 Nataliya V. Melnikova1
Nataliya V. Melnikova1 Alexander A. Arkhipov1
Alexander A. Arkhipov1 Yury A. Zubarev2
Yury A. Zubarev2 Roman O. Novakovskiy1
Roman O. Novakovskiy1 Anastasia A. Turba1
Anastasia A. Turba1 Elena N. Pushkova1
Elena N. Pushkova1 Daiana A. Zhernova1,3
Daiana A. Zhernova1,3 Anna S. Mazina1,4
Anna S. Mazina1,4 Ekaterina M. Dvorianinova1,5
Ekaterina M. Dvorianinova1,5 Elizaveta A. Sigova1,5
Elizaveta A. Sigova1,5 George S. Krasnov1
George S. Krasnov1 Chengjiang Ruan6
Chengjiang Ruan6 Elena V. Borkhert1
Elena V. Borkhert1 Alexey A. Dmitriev1*
Alexey A. Dmitriev1*- 1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow, Russia
- 2Federal Altai Scientific Center of Agrobiotechnologies, Barnaul, Russia
- 3Faculty of Biology, Lomonosov Moscow State University, Moscow, Russia
- 4Lomonosov Institute of Fine Chemical Technologies, MIREA—Russian Technological University, Moscow, Russia
- 5Moscow Institute of Physics and Technology, Moscow, Russia
- 6Key Laboratory of Biotechnology and Bioresources Utilization, Ministry of Education, Institute of Plant Resources, Dalian Minzu University, Dalian, China
1 Introduction
Sea buckthorn (Hippophae rhamnoides L.) is a woody oil tree known for its fruits, which are a rich source of bioactive compounds, including carotenoids and flavonoids (Ciesarova et al., 2020; Mihal et al., 2023). In addition, the unique fatty acid composition of the fruit pulp oil, especially the high content of omega-7 monounsaturated palmitoleic acid, which is rare in plants, contributes to the nutritional benefits of its products (Sola Marsinach and Cuenca, 2019). In this regard, sea buckthorn products are used in medicine, cosmetics, and nutraceuticals (Gatlan and Gutt, 2021; Wang et al., 2022; Zuchowski, 2023). In addition to cultivation for fruit production, sea buckthorn is also used for ecological restoration due to its high resistance to extreme conditions (Ruan et al., 2013).
Sea buckthorn is mainly cultivated in China (2.07 million ha), India (0.02 million ha), Romania (0.02 million ha), Mongolia (0.02 million ha), Russia (0.01 million ha), and Pakistan (0.01 million ha) (Nybom et al., 2023). Thus, 90% of sea buckthorn resources are located in China (Singh, 2022). However, the pioneer in sea buckthorn breeding was Russia, where selection of H. rhamnoides ssp. mongolica Rousi started in 1933 and allowed the development of a wide range of high-yield varieties with high-quality fruits (Singh and Zubarev, 2014). In contrast, breeding of sea buckthorn in China started later, mainly with H. rhamnoides ssp. sinensis Rousi (Nybom et al., 2023). Varieties of H. rhamnoides ssp. mongolica are characterized by large fruits, high yield, high oil content, and lower acidity compared to H. rhamnoides ssp. sinensis varieties, which are better adapted to abiotic and biotic stresses (Nybom et al., 2023). Sea buckthorn breeding does not stand still, new improved varieties are being developed and genetic data can contribute to this. However, only a few DNA markers potentially useful for sea buckthorn breeding are known. Markers were proposed to distinguish Hippophae species and subspecies, including H. rhamnoides ssp. sinensis and H. rhamnoides ssp. mongolica (Liu et al., 2015, 2016, 2018; Piao et al., 2022). Hippophae species are dioecious, and attempts were made to develop DNA markers to identify sex, but these markers do not always work in genetically diverse material (Korekar et al., 2012; Das et al., 2017; Zhou et al., 2018; Zeng et al., 2024a, b). Markers associated with oil content in fruits (Ding et al., 2016) and genes involved in flavonoid biosynthesis (Zhang et al., 2024) were identified. Several works were performed to search for genes associated with resistance of Hippophae species to biotic and abiotic stressors (Nybom et al., 2023). In recent years, high-quality genome assemblies of H. rhamnoides (with sizes of 849, 730, and 919 Mb) (Wu et al., 2022; Yu et al., 2022; Yang et al., 2024), Hippophae tibetana (957 and 1453 Mb) (Wang et al., 2022b; Zhang et al., 2024), and Hippophae gyantsensis (716 Mb) (Chen et al., 2024) were obtained. However, a very limited number of sea buckthorn genotypes were studied using whole-genome sequencing. Whole-genome sequencing of only one set of 40 wild H. rhamnoides ssp. mongolica and H. rhamnoides ssp. sinensis representatives and 15 cultivated H. rhamnoides ssp. mongolica varieties was performed by Yu et al. (Yu et al., 2022). Therefore, there is a lack of genomic data for varieties of sea buckthorn. The aim of the present study was to fill this gap by performing whole-genome sequencing of the unique set of 55 varieties of Russian breeding, which are likely to be significantly different from the Chinese varieties and characterized by valuable traits. These data can significantly expand the knowledge of the diversity of H. rhamnoides at the whole-genome level and provide the necessary data for the development of genetic technologies for sea buckthorn breeding.
2 Materials and methods
2.1 Plant material
To cover the diversity of sea buckthorn cultivated in Russia, a set of 56 accessions representing 55 varieties of H. rhamnoides L. was formed: one replicate for 54 varieties (one tree for each variety) and two biological replicates for the variety Elizaveta (two different trees). The following valuable characteristics were considered: weight, flavor, shape, and color of the fruits and differences in origin (Table 1). Characteristics of sea buckthorn varieties were assessed according to Kondrashov et al. (Kondrashov et al., 1999). Dormant shoots of the selected genotypes were collected at the Federal Altai Scientific Center of Agrobiotechnologies (Barnaul, Russia) in April 2023. The shoots were placed in containers with water in a room with a temperature of ~22°C for one week. When the leaves appeared, they were collected in tubes, frozen in liquid nitrogen, and stored in a low-temperature freezer until DNA extraction.
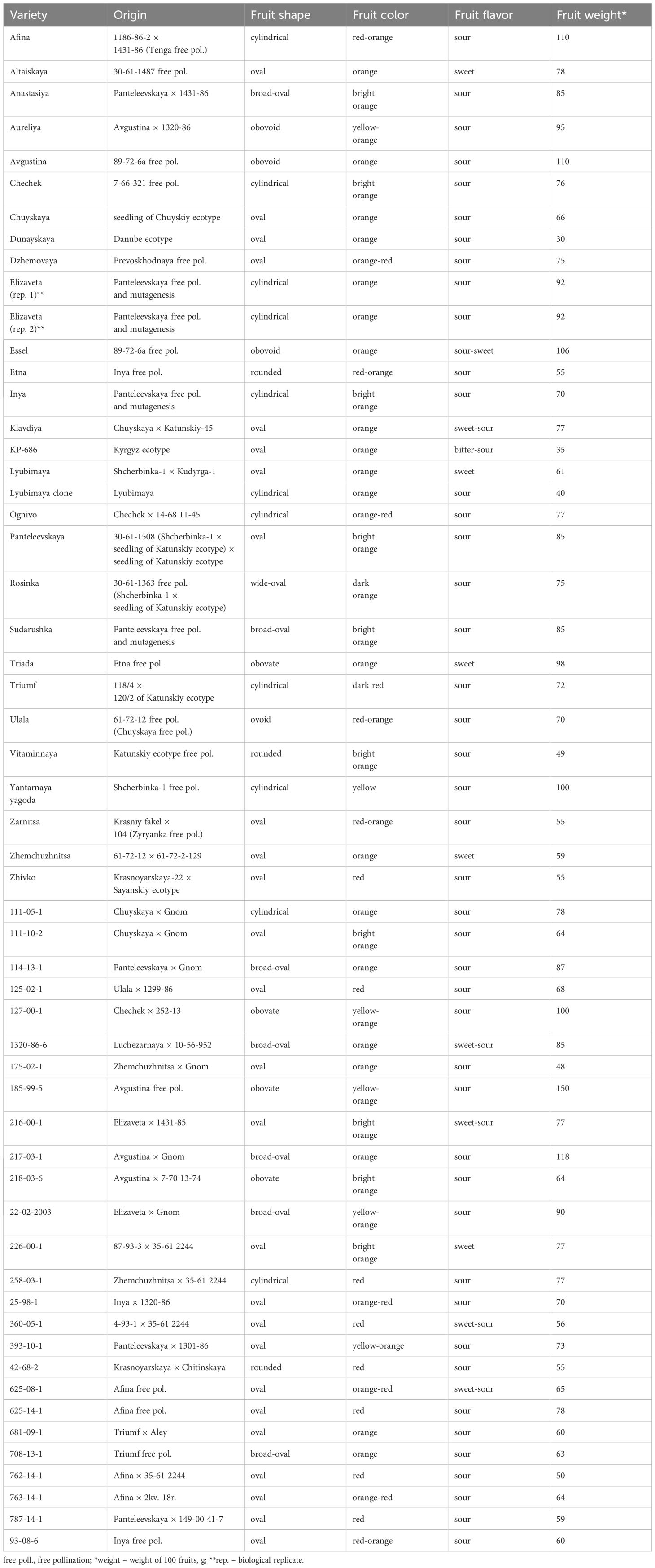
Table 1. Characteristics of the 56 sea buckthorn accessions analyzed in the study.
2.2 DNA extraction
DNA was extracted using the Magen HiPure Plant DNA Mini Kit (Magen, Guangzhou, China). The quality and quantity of DNA were evaluated using NanoDrop 2000C (Thermo Fisher Scientific, Waltham, MA, USA), Qubit 4.0 (Thermo Fisher Scientific), and agarose gel electrophoresis (2% agarose).
2.3 Whole-genome sequencing
The QIAseq FX DNA Library UDI Kit (Qiagen, Chatsworth, CA, USA) was used for DNA library preparation. Quantity and quality of DNA libraries were assessed using Qubit 4.0 (Thermo Fisher Scientific) and Qsep1-Plus (Bi-Optic, New Taipei City, Taiwan). Genome sequencing was performed on a NovaSeq 6000 (Illumina, San Diego, CA, USA) with a read length of 150 + 150 bp.
2.4 Sequencing data analysis
The obtained Illumina reads were processed with Trimmomatic 0.39 (TRAILING:28, SLIDINGWINDOW:4:17, MINLEN:40) (Bolger et al., 2014). The processed reads were mapped to the annotated H. rhamnoides genome from the CNGB Nucleotide Sequence Archive (https://db.cngb.org/cnsa), project ID CNP0001846 (Wu et al., 2022), and VAF (Variant Allele Frequencies) values were calculated for genome regions corresponding to genes (exons and introns) using PPLine (Krasnov et al., 2015). Genetic distances between sea buckthorn accessions were calculated and clustered with Ward’s method (ward.D2) in PPLine (Krasnov et al., 2015).
3 Preliminary data analysis
A representative set of 56 accessions comprising 55 sea buckthorn varieties (for the variety Elizaveta, two different trees were analyzed) was formed from the unique collection of the Federal Altai Scientific Center of Agrobiotechnologies (Barnaul, Russia). The selected varieties had different fruit characteristics and different origins in order to maximize the diversity of the analyzed set (Table 1).
Whole-genome sequencing was performed and at least 23 Gbases of raw Illumina data were obtained for each accession, which corresponded to more than 25× genome coverage (raw Illumina reads were deposited to NCBI SRA, BioProject PRJNA1177110). After mapping the reads to the annotated H. rhamnoides reference genome, data on about 4 million DNA polymorphisms in genes were obtained (lists of DNA polymorphisms were deposited to Zenodo, https://zenodo.org/records/13999625). These data are useful for studying the diversity of allelic variants for specific genes, especially those that may be associated with valuable traits, such as the content of bioactive compounds and other fruit characteristics and resistance to stressors. It is worth noting that a significant part of the identified DNA polymorphisms was present in all analyzed sea buckthorn varieties, indicating that they are genetically distinct from the used reference genome. In addition, genetic distances between the accessions were calculated to evaluate their relationships (Supplementary Table S1).
To visualize the relationships of the studied varieties based on DNA polymorphisms in gene sequences, a dendrogram was constructed (Figure 1). Cluster I was the most distinct and included KP-686 (Kyrgyz ecotype), Dunayskaya, and Yantarnaya Yagoda, which are not varieties of Altai breeding and probably have significant differences at the genome level from the other studied accessions. The same cluster included 175-02-1, obtained by crossing varieties of Altai breeding, and its position in the dendrogram is not expected and requires additional research.
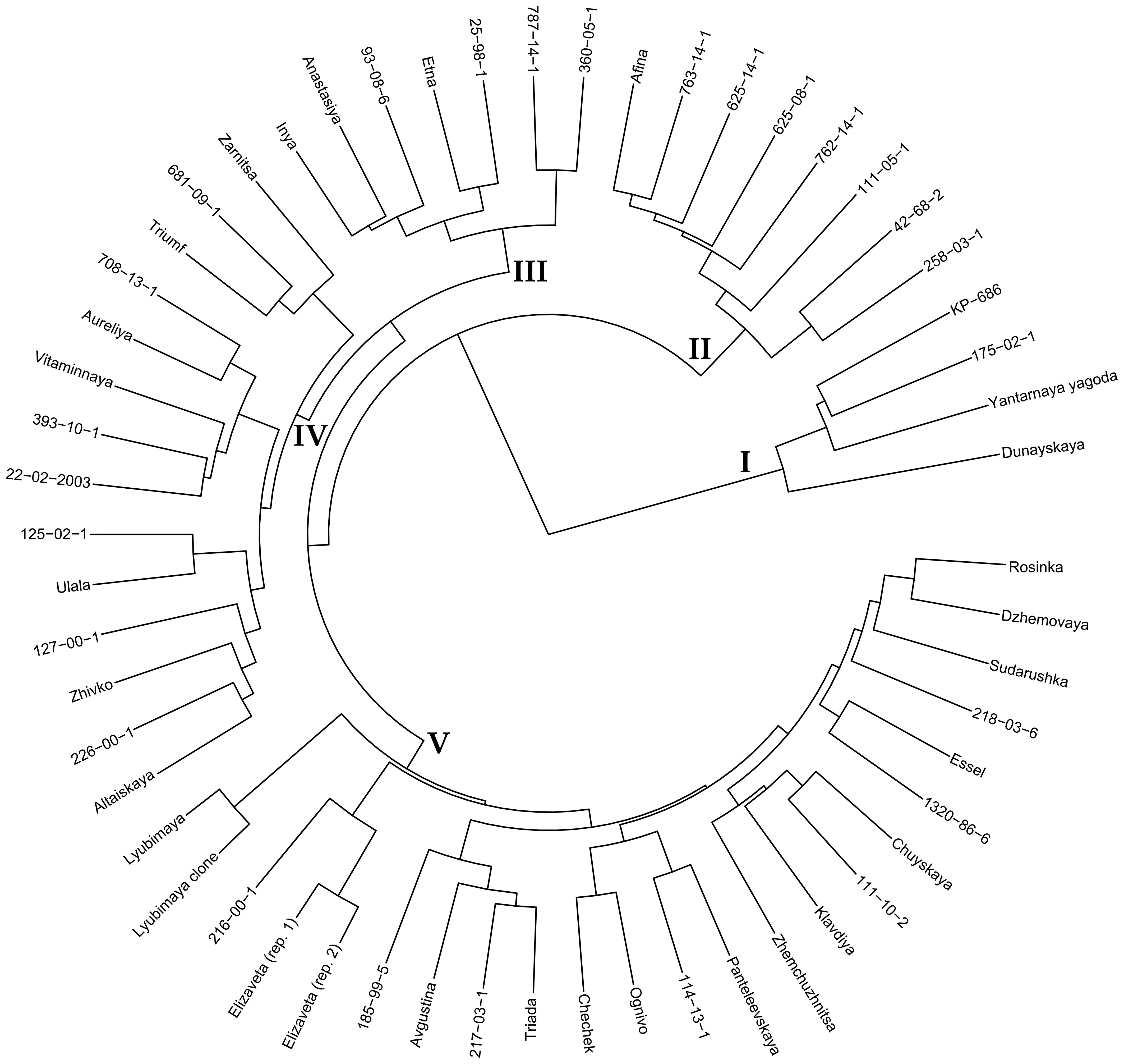
Figure 1. Dendrogram for 56 sea buckthorn accessions based on the obtained whole-genome sequencing data. DNA polymorphisms (VAF values) in gene sequences were analyzed. Ward’s method of cluster analysis.
The remaining sea buckthorn varieties were divided into four clusters. Cluster II included Afina and all studied progeny of this variety, namely 625-08-1, 625-14-1, 762-14-1, and 763-14-1. It can be assumed that Afina and sea buckthorn genotypes obtained with its participation are genetically quite different from the other studied varieties of Altai breeding. In addition, 111-05-1, 258-03-1, and 42-68-2, which are believed to be unrelated to Afina, were in Cluster II, which is difficult to explain from a genealogical point of view.
Cluster III clearly distinguished a group of sea buckthorn varieties with Panteleevskaya in their lineages. Thus, this group is likely to be significantly different from other studied sea buckthorn genotypes at the genome level.
Cluster IV included 14 varieties, among which the genetic relationships were not as clear as in the first three clusters, but they were still present. Thus, a group of four Novosibirsk accessions was isolated: Triumf, Zarnitsa, 681-09-1, and 708-13-1, with Triumf being the parental form for 681-09-1 and 708-13-1. Ulala and its progeny 125-02-1 were also in this cluster. Two varieties with Panteleevskaya in their lineages were also in Cluster IV: 22-02-2003 and 226-00-1. In general, however, this cluster contained a mixture of quite different sea buckthorn varieties.
Cluster V contained 23 accessions. In this cluster, as in other clusters, some relationships corresponding to lineages were observed. For example, Rosinka and Sudarushka, which entered this cluster, have common roots. In addition, varieties Essel and 218-03-6 have the genotype 89-72-6a in their lineages. 89-72-6a is very interesting in terms of strong inheritance of large fruit size. In this respect, it is the progenitor of many varieties, most of which were present in Cluster V. The exception was the variety Aureliya, which was in Cluster IV. Other relationships can also be traced in Cluster V. For example, Lyubimaya clone is a seedless mutant of the variety Lyubimaya. Several closely related groups were also isolated: Elizaveta (two biological replicates) and its progeny 2016-00-1, Chechek and its progeny Ognivo, Chuyskaya and its progeny Klavdiya and 111-10-2, and Panteleevskaya and its progeny 114-13-1.
In general, the dendrogram obtained by us on the basis of DNA polymorphisms in all sea buckthorn genes annotated in the used reference genome (Wu et al., 2022) reflected well the known data on the relationship of the studied genotypes. The research on H. rhamnoides performed by Yu et al. using whole-genome sequencing allowed the authors to separate wild genotypes of H. rhamnoides ssp. mongolica from cultivated ones, as well as to separate H. rhamnoides ssp. sinensis accessions into a separate group (Yu et al., 2022). However, we were unable to find any other work that characterized representative sets of sea buckthorn genotypes using whole-genome sequencing (NCBI PubMed, https://pubmed.ncbi.nlm.nih.gov/; Google Scholar, https://scholar.google.com; accessed October 28, 2024). Meanwhile, whole-genome sequencing and linkage mapping is an urgent need for sea buckthorn studies (Sharma, 2022).
Data on the diversity of sea buckthorn varieties at the genomic level are of great value in understanding the extent to which selection has affected the gene pool of this crop and what patterns can be traced by analyzing the genetic data. We studied the sea buckthorn varieties of Russian breeding, which has a long history. The forms with valuable traits created by Russian breeders became the progenitors of many varieties all over the world (Singh, 2022), so the obtained by us data are of special value. In addition, the evaluation of genetic relationships of different accessions is important for breeders when selecting parental forms for crosses.
Recently, there has been an increasing number of articles devoted to the beneficial properties of sea buckthorn (Wang et al., 2022a; Chen et al., 2023; Mihal et al., 2023; Nybom et al., 2023; Teng et al., 2024; Xu et al., 2024), but in terms of genetics, this crop is still relatively understudied (Sharma, 2022). Indeed, several high-quality genome assemblies of H. rhamnoides were obtained (Wu et al., 2022; Yu et al., 2022; Yang et al., 2024) and some transcriptome studies were performed (Bansal et al., 2018; Ye et al., 2018; Liang et al., 2022; Lyu et al., 2022; Yu et al., 2022). A number of works were also devoted to fatty acid synthesis in sea buckthorn and genes/microRNAs involved in this process (Ding et al., 2018, 2019, 2022; Yu et al., 2022; Arkhipov et al., 2024). However, the genetic determinants and their diversity remain unknown for most of the key traits that define the value of sea buckthorn varieties, including carotenoid content, fruit shape and flavor. In this context, data on DNA polymorphisms in gene sequences obtained for a representative set of accessions characterized by phenotype will allow the search for associations between allelic variants of genes and valuable traits. These data are the basis for the development of marker-assisted and genomic selection of sea buckthorn, which are increasingly used in breeding practice for other agricultural plants (Xu et al., 2020; Hasan et al., 2021; Thudi et al., 2021; Dmitriev et al., 2022; Werner et al., 2023; Mangal et al., 2024).
4 Conclusions
H. rhamnoides is a valuable crop whose fruits are rich in bioactive compounds with health benefits. To date, there is a lack of genetic data for varieties of sea buckthorn. This fact hinders the identification of genetic determinants of valuable traits and limits the efficiency of breeding. In the present study, we analyzed a representative set of 55 valuable H. rhamnoides varieties of Russian breeding with different fruit characteristics and diverse lineages. Whole-genome sequencing was performed on the Illumina platform, and at least 25× genome coverage was obtained for each accession. Based on the sequencing data, DNA polymorphisms were identified in genomic regions corresponding to genes. These polymorphisms were used to evaluate the genetic relationships of the studied sea buckthorn varieties. We revealed genetically distinct groups of accessions that mostly corresponded to the lineages of the genotypes. Our data are important for assessing the effect of selection on sea buckthorn diversity and for evaluating the genetic relationship of different varieties, which is useful for breeders when selecting parental forms for crosses. The obtained data on genomic sequences of 55 H. rhamnoides varieties in combination with information on valuable traits of their fruits are the basis for identification of quantitative trait loci (QTL) and quantitative trait nucleotides (QTN) for further development of DNA tests. This will be the basis for marker-assisted selection of sea buckthorn. The obtained information on DNA polymorphisms is also necessary to study the diversity of genes, including those that may determine valuable traits, such as fruit characteristics. This will help to promote genomic breeding of H. rhamnoides. Thus, our data can benefit both basic and applied research on sea buckthorn.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA1177110.
Author contributions
NM: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. AA: Investigation, Writing – review & editing. YZ: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. RN: Investigation, Writing – review & editing. AT: Investigation, Writing – review & editing. EP: Investigation, Writing – review & editing. DZ: Investigation, Writing – review & editing. AM: Investigation, Writing – review & editing. ED: Investigation, Writing – review & editing. ES: Investigation, Writing – review & editing. GK: Investigation, Writing – review & editing. CR: Investigation, Writing – review & editing. EB: Investigation, Writing – review & editing. AD: Conceptualization, Investigation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was financially supported by the Russian Science Foundation, grant 23-46-00026, https://rscf.ru/project/23-46-00026/ (genome sequencing and analysis) and National Natural Science Foundation of China, grant 32261133521 (genome analysis).
Acknowledgments
This work was performed using the equipment of the EIMB RAS “Genome” center (http://www.eimb.ru/ru1/ckp/ccu_genome_ce.php).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1542552/full#supplementary-material
Supplementary Table 1 | Matrices of genetic distances between 56 sea buckthorn accessions based on the analysis of DNA polymorphisms in gene sequences at the whole-genome level.
References
Arkhipov, A. A., Dvorianinova, E. M., Turba, A. A., Novakovskiy, R. O., Zubarev, Y. A., Predushchenko, P. A., et al. (2024). Identification and analysis of KAS II, FAT, SAD, and FAD gene families in Hippophae rhamnoides. Plants (Basel) 13, 3486. doi: 10.3390/plants13243486
Bansal, A., Salaria, M., Sharma, T., Stobdan, T., Kant, A. (2018). Comparative de novo transcriptome analysis of male and female Sea buckthorn. 3 Biotech. 8, 96. doi: 10.1007/s13205-018-1122-5
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Chen, M., Yang, D., Yang, S., Yang, X., Chen, Z., Yang, T., et al. (2024). Chromosome-level genome assembly of Hippophae gyantsensis. Sci. Data 11, 126. doi: 10.1038/s41597-024-02909-w
Chen, Y., Cai, Y., Wang, K., Wang, Y. (2023). Bioactive compounds in sea buckthorn and their efficacy in preventing and treating metabolic syndrome. Foods 12, 1985. doi: 10.3390/foods12101985
Ciesarova, Z., Murkovic, M., Cejpek, K., Kreps, F., Tobolkova, B., Koplik, R., et al. (2020). Why is sea buckthorn (Hippophae rhamnoides L.) so exceptional? A review. Food Res. Int. 133, 109170. doi: 10.1016/j.foodres.2020.109170
Das, K., Ganie, S. H., Mangla, Y., Dar, T. U., Chaudhary, M., Thakur, R. K., et al. (2017). ISSR markers for gender identification and genetic diagnosis of Hippophae rhamnoides ssp. turkestanica growing at high altitudes in Ladakh region (Jammu and Kashmir). Protoplasma 254, 1063–1077. doi: 10.1007/s00709-016-1013-8
Ding, J., Ruan, C., Du, W., Guan, Y. (2019). RNA-seq data reveals a coordinated regulation mechanism of multigenes involved in the high accumulation of palmitoleic acid and oil in sea buckthorn berry pulp. BMC Plant Biol. 19, 207. doi: 10.1186/s12870-019-1815-x
Ding, J., Ruan, C., Guan, Y., Krishna, P. (2018). Identification of microRNAs involved in lipid biosynthesis and seed size in developing sea buckthorn seeds using high-throughput sequencing. Sci. Rep. 8, 4022. doi: 10.1038/s41598-018-22464-w
Ding, J., Ruan, C., Guan, Y., Li, H., Du, W., Lu, S., et al. (2022). Nontargeted metabolomic and multigene expression analyses reveal the mechanism of oil biosynthesis in sea buckthorn berry pulp rich in palmitoleic acid. Food Chem. 374, 131719. doi: 10.1016/j.foodchem.2021.131719
Ding, J., Ruan, C. J., Guan, Y., Shan, J. Y., Li, H., Bao, Y. H. (2016). Characterization and identification of ISSR markers associated with oil content in sea buckthorn berries. Genet. Mol. Res. 15, gmr.15038278. doi: 10.4238/gmr.15038278
Dmitriev, A. A., Pushkova, E. N., Melnikova, N. V. (2022). Plant genome sequencing: modern technologies and novel opportunities for breeding. Mol. Biol. 56, 495–507. doi: 10.1134/S0026893322040045
Gatlan, A. M., Gutt, G. (2021). Sea buckthorn in plant based diets. An analytical approach of sea buckthorn fruits composition: nutritional value, applications, and health benefits. Int. J. Environ. Res. Public Health 18, 8986. doi: 10.3390/ijerph18178986
Hasan, N., Choudhary, S., Naaz, N., Sharma, N., Laskar, R. A. (2021). Recent advancements in molecular marker-assisted selection and applications in plant breeding programmes. J. Genet. Eng. Biotechnol. 19, 128. doi: 10.1186/s43141-021-00231-1
Kondrashov, V. T., Panteleeva, E. I., Kalinina, I. P., Griuner, L. A. (1999). “Sea buckthorn,” in Program and methodology of variety studies of fruit, berry, and nut crops. Eds. Sedova, E. N., Ogoltsova, T. P. (VNIISPK, Orel, Russia), 404–416.
Korekar, G., Sharma, R. K., Kumar, R., Meenu, Bisht, N. C., Srivastava, R. B., et al. (2012). Identification and validation of sex-linked SCAR markers in dioecious Hippophae rhamnoides L. (Elaeagnaceae). Biotechnol. Lett. 34, 973–978. doi: 10.1007/s10529-012-0852-4
Krasnov, G. S., Dmitriev, A. A., Kudryavtseva, A. V., Shargunov, A. V., Karpov, D. S., Uroshlev, L. A., et al. (2015). PPLine: an automated pipeline for SNP, SAP, and splice variant detection in the context of proteogenomics. J. Proteome Res. 14, 3729–3737. doi: 10.1021/acs.jproteome.5b00490
Liang, J., Zhang, G., Song, Y., He, C., Zhang, J. (2022). Targeted metabolome and transcriptome analyses reveal the pigmentation mechanism of Hippophae (sea buckthorn) fruit. Foods 11, 3278. doi: 10.3390/foods11203278
Liu, Y., Liu, C., Tan, E., Fan, G., Xiang, L., Li, X. D., et al. (2016). Genetic and chemical discrimination of traditional Tibetan medicine seabuckthorn based on DNA barcode and (1)H-NMR metabolic method. Zhongguo Zhong Yao Za Zhi 41, 578–585. doi: 10.4268/cjcmm20160405
Liu, Y., Sun, W., Liu, C., Zhang, Y., Chen, Y., Song, M., et al. (2015). Identification of Hippophae species (Shaji) through DNA barcodes. Chin. Med. 10, 28. doi: 10.1186/s13020-015-0062-9
Liu, Y., Xiang, L., Zhang, Y., Lai, X., Xiong, C., Li, J., et al. (2018). DNA barcoding based identification of Hippophae species and authentication of commercial products by high resolution melting analysis. Food Chem. 242, 62–67. doi: 10.1016/j.foodchem.2017.09.040
Lyu, Z., Zhang, G., Song, Y., Diao, S., He, C., Zhang, J. (2022). Transcriptome and DNA methylome provide insights into the molecular regulation of drought stress in sea buckthorn. Genomics 114, 110345. doi: 10.1016/j.ygeno.2022.110345
Mangal, V., Verma, L. K., Singh, S. K., Saxena, K., Roy, A., Karn, A., et al. (2024). Triumphs of genomic-assisted breeding in crop improvement. Heliyon 10, e35513. doi: 10.1016/j.heliyon.2024.e35513
Mihal, M., Roychoudhury, S., Sirotkin, A. V., Kolesarova, A. (2023). Sea buckthorn, its bioactive constituents, and mechanism of action: potential application in female reproduction. Front. Endocrinol. (Lausanne) 14, 1244300. doi: 10.3389/fendo.2023.1244300
Nybom, H., Ruan, C., Rumpunen, K. (2023). The systematics, reproductive biology, biochemistry, and breeding of sea buckthorn-A review. Genes (Basel) 14, 2120. doi: 10.3390/genes14122120
Piao, X., Mohanan, P., Anandhapadmanaban, G., Ahn, J. C., Park, J. K., Yang, D. C., et al. (2022). Authentication of Hippophae rhamnoides ssp. sinensis and ssp. mongolica based on single nucleotide polymorphism at ribosomal DNA and their vitamin content analysis. Plants (Basel) 11, 1843. doi: 10.3390/plants11141843
Ruan, C. J., Rumpunen, K., Nybom, H. (2013). Advances in improvement of quality and resistance in a multipurpose crop: sea buckthorn. Crit. Rev. Biotechnol. 33, 126–144. doi: 10.3109/07388551.2012.676024
Singh, V. (2022). “Global distribution of seabuckthorn (Hippophae sp.) resources and their utilization,” in The Seabuckthorn Genome. Ed. Sharma, P. C. (Springer International Publishing, Cham), 345–368.
Singh, V., Zubarev, Y. (2014). “Breeding strategies of Russian seabuckthorn (Hippophae rhamnoides ssp. mongolica) varieties and their global introduction,” in Seabuckthorn (Hippophae L.): A Multipurpose Wonder Plant, Vol. IV: Emerging Trends in Research and Technologies. Ed. Singh, V. (New Delhi, India: Daya Publishing House), 71–88.
Sola Marsinach, M., Cuenca, A. P. (2019). The impact of sea buckthorn oil fatty acids on human health. Lipids Health Dis. 18, 145. doi: 10.1186/s12944-019-1065-9
Teng, H., He, Z., Hong, C., Xie, S., Zha, X. (2024). Extraction, purification, structural characterization and pharmacological activities of polysaccharides from sea buckthorn (Hippophae rhamnoides L.): A review. J. Ethnopharmacol. 324, 117809. doi: 10.1016/j.jep.2024.117809
Thudi, M., Palakurthi, R., Schnable, J. C., Chitikineni, A., Dreisigacker, S., Mace, E., et al. (2021). Genomic resources in plant breeding for sustainable agriculture. J. Plant Physiol. 257, 153351. doi: 10.1016/j.jplph.2020.153351
Wang, K., Xu, Z., Liao, X. (2022a). Bioactive compounds, health benefits and functional food products of sea buckthorn: a review. Crit. Rev. Food Sci. Nutr. 62, 6761–6782. doi: 10.1080/10408398.2021.1905605
Wang, R., Wu, B., Jian, J., Tang, Y., Zhang, T., Song, Z., et al. (2022b). How to survive in the world’s third poplar: Insights from the genome of the highest altitude woody plant, Hippophae tibetana (Elaeagnaceae). Front. Plant Sci. 13, 1051587. doi: 10.3389/fpls.2022.1051587
Wang, Z., Zhao, F., Wei, P., Chai, X., Hou, G., Meng, Q. (2022). Phytochemistry, health benefits, and food applications of sea buckthorn (Hippophae rhamnoides L.): A comprehensive review. Front. Nutr. 9, 1036295. doi: 10.3389/fnut.2022.1036295
Werner, C. R., Gaynor, R. C., Sargent, D. J., Lillo, A., Gorjanc, G., Hickey, J. M. (2023). Genomic selection strategies for clonally propagated crops. Theor. Appl. Genet. 136, 74. doi: 10.1007/s00122-023-04300-6
Wu, Z., Chen, H., Pan, Y., Feng, H., Fang, D., Yang, J., et al. (2022). Genome of Hippophae rhamnoides provides insights into a conserved molecular mechanism in actinorhizal and rhizobial symbioses. New Phytol. 235, 276–291. doi: 10.1111/nph.18017
Xu, X., Liu, X., Yu, S., Wang, T., Li, R., Zhang, Y., et al. (2024). Medicinal and edible polysaccharides from ancient plants: extraction, isolation, purification, structure, biological activity and market trends of sea buckthorn polysaccharides. Food Funct. 15, 4703–4723. doi: 10.1039/d3fo04140a
Xu, Y., Liu, X., Fu, J., Wang, H., Wang, J., Huang, C., et al. (2020). Enhancing genetic gain through genomic selection: from livestock to plants. Plant Commun. 1, 100005. doi: 10.1016/j.xplc.2019.100005
Yang, X., Luo, S., Yang, S., Duoji, C., Wang, Q., Chen, Z., et al. (2024). Chromosome-level genome assembly of Hippophae rhamnoides variety. Sci. Data 11, 776. doi: 10.1038/s41597-024-03549-w
Ye, G., Ma, Y., Feng, Z., Zhang, X. (2018). Transcriptomic analysis of drought stress responses of sea buckthorn (Hippophae rhamnoides subsp. sinensis) by RNA-Seq. PloS One 13, e0202213. doi: 10.1371/journal.pone.0202213
Yu, L., Diao, S., Zhang, G., Yu, J., Zhang, T., Luo, H., et al. (2022). Genome sequence and population genomics provide insights into chromosomal evolution and phytochemical innovation of Hippophae rhamnoides. Plant Biotechnol. J. 20, 1257–1273. doi: 10.1111/pbi.13802
Zeng, Z., Wang, J., Tian, Z., Norbu, N., Chen, Y., Chen, J., et al. (2024a). Development of sex-specific molecular markers for early sex identification in Hippophae gyantsensis based on whole-genome resequencing. BMC Plant Biol. 24, 1187. doi: 10.1186/s12870-024-05978-6
Zeng, Z., Wang, R., Wang, J., Chen, Y., Wang, Y., Song, Z., et al. (2024b). Development and validation of sex-linked molecular markers for rapid and accurate identification of male and female Hippophae tibetana plants. Sci. Rep. 14, 19243. doi: 10.1038/s41598-024-69918-y
Zhang, G., Song, Y., Chen, N., Wei, J., Zhang, J., He, C. (2024). Chromosome-level genome assembly of Hippophae tibetana provides insights into high-altitude adaptation and flavonoid biosynthesis. BMC Biol. 22, 82. doi: 10.1186/s12915-024-01875-4
Zhou, W., Wang, Y., Zhang, G., Luan, G., Chen, S., Meng, J., et al. (2018). Molecular sex identification in dioecious Hippophae rhamnoides L. via RAPD and SCAR markers. Molecules 23, 1048. doi: 10.3390/molecules23051048
Keywords: sea buckthorn, Hippophae rhamnoides, varieties, fruit characteristics, whole-genome sequencing, genetic diversity, DNA polymorphisms
Citation: Melnikova NV, Arkhipov AA, Zubarev YA, Novakovskiy RO, Turba AA, Pushkova EN, Zhernova DA, Mazina AS, Dvorianinova EM, Sigova EA, Krasnov GS, Ruan C, Borkhert EV and Dmitriev AA (2025) Genetic diversity of Hippophae rhamnoides varieties with different fruit characteristics based on whole-genome sequencing. Front. Plant Sci. 16:1542552. doi: 10.3389/fpls.2025.1542552
Received: 10 December 2024; Accepted: 13 February 2025;
Published: 04 March 2025.
Edited by:
Gulmira Khassanova, S.Seifullin Kazakh AgroTechnical Research University, KazakhstanReviewed by:
Siddanna Savadi, Directorate of Cashew Research (ICAR), IndiaMyriam Lamine, Center of Biotechnology of Borj Cedria (CBBC), Tunisia
Copyright © 2025 Melnikova, Arkhipov, Zubarev, Novakovskiy, Turba, Pushkova, Zhernova, Mazina, Dvorianinova, Sigova, Krasnov, Ruan, Borkhert and Dmitriev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexey A. Dmitriev, YWxleF8yNDVAbWFpbC5ydQ==