 Sehrish Akbar1
Sehrish Akbar1 Xuiting Hua2
Xuiting Hua2 Yingying Zhang1Gang Liu1
Yingying Zhang1Gang Liu1 Tianyou Wang2Huihong Shi1Zhen Li2Yiying Qi1
Tianyou Wang2Huihong Shi1Zhen Li2Yiying Qi1 Habiba Habiba3
Habiba Habiba3 Wei Yao2Mu-Qing Zhang2
Wei Yao2Mu-Qing Zhang2 Jisen Zhang2*†
Jisen Zhang2*†- 1Center for Genomics and Biotechnology, National Sugarcane Engineering Technology Research Center, Fujian Provincial Key Laboratory of Haixia Applied Plant Systems Biology, College of Agriculture, Fujian Agriculture and Forestry University, Fuzhou, China
- 2State Key Laboratory for Conservation and Utilization of Subtropical Agro-bioresources, Guangxi Key Laboratory of Sugarcane Biology, Guangxi University, Guangxi, China
- 3Department of Biological Science, Lehman College, City University of New York, Bronx, NY, United States
Sugar, the primary product of photosynthesis, is a vital requirement for cell activities. Allocation of sugar from source to sink tissues is facilitated by sugar transporters (ST). These STs belong to the Major Facilitator Superfamily (MFS), the largest family of STs in plants. In this study, we performed genome wide and gene expression data analysis to identify the putative ST genes in Erianthus rufipilus (E. rufipilus) and in Saccharum officinarum (S. officinarum). We identified 78 ST gene families in E. rufipilus and 86 ST gene families in S. officinarum. Phylogenetic analysis distributed the ST genes into eight distinct subfamilies (INT, MST, VGT, pGlcT, PLT, STP, SFP and SUT). Chromosomal distribution of ST genes clustered them on 10 respective chromosomes. Furthermore, synteny analysis with S. spontaneum and Sorghum bicolor (S. bicolor) revealed highly colinear regions. Synonymous and non-synonymous ratio (Ka/Ks) showed purifying selection in gene evolution. Promoter analysis identified several cis-regulatory elements, mainly associated with light responsiveness. We also examined the expression pattern of ST genes in different developing tissues (mature leaf, pre-mature stem, mature stem and seedling stem). Under sugar stress, we identified the significant ST genes showing differential expression patterns. Moreover, our yeast one-hybrid (Y1H) assays identified NAM, ATAF and CUC (NAC) and Lesion Simulating Disease (LSD) potential transcription factors (TFs) that may bind to the SUT1-T1 promoter in S. officinarum, showing negative correlation pattern with SUT1-T1. Our results deepen our understanding of ST gene evolution in Saccharum species and will facilitate the future investigation of functional analysis of the ST gene family.
Introduction
Sugarcane, from the genus Saccharum within the tribe Andropogoneae, stands as the world’s leading crop, thriving in both tropical and temperate zones (D’Hont, 2005; Mulyono, 2016). The genus encompasses around 90 genera and about 1270 species, showcasing a wide geographical spread (Kellogg, 2015). Among these, six species (Saccharum robustum, S. spontaneum, S. officinarum, Saccharum sinense, Saccharum barberi, and Saccharum edule) are recognized within the Saccharum L. genus, which is further categorized into two major clades: Saccharum S. str. and Erianthus, with the former including only Old-World taxa (Mukherjee, 1957; Daniels et al., 1987; Besse et al., 1997, 1998).
S. officinarum, evolving from S. robustum, is noted for its high sugar content and substantial biomass, characterized by a basic chromosome number of 2n = 8x = 80 (Lu et al., 1994; D’Hont et al., 1996; Schenck et al., 2004). Historically, the Saccharum genus was considered to include only polyploid species (Wang et al., 2023). However, E. rufipilus, a diploid species within the genus, demonstrates significant cold, drought, and disease resistance, making it valuable for sugarcane breeding through interspecific hybridization (Wang et al., 2010, 2023).
Sucrose serves as a main photosynthetic product in sugarcane and is the primary sugar transported from source to sink and storage tissues. The synthesis, transportation, and metabolism of sucrose involve a network of enzymes [such as Sucrose Phosphate Synthase (SPS), Sucrose Synthase (SuSy), invertases], sugar transporters (STs), transcription factor (TFs), protein kinases, and hormones (Khan et al., 2023). STs have a vital role in coordinating carbon efflux, facilitating the transport of sugars from photosynthetic organs (source) to storage organs (sink) (Lemoine, 2000). These STs are found in various plant species and consist of different gene families such as monosaccharide transporters (MSTs) and sucrose transporters (SUTs); the sugars will eventually be exported transporters (SWEET) and sucrose carriers (SUCs) (Slewinski et al., 2009; Lin et al., 2014). Additionally, TFs like AP2/ERF, NAC, GRF, and bZIP are known to play a significant role in sucrose synthesis and transport (Ma et al., 2019; Stein and Granot, 2019; Wang et al., 2022).
Research has identified ST gene families across various species, including dicots and monocots like Arabidopsis (Arabidopsis thaliana) (Büttner, 2007; Sauer, 2007), rice (Oryza sativa) (Aoki et al., 2003), tomato (Solanum lycopersicum) (Reuscher et al., 2014), pear (Pyrus) (Li et al., 2015), and grapes (Vitis vinifera) (Afoufa-Bastien et al., 2010). The Saccharum spontaneum genome has revealed the presence of the ST gene family, associated with the MFS, which plays a pivotal role in sugar transport (Zhang et al., 2018, 2021). The SUT and MST transporter families, powered by H+ ATPase pumps, regulate carbon allocation within plants, significantly impacting crop yield and nutritional value (Eom et al., 2015; Jeena et al., 2019).
This study marks the first identification of ST genes in E. rufipilus (diploid) and S. officinarum (polyploid) genomes, employing a bioinformatics approach to explore their physiochemical properties, chromosomal distribution, and evolutionary relationships. By analyzing the TFs that regulate the SUT1-T1 gene in S. officinarum using Y1H assay, we identify potential TFs for improving sugarcane breeding. Additionally, investigating ST genes in E. rufipilus offers fundamental insights into gene evolution and the genetic framework of sugar transport and allocation, underscoring the complexity and potential within sugarcane genetics for agricultural advancement.
Materials and methods
Plant materials
Saccharum species, E. rufipilus and S. officinarum, studied as an experimental material. While, two species from the Andropogonae tribe, S. spontaneum and S. bicolor, are used as a reference species.
Identification of ST protein in E. rufipilus and S. officinarum
ST genes in E. rufipilus and S. officinarum were initially identified by performing BLASTP (with a cutoff e-value 1e−5) search with already reported ST genes from S. spontaneum (Zhang et al., 2018). Distinguished ST genes were further annotated using the CDD batch search (Marchler-Bauer et al., 2015). Then, the candidate ST genes were confirmed through the PFAM (with e value <1e−5) database (Mistry et al., 2021), using HMMER software v3.2.1.
Evolutionary relationship of ST gene family
ST gene sequences of E. rufipilus and S. officinarum were aligned with S. spontaneum and S. bicolor ST gene sequences using the maximum likelihood (ML) method. The ML tree was generated using MEGA, version 7.0, with a bootstrap value of 1,000 replicates and a “Poisson correction” model (Tamura et al., 2021). The results were then visualized in the interactive tree of life (iTOL) program for generating a phylogenetic tree (Letunic and Bork, 2019).
Physical properties, conserved motifs, and gene structure analysis
To analyze the conserved motifs among all the ST genes of E. rufipilus and S. officinarum, protein sequences were submitted to the online MEME (motif-based sequence analysis tools) suite 4.11.1 program (http://meme.nbcr.net/mem/cgi-bin/mem.cgi) (Bailey et al., 2015). The following parameters were adjusted: maximum number of motifs 20, minimum and maximum length between 15 and 60, number of repetitions, any. The results were then visualized in the TBtools program (Chen et al., 2018). Gene structures were identified using DNA sequences and exons of ST genes displayed using the Gene Structure Display Server (GSDS) (Hu et al., 2015). Molecular weight (MW) and isoelectric point (pI) of each ST gene were determined using the ExPASy online tool (http://web.expasy.org/compute_pi/). Subcellular localization of ST proteins was predicted through WoLF PSORT (http://wolfpsort.hgc.jp) (Horton et al., 2007). The transmembrane domain from the amino acid sequences of ST proteins were predicted using TMHMM Server v.2.0 (http://www.cbs.dtu.dk/services/TMHMM/).
Identification of cis-regulatory elements in the promoter region
The promoter region upstream of 2500 bp of transcription start site of each ST gene was extracted using TBtools. cis-regulatory elements (CRE) located in ST promoters were predicted using PlantCARE online database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/). The identified cis-regulatory elements were analyzed using the Simple Biosequence viewer function on TBtools.
Duplication and chromosomal location of ST genes
ST gene positions on chromosomes were detected from the General Feature Format Files (GFF3), and karyotyping was executed in TBtools (Chen et al., 2018). The duplication pattern for ST genes and protein-coding genes was investigated by an all-vs-all local BLAST with an E-value <1e−5. BLAST results were imported into MCScanX (v0.8) software (http://chibba.pgml.uga.edu/mcscan2/). Whole Genome Duplication (WGD)/Segmental Duplication (SD), and Tandem Duplication (TD) were identified with default parameters (Wang et al., 2012). Orthologous ST genes between S. spontaneum, E. rufipilus, S. officinarum and S. bicolor were identified using the Dual Synteny Plot tool in TBtools. The coding sequences of ST gene pairs were subjected to synonymous (ks) and non-synonymous (ka) substitution ratio according to the Nei-Gojobori method (Kumar et al., 2016). If the ratio of Ka/Ks is greater than 1, it shows positive selection. When the value of Ka/Ks is equal to 1, it represents neutral selection. However, when the ratio of Ka/Ks is less than 1, it suggests negative or purifying selection. Additionally, we used the Ks values for estimation of duplication event time (T) in MYA (Million Years Ago). Here, T = Ks/(2 × 6.1 × 10−9) × 10−6 Mya (Gaut et al., 1996).
Plant material and RNA extraction
We utilized different samples from 4–6 months old E. rufipilus and S. officinarum (B-48) tissues, including mature leaf, seedling stem, pre-mature stem, and mature stem. Total RNA from these harvested samples was extracted using a Quick-RNA™ Miniprep kit (Zymo Research, USA), according to the manufacturer’s recommendations.
Transcriptome analysis by RNA-sequencing
We used RNA-seq data for E. rufipilus and S. officinarum, which included transcript abundance [Transcript Per Million (TPM)] from various developmental stages from our previous studies. Additionally, we incorporated RNA-seq data for S. officinarum that analyzed circadian rhythms, gradients of leaf development, and the effects of different hormones such as abscisic acid (ABA), gibberellin (GA), indole acetic acid (IAA), and ethylene (ETH) (Zhang et al., 2018, 2021; Hua et al., 2022; Wang et al., 2023).
Trimmed reads were aligned with the reference gene model of the S. spontaneum genome by Trinity with default settings (Grabherr et al., 2011). Transcript assembly was performed by Stringtie (Pertea et al., 2015), and TPM values were estimated by RSEM method (Li and Dewey, 2011).
Plant growth and sugar treatment
E. rufipilus and S. officinarum stalks were grown in a greenhouse under growth conditions of 16h 30°C, 8h/22°C, and a relative humidity of 75%. Three sugar treatments (1% Sucrose, 1% Glucose, and 1% Fructose) were applied uniformly to the 6 months old seedlings. Each experiment was performed in triplicate. Leaf samples were collected after 8h of treatments and stored at −80°C for further processing.
Confirmation of ST gene expression by quantitative RT-PCR
For quantitative RT-qPCR, about ≤1µg RNA was obtained from seedling stem, pre-mature stem, mature stem, and mature leaf samples. 5xPrimescript RT master mix (Takara Bio) was used for cDNA synthesis. RT-qPCR reaction comprising of the following reaction mixture in 20 µL solution: 10 µL of Master Mix (SYBR Green; Roche, Germany), 1 µL of template cDNA, 1 µL of forward and reverse primers each (10 µM), and water up to the final volume. Thermal cycling was as follows: initial denaturation at 95°C for 10 min, subjected by 40 cycles of denaturation at 95°C for 15 s, further annealing, and extension at 60°C for 60 s. For normalization of expression data, Actin and eEF-1a were used as reference genes. Each experiment was performed in three technical replicates. The relative expression level of each sample was calculated with the 2-ΔΔCq method, as mentioned earlier (Akbar et al., 2021). A list of primers is mentioned in Supplementary Table S1.
Transient protein expression and confocal microscopy
Full length CDS (SUT1-T1) without stop codon was amplified from S. officinarum cDNA (Supplementary Table S1). Amplified fragment of SUT1-T1 was inserted into pCAMBIA1300-GFP vector. The vectors were then transformed into the Agrobacterium tumefaciens GV3101 strain. The bacterial suspensions with OD600 = 0.5 were inoculated into Nicotiana benthamiana leaves. FM™4-64 (N-(3-Triethylammoniumpropyl) was used as fluorescent dye. After 2–3 days of inoculation, leaf portions were excised from the injected area and observed under GFP fluorescence signals using a laser confocal microscope (LEICA TCS SP8).
Yeast One-hybrid Assay
Yeast One-hybrid Assay (Y1H) screening was conducted using the Matchmaker Gold Yeast One-Hybrid Screening System (Clontech, 630489). The targeted sequences (bait) were cloned into the pAbAi vector. SUT1-T1 promoter region (2000 bp upstream region from start codon) was amplified in four fragments [−1 to −500 (cis-1), −501 to −1000 (cis-2), −1001 to −1500 (cis-3), −1501 to −2000 (cis-4)] and cloned into pAbAi vector. The pAbAi-bait plasmids were then linearized with BstB1 restriction enzyme and transformed into Y1H Gold yeast-competent cells. The colonies were screened on synthetic dextrose medium lacking uracil. The bait strains were then tested for Aureobasidin A (AbA) resistance, and the minimal inhibitory concentration of AbA was determined for bait strains. A cDNA library (prey) was generated by Ouyi Biomedical Technology Co., Ltd. (Shangai, China). The AD-prey vectors were co-transformed with bait-pAbAi plasmids and screened on SD/-Leu/AbA media. Potential binding partners were confirmed through sequencing. TFs were predicted through the Plant transcription factor database (PlantTFDB) (https://planttfdb.gao-lab.org/prediction.php). The primers used in Y1H assay are mentioned in Supplementary Table S1.
For the one-to-one interaction of SUT1-T1 with LSD and NAC TFs, we extracted yeast plasmids from colonies confirmed by sequencing. The colonies were grown in YPDA medium, and plasmids were extracted using a yeast plasmid extraction kit (Solarbio, D1160). Due to the low copy number of yeast cells, we initially transformed 5 μL of the extracted yeast plasmids into Escherichia coli (E.coli) DH5α competent cells. Next, we transformed 100 ng of prey plasmid into yeast strain containing the corresponding bait plasmid to verify one-to-one interaction. The growth of the transformed yeast was analyzed on the SD/-Leu/AbA* selection medium.
Results
Identification of ST genes and their physical attributes
A genome-wide search was performed using E. rufipilus and S. officinarum amino acid sequences as a query with BLASTp analysis, against the previously published ST genes in S. spontaneum (Zhang et al., 2021; Xiao et al., 2022). Query sequences were further confirmed through batch CDD search and PFAM. Subsequently, we identified 78 reliable ST genes from E. rufipilus and 86 ST genes from S. officinarum. These ST genes were further divided into eight subfamilies, including seven monosaccharide transporter families (VGT, INT, SFP, PLT, STP, MST, and pGlcT) and one sucrose transporter family (SUT). Consistent with earlier findings, phylogenetic analysis distributed the ST genes into eight distinct subfamilies (Figure 1) (Zhang et al., 2021; Xiao et al., 2022). All these ST genes were numbered according to their designated position in the evolutionary tree, with respective of S. spontaneum and S. bicolor ST genes. Using S. spontaneum ST genes as a reference, phylogenetic tree distinguished the 4 members in INT, 5 members each in MST and SUT, 2 members in pGlcT and VGT each, 25 members in PLT, 8 members in SFP, and E. rufipilus genome. In S. officinarum genome, 4 members were found in INT and VGT each, 7 members each in MST and SUT, 3 members in pGlcT, 38 members in PLT, and 8 members in SFP. Noticeably, the highest number of ST genes were found in the STP subfamily in E. rufipilus, which were 27 in numbers while in S. officinarum PLT subfamily contained the highest gene numbers, with 38 members.
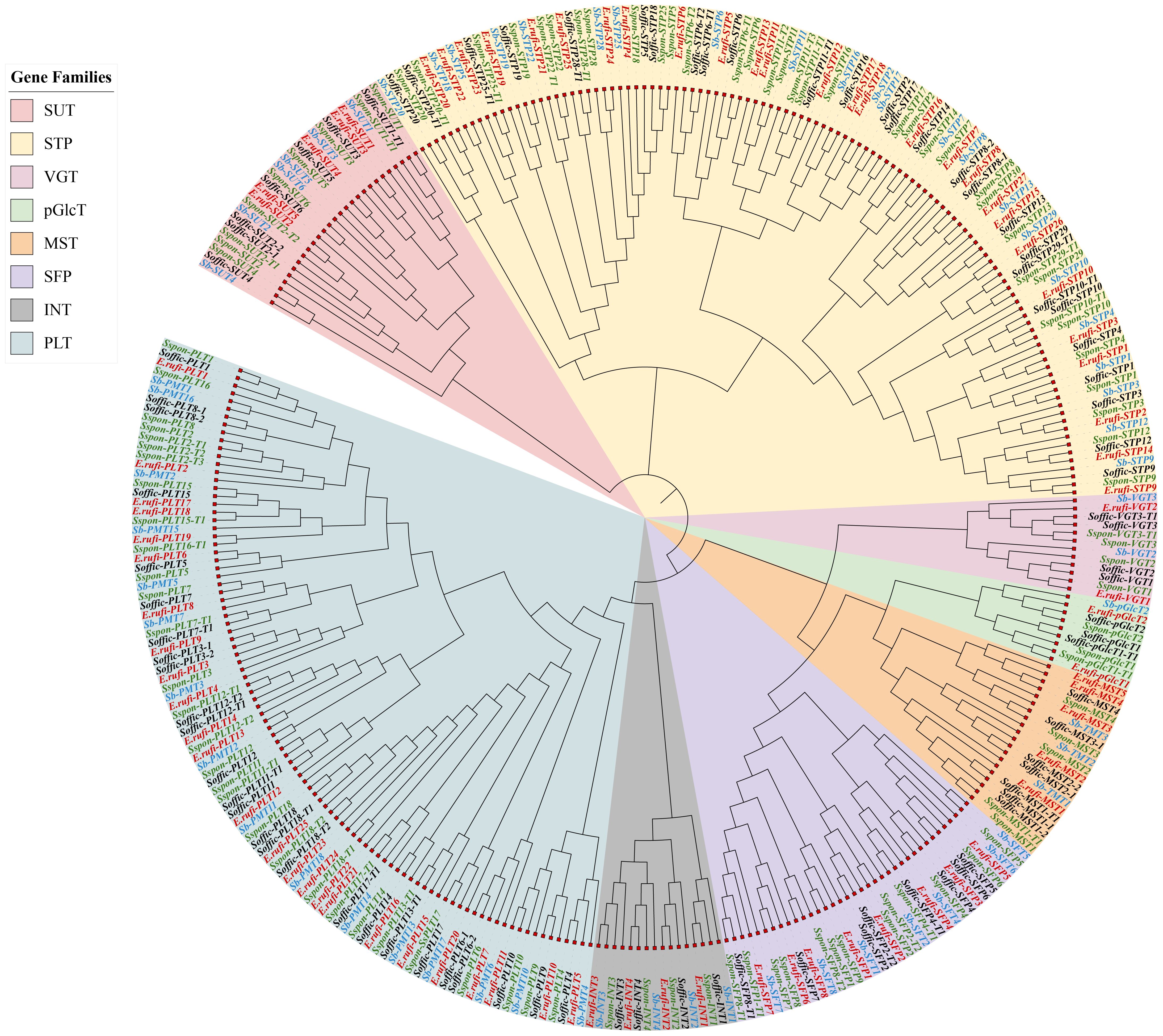
Figure 1. Phylogenetic tree of ST gene family of S. officinarum, E. rufipilus, S. spontaneum and S. bicolor.
ST gene families in E. rufipilus encode proteins with a length ranging from 348 amino acids (Erufi. 02G037360-PLT23) to 761 amino acids (Erufi.10G030260-MST2). Their molecular weight ranges from 37.14 KDa (Erufi.02G037360-PLT23) to 80.91 KDa (Erufi.10G030260-MST2). Isoelectric point (pI) predicted in the range of 4.74 pH (Erufi.10G030260-MST2) to 10 pH (Erufi.09G014150-STP27). Several trans-membrane domains are found to be between 6 and 12. Subcellular localization predicted by WoLF PSORT identified 40 ST genes located in plasmalemma, 27 in vacuole, 6 in chloroplast, and 5 in cytoplasm (Supplementary Table S2A). In S. officinarum, ST proteins range in length from 310 amino acids (Soffic.09G0019430-6P-SFP7) to 760 amino acids (Soffic.06G0004520-3E-MST2). Their molecular weight ranges from 33.109 KDa (Soffic.09G0019430-6P-SFP7) to 80.687 KDa (Soffic.06G0004520-3E-MST2). Iso-electric point (pI) is in the range of 4.76 pH (Soffic.06G0004520-3E-MST2) to 10.3 pH (LAp.01H0033380-SFP4). The number of transmembrane domain ranges between 6 and 12. Subcellular localization detected 45 ST genes in plasmalemma, 27 in vacuole, 7 in chloroplast, and 7 in cytoplasm (Supplementary Table S2B).
Analysis of conserved motifs, coding sequences, and promoter sequences
Each member of the ST gene family has a unique sequence and distinct structural features. We identified conserved motifs to comprehend the structural diversity and evolutionary relationship. Importantly, few motifs were detected only in an individual family, which depicts that they are associated with specific functions. We detected 15 conserved motifs in ST genes of both species using the MEME suite (Figures 2A, B). In E. rufipilus and S. officinarum, commonly found motif at the N terminal is motif 7, except in SUT, while at C-terminal motif 2 is most common. Evidently, in the monosaccharide family number, the specificity of conserved motifs is highly comparable. However, some specific motifs are present or absent in each subfamily. Moreover, the number of conserved motifs in STP genes is 13 in both E. rufipilus and S. officinarum. Similarly, in the PLT subfamily conserved motifs are also found to be 13 in number in most of ST genes in both species. Furthermore, the number of conserved motifs in SUT genes ranges from 3 to 6 in E. rufipilus and 3 to 5 in S. officinarum. Conserved motifs depicted the origination of subfamilies from a common ancestor. However, slight differences showed the specificity and functional divergence of each subfamily.
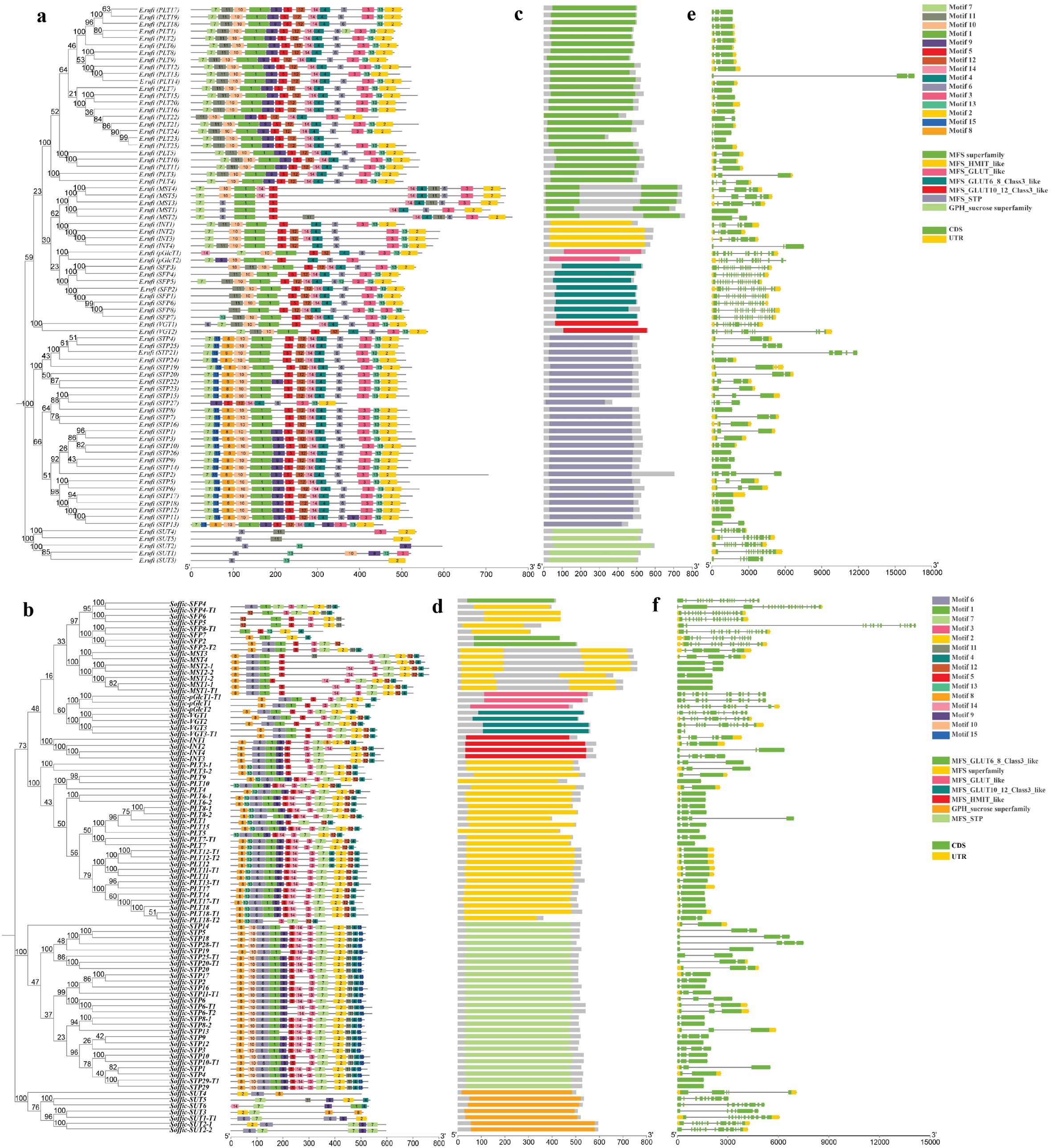
Figure 2. Phylogenetic tree, motif pattern, superfamilies, and gene structure of ST genes in E rufipilus and S. officinarum (A, B) The phylogenetic tree is contructed with Neighbor joining method with 1,000 replicates on each node, using full-length sequences of E rufipilus and S. officinarum ST proteins (number represents nucleotide substitutions per site). The amino acid motifs (numbered 1–15) are demonstrated in colored boxes, with black lones represented the protein length (C, D) Colored bars represent the distinct superfamilies in each sub family (E, F) Green and yellow rectangles represent the UTR (untranslated region) and exon, respectively.
Next, we detected seven superfamilies (MFS superfamily, MFS-GLUT10-12_Class3_like, MFS_GLUT_like, MFS_GLUT6_8_Class3-like, MFS_HMIT_like, MFS_STP, GPH_sucrose superfamily) in ST genes (Figures 2C, D). Each subfamily possesses a distinct superfamily. The PLT family was found to have MFS superfamily, VGT has MFS-GLUT10-12_Class3_like superfamily, MST also contained MFS superfamily, pGlcT is composed of MFS_GLUT_like superfamily, SFP has MFS_GLUT6_8_Class3-like superfamily, and INT encodes MFS_HMIT_like superfamily. Moreover, STP contained MFS_STP and SUT composed of the GPH_sucrose superfamily. The occurrence of these superfamilies contributed to the uniqueness and specific functionality of ST genes.
Regarding ST gene conservation, gene structures were illustrated with Gene Structure Display Server (GSDS). Each gene family was observed to display conserved exon and intron positions relative to domain position and structure (Figures 2E, F). To comprehend the evolutionary relationships and characteristics of ST sequences, the cDNA sequence of each ST gene was aligned with the genomic sequence. The majority of ST genes comprises only exons. In E. rufipilus, a total of 23 ST genes contain only exons, while 55 genes are composed of exons and untranslated regions (UTRs) as well. The number of exons per gene ranges from 2 to 18. Among 55 genes containing UTRs, only 16 genes have 3′ UTR and 39 have both 5′ and 3′ UTRs. In S. officinarum, 43 ST genes composed of only exons, with the number of exons per gene ranging from one to as many as 20 exons per gene.
For identification of cis-regulatory elements in the region, upstream 2.5 kb sequences from the start codon of each gene were submitted to PlantCARE. Promoter regions from ST genes revealed a number of cis-acting elements related to different functions (Supplementary Figures S1A, B). The majority of identified cis-acting elements were grouped into cellular function, stress response, light response, and hormonal regulation categories (Table 1). A number of cis elements are related to light responses such as ACE and AE box. Box4, GT1 motif, I-box, and Sp1 are present in both S. officinarum and E. rufipilus.
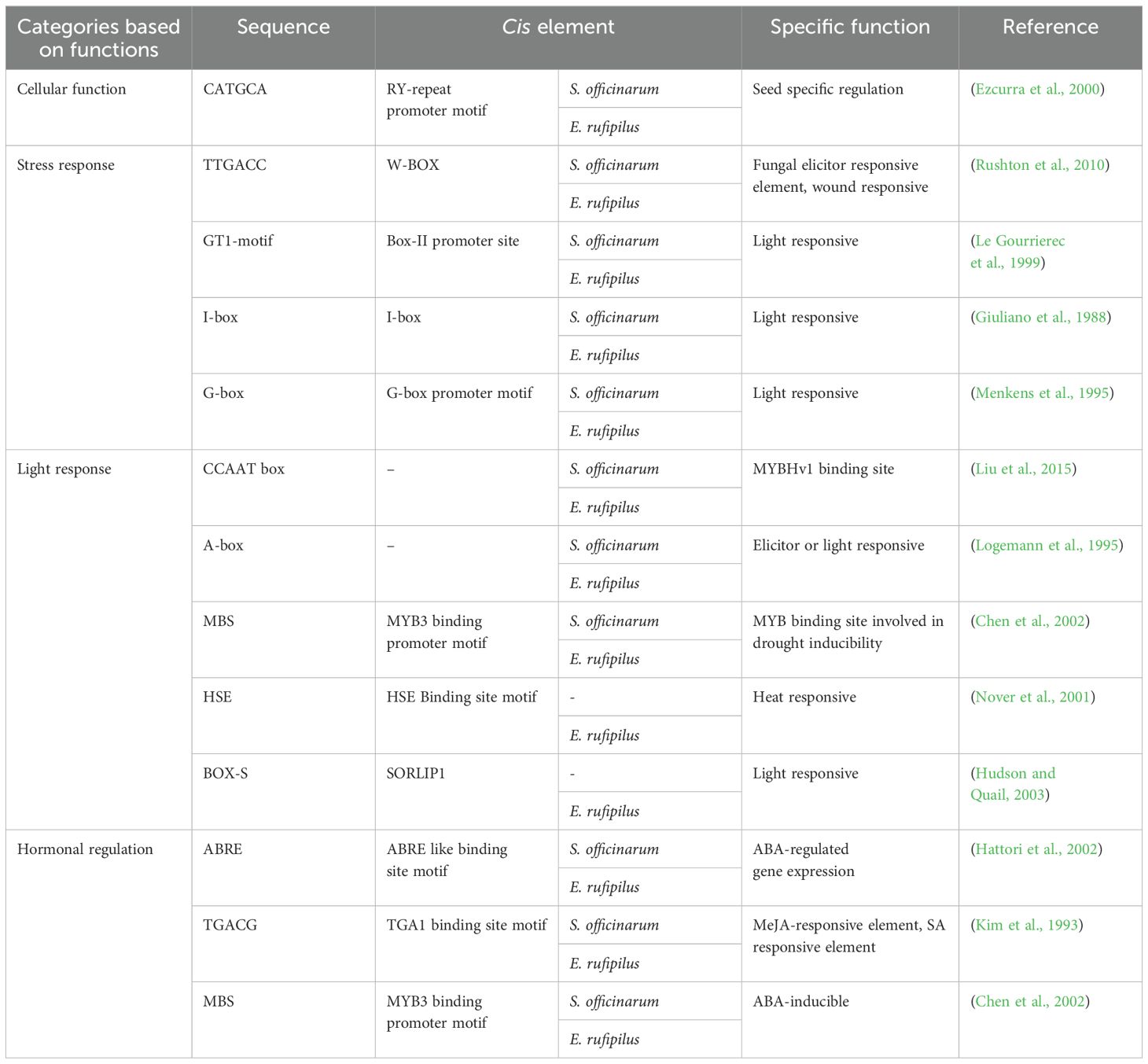
Table 1. Category wise list of cis-elements identified from 2.5kb upstream region of ST gene from S. officinarum and E. rufipilus.
Chromosomal distribution and synteny analysis of ST genes
We investigated the chromosomal distribution of ST genes on 10 chromosomes of E. rufipilus and S. officinarum (Figures 3A, B). We found that there is no substantial co-relation between the number of genes and chromosome length. The majority of the genes were found to be concentrated on Chr01 and Chr02 in S. officinarum while on Chr02 in E. rufipilus. All genes were indiscriminately distributed on chromosomes, with most genes located at the lower telomere.
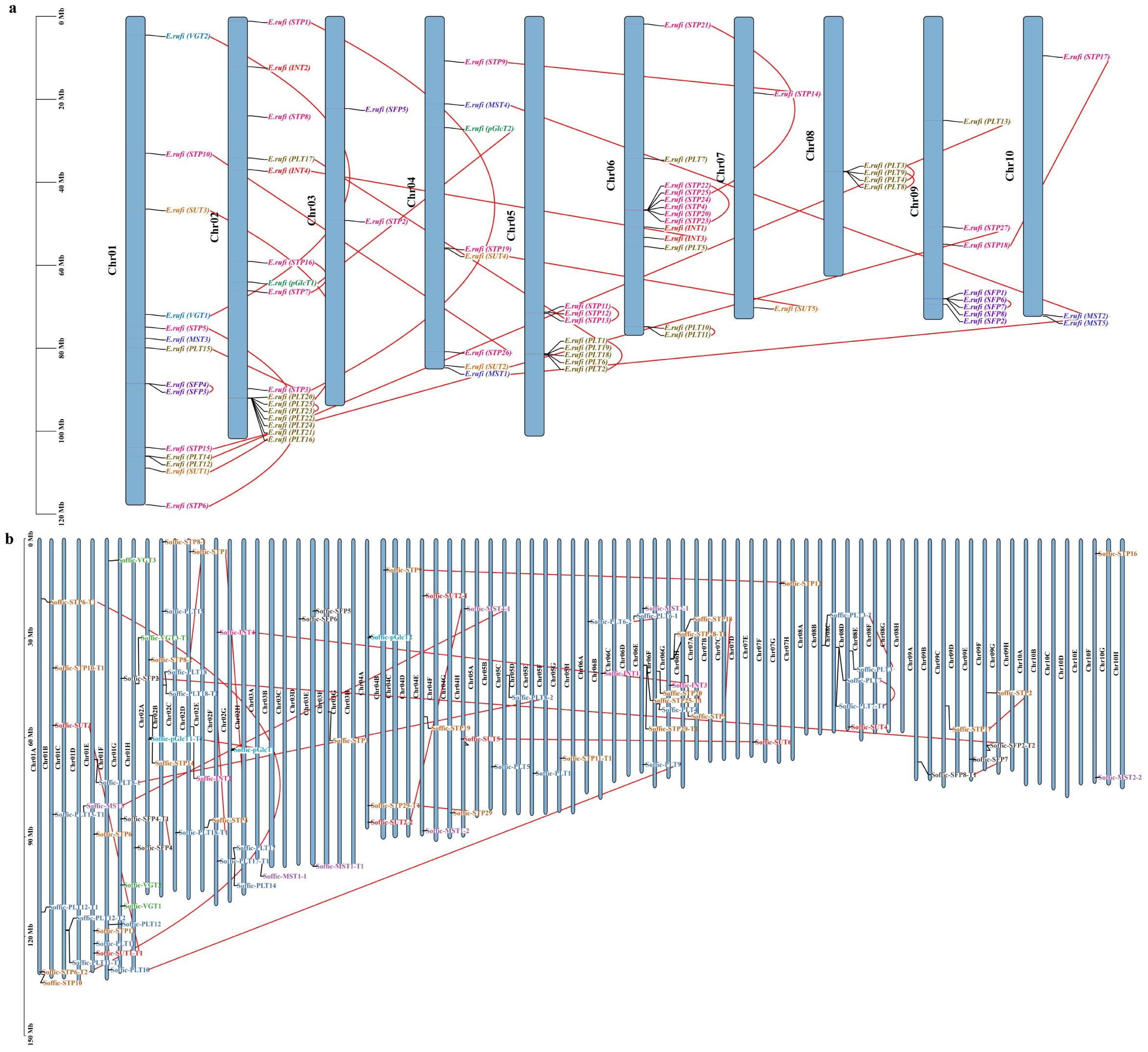
Figure 3. Chromosomal location of ST genes in (A) E rufipilus (B) S. officinarum. The colored rectangle bars represent the chromosomes of E rufipilus and S. officinarum and scale represent the chromosome length, the Chr 1–10 represent each corresponding chromosome, each gene sub family is represented by distinct color and red line represent the gene pairing.
Genomic sequence duplications, including WGD, TD, and SD, provide a genetic link for evolution (Xiao et al., 2022). The ST gene family duplication event was performed by MCScanX. Duplication analysis identified dispersed duplication (DD), WGD, or SD events in both genomes. WGD or SDs was found to be the primary reason for ST gene family expansion (Supplementary Tables S3A, B). In E. rufipilus, a total of 52 genes displayed WGD, or SD, and 26 genes were found to be DD. However, in S. officinarum, 58 genes depicted WGD or SD and 29 genes showed SD. This indicates that WGD or SD might be the major driving force in the evolution of the ST gene family. Additionally, 25 ST gene pairs were identified in E. rufipilus while 24 gene pairs were found in S. officinarum. These gene pairs were distributed on all 10 chromosomes (Supplementary Figure S2).
Estimation of evolutionary rate and collinearity analysis
To further infer the evolutionary rate of ST genes, we calculated Ka/Ks (the ratio of synonymous and non-synonymous substitution rate). Ks values investigated positive (Darwinian) selection or negative (purifying) selection and duplication dates. ST gene pairs estimated to have undergone purifying selection. Duplication time was estimated to be in the range between 8.802 and 277.38 Mya in E. rufipilus and 0.182-90.444 Mya in S. officinarum (Supplementary Tables S4A, B). We performed collinearity analysis with S. spontaneum and S. bicolor to further identify homology between related species. All the ST genes belonging to E. rufipilus and S. officinarum showed collinearity with the syntenic region in S. spontaneum and S. bicolor (Supplementary Figure S3).
Expression pattern of ST genes during different developmental stages
To gain insights into the role of ST genes in sugar transport and mobilization, we analyzed the transcriptome profiles of ST gene expression in various tissues of E. rufipilus and S. officinarum. We found that the ST genes exhibited different expression patterns in different tissues. TPM values of selected ST genes were visualized using heatmaps. Specifically, we focused on the highly expressing genes in the mature leaf zone. In E. rufipilus, the following genes were up-regulated in the mature leaf zone: SUT2, STP18, STP15, STP4, STP17, STP1, STP8, PLT2, PLT7, PLT18, PLT15, PLT9, PLT8, PLT14, PLT22, PLT19, SFP3, INT1, INT2, and MST4 (Figure 4A). However, in S. officinarum, the undermentioned genes were enhanced in the mature leaf zone: SUT1-T1, SUT2, STP16, STP11-T1, STP17, STP2, STP1, STP13, STP8-2, PLT9, PLT18, PLT3-2, PLT17-T1, PLT7, PLT12-T2, PLT12-T1, PLT17, PLT12, SFP4-T1, INT3, VGT2, VGT3, VGT3-T1 (Figure 4B).
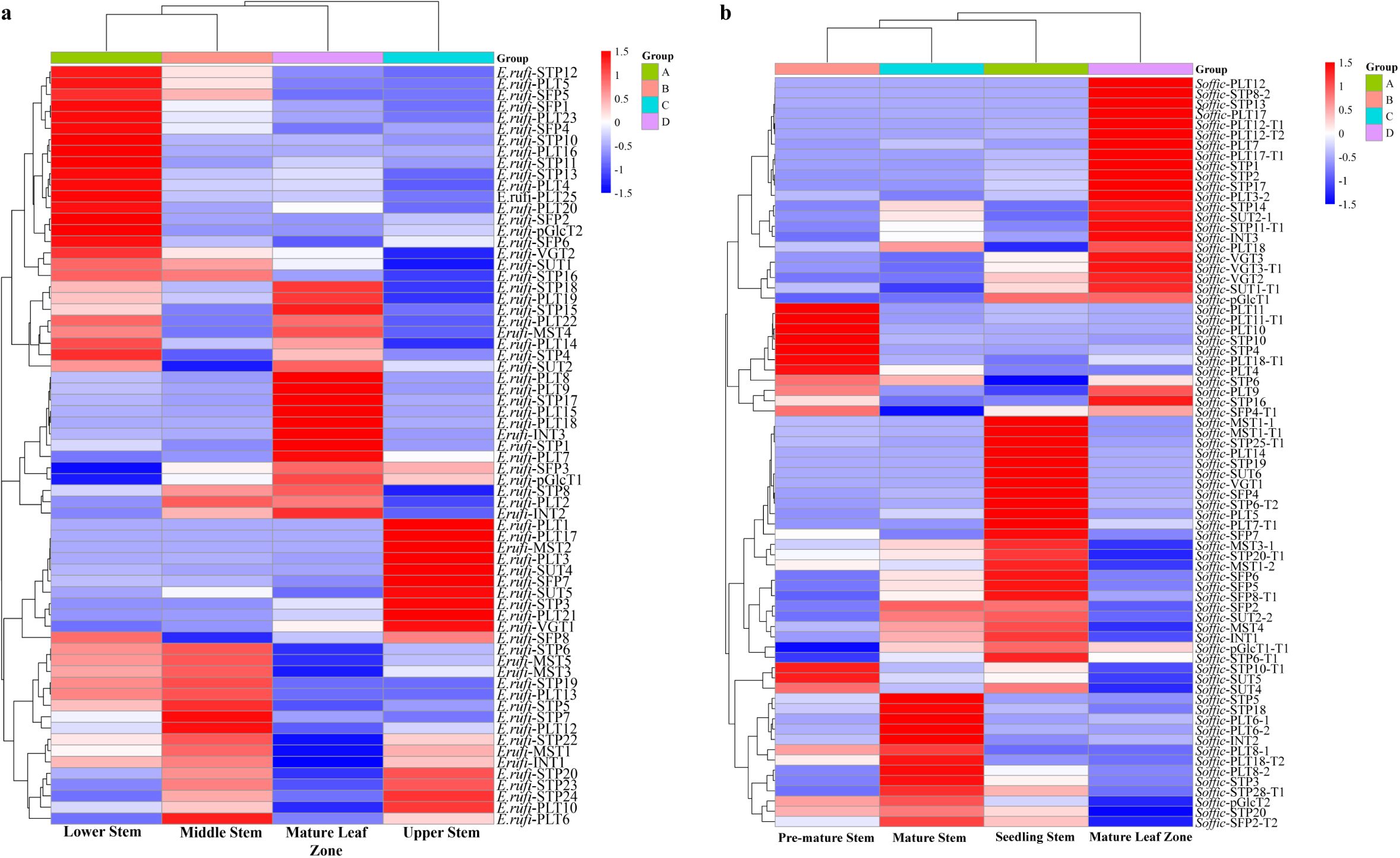
Figure 4. Gene expression analysis of ST genes (A) in different tissues (seedling stem, mature leaf, mature stem and pre-mature stem) of E rufipilus (B) in different tissues (seedling stem, pre-mature stem, mature stem, mature leaf) of S. officinarum.
Validation of gene expression pattern through RT-qPCR
To verify the accuracy of the RNA seq data, we conducted RT-qPCR on different tissue samples from E. rufipilus and S. officinarum. The RT-qPCR results showed that SUT1 is highly expressed in the mature stem of E. rufipilus and mature leaf of S. officinarum. However, INT1 exhibits high expression in the pre-mature stem of E. rufipilus and mature leaf of S. officinarum. Additionally, in S. officinarum, there is higher expression of STP4 and PLT11 in pre-mature stem. In E. rufipilus, PLT15 and STP23 depicted enhanced expression in mature leaf. Conclusively, we observed consistent trends in the relative expression of selected ST genes and their corresponding FPKM values (Supplementary Figure S4).
Expression pattern of ST genes during different sugar stresses and subcellular localization of SUT1-T1
To better understand the role of ST genes, we treated the S. officinarum and E. rufipilus leaves with different sugar solutions and analyze their expression pattern through RT-qPCR. Expression data was obtained from 4 to 6 months old leaves of S. officinarum and E. rufipilus, treated with 1% solution of sucrose, glucose, and fructose. The expression level of ST genes showed variation during different sugar treatment (Figure 5).
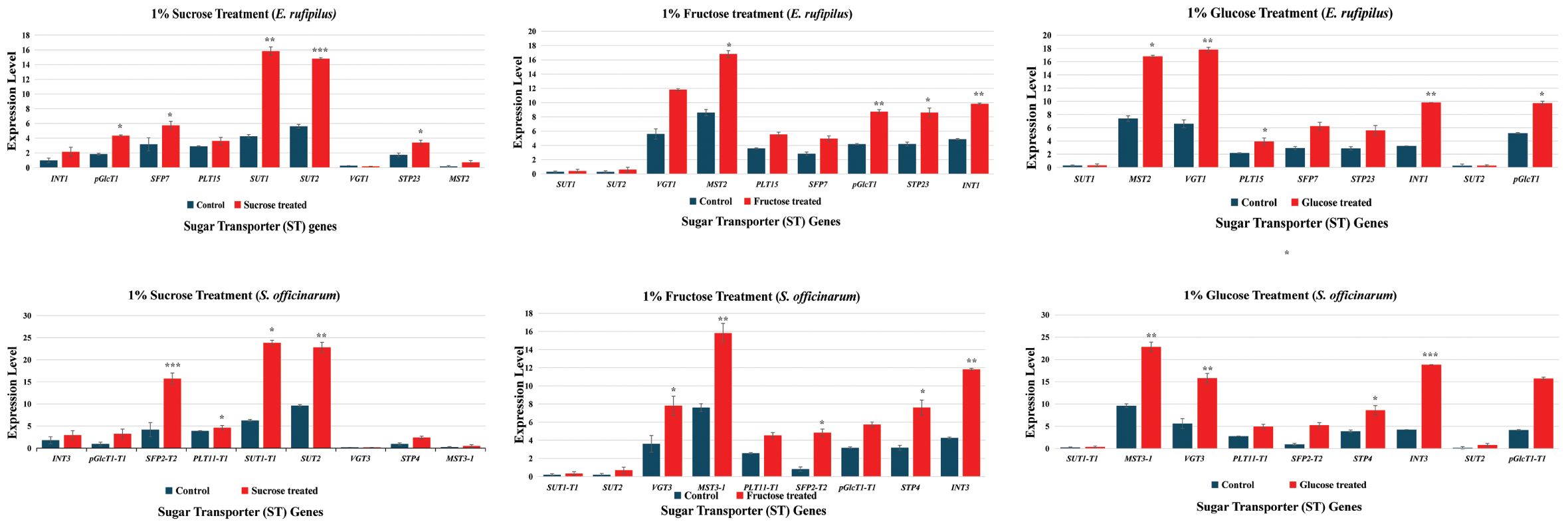
Figure 5. Relative gene expression level of nine selected ST genes (INT1, pGlcT1, SFP7, PLT15, SUT1, SUT2, VGT1, STP23, MST5) in E. rufipilus and S. officinarum, under different sugar stress (1% sucrose, 1% glucose, 1% fructose). * (p-value <0.05), ** (p-value <0.01), *** (p-value <0.001).
During 1% sucrose treatment, SUT1 and SUT2 showed enhanced expression in both E. rufipilus and S. officinarum leaves. Additionally, SFP expression was also found to be relatively higher in sucrose-treated S. officinarum leaves. Furthermore, the 1% glucose treatment regulates expression of MST, VGT, INT, and pGlcT gene levels in both studied species as compared to their wild-type counterparts. Interestingly, the 1% fructose treatment resulted in increased expression of MST, VGT, and INT in both E. rufipilus and S. officinarum. We also observed moderately higher expression levels of PLT, SFP, pGlcT, and STP in both species compared to their wild-type counterparts. Notably, the expression levels of SUT1 and SUT2 were remarkably low during the glucose and fructose treatments.
Based on the analysis of expression data, we identified SUT1-T1 with high expression in the mature leaf zone of S. officinarum. We selected this gene to investigate its subcellular location. Confocal microscopy determines that SUT1-T1 is located in the plasma membrane (Figures 6A, B). This finding is consistent with the prediction of the WoLF PSORT tool, which also identified the plasma membrane as the location of the SUT1-T1 gene.
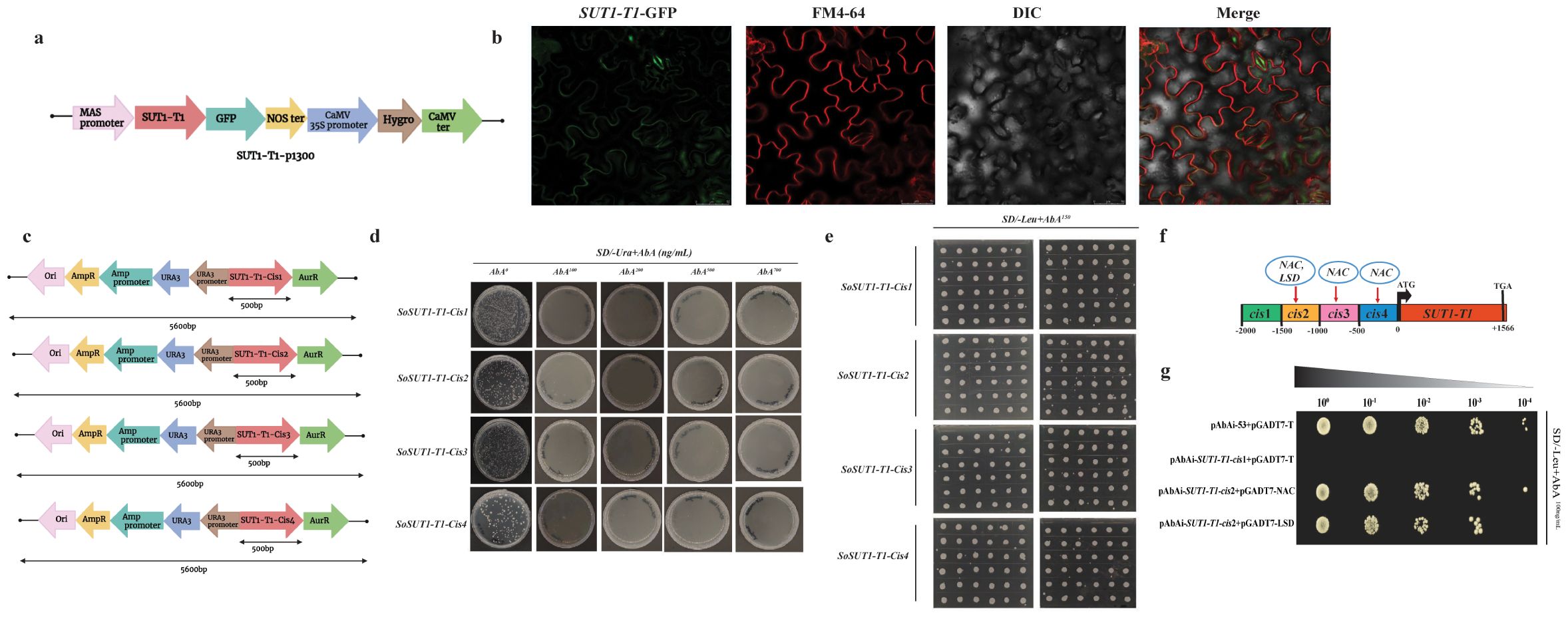
Figure 6. Sub cellular localization of SUT1-T1 gene in Nicotiana benthamiana leaves (A) Diagrammatic representation of SUT1-T1 gene in p1300 binary vector (B) Confocal microscopy of SUT1-T1 gene in N. benthamiana leaf epidermis. Scale bar used 50μm (C) Schematic representation of four cis-SUT1-T1 fragments in AbAi vector that were used as baits in yeast one hybrid (Y1H) screening (D) Growth of yeast on SD/-Ura+AbA medium for primary screening. AbA0 without AbA antibiotic, AbA100: 100ng/mL, AbA200: 200 ng/mL, AbA500: 500 ng/mL, AbA700: 700 ng/mL (E) Growth of yeast on SD/-Leu+AbA150 medium for secondary screening (F) Schematic representation of the identified TFs in the promoter region of the SUT1-T1 gene (G) Yeast one to one verification. PAbA53_pGADT7-T is a positive control and pAbAi-SUT1-T1-cis1+pGADT7-T is a negative control. Top bar indicates the serial dilutions.
Identification of TFs in SUT1-T1 promoter region of S. officinarum
In order to identify the potential TFs that bind to the SUT1-T1 promoter region, we divided the 2000 bp promoter region into smaller fragments of 500 bp (Figure 6C). These fragments were referred to as bait and were used to screen sugarcane cDNA libraries using the Y1H system. The activity of the bait fragments was repressed using 100 ng ml−1 of AbA (Figure 6D). We determined that the minimal inhibitory concentration of AbA for screening the sugarcane cDNA library was 100 ng ml−1. A total of 100 clones for each fragment were grown on SD/-Leu/+AbA (150 ng ml−1) medium and screened for TFs prediction (Figure 6E). Of the total clones screened, only four showed potential TFs. According to Plant TFDB, NAC (Soffic.10G0023830-1A) and LSD (Soffic.07G0003300-4D) were identified as potential TFs (Figure 6F). Next, the one-to-one interaction between NAC and LSD TFs with the SUT-T1-cis2 promoter region confirms that both TFs interact with the gene promoter region (Figure 6G).
To investigate the expression patterns of TFs identified in the Y1H experiment, RNA-seq analyses were performed in S. officinarum, focusing on various developmental stages, diurnal cycles, and leaf development. We compared the expression patterns of NAC and LSD TFs with SUT1-T1 by analyzing the seedling leaves. Seedling leaves, measuring 15 cm in length, were divided into 15 equal segments. The expression of SUT1-T1 consistently increased from the basal to the mature zone. LSD exhibited a positive correlation with SUT1-T1, while NAC depicted a negative expression pattern (Figure 7).
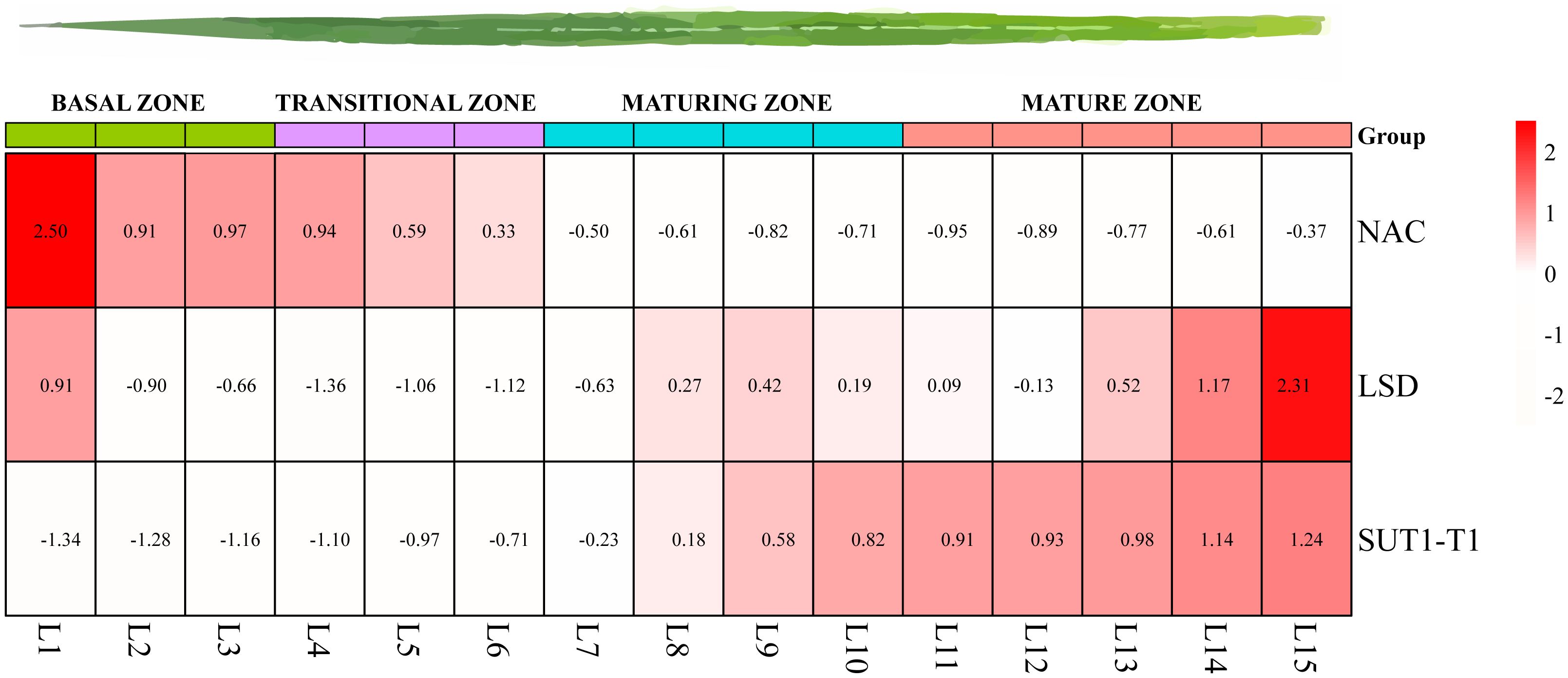
Figure 7. Expression patterns of SUT1-T1 and its potential upstream TFs across gradient developmental leaves based on TPM.
To investigate how TFs regulate SUT1-T1 at various developmental stages, we measured their transcript abundance in mature tissues, specifically in leaves and stems. Both TFs, NAC and LSD, exhibited higher expression levels in mature stem-internode 9. In contrast, SUT1-T1 was expressed in seedling leaves. This indicates that NAC and LSD TFs negatively regulate the SUT1-T1 gene (Supplementary Figure S5A). Next, we compared the expression pattern of TFs on SUT1-T1 during day-night rhythm in S. officinarum (Supplementary Figure S5B). SUT1-T1 showed higher expression levels during the daytime (from 6:00 to 10:00) and decreased expression for the reminder of day. LSD displayed enhanced expression in the morning (at 10:00) while NAC showed increased expression from 14:00 until midnight.
The expression patterns of NAC in the leaves and stems of S. officinarum were assessed following treatments with different hormones (IAA, ETH, GA, ABA) at various time points (Supplementary Figure S5C). NAC was found to be highly expressed in the leaves after ABA treatment and in the stem after 24h. However, NAC expression was higher in the stem at 48h and 96h after ETH treatment. GA increased NAC expression at all time points in the stem, while in the leaves, it was only elevated after 96h. IAA induced a strong NAC expression in the leaves after 48h of treatment. However, ETH and ABA had no effect on LSD expression, whereas GA and IAA enhanced LSD expression in all treated samples. SUT1-T1 expression is high in stems treated with ABA for 48 and 96 h. ETH increases SUT1-T1 expression in the 48h leaf, 24h stem, and 96h stem samples. GA enhances SUT1-T1 expression in the 24h and 96h leaf samples, as well as the 24h stem. Conversely, IAA enhanced the expression of SUT1-T1 in all treated leaf samples. Overall, there is no correlation observed in the expression patterns of NAC, LSD, and SUT1-T1 under various hormone treatments. Additionally, we have found that the expression pattern of the selected genes depicted a consistent trend between transcriptome data and RT-qPCR values (Supplementary Figure S6).
Discussion
Whole genome sequencing (WGS) offers opportunities to identify and functionally analyze the gene families in plants. However, it is troublesome to study polyploid genomes, as they are composed of homeologous sub-genomes, and allocating each sequence to respective chromosomes is challenging. Previously in the genus Saccharum, genomes of S. spontaneum and E. rufipilus have been published, providing detailed information about their genome architecture and origins (Zhang et al., 2018; Wang et al., 2023). Additionally, E. rufipilus (diploid) genome could be used as a reference for genomics analysis in sugarcane (Wang et al., 2023).
Gene duplication events are important for evolution and the expansion of gene families (Moore and Purugganan, 2003). Previous studies have shown that multigene families have evolved in the Saccharum genome by two WGD events (Zhang et al., 2019). WGD or SD suggests that a gene might arise from WGD or SD. This is one of the highest duplication events in angiosperms, which have underwent through minimum one WGD event (Jiao et al., 2011). During DD events, genes might arise from transposition such as replicative transposition, non-replicative transposition, or conservative transposition (Taylor and Raes, 2005). In our study, WGD/SD occurred in 52 and 58 ST genes of E. rufipilus and S. officinarum, respectively. Meanwhile, 26 and 29 ST genes showed DD events in E. rufipilus and S. officinarum, respectively. This suggests that WGD/SD is the major driving force behind the evolution of ST gene families in both studied species.
Sugars are transported from leaves to stems or sink tissues with various ST proteins or genes. Various STs are located inside the cell to distribute sugar to different compartments. We compared the expression pattern of various ST genes across different tissues in both species (Figure 4). Plastid sugar transporters (pGlcT) are located on the inner-envelop membrane of plastid, which function in starch mobilization. Previously, pGlcT2 expression was observed in the seedlings and early stages of plant growth (Valifard et al., 2023). However, analysis of RNA-seq data revealed that pGlcT2 exhibits a high expression level in the mature stem of S. officinarum and E. rufipilus. PLT members are located on the plasma membrane and are responsible for the transportation of hexoses and various sugar alcohols (Klepek et al., 2005). Previously, PLT11 and PLT11_T1 showed high expression levels in sclerenchyma and parenchyma cells of mature stalk and resulted in high sugar content in S. officinarum (Zhang et al., 2021). Based on the RNA-expression data, it has been determined that PLT11 and PLT11-T1 are found to be higher in the pre-mature stem of S. officinarum.
Glucose and fructose molecules are transported inside vacuoles with VGT genes. In both studied species, VGT1 has been found to have high expression levels in the seedling stems. Earlier research has shown that VGT3 exhibits high expression level in the leaves of S. officinarum and S. lycopersicum (Reuscher et al., 2014). SUTs, belonging to the MFS, are responsible for the long-distance transport of sucrose in plants. In Saccharum, SUT proteins primarily bind with the disaccharide sucrose (Wendler et al., 1991). In S. officinarum, the expression of SUT2 and SUT1-T1 is high in mature leaf, while in E. rufipilus, SUT2 is specifically enhanced in mature leaf. Consistently, transcription activity of SUT1 and SUT2 was also found to be higher in mature leaves during different sugar treatment (Figure 5).
TFs play a crucial role in regulating various physiological activities in plants by binding to conserved sites within promoter regions of target genes. In the upstream 2 kb promoter sequence of SUT1-T1, cis-element prediction has revealed the presence of binding sites for NAC and LSD TFs. NAC TFs play a role in various biological processes that control plant growth and development, such as responding to external stress (Nuruzzaman et al., 2013), forming flower organs (Sablowski and Meyerowitz, 1998), forming shoot and root apical meristems (Souer et al., 1996), developing lateral roots, and regulating senescence (Xie et al., 2000; Podzimska-Sroka et al., 2015). The role of LSD TFs in Arabidopsis is to negatively regulate a plant cell death pathway. LSD1 triggers the systemic acquired resistance (SAR) response to both biotic and abiotic stresses (Dietrich et al., 1997). In S. spontaneum, 115 NAC genes have been identified that are involved in responding to biotic and abiotic stresses (Shen et al., 2022). However, the role of LSD in Saccharum has not yet been studied.
Based on the TFs identified in the Y1H assay, a series of transcriptome analyses were conducted to investigate their relationship with SUT1-T1. In this study, SUT1-T1 is found to be highly expressed in seedling leaves (Supplementary Figure S5). The expression patterns of NAC and LSD, in conjuction with the leaf gradient, exhibited an inverse relationship with that of SUT1-T1. Expression level of SUT1-T1 is peaked at 08:00 and begins to decline by 10:00; Wile LSD expression reaches its peak at 10:00. Additionally, the diurnal expression pattern of NAC is negatively correlated with that of SUT1-T1. Therefore, we hypothesize that NAC and LSD in S. officinarum respond to sucrose transport by exhibiting a negative regulatory relationship with SUT1-T1.
Overall, regarding the function of SUT1-T1 and its potential TFs, we proposed a regulatory network for SUT1-T1 based on gene expression profiles (Supplementary Figure S7). SUT1-T1 is primarily active in mature leaves and seedling leaves, playing a role in sucrose transportation from source to sink tissues. In the source tissue, SUT1-T1 is highly active but negatively regulated by NAC and LSD. In contrast, SUT1-T1 is less active in sink tissues, where NAC and LSD are highly expressed. Additionally, SUT1-T1 shows increased activity during daytime, as part of the circadian rhythm. However, this speculation still requires verification through additional approaches.
Conclusion
In this study, we performed a comprehensive analysis of the ST gene family in E. rufipilus and S. officinarum. This study highlights the key genes, their location, interaction, and expression pattern. Additionally, our transcriptome analysis of different tissues and sugar stress provided insights into the key genes involved in the sugar transportation pathway. We also discovered that NAC and LSD TFs have the ability to bind with the SUT1-T1 promoter. Transcriptome analyses of TFs across developmental gradient leaves, various time points during circadian cycles, and stems, and leaves at different growth stages reveal potential expression patterns and regulatory networks between these TFs and SUT1-T1. The data we generated will be valuable in understanding the ST gene families in the Saccharum genus. These fundamental results will be beneficial in identifying the key ST genes in other monocots, which can be utilized in plant engineering strategies.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here:The RNA-seq data for E. rufipilus is available in the National Genomics Data Center (NGDC) under Bioproject accession PRJCA014818. The RNA-seq data for S. officinarum can be accessed through the National Center for Biotechnology Information (NCBI) under Bioproject ID PRJNA1200917.
Author contributions
SA: Writing – original draft, Conceptualization, Data curation, Formal analysis, Methodology. XH: Writing – review & editing. YZ: Writing – review & editing, Methodology. GL: Writing – review & editing, Software. TW: Writing – review & editing, Data curation. HS: Methodology, Writing – review & editing. ZL: Data curation, Writing – review & editing. YQ: Data curation, Writing – review & editing. HH: Formal analysis, Writing – review & editing. WY: Investigation, Writing – review & editing. M-QZ: Investigation, Writing – review & editing. JZ: Conceptualization, Funding acquisition, Investigation, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We thank the National Key Research and Development program (2021YFF1000101), the National Natural Science Foundation of China (32272196) and Guangxi Science and Technology Major Program (Guike AA24206023) for supporting this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1502649/full#supplementary-material
Supplementary Figure S1 | Cis-acting elements of the promoter region (upstream 2500 bp) of ST genes in (A) E. rufipilus and (B) S. officinarum.
Supplementary Figure S2 | Collinearity of ST genes in (A) E. rufipilus and (B) S. officinarum. The red lines indicate the duplicated ST gene pairs. The chromosome number is shown inside each chromosome.
Supplementary Figure S3 | Synteny analysis of ST genes between E. rufipilus, S. officinarum, S. bicolor and S. spontaneum. The gray region represents synteny blocks in the wide genome of four species, while colored lines represent the orthologous relationship of ST genes.
Supplementary Figure S4 | Co-relation analysis of RT-qPCR data and gene expression data (A) in E. rufipilus (B) S. officinarum. Green bars represent the relative expression level of each gene in various tissues and red line indicates the positive co-relation of RNA-seq data with RT-qPCR data.
Supplementary Figure S5 | Expression pattern of SUT1-T1 and its potential upstream TFs were analyzed across multiple tissues, circadian rhythms and hormonal treatments, based on TPM.
Supplementary Figure S6 | RT-qPCR verification of LSD (A) NAC (B) SUT1-T1 (C) expression pattern in gradient developing leaves segments.
Supplementary Figure S7 | Schematic representation of the expression pattern of LSD, NAC and SUT1-T1 at different stages of S. officinarum, during both daytime and night time. Red indicates higher expression levels, white and yellow indicates lower expression levels.
References
Afoufa-Bastien, D., Medici, A., Jeauffre, J., Coutos-Thévenot, P., Lemoine, R., Atanassova, R., et al. (2010). The Vitis vinifera sugar transporter gene family: phylogenetic overview and macroarray expression profiling. BMC Plant Biol. 10, 1–22. doi: 10.1186/1471-2229-10-245
Akbar, S., Yao, W., Qin, L., Yuan, Y., Powell, C. A., Chen, B., et al. (2021). Comparative analysis of sugar metabolites and their transporters in sugarcane following Sugarcane mosaic virus (SCMV) Infection. Int. J. Mol. Sci. 22, 13574. doi: 10.3390/ijms222413574
Aoki, N., Hirose, T., Scofield, G. N., Whitfeld, P. R., Furbank, R. T. (2003). The sucrose transporter gene family in rice. Plant Cell Physiol. 44, 223–232. doi: 10.1093/pcp/pcg030
Bailey, T. L., Johnson, J., Grant, C. E., Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43, 39–49. doi: 10.1093/nar/gkv416
Besse, P., McIntyre, C. L., Berding, N. (1997). Characterisation of Erianthus sect. Ripidium and Saccharum germplasm (Andropogoneae-Saccharinae) using RFLP markers. Euphytica 93, 283–292. doi: 10.1023/A:1002940701171
Besse, P., Taylor, G., Carroll, B., Berding, N., Burner, D., McIntyre, C. (1998). Assessing genetic diversity in a sugarcane germplasm collection using an automated AFLP analysis. Genetica 104, 143–153. doi: 10.1023/A:1003436403678
Büttner, M. (2007). The monosaccharide transporter (-like) gene family in Arabidopsis. FEBS Lett. 581, 2318–2324. doi: 10.1016/j.febslet.2007.03.016
Chen, C., Chen, H., He, Y., Xia, R. (2018). TBtools, a toolkit for biologists integrating various biological data handling tools with a user-friendly interface. BioRxiv 289660, 289660. doi: 10.1101/289660
Chen, W., Provart, N. J., Glazebrook, J., Katagiri, F., Chang, H.-S., Eulgem, T., et al. (2002). Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. Plant Cell 14, 559–574. doi: 10.1105/tpc.010410
D’Hont, A. (2005). Unraveling the genome structure of polyploids using FISH and GISH; examples of sugarcane and banana. Cytogenet. Genome Res. 109, 27–33. doi: 10.1159/000082378
D’Hont, A., Grivet, L., Feldmann, P., Glaszmann, J., Rao, S., Berding, N. (1996). Characterisation of the double genome structure of modern sugarcane cultivars (Saccharum spp.) by molecular cytogenetics. Mol. Gen. Genet. 250, 405–413. doi: 10.1007/BF02174028
Daniels, J., Roach, B., Heinz, D. (1987). Developments in Crop Science (Amsterdam, Netherlands: Elsevier), 143–210.
Dietrich, R. A., Richberg, M. H., Schmidt, R., Dean, C., Dangl, J. L. (1997). A novel Zinc finger protein encoded by the Arabidopsis LSD1 gene and functions as a negative regulator of plant cell death. Cell 88, 685–694. doi: 10.1016/S0092-8674(00)81911-X
Eom, J.-S., Chen, L.-Q., Sosso, D., Julius, B. T., Lin, I., Qu, X.-Q., et al. (2015). SWEETs, transporters for intracellular and intercellular sugar translocation. Curr. Opin. Plant Biol. 25, 53–62. doi: 10.1016/j.pbi.2015.04.005
Ezcurra, I., Wycliffe, P., Nehlin, L., Ellerström, M., Rask, L. (2000). Transactivation of the Brassica napus napin promoter by ABI3 requires interaction of the conserved B2 and B3 domains of ABI3 with different cis-elements: B2 mediates activation through an ABRE, whereas B3 interacts with an RY/G-box. Plant J. 24, 57–66. doi: 10.1046/j.1365-313x.2000.00857.x
Gaut, B. S., Morton, B. R., McCaig, B. C., Clegg, M. T. (1996). Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc. Natl. Acad. Sci. U.S.A. 93, 10274–10279. doi: 10.1073/pnas.93.19.10274
Giuliano, G., Pichersky, E., Malik, V., Timko, M., Scolnik, P., Cashmore, A. (1988). An evolutionarily conserved protein binding sequence upstream of a plant light-regulated gene. Proc. Natl. Acad. Sci. 85, 7089–7093. doi: 10.1073/pnas.85.19.7089
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644. doi: 10.1038/nbt.1883
Hattori, T., Totsuka, M., Hobo, T., Kagaya, Y., Yamamoto-Toyoda, A. (2002). Experimentally determined sequence requirement of ACGT-containing abscisic acid response element. Plant Cell Physiol. 43, 136–140. doi: 10.1093/pcp/pcf014
Horton, P., Park, K.-J., Obayashi, T., Fujita, N., Harada, H., Adams-Collier, C., et al. (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, 585–587. doi: 10.1093/nar/gkm259
Hu, B., Jin, J., Guo, A.-Y., Zhang, H., Luo, J., Gao, ,. G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinform 31, 1296–1297. doi: 10.1093/bioinformatics/btu817
Hua, X., Shen, Q., Li, Y., Zhou, D., Zhang, Z., Akbar, S., et al. (2022). Functional characterization and analysis of transcritpional regulation of sugar transporter SWEET13c in sugarcane Saccharum spontaneum. BMC Plant Biol. 22, 363. doi: 10.1186/s12870-022-03749-9
Hudson, M. E., Quail, P. H. (2003). Identification of promoter motifs involved in the network of phytochrome A-regulated gene expression by combined analysis of genomic sequence and microarray data. Plant Physiol. 133, 1605–1616. doi: 10.1104/pp.103.030437
Jeena, G. S., Kumar, S., Shukla, R. K. (2019). Structure, evolution and diverse physiological roles of SWEET sugar transporters in plants. Plant Mol. Biol. 100, 351–365. doi: 10.1007/s11103-019-00872-4
Jiao, Y., Wickett, N. J., Ayyampalayam, S., Chanderbali, A. S., Landherr, L., Ralph, P. E., et al. (2011). Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100. doi: 10.1038/nature09916
Khan, Q., Qin, Y., Guo, D.-J., Yang, L.-T., Song, X.-P., Xing, Y.-X., et al. (2023). A review of the diverse genes and molecules involved in sucrose metabolism and innovative approaches to improve sucrose content in sugarcane. Agronomy 13, 2957. doi: 10.3390/agronomy13122957
Kim, S.-R., Kim, Y., An, G. (1993). Identification of methyl jasmonate and salicylic acid response elements from the nopaline synthase (nos) promoter. Plant Physiol. 103, 97–103. doi: 10.1104/pp.103.1.97
Klepek, Y.-S., Geiger, D., Stadler, R., Klebl, F., Landouar-Arsivaud, L., Lemoine, R., et al. (2005). Arabidopsis POLYOL TRANSPORTER5, a new member of the monosaccharide transporter-like superfamily, mediates H+-symport of numerous substrates, including myo-inositol, glycerol, and ribose. Plant Cell 17, 204–218. doi: 10.1105/tpc.104.026641
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Le Gourrierec, J., Li, Y. F., Zhou, D. X. (1999). Transcriptional activation by Arabidopsis GT-1 may be through interaction with TFIIA–TBP–TATA complex. Plant J. 18, 663–668. doi: 10.1046/j.1365-313x.1999.00482.x
Lemoine, R. (2000). Sucrose transporters in plants: update on function and structure. Biochim. Biophys. Acta Biomembr. 1465, 246–262. doi: 10.1016/S0005-2736(00)00142-5
Letunic, I., Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, 256–259. doi: 10.1093/nar/gkz239
Li, B., Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinfo. 12, 323. doi: 10.1186/1471-2105-12-323
Li, J.-M., Zheng, D.-M., Li, L.-T., Qiao, X., Wei, S.-W., Bai, B., et al. (2015). Genome-wide function, evolutionary characterization and expression analysis of sugar transporter family genes in pear (Pyrus bretschneideri Rehd). Plant Cell Physiol. 56, 1721–1737. doi: 10.1093/pcp/pcv090
Lin, I. W., Sosso, D., Chen, L. Q., Gase, K., Kim, S. G., Kessler, D., et al. (2014). Nectar secretion requires sucrose phosphate synthases and the sugar transporter SWEET9. Nature 50, 546–549. doi: 10.1038/nature13082
Liu, J., Wang, F., Yu, G., Zhang, X., Jia, C., Qin, J., et al. (2015). Functional analysis of the maize C-repeat/DRE motif-binding transcription factor CBF3 promoter in response to abiotic stress. Intl J. Mol. Sci. 16, 12131–12146. doi: 10.3390/ijms160612131
Logemann, E., Parniske, M., Hahlbrock, K. (1995). Modes of expression and common structural features of the complete phenylalanine ammonia-lyase gene family in parsley. Proc. Natl. Acad. Sci. 92, 5905–5909. doi: 10.1073/pnas.92.13.5905
Lu, Y., D’Hont, A., Walker, D., Rao, P., Feldmann, P., Glaszmann, J.-C. (1994). Relationships among ancestral species of sugarcane revealed with RFLP using single copy maize nuclear probes. Euphytica 78, 7–18. doi: 10.1007/BF00021393
Ma, S., Li, Y., Li, X., Sui, X., Zhang, Z. (2019). Phloem unloading strategies and mechanisms in crop fruits. J. Plant Growth Regul. 38, 494–500. doi: 10.1007/s00344-018-9864-1
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–D226. doi: 10.1093/nar/gku1221
Menkens, A. E., Schindler, U., Cashmore, A. R. (1995). The G-box: a ubiquitous regulatory DNA element in plants bound by the GBF family of bZIP proteins. Trends Biochem. Sci. 20, 506–510. doi: 10.1016/S0968-0004(00)89118-5
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419. doi: 10.1093/nar/gkaa913
Moore, R. C., Purugganan, M. D. (2003). The early stages of duplicate gene evolution. Proc. Natl. Acad. Sci. 100, 15682–15687. doi: 10.1073/pnas.2535513100
Mukherjee, S. K. (1957). Origin and distribution of saccharum. Bot. Gaz. 119, 55–61. doi: 10.1086/335962
Mulyono, S. (2016). Identifying sugarcane plantation using LANDSAT-8 images with support vector machines. IOP Conf. Ser. Earth Environ. Sci. 47, 012008. doi: 10.1088/1755-1315/47/1/012008
Nover, L., Bharti, K., Döring, P., Mishra, S. K., Ganguli, A., Scharf, K.-D. (2001). Arabidopsis and the heat stress transcription factor world: how many heat stress transcription factors do we need? Cell Stress Chaperones 6, 177. doi: 10.1379/1466-1268(2001)006<0177:AATHST>2.0.CO;2
Nuruzzaman, M., Sharoni, A. M., Kikuchi, S. (2013). Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 47(1), 248. doi: 10.3389/fmicb.2013.00248
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T.-C., Mendell, J. T., Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Podzimska-Sroka, D., OShea, C., Gregersen, P. L., Skriver, K. (2015). NAC transcription factors in senescence: From molecular structure to function in crops. Plants 4, 412–448. doi: 10.3390/plants4030412
Reuscher, S., Akiyama, M., Yasuda, T., Makino, H., Aoki, K., Shibata, D., et al. (2014). The sugar transporter inventory of tomato: genome-wide identification and expression analysis. Plant Cell Physiol. 55, 1123–1141. doi: 10.1093/pcp/pcu052
Rushton, P. J., Somssich, I. E., Ringler, P., Shen, Q. J. (2010). WRKY transcription factors. Trends Plant Sci. 15, 247–258. doi: 10.1016/j.tplants.2010.02.006
Sablowski, R. W. M., Meyerowitz, E. M. (1998). A homolog of NO APICAL MERISTEM is an immediate target of the floral homeotic genes APETALA3/PISTILLATA. Cell 92, 93–103. doi: 10.1016/S0092-8674(00)80902-2
Sauer, N. (2007). Molecular physiology of higher plant sucrose transporters. FEBS Lett. 581, 2309–2317. doi: 10.1016/j.febslet.2007.03.048
Schenck, S., Crepeau, M., Wu, K., Moore, P., Yu, Q., Ming, R. (2004). Genetic diversity and relationships in native Hawaiian Saccharum officinarum sugarcane. J. Hered. 95, 327–331. doi: 10.1093/jhered/esh052
Shen, Q., Qian, Z., Wang, T., Zhao, X., Gu, S., Rao, X., et al. (2022). Genome-wide identification and expression analysis of the NAC transcription factor family in Saccharum spontaneum under different stresses. Plant Signal Behav. 17, 2088665. doi: 10.1080/15592324.2022.2088665
Slewinski, T. L., Meeley, R., Braun, D. M. (2009). Sucrose transporter1 functions in phloem loading in maize leaves. J. Exp. Bot. 60, 881–892. doi: 10.1093/jxb/ern335
Souer, E., Van Houwelingen, A., Kloos, D., Mol, J., Koes, R. (1996). The no apical meristem gene of Petunia is required for pattern formation in embryos and flowers and is expressed at meristem and primordia boundaries. Cell 85, 159–170. doi: 10.1016/S0092-8674(00)81093-4
Stein, O., Granot, D. (2019). An overview of sucrose synthases in plants. Front. Plant Sci. 10, 95. doi: 10.3389/fpls.2019.00095
Tamura, K., Stecher, G., Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Taylor, J. S., Raes, J. (2005). “Small-scale gene duplications,” in The Evolution of the Genome (Amsterdam, The Netherlands: Elsevier), 289–327.
Valifard, M., Fernie, A. R., Kitashova, A., Nägele, T., Schröder, R., Meinert, M., et al. (2023). The novel chloroplast glucose transporter pGlcT2 affects adaptation to extended light periods. J. Biol. Chem. 299, 104741. doi: 10.1016/j.jbc.2023.104741
Wang, M., Li, A. M., Liao, F., Qin, C. X., Chen, Z. L., Zhou, L., et al. (2022). Control of sucrose accumulation in sugarcane (Saccharum spp. hybrids) involves miRNA-mediated regulation of genes and transcription factors associated with sugar metabolism. GCB Bioener. 14, 173–191. doi: 10.1111/gcbb.12909
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, 49–49. doi: 10.1093/nar/gkr1293
Wang, T., Wang, B., Hua, X., Tang, H., Zhang, Z., Gao, R., et al. (2023). A complete gap-free diploid genome in Saccharum complex and the genomic footprints of evolution in the highly polyploid Saccharum genus. Nat. Plants. 9, 554–571. doi: 10.1038/s41477-023-01378-0
Wang, X. H., Yang, Q. H., Li, F. S., He, L. L., He, S. C. (2010). Characterization of the chromosomal transmission of intergeneric hybrids of Saccharum spp. and Erianthus fulvus by genomic in situ hybridization. Crop Sci. 50, 1642–1648. doi: 10.2135/cropsci2010.01.0004
Wendler, R., Veith, R., Dancer, J., Stitt, M., Komor, E. (1991). Sucrose storage in cell suspension cultures of Saccharum sp.(sugarcane) is regulated by a cycle of synthesis and degradation. Planta 183, 31–39. doi: 10.1007/BF00197564
Xiao, Q.-L., Zhen, L., Wang, Y.-Y., Hou, X.-B., Wei, X.-M., Xiao, Z., et al. (2022). Genome-wide identification, expression and functional analysis of sugar transporters in sorghum (Sorghum bicolor L.). J. Integr. Agric. 21, 2848–2864. doi: 10.1016/j.jia.2022.07.034
Xie, Q., Frugis, G., Colgan, D., Chua, N.-H. (2000). Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev. 14, 3024–3036. doi: 10.1101/gad.852200
Zhang, Q., Hua, X., Liu, H., Yuan, Y., Shi, Y., Wang, Z., et al. (2021). Evolutionary expansion and functional divergence of sugar transporters in Saccharum (S. spontaneum and S. officinarum). Plant J. 105, 884–906. doi: 10.1111/tpj.v105.4
Zhang, J., Zhang, Q., Li, L., Tang, H., Zhang, Q., Chen, Y., et al. (2019). Recent polyploidization events in three Saccharum founding species. Plant Biotechnol. J. 17, 264–274. doi: 10.1111/pbi.2019.17.issue-1
Keywords: sugar transporter, genome wide analysis, Erianthus rufipilus, gene family analysis, Saccharum officinarum, yeast-1-hybrid (Y1H) assay
Citation: Akbar S, Hua X, Zhang Y, Liu G, Wang T, Shi H, Li Z, Qi Y, Habiba H, Yao W, Zhang M-Q and Zhang J (2025) Genome-wide analysis of sugar transporter gene family in Erianthus rufipilus and Saccharum officinarum, expression profiling and identification of transcription factors. Front. Plant Sci. 15:1502649. doi: 10.3389/fpls.2024.1502649
Received: 27 September 2024; Accepted: 29 November 2024;
Published: 09 January 2025.
Edited by:
Xiangshu Dong, Yunnan University, ChinaReviewed by:
Chang-Cao Peng, South China Agricultural University, ChinaFan Chengming, Chinese Academy of Sciences (CAS), China
Copyright © 2025 Akbar, Hua, Zhang, Liu, Wang, Shi, Li, Qi, Habiba, Yao, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jisen Zhang, emppc2VuQDEyNi5jb20=
†ORCID: Jisen Zhang, orcid.org/0000-0003-1041-2757