 Dongtian Zang
Dongtian Zang Wenjia You1
Wenjia You1 Peisen Su
Peisen Su- 1College of Agriculture and Biology, Liaocheng University, Liaocheng, China
- 2Institute of Huanghe Studies, Liaocheng University, Liaocheng, China
Some peroxidases (PRXs) are involved in abiotic stress response. However, to the best of our knowledge, the effects of PRXs on agronomic traits including grain number per spike (GNS), spikelet number per spike (SNS) and spike length (SL) are also largely unknown. In our study, we cloned a wheat PRX gene TaPRX-2A and identified its function in controlling GNS by generating transgenic overexpression lines. The results showed that TaPRX-2A overexpression displayed lower GNS and shorter SL, compared with the wild-type plants. RNA-seq analysis indicated alterations in various pathways including flavonoid biosynthesis, lignin biosynthesis, phytohormone signaling, as well as sucrose and starch biosynthesis. Co-expression analysis showed that transcription factors, such as bHLH, WRKY, and bZIP may be involved in the regulation of various genes associated with these pathways. Our findings provide insights into the mechanisms by which PRXs regulate agronomic traits, illustrating potential applicability in crop improvement programs.
Introduction
Common wheat (Triticum aestivum L.) is a major cereal crop worldwide, providing ~20% of dietary calories to humans and constitutes 30% of global grain production (Shiferaw et al., 2013; Chand, 2009). Yield in wheat is a complex quantitative trait determined by three components: kernel weight, spike number per unit area, and grain number per spike (GNS) (Ma et al., 2018). Spike length (SL) is another crucial component primarily affecting GNS (Wu et al., 2014). Up to now, numerous genes, such as TaSPL17, TaAIRP2-1B, TaSus1, TaWRKY37-A1, TaMYC2-A1, TaMYB30-A1 have been identified that regulate GNS and SL in wheat (Liu et al., 2023; Wu et al., 2017; Zhang et al., 2023; Lin et al., 2024)
PRXs are antioxidant enzymes that catalyze the oxidation of many substrates and are widespread in several organisms, such as plants, animals, and microbes. PRXs are structurally categorized into nonhaem and haem (animal and nonanimal) (Welinder, 1992; Taurog, 1999). The PRX superfamily is functionally divided into three classes: I (e.g., APXs), II (lignin PRXs), and III (secretory PRXs) (Bhatt and Tripathi, 2011). Several studies have reported the class III families in plants to be multigenic. For example, 374, 159 and 169 class III PRXs exist in Triticum aestivum, T. urartu and Aegilops tauschii, respectively (Yan et al., 2019). Additionally, 119 members in maize, 75 class in Arabidopsis thaliana, 151 in Brachypodium distachyon and 155 in rice have been identified (Fawal et al., 2013).
In recent years, numerous class III PRXs have been functionally annotated, with many of them being involved in tolerance to various adverse stresses, especially abiotic stresses (drought and salt), and pathogen defense (Cosio and Dunand, 2009; Su et al., 2020, Su et al., 2023). For example, the overexpression of OsPrx30 enhanced the susceptibility of rice to bacterial blight by reducing H2O2 contents (Liu et al., 2021). In Arabidopsis thaliana, AtPrx64 overexpression improved the tolerance to aluminum-induced stress by suppressing ROS accumulation and enhancing lignin content (Wu et al., 2017). In previous study, we found that TaPRX-2A overexpression improves drought and salt tolerance in transgenic lines by activating ABA pathway and enhancing oxidative stress tolerance, such as higher antioxidant activities of peroxidase (POD), superoxide dismutase (SOD) and catalase (CAT) enzymes, and reduction of reactive oxygen species (ROS) accumulation, and lower levels of MDA content (Su et al., 2020, Su et al., 2023). Meanwhile, class III PRXs are involved in developmental processes. For example, AtPrx71 is involved in cell growth and inhibition of cell expansion through H2O2 accumulation (Raggi et al., 2015). At low temperatures, the apoplastic class III peroxidases PRX62 and PRX69 promote root hair growth in Arabidopsis thaliana (Pacheco et al., 2022). However, it remains to be elucidated whether PRXs are involved in regulating spike agronomic traits in wheat.
Common wheat is a major cereal crop worldwide. Its yield is determined by KNS, which is associated with SL. In this study, we functionally characterized a class III PRX gene TaPRX-2A which regulates SL. TaPRX-2A overexpression lines displayed lower GNS and shorter SL, which occurred through activating flavonoids, sucrose and starch biosynthesis as well as phytohormone (ABA, IAA and JA) pathways. Our findings revealing the function of TaPRX-2A in regulating GNS can provide researchers with new insights into the class III PRXs-related molecular mechanisms underlying GNS formation in wheat.
Materials and methods
Plant materials and growing conditions
The common wheat (T. aestivum cultivars “KN199”) and TaPRX-2A overexpression transgenic lines were used in this study and obtained from Shandong Agricultural University. The TaPRX-2A overexpression transgenic lines and WT “KN199” were as grown in the field at Liaocheng, Shandong, China in 2022 and 2023. Each transgenic line and WT plants were planted in a ten-row plot. Each row was 2.0 m in length with 20 plants, and the row spacing was 10 cm. The spikes from main shoots of 5 individual plants were randomly selected in each transgenic line and WT plants to measure spike length (SL), grain number per spike (GNS), plant height (PH), and thousand kernel
weight (TKW).
Isolation and cloning of TaPRX-2A and transgenic plant generation
TaPRX-2A was cloned from the wheat cultivar ‘Sumai 3’. The leaves of seedlings were harvested and ground into a powder in liquid nitrogen. For total RNA extraction, the TRIzol reagent (TransGen, Beijing) was used. It was reverse-transcribed into first-strand cDNA by HiScript II Q RT SuperMix (Vazyme, Nanjing). The full-length cDNA sequence of TaPRX-2A was obtained from Ensembl plants (http://plants.ensembl.org/index.html ; ID: TraesCS2A02G573900). We designed the gene-specific primers based on the TaPRX-2A sequence using Primer 5. Subsequently, TaPRX-2A was cloned into the vector PC414C and then ligated into the OE vector PC186 (pUbi::GWOE::Nos). The resulting vector was used to transform plants of the wheat cultivar ‘KN199’ using particle gun-mediated gene transformation (Yao et al., 2006).
Transcriptional profiling
The spikes of TaPRX-2A overexpression transgenic lines and WT plants at booting stage were harvested and immediately placed in liquid nitrogen for transcriptome analysis. The total RNA were extracted with RNAprep Pure Plant Kit (TIANGEN), then were sent to Metware Corporation (Wuhan) for transcriptional profiling. The transcriptome analysis was performed by Illumina HiSeq™ 2000 instrument. We removed the adapter sequences and low sequencing quality reads using Fastp (Chen et al., 2018). The paired-end reads from transcriptome sequencing data were mapped onto reference genome of wheat (http://plants.ensembl.org/Triticum_aestivum/Info/Index) by HISAT2 (Kim et al., 2015), then were assembled into transcripts using StringTie (Pertea et al., 2015). We normalized the expression data for each gene by the featureCounts (Liao et al., 2014). Then, we analyzed differentially expressed genes (DEGs) using the DEGseq R package with the default parameters (false discovery rate (FDR) < 0.05, |log2(fold change)|>= 1). Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of DEGs were performed by online website (https://www.genome.jp/kegg/). We performed the analysis of GO terms and KEGG pathways with the default parameters (Q-values <0.05).
Gene co-expression networks
To better elucidate functional relationships of DEGs in flavonoid biosynthesis, lignin biosynthesis, phytohormone signaling, sucrose and starch biosynthesis, we constructed protein interaction network of the Inter- and intra-pathways and the visualization of gene co-expression networks were performed through software Cytoscape (Shannon et al., 2003).
Co-expression pattern cluster and transcription factor binding site analysis
To explore the binding sites of TFs to the promoters of the genes involved in flavonoid biosynthesis, lignin biosynthesis, phytohormone signaling as well as sucrose and starch biosynthesis, 2000 bp sequences upstream of the initiation codon were retrieved. The binding sites of the TFs were analyzed using the TBtools software (Chen et al., 2020). The co-expression pattern cluster of the TFs and these genes were analyzed utilizing R (base package). We grouped the DEGs with the same expression trends into a data set by analyzing the variations in the patterns of mRNA expression abundance among the TaPRX-2A overexpression lines and WT plants. The data set was then drawn.
Expression pattern analysis
The leaves of “Sumai 3” were harvested from seedling and ground into powder in liquid nitrogen for RNA extraction. We used the TRIzol reagent (TransGen, Beijing) to extract the total RNA. The total RNA was reversed into First strand cDNA by HiScript II Q RT SuperMix (Vazyme, Nanjing), which was used as the template for expression analysis of these genes in different pathways. The expression levels of the genes were performed by quantitative real time PCR using the Roche LightCycler ®480 system (Roche, Germany). The wheat 18SrRNA gene was utilized as internal controls for normalization. The quantifications of these genes were calculated using the 2^(−ΔΔCT) method. Each experiment collected at least three independent biological replicates. All the qRT–PCR primers used in this study are listed in Supplementary Table S4.
Results
Overexpression of TaPRX-2A exhibits negative effects on GNS
In a previous study, we analyzed the expression levels of TaPRX-2A, TaPRX-2B and TaPRX-2C based on transcripts per kilobase million (TPM) values collected from the WheatOmics site (Seifert et al., 2016; Li et al., 2018). TaPRX-2A was highly expressed in the double ridge stage (KNIV) and microspore embryogenesis S3 (Supplementary Figure S1). To understand the association between the alterations in TaPRX-2A expression and spike development, we generated TaPRX-2A-overexpression lines in the common wheat ‘KN199’ background and successfully genotyped three independent homozygous lines. The TaPRX-2A-overexpression lines and WT plants were planted in a greenhouse and their agronomic traits were observed. Plant height did not differ significantly between the transgenic lines and WT plants (Figure 1A, B). The spike morphology differed in SL and GNS between the two. Compared with WT (8 cm), the SL (5.89 cm) was substantially shortened in the transgenic lines (Figure 1C, D) and the GNS (31.25) decreased markedly (45.6) (Figure 1E). These agronomic traits, including kernel length, kernel width, and thousand-grain weight did not vary conspicuously between the transgenic lines and the WT (Figures 1F–J). These results confirmed the function of TaPRX-2A in regulating GNS.
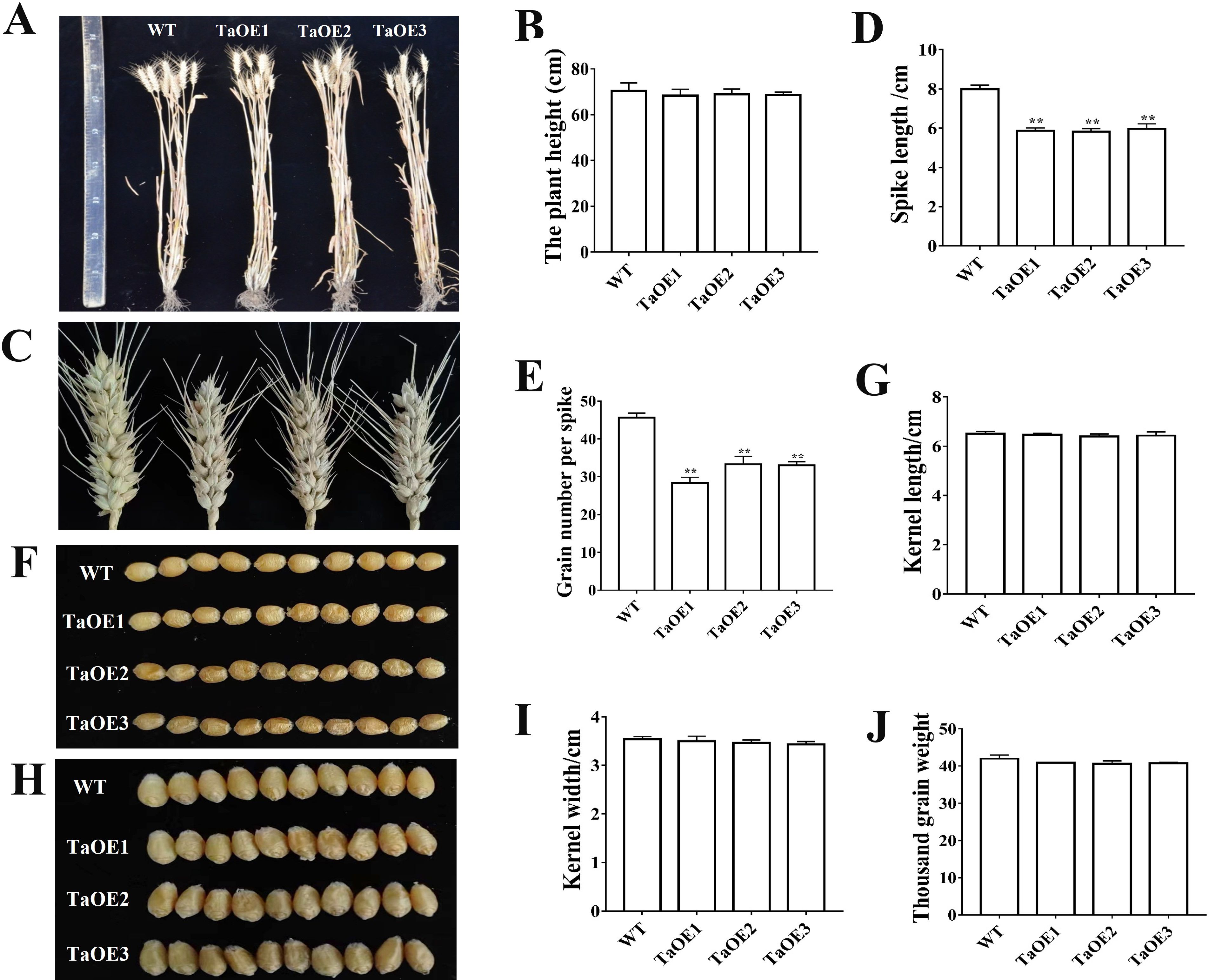
Figure 1. Phenotypic characterization of spike traits in TaPRX-2A overexpression transgenic lines and WT plants. (A) Representative images of plant height between TaPRX-2A overexpression transgenic lines and WT plants. (B) Plant height. (C) Representative images of spikes between TaPRX-2A overexpression transgenic lines and WT plants. (D) Spike length (SL). (E) Grain number per spike (GNS). (F-I) Representative images showing length and width of kernels from TaPRX-2A overexpression transgenic lines and WT plants. (J) Thousand grain weight. Each experiment was conducted in three replicates and values are means ± SD (n = 6). ** indicates a significant difference at P < 0.01.
TaPRX-2A overexpression alters gene expression profiles in wheat
To further elucidate the regulatory mechanisms by which TaPRX-2A affects GNS in wheat, the transcriptomes of TaPRX-2A overexpression lines and WT plants were compared. We found 1317 DEGs between the two, including 548 down and 769 upregulated ones (Figure 2A, Supplementary Figure S2, Supplementary Table S1). In addition, GO classification and KEGG analyses revealed that biological processes (biological regulation, response to stimulus, developmental processes, growth and reproductive processes), molecular functions (catalytic, antioxidant, binding, and structural molecule activities) and cell components (protein-containing complex and cellular anatomical entity) were highly enriched terms (Figure 2B, Supplementary Table S2). KEGG enrichment analyses indicated the DEGs to be mainly enriched in hormone signal transduction, starch and sucrose metabolism, phenylpropanoid biosynthesis, and MAPK signaling pathway (Figure 2C, Supplementary Table S2).
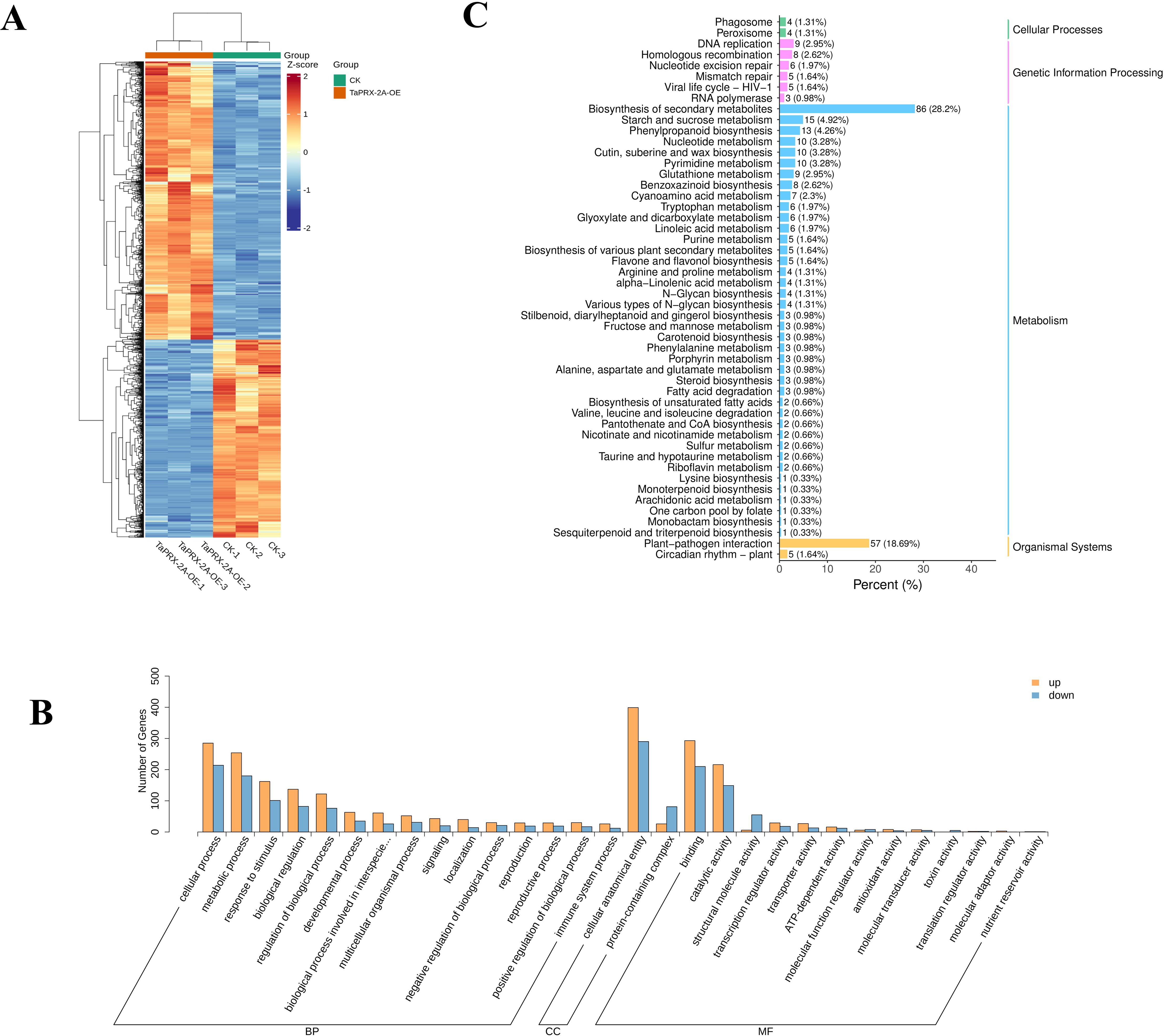
Figure 2. Transcriptome analysis in TaPRX-2A overexpression transgenic lines and WT plants. (A) The heatmap of the DEGs between TaPRX-2A overexpression transgenic lines and WT plants. The heatmap was created based on FPKM expression values. (B) GO classification of DEGs between TaPRX-2A overexpression transgenic lines and WT plants. (C) KEGG pathway enrichment between TaPRX-2A overexpression transgenic lines and WT plants. Each sample was conducted in three replicates and named “CK-1”, “CK-2”, and “CK-3”.
Enriched DEGs in TaPRX-2A overexpression lines in flavonoid and lignin biosynthesis pathways
We generated a network illustrating the relationships between TaPRX-2A, flavonoid and lignin biosynthesis to further study how TaPRX-2A affects GNS. The expression levels of 19 genes associated with the flavonoid and lignin biosynthesis pathway were altered, among which 4-coumarate-CoA ligase (4CL), flavonol synthase (FLS), and flavonoid 3’-monooxygenase (CYP75B1) were upregulated compared to the WT plants, while anthocyanidin reductase (ANR), flavonoid 3’,5’-hydroxylase (CYP75A), and flavonol-3-O-L-rhamnoside-7-O-glucosyltransferase (UGT73C6) were downregulated (Figure 3A, Supplementary Table S3). In addition, the expression profiles of PRX (TraesCS6D02G054600(1.79326532777333), TraesCS2A02G573900(3.1764958969293), TraesCS6A02G047400(1.22826501558943), TraesCS2B02G613900(1.08348514467672), TraesCS6D02G127100(1.10257940548889), TraesCS3B02G209700(1.63944763210169), TraesCS6D02G108400(1.11318941408978), TraesCS5A02G323200(-1.05843591801709), TraesCS5D02G330300(-1.27076575042592), TraesCS7A02G452900(-1.44407938464799), TraesCS3B02G578000(-1.19525771883893)) and flavonol-3-O-glucoside (FG2) (TraesCS4A02G385500(1.71126108560809), TraesCS5A02G325200(-1.24485547304725)) were differed. Some genes are upregulated in the overexpression lines, including TraesCS6D02G054600, TraesCS2A02G573900, TraesCS6A02G047400, TraesCS2B02G613900, TraesCS6D02G127100, TraesCS3B02G209700, TraesCS6D02G108400, TraesCS4A02G385500, while others are downregulated, such as TraesCS5A02G323200, TraesCS5D02G330300, TraesCS7A02G452900, TraesCS3B02G578000, TraesCS5A02G325200 (Supplementary Table S3). In addition, the expression levels of the flavonoid and lignin biosynthesis pathways genes were measured, which supported the transcriptome data set (Supplementary Figure S3). Detailed information is displayed in Supplementary Table S3.
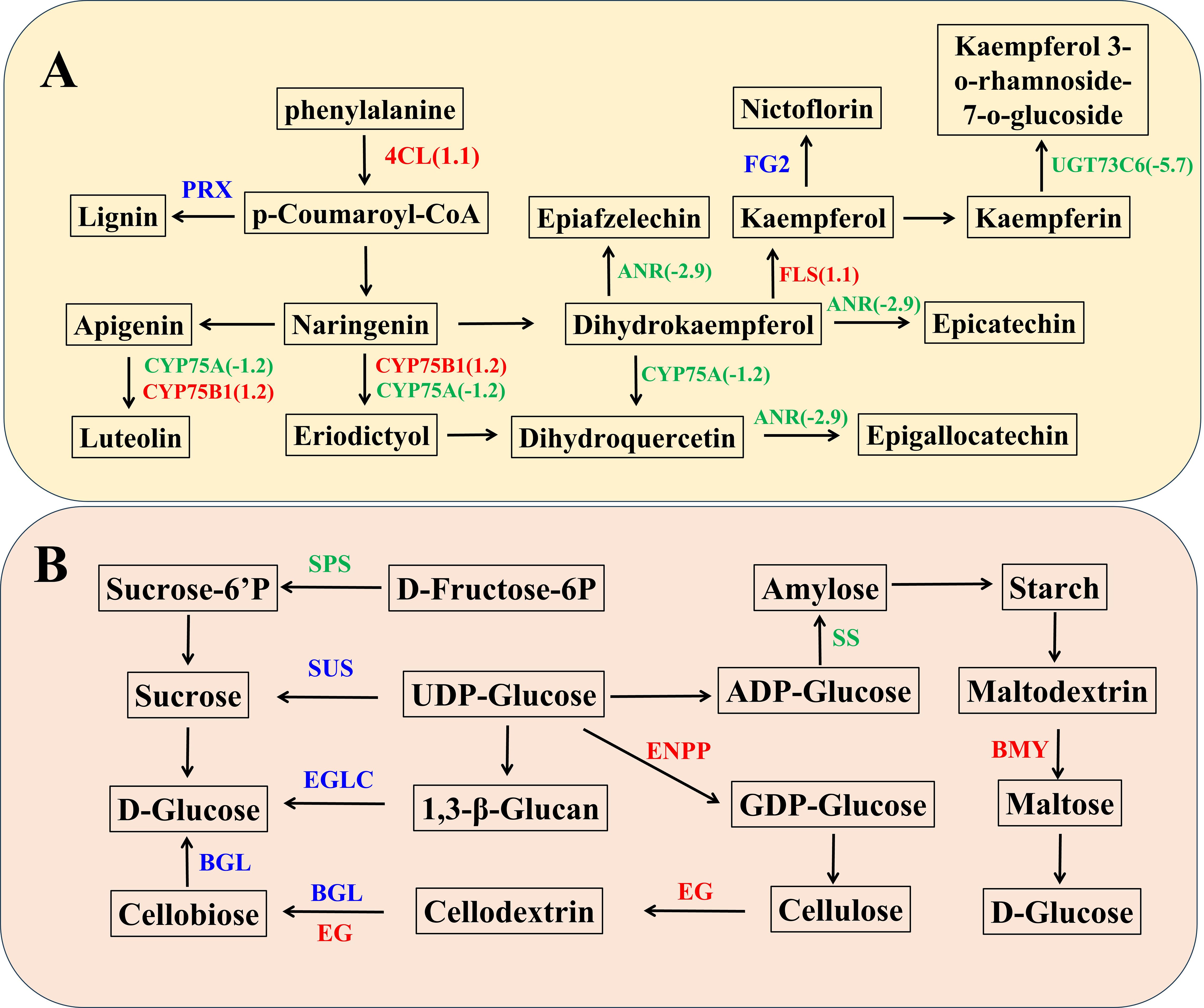
Figure 3. TaPRX-2A overexpression alters flavonoid, sucrose and starch biosynthesis pathway. (A) The network was constructed based on known genes expression levels involved in flavonoid pathway. (B) The network was constructed based on known genes expression levels involved in sucrose and starch biosynthesis pathway. The blue color represents gene families encoding the same group of proteins with similar functions, where individual genes within these families show either up-regulation or down-regulation, while red represented upregulated and green represented downregulated in TaPRX-2A overexpression transgenic lines compared to WT plants, respectively. 4CL, 4-coumarate–CoA ligase; FLS, flavonol synthase; CYP75B1, flavonoid 3’-monooxygenase; ANR, anthocyanidin reductase; UGT73C6, flavonol-3-O-L-rhamnoside-7-O-glucosyltransferase; CYP75B1, flavonoid 3’-monooxygenase; ENPP, ectonucleotide pyrophosphatase; BMY, beta-amylase; EG, endoglucanase; SPS, sucrose-phosphate synthase; SS, starch synthase; BGL, beta-glucosidase; EGLC, glucan endo-1,3-beta-D-glucosidase.
Enriched DEGs in TaPRX-2A overexpression lines in sucrose and starch biosynthesis pathways
TaPRX-2A overexpression modulated the sucrose and starch biosynthesis pathway. The transcriptomic data revealed significant differences in the expression profiles of many sucrose and starch biosynthesis-related genes. Among them, ectonucleotide pyrophosphatase (ENPP), beta-amylase (BMY) and endoglucanase (EG) were upregulated, while sucrose-phosphate synthase (SPS) and starch synthase (SS) were downregulated compared to the WT plants (Figure 3B, Supplementary Table S3). In addition, the expression profiles of sucrose synthase (SUS) (TraesCS4B02G167500(1.27482309742693), TraesCS4D02G169800(-6.32054333672797)), β-glucosidase (BGL) (TraesCSU02G036600(1.05689194722632), TraesCS4D02G038500(9.09766571668372), TraesCS2B02G550300(-1.17328950677694), TraesCS2B02G401500(-1.47248308622032)) and glucan endo-1,3-β-D-glucosidase (EGLC) (TraesCS6D02G099100(1.1146060158768), TraesCS4D02G191900(-1.14398141299911)) were varied. Some genes are upregulated in the overexpression lines, including TraesCS4B02G167500, TraesCSU02G036600, TraesCS4D02G038500, TraesCS6D02G099100, while others are downregulated, such as TraesCS4D02G169800, TraesCS2B02G550300, TraesCS2B02G401500, TraesCS4D02G191900 (Supplementary Table S3). In addition, the expression levels of these sucrose and starch biosynthesis pathway-associated genes were ascertained, which supported the transcriptome data set. Detailed information is displayed in Supplementary Table S3.
Enriched DEGs in TaPRX-2A overexpression lines in hormone-related pathways
Our transcriptome data indicated that TaPRX-2A overexpression also modulated hormone pathways, including ABA, IAA, JA and GA. Therefore, we generated a network to explore the relationships between TaPRX-2A overexpression and hormone-associated pathways. The results showed that several genes in the plant hormone pathways were enriched: six ABA-related genes were identified, among which 15-cis-phytoene synthase (ctrB), protein phosphatase 2C (PP2C) and serine/threonine-protein kinase SRK2 (SnRK2) were upregulated in the transgenic lines, while abscisic acid receptor PYR/PYL family (PYR/PYL) and xanthoxin dehydrogenase (ABA2) were downregulated compared to the WT plants. Further, ten auxin-related genes were found, among which indole-3-pyruvate monooxygenase (YUCCA), acetylserotonin O-methyltransferase (ASMT), auxin response factor (ARF) and SAUR family protein (SAUR) were upregulated, while amidase (AMI) and auxin-responsive protein (AUX/IAA) were downregulated in the transgenic lines compared to the WT plants. In addition, five JA-related genes lipoxygenase (LOX), hydroperoxide dehydratase (AOS), MFP2 and MYC2, as well as five GA-related genes (CYP714B and TF) were altered upon TaPRX-2A overexpression. The detailed information is displayed in Figure 4 and Supplementary Table S3.
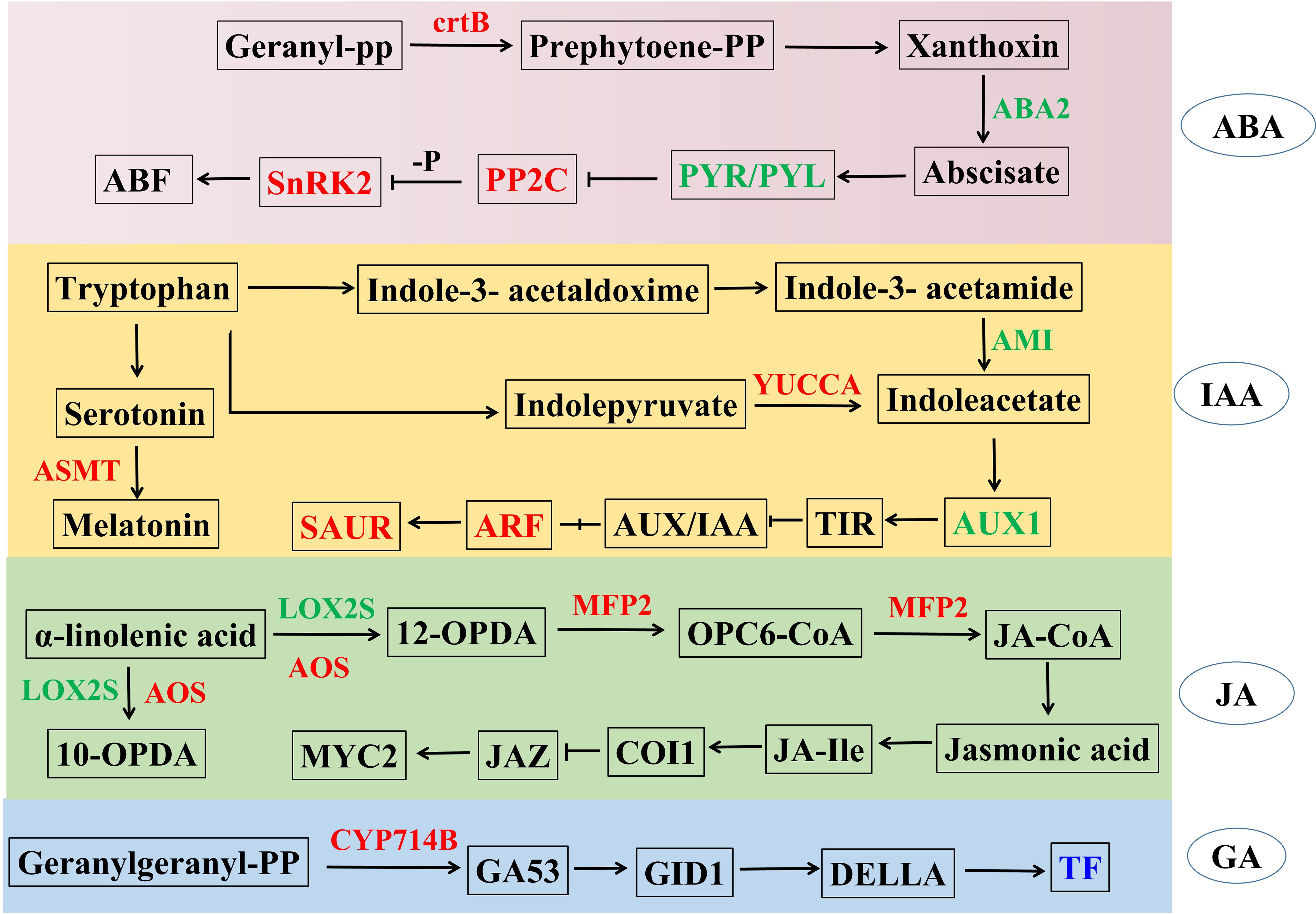
Figure 4. TaPRX-2A overexpression alters plant hormone pathways including ABA, IAA, JA and GA. The network was constructed based on known genes expression levels involved in plant hormone pathways. The blue color represents gene families encoding the same group of proteins with similar functions, where individual genes within these families show either up-regulation or down-regulation, while red represented upregulated and green represented downregulated in TaPRX-2A overexpression transgenic lines compared to WT plants, respectively. ctrB, 15-cis-phytoene synthase; ABA2, xanthoxin dehydrogenase; PP2C, protein phosphatase 2C; PYL, abscisic acid receptor PYR/PYL family; SnRK2, serine/threonine-protein kinase SRK2; YUCCA, indole-3-pyruvate monooxygenase; ASMT, acetylserotonin O-methyltransferase, AUX1, auxin influx carrier; ARF, auxin response factor; SAUR, SAUR family protein; LOX, lipoxygenase; AOS, hydroperoxide dehydratase; MFP2, enoyl-CoA hydratase/3-hydroxy acyl-CoA dehydrogenase; IF, phytochrome-interacting factor; CYP714B, gibberellin 13-oxidase.
To further elucidate the relevant functional relationships, the gene–gene relationships focusing on the flavonoid and lignin biosynthesis, sucrose and starch biosynthesis as well as hormone-related pathways upon TaPRX-2A overexpression were analyzed employing STRING (http://string-db.org) (Supplementary Figure S4). These genes encompassed 4CL, CAT, ABA2, MFP2, BGL, PP2C, EG, ANR, PP2C, PYR/PYL, AOS, LOX, GST, CYP75A, CYP75B1, SUS, SPS, SS and ctrB. They were involved in the same biosynthesis pathway and interacted with each other. For example, CYP75A (TraesCS4A02G446400) with CYP75B1 (TraesCS6D02G015200); BGL (TraesCS4D02G038500, TraesCS2B02G550300 and TraesCS2B02G401500) with EG (TraesCS6A02G093200 and TraesCS6D02G112100); and AOS (TraesCS6D02G172200) with LOX (TraesCS5D02G013400) and TraesCS5B02G006500). In addition, the crosstalk between different pathways was also elucidated, among which MFP2 (TraesCS6D02G116200) in the JA pathway interacted with ABA2 (TraesCS2B02G143900 and TraesCS2D02G124800) in the ABA pathway, CAT (TraesCS6D02G048300) in the antioxidant pathway and 4CL (TraesCS6D02G141700) in the flavonoid pathway. ANR (TraesCS2D02G483100) in the flavonoid pathway interplayed with ctrB (TraesCS5D02G365100) in the ABA pathway. The protein–protein interactive relationships are detailed in Supplementary Figure S4.
DEGs related to flavonoid, phytohormon, sucrose and starch biosynthesis pathways are more abundant in TaPRX-2A overexpression lines
To further elucidate the regulatory relationships between the different pathways, we performed a co-expression cluster analysis of the genes associated with them and all differentially expressed TFs. The Fragments Per Kilobase of exon model per Million mapped fragments (FPKM) values of all DEGs were standardized using R language functions (scale ()), and then cluster analysis was carried out. We divided all DEGs into ten clusters based on different expression profile (Figure 5A). Further analysis of the various types of TFs in the different clusters showed that NAC, WRKY, bHLH, FAR1, AP2/ERF and MYB were more abundant in TaPRX-2A overexpression lines (Figure 5B). The promoter binding sites (sequences 2000 bp upstream of the start site) of these genes from various pathways were analyzed. Compared to the promoters of 4CL, CYP75A, CYP75B1, ANR, UGT73C6, FLS, PRX, SPS, SUS, BGL, EG, EGLC, ENPP, SS, GST, RRM1, GGCT, CAT, ABA2, PP2C, PYR/PYL, SnRK2, YUCCA, AMST, LOX, and TF, more binding sites were identified for B3, bHLH, WRKY, C2H2, and bZIP (Figure 5C, D). TaPRX-2A overexpression enhanced the expression of these genes belonging to different pathways by regulating the expression of TFs, such as B3, bHLH, WRKY, and bZIP.
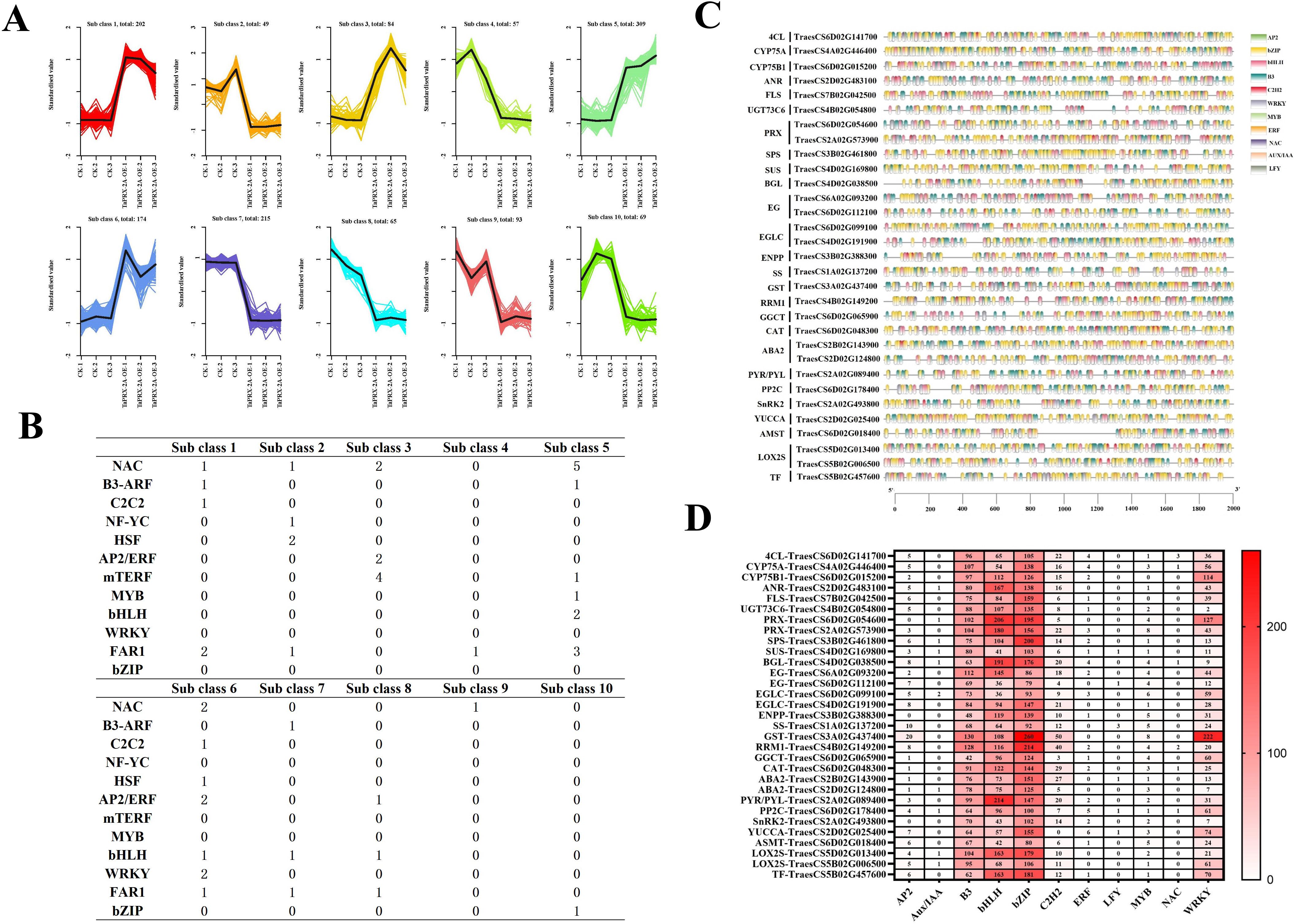
Figure 5. (A) The cluster analysis of co-expression patterns of all DEGs. (B) Quantity statistics of the transcription factors in different subclasses. (C) Promoter binding site analysis of these genes in flavonoid biosynthesis, lignin biosynthesis, phytohormone, sucrose and starch biosynthesis pathways. (D) The cis-elements distribution of genes in flavonoid biosynthesis, lignin biosynthesis, phytohormone, sucrose and starch biosynthesis pathways. The upstream (2000 bp DNA sequence) of these genes in different pathways was used to analyze IF binding site and were represented with different colors.
Discussion
Common wheat (Triticum aestivum L.) is a major cereal crop worldwide. Its yield is determined by three components: kernel weight, spike number per unit area, and GNS (Ma et al., 2018). SL is a crucial component of yield and primarily affects KNS (Wu et al., 2014). So far, a few wheat SL and GNS-related genes, such as TaSPL17, TaAIRP2-1B, TaSus1, TaWRKY37-A1, TaMYC2-A1 and TaMYB30-A1 have been identified (Liu et al., 2023; Wu et al., 2017; Zhang et al., 2023; Lin et al., 2024). Here, we functionally characterized the role of the class III PRX gene, TaPRX-2A in controlling wheat GNS by activating various metabolic pathways.
GNS is a critical agronomic trait that affects wheat yield strongly. Several studies have reported the control of GNS by various genes. For example, triple mutants of the wheat sucrose synthase gene TaSus1 displayed lower than the wild-type plants (Shen et al., 2024). The overexpression of TaSPL17 enhanced GNS and SL as indicated by a high−resolution genotype–phenotype map in wheat (Liu et al., 2023). A mutation in the TF bZIPC and upon interaction with FT2 reduced the SNS in tetraploid wheat (Glenn et al., 2023). TaMYC2-A1 and TaMYB30-A1 are involved in wheat spike development as evidenced by integrated multiomics, transcriptional networks, GWAS, and genetic analyses (Lin et al., 2024). However, research on the function of the wheat PRX in the context of GNS and SL has been limited. This study validated the effects of TaPRX-2A by the phenotypic analysis of newly generated overexpression transgenic lines. The results indicated that TaPRX-2A overexpression displayed lower GNS and shorter SL. Further study will be explored the effect of TaPRX-2A mutation on GNS and provide materials for crop gene pyramiding molecular breeding.
Extensive research has reported the functional role of starch and sucrose biosynthesis pathways as GNS determinants. For example, the wheat sucrose synthase gene TaSus1 controlled grain number per spike by influencing the fructose contents. The result showed that expression levels of TaSus-A1 was higher than TaSus-B1 or TaSus-D1 during early carpel development and two single-nucleotide polymorphisms in TaSus-A1 contributed differently to GNS (Shen et al., 2024). Additionally, suppressed ABA signal transduction reduced grain number by promoting sucrose use in wheat under drought conditions (Zhang et al., 2020). The MATE transporter GFD1 in rice controls the grain-filling duration, grain size and number by interacting with two sugar transporters, OsSWEET4 and OsSUT2 (Sun et al., 2023). These studies suggest that starch and sucrose biosynthesis is involved in wheat spike development. They are consistent with the present findings, wherein TaPRX-2A overexpression activated the expression levels of genes including SPS, SUS, EGLC, ENPP, EG, SS and BGL involved in the starch and sucrose biosynthesis pathway. Overall, its potential application for wheat improvement to explore the contribution of TaPRX-2A, TaPRX-2B, and TaPRX-2D, interaction between PRXs and starch and sucrose biosynthesis-related genes, and examine different haplotypes of TaPRX-2A.
Some study showed that PRX enzymes regulate the polymerization of lignin monomers (Liu et al., 2018). From example, the previous study showed that ZmWRKY86 regulated the peroxidase gene ZmPRX1 to control drought tolerance by promoting root development and lignification in maize (Zhai et al., 2024). Overexpression of swpa4 peroxidase increases the lignin content in sweet potato (Kim et al., 2008). Consistent with these studies, we also found that TaPRX-2A overexpression alters the lignin biosynthesis and expression levels of some WRKYs. In addition, our previous research found that TaPRX-2A overexpression reduced ROS levels by enhancing oxidative stress tolerance, such as superoxide dismutase (SOD), peroxidase (POD) and catalase (CAT) enzymes (Su et al., 2020, Su et al., 2023). In Arabidopsis, the class III peroxidases PRX62 and PRX69 promote root hair growth by modulating ROS-homeostasis at low temperature (Pacheco et al., 2022). In our study, TaPRX-2A overexpression alters the expression levels of antioxidant-related genes, such as CAT、GST、RRM1. Taken together, we predicted that TaPRX-2A may control grain number per spike by WRKYs regulation and controlling ROS levels.
Some previous studies have shown that FTs affected SNS. For example, FT2 overexpression reduced spikelet number in Brachypodium distachyon and barley, and its loss-of-function mutations increased SNS (Shaw et al., 2019; Glenn et al., 2022). Consistent with these studies, we identified that TaPRX-2A overexpression altered the expression levels of FT. These results can be explained by TaPRX-2A effectively influencing GNS. In addition, several pieces of evidence indicate the crosstalk between starch and sucrose biosynthesis, flavonoids and hormone-related pathways. For example, the CIRCADIAN CLOCK ASSOCIATED1 (OsCCA1) mediated panicle development and tiller growth through strigolactone signaling and sugar sensing (Wang et al., 2020). ABA signal transduction regulates grain number by promoting sucrose catabolism (Zhang et al., 2020). ABA is involved in tuning stem elongation and ear development by collaborating with lignin and flavonoid biosynthesis in maize (Zea mays L.) (Gao et al., 2023). Our previous studies found that TaPRX-2A conferred drought tolerance in wheat via the ABA pathway. This study also identified that TaPRX-2A controlled GNS via the ABA pathway based on transcriptomics data. Thus, our evidence combined with those from previous studies suggests that TaPRX-2A regulated drought tolerance and developmental balance by activating the ABA signaling pathway. Further study is warranted to explore the networks of growth regulation and stress response-related crosstalk between TaPRX-2A and ABA and elucidate critical links, providing a high potential for application in wheat improvement.
Conclusion
In this study, we characterized the function of the class III peroxidase gene TaPRX-2A in controlling grain number per spike in wheat. The results demonstrated that TaPRX-2A overexpression significantly decreased grain number per spike in transgenic wheat by activating the starch and sucrose biosynthesis, flavonoid biosynthesis, lignin biosynthesis, and phytohormone signaling pathways. This work and its finding will deepen the understanding of wheat GNS development mechanisms and have high potential application value for wheat improvement.
Data availability statement
All data generated or analyzed during this study are included in this published article and its Supplementary Information Files. The raw RNA-Seq data in this study were submitted to the NCBI Sequence ReadArchive (accession numbers: PRJNA1206736).
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
DZ: Formal analysis, Writing – original draft. WY: Software, Writing – original draft. YW: Software, Writing – original draft. PW: Formal analysis, Writing – original draft. ZW: Formal analysis, Writing – original draft. QY: Formal analysis, Writing – original draft. SC: Formal analysis, Writing – original draft. PS: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by Natural Science Foundation of Shandong Province (ZR2022QC129); Doctoral research start-up funds, Liaocheng University (318052018); Youth innovation team project of Shandong Province (2023KJ208); Transgenic and phenotypic identification of wheat, Liaocheng University (318/K24LD76); Introduction of fresh corn varieties and determination of nutritional constituents, Liaocheng University (318/K24LD75).
Acknowledgments
We thank all contributors for their work and would like to thank the editor and reviewers for their valuable comments and suggestions. We would like to thank EditChecks (https://editchecks.com.cn/) for providing linguistic assistance during the preparation of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1501029/full#supplementary-material
Supplementary Figure 1 | The expression levels of TaPRX-2A, TaPRX-2B, TaPRX-2C based on transcripts per kilobase million (TPM) values collected from the WheatOmics site.
Supplementary Figure 2 | The count of all detected DEGs in TaPRX-2A overexpression lines, compared with WT plants.
Supplementary Figure 3 | The relative expression of these genes in flavonoid biosynthesis, lignin biosynthesis, phytohormone, sucrose and starch biosynthesis pathways and TFs. The wheat gene 18SrRNA was as an endogenous control. The relative expression of these genes was calculated by the cycle threshold (Ct) values using formula 2–ΔΔCT. Each experiment was conducted in three replicates and values are means ± SD.
Supplementary Figure 4 | The interaction networks of differentially expressed genes in flavonoid biosynthesis, lignin biosynthesis, phytohormone, sucrose and starch biosynthesis pathways.
Supplementary Table 3 | DEGs involved in flavonoid biosynthesis, lignin biosynthesis, phytohormone, sucrose and starch biosynthesis pathways.
References
Bhatt, I., Tripathi, B. N. (2011). Plant peroxiredoxins: catalytic mechanisms, functional significance and future perspectives. Biotechnol. Adv. 29, 850–859. doi: 10.1016/j.bioteChadv.2011.07.002
Chand, R. R. (2009). “Challenges to ensuring food security through wheat,” in CAB reviews perspectives in agriculture veterinary science nutrition and natural resources, vol. 4. . doi: 10.1079/PAVSNNR20094065
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. . Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, S., Zhou, Y., Chen, Y., Gu, J. (2018). Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cosio, C., Dunand, C. (2009). Specific functions of individual class III peroxidase genes. J. Exp. Bot. 60, 391–408. doi: 10.1093/jxb/ern318
Fawal, N., Li, Q., Savelli, B., Brette, M., Passaia, G., Fabre, M., et al. (2013). PeroxiBase: a database for large-scale evolutionary analysis of peroxidases. Nucleic Acids Res. 41, D441–D444. doi: 10.1093/nar/gks1083
Gao, J., Zhang, Y., Xu, C., Wang, X., Wang, P., Huang, S. (2023). Abscisic acid collaborates with lignin and flavonoid to improve pre-silking drought tolerance by tuning stem elongation and ear development in maize (Zea mays L.). Plant J. 114, 437–454. doi: 10.1111/tpj.16147
Glenn, P., Woods, D. P., Zhang, J., Gabay, G., Odle, N., Dubcovsky, J. (2023). Wheat bZIPC1 interacts with FT2 and contributes to the regulation of spikelet number per spike. Theor. Appl. Genet. 136, 237. doi: 10.1007/s00122-023-04484-x
Glenn, P., Zhang, J., Brown-Guedira, G., DeWitt, N., Cook, J. P., Li, K., et al. (2022). Identification and characterization of a natural polymorphism in FT-A2 associated with increased number of grains per spike in wheat. Theor. Appl. Genet. 135, 679–692. doi: 10.1007/s00122-021-03992-y
Kim, Y., Kim, C., Song, W., Park, D., Kwon, S., Lee, H., et al. (2008). Overexpression of sweet potato swpa4 peroxidase results in increased hydrogen peroxide production and enhances stress tolerance in tobacco. Planta. 227, 867–881. doi: 10.1007/s00425-007-0663-3
Kim, D., Langmead, B., Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Li, Y., Fu, X., Zhao, M., Zhang, W., Li, B., An, D., et al. (2018). A genome-wide view of transcriptome dynamics during early spike development in bread wheat. Sci. Rep. 8, 15338. doi: 10.1038/s41598-018-33718-y
Liao, Y., Smyth, G. K., Shi, W. (2014). Feature Counts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 30, 923–930. doi: 10.1093/bioinformatics/btt656
Lin, X., Xu, Y., Wang, D., Yang, Y., Zhang, X., Bie, X., et al. (2024). Systematic identification of wheat spike developmental regulators by integrated multi-omics and genetic analyses. Mol. Plant 17, 438–459. doi: 10.1016/j.molp.2024.01.010
Liu, Y., Chen, J., Yin, C., Wang, Z., Wu, H., Shen, K., et al. (2023). A high-resolution genotype-phenotype map identifies the TaSPL17 controlling grain number and size in wheat. Genome Biol. 24, 196. doi: 10.1186/s13059-023-03044-2
Liu, H., Dong, S., Li, M., Gu, F., Yang, G., Guo, T., et al. (2021). The Class III peroxidase gene OsPrx30, transcriptionally modulated by the AT-hook protein OsATH1, mediates rice bacterial blight-induced ROS accumulation. J. Integr. Plant Biol. 63, 393–408. doi: 10.1111/jipb.13040
Liu, Q., Luo, L., Zheng, L. (2018). Lignins: biosynthesis and biological functions in plants. Int. J. Mol. Sci. 19, 335. doi: 10.3390/ijms19020335
Ma, F., Xu, Y., Ma, Z., Li, L., An., D. (2018). Genome-wide association and validation of key loci for yield-related traits in wheat founder parent Xiaoyan 6. Mol. Breeding. 38, 91. doi: 10.1007/s11032-018-0837-7
Pacheco, J. M., Ranocha, P., Kasulin, L., Fusari, C. M., Servi, L., Aptekmann, A. A., et al. (2022). Apoplastic class III peroxidases PRX62 and PRX69 promote Arabidopsis root hair growth at low temperature. Nat. Commun. 13. doi: 10.1038/s41467-022-28833-4
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Raggi, S., Ferrarini, A., Delledonne, M., Dunand, C., Ranocha, P., De Lorenzo, G., et al. (2015). The arabidopsis class III peroxidase atPRX71 negatively regulates growth under physiological conditions and in response to cell wall damage. Plant Physiol. 169, 2513–2525. doi: 10.1104/pp.15.01464
Seifert, F., Bössow, S., Kumlehn, J., Gnad, H., Scholten, S. (2016). Analysis of wheat microspore embryogenesis induction by transcriptome and small RNA sequencing using the highly responsive cultivar “Svilena. BMC Plant Biol. 16, 97. doi: 10.1186/s12870-016-0782-8
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shaw, L. M., Lyu, B., Turner, R., Li, C., Chen, F., Han, X., et al. (2019). FLOWERING LOCUS T2 regulates spike development and fertility in temperate cereals. J. Exp. Bot. 70, 193–204. doi: 10.1093/jxb/ery350
Shen, L., Zhang, L., Yin, C., Xu, X., Liu, Y., Shen, K., et al. (2024). The wheat sucrose synthase gene TaSus1 is a determinant of grain number per spike. Crop J. 12, 295–300. doi: 10.1016/j.cj.2023.11.007
Shiferaw, B., Smale, M., Braun, H. J., Duveiller, E., Muricho, G. (2013). Crops that feed the world 10 Past successes and future challenges to the role played by wheat in global food security. Food Secur. 5, 291–317. doi: 10.1007/s12571-013-0263-y
Su, P., Sui, C., Niu, Y., Li, J., Wang, S., Sun, F., et al. (2023). Comparative transcriptomic analysis and functional characterization reveals that the class III peroxidase gene TaPRX-2A regulates drought stress tolerance in transgenic wheat. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1119162
Su, P., Yan, J., Li, W., Wang, L., Zhao, J., Ma, X., et al. (2020). A member of wheat class III peroxidase gene family, TaPRX-2A, enhanced the tolerance of salt stress. BMC Plant Biol. 20, 392. doi: 10.1186/s12870-020-02602-1
Sun, C., Wang, Y., Yang, X., Tang, L., Wan, C., Liu, J., et al. (2023). MATE transporter GFD1 cooperates with sugar transporters, mediates carbohydrate partitioning and controls grain-filling duration, grain size and number in rice. Plant Biotechnol. J. 21, 621–634. doi: 10.1111/pbi.13976
Taurog, A. (1999). Molecular evolution of thyroid peroxidase. Biochimie. 81, 557–562. doi: 10.1016/s0300-9084(99)80110-2
Wang, F., Han, T., Song, Q., Ye, W., Song, X., Chu, J., et al. (2020). The rice circadian clock regulates tiller growth and panicle development through strigolactone signaling and sugar sensing. Plant Cell. 32, 3124–3138. doi: 10.1105/tpc.20.00289
Welinder, K. G. (1992). Superfamily of plant, fungal and bacterial peroxidases. Curr. Opin. Struct. Biol. 2, 388–393. doi: 10.1016/0959-440X(92)90230-5
Wu, X., Cheng, R., Xue, S., Kong, Z., Ma, Z. (2014). Precise mapping of a quantitative trait locus interval for spike length and grain weight in bread wheat (Triticum aestivum L.). Mol. Breeding. 33, 129–138. doi: 10.1007/s11032-013-9939-4
Wu, Y., Yang, Z., How, J., Xu, H., Chen, L., Li, K., et al (2017). Overexpression of a peroxidase gene (AtPrx64) of Arabidopsis thaliana in tobacco improves plant's tolerance to aluminum stress. Plant Mol Biol. 95, 157–168. doi: 10.1007/s11103-017-0644-2
Yan, J., Su, P., Li, W., Xiao, G., Zhao, Y., Ma, X., et al. (2019). Genome-wide and evolutionary analysis of the class III peroxidase gene family in wheat and Aegilops tauschii reveals that some members are involved in stress responses. BMC Genomics 20, 666. doi: 10.1186/s12864-019-6006-5
Yao, Q., Cong, L., Chang, J. L., Li, K. X., Yang, G. X., He, G. Y. (2006). Low copy number gene transfer and stabl expression in a commercial wheat cultivar via particle bombardment. J. Exp. Bot. 57, 3737–3746. doi: 10.1093/jxb/erl145
Zhai, X., Yan, X., Zenda, T., Wang, N., Dong, A., Yang, Q., et al. (2024). Overexpression of the peroxidase gene ZmPRX1 increases maize seedling drought tolerance by promoting root development and lignification. Crop J. doi: 10.1016/j.cj.2024.04.008
Zhang, Z., Huang, J., Gao, Y., Liu, Y., Li, J., Zhou, X., et al. (2020). Suppressed ABA signal transduction in the spike promotes sucrose use in the stem and reduces grain number in wheat under water stress. J. Exp. Bot. 71, 7241–7256. doi: 10.1093/jxb/eraa380
Keywords: wheat, peroxidases (PRXs), TaPRX-2A, grain number per spike (GNS), co-expression analysis
Citation: Zang D, You W, Wu Y, Wang P, Wang Z, Yang Q, Chi S and Su P (2025) The Class III Peroxidase gene TaPRX-2A controls grain number per spike in common wheat (Triticum aestivum L.). Front. Plant Sci. 15:1501029. doi: 10.3389/fpls.2024.1501029
Received: 24 September 2024; Accepted: 19 December 2024;
Published: 06 February 2025.
Edited by:
Jingjing Huang, Ghent University, BelgiumCopyright © 2025 Zang, You, Wu, Wang, Wang, Yang, Chi and Su. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peisen Su, cHNzdTIwMTRAMTYzLmNvbQ==