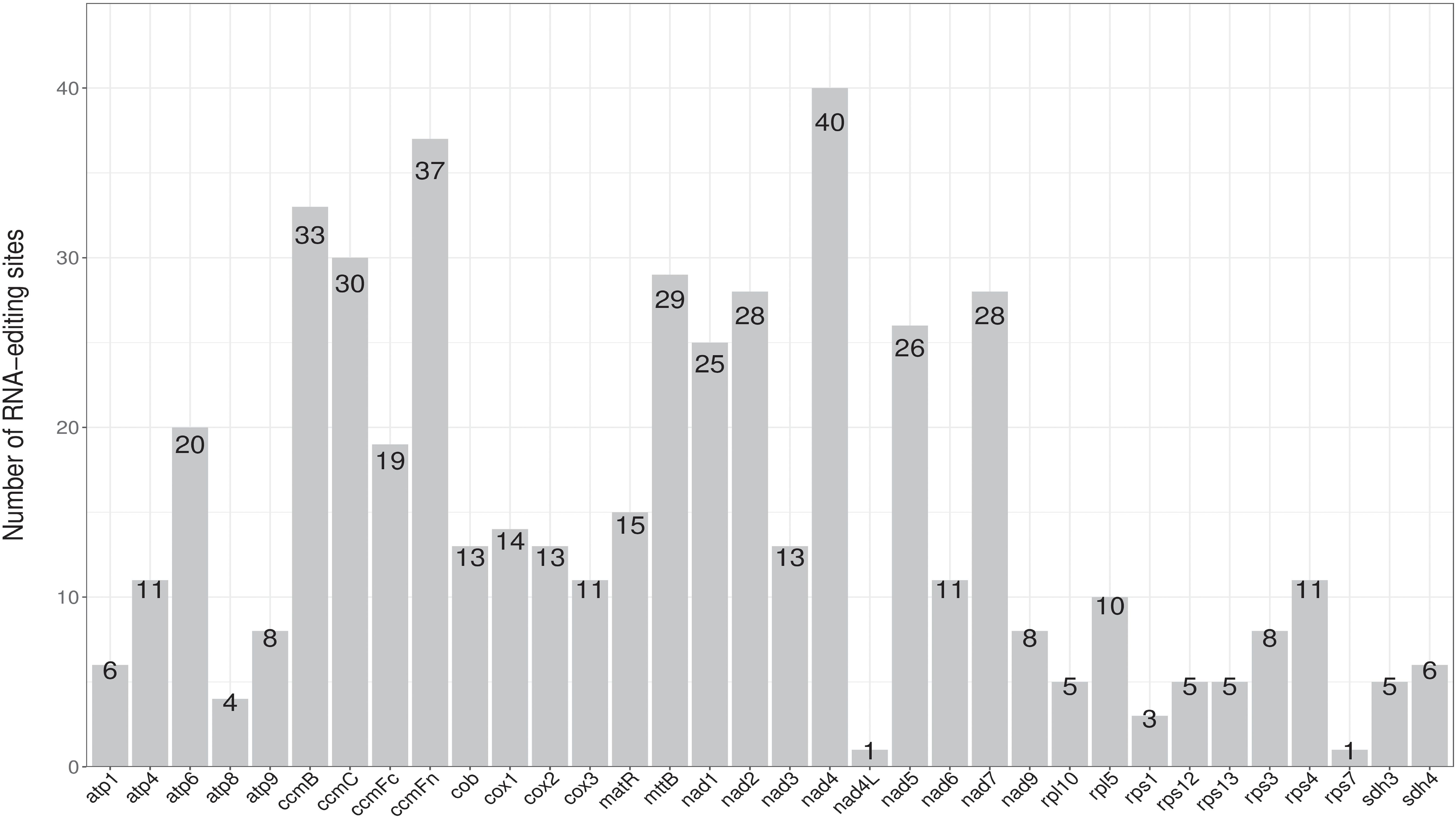
Figure 4. The RNA editing sites distribution in the P. davidiana mitogenome. The number shown by grey box represents the RNA editing sites of each gene.
Repeated sequences are widespread in the mitochondrial genome and mainly include SSRs, tandem repeats and dispersed repeats (Koren et al., 2017). SSRs are tandemly repeated motifs of one to six bases, that are commonly used as molecular markers in the study and identification of species and the analysis of genetic diversity (Langmead and Salzberg, 2012). In the present study, a total of 132 SSRs were identified in the P. davidiana mitogenome, including 56 (38.62%) mono-, 21 (14.48%) di-, 11 (7.59%) tri-, 46 (31.72%) tetra-, 7 (4.83%) penta- and 4 (2.76%) hexanucleotide repeats (Table 2). More than 70.34% of the 132 SSRs belonged to monomers and tetramers. In addition, the results showed that 67.86% of the monomers were A/T contents. In previous studies, the proportion of A/T in SSRs contributed to the proportion of A/T in the whole mitogenome (Schattner et al., 2005; Langmead and Salzberg, 2012).
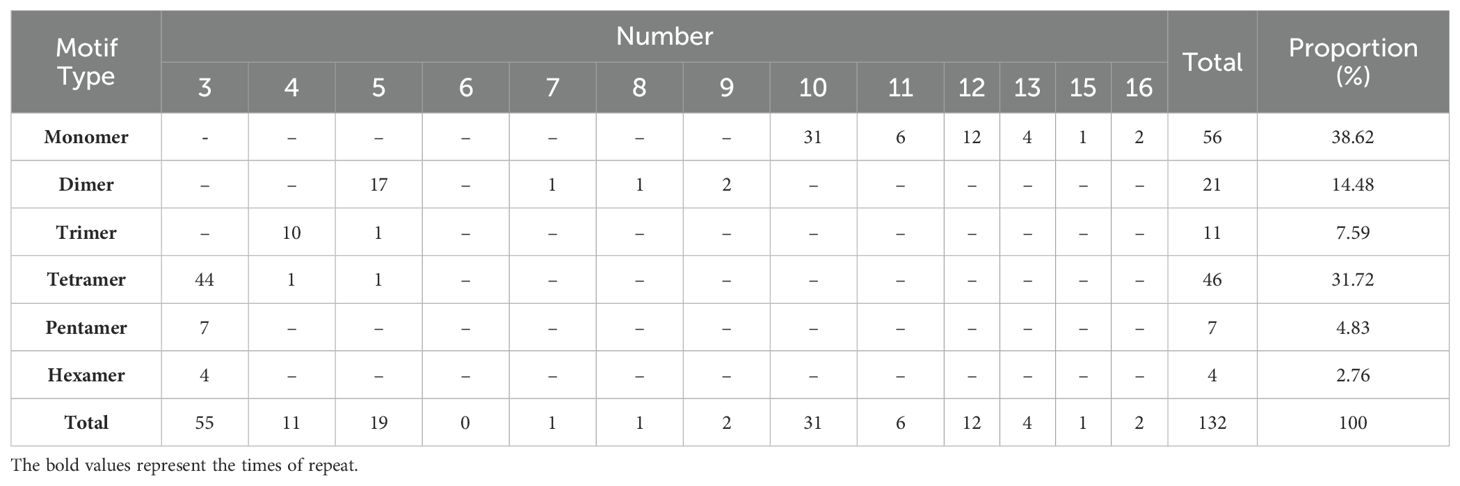
Table 2. The SSR motifs frequency in the P. davidiana mitogenome.
In addition to the SSRs, 177 forward, 201 palindromic and 24 tandem repeats were detected in the P. davidiana mitogenome. The total repeat length was 59, 637 bp, representing 14.63% of the total P. davidiana mitogenome. The majority of the forward and palindromic repeats were between 29 and 60 bp, the longest being 20, 216 bp, whereas the tandem repeats were shorter than 39 bp (Supplementary Table S2, Supplementary Table S3).
The largest co-linear blocks of 35.0 and 34.9 kb were both identified in the dot plot with P. avium and P. salicina, respectively. In addition, the co-linear blocks were not arranged in the same order, a larger number of homologous co-linear blocks were detected between P. davidiana and the closely related species (Figure 5). A total of 74 homologous co-linear blocks (>500 bp) were identified between P. davidiana and P. avium, the largest block being 29,172 bp. Similarly, a total of 85 homologous co-linear blocks (>500 bp) were identified between P. davidiana and P. salicina, with the largest block being 8, 728 bp in length. The results also showed that the mitogenomes are highly unconserved in structure.
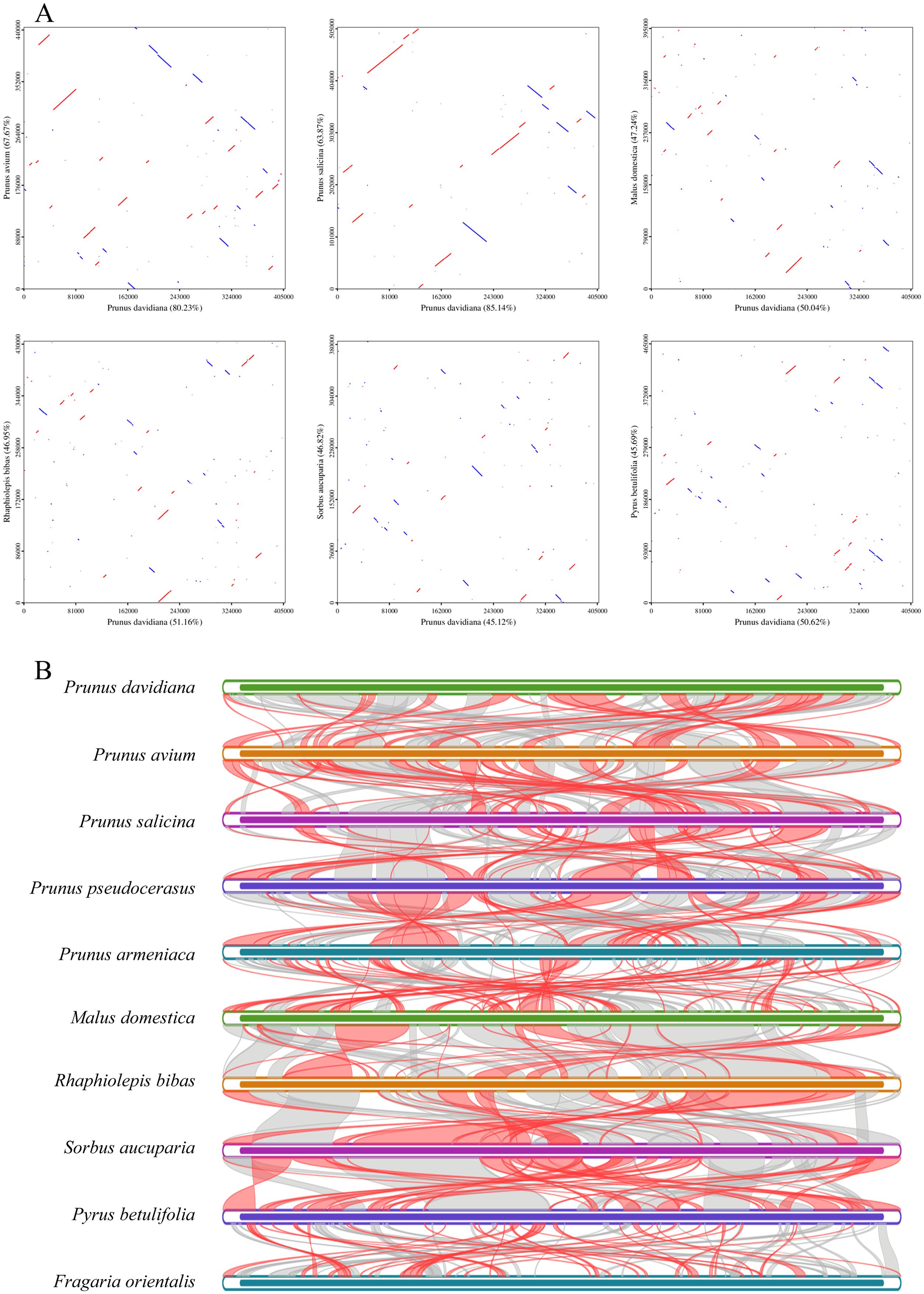
Figure 5. (A) Dot-plots analysis of P. davidiana with closely related species. (B) Synteny analysis of ten mitogenomes. Bars indicated the mitogenomes, and the ribbons showed the homologous sequences between the adjacent species. The common blocks less than 0.5 kb in length are not retained.
The mitogenomes provide an opportunity to confirm the phylogenetic positions of plants. In the present study, to further explore the evolutionary relationships of P. davidiana mitochondria, 31 plant mitogenomes were downloaded from the GenBank database (https://www.ncbi.nlm.nih.gov/genome/browse/#!/overview/). The 31 conserved single-copy orthologous genes present in all 32 mitogenomes were selected to construct a phylogenetic tree. Five monocotyledonous species were used as an outgroup. As shown in Figure 6, 25 out of 27 nodes in the generated tree had bootstrap support values > 70%, and 17 nodes were 100% supported. The phylogenetic tree strongly supports (bootstrap support = 100%) the close phylogenetic relationship between P. davidiana and P. avium and P. salicina (Figure 6). Overall, the results provide a valuable basis for future analyses of the phylogenetic affinities of Rosaceae species.
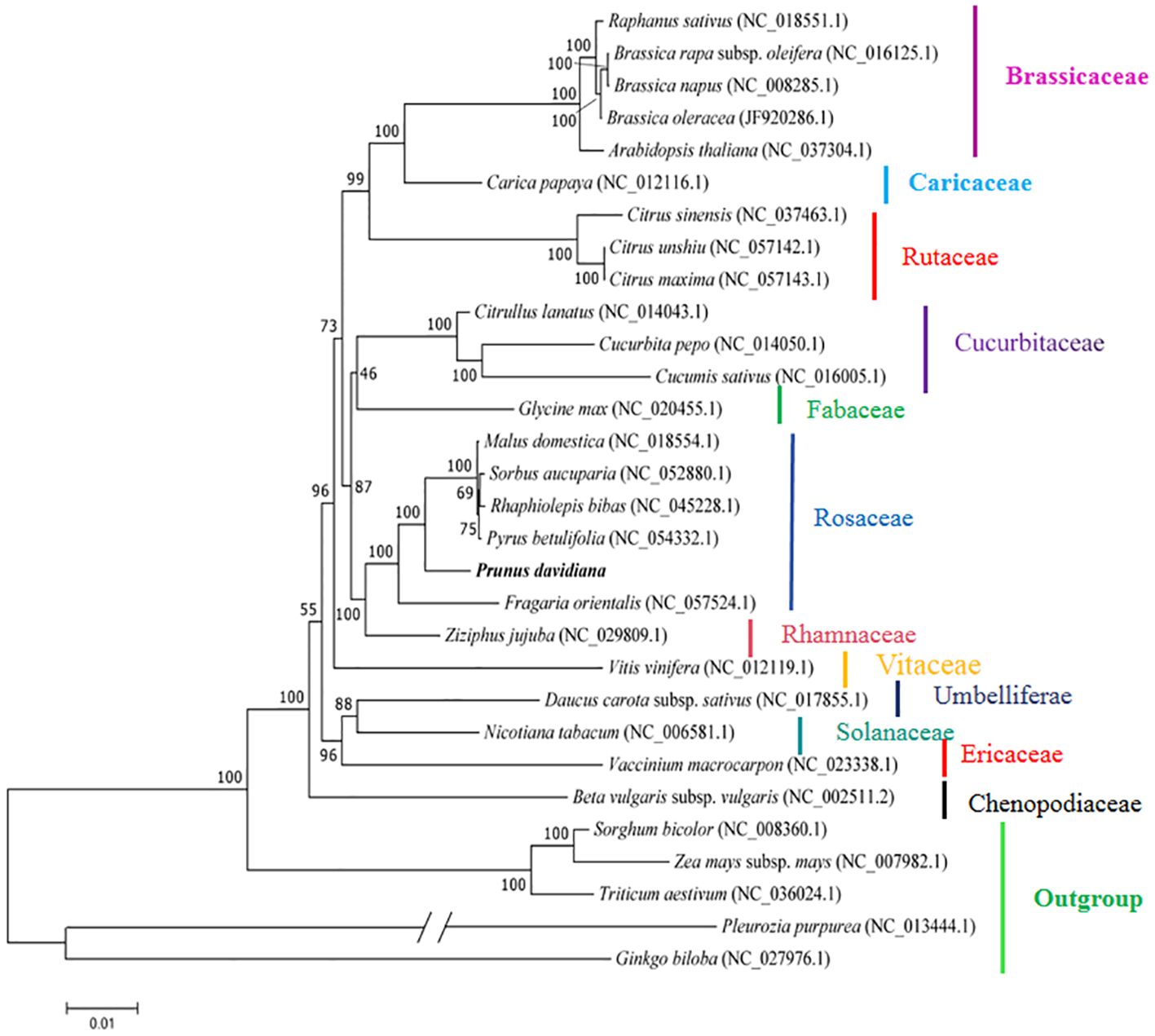
Figure 6. The phylogenetic tree based on 31 single-copy orthologous genes shared among 32 species. The bootstrap support values show at nodes. Sorghum bicolour, Zea mays, Triticum aestivum, Pleurozia purpurea, and Ginkgo biloba served as outgroups.
DNA fragment transfers are common events in plant evolution between nuclear and organellar genomes. The nuclear and cp genomes of P. davidiana were searched using its mitogenome sequences as queries to further understand the characteristics of the sequence transfer event. The 355.99 kb sequences were obtained from the nuclear genome transferred to the mitogenome. In addition, the largest length was 24, 359 bp, and they were mainly between 200 bp and 400 bp. The 27 complete genes (rps13, trnP-TGG, trnF-GAA, trnS-GCT, cob, rpl5, trnH-GTG, trnK-TTT, ccmB, trnN-ATT, ccmC, trnD-GTC, rps3, trnN-GTT, rps1, trnM-CAT, ccmFc, trnS-TGA, atp4, nad4L, mttB, rps1, cox2, sdh4, cox3, atp8 and rps4) were included in the common sequences (Supplementary Table S4).
The P. davidiana mitogenome sequence (407,608 bp) was approximately 2.58 times longer than the cp genome sequence (158,055 bp). The results showed that 16 fragments with a total length of 4, 238 bp had migrated from the cp genome to the mitogenome in P. davidiana (Table 3). Three of these fragments were longer than 500 bp and the longest was 892 bp. Six intact cp genes (psaJ, petN, trnD-GUC, trnI-CAU, trnN-GUU, and trnM-CAU) were identified in the fragments. Others were partial sequences of transferred genes. The transferred genes would have facilitated the movement of genetic material throughout Prunus.
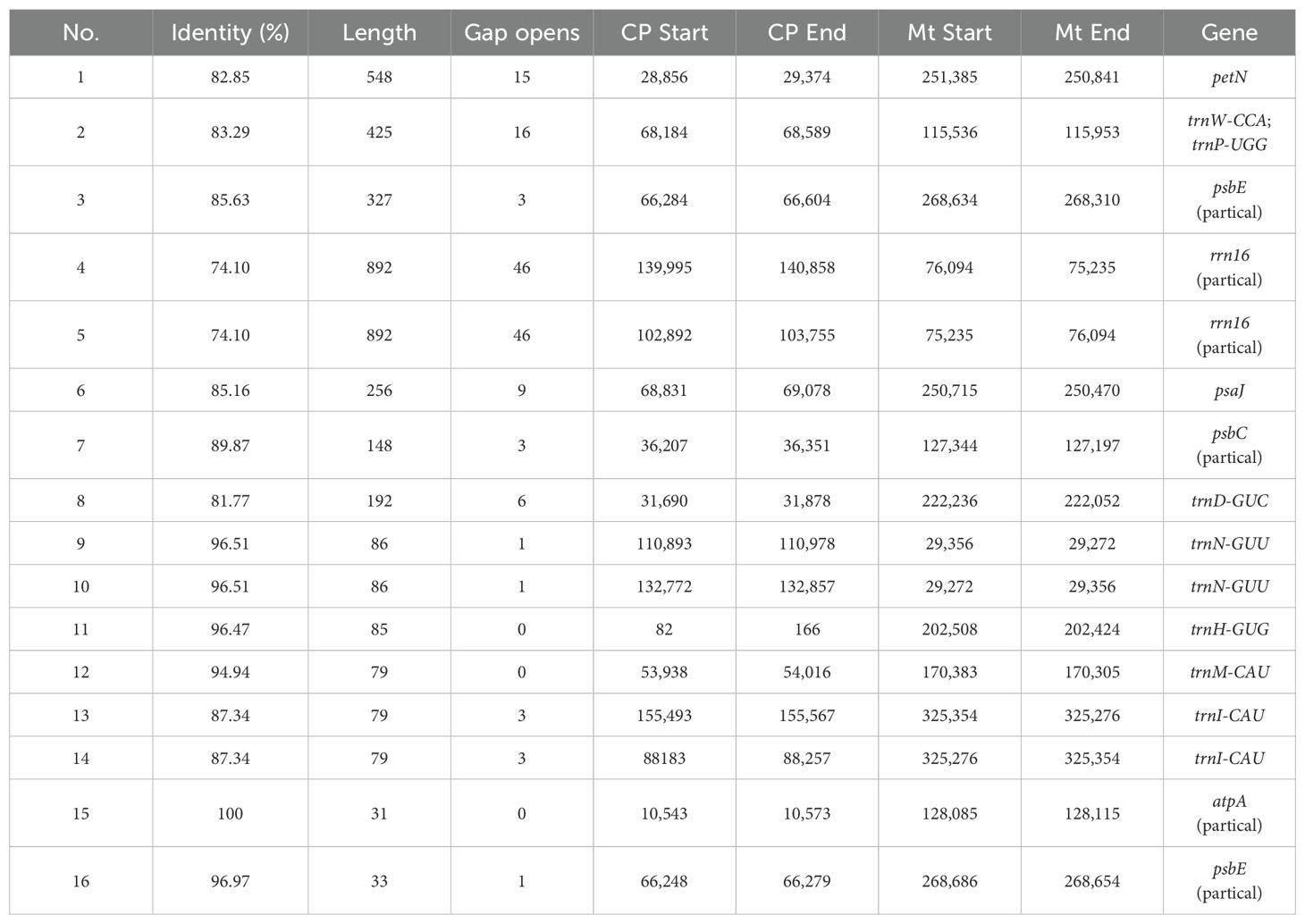
Table 3. Fragments transferred from chloroplasts to mitochondria in P. davidiana..
Plant mitochondrial markers are an important complement to the development of plastid DNA markers (Duminil and Besnard, 2021). Indel regions are relatively easy to detect and are often used to develop markers for species identification (Ma et al., 2019). To develop indel markers for Prunus, the mitochondrial genome sequences of four Prunus species (P. pseudocerasus, P. armeniaca, P. salicina and P. davidiana) were aligned, and two detections were detected in the rrn18-rrn5 (61bp) and rrn5-Nad1 (31bp) regions. To develop indel markers, the sequence-specific primers were designed and the predicted products were successfully amplified with the conserved regions of flanking rrn18-rrn5 and rrn5-Nad1 (Figure 7A, Supplementary Table S5). The length of the amplified rrn18-rrn5 sequence was similar for P. pseudocerasus, P. armeniaca and P. davidiana. In contrast, the corresponding sequence in P. salicina was shorter due to the 61 bp deletion. Similarly, a 31 bp deletion of rrn5-Nad1 was amplified in P. armeniaca (Figure 7B). The predicted sizes of the indels were consistent with the sizes of the fragments amplified from the samples analysed in this study. Indel markers are commonly used to distinguish closely related species (Ma et al., 2019; Grosser et al., 2023). However, Prunus species have not been identified using this approach.

Figure 7. Schematic diagram of the development of the indel markers in four Prunus species. (A) Results of the PCR amplification of the indel marker in the follow Prunus species: rrn18-rrn5 (1) P. pseudocerasus, (2) P. salicina, (3) P. armeniaca, (4) P. davidiana; rrn5-NAD1 (1) P. salicina, (2) P. pseudocerasus, (3) P. armeniaca, (4) P. davidiana; Marker: 2,000 bp ladder. (B): Alignment of the indel markers sequence with MEGA 6.0.
Mitochondria are vital organelles in organisms that provide energy for the physiological activities of various cells. Studies have reported that plant mitogenomes are more complex than animal mitogenomes in terms of size variation, repetitive content, protein coding sequences and structure (Lewis et al., 2002; Han et al., 2022). Previous studies have reported the circular, linear, branched and numerous smaller circular molecule structures of mitogenomes (Sloan, 2013). In the present study, we have assembled and characterised the P. davidiana mitogenome in detail. The size of the P. davidiana mitogenome was 407,608 bp, which is shorter than that of P. salicina (Fang et al., 2021) and P. armeniaca (510, 342 bp; PQ374427), and longer than that of P. pseudocerasus (387, 658 bp; PP968944), similar to the previously reported mitogenome of M. domestica (396, 947 bp) (Kumar et al., 2016). In the previous studies, the gene order and structural features are relatively conservative, while the number genes of P. armeniaca (57) and P. pseudocerasus (55) are less than P. davidiana (64) mitgenome. GC content is important for species assessment, and the GC content of P. davidiana was 45.5%, which is similar to the typically stable 43%-46% in higher plants (Schattner et al., 2005; Ma et al., 2022). This supports that GC content is highly conserved in higher plants. In the published mitogenomes of Rosaceae, some multiple copy genes have been found, such as atp8, cox2, nad4. Here, a three-copy gene (nad5) was identified, which has hardly been reported before.
Repeated sequences are widespread in the mitogenome and include two main categories: tandem and interspersed repeats (Cheng et al., 2021; Bi et al., 2022). They are important for the development of markers for population and evolutionary studies (Kurtz et al., 2001; Thiel et al., 2003; Librado, 2009). In the present study, a total of 59, 637 bp repeats were identified in the P. davidiana mitogenome, representing 14.63% of the total P. davidiana mitogenome. Such repeats would accelerate intermolecular recombination in mitogenomes during Rosaceae evolution.
In plants, RNA editing plays an important role in protein folding and is enriched in mitochondrial and cp genomes, especially cytidine (C)-to-uridine (U) RNA editing sites (Langmead and Salzberg, 2012; Koren et al., 2017; Sun et al., 2024). Previous studies have identified approximately 491 RNA editing sites in 34 genes in Oryza sativa L (Cheng et al., 2021), 486 RNA editing sites in 31 genes in Phaseolus vulgaris L (Koren et al., 2017), and 421 RNA editing sites in 26 genes in Acer truncatum (Ma et al., 2022). In the present study, 502 RNA editing sites were identified in 32 PCGs in the P. davidiana mitogenome. The results showed that the number of RNA editing sites varies greatly between mitogenomes of plant species. Interestingly, all were C-U RNA editing sites. However, the majority of RNA editing sites were associated with cytochrome c biogenesis and NADH dehydrogenase genes, a trend similar to A. truncatum (Ma et al., 2022) and Clematis acerifolia (Librado, 2009).
DNA fragment transfer events are common in mitochondria, nuclei and cp of angiosperms. Furthermore, previous studies have suggested that the most prominent direction of transfer is from organellar genomes to nuclear genomes, and then from chloroplast genomes and nuclei to the mitogenomes (Edgar, 2004; Wang et al., 2012; Koren et al., 2017). According to the results of the present study, we found that the 303.0 kb sequences of nuclear DNA were transferred into the 15 molecules in the P. davidiana mitogenome. In addition, 16 fragments were identified that were transferred from the cp genome to the mitogenome.
To explore homologous covariance blocks between the related species, the arrangement of homologous genes or sequences was determined using a covariance study. According to the results, the largest co-linear blocks of 35.0 and 34.9 kb were identified in the dot plot with P. avium and P. salicina, respectively. However, an inconsistent order of co-linear block arrangement was observed. The results suggest that the P. davidiana mitogenome has undergone extensive genomic rearrangements.
Plant plastid genomes are relatively more conserved in gene order or sequence than in intergenic spacers. The intergenic and intron regions tend to be high polymorphism. Many SSRs or indels markers in the mitogenome have been developed to distinguishing plant species (Grosser et al., 2023). In this study, the complete P. davidiana mitogenome was assembled and analysed, and two indel loci in the rrn18-rrn5 and rrn5-Nad1 (31bp) regions were amplified and analysed. The Nad1 intergenic region has been commonly developed for markers (Grosser et al., 2023). Although mitogenomes of several Prunus species such as P. pseudocerasus, P. armeniaca and P. salicina have been assembled, the molecular markers were not reported in previous studies. Therefore, the two developed indel markers may be applicable for species classification and identification of Prunus species.
In this study, we sequenced, assembled and annotated the mitogenome of P. davidiana. Detailed analyses of the mitogenome were performed based on DNA and amino acid sequences. The total length of the P. davidiana mitogenome is 375,671 bp, including 15 linear molecules, with a GC content of 45.5%. We annotated 64 genes, including 39 PCGs, 22 tRNA genes and 3 rRNA genes. Codon usage, repetitive sequences, Ka/Ks, RNA editing, synteny and DNA fragment transfer events were also analysed. To confirm the evolutionary status, we performed a phylogenetic analysis of the P. davidiana mitogenome and 31 other plants. We also developed two indel markers for Prunus. The results of this study provide valuable information and a theoretical basis for future P. davidiana research and breeding activities.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethical approval and consent to participate in our experimental research on the collection and handling of ‘DXS1’ in accordance with relevant national/institutional guidelines.
YZ: Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Writing – original draft, Writing – review & editing. XG: Formal Analysis, Software, Writing – review & editing. RM: Investigation, Methodology, Resources, Writing – review & editing. MY: Methodology, Software, Investigation, Writing – review & editing. JX: Formal Analysis, Methodology, Resources, Software, Writing – review & editing. ZS: Investigation, Methodology, Software, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Jiangsu Key Research and Development Program (Modern Agriculture) (BE2022381), the National Natural Science Foundation of China (31901979), and the National Key Research and Development Program Subproject of China (2019YFD1000801-02).
Thanks to all members of the Institute of Pomology team for their contributions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1558619/full#supplementary-material
PCR, Polymerase chain reaction; rRNA, ribosomal RNA; Ka/Ks, Snonsynonymous-to-synonymous substitution ratio; Leu, Leucine; DNA, Deoxyribonucleic acid; tRNA:transfer RNA; PCGs:protein-coding genes; Ile, Ileucine; Ser, Serine Thr, Threonine; SSRs, Simple Sequence Repeat; PCGs, protein-coding genes; RSCU, relative synonymous codon utilization; mitogenomes, mitochondrial genomes; cp gemome, chloroplast genome; Arg, Arginine; Cys, Cysteine.
Arseneau, J. R., Steeves, R., Laflamme, M. (2017). Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol. Ecol. Resour. 17, 686–693. doi: 10.1111/1755-0998.12616
Bergthorsson, U., Adams, K., Thomason, B., Palmer, J. (2003). Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature 424, 197–201. doi: 10.1038/nature01743
Bi, C., Lu, N., Xu, Y., He, C., Lu, Z. (2020). Characterization and analysis of the mitochondrial genome of common bean (Phaseolus vulgaris) by comparative genomic approaches. Int. J. Mol. Sci. 21, 3778. doi: 10.3390/ijms21113778
Bi, C., Qu, Y., Hou, J., Wu, K., Ye, N., Yin, T. (2022). Deciphering the multi-chromosomal mitochondrial genome of populus simonii. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.914635
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinf. (Oxford England). 30, 2114–2120. doi: 10.1007/s11515-015-1364-0
Cheng, Y., He, X., Priyadarshani, S. V. G. N., Wang, Y., Ye, L., Shi, C., et al. (2021). Assembly and comparative analysis of the complete mitogenomes of Suaeda Glauca. BMC Genom. 22, 167. doi: 10.1186/s12864-021-07490-9
Christensen, A. C. (2013). Plant mitochondrial genome evolution can be explained by DNA repair mechanisms. Genome Biol. Evol. 5, 1079–1086. doi: 10.1093/gbe/evt069
Clifton, S. W., Minx, P., Fauron, C. M.-R., Gibson, M., Allen, J. O., Sun, H., et al. (2004). Sequence and comparative analysis of the maize NB mitochondrial genome. Plant Physiol. 136, 3486–3503. doi: 10.2307/4356703
Duminil, J., Besnard, G. (2021). Utility of the mitochondrial genome in plant taxonomic studies. Methods Mol. Biol. 2222, 107–108. doi: 10.1007/978-1-0716-0997-2_6
Edgar, R. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Fang, B., Li, J., Zhao, Q., Liang, Y., Yu, J. (2021). Assembly of the complete mitochondrial genome of Chinese plum (Prunus salicina): characterization of genome recombination and RNA editing sites. Genes (Basel) 12 (12), 1970. doi: 10.3390/genes12121970
Goremykin, V. V., Lockhart, P. J., Viola, R., Velasco, R. (2012). The mitochondrial genome of Malus domestica and the import-driven hypothesis of mitochondrial genome expansion in seed plants. Plant J. 71, 615–626. doi: 10.1111/j.1365-313X.2012.05014.x
Grosser, M. R., Sites, S. K., Murata, M. M., Lopez, Y., Chamusco, K. C., Love, Harriage, K., et al. (2023). Plant mitochondrial introns as genetic markers - conservation and variation. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1116851
Gualberto, J. M., Mileshina, D., Wallet, C., Niazi, A. K., Weber-Lotfi, F., Dietrich, A. (2014). The plant mitochondrial genome: dynamics and maintenance. Biochimie 100, 107–120. doi: 10.1016/j.biochi.2013.09.016
Han, F., Qu, Y., Chen, Y., Xu, L., Bi, C. (2022). Assembly and comparative analysis of the complete mitochondrial genome of Salix wilsonii using PacBio HiFi sequencing. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1031769
Hesse, C. O. (1975). Advances in fruit breeding Peaches (West Lafayette, Indiana: Purdue University Press), 325–326. doi: 10.1016/S0304-4238(76)80013-1
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.11
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7. 0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kurtz, S., Choudhuri, J., Enno, O., Chris, S., Jens, S., Robert, G. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lewis, S. E., Searle, S., Harris, N., Gibson, M., Iyer, V., Richter, J., et al. (2002). Apollo: A sequence annotation editor. Genome Biol. 3, research0082. doi: 10.1093/nar/gki366
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, J., Li, J., Ma, Y., Kou, L., Wei, J., Wang, W. (2022). The complete mitochondrial genome of okra (Abelmoschus esculentus): using nanopore long reads to investigate gene transfer from chloroplast genomes and rearrangements of mitochondrial DNA molecules. BMC Genomics 23, 1–14. doi: 10.1186/s12864-022-08706-2
Librado, R.J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Liu, J., Liu, Y. Q., Jing, L. I., Sun, S. X., Wang, Y. Q. (2017). Effects of alkali stress on Prunus davidiana (Carr.) leaf morphological structure and photosynthetic characteristics. Southwest China J. Agric. Sci. 30, 327–332. doi: 10.16213/j.cnki.scjas.2017.2.015
Liu, D., Qu, K., Yuan, Y. C., Zhao, Z. H., Chen, Y., Han, B., et al. (2023). Complete sequence and comparative analysis of the mitochondrial genome of the rare and endangered Clematis acerifolia, the first clematis mitogenome to provide new insights into the phylogenetic evolutionary status of the genus. Front. Genet. 13. doi: 10.3389/fgene.2022.1050040
Ma, Q., Li, S., Bi, C., Hao, Z., Sun, C., Ye, N. (2017). Complete chloroplast genome sequence of a major economic species, Ziziphus jujuba (Rhamnaceae). Curr. Genet. 63, 117–129. doi: 10.1007/s00294-016-0612-4
Ma, Q., Wang, Y., Li, S., Wen, J., Zhu, L., Yan, K., et al. (2022). Assembly and comparative analysis of the first complete mitochondrial genome of Acer truncatum Bunge: a woody oil-tree species producing nervonic acid. BMC Plant Biol. 22, 29. doi: 10.1186/s12870-021-03416-5
Ma, Q., Wang, Y., Zhu, L., Bi, C., Li, S., Li, S. (2019). Characterization of the complete chloroplast genome of acer truncatum bunge (Sapindales: aceraceae): A new woody oil tree species producing nervonic acid. Biomed. Res. Int. 2019, 7417239. doi: 10.1155/2019/7417239
Ogihara, Y., Yamazaki, Y., Murai, K., Kanno, A., Terachi, T., Shiina, T., et al. (2005). Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33, 6235–6250. doi: 10.1093/nar/gki925
Rice, D., Alverson, A., Richardson, A., Young, G., Sanchez-Puerta, M., Munzinger, J., et al. (2013). Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 342, 1468–1473. doi: 10.1126/science.1246275
Schattner, P., Brooks, A. N., Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Sebbenn, A., Blanc-Jolivet, C., Mader, M., Meyer-Sand, B., Degen, B. (2019). Nuclear and plastidial snp and indel markers for genetic tracking studies of jacaranda copaia. Conserv. Genet. Resour 2019, 341–343. doi: 10.1007/s12686-019-01097-9
Skippington, E., Barkman, T., Rice, D., Palmer, J. (2015). Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. P. Natl. Acad. Sci. U.S.A. 112, E3515. doi: 10.1073/pnas.1504491112
Sloan, D. B. (2013). One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 200, 978–985. doi: 10.1111/nph.12395
Sloan, D., Alverson, A., Chuckalovcak, J., Wu, M., Mccauley, D. (2012). Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PloS Biol. 10, e1001241. doi: 10.1371/journal.pbio.1001241
Sun, N., Han, F., Wang, S., Shen, F., Liu, W., Fan, W., et al. (2024). Comprehensive analysis of the Lycopodium japonicum mitogenome reveals abundant tRNA genes and cis-spliced introns in Lycopodiaceae species. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1446015
Thiel, T., Michalek, W., Varshney, R., Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Varré, J. S., D’Agostino, N., Touzet, P., Gallina, S., Tamburino, R., Cantarella, C., et al. (2019). Complete sequence, multichromosomal architecture and transcriptome analysis of the Solanum tuberosum mitochondrial genome. Int. J. Mol. Sci. 20, 4788. doi: 10.3390/ijms20194788
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. doi: 10.1093/nar/gkr1293
Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. (2017). Unicycler. Resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 13, e1005595. doi: 10.1371/journal.pcbi.1005595
Wynn, E., Christensen, A. (2019). Repeats of unusual size in plant mitochondrial genomes: identification, incidence and evolution. G3: Genes Genomes Genet. 9, 549–559. doi: 10.1534/g3.118.200948
Xiong, Y., Lei, X., Bai, S., Liu, W., Wu, W., et al. (2021). Genomic survey sequencing, development and characterization of single- and multi-locus genomic SSR markers of Elymus sibiricus L. BMC Plant Biol. 21, 3. doi: 10.1186/s12870-020-02770-0
Xu, D., Li, H., Lin, L., Liao, M., Xia, H. (2020). Effects of carboxymethyl chitosan on the growth and nutrient uptake in Prunus davidiana seedlings. Physiol. Mol. Biol. Plants. 26, 661–668. doi: 10.1007/s12298-020-00791-5
Yan, M., Zhang, X., Zhao, X., Yuan, Z. (2019). The complete mitochondrial genome sequence of sweet cherry (Prunus avium cv. ‘summit’). Mitochondrial DNA Part B 4, 1996–1997. doi: 10.1080/23802359.2019.1617082
Keywords: Prunus davidiana, mitochondrial genome, repeats, phylogenetic analysis, indel marker
Citation: Zhang Y, Guo X, Ma R, Yu M, Xu J and Shen Z (2025) Assembly and analysis of the complete mitochondrial genome of Prunus davidiana (Rosaceae). Front. Plant Sci. 16:1558619. doi: 10.3389/fpls.2025.1558619
Received: 10 January 2025; Accepted: 28 February 2025;
Published: 27 March 2025.
Edited by:
Changwei Bi, Nanjing Forestry University, ChinaReviewed by:
Zhechen Qi, Zhejiang Sci-Tech University, ChinaCopyright © 2025 Zhang, Guo, Ma, Yu, Xu and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuyan Zhang, emhhbmd5dXlhbkBqYWFzLmFjLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.