
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Plant Sci. , 20 December 2024
Sec. Plant Membrane Traffic and Transport
Volume 15 - 2024 | https://doi.org/10.3389/fpls.2024.1515163
 Alessia Gallucci1†
Alessia Gallucci1† Deborah Giordano2†
Deborah Giordano2† Angelo Facchiano2
Angelo Facchiano2 Clizia Villano1*
Clizia Villano1* Domenico Carputo1
Domenico Carputo1 Riccardo Aversano1
Riccardo Aversano1Transmembrane proteins (TMPs) are pivotal components of plant defence mechanisms, serving as essential mediators in the response to biotic stresses. These proteins are among the most complex and diverse within plant cells, making their study challenging. In spite of this, relatively few studies have focused on the investigation and characterization of TMPs in plants. This is particularly true for grapevine. This review aims to provide a comprehensive overview of TMP-encoding genes involved in grapevine immunity. These genes include Lysin Motif Receptor-Like Kinases (LysM-RLKs), which are involved in the recognition of pathogens at the apoplastic level, Plant Respiratory Burst Oxidase Homologs (Rbohs), which generate reactive oxygen species (ROS) for host defense, and Sugars Will Eventually be Exported Transporters (SWEETs), which play a role in nutrient allocation and stress responses. Furthermore, the review discusses the methodologies employed to study TMPs, including in vivo, in vitro and in silico approaches, highlighting their strengths and limitations. In vivo studies include the assessment of TMP function in whole plants or plant tissues, while in vitro experiments focus on isolating and characterizing either specific TMPs or their components. In silico analyses utilize computational tools to predict protein structure, function, and interactions. By identifying and characterizing genes encoding TMPs involved in grapevine immunity, researchers can develop strategies to enhance grapevine resilience and lead to more sustainable viticulture.
Transmembrane proteins (TMPs) are integral components of lipid bilayer membranes in living organisms that function as first barriers to the outside world. They constitute half of the components in cell membranes (Goossens and De Winter, 2018) and are among the most complex and diverse proteins within plant cells, making them difficult to study (Corradi et al., 2019; Barrera et al., 2019). From a structural point of view, TMP differ from other membrane proteins in their folding, which allocates them across the membrane. The protein can cross the membrane with a single passage (bitopic) or a multiple passage (polytopic). The segments that cross the membrane are folded with the typical alpha-helix structure, with the peculiarity of having the side chains of amino acids in contact with the lipid portion of the membrane. Another structural organization of the transmembrane portion, although less frequent, is the beta-barrel type architecture, observed in mitochondria, chloroplasts, and Gram-negative bacteria. In any case, the hydrophobicity of the amino acid side chain is an expected feature in the transmembrane regions, related to the need to adapt to the apolar environment of the lipid region of the membrane. Transmembrane regions are alternated in the amino acid sequence by polar regions that remain in the polar environment outside the membrane, and function as simple connection loops as independent domains with a fold like water-soluble proteins. The alternation of polar and apolar regions is a typical feature that may allow integral membrane proteins to be recognized by the analysis of chemical-physical properties along the amino acid sequence. Typical signal peptides can also be recognized. The mechanism of the embedding in the lipid membrane is the object of many studies and is still poorly understood (Argudo, 2024).
In plants, TMPs take over numerous critical functions like solute transport (active/passive) (Pellizzaro et al., 2015), signal transduction (Kemmerling et al., 2011; Mohd-Radzman et al., 2015; Song et al., 2015; Imin et al., 2018) and cell-cell recognition (Langhans et al., 2017). Because of these functions, they participate in many physiological and pathological processes such as growth, development (Clark et al., 1997; van der Knaap et al., 1999; Osakabe et al., 2005; ten Hove et al., 2011) and photosynthesis (Liu and Last, 2015; Okumura et al., 2016). TMPs can also respond to environmental stresses by triggering physiological adaptation that enhance plant resilience (Kraffe et al., 2007). Over recent years, researcher has focused on elucidating TMPs roles in plant immunity, particularly in pathogen detection, signal transduction, and defense activation (Gull et al., 2019). Extensive studies have now identified and characterized numerous TMPs involved in plant-pathogen interactions (Table 1) (Kumara et al., 2022). These TMPs function as Pattern Recognition Receptors (PRRs) on the plant membrane, detecting microbial molecules termed Microbe/Pathogen-Associated Molecular Patterns (MAMPs/PAMPs) — including bacterial flagellin, EF-Tu, and fungal chitin — and plant-derived molecules such as oligogalacturonides, categorized as Damage-Associated Molecular Patterns (DAMPs). This detection trigger Pattern-Triggered Immunity (PTI). The predominant classes of plant PRRs are the cell surface Leucine-Rich Repeat domain (LRR) Receptor Kinases (LRKs) and LRR Receptor Proteins (LRPs), which feature ligand-binding (e.g., LRR) and transmembrane domains (e.g., LysM). Additionally, Receptor-Like Kinase (RLKs) feature an intercellular kinase domain essential for signal transduction that leads to robust antimicrobial responses through mitogen-activated protein kinase (MAPK) cascades (Yamada et al., 2017). Pathogens can suppress the PTI by releasing effector proteins into the host cells. Additionally, in Effector-Triggered Immunity (ETI), plants counteract pathogen effector proteins through intracellular nucleotide-binding leucine-rich repeat receptors (NB-LRR), leading to defensive responses like leaf necrosis, cell death, and reactive oxygen species (ROS) release – symptoms of a hypersensitive response (HR) (Yuan et al., 2021b). TMPs enhance ETI by amplifying signal transduction and modifying ion fluxes, underscoring their vital role in plant defense mechanisms. Recent studies support the inclusion of TMPs in sustainable disease control strategies of crops, such as grapevine (Vitis vinifera). It is the most economically significant fruit crop cultivated worldwide that is threatened by several pathogens, such as powdery mildew (Erysiphe necator) and botrytis (Botrytis cinerea), which not only significantly reduce fruit yield and quality but also impact the global viticulture sector. It is indicative that viticulture consumes 70% of all agrochemicals used in the European Union, with associated environmental and health risks (Hollomon, 2015). Despite the challenges in identifying TMPs due to their high hydrophobicity (Tusnády et al., 2004; Nakao et al., 2020), emerging techniques are enhancing our understanding of grapevine immunity. This could have profound implications for viticulture, offering strategies to improve grapevine resilience against pathogens, supporting sustainable agriculture and crop protection efforts.
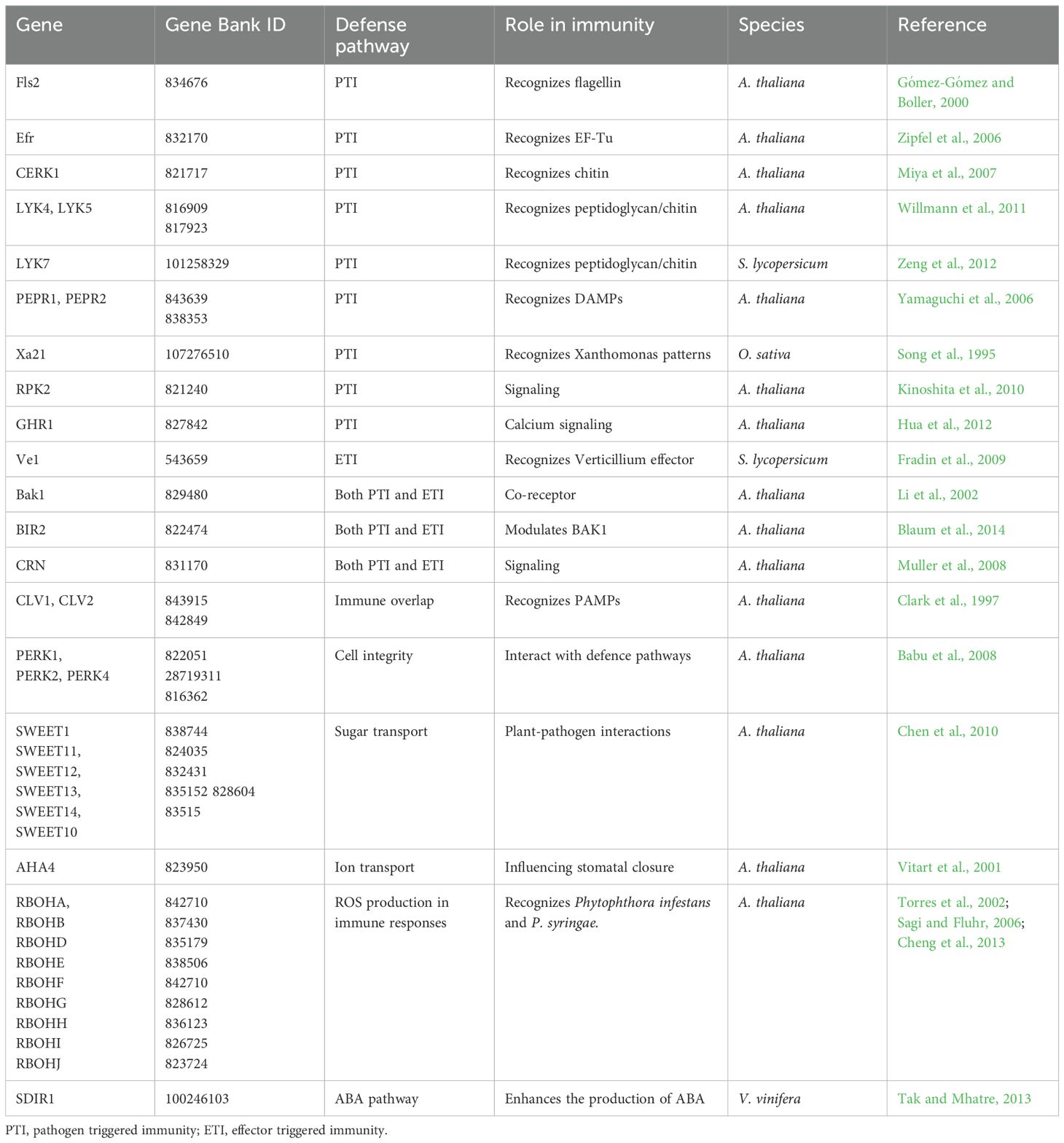
Table 1. List of transmembrane genes characterized in plants and involved in defense pathways.
This review synthesizes the current research on TMPs identified in V. vinfera and knowledge on their role in response to the most significant threats to grapevine. We first provide an overview of these genes and then examine various methodologies used to investigate TMP structure, function and regulation, highlighting their advantages and limitations. Our aim is to identify research gaps and suggest future research directions, providing a robust framework to enhance our understanding of crop immunity and promote sustainable agricultural practices.
To date, our understanding of TMPs involved in biotic stress in grapevines remains limited. Notably, only three of them have been studied for their involvement in pathogen infection reaction, namely the Lysin-Motif Receptor-Like Kinase (LysM-RLK)(Table 2), the plant respiratory burst oxidase homolog (Rboh) (Table 3), and the Sugar Will Eventually Be Exported Transporter (SWEET) (Table 4; Figure 1) (Baker et al., 2012; Cheng et al., 2013; Brulé et al., 2019). The following paragraph will delve into the current knowledge on each of these gene families, examining their specific roles in plant defence mechanisms and their potential applications in enhancing grapevine resistance to biotic stresses.
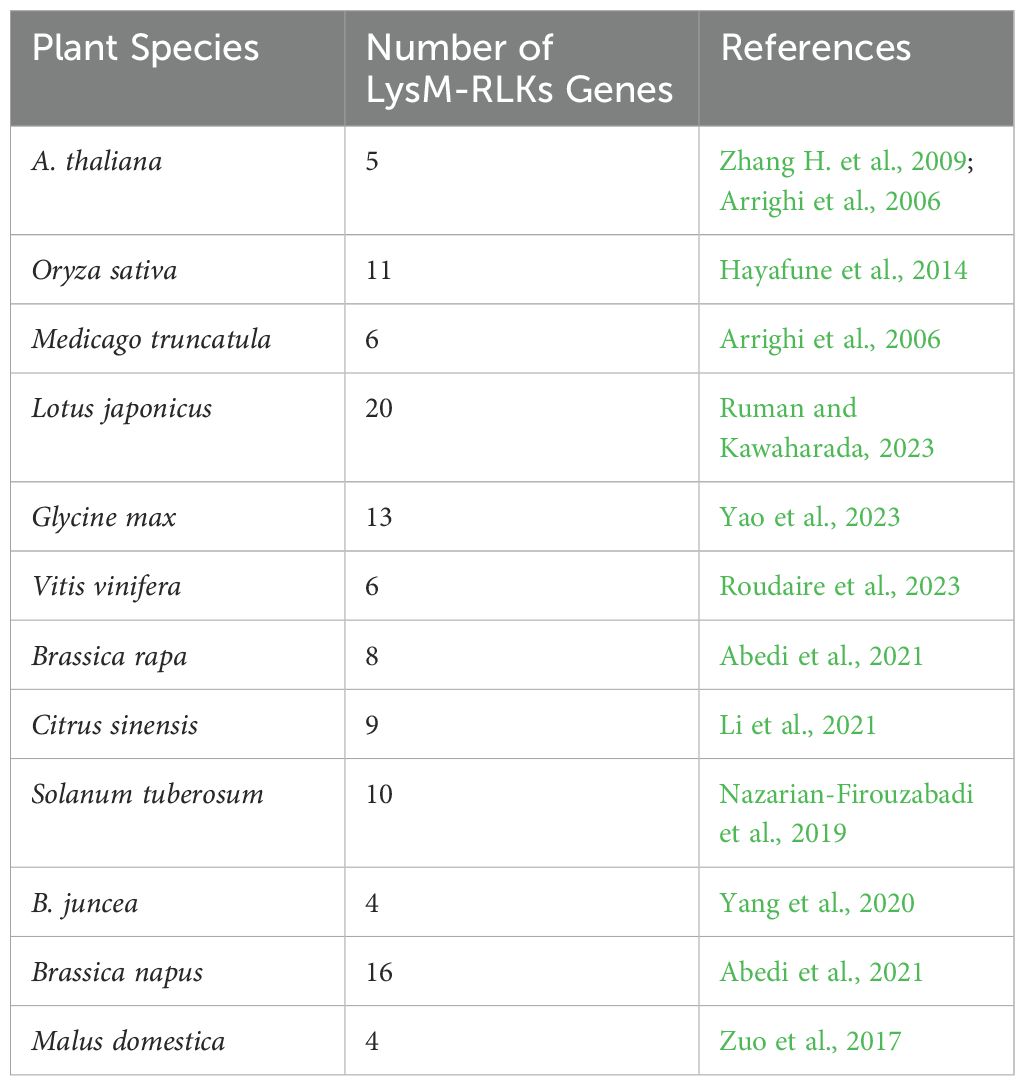
Table 2. Number of LysM-RLK family members characterized in plant species.
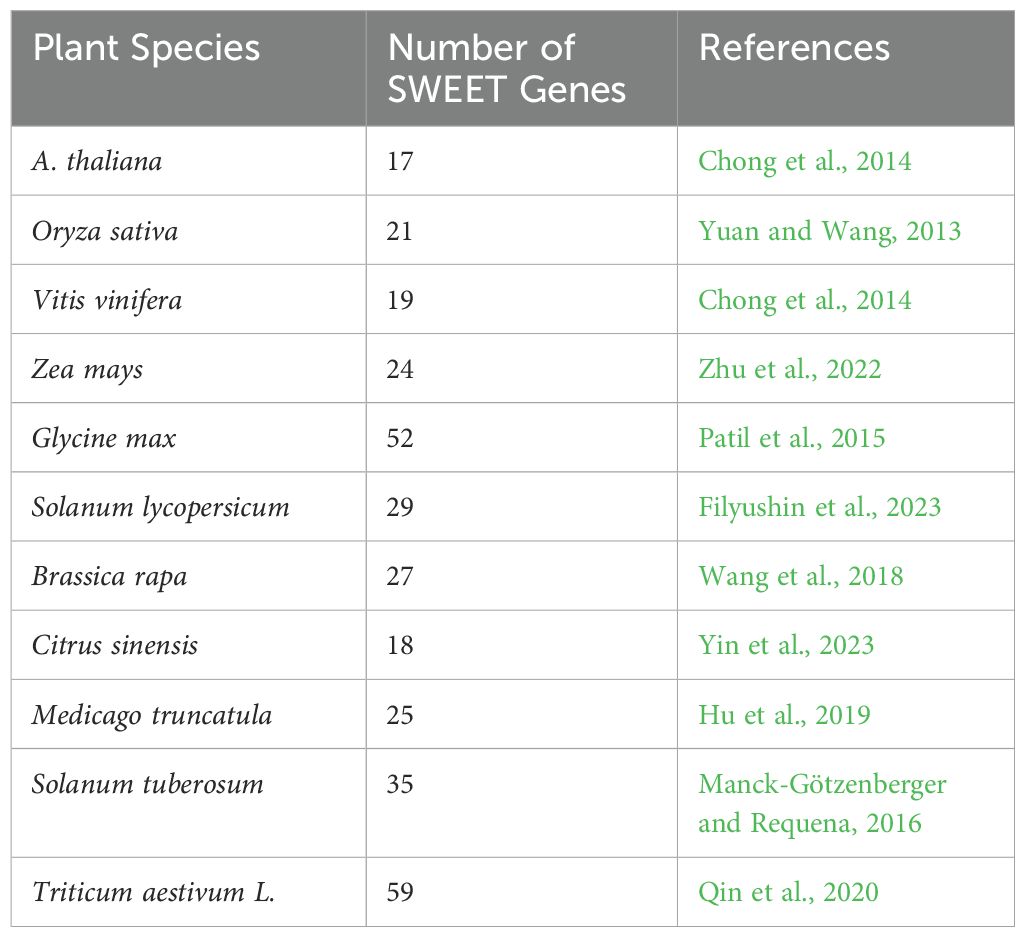
Table 3. Number of Rboh family members characterized in plant species.
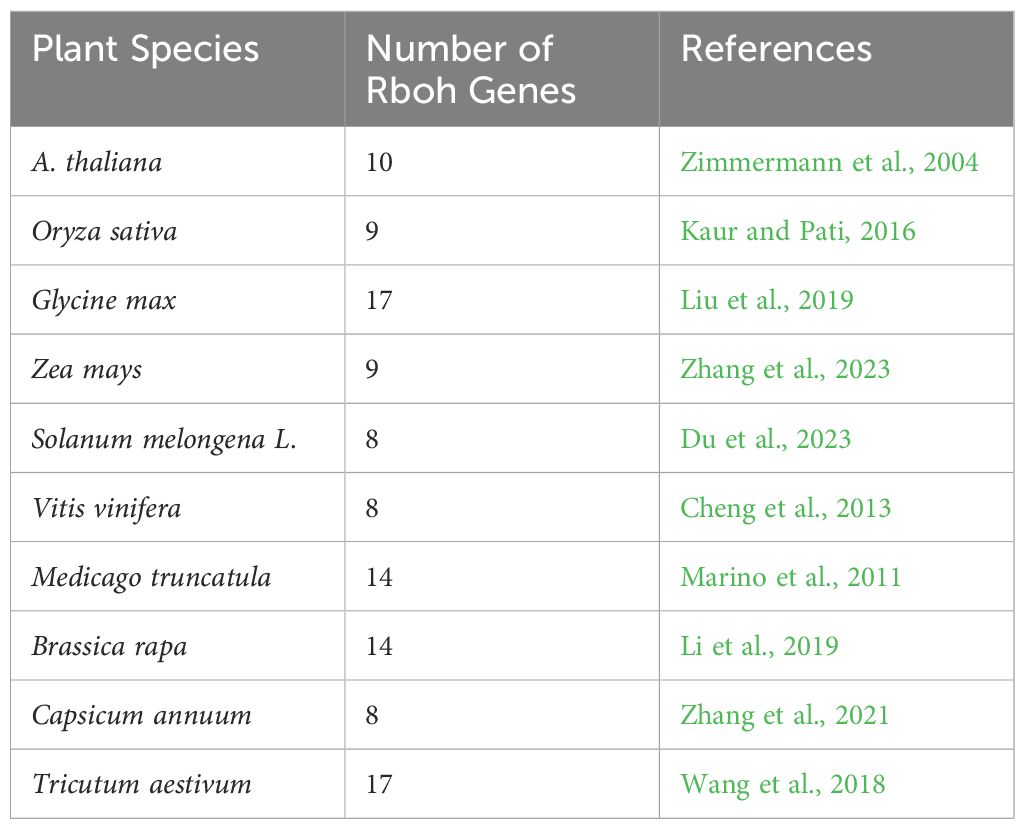
Table 4. Number of SWEET family members characterized in plant species.
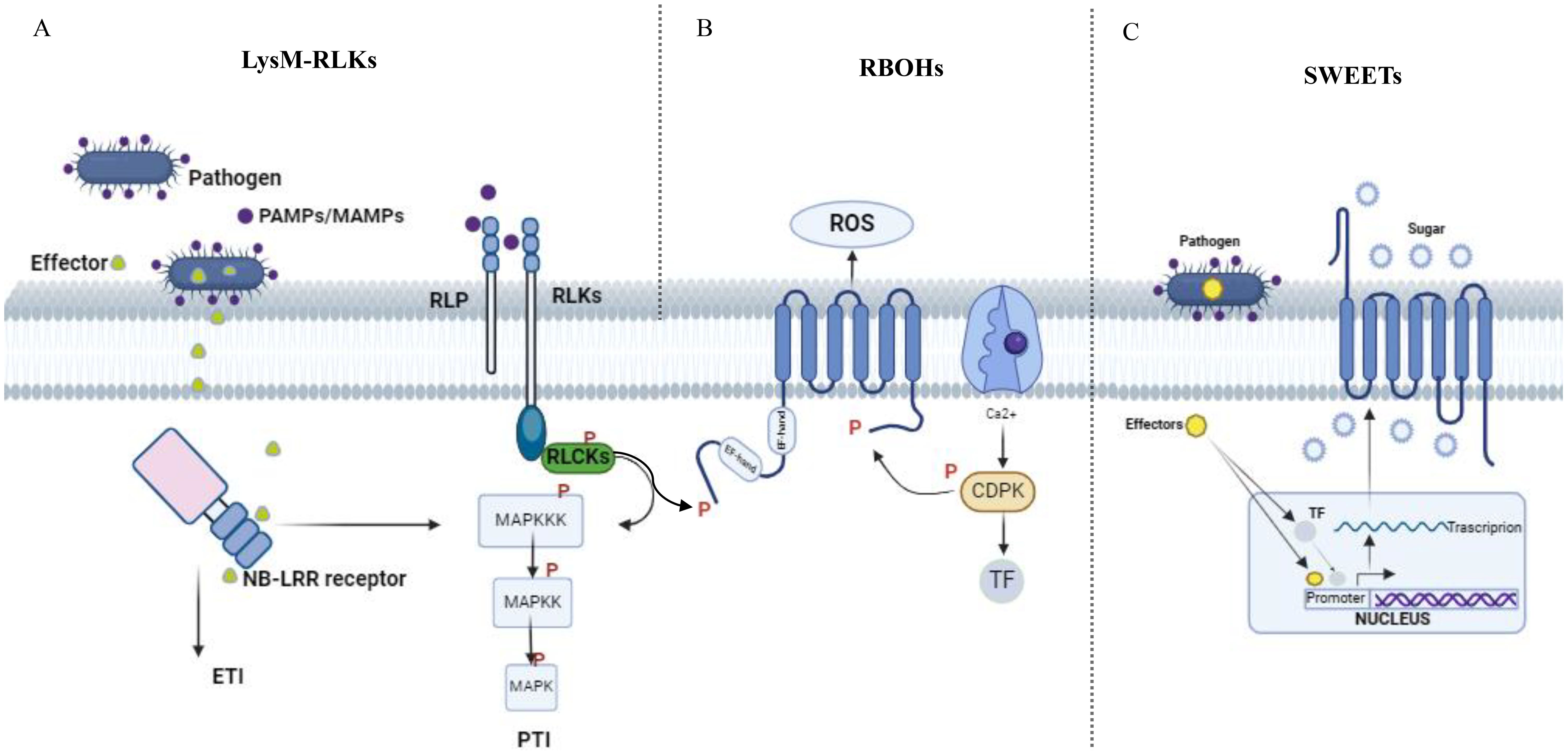
Figure 1. Overview of signalling mediated by plant after pathogen infection. After a plant is infected by a pathogen, triggers a signaling process. This begins with the recognition of Pathogen/Microbe-Associated Molecular Patterns (PAMPs/MAMPs) by pattern recognition receptors (PRRs) (A), which interact dynamically with co-receptors and receptor-like cytoplasmic kinases (RLCKS). Transphosphorylation occurs within the PRR complexes, initiating downstream signaling. These signals, originating from PRRs, are transmitted through phosphorylation cascades involving mitogen-activated protein kinases (MAPKs) and calcium-dependent protein kinases (CDPKs). This signaling pathway ultimately affects downstream targets like the NADPH oxidase RBOHD (B) during Pattern-Triggered Immunity (PTI). Additionally, many pathogens rely on glucose from host plants as a carbon source for their growth before they can successfully invade. Upon invading plants, pathogens release TAL effectors into host plant cells. These effectors prompt the expression of plant SWEETS (C), either directly or indirectly through the activation of transcription factors. As a result, sugar flows into the apoplast, providing nutrition for the pathogens.
Lysin Motif Receptor Like Kinases (LysM-RLKs) represent one of the major classes of PRRs (Gust et al., 2012). Upon binding to PAMPs/MAMPs, LysM-RLKs initiate signalling cascades that lead to the activation of defense responses in plants. These responses may include the production of ROS, activation of defense genes, and reinforcement of the PTI (Buendia et al., 2018) (Figure 1). Beyond their function in immunity, they participate in symbiotic interactions, specifically recognizing signals from beneficial microbes like mycorrhizal fungi and rhizobia. Through this recognition, LysM-RLKs facilitate the formation of symbiotic partnerships, resulting in increased nutrient absorption, heightened stress resilience and overall enhancement of plant growth and development (Chiu and Paszkowski, 2020; Cope et al., 2023). Their proteins contain an extracellular ligand-binding domain, a transmembrane domain, and an intracellular kinase domain (Figure 2). The extracellular domain includes one to three LysM domains, each approximately forty amino acids long, with a conserved βααβ structure, crucial in detecting and responding to external stimuli (Bellande et al., 2017, Zhang X.C. et al., 2009). Between the LysMs of all plants are found highly conserved cysteine pairs separated by one amino acid (CXC) that participates to the structural stability of the domain (Figure 2). The transmembrane region ensures the protein correct positioning within the cell membrane, supporting its overall functionality (Tombuloglu et al., 2019). Lastly, the intracellular kinase domain typically encompasses conserved motifs or residues necessary for ATP hydrolysis (Tanaka et al., 2013) and catalyzes reactions crucial for downstream signalling (Zeng et al., 2012). LysM-RLKs were first discovered in A. thaliana (Shiu and Bleecker, 2001) and subsequently in other plant species, including V. vinifera (Table 2). LysM-RLKs can be divided into two major classes, the LYKs (Lysin Motif Receptor Kinase) in which the kinase domain is active and the LYRs (LysM-containing Receptor-like Protein) with an inactive pseudokinase domain (Schwessinger et al., 2011). In 2007, Miya et al. discovered CERK1, an RLK that is essential in the recognition and transmission signals from the chitin oligosaccharide elicitor in A. thaliana. Subsequent studies have highlighted LYK4 and LYK5 as pivotal for chitin signalling in this species (Wan et al., 2008; Shimizu et al., 2010; Cao et al., 2014; Xue et al., 2019). In particularly, AtLYK5 binds strongly to chitin and is proposed to form dimers with AtLYK4 upon ligand binding (Cao et al., 2014). However, neither receptors can transduce signals independently; AtLYK5 must interact with AtCERK1/LYK1, which carries a functional kinase domain (Xue et al., 2019). A comparable molecular complex model involving the LysM-RLK OsCERK1 and the LysM proteins OsCEBIP, OsLYP4, and OsLYP6 has been suggested for chitin perception in rice (Shimizu et al., 2010). Recently, research on grapevine has confirmed the involvement of LysM-RLKs in immune response activation (Brulé et al., 2019; Roudaire et al., 2023). The latest annotation of the grapevine genome indicates the presence of 16 VvLYK genes, which encode 16 LysM-RLKs proteins (Roudaire et al., 2023). Among the orthologs of AtCERK1/LYK1, only VvLYK1-1 and VvLYK1-2 have been demonstrated to facilitate the perception of PAMPs/MAMPs in grapevine. As in A. thaliana and rice, VvLYK1-1 and VvLYK5-1 interact only after chitin recognition (Brulé et al., 2019). Roudaire et al. (2023) observed an up-regulation of VvLYK5-1 in grapevine leaves after E. necator attack, indicating its involvement in chitin perception. Interestingly, both VvLYK5-1 and VvLYK5-2 lack most of the amino acids necessary for kinase activity, like AtLYK5, implying that the functional kinase domain is absent in VvLYK5-1 and VvLYK5-2. Consequently, it is proposed that VvLYK5 may interact with a co-receptor to transmit signals, thereby activating chitin-induced immunity (Roudaire et al., 2023). Chitin is a well-known PAMP recognized by LYK proteins in plant cells infected by fungi. However, chitin perception mechanisms in grapevine remain unexplored, particularly regarding the genetic diversity of LysM-RLK genes among various grapevine cultivars. Xue et al. (2019) demonstrated that A. thaliana perceives chitin via a receptor complex composed of LYK1, LYK4 and LYK5. It would be interesting to investigate if a similar tripartite chitin receptor complex involving VvLYK1-1, VvLYK4-2 and VvLYK5-2 exists in V. vinifera. Resolving the entire mechanism of chitin and chitosan perception in grapevine holds significant agricultural importance, as this knowledge, could enhance disease resistance breeding program, promoting sustainable grape production practices.
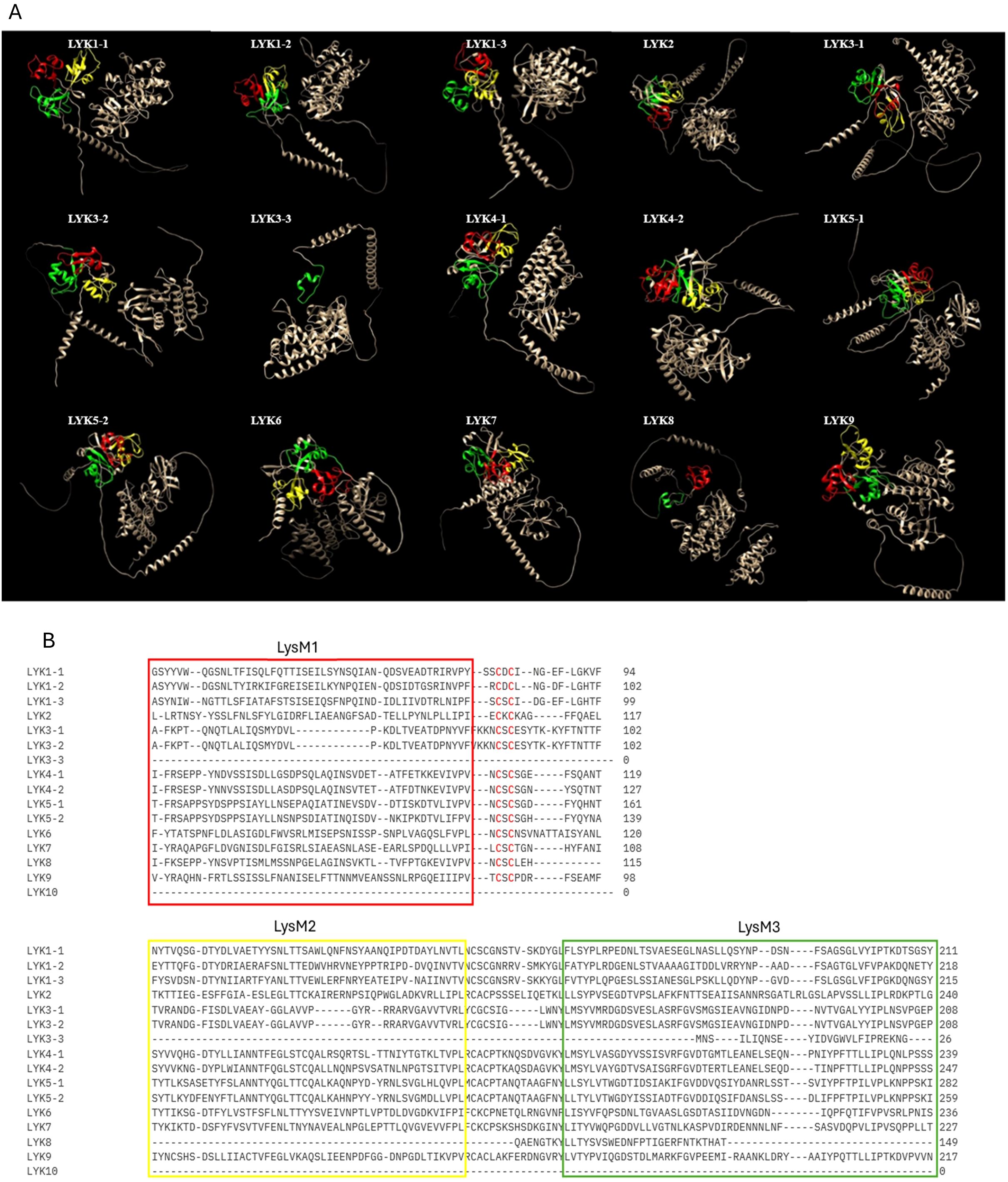
Figure 2. (A) AlphaFold prediction of the structure of the LysM-RLKS (LYK) proteins performed with ChimeraX AlphaFold, each LysM domain is represented in different colour: LysM1 (red), LysM2 (yellow) and LysM3 (green). (B) Sequence alignment of conserved LysM domains located within N-terminal domain of LYKs protein.
Plant cells respond to biotic stress by accumulating reactive oxygen species (ROS), a critical component of PTI and ETI. One key ROS, hydrogen peroxide (H2O2), primarily forms through the conversion of superoxyde, a reaction catalysed by the enzyme respiratory burst oxidase homolog (Rboh) protein in the apoplast (Liu et al., 2016; Navathe et al., 2019). Rboh proteins are integral membrane proteins characterized by six conserved transmembrane helices. They feature two EF-hand calcium-binding domains in their N-terminal region, regulated directly by Ca²+ ions. The C-terminal region consists of a hydrophilic domain with binding sites for flavin adenine dinucleotide (FAD) and NADPH, oriented towards the cytosol. In the apoplast, heme groups facilitate electron transport across the membrane to O2, the electron acceptor, through FAD (Mahalingam et al., 2021) (Chapman et al., 2019) (Figure 1). When plants encounter pathogens, they produce ROS via Rboh activity as part of their defense mechanisms (Mittler, 2017; Mansoor et al., 2022; Ali et al., 2023). The Rboh gene family, which is conserved across both angiosperms and gymnosperms, include several subgroups that have been well-characterized (Zhang et al., 2023). The rice rbohA gene, the first Rboh gene identified in plant, shares homology with the mammalian gene gp91phox (Groom et al., 1996). Since this discovery, Rbohs genes have been identified across various plant species, with a variable number of family members (Table 3). In grapevine, 7 Rboh proteins have been identified, with VvRbohA, VvRbohC1 and VvRbohD localized in the plasma membrane and the others predicted to be in the chloroplast thylakoid membrane. These proteins have N-terminal sequences containing two potential Ca2+-binding EF-hand motifs, critical for their regulation (Figure 3) (Cheng et al., 2013). Notably, VvRbohD shows significant up-regulation after powdery mildew inoculation, highlighting its potential role in biotic stress responses (Cheng et al., 2013; Rivas et al., 2023). In other species, such as eggplant, it has been shown that SmRbohB significantly increases ROS production upon Verticillium dahliae treatment, restricting pathogen growth and highlighting its potential as a gene for stress tolerance (Du et al., 2023). These results mark early steps in studying Rboh gene functions in grape, pointing to the need for further research to fully understand their contributions to stress resistance.
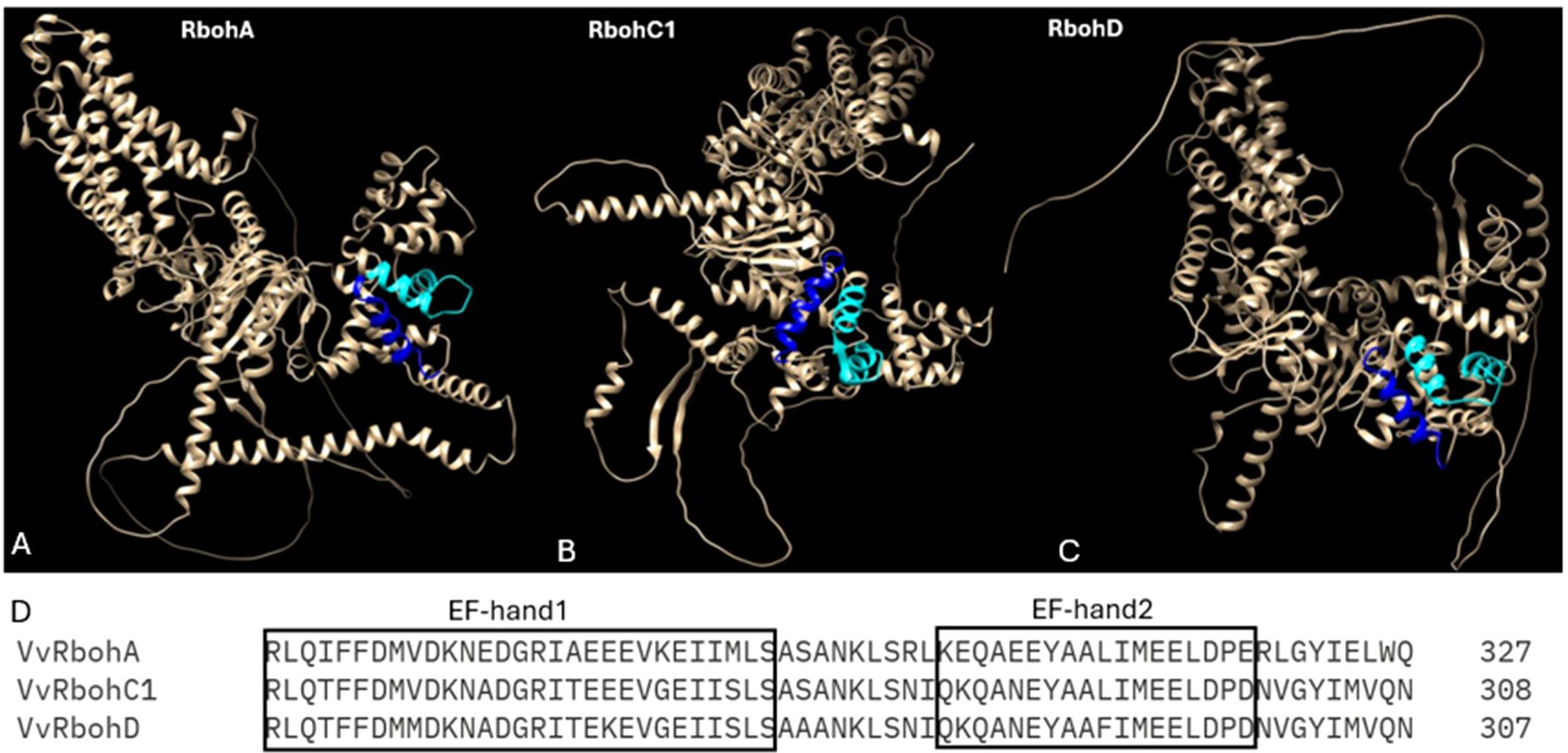
Figure 3. AlphaFold prediction of the structure of the RBOHs proteins performed with ChimeraX AlphaFold, each EF-hand domain is represented in different colour: EF-domain1 (cyan) and EF-hand domain2 (blue). (A) The 3D model of VvRbohA (B) the 3D model of VvRbohC1 (C) the 3D model of VvRbohD (D) Sequence alignment of conserved two EF-hand domains located within N-terminal extension.
The Sugar Will Eventually be Exported Transporter (SWEET) genes engage in sugar transport and distribution in plants, playing a significant role in plant physiology (Eom et al., 2015). These genes are part of the MtN3/saliva family and consist of seven transmembrane α-helices (TM1 to TM7). An ancient duplication resulted in their unique 3-1-3 configuration, forming a central channel for sugar transport. Both the N-terminal and C-terminal ends of the SWEET protein are cytoplasmatic (Chen et al., 2010) (Figure 4). SWEET genes are highly conserved across different plant species and categorized into four clades, each specializing in the transport of specific sugars: hexoses (Clades I and II), sucrose (Clade III), and fructose (Clade IV). These proteins are expressed in various tissues – leaves, seeds, roots, and flowers – and regulate important physiological processes such as pollen development, fruit ripening, and leaf senescence (Chen et al., 2012; Gautam et al., 2022; Ko et al., 2022). Their expression can be induced by biotic stresses (Zhu and Gong, 2014; Zhu et al., 2019). In various plant species, including V. vinifera, SWEET homologs have been identified as susceptibility genes, acting as targets of effector proteins during host-microbe interactions. Indeed, some pathogens hijack SWEET genes to manipulate sugar flow and create nutrient-rich environments for their invasion and growth (Baker et al., 2012). On the other hand, some SWEETs can also function as resistance genes, promoting resistance to certain pathogens (Kang et al., 2023). AtSWEET1 from A. thaliana was the first SWEET protein to be identified in plants (Chong et al., 2014). Later, SWEETs were identified in many other plant species (Table 4) such as M. truncatula (Hu et al., 2019), O. sativa (Yuan and Wang, 2013), S. tuberosum (Manck-Götzenberger and Requena, 2016), T. aestivum (Qin et al., 2020) and G. max (Patil et al., 2015). Research has shown that different SWEET genes in plants are upregulated in response to powdery mildew infection, indicating their crucial role in biotic stress resistance. Chong et al. (2014) characterized the SWEET family in grape genome with VvSWEET4 particularly upregulated after B. cinerea infection. More recently, Zhong et al. (2024) identified notable differences in VvSWEET gene expression between resistant (Shangyou, Beihong, Beimei and Gold Finger) and susceptible (Red Globe, Gebixinxiu, Thompson Seedless and JingXiangyu) grape varieties. VvSWEET1 and VvSWEET10b exhibited elevated expressions in the latter, while VvSWEET3 was more expressed in the former, suggesting a potential role for these genes in the response to B. cinerea infection. Chen et al. (2010) found an induction in AtSWEET12 during powdery mildew infection in A. thaliana, while Pan et al. (2024) observed significant upregulation of AsSWEET1a, 3a, 11, and 16 in Avena sativa. Additionally, Chong et al. (2014) highlighted thew role of AtSWEET4 in resistance to B. cinerea, demonstrating that atsweet4 knockout mutants exhibited resistance. Given the significant role of soluble sugar content in determining the yield and economic value of grape berries, the efficient sugar partitioning is crucial in grapevine. These findings underline the potential of SWEET genes in crop improvement strategies aimed at enhancing yield and stress resistance through biotechnological manipulation of sugar transport mechanisms.
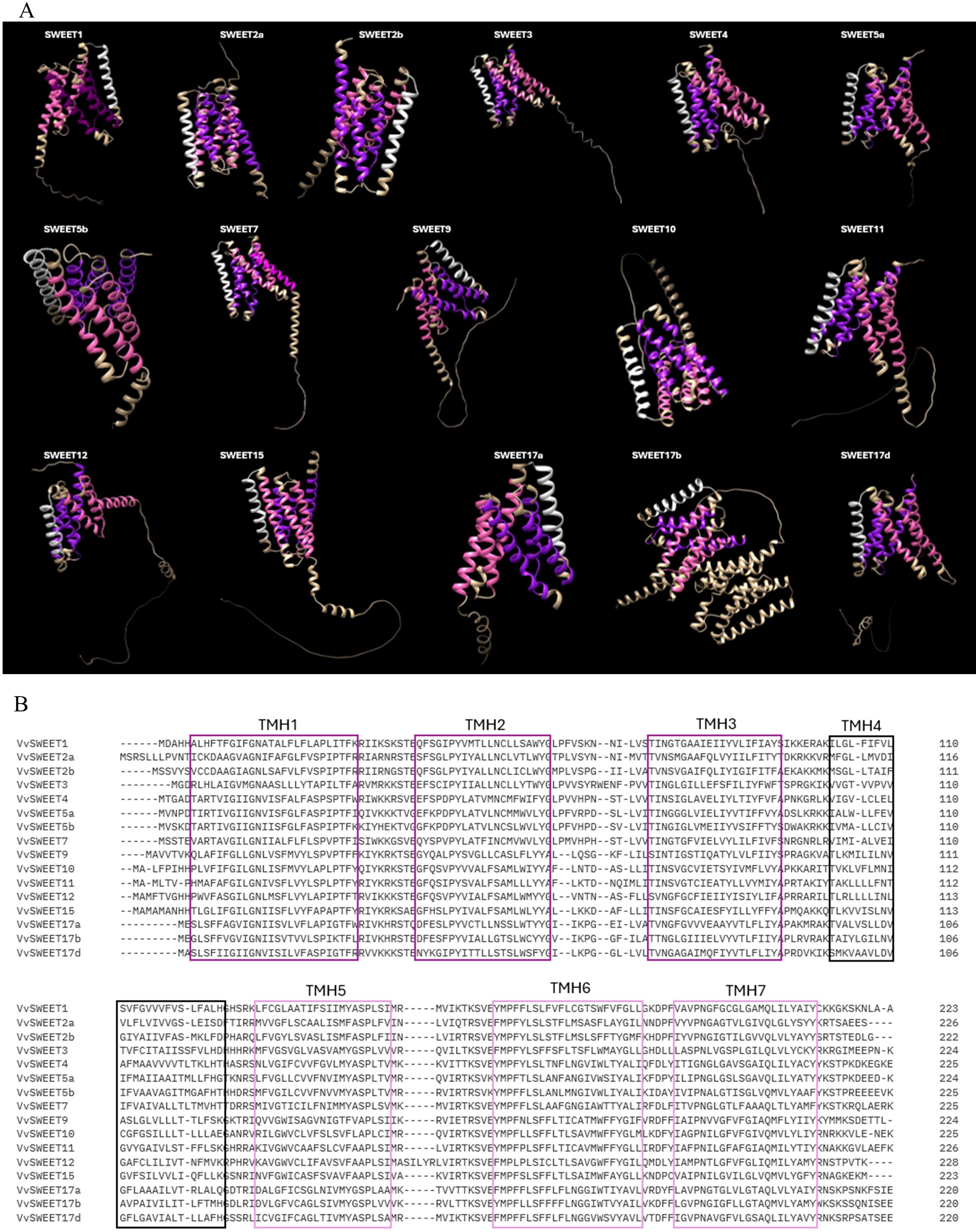
Figure 4. (A) AlphaFold prediction of the structure of the SWEETS proteins performed with ChimeraX AlphaFold, Each domain which comprises three transmembrane helices (3-TMs) is represented in different colour: The first domain is purple, the second domain is pink and the fourth transmembrane helix (TMH), which is less conserved and divides the SWEET protein into two domains is white. (B) Sequence alignment of the seven transmembrane domains of SWEET proteins.
Understanding the structure and function of TMPs is crucial for unraveling their roles in various biological processes and guiding targeted breeding programs. However, studying these integral membrane proteins presents unique challenges, such as low expression levels and the difficulties associated with detergent-based structural studies (Thoma et al., 2018). To overcome these hurdles, researchers have developed a suite of assays that, when used in combination, can provide a comprehensive understanding of TMP structure, function, and regulation across various physiological contexts (Boulos et al., 2023). In this section, we delve into in vitro, in vivo and in silico techniques that can be employed to study TMPs in plants offering researchers a valuable resource for selecting the most appropriate strategies in TMP research. A summary of the techniques considered with strengths and limitations is reported in Table 5.
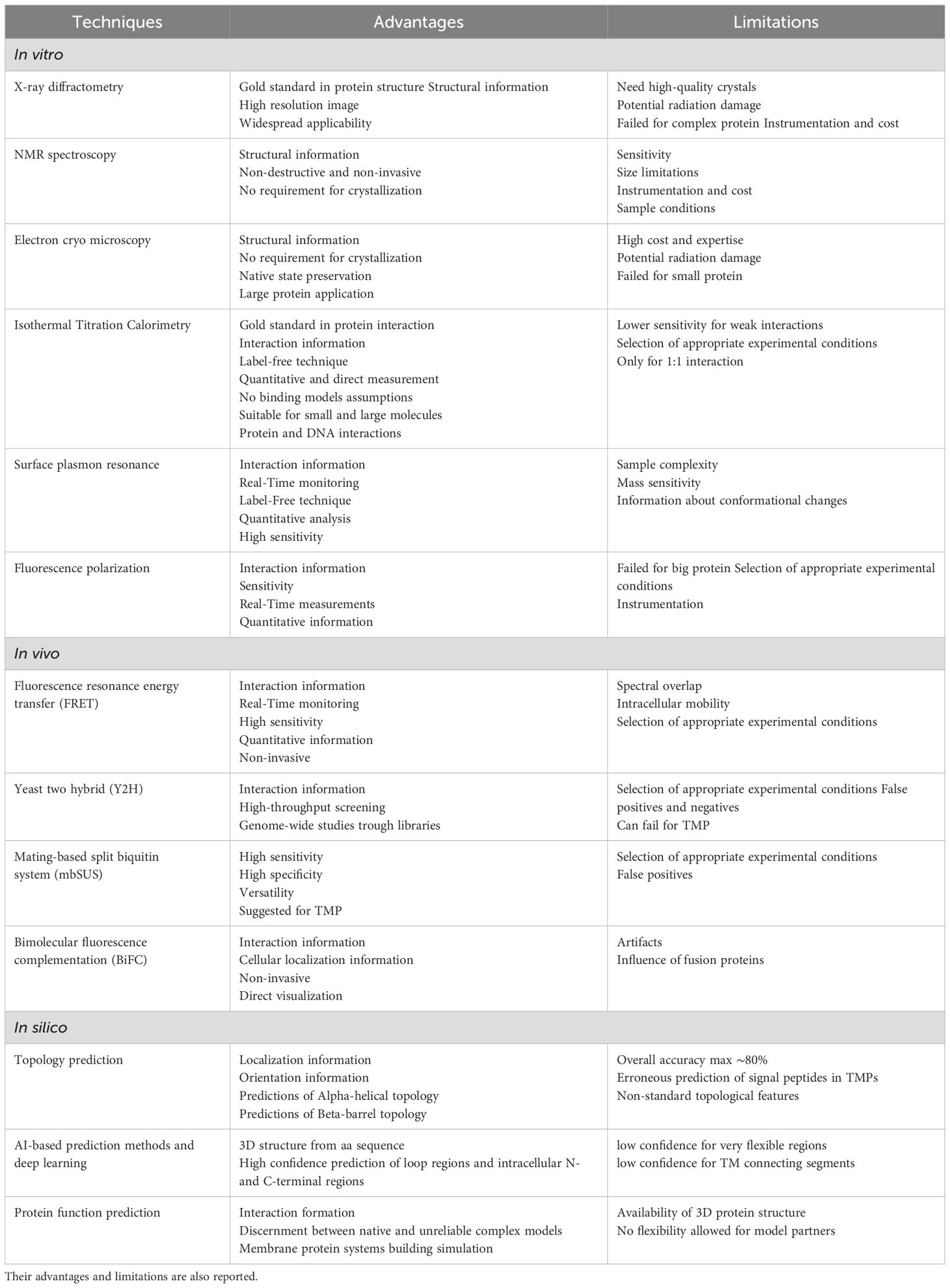
Table 5. In vitro, in vivo and in silico techniques mainly used for protein structure and interaction studies.
In vitro techniques are experimental methods conducted outside of a living organism, typically in a controlled laboratory setting, used to simplify complex systems and make it easier to identify and analyze specific structures or interactions. The main in vitro techniques for TMP studies include the X-ray diffractometry, the Nuclear Magnetic Resonance (NMR), and the electron cryo-microscopy (Cryo-EM). Additionally, other in vitro techniques can be used to study protein functions and interactions, such as the isothermal titration calorimetry, the surface plasmon resonance and the fluorescence polarization. Hereunder, we provide a critical analysis of their principles, applications, strengths, and limitations for TMPs investigation.
X-ray diffractometry is a technique used to determine the 3D structure of proteins by diffracting X-rays through protein crystals. The diffraction of the X-rays provides exceptional resolution (<1.5 Å), enabling precise determination of atomic positions. While this technique is widely employed in grape protein research, it has not been applied to the study of grape TMPs yet. One of the primary limitations of X-ray diffractometry is the difficulty in obtaining highly purified proteins in crystalline form, especially for TMPs. The main challenge is that purifying and crystallizing TMPs is difficult because they do not fold correctly in normal aqueous (water-based) environments, which traditional crystallization methods depend on. Special approaches are required to work with TMPs since they behave differently from water-soluble proteins. A successful strategy for determining the structure of TMPs is the use of detergents (Tate, 2010). This approach has been effective in determining the crystallographic structure of a CMP-sialic acid transporter from Zea mays (Nji et al., 2019) and a multidrug efflux transporter from Camelina sativa (Tanaka et al., 2017).
NMR can determine the 3D structure of proteins in solution, revealing the arrangement of atoms and chemical bonds. Proteins are typically dissolved in a suitable solvent and placed in a strong magnetic field, causing the nuclei of atoms to align. Radio frequency (RF) pulses are then applied to the sample, perturbing the alignment of the nuclei. As the nuclei relax back to their equilibrium state, they emit detectable NMR signals, which are processed and analyzed to reconstruct the protein structure. While NMR is widely used for studying proteins, its application to transmembrane proteins (TMPs) is limited (Yeh et al., 2020), similar to X-ray diffractometry. This limitation arises from the size and structural characteristics of TMPs, as NMR typically requires smaller proteins and a significant amount of sample due to its sensitivity constraints (Xu et al., 2007). These factors make studying large TMPs challenging. However, NMR has the advantage of being able to simulate the membrane environment in solution, making it potentially valuable for TMP research. Despite these advantages, no NMR studies have been published specifically on grape TMPs. However, a few studies have focused on portions of TMPs in plants. For instance, a fully functional C-terminally truncated twin-arginine translocase (Tat) from A. thaliana, consisting of 53 amino acids, was investigated using solution-state NMR in micelles and lipid bilayers (Pettersson et al., 2018). Another example is the chloroplast outer envelope channel OEP21 from Pisum sativum, a protein of 187 amino acids, whose structure was solved using solution NMR, revealing a beta-barrel pore structure (Günsel et al., 2023).
In the past 10 years, cryo-EM has emerged as a revolutionary technique in structural biology, enabling researchers to visualize biological molecules, including TMPs, at near-atomic resolution (Wang, 2022; Castells-Graells et al., 2023). This technique involve the use of biological samples rapidly frozen in a thin layer of vitreous ice to preserve their native state and the frozen sample is then imaged using an electron microscope. Electrons interact with the sample, creating multiple 2D projection images that are collected from different orientations of the sample to reconstruct the 3D structure of the molecule. In 2019, the number of membrane proteins structurally resolved by cryo-EM surpassed those resolved by X-ray diffractometry (García-Nafría and Tate, 2021), indicating that cryo-EM has become the most effective technique for resolving the 3D structures of even large proteins, typically those exceeding 50 kDa in size. In relation to plant TMPs, over 200 structures have been deposited in databases in the past five years. The majority of these involve channel structures, such as potassium, calcium, chloride, and glutamate receptor-like channels (Dickinson et al., 2021), as well as transporters, including ABC and sodium transporters (Huang et al., 2024; Ying et al., 2024).
Isothermal titration calorimetry (ITC) is widely used for characterizing the binding of small compounds to larger macromolecules as DNA or proteins. It has become the gold standard for studying molecular interactions in solution measuring the heat released or absorbed during protein-ligand interactions by a microcalorimeter. This instrument has two cells that need to be maintained at the same temperature: one containing only buffer and the other containing the sample. By gradually adding small amounts of ligand, any interaction with the target sample will cause a detectable heat variation. This method allows researchers to define a complete thermodynamic profile of the interaction, providing data on enthalpy and entropy changes, binding constants, and reaction stoichiometry. Overall, ITC has the advantage of providing direct and quantitative measurements and is highly versatile in the types of reactions it can study. However, it has lower sensitivity for weak interactions and is most applicable to 1:1 interactions (Falconer, et al., 2021). ITC has already been used to study the interactions of medical compounds with three major classes of membrane proteins: G-protein coupled receptors, ion channels, and transporters (Draczkowski et al., 2014). In plants, it has been employed for several purposes, for example to test the binding capability of the Ferredoxin-NADP+ reductase to the cytochrome b6f complex from Spinacia oleracea and Z. mays in the electron transport chain of oxygenic photosynthesis (Cramer, 2022) or to test the interaction between the alkaline isoform of the PR-5 protein, purified from soybean hulls, and the CM-pachyman or bilayer vesicle to explain its antimicrobial activity (Liu et al., 2016).
Surface plasmon resonance (SPR) spectroscopy enables the real-time characterization of binding affinity and kinetics for membrane protein-ligand interactions, using relatively small amounts of membrane protein in a native or native-like environment. The SPR technique typically requires the target molecule to be immobilized on a sensor chip while the analyte in solution flows over the sensor surface. The binding of biomolecules results in a signal that depends on changes in the refractive index at the sensor surface (Patching, 2014). The advantages of SPR include real-time kinetic measurements, high sensitivity, and the ability to provide binding affinities and association/dissociation rates for label-free molecules. However, limitations include weak signals when analyzing small molecules, the need for high-purity analytes, and complications from protein conformational changes (Dobrovodský and Di Primo, 2023). Additionally, non-specific signals may require inhibitors or competitive molecules, complicating data analysis, and careful experimental control is needed to prevent unwanted analyte binding to the sensor surface. SPR is widely applied to study a broad range of membrane proteins and can also be used for various biomolecular interactions, including protein-DNA, protein-protein, protein-carbohydrate, protein-RNA, and protein-lipid interactions. In plants, SPR has been employed to investigate G-protein-coupled receptor (GPCR) transmembrane proteins and molecular interactions involved in various aspects of plant development, such as protein-carbohydrate interactions, protein-chaperone interactions, phytohormone signaling detection, and plant-virus diagnostics (Jain et al., 2016).
Fluorescence polarization (FP) is a non-disruptive technique that allows rapid, quantitative analysis of enzyme activities and molecular interactions by measuring the binding of a fluorescently labeled ligand to a larger molecule. A freely rotating small molecule with a fluorescent probe emits depolarized light. When it binds to a larger target, its rotation slows, causing the emitted light to become more polarized. FP detects these changes in light polarization (Hall et al., 2016). In its variant, single-molecule fluorescence polarization (SMFP), the technique also reveals the structural basis of protein activity and tracks conformational changes between distinct states (Forkey et al., 2000). The ability to study molecular processes in solution, along with time-course readability and a homogeneous assay format, makes FP particularly suitable for high-throughput screening. However, FP has limitations. It is highly dependent on the size difference between the interacting molecules and on the choice of fluorescent label. If the size difference is too small, the method may not perform reliably. Additionally, the fluorescent probe can interfere with ligand binding or create nonspecific interactions. Low-affinity interactions can also skew results, as high concentrations of unlabelled protein can lead to artificial crowding effects (Dharadhar et al., 2019). FP has been used to study transmembrane GPCRs, avoiding the need for radioactive methods previously required to monitor binding events (Burke et al., 2003). High-throughput FP assays have also been developed to quantify biotin and biotin-binding proteins in Nicotiana tabacum leaf extracts (Martin et al., 2008) and to screen ligands of the jasmonate COI1–JAZ co-receptor in Arabidopsis thaliana (Takaoka et al., 2019).
Unlike in vitro methods, in vivo approaches enable for observation of proteins in their natural state within living cells, revealing their dynamic interactions and functional roles. The techniques we have explored in depth include Fluorescence Resonance Energy Transfer (FRET), the Yeast Two-Hybrid (Y2H) system, and Bimolecular Fluorescence Complementation (BiFC). These methods are emphasized due to their accessibility and their effective combination for achieving optimal results, marking considerable progress in structural biology.
FRET is a powerful technique used to study interactions proteins (Förster, 1948). It relies on the non-radiative energy transfer between a donor and an acceptor fluorophore, which occurs when they are in proximity (typically within 1-10 nm). When the donor fluorophore, absorbing light at a specific wavelength, is excited, it can transfer energy to the acceptor fluorophores if they are within the critical FRET distance; this leads to fluorescence emission from the acceptor while the donor fluorescence intensity decreases (Periasamy, 2001). FRET is a versatile technique widely used across various biological systems such as plant (Bücherl et al., 2014), mammalian (Mitchell et al., 2017), yeast (Skruzny et al., 2019) and bacteria (Majoul, 2004). Among numerous studies, Fliegmann et al. (2016) employed FRET to reveal the formation of a complex between MtLYR3 and MtLYK3 at the plasma membrane in N. benthamiana leaf cells. Similarly, Roudaire et al. (2023) used FRET to demonstrate a chitin-dependent interaction between VvLYK5-1 and VvLYK1-1. Despite FRET efficiency decreasing rapidly as the distance between the donor and acceptor increases, the popularity of FRET is due to its advantages. These include high sensitivity, real-time analysis capabilities, and its non-invasive nature, eliminating the need for disrupting cellular processes.
Y2H system is a method used for the analysis of protein-protein interactions in yeast. This assay is based on the ability of transcription factors to positively regulate a responsive gene for the growth on selective medium and to be organized into two separable functional domains such as the DNA-binding domain (BD) and the activation domain (AD) (Bai and Elledge, 1997). These domains can be separated and combined in a reversible way. If physically separated, the BD and AD cannot transcript the responsive gene (Causier, 2004); when recombined, they reconstitute an active transcription factor (Paiano et al., 2018). In the Y2H, given two proteins of interest, named “bait” and “prey”, two vectors are produced fusing the BD to the bait and the AD to the prey. After expression of the two proteins in yeast cells, if the pray and bait interact, BD and AD will combine reconstituting an active transcription factor and the yeast will grow on the selective media (Paiano et al., 2018). The Y2H technique is a versatile and widely used method for studying protein-protein interactions in plant (Shimizu et al., 2010) and human (Smirnova et al., 2022). However, it has limitations, particularly with membrane proteins due to their localization. Furthermore, Y2H may not be suitable for analyzing interactions that require specific cofactors or post-translational modifications not present in the yeast system. Additionally, it can be sensitive to false positives or negatives. A strategy to overcome the challenges associated with studying TMPs is to use their N-terminal and C-terminal portions as reported by Smirnova et al. (2022) in a study about human protein interaction. Another adaptation of Y2H for the study of TMPs is the split-ubiquitin system. In this method, ubiquitin is split into two halves and the bait and prey are fused to them. Their interaction reassembles ubiquitin, triggering the release of a transcription factor that then activates the expression of a reporter gene (Thaminy et al., 2004). Through this method, Wang et al. (2020) highlighted the interaction of VviABCG14 with VviABCG7, suggesting a role in cytokinin transport, important hormones involved in many aspects of plant growth and development. Y2H is advantageous for studying protein-protein interactions by enabling the screening of large protein libraries to identify new protein partners. For example, Liu et al. (2020) used Y2H to construct a cDNA library from grape leaves infected with P. viticola, the causal agent of downy mildew. They screened for the targets of pathogenic factors in host grapes, providing insight into the molecular mechanisms of P. viticola infection. In conclusion, Y2H is known for its user-friendly nature and its ability to detect protein-protein interactions. When coupled with a complementary technique (co-immunoprecipitation, BiFC, FRET), it becomes even more powerful in providing detailed insights into the biological processes under study.
The mbSUS system is a powerful tool for studying protein-protein interactions similar to Y2H system and is particularly used for membrane proteins. It involves splitting the ubiquitin protein into two parts: Cub (C-terminal ubiquitin) and Nub (N-terminal ubiquitin). One protein of interest is fused to Cub, while the other is fused to Nub. When these proteins interact, the two halves of ubiquitin reassemble, triggering the expression of a reporter gene. This system has been set up in Arabidopsis to study the interactome of K+ channels (Obrdlik et al., 2004), afterwards it has been successfully employed to study protein-protein interactions in various plant species such as grape, as exhaustively reviewed by Xing et al., 2016. In grapevine, mbSUS has effectively been used to investigate the interactions of two VpCDPKs that directly interact with VpMAPK3/6 and VpACS1/2, which promot the expression of many defense-associated genes in response to powdery mildew infection (Hu et al., 2021). While mbSUS is a robust tool, it is important to note that its limitations, including false positives and negatives, and the fact that the yeast environment may not perfectly mimic plant cell conditions. Furthermore, Y2H-like assays face significant challenges when studying cytoplasmic proteins that require nuclear translocation for their function (Cuadrado and Van Damme, 2024). To overcome these limitations, careful experimental design and validation of results are crucial. By combining mbSUS with other techniques, such as co-immunoprecipitation, BiFC or the cytoplasmic specific Cytotrap (Mohan et al., 2023), researchers can gain a more comprehensive understanding of protein-protein interactions in grapevine.
BiFC is a method used to detect protein-protein interactions in living cells. This assay is based on the reconstitution of a fluorescent protein in vivo, named reporter, that is truncated in two non-fluorescent halves. These halves are then fused to the proteins of interest (the bait and the prey). When these proteins interact, the two halves of the fluorescent protein reconstitute into a functional fluorescent protein, emitting fluorescence that can be detected using fluorescence microscopy. The inverted fluorescence microscope enables the detection and localization of the fluorescent signal within the cell. Moreover, the emitted fluorescence intensity corresponds proportionally to the strength of the interaction. Higher fluorescence levels signify close or direct interactions, while lower levels suggest interactions within a complex (Morell et al., 2008). BiFC assays have been employed in high-throughput screens to reveal new protein-protein interactions in yeast (Sung and Huh, 2007), plant (Peiró et al., 2014) and mammalian cells (Grau et al., 2017). Utsugi et al. (2015) used BiFC to confirm the interaction between HvTIP1-2 and HvTIP3 in onion. They showed that when HvTIP1-2, which has established water transport capability, interacts with HvTIP31, which does not exhibit such activity, they form heterotetramers that enhance water permeability. Xue et al. (2019) used BiFC to evidence a direct physical interaction between LYK1 and LYK4 in A. thaliana. BiFC offers several advantages, including its ability to provide real-time, spatially resolved information about interactions in living cells without the need for complex biochemical assays. The simplicity and adaptability of BiFC make it suitable for high-throughput screening applications, allowing researchers to analyze multiple protein interactions all together. It is also a valuable tool for the study of membrane protein interactions (García-Murria et al., 2020; Barriga et al., 2021; Duart et al., 2021).
In silico methodologies are a useful tool to work around the difficulties associated with traditional in vitro or in vivo approaches and to bridge the gap between knowledge of TMPs structures and properties. In silico techniques can be applied on two diverse levels: directly on the protein sequence, to predict the most probable topology or the most favored 3-D structure, or directly to the structure (experimental or predicted). In this latter case, the in silico methodologies are employed principally to predict protein function or protein behavior; therefore, they make it possible to simulate protein-protein interactions, protein-ligand interactions or generally membrane system model behaviors. In the following paragraphs, a description of the principal methods applied for in silico predictions and of their applicability is presented.
Prediction methods have been developed to provide valuable structural insights into TMPs, addressing their biological roles and the challenges associated with experimentally determining their three-dimensional structures. Topology prediction methods focus on two key aspects: the localization of transmembrane regions and their orientation. The hydrophobicity of buried amino acids is used to predict transmembrane regions, while the “positive-inside” rule aids in determining the orientation of these segments (Von Heijne, 1992). Various networks have been designed to predict TMPs, each employing different approaches. Some networks, such as TMpred and DAS, analyze local properties of amino acid sequences using a sliding window approach to detect sub-sequences that span the membrane. Others, like PHD, utilize a neural network and can also be classified as local methods. In contrast, global prediction methods such as TMHMM 2.0 and HMTOP employ Hidden Markov Models to determine the most statistically probable topology for the entire protein. Additionally, hybrid approaches, including MEMSAT 1.5, Toppred22, and TMAP, combine elements of both local and global prediction strategies (Möller et al., 2001). Before the advent of AI, several limitations in prediction techniques were identified (Tsirigos et al., 2018). However, recent advancements in AI-based approaches have increased the accuracy of these predictions. For instance, MemBrain 3.0 integrates a transmembrane helix prediction module with an orientation prediction module, utilizing a support vector machine classifier and an MMA strategy (Feng et al., 2020). These methods generally perform well for predicting alpha-helical or beta-barrel topologies but still struggle with non-standard topological features. Martinez-Navarro et al. (2024) analyzed full-length amino acid sequences using hybrid approaches to study the domain architecture of the TMP LecRLKs in Vitis vinifera. These methodologies are also applied to indirectly determine the cellular localization of gene products. For example, in a study on the LAR protein, which plays a key role in flavonoid synthesis in A. thaliana, the absence of a transmembrane region suggested a cytoplasmic localization of the protein (Han et al., 2018). To gain a deeper understanding of protein function, various computational and experimental approaches have been employed. Among these, the assessment of Gene Ontology (GO) terms has been extensively used to functional similarity between proteins. GO terms provide insights into a protein cellular localization, involvement in biological processes, and molecular functions. Since interacting proteins often share similar pathways, processes, functions, or cellular compartments these methods are particularly effective for categorizing interactions based on the similarity of their associated GO terms (Grassmann et al., 2024). Protein-protein interaction networks further provide valuable insights into the functional relationships between proteins of the same family and with other cellular proteins. For example, the probable functions of Rboh proteins in Aquilaria species were predicted on the basis of assignment of GO terms. These terms revealed interactions with various signaling components, such as receptor kinases, calcium sensors, and transcription factors (Begum et al., 2024). Overall, these prediction approaches offer several benefits, such as determining protein localization and orientation, along with accurate predictions for both alpha-helical and beta-barrel topologies. However, they are also prone to certain limitations, particularly the frequent misprediction of signal peptides in TMPs.
Shortly after the first experimental determinations of protein three-dimensional structures, computational methods were developed to predict protein folding based exclusively on amino acid sequences. Over the past 70 years, continuous advancements in these methods have been made, particularly with the integration of AI and deep learning techniques, which have led to significant innovations. These advancements have garnered recognition within the scientific community for producing predictions that closely resemble the quality of experimentally determined structures (Bordin et al., 2023). Among the leading deep learning methods for protein structural modelling are AlphaFold2 (Jumper et al., 2021) and ESMFold, a large language model for protein sequences (Lin et al., 2023). Nguyen et al. (2024) compared structures resolved through experimental methods with predictions obtained from these deep learning approaches. The findings indicate that high-confidence predictions are achieved in many regions of the proteins, while lower confidence is observed in areas lacking a defined structure and exhibiting significant flexibility, such as the connecting segments between TMPs. Specifically, the study evaluated the structural modeling of ion channels generated by these methods against experimental Cryo-EM structures. It was found that AlphaFold2 successfully predicted the majority of the hNaV1.8 domains with very high confidence scores (pLDDT > 90). In contrast, RoseTTAFold2 predicted most TMP regions with lower confidence (50 < pLDDT < 70) but accurately modeled the pore domain. Meanwhile, ESMFold demonstrated good confidence in predicting the voltage-sensing domains, pore domain, and extracellular loop regions (70 < pLDDT < 90). Currently, a search for protein models from V. vinifera in the AlphaFold database yields 183 modeled structures corresponding to sequences reviewed in SwissProt. In contrast, the Transmembrane Protein Structure Database (TmAlphaFold) (Dobson et al., 2023) provides 64 results, of which 51 exhibit excellent evaluations of their corresponding models. Overall, these methodologies bridge the gap between available sequences and resolved structures, enabling the prediction of protein 3D structures directly from amino acid sequences. While these methods offer several advantages, including the ability to simulate interaction formation and effectively build TMP systems, they also face limitations, such as the availability of 3D protein structures and challenges in predicting TMPs.
Computational methods for studying proteins are invaluable for predicting three-dimensional structures and simulating protein-protein interactions, protein-ligand interactions, and molecular dynamics. Reliable molecular interaction simulations require that a protein’s three-dimensional structure be either experimentally determined or predicted with high confidence. Various docking software is available for investigating protein interactions, including GRAMM-X (Tovchigrechko and Vakser, 2006), ClusPro (Comeau et al., 2004), HDOCK (Yan et al., 2020), ZDOCK (Pierce et al., 2014), LZerD (Christoffer et al., 2021), and HADDOCK (Van Zundert et al., 2016). While these tools facilitate protein-protein docking, they typically do not account for the explicit flexibility of the modelled partners, although some can address this limitation through energy minimization. In silico methods are particularly useful for predicting membrane-associated protein assemblies. For instance, LightDock combines efficient rigid-body docking with artificial intelligence algorithms and employs flexible refinement using HADDOCK to resolve potential clashes at the interface (Roel-Touris et al., 2020). Other approaches, like JabberDock (Rudden and Degiacomi, 2021), utilize spatial and temporal influence density maps derived from short molecular dynamics simulations, while MPDock (Alford et al., 2015) leverages existing Rosetta sampling and scoring methods. A novel approach, AlphaFold-Multimer (Evans et al., 2021), is specifically trained for multimeric inputs with known stoichiometry, significantly improving the accuracy of predicted interfaces compared to the original single-chain AlphaFold. Selecting the most suitable method for protein interaction predictions is closely tied to the specific properties of the proteins being studied. The CAPRI competition, which stands for Critical Assessment of PRedicted Interactions, provides a valuable resource for guiding this choice by evaluating the success rates of various methods (Vakser, 2014). A notable example is Voss et al. (2023), who utilized AlphaFold-Multimer to predict the dimerization of EDS1 in Vitis vinifera. Their findings suggest that VvEDS1 dimers may serve as reservoirs for forming heterodimers with other proteins crucial for immune signaling in plants, demonstrating the significant impact of AI on understanding host-microbe interactions. Simulation tools offer several advantages, including tailored options for membrane-protein interactions and membrane remodeling (Goossens and De Winter, 2018). However, the lack of a uniform simulation method and the reliance on rigid body models can limit the accuracy of the resulting predictions.
Despite significant advancements in the identification and study of TMPs in plant growth, development, and stress responses, further research is necessary to fully understand their roles in grapevine defense against biotic stress. Existing studies have underscored the importance of specific TMPs such as LYKs, Rboh and SWEET in this context. However, the precise mechanisms underlying TMP-mediated intracellular signal transduction remain unclear, particularly in terms of how receptors transmit signals and the detailed pathways involved. Additionally, different TMPs involved in defense mechanisms have been identified in other plant species (see Zhou et al., 2022), and expanding research to them in grapevine could help uncover conserved mechanisms and contribute to a broader understanding of TMP functions in grape immunity. Emerging technologies present unique opportunities to resolve these gaps with greater accuracy. CRISPR/Cas9 technology, for instance, offers a promising, transgene-free method for precisely editing TMP-related genes, and has already been successfully applied in grapevine research. Furthermore, advanced molecular imaging techniques and high-resolution mass spectrometry can provide critical insights into the mechanisms of TMP action and interaction dynamics, shedding light on their functional roles within plant cells. The study of TMPs would also benefit from the integration of multi-omics approaches, such as proteomics and transcriptomics (such as single-cell RNASeq), to construct a more comprehensive understanding of the signaling pathways and interaction networks involved in biotic stress responses. As more TMPs are discovered, deeper exploration of their structural, regulatory, and functional diversity will be essential for advancing our understanding of their multifaceted roles in grape biology.
AG: Writing – original draft, Writing – review & editing. DG: Writing – review & editing. AF: Writing – review & editing. CV: Conceptualization, Writing – original draft, Writing – review & editing. DC: Writing – review & editing. RA: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the Italian Ministry of University and Research (MUR) under the PRIN 2022 project "DEMETRA: Deciphering the molecular MEchanism of powdery mildew defense response in grapevine ThRrough a multi-omic Approach" (Grant Prot. P2022RWYNM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abedi, A., Hajiahmadi, Z., Kordrostami, M., Esmaeel, Q., Jacquard, C. (2021). Analyses of lysin-motif receptor-like kinase (LysM-RLK) gene family in allotetraploid brassica napus L. and its progenitor species: an in-silico study. Cells 11, 37. doi: 10.3390/cells11010037
Alford, R. F., Koehler Leman, J., Weitzner, B. D., Duran, A. M., Tilley, D. C., Elazar, A., et al. (2015). An integrated framework advancing membrane protein modeling and design. PloS Comput. Biol. 11, e1004398. doi: 10.1371/journal.pcbi.1004398
Ali, S., Tyagi, A., Bae, H. (2023). ROS interplay between plant growth and stress biology: Challenges and future perspectives. Plant Physiol. Biochem. 203, 108032. doi: 10.1016/j.plaphy.2023.108032
Argudo, P. G. (2024). Lipids and proteins: Insights into the dynamics of assembly, recognition, condensate formation. What is still missing? Biointerphases 19, 1–9. doi: 10.1116/6.0003662
Arrighi, J. F., Barre, A., Ben Amor, B., Bersoult, A., Soriano, L. C., Mirabella, R., et al. (2006). The Medicago truncatula lysine motif-receptor-like kinase gene family includes NFP and new nodule-expressed genes. Plant Physiol. 142, 265–279. doi: 10.1104/pp.106.084657
Babu, C. S., Babar, S. M. E., Song, E. J., Oh, E., Yoo, Y. S. (2008). Kinetic analysis of the MAPK and PI3K/Akt signaling pathways. Mol. Cells 25, 397–406. doi: 10.1016/S1016-8478(23)17598-4
Bai, C., Elledge, S. J. (1997). “Gene identification using the yeast two-hybrid system,” in Methods in enzymology, vol. 283. (United Kingdom: Academic Press), 141–156).
Baker, R.F., Leach, K. A., Braun, D. M. (2012). SWEET as sugar: new sucrose effluxers in plants. Mol. Plant 5.4, 766–768. doi: 10.1093/mp/SSS054
Barrera, E. E., Machado, M. R., Pantano, S. (2019). Fat SIRAH: coarse-grained phospholipids to explore membrane–protein dynamics. J. Chem. Theory Compt. 15 (10), 5674–5688. doi: 10.1021/acs.jctc.9b00435
Barriga, A., Morán-Lalangui, M., Castillo-Sánchez, J. C., Mingarro, I., Pérez-Gil, J., García-Álvarez, B. (2021). Role of pulmonary surfactant protein Sp-C dimerization on membrane fragmentation: An emergent mechanism involved in lung defense and homeostasis. Biochim. Biophys. Acta (BBA)-Biomembr. 1863, 183572. doi: 10.1016/j.bbamem.2021.183572
Begum, K., Das, A., Ahmed, R., Akhtar, S., Kulkarni, R., Banu, S. (2024). Genome-wide analysis of respiratory burst oxidase homolog (Rboh) genes in Aquilaria species and insight into ROS-mediated metabolites biosynthesis and resin deposition. Front. Plant Sci. 14, 1326080. doi: 10.3389/fpls.2023.1326080
Bellande, K., Bono, J. J., Savelli, B., Jamet, E., Canut, H. (2017). Plant lectins and lectin receptor-like kinases: how do they sense the outside? Int. J. Mol. Sci. 18, 1164. doi: 10.3390/ijms18061164
Blaum, B. S., Mazzotta, S., Nöldeke, E. R., Halter, T., Madlung, J., Kemmerling, B., et al. (2014). Structure of the pseudokinase domain of BIR2, a regulator of BAK1-mediated immune signaling in Arabidopsis. J. Struct. Biol. 186, 112–121. doi: 10.1016/j.jsb.2014.02.005
Bordin, N., Dallago, C., Heinzinger, M., Kim, S., Littmann, M., Rauer, C., et al. (2023). Novel machine learning approaches revolutionize protein knowledge. Trends Biochem. Sci. 48, 345–359. doi: 10.1016/j.tibs.2022.11.001
Boulos, I., Jabbour, J., Khoury, S., Mikhael, N., Tishkova, V., Candoni, N., et al. (2023). Exploring the world of membrane proteins: techniques and methods for understanding structure, function, and dynamics. Molecules 28, 7176. doi: 10.3390/molecules28207176
Brulé, D., Villano, C., Davies, L. J., Trdá, L., Claverie, J., Héloir, M. C., et al. (2019). The grapevine (Vitis vinifera) LysM receptor kinases Vv LYK 1-1 and Vv LYK 1-2 mediate chitooligosaccharide-triggered immunity. Plant Biotechnol. J. 17, 812–825. doi: 10.1111/pbi.13017
Bücherl, C. A., Bader, A., Westphal, A. H., Laptenok, S. P., Borst, J. W. (2014). FRET-FLIM applications in plant systems. Protoplasma 251, 383–394. doi: 10.1007/s00709-013-0595-7
Buendia, L., Girardin, A., Wang, T., Cottret, L., Lefebvre, B. (2018). LysM receptor-like kinase and LysM receptor-like protein families: an update on phylogeny and functional characterization. Front. Plant Sci. 9, 409605. doi: 10.3389/fpls.2018.01531
Burke, T. J., Loniello, K. R., Beebe, J. A., Ervin, K. M. (2003). Development and application of fluorescence polarization assays in drug discovery. Combinatorial Chem. High throughput screen. 6, 183–194. doi: 10.2174/138620703106298365
Cao, Y., Liang, Y., Tanaka, K., Nguyen, C. T., Jedrzejczak, R. P., Joachimiak, A., et al. (2014). The kinase LYK5 is a major chitin receptor in A. and forms a chitin-induced complex with related kinase CERK1. elife 3, e03766. doi: 10.7554/eLife.03766
Castells-Graells, R., Meador, K., Arbing, M. A., Sawaya, M. R., Gee, M., Cascio, D., et al. (2023). Cryo-EM structure determination of small therapeutic protein targets at 3 Å-resolution using a rigid imaging scaffold. Proc. Natl. Acad. Sci. 120, e2305494120. doi: 10.1073/pnas.2305494120
Causier, B. (2004). Studying the interactome with the yeast two-hybrid system and mass spectrometry. Mass spectrom. Rev. 23, 350–367.). doi: 10.1002/mas.10080
Chapman, J. M., Muhlemann, J. K., Gayomba, S. R., Muday, G. K. (2019). RBOH-dependent ROS synthesis and ROS scavenging by plant specialized metabolites to modulate plant development and stress responses. Chem. Res. Toxicol. 32, 370–396. doi: 10.1021/acs.chemrestox.9b00028
Chen, L.-Q., Hou, B.-H., Lalonde, S., Takanaga, H., Hartung, M. L., Qu, X.-Q., et al. (2010). Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527–532. doi: 10.1038/nature09606
Chen, L.-Q., Qu, X.-Q., Hou, B.-H., Sosso, D., Osorio, S., Fernie, A. R., et al. (2012). Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 335, 207–211. doi: 10.1126/science.1213351
Cheng, C., Xu, X., Gao, M., Li, J., Guo, C., Song, J., et al. (2013). Genome-wide analysis of respiratory burst oxidase homologs in grape (Vitis vinifera L.). Int. J. Mol. Sci. 2013 14, 24169–24186. doi: 10.3390/ijms141224169
Chiu, C. H., Paszkowski, U. (2020). Receptor-like kinases sustain symbiotic scrutiny. Plant Physiol. 182, 1597–1612. doi: 10.1104/pp.19.01341
Chong, J., Piron, M. C., Meyer, S., Merdinoglu, D., Bertsch, C., Mestre, P. (2014). The SWEET family of sugar transporters in grapevine: VvSWEET4 is involved in the interaction with Botrytis cinerea. J. Exp. Bot. 65, 6589–6601. doi: 10.1093/jxb/eru375
Christoffer, C., Chen, S., Bharadwaj, V., Aderinwale, T., Kumar, V., Hormati, M., et al. (2021). LZerD webserver for pairwise and multiple protein–protein docking. Nucleic Acids Res. 49, W359–W365. doi: 10.1093/nar/gkab336
Clark, S. E., Williams, R. W., Meyerowitz, E. M. (1997). The CLAVATA1gene encodes a putative receptor kinase that controls shoot and floral meristem size in Arabidopsis. Cell 89, 575–585. doi: 10.1016/S0092-8674(00)80239-1
Comeau, S. R., Gatchell, D. W., Vajda, S., Camacho, C. J. (2004). ClusPro: a fully automated algorithm for protein–protein docking. Nucleic Acids Res. 32, W96–W99. doi: 10.1093/nar/gkh354
Cope, K. R., Prates, E. T., Miller, J. I., Demerdash, O. N., Shah, M., Kainer, D., et al. (2023). Exploring the role of plant lysin motif receptor-like kinases in regulating plant-microbe interactions in the bioenergy crop Populus. Comput. Struct. Biotechnol. J. 21, 1122–1139. doi: 10.1016/j.csbj.2022.12.052
Corradi, V., Sejdiu, B. I., Mesa-Galloso, H., Abdizadeh, H., Noskov, S. Y., Marrink, S. J., et al. (2019). Emerging diversity in lipid–protein interactions. Chem. Rev. 119 (9), 5775–5848. doi: 10.1021/acs.chemrev.8b00451
Cramer, W. A. (2022). Isothermal titration calorimetry of membrane protein interactions: FNR and the cytochrome b6f complex. Biophys. J. 121, 342a. doi: 10.1016/j.bpj.2021.11.1046
Cuadrado, A. F., Van Damme, D. (2024). Unlocking protein–protein interactions in plants: a comprehensive review of established and emerging techniques. J. Exp. Bot. 75, erae088. doi: 10.1093/jxb/erae088
Dharadhar, S., Kim, Q. R., Uckelmann, M., Sixma, T. K. (2019). “Quantitative analysis of USP activity in vitro,” in Methods in enzymology. Ed. Hochstrasser, M. (Cambridge: Academic Press), 281–319.
Dickinson, M. S., Pourmal, S., Gupta, M., Bi, M., Stroud, R. M. (2021). Symmetry reduction in a hyperpolarization-activated homotetrameric ion channel. Biochemistry 61, 2177–2181. doi: 10.1021/acs.biochem.1c00654
Dobrovodský, D., Di Primo, C. (2023). Do conformational changes contribute to the surface plasmon resonance signal? Biosensors Bioelectron. 232, 115296. doi: 10.1016/j.bios.2023.115296
Dobson, L., Szekeres, L. I., Gerdán, C., Langó, T., Zeke, A., Tusnády, G. E. (2023). TmAlphaFold database: membrane localization and evaluation of AlphaFold2 predicted alpha-helical transmembrane protein structures. Nucleic Acids Res. 51, D517–D522. doi: 10.1093/nar/gkac928
Draczkowski, P., Matosiuk, D., Jozwiak, K. (2014). Isothermal titration calorimetry in membrane protein research. J. Pharm. Biomed. Anal. 87, 313–325. doi: 10.1016/j.jpba.2013.09.003
Du, L., Jiang, Z., Zhou, Y., Shen, L., He, J., Xia, X., et al. (2023). Genome-wide identification and expression analysis of respiratory burst oxidase homolog (RBOH) gene family in eggplant (Solanum melongena L.) under abiotic and biotic stress. Genes 14, 1665. doi: 10.3390/genes14091665
Duart, G., Grau, B., Mingarro, I., Martinez-Gil, L. (2021). Methodological approaches for the analysis of transmembrane domain interactions: A systematic review. Biochim. Biophys. Acta (BBA)-Biomembr. 1863, 183712. doi: 10.1016/j.bbamem.2021.183712
Eom, J.-S., Chen, L.-Q., Sosso, D., Julius, B. T., Lin, I. W., Qu, X.-Q., et al. (2015). SWEETs, transporters for intracellular and intercellular sugar translocation. Curr. Opin. Plant Biol. 25, 53–62. doi: 10.1016/j.pbi.2015.04.005
Evans, R., O’Neill, M., Pritzel, A., Antropova, N., Senior, A., Green, T., et al. (2021). Protein complex prediction with AlphaFold-Multimer. biorxiv, 1-25. doi: 10.1101/2021.10.04.463034
Falconer, R. J., Schuur, B., Mittermaier, A. K. (2021). Applications of isothermal titration calorimetry in pure and applied research from 2016 to 2020. J. Mol. Recogn. 34, e2901. doi: 10.1002/jmr.v34.10
Feng, S. H., Zhang, W. X., Yang, J., Yang, Y., Shen, H. B. (2020). Topology prediction improvement of α-helical transmembrane proteins through helix-tail modeling and multiscale deep learning fusion. J. Mol. Biol. 432, 1279–1296. doi: 10.1016/j.jmb.2019.12.007
Filyushin, M. A., Slugina, M. A., Shchennikova, A. V., Kochieva, E. Z. (2023). Differential expression of sugar uniporter genes of the SWEET family in the regulation of qualitative fruit traits in tomato species (Solanum section lycopersicon). Russian J. Plant Physiol. 70, 70. doi: 10.1134/S102144372360023X
Fliegmann, J., Jauneau, A., Pichereaux, C., Rosenberg, C., Gasciolli, V., Timmers, A. C. J., et al. (2016). LYR 3, a high-affinity LCO-binding protein of Medicago truncatula, interacts with LYK 3, a key symbiotic receptor. FEBS Lett. 590, 1477–1487. doi: 10.1002/1873-3468.12191
Forkey, J. N., Quinlan, M. E., Goldman, Y. E. (2000). Protein structural dynamics by single-molecule fluorescence polarization. Prog. biophys. Mol. Biol. 74, 1–35. doi: 10.1016/S0079-6107(00)00015-8
Förster, T. (1948). Zwischenmolekulare energiewanderung und fluoreszenz. Annalen der physik 437, 55–75. doi: 10.1002/andp.19484370105
Fradin, E. F., Zhang, Z., Juarez Ayala, J. C., Castroverde, C. D., Nazar, R. N., Robb, J., et al. (2009). Genetic dissection of Verticillium wilt resistance mediated by tomato Ve1. Plant Physiol. 150, 320–332. doi: 10.1104/pp.109.136762
García-Murria, M. J., Duart, G., Grau, B., Diaz-Beneitez, E., Rodríguez, D., Mingarro, I., et al. (2020). Viral Bcl2s’ transmembrane domain interacts with host Bcl2 proteins to control cellular apoptosis. Nat. Commun. 11, 6056. doi: 10.1038/s41467-020-19881-9
García-Nafría, J., Tate, C. G. (2021). Structure determination of GPCRs: cryo-EM compared with X-ray crystallography. Biochem. Soc. Trans. 49, 2345–2355. doi: 10.1042/BST20210431
Gautam, T., Dutta, M., Jaiswal, V., Zinta, G., Gahlaut, V., Kumar, S. (2022). Emerging roles of SWEET sugar transporters in plant development and abiotic stress responses. Cells 11, 1303. doi: 10.3390/cells11081303
Gómez-Gómez, L., Boller, T. (2000). FLS2: an LRR receptor–like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol. Cell 5, 1003–1011. doi: 10.1016/S1097-2765(00)80265-8
Goossens, K., De Winter, H. (2018). Molecular dynamics simulations of membrane proteins: an overview. J. Chem. Inf. Model. 58, 2193–2202. doi: 10.1021/acs.jcim.8b00639
Grassmann, G., Miotto, M., Desantis, F., Di Rienzo, L., Tartaglia, G. G., Pastore, A., et al. (2024). Computational approaches to predict protein–protein interactions in crowded cellular environments. Chem. Rev. 124, 3932–3977. doi: 10.1021/acs.chemrev.3c00550
Grau, B., Javanainen, M., García-Murria, M. J., Kulig, W., Vattulainen, I., Mingarro, I., et al. (2017). The role of hydrophobic matching on transmembrane helix packing in cells. Cell Stress 1, 90. doi: 10.15698/cst2017.11.111
Groom, Q. J., Torres, M. A., Fordham-Skelton, A. P., Hammond-Kosack, K. E., Robinson, N. J., Jones, J. D. (1996). rbohA, a rice homologue of the mammalian gp91phox respiratory burst oxidase gene. Plant J. 10, 515–522. doi: 10.1046/j.1365-313X.1996.10030515.x
Gull, A., Lone, A. A., Wani, N. U. I. (2019). Biotic and abiotic stresses in plants. Abiotic biotic Stress Plants 1–19.
Günsel, U., Klöpfer, K., Häusler, E., Hitzenberger, M., Bölter, B., Sperl, L. E., et al. (2023). Structural basis of metabolite transport by the chloroplast outer envelope channel OEP21. Nat. Struct. Mol. Biol. 30, 761–769. doi: 10.1038/s41594-023-00984-y
Gust, A. A., Willmann, R., Desaki, Y., Grabherr, H. M., Nürnberger, T. (2012). Plant LysM proteins: modules mediating symbiosis and immunity. Trends Plant Sci. 17 (8), 495–502. doi: 10.1016/j.tplants.2012.04.003
Hall, M. D., Yasgar, A., Peryea, T., Braisted, J. C., Jadhav, A., Simeonov, A., et al. (2016). Fluorescence polarization assays in high-throughput screening and drug discovery: a review. Methods Appl. fluorescence 4, 022001. doi: 10.1088/2050-6120/4/2/022001
Han, Y., Ma, L., Zhao, L., Feng, W., Zheng, X. (2018). “Bioinformatics analysis of BAN gene in Arabidopsis thaliana,” in IOP Conference Series: Earth and Environmental Science, vol. 170. (Bristol: IOP Publishing), 052022.
Hayafune, M., Berisio, R., Marchetti, R., Silipo, A., Kayama, M., Desaki, Y., et al. (2014). Chitin-induced activation of immune signaling by the rice receptor CEBiP relies on a unique sandwich-type dimerization. Proc. Natl. Acad. Sci. 111, E404–E413. doi: 10.1073/pnas.1312099111
Hollomon, D. W. (2015). Fungicide resistance: 40 years on and still a major problem. Fungicide Resist. Plant Pathog., 3–11. doi: 10.1007/978-4-431-55642-8_1
Hu, Y., Cheng, Y., Yu, X., Liu, J., Yang, L., Gao, Y., et al. (2021). Overexpression of two CDPKs from wild Chinese grapevine enhances powdery mildew resistance in Vitis vinifera and Arabidopsis. New Phytol. 230, 2029–2046. doi: 10.1111/nph.v230.5
Hu, B., Wu, H., Huang, W., Song, J., Zhou, Y., Lin, Y. (2019). SWEET gene family in Medicago truncatula: genome-wide identification, expression and substrate specificity analysis. Plants 8, 338. doi: 10.3390/plants8090338
Hua, D., Wang, C., He, J., Liao, H., Duan, Y., Zhu, Z., et al. (2012). A plasma membrane receptor kinase, GHR1, mediates abscisic acid-and hydrogen peroxide-regulated stomatal movement in Arabidopsis. Plant Cell 24, 2546–2561. doi: 10.1105/tpc.112.100107
Huang, Y., Xue, C., Bu, R., Wu, C., Li, J., Zhang, J., et al. (2024). Inhibition and transport mechanisms of the ABC transporter hMRP5. Nat. Commun. 15, 4811. doi: 10.1038/s41467-024-49204-1
Imin, N., Patel, N., Corcilius, L., Payne, R. J., Djordjevic, M. A. (2018). CLE peptide tri-arabinosylation and peptide domain sequence composition are essential for SUNN-dependent autoregulation of nodulation in medicago truncatula. New Phytol. 218, 73–80. doi: 10.1111/nph.15019
Jain, P., Arora, D., Bhatla, S. C. (2016). Surface plasmon resonance based recent advances in understanding plant development and related processes. Biochem. Anal. Biochem. 5, 2161–1009. doi: 10.4172/2161-1009.1000300
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. nature 596, 583–589. doi: 10.1038/s41586-021-03819-2
Kang, X., Huang, S., Feng, Y., Fu, R., Tang, F., Zheng, L., et al. (2023). SWEET transporters and their potential roles in response to abiotic and biotic stresses in mulberry. Beverage Plant Res. 3, 1–13. doi: 10.48130/BPR-2023-0006
Kaur, G., Pati, P. K. (2016). Analysis of cis-acting regulatory elements of Respiratory burst oxidase homolog (Rboh) gene families in Arabidopsis and rice provides clues for their diverse functions. Comput. Biol. Chem. 62, 104–118. doi: 10.1016/j.compbiolchem.2016.04.002
Kemmerling, B., Halter, T., Mazzotta, S., Mosher, S., Nurnberger, T. (2011). A genome-wide survey for arabidopsis leucine-rich repeat receptor kinases implicated in plant immunity. Front. Plant Sci. 2, 88. doi: 10.3389/fpls.2011.00088
Kinoshita, A., Betsuyaku, S., Osakabe, Y., Mizuno, S., Nagawa, S., Stahl, Y., et al. (2010). RPK2 is an essential receptor-like kinase that transmits the CLV3 signal in Arabidopsis. Development 137, 3911–3920. doi: 10.1242/dev.048199
Ko, H. Y., Tseng, H. W., Ho, L. H., Wang, L., Chang, T. F., Lin, A., et al. (2022). Hexose translocation mediated by Sl SWEET5b is required for pollen maturation in Solanum lycopersicum. Plant Physiol. 189, 344–359. doi: 10.1093/plphys/kiac057
Kraffe, E., Marty, Y., Guderley, H. (2007). Changes in mitochondrial oxidative capacities during thermal acclimation of rainbow trout Oncorhynchus mykiss: roles of membrane proteins, phospholipids and their fatty acid compositions. J. Exp. Biol. 210, 149–165. doi: 10.1242/jeb.02628
Kumara, U. A., Cooray, P. L. V. N., Ambanpola, N., Thiruchchelvan, N. (2022). “Plant-pathogen interaction: Mechanisms and evolution,” in Trends of Applied Microbiology for Sustainable Economy (United Kingdom: Academic Press), 655–687.
Langhans, M., Weber, W., Babel, L., Grunewald, M., Meckel, T. (2017). The right motifs for plant cell adhesion: what makes an adhesive site? Protoplasma 254, 95–108. doi: 10.1007/s00709-016-0970-2
Li, Q., Qi, J., Qin, X., Hu, A., Fu, Y., Chen, S., et al. (2021). Systematic identification of lysin-motif receptor-like kinases (LYKs) in Citrus sinensis, and analysis of their inducible involvements in citrus bacterial canker and phytohormone signaling. Sci. Hortic. 276, 109755. doi: 10.1016/j.scienta.2020.109755
Li, J., Wen, J., Lease, K. A., Doke, J. T., Tax, F. E., Walker, J. C. (2002). BAK1, an Arabidopsis LRR receptor-like protein kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell 110, 213–222. doi: 10.1016/S0092-8674(02)00812-7
Li, D., Wu, D., Li, S., Dai, Y., Cao, Y. (2019). Evolutionary and functional analysis of the plant-specific NADPH oxidase gene family in Brassica rapa L. R. Soc. Open Sci. 6, 181727. doi: 10.1098/rsos.181727
Lin, Z., Akin, H., Rao, R., Hie, B., Zhu, Z., Lu, W., et al. (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130. doi: 10.1126/science.ade2574
Liu, C., Cheng, F., Sun, Y., Ma, H., Yang, X. (2016). Structure–function relationship of a novel pr-5 protein with antimicrobial activity from soy hulls. J. Agric. Food Chem. 64, 948–959. doi: 10.1021/acs.jafc.5b04771
Liu, J., Last, R. L. (2015). A land plant-specific thylakoid membrane protein contributes to photosystem II maintenance in arabidopsis thaliana. Plant J. 82, 731–743. doi: 10.1111/tpj.12845
Liu, J., Lu, H., Wan, Q., Qi, W., Shao, H. (2019). Genome-wide analysis and expression profiling of respiratory burst oxidase homologue gene family in Glycine max. Environ. Exp. Bot. 161, 344–356. doi: 10.1016/j.envexpbot.2018.07.015
Liu, L. L., Qu, J. J., Guo, Z. X., Sun, D. Y., Pan, F. Y., Yin, L. (2020). Construction of a yeast two-hybrid cDNA library from Vitis vinifera leaves infected by downy mildew. J. South. Agric. 51, 829–835. doi: 10.3969/j.issn.2095-1191.2020.04.013
Mahalingam, R., Graham, D., Walling, J. G. (2021). The barley (Hordeum vulgare ssp. vulgare) respiratory burst oxidase homolog (HvRBOH) gene family and their plausible role on malting quality. Front. Plant Sci. 12, 608541. doi: 10.3389/fpls.2021.608541
Majoul, I. (2004). Analysing the action of bacterial toxins in living cells with fluorescence resonance energy transfer (FRET). Int. J. Med. Microbiol. 293, 495–503. doi: 10.1078/1438-4221-00307
Manck-Götzenberger, J., Requena, N. (2016). Arbuscular mycorrhiza symbiosis induces a major transcriptional reprogramming of the potato SWEET sugar transporter family. Front. Plant Sci. 7, 191086. doi: 10.3389/fpls.2016.00487
Mansoor, S., Ali Wani, O., Lone, J. K., Manhas, S., Kour, N., Alam, P., et al. (2022). Reactive oxygen species in plants: from source to sink. Antioxidants. 11 (2), 225. doi: 10.3390/antiox11020225
Marino, D., Andrio, E., Danchin, E. G., Oger, E., Gucciardo, S., Lambert, A., et al. (2011). A Medicago truncatula NADPH oxidase is involved in symbiotic nodule functioning. New Phytol. 189, 580–592. doi: 10.1111/j.1469-8137.2010.03509.x
Martin, H., Murray, C., Christeller, J., McGhie, T. (2008). A fluorescence polarization assay to quantify biotin and biotin-binding proteins in whole plant extracts using Alexa-Fluor 594 biocytin. Anal. Biochem. 381, 107–112. doi: 10.1016/j.ab.2008.06.025
Martínez-Navarro, S. M., de Iceta Soler, X., Martínez-Martínez, M., Olazábal-Morán, M., Santos-Moriano, P., Gómez, S. (2024). Structural and phylogenetic in silico characterization of vitis vinifera PRR protein as potential target for plasmopara viticola infection. Int. J. Mol. Sci. 25, 9553. doi: 10.3390/ijms25179553
Mitchell, A. L., Addy, P. S., Chin, M. A., Chatterjee, A. (2017). A unique genetically encoded FRET pair in mammalian cells. ChemBioChem 18, 511–514. doi: 10.1002/cbic.201600668
Mittler, R. (2017). ROS are good. Trends Plant Sci. 22 (1), 11–19. doi: 10.1016/j.tplants.2016.08.002
Miya, A., Albert, P., Shinya, T., Desaki, Y., Ichimura, K., Shirasu, K., et al. (2007). CERK1, a LysM receptor kinase, is essential for chitin elicitor signaling in A. Proc. Natl. Acad. Sci. 104, 19613–19618. doi: 10.1073/pnas.0705147104
Mohan, B., Thingujam, D., Pajerowska-Mukhtar, K. M. (2023). “Cytotrap: an innovative approach for protein–protein interaction studies for cytoplasmic proteins,” in Protein-Protein Interactions: Methods and Protocols (Springer US, New York, NY), 9–22.
Mohd-Radzman, N. A., Binos, S., Truong, T. T., Imin, N., Mariani, M., Djordjevic, M. A. (2015). Novel MtCEP1 peptides produced in vivo differentially regulate root development in medicago truncatula. J. Exp. Bot. 66, 5289–5300. doi: 10.1093/jxb/erv008
Möller, S., Croning, M. D., Apweiler, R. (2001). Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 17, 646–653. doi: 10.1093/bioinformatics/17.7.646
Morell, M., Espargaro, A., Aviles, F. X., Ventura, S. (2008). Study and selection of in vivo protein interactions by coupling bimolecular fluorescence complementation and flow cytometry. Nat. Protoc. 3, 22–33. doi: 10.1038/nprot.2007.496
Muller, R., Bleckmann, A., Simon, R. (2008). The receptor kinase CORYNE of Arabidopsis transmits the stem cell–limiting signal CLAVATA3 independently of CLAVATA1. Plant Cell 20, 934–946. doi: 10.1105/tpc.107.057547
Nakao, H., Sugimoto, Y., Ikeda, K., Saito, H., Nakano, M. (2020). Structural feature of lipid scrambling model transmembrane peptides: Same-side positioning of hydrophilic residues and their deeper position. J. Phys. Chem. Lett. 11, 1662–1667. doi: 10.1021/acs.jpclett.0c00175
Navathe, S., Singh, S., Singh, V. K., Chand, R., Mishra, V. K., Joshi, A. K. (2019). Genome-wide mining of respiratory burst homologs and its expression in response to biotic and abiotic stresses in triticum aestivum. Genes Genom. 41, 1027–1043. doi: 10.1007/s13258-019-00821-x
Nazarian-Firouzabadi, F., Joshi, S., Xue, H., Kushalappa, A. C. (2019). Genome-wide in silico identification of LysM-RLK genes in potato (Solanum tuberosum L.). Mol. Biol. Rep. 46, 5005–5017. doi: 10.1007/s11033-019-04951-z
Nguyen, P. T., Harris, B. J., Mateos, D. L., González, A. H., Murray, A. M., Yarov-Yarovoy, V. (2024). Structural modeling of ion channels using AlphaFold2, RoseTTAFold2, and ESMFold. Channels 18, 2325032. doi: 10.1080/19336950.2024.2325032
Nji, E., Gulati, A., Qureshi, A. A., Coincon, M., Drew, D. (2019). Structural basis for the delivery of activated sialic acid into Golgi for sialyation. Nat. Struct. Mol. Biol. 26, 415–423. doi: 10.1038/s41594-019-0225-y
Obrdlik, P., El-Bakkoury, M., Hamacher, T., Cappellaro, C., Vilarino, C., Fleischer, C., et al. (2004). K+ channel interactions detected by a genetic system optimized for systematic studies of membrane protein interactions. Proc. Natl. Acad. Sci. 101, 12242–12247. doi: 10.1073/pnas.0404467101
Okumura, M., Inoue, S., Kuwata, K., Kinoshita, T. (2016). Photosynthesis activates plasma membrane h+-ATPase via sugar accumulation. Plant Physiol. 171, 580–589. doi: 10.1104/pp.16.00355
Osakabe, Y., Maruyama, K., Seki, M., Satou, M., Shinozaki, K., Yamaguchi-Shinozaki, K. (2005). Leucine-rich repeat receptor-like Kinase1 is a key membrane-bound regulator of abscisic acid early signaling in arabidopsis. Plant Cell. 17, 1105–1119. doi: 10.1105/tpc.104.027474
Paiano, A., Margiotta, A., De Luca, M., Bucci, C. (2018). Yeast two-hybrid assay to identify interacting proteins. Curr. Protoc. Protein Sci. 95, e70. doi: 10.1002/cpps.70
Pan, Y., Niu, K., Miao, P., Zhao, G., Ju, Z., Chai, J., et al. (2024). Genome-wide analysis of the SWEET gene family and its response to powdery mildew and leaf spot infection in the common oat (Avena sativa L.). BMG Geno. 25, 1–13. doi: 10.1186/s12864-024-10933-8
Patching, S. G. (2014). Surface plasmon resonance spectroscopy for characterisation of membrane protein–ligand interactions and its potential for drug discovery. Biochim. Biophys. Acta (BBA)-Biomembr. 1838, 43–55. doi: 10.1016/j.bbamem.2013.04.028
Patil, G., Valliyodan, B., Deshmukh, R., Prince, S., Nicander, B., Zhao, M., et al. (2015). Soybean (Glycine max) SWEET gene family: insights through comparative genomics, transcriptome profiling and whole genome re-sequence analysis. BMC Genomics 16, 1–16. doi: 10.1186/s12864-015-1730-y
Peiró, A., Martínez-Gil, L., Tamborero, S., Pallás, V., Sánchez-Navarro, J. A., Mingarro, I. (2014). The Tobacco mosaic virus movement protein associates with but does not integrate into biological membranes. J. Virol. 88, 3016–3026. doi: 10.1128/JVI.03648-13
Pellizzaro, A., Clochard, T., Planchet, E., Limami, A. M., Morère-Le Paven, M. C. (2015). Identification and molecular characterization of medicago truncatula NRT2 and NAR2 families. Physiologia Plantarum. (2), 256–269. doi: 10.1111/ppl.12314
Periasamy, A. (2001). Fluorescence resonance energy transfer microscopy: a mini review. J. Biomed. optics 6, 287–291. doi: 10.1117/1.1383063
Pettersson, P., Ye, W., Jakob, M., Tannert, F., Klösgen, R. B., Mäler, L. (2018). Structure and dynamics of plant TatA in micelles and lipid bilayers studied by solution NMR. FEBS J. 285, 1886–1906. doi: 10.1111/febs.2018.285.issue-10
Pierce, B. G., Wiehe, K., Hwang, H., Kim, B. H., Vreven, T., Weng, Z. (2014). ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773. doi: 10.1093/bioinformatics/btu097
Qin, J. X., Jiang, Y. J., Lu, Y. Z., Peng, Z. H. A. O., Wu, B. J., Li, H. X., et al. (2020). Genome-wide identification and transcriptome profiling reveal great expansion of SWEET gene family and their wide-spread responses to abiotic stress in wheat (Triticum aestivum L.). J. Integr. Agric. 19, 1704–1720. doi: 10.1016/S2095-3119(19)62761-9
Rivas, F. J. M., Fernie, A. R., Aarabi, F. (2023). Roles and regulation of the RBOHD enzyme in initiating ROS-mediated systemic signaling during biotic and abiotic stress. Plant Stress 11, 100327. doi: 10.1016/j.stress.2023.100327
Roel-Touris, J., Jiménez-García, B., Bonvin, A. M. (2020). Integrative modeling of membrane-associated protein assemblies. Nat. Commun. 11, 6210. doi: 10.1038/s41467-020-20076-5
Roudaire, T., Marzari, T., Landry, D., Löffelhardt, B., Gust, A. A., Jermakow, A., et al. (2023). The grapevine LysM receptor-like kinase VvLYK5-1 recognizes chitin oligomers through its association with VvLYK1-1. Front. Plant Sci. 14, 1130782. doi: 10.3389/fpls.2023.1130782
Rudden, L. S., Degiacomi, M. T. (2021). Transmembrane protein docking with jabberdock. J. Chem. Inf. Model. 61, 1493–1499. doi: 10.1021/acs.jcim.0c01315
Ruman, H., Kawaharada, Y. (2023). A new classification of Lysin motif receptor-like kinases in Lotus japonicus. Plant Cell Physiol. 64, 176–190. doi: 10.1093/pcp/pcac156
Sagi, M., Fluhr, R. (2006). Production of reactive oxygen species by plant NADPH oxidases. Plant Physiol. 141, 336–340. doi: 10.1104/pp.106.078089
Schwessinger, B., Roux, M., Kadota, Y., Ntoukakis, V., Sklenar, J., Jones, A., et al. (2011). Phosphorylation-dependent differential regulation of plant growth, cell death, and innate immunity by the regulatory receptor-like kinase BAK1. PloS Genet. 7, e1002046. doi: 10.1371/journal.pgen.1002046
Shimizu, T., Nakano, T., Takamizawa, D., Desaki, Y., Ishii-Minami, N., Nishizawa, Y., et al. (2010). Two LysM receptor molecules, CEBiP and OsCERK1, cooperatively regulate chitin elicitor signaling in rice. Plant J. 64, 204–214. doi: 10.1111/j.1365-313X.2010.04324.x
Shiu, S. H., Bleecker, A. B. (2001). Receptor-like kinases from A. form a monophyletic gene family related to animal receptor kinases. Proc. Natl. Acad. Sci. United States America 98, 10763–10768. doi: 10.1073/pnas.181141598
Skruzny, M., Pohl, E., Abella, M. (2019). FRET microscopy in yeast. Biosensors 9, 122. doi: 10.3390/bios9040122
Smirnova, E. V., Rakitina, T. V., Saratov, G. A., Kudriaeva, A. A., Belogurov, A. A., Jr (2022). Deconvolution of the MBP-bri2 interaction by a yeast two hybrid system and synergy of the alphaFold2 and high ambiguity driven protein-protein docking. Crystals 12, 197. doi: 10.3390/cryst12020197
Song, W., Wang, B., Li, X., Wei, J., Chen, L., Zhang, D., et al. (2015). Identification of immune related LRR-containing genes in maize (Zea mays L.) by genome-wide sequence analysis. Int. J. Genom. 2015, 231358. doi: 10.1155/2015/231358
Song, W. Y., Wang, G. L., Chen, L. L., Kim, H. S., Pi, L. Y., Holsten, T., et al. (1995). A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science 270, 1804–1806. doi: 10.1126/science.270.5243.1804
Sung, M. K., Huh, W. K. (2007). Bimolecular fluorescence complementation analysis system for in vivo detection of protein–protein interaction in Saccharomyces cerevisiae. Yeast 24, 767–775. doi: 10.1002/yea.v24:9
Tak, H., Mhatre, M. (2013). Molecular characterization of VvSDIR1 from Vitis vinifera and its functional analysis by heterologous expression in Nicotiana tabacum. Protoplasma 250, 565–576. doi: 10.1007/s00709-012-0442-2
Takaoka, Y., Nagumo, K., Azizah, I. N., Oura, S., Iwahashi, M., Kato, N., et al. (2019). A comprehensive in vitro fluorescence anisotropy assay system for screening ligands of the jasmonate COI1-JAZ co-receptor in plants. J. Biol. Chem. 294, 5074–5081. doi: 10.1074/jbc.RA118.006639
Tanaka, Y., Iwaki, S., Tsukazaki, T. (2017). Crystal structure of a plant multidrug and toxic compound extrusion family protein. Structure 25, 1455–1460. doi: 10.1016/j.str.2017.07.009
Tanaka, K., Nguyen, C. T., Liang, Y., Cao, Y., Stacey, G. (2013). Role of LysM receptors in chitin-triggered plant innate immunity. Plant Signaling Behav. 8, e22598. doi: 10.4161/psb.22598
Tate, C. G. (2010). Practical considerations of membrane protein instability during purification and crystallisation. Heterol. Expression Membrane Proteins: Methods Protoc. 601, 187–203. doi: 10.1007/978-1-60761-344-2_12
ten Hove, C. A., Bochdanovits, Z., Jansweijer, V. M. A., Koning, F. G., Berke, L., Sanchez-Perez, G. F., et al. (2011). Probing the roles of LRR RLK genes in arabidopsis thaliana roots using a custom T-DNA insertion set. Plant Mol. Biol. 76, 69–83. doi: 10.1007/s11103-011-9769-x
Thaminy, S., Miller, J., Stagljar, I. (2004). The split-ubiquitin membrane-based yeast two-hybrid system. Protein-Protein Interactions: Methods Appl. 261, 297–312. doi: 10.1385/1-59259-762-9:297
Thoma, J., Manioglu, S., Kalbermatter, D., Bosshart, P. D., Fotiadis, D., Müller, D. J. (2018). Protein-enriched outer membrane vesicles as a native platform for outer membrane protein studies. Commun. Biol. 1, 23. doi: 10.1038/s42003-018-0027-5
Tombuloglu, G., Tombuloglu, H., Cevik, E., Sabit, H. (2019). Genome-wide identification of Lysin-Motif Receptor-Like Kinase (LysM-RLK) gene family in Brachypodium distachyon and docking analysis of chitin/LYK binding. Physiol. Mol. Plant Pathol. 106, 217–225. doi: 10.1016/j.pmpp.2019.03.002
Torres, M. A., Dangl, J. L., Jones, J. D. (2002). Arabidopsis gp91phox homologues AtrbohD and AtrbohF are required for accumulation of reactive oxygen intermediates in the plant defense response. Proc. Natl. Acad. Sci. 99, 517–522. doi: 10.1073/pnas.012452499
Tovchigrechko, A., Vakser, I. A. (2006). GRAMM-X public web server for protein–protein docking. Nucleic Acids Res. 34, W310–W314. doi: 10.1093/nar/gkl206
Tsirigos, K. D., Govindarajan, S., Bassot, C., Västermark, Å., Lamb, J., Shu, N., et al. (2018). Topology of membrane proteins—predictions, limitations and variations. Curr. Opin. Struct. Biol. 50, 9–17. doi: 10.1016/j.sbi.2017.10.003
Tusnády, G. E., Dosztányi, Z., Simon, I. (2004). Transmembrane proteins in the Protein Data Bank: identification and classification. Bioinformatics 20, 2964–2972. doi: 10.1093/bioinformatics/bth340
Utsugi, S., Shibasaka, M., Maekawa, M., Katsuhara, M. (2015). Control of the water transport activity of barley HvTIP3; 1 specifically expressed in seeds. Plant Cell Physiol. 56, 1831–1840. doi: 10.1093/pcp/pcv104
Vakser, I. A. (2014). Protein-protein docking: From interaction to interactome. Biophys. J. 107, 1785–1793. doi: 10.1016/j.bpj.2014.08.033
van der Knaap, E., Song, W. Y., Ruan, D. L., Sauter, M., Ronald, P. C., Kende, H. (1999). Expression of a gibberellin-induced leucine-rich repeat receptor-like protein kinase in deepwater rice and its interaction with kinase-associated protein phosphatase. Plant Physiol. 120, 559–570. doi: 10.1104/pp.120.2.559
Van Zundert, G. C. P., Rodrigues, J. P. G. L. M., Trellet, M., Schmitz, C., Kastritis, P. L., Karaca, E., et al. (2016). The HADDOCK2. 2 web servers: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 428, 720–725. doi: 10.1016/j.jmb.2015.09.014
Vitart, V., Baxter, I., Doerner, P., Harper, J. F. (2001). Evidence for a role in growth and salt resistance of a plasma membrane H+-ATPase in the root endodermis. Plant J. 27, 191–201. doi: 10.1046/j.1365-313x.2001.01081.x
Von Heijne, G. (1992). Membrane protein structure prediction: hydrophobicity analysis and the positive-inside rule. J. Mol. Biol. 225, 487–494. doi: 10.1016/0022-2836(92)90934-C
Voss, M., Cseke, L. J., Gassmann, W., Niefind, K. (2023). A splicing variant of EDS1 from Vitis vinifera forms homodimers but no heterodimers with PAD4. Protein Sci. 32, e4624. doi: 10.1002/pro.v32.4
Wan, J., Zhang, X. C., Neece, D., Ramonell, K. M., Clough, S., Kim, S. Y., et al. (2008). A LysM receptor-like kinase plays a critical role in chitin signaling and fungal resistance in A. Plant Cell 20, 471–481. doi: 10.1105/tpc.107.056754
Wang, H. W. (2022). A commentary of “Cryo-EM achieves atomic resolution” in 10 remarkable discoveries from 2020 in Nature. Fundam. Res. 2, 349–350. doi: 10.1016/j.fmre.2022.01.014
Wang, W., Chen, D., Zhang, X., Liu, D., Cheng, Y., Shen, F. (2018). Role of plant respiratory burst oxidase homologs in stress responses. Free Radical Res. 52, 826–839. doi: 10.1080/10715762.2018.1473572
Wang, L., Xue, J., Yan, J., Liu, M., Tang, Y., Wang, Y., et al. (2020). Expression and functional analysis of VviABCG14 from Vitis vinifera suggest the role in cytokinin transport and the interaction with VviABCG7. Plant Physiol. Biochem. 153, 1–10. doi: 10.1016/j.plaphy.2020.05.011
Willmann, R., Lajunen, H. M., Erbs, G., Newman, M. A., Kolb, D., Tsuda, K., et al. (2011). Arabidopsis lysin-motif proteins LYM1 LYM3 CERK1 mediate bacterial peptidoglycan sensing and immunity to bacterial infection. Proc. Natl. Acad. Sci. 108, 19824–19829. doi: 10.1073/pnas.1112862108
Xing, S., Wallmeroth, N., Berendzen, K. W., Grefen, C. (2016). Techniques for the analysis of protein-protein interactions in vivo. Plant Physiol. 171, 727–758. doi: 10.1104/pp.16.00470
Xu, Y., Long, D., Yang, D. (2007). Rapid data collection for protein structure determination by NMR spectroscopy. J. Am. Chem. Soc. 129, 7722–7723. doi: 10.1021/ja071442e
Xue, D. X., Li, C. L., Xie, Z. P., Staehelin, C. (2019). LYK4 is a component of a tripartite chitin receptor complex in A. thaliana. J. Exp. Bot. 70, 5507–5516. doi: 10.1093/jxb/erz313
Yamada, K., Yamaguchi, K., Yoshimura, S., Terauchi, A., Kawasaki, T. (2017). Conservation of chitin-induced MAPK signaling pathways in rice and A. Plant Cell Physiol. 58, 993–1002. doi: 10.1093/pcp/pcx042
Yamaguchi, Y., Pearce, G., Ryan, C. A. (2006). The cell surface leucine-rich repeat receptor for At Pep1, an endogenous peptide elicitor in Arabidopsis, is functional in transgenic tobacco cells. Proceedings of the National Academy of Sciences 103 (26), 10104–10109.
Yan, Y., Tao, H., He, J., Huang, S. Y. (2020). The HDOCK server for integrated protein–protein docking. Nat. Protoc. 15, 1829–1852. doi: 10.1038/s41596-020-0312-x
Yang, H., Bayer, P. E., Tirnaz, S., Edwards, D., Batley, J. (2020). Genome-wide identification and evolution of receptor-like kinases (RLKs) and receptor like proteins (RLPs) in Brassica juncea. Biology 10, 17. doi: 10.3390/biology10010017
Yao, K., Wang, Y., Li, X., Ji, H. (2023). Genome-wide identification of the soybean LysM-RLK family genes and its nitrogen response. Int. J. Mol. Sci. 24, 13621. doi: 10.3390/ijms241713621
Yeh, V., Goode, A., Bonev, B. B. (2020). Membrane protein structure determination and characterisation by solution and solid-state nmr. Biology 9, 396. doi: 10.3390/biology9110396
Yin, T., Han, P., Xi, D., Yu, W., Zhu, L., Du, C., et al. (2023). Genome-wide identification, characterization, and expression profile of NBS-LRR gene family in sweet orange (Citrus sinensis). Gene 854, 147117. doi: 10.1016/j.gene.2022.147117
Ying, W., Wang, Y., Wei, H., et al. (2024). Structure and function of the Arabidopsis ABC transporter ABCB19 in brassinosteroid export. Science 383, eadj4591. doi: 10.1126/science.adj4591
Yuan, M., Ngou, B. P. M., Ding, P., Xin, X. F. (2021). PTI-ETI crosstalk: an integrative view of plant immunity. Curr. Opin. Plant Biol. 62, 102030. doi: 10.1016/j.pbi.2021.102030
Yuan, M., Wang, S. (2013). Rice MtN3/saliva/SWEET family genes and their homologs in cellular organisms. Mol. Plant 6, 665–674. doi: 10.1093/mp/sst035
Zeng, L., Velásquez, A. C., Munkvold, K. R., Zhang, J., Martin, G. B. (2012). A tomato LysM receptor-like kinase promotes immunity, and its kinase activity is inhibited by AvrPtoB. Plant J. 69, 92–103. doi: 10.1111/j.1365-313X.2011.04773.x
Zhang, X. C., Cannon, S. B., Stacey, G. (2009). Evolutionary genomics of LysM genes in land plants. BMC Evol. Biol. 9, 1–13. doi: 10.1186/1471-2148-9-183
Zhang, H., Fang, Q., Zhang, Z., Wang, Y., Zheng, X. (2009). The role of respiratory burst oxidase homologues in elicitor-induced stomatal closure and hypersensitive response in Nicotiana benthamiana. J. Exp. Bot. 60, 3109–3122. doi: 10.1093/jxb/erp146
Zhang, H., Wang, X., Yan, A., Deng, J., Xie, Y., Liu, S., et al. (2023). Evolutionary analysis of respiratory burst oxidase homolog (RBOH) genes in plants and characterization of ZmRBOHs. Int. J. Mol. Sci. 24, 3858. doi: 10.3390/ijms24043858
Zhang, J., Xie, Y., Ali, B., Ahmed, W., Tang, Y., Li, H. (2021). Genome-wide identification, classification, evolutionary expansion and expression of RbOH family genes in pepper (Capsicum annuum L.). Trop. Plant Biol. 14, 251–266. doi: 10.1007/s12042-021-09286-3
Zhong, L., Xu, S., Song, C., Zhao, N., Yang, Z., Liu, Y., et al. (2024). Genome-wide identification, characterization, and expression profile of SWEETs gene family in grapevine (Vitis vinifera L.). Horticulturae 10, 428. doi: 10.3390/horticulturae10050428
Zhou, Y., Wang, B., Yuan, F. (2022). The role of transmembrane proteins in plant growth, development, and stress responses. Int. J. Mol. Sci. 23, 13627. doi: 10.3390/ijms232113627
Zhu, Y., Gong, H. (2014). Beneficial effects of silicon on salt and drought tolerance in plants. Agron. Sustain. Dev. 34, 455–472. doi: 10.1007/s13593-013-0194-1
Zhu, Y. X., Gong, H. J., Yin, J. L. (2019). Role of silicon in mediating salt tolerance in plants: a review. Plants 8, 147. doi: 10.3390/plants8060147
Zhu, J., Zhou, L., Li, T., Ruan, Y., Zhang, A., Dong, X., et al. (2022). Genome-wide investigation and characterization of SWEET gene family with focus on their evolution and expression during hormone and abiotic stress response in maize. Genes 13, 1682. doi: 10.3390/genes13101682
Zimmermann, P., Hirsch-Hoffmann, M., Hennig, L., Gruissem, W. (2004). GENEVESTIGATOR. Arabidopsis microarray database and analysis toolbox. Plant Physiol. 136, 2621–2632. doi: 10.1104/pp.104.046367
Zipfel, C., Kunze, G., Chinchilla, D., Caniard, A., Jones, J. D., Boller, T., et al. (2006). Perception of the bacterial PAMP EF-Tu by the receptor EFR restricts Agrobacterium-mediated transformation. Cell 125, 749–760. doi: 10.1016/j.cell.2006.03.037
Keywords: Vitis, receptor, LysM-RLKs, SWEETs, RBOH, NMR, FRET, topology
Citation: Gallucci A, Giordano D, Facchiano A, Villano C, Carputo D and Aversano R (2024) Transmembrane proteins in grape immunity: current knowledge and methodological advances. Front. Plant Sci. 15:1515163. doi: 10.3389/fpls.2024.1515163
Received: 22 October 2024; Accepted: 30 November 2024;
Published: 20 December 2024.
Edited by:
Gerald Alan Berkowitz, University of Connecticut, United StatesReviewed by:
Rucha Karnik, University of Glasgow, United KingdomCopyright © 2024 Gallucci, Giordano, Facchiano, Villano, Carputo and Aversano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clizia Villano, Y2xpemlhLnZpbGxhbm9AdW5pbmEuaXQ=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.