
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 06 January 2025
Sec. Plant Pathogen Interactions
Volume 15 - 2024 | https://doi.org/10.3389/fpls.2024.1496770
This article is part of the Research Topic Genetics and Genomics of Emerging and Multifactorial Stresses Affecting Plant Survival and Associated Plant Microbiomes View all 14 articles
 Yue Deng1,2
Yue Deng1,2 Wenxian Wu2Xiaoqing Huang1,2Xiaoxiang Yang1,2Yaoyin Yu1,2Zhongmei Zhang1,2Zijin Hu1Xiquan Zhou1
Wenxian Wu2Xiaoqing Huang1,2Xiaoxiang Yang1,2Yaoyin Yu1,2Zhongmei Zhang1,2Zijin Hu1Xiquan Zhou1 Kang Zhou3,4*
Kang Zhou3,4* Yong Liu1,2*
Yong Liu1,2* Lei Zhang1,2*
Lei Zhang1,2*Rhizosphere microbiomes are constantly mobilized during plant–pathogen interactions, and this, in turn, affects their interactions. However, few studies have examined the activities of rhizosphere microbiomes in plants with different susceptibilities to soil-borne pathogens, especially those that cause clubroot disease. In this study, we compared the rhizosphere bacterial community in response to infection of Plasmodiophora brassicae among the four different clubroot susceptibility cultivars of oilseed rape (Brassica napus). Our results revealed obvious differences in the responses of rhizosphere bacterial community to the P. brassicae infection between the four cultivars of oilseed rape. Several bacterial genera that are associated with the nitrogen cycle, including Limnobacter, Thiobacillus, Anaeromyxobacter, Nitrosomonas, Tumebacillus, and Halomonas, showed significantly different changes between susceptible and resistant cultivars in the presence of P. brassicae infection. Moreover, increased connectedness and robustness were exhibited in the rhizosphere bacterial community co-occurrence network in clubroot-susceptible cultivars that were infected with P. brassicae, while only slight changes were observed in clubroot-resistant cultivars. Metagenomic analysis of microbial metabolism also indicated differences in the rhizosphere bacterial community between susceptible and resistant cultivars that were infected with P. brassicae. Functional analysis of the nitrogen cycle showed that genes related to nitrification (nxrB) were upregulated in susceptible cultivars, while genes related to assimilatory nitrate reduction (nasA, narB, and nirA) were upregulated in resistant cultivars that were infected with P. brassicae. These findings indicate that the synthesis and assimilation process of NO3- content were promoted in susceptible and resistant cultivars, respectively. Our study revealed differences in the characteristics of rhizosphere bacterial communities in response to P. brassicae infection between clubroot-susceptible and clubroot-resistant cultivars as well as the potential impact of these differences on the plant–P. brassicae interaction.
Plant rhizosphere microbiomes are increasingly being studied in recent years in relation to their contributions to various aspects of plant growth, development, and health (Berendsen et al., 2012; Bardgett and van der Putten, 2014; Saleem et al., 2019). Rhizosphere microbiomes are sensitive to alterations in biotic and abiotic factors, including plant development, infection by pathogens, and disorders in soil properties, resulting in a highly dynamic and diverse microbial community throughout the entire life cycle of plants (Zegeye et al., 2019; Panke-Buisse et al., 2015; Kwak et al., 2018; Sun et al., 2020). Moreover, rhizosphere microbiomes are responses to plant/soil-borne pathogen interactions and have been widely studied in recent years. Furthermore, there is a two-way effect between microbial community assemblage and plant/pathogen interaction. In the plant/pathogen systems, plants secrete root exudates and recruit specific microbial communities that confer them with disease resistance according to the “cry for help” theory (Hu et al., 2020, 2018). Similarly, some microbes are conducive to pathogen invasion through nutritional complementarity feedback mechanisms (Li et al., 2019; Pacheco et al., 2019; Kramer et al., 2020). Changes in the microbial community of diseased plants compared with those of healthy plants are external manifestations of disease processes, and this contributes to variations in microbial metabolisms associated with energy and material metabolic cycles, such as carbon and nitrogen cycles (Cao et al., 2024). Therefore, recent studies have focused mainly on identifying certain microbial taxa in rhizosphere microbiomes that contribute to crop health by affecting microbial metabolism or inhibiting pathogen growth. These microorganisms are considered potential tools for soil-borne disease control and sustainable farming and have been widely studied in multiple crops (Sun et al., 2022; Zhang et al., 2022; Compant et al., 2019). However, relatively limited studies have focused on the impact of changes in rhizosphere microbiomes in response to plant/pathogen interactions on the development of crop disease.
Clubroot, caused by the obligate protist, Plasmodiophora brassicae (P. brassicae), is a soil-borne disease that threatens the production of Cruciferous crops worldwide, as it results in a significant reduction of 40% to 60% in both crop yield and quality (Javed et al., 2023; Chai et al., 2014). The typical symptoms of clubroot disease are the presence of root galls as well as wilting and stunting of the above-ground parts of the plant (Schuller and Ludwig-Müller, 2016; Kageyama and Asano, 2009). Traditional measures for clubroot control cannot achieve the goals of eradication due to the long survival times (5–20 years) of resting spores in the soil (Donald and Porter, 2014). Moreover, severely affected fields are unsuitable for crop cultivation for extended periods of time (Dixon, 2009). Genetic resistance is presently being considered the most economical and effective approach for clubroot control worldwide (Diederichsen et al., 2009; Rahman et al., 2014). Multiple potential clubroot resistance genes (CR gene) in Brassica crops that are involved in modulating disease resistance responses to P. brassicae infections have been identified using next-generation sequencing (NGS) and other “omics”-based methods (Nagaoka et al., 2010; Hasan et al., 2021). Furthermore, numerous clubroot-resistant cultivars of Brassica crops have been bred and promoted commercially, including Huashuang 5R and Huayouza 62R (Zhan et al., 2015; Shah et al., 2019). Brassica crops with CR loci have been shown to successfully resist P. brassicae infection through regulating the plant innate immunity (Zhou et al., 2020). Differences in clubroot resistance between susceptible and resistant cultivars were directly reflected in plant roots, including transcription, proteins, and metabolism (Chen et al., 2015; Zhang et al., 2016; Pedras et al., 2008; Cao et al., 2008; Li et al., 2022). However, studies on whether these CR genes participate in interactions between host root and soil microbiome during the clubroot disease process are limited.
Recent studies on clubroot have indicated a distinct shift in microbial communities of Brassica crops when infected with P. brassicae (Kang et al., 2024; Wu et al., 2020). Moreover, variations in microbial community diversity have been shown to be correlated with clubroot disease severity and are highly sensitive as indicated by the microbial community in response to infections with P. brassicae (Lebreton et al., 2019; Liu et al., 2024). Changes in the microbial community due to host/P. brassicae interactions also differ in multiple situations, including disease resistance, pathotype, soil property, and fertilization (Lebreton et al., 2019; Liu et al., 2024; Cordero-Elvia et al., 2024; Gazengel et al., 2021). A more diverse microbial community also appeared to have a more obvious effect in promoting clubroot occurrence; however, this effect varied between susceptible and resistant cultivars (Wang et al., 2023; Daval et al., 2020). Moreover, the positive impact of nitrogen supply on clubroot occurrence also varies between susceptible and resistant cultivars (Gazengel et al., 2021). These phenomena suggest that clubroot resistance mechanisms in resistant cultivar may participate in the interaction within plants, pathogens, and soil microbiomes and may play an important role in shaping microbial communities. Although several studies have shown significant differences in root performance between clubroot-susceptible and clubroot-resistant cultivars when infected with P. brassicae, it is still unclear how clubroot-resistant cultivars manipulate the shaping of microbial communities based on their resistance mechanisms when infected with P. brassicae.
Additionally, we selected four cultivars of Brassica napus with different clubroot resistance levels to investigate whether clubroot resistance mechanisms affect the response of the rhizosphere microbiomes to plant/P. brassicae interactions. Based on 16S rRNA and metagenomic sequencing, we revealed the differences in microbial community, interaction within microbial communities, and microbial metabolisms of rhizosphere microbiomes between clubroot-susceptible and clubroot-resistant cultivars with or without P. brassicae infection. Our results suggest that those obvious differences in the rhizosphere microbiomes between two type cultivars may be caused by a resistant mechanism based on CR genes, which further affects the plant/P. brassicae interactions. Our study will help broaden the strategies for clubroot resistance breeding of oilseed rape and lay the foundation for utilizing soil microbial communities to control the occurrence of clubroot disease.
The cultivars of oilseed rape (Brassica napus subs. napus, hybrid) C36, H62, H62R, and Menh, as well as P. brassicae pathotype 4 isolate (Williams, 1966) were used in this study (Table 1). C36, also referred to as Chuanyou 36, was provided by the Crop Research Institute, Sichuan Academy of Agricultural Sciences (China) (Jiang et al., 2011). H62 and H62R, referred to as Huayouza62 (clubroot susceptible) and Huayouza62R (clubroot resistant), were provided by Prof. Chunyu Zhang of the College of Plant Science and Technology, Huazhong Agricultural University (China) (Li et al., 2021). Menh, referred to as Menhir (clubroot resistant), was provided by Norddeutsche Pflanzenzucht Hans Georg Lembke KG (NPZ) (Germany, https://www.proplanta.de/pflanzenbauberater/sorten/menhir-winterraps-hauptfruchtanbau_sks_4351raw1.html). Meanwhile, cultivars C36 and H62 are conventional hybrids without any clubroot resistance genes. H62R was generated by crossbreeding with H62 (Brassica napus, recipient parent) and CR Shinki (Chinese cabbage, CRb, donor parent). Menh was generated by crossbreeding with clubroot-resistant (P. brassicae pathotype 3) Mendel. Resting spores were extracted from galled root tissue collected from Brassica napus ‘Chuanyou 81’ in Shifang City, Sichuan Province. Resting spore suspensions were prepared as described in a previous study (Strelkov et al., 2006). The spore suspension was adjusted to a final concentration of 107 spores/mL. Each plant was inoculated with 10 mL of resting spore suspension. The resting spore suspension was injected into the soil close to the plant using a 10-mL syringe to ensure successful infection.
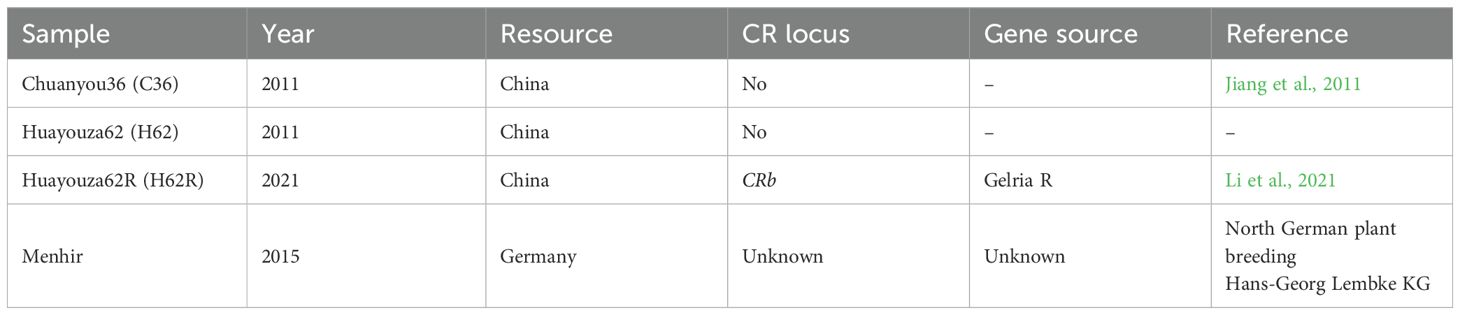
Table 1. Summary of oilseed rape cultivars used in this study.
All experiments were conducted in a greenhouse (23°C, 16-h light/8-h dark). Organic matter soil was purchased from a local market and directly used for plant cultivation without sterilization. Four cultivars of oilseed rape were also planted separately in organic matter soil in plugs (size: 540 mm × 280 mm), with each plug containing 21 holes for sowing oilseed rape. Each cultivar of oilseed rape was planted with a total of six plugs. Three plugs of plants per cultivar with a total of 63 plants were inoculated by resting spore suspensions of P. brassicae at 7 days; the rest of the three plugs of plants were treated with sterile water as the control treatment. For the P. brassicae-treated group, clubroot incidence (CI) and the disease severity index (DSI) of clubroot in each plug were calculated after 40 days of inoculation, and the rhizosphere soil of all diseased plants in the same plug were collected simultaneously into a single bag (Kuginuki et al., 1999). Regarding the control treatment, the rhizosphere soil of all plants in the same plug was also collected simultaneously into a single bag. Finally, a total of 24 rhizosphere soil samples were collected, and each treatment contained three replicates for the subsequent sequencing.
Collection and pretreatment of rhizosphere soil were done as described in a previous study (Edwards et al., 2015). The soil that remained tightly adhered to the roots after intense shaking was used as the rhizosphere soil sample. Root samples were collected into a 50-mL centrifuge tube with 25 mL of 1× PBS solution (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4·12H2O, and 2 mM KH2PO4). The mixture was sonicated at 40 Hz for 1 min and then shaken to separate rhizosphere soil from the roots. The rhizosphere soil was then transferred to a new sterile 50-mL centrifuge tube and centrifuged at 9,000 rpm for 5 min, after which the precipitated rhizosphere soil was subjected to freeze drying (BILON-FD80AD, Shanghai Bilang Instrument Manufacturing Co., Ltd., China). The dry rhizosphere soil was then homogenized by grinding (Tissuelyser-48, Jingxin, China). Finally, the processed samples were stored at -80°C for the subsequent 16S rRNA and metagenome sequencing.
Microbial DNA was extracted from 5 g of rhizosphere soil samples of oilseed rape using the E.Z.N.A.® stool DNA Kit (Omega Bio-tek, Norcross, GA, U.S.) according to the manufacturer’s instructions. DNA samples were prepared and stored at -80°C for the subsequent 16S rRNA and metagenomic sequencing. The V3–V4 region of the 16S rRNA gene was PCR-amplified (95°C for 2 min, followed by 25 cycles at 95°C for 30 s, 55°C for 30 s, 72°C for 30 s, and a final extension at 72°C for 5 min) to investigate bacterial communities using the primers 338F (5′-ACTCCTACGGGAGGCAGCA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) (Mori et al., 2013). PCR reactions were performed in triplicate 20-μL mixtures containing 4 μL of 5× FastPfu buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu Polymerase, and 10 ng of template DNA. The PCR products were detected using 1.5% agarose gel electrophoresis and further purified using an AxyPrepTM DNA Gel Extraction Kit (Axygen Scientific, USA). PCR products were quantified using Qubit®3.0 (Life Invitrogen) and pooled in equimolar concentrations of 10 ng/μL. Paired-end sequencing was performed on an Illumina HiSeq 2500 platform at Beijing Biomarker Technologies Co., Ltd., Beijing, China. Microbial bioinformatic analysis was performed using QIIME 2 2021.11 (Bolyen et al., 2019). The raw sequencing data was demultiplexed and filtered using the q2-demux plugin followed by denoising with DADA2 (Callahan et al., 2016). The phylogenetic affiliation of each 16S rRNA gene sequence was analyzed using RDP Classifier (http://rdp.cme.msu.edu/) against the silva (SSU132) 16S rRNA database using a confidence threshold of 70% (Amato et al., 2013).
Total DNA was also extracted from the above-mentioned rhizosphere soil samples using the E.Z.N.A.® Viral DNA Kit (Omega Bio-tek, Norcross, GA, USA) according to the manufacturer’s protocols. High-quality DNA sample (OD260/280 = 1.8–2.2, OD260/230 ≥ 2.0) was used to construct a sequencing library. Metagenomic shotgun sequencing libraries were constructed and sequenced at Shanghai Biozeron Biological Technology Co., Ltd. Briefly, for each sample, 1 μg of genomic DNA was sheared by Covaris S220 Focused-ultrasonicator (Woburn, MA, USA), and sequencing libraries were prepared with a fragment length of approximately 450 bp. All samples were sequenced using the Illumina NovaSeq 6000 platform at Shanghai Biozeron Biotechnology Co., Ltd., Shanghai, China.
Raw sequence reads underwent quality trimming using Trimmomatic v0.36 to remove adaptor contaminants and low-quality (quality below 20 and shorter than 50 bp) reads (Bolger et al., 2014). The taxonomy of clean reads for each sample was determined by Kraken2 using the customized kraken database. The abundances of taxonomy were estimated using Bracken (https://ccb.jhu.edu/software/bracken/) which can produce accurate species- and genus-level abundance even in multiple near-identical species. Clean sequence reads were assembled with MegaHit (v1.1.1-2-g02102e1). Assembled contigs were predicted using METAProdigal (v2.6.3), and a set of unique genes were generated using CD-HIT. Gene prediction was generated using MetaGeneMark software to identify coding regions in the genome. A non-redundant genome set (95% similarity threshold, 90% coverage threshold) was conducted using MMseq2 software. GO (Gene Ontology) annotation was performed using the goatools package. The unique gene set was first translated into protein sequences and then searched against the NCycle database (Tu et al., 2019) using DIAMOND (Buchfink et al., 2015) to identify the gene functions with the following filter parameters: evalue 0.00001, identity 90%. CAZymes were annotated using HMMER (v.3.2.1) to match the protein sequences to entries in the hidden Markov model (HMM) libraries of CAZyme (carbohydrate-active enzymes database) families downloaded from the CAZy (Lombard et al., 2014) database (http://www.cazy.org/, v12). KEGG (Kyoto Encyclopedia of Genes and Genomes) ortholog annotation was performed using KofamScan (https://www.genome.jp/tools/kofamkoala/) with the HMMSEARCH package (https://www.ebi.ac.uk/Tools/hmmer/search/hmmsearch).
The composition of the rhizosphere bacterial community at the phylum level based on the OTUs (operational taxonomic units) data was generated using the ggplot2 package in R (v 4.3.1). The α-diversity of the bacterial community was estimated using the non-parametric Shannon and Chao1 indices. A principal coordinate analysis (PCoA) based on Bray–Curtis distance metrics was performed with R (version 4.3.1) using the vegan package to explore differences in bacterial community compositions between clubroot-susceptible and clubroot-resistant cultivars of oilseed rape. The Bray–Curtis distance was generated based on OUT datasets at the genus level. Multivariate analysis of variance (MANOVA) was conducted based on Bray–Curtis distance metrics to further confirm the observed differences. The heatmap of the down- and upregulation of bacterial genera in the four cultivars of oilseed rape was calculated based on the relative abundance of each bacterial genera data in all the sequencing samples. Significance analysis of bacterial genera in the four cultivars of oilseed rape was generated using the STAMP software. Commonality analysis of variation in bacterial genera between the four cultivars was performed using an upset-venn diagram. The upset-venn diagram was completed using Wekomo Bioincloud (https://www.bioincloud.tech) (Gao et al., 2024).
Based on the disease index of clubroot and commonality analysis of variation in bacterial genera between the four cultivars, C36 and H62 were classified as clubroot-susceptible types, and H62R and Menhir were classified as clubroot-resistant types (Table 2; Figure 1F). The averages of sequencing data from two cultivars representing the clubroot-susceptible or clubroot-resistant type data were used for the subsequent analysis. The bubble diagram was completed using Wekomo Bioincloud. Microbial co-occurrence networks were used to uncover the potential interactions between rhizosphere microbiomes for clubroot-susceptible and clubroot-resistant oilseed rape cultivars with or without P. brassicae treatment. For each treatment, we constructed one network to display the co-occurrence patterns of bacterial ASVs (amplicon sequence variants) in the rhizosphere with or without exposure to P. brassicae infection. Bacterial ASVs (with a relative abundance >0.1% for at least one sample) in the rhizosphere were selected for network construction. A pairwise Spearman correlation matrix was calculated with the “corr.test” function in the psych package in R (version 4.3.1). Robust correlations with Spearman’s correlation coefficients (p) > 0.6 or < -0.6 and p < 0.01 were used to construct networks. Network properties were performed in the igraph package in R (version 4.3.1). SIMPER (similarity percentages) analysis, which was completed based on the abundance data of bacterial ASVs using Wekomo Bioincloud, was used to identify the key bacterial genera that contribute significant differences in the rhizosphere bacterial co-occurrence network between susceptible and resistant cultivars. The mean relative abundance of bacterial genera was generated using the ggplot2 package in R (version 4.3.1). Alpha and beta diversity of GO, KEGG, and CAZy pathways were performed using the ggplot2 package in R (version 4.3.1). Significant difference analyses of the relative abundance of the KEGG pathway were completed using the STAMP software.
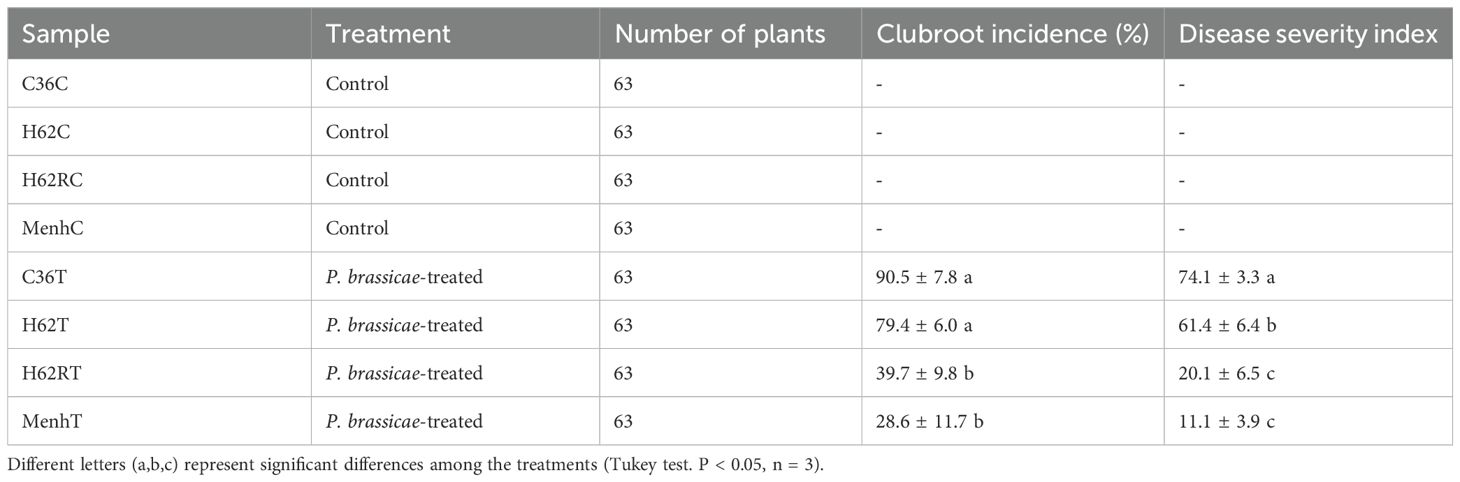
Table 2. Summary of clubroot disease indices in all the treatment groups in this study.
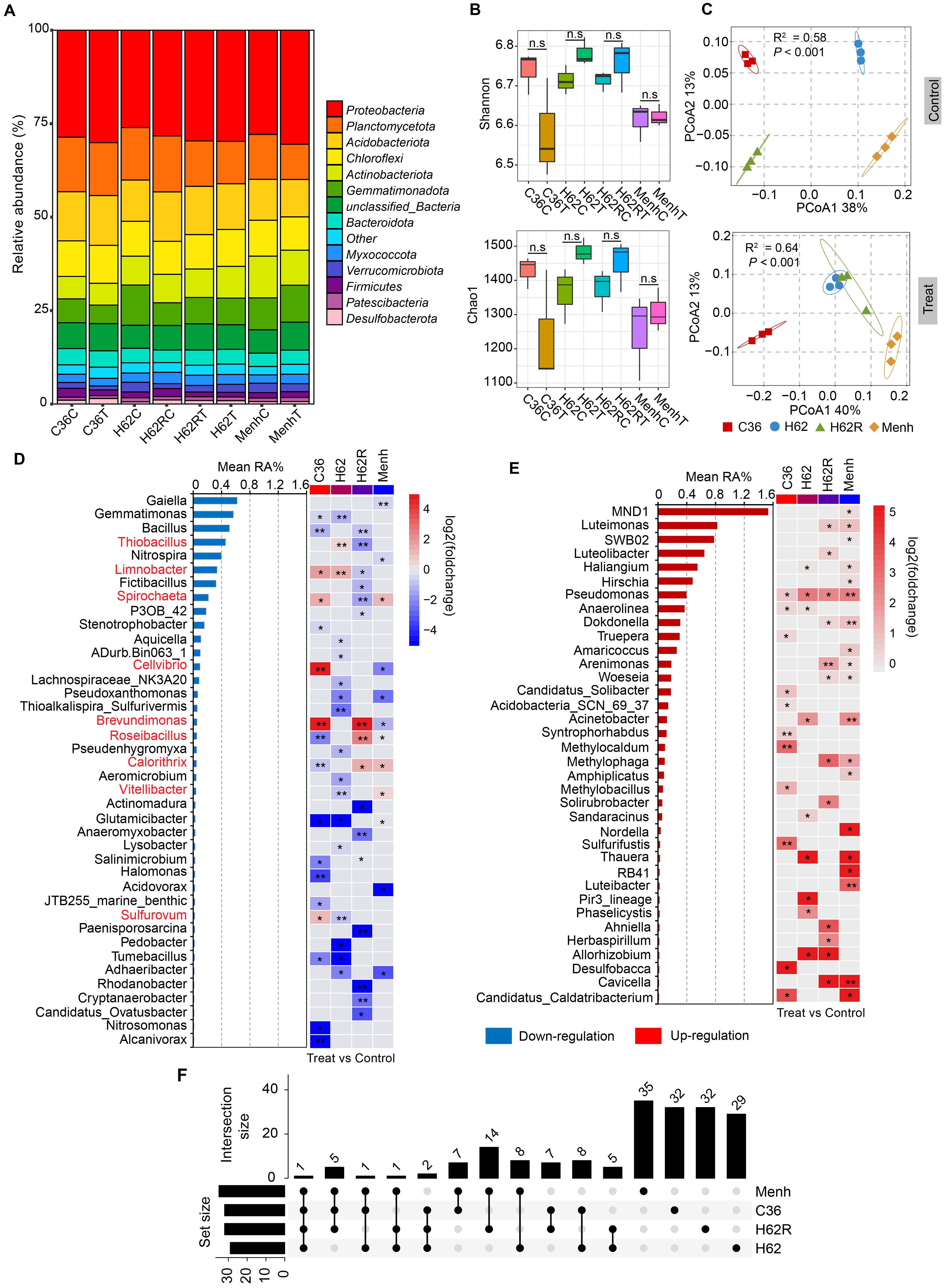
Figure 1. Comparison of rhizosphere bacterial diversity among the four of oilseed rape cultivars with or without P. brassicae. (A) Composition of rhizosphere bacterial community in the phylum level in all samples. C36C, H62C, H62RC, and MenhC means cultivars Chuanyou36, Huayouza62, Huayouza62R, and Menhir treated with water (Control). C36T, H62T, H62RT, and MenhT means four oilseed rape cultivars treated with resting spores of P. brassicae. (B) Rhizosphere bacterial α-diversity (Shannon and Chao1 indices) in all samples. “n.s.” means no significant difference between two samples (P > 0.05, n = 3, Student's t-test). (C) PCoA of rhizosphere bacterial community in C36, H62, H62R, and Menh of oilseed rape cultivars between the P. brassicae-treated and control groups. (D) Downregulation of bacterial genera in the four cultivars with P. brassicae-treated group compared with those of controls. “*, **” indicate significant differences among the samples (P < 0.05 and P < 0.01, n = 3, Student's t-test). (E) Upregulation of bacterial genera in the four cultivars with P. brassicae treatment compared with the controls. “*, **” indicate significant differences among the samples (P < 0.05 and P < 0.01, n = 3, Student's t-test). (F) Commonality analysis of variation in bacterial genera among the four cultivars based on the statistical results shown in (E, F).
The 16S rRNA amplicon data (SAMN40276299-SAMN40276322) and metagenome data (SAMN40350186-SAMN40350209) associated with this study have been deposited in the NCBI sequence read archive (SRA) under project accession PRJNA1084241. Source data have been provided in this article.
The greenhouse experiment revealed that the four oilseed rape cultivars had varying susceptibility to P. brassicae infection. Data of CI and DSI showed that C36 and H62 cultivars were more susceptible than H62R and Menhir cultivars to P. brassicae infection (Table 2). We also tested rhizosphere bacterial community diversity in all treatments through 16S rRNA sequencing. A total of 794,568 16S rRNA gene reads were obtained from 16S rRNA sequencing data of all samples, with 2,277 bacterial zero-radius OTUs identified. The rhizosphere bacterial community in all samples was mainly dominated by phylum Proteobacteria (reads: 229,250), Planctomycetota (reads: 102,023), Acidobacteriota (reads: 96,102), Chloroflexi (reads: 68,968), Actinobacteriota (reads: 63,380), Gemmatimonadota (reads: 60,356), and Bacteroidota (reads: 31,054) (Figure 1A). The infection of P. brassicae did not significantly affect the rhizosphere bacterial α-diversity of oilseed rape at 40 dpi. Compared with the control group, the bacterial Shannon (6.58–6.78) and Chao1 (1,237–1,483) indices showed no significant difference in the four cultivars that were treated for P. brassicae (P > 0.05, Figure 1B). However, shifts in the rhizosphere bacterial communities were varied in the four cultivars under P. brassicae infection. PCoA analysis at the genus level indicated a significant difference in the structure of the rhizosphere bacterial community between C36 and Menhir cultivars in the control treatment, excluding H62 and H62R (Figure 1C). Consistently, MANOVA analysis confirmed that the cultivar type was the main driver of rhizosphere bacterial β-diversity under normal growth conditions (R2 = 0.58, P < 0.001). However, compared with the control, the responses among the four cultivars differed when infected with P. brassicae, suggesting that the rhizosphere bacterial community changed in response to P. brassicae infection among the four cultivars (R2 = 0.64, P < 0.001, Figure 1C).
Compared to the controls, the relative abundance of 76 (76/443, 17.2%) bacterial genera was significantly changed in the four cultivars when infected with P. brassicae (P < 0.05). A relative abundance of 40 bacterial genera was also significantly downregulated in the four cultivars when infected with P. brassicae, wherein nine bacterial genera underwent different changes in the four cultivars (Figure 1D). The rest of the 36 bacterial genera in the relative abundance level was significantly upregulated in the four cultivars (Figure 1E). The commonality analysis of variation in bacterial genera based on the upset-venn diagram showed that the four cultivars could be classified into two categories, C36 and H62 (top 2 intersection size: 8) and H62R and Menh (top 1 intersection size: 14) (Figure 1F)—for example, the relative abundance of Gemmatimonas, Glutamicibacter, and Tumebacillus, respectively, were significantly downregulated in the C36 and H62 cultivars. The relative abundance of Limnobacter was significantly upregulated in the C36 and H62 cultivars, while it was significantly downregulated in the H62R cultivar (Figure 1D). The relative abundance of Luteimonas, Dokdonella, Arenimonas, Woeseia, Methylophaga, and Cavicella were only significantly upregulated in the H62R and Menh cultivars (Figure 1E).
Two categories, clubroot-susceptible (C36 and H62) and clubroot-resistant cultivars (H62R and Menh), were classified based on DSI and commonality analysis of variation in rhizosphere bacterial genera for subsequent analysis (Table 2; Figure 1F). Compared with the control groups, the bacterial Shannon index showed no significant differences in both clubroot-susceptible and clubroot-resistant cultivars infected with P. brassicae (P > 0.05, Figure 2A). However, the bacterial β-diversity showed significant differences among the four groups (R-C, R-T, S-C, and S-T), indicating an obvious difference in bacterial community composition between them (R2 = 0.15, P < 0.05, Figure 2B). Compared to the control group, a total of 49 bacterial genera with relative abundance levels were significantly changed in the clubroot-susceptible (N = 22) or clubroot-resistant (N = 24) cultivars under P. brassicae infestation (Figure 2C). Meanwhile, the relative abundance of Pseudomonas and Amaricoccus were significantly upregulated in both susceptible and resistant cultivars. Some bacterial genera, such as Limnobacter, Thiobacillus, and Anaeromyxobacter, were significantly upregulated in susceptible cultivars, while thy were downregulated in resistant cultivars. Furthermore, Nitrosomonas, Tumebacillus, and Halomonas were significantly downregulated in susceptible cultivars; however, these were upregulated in resistant cultivars.
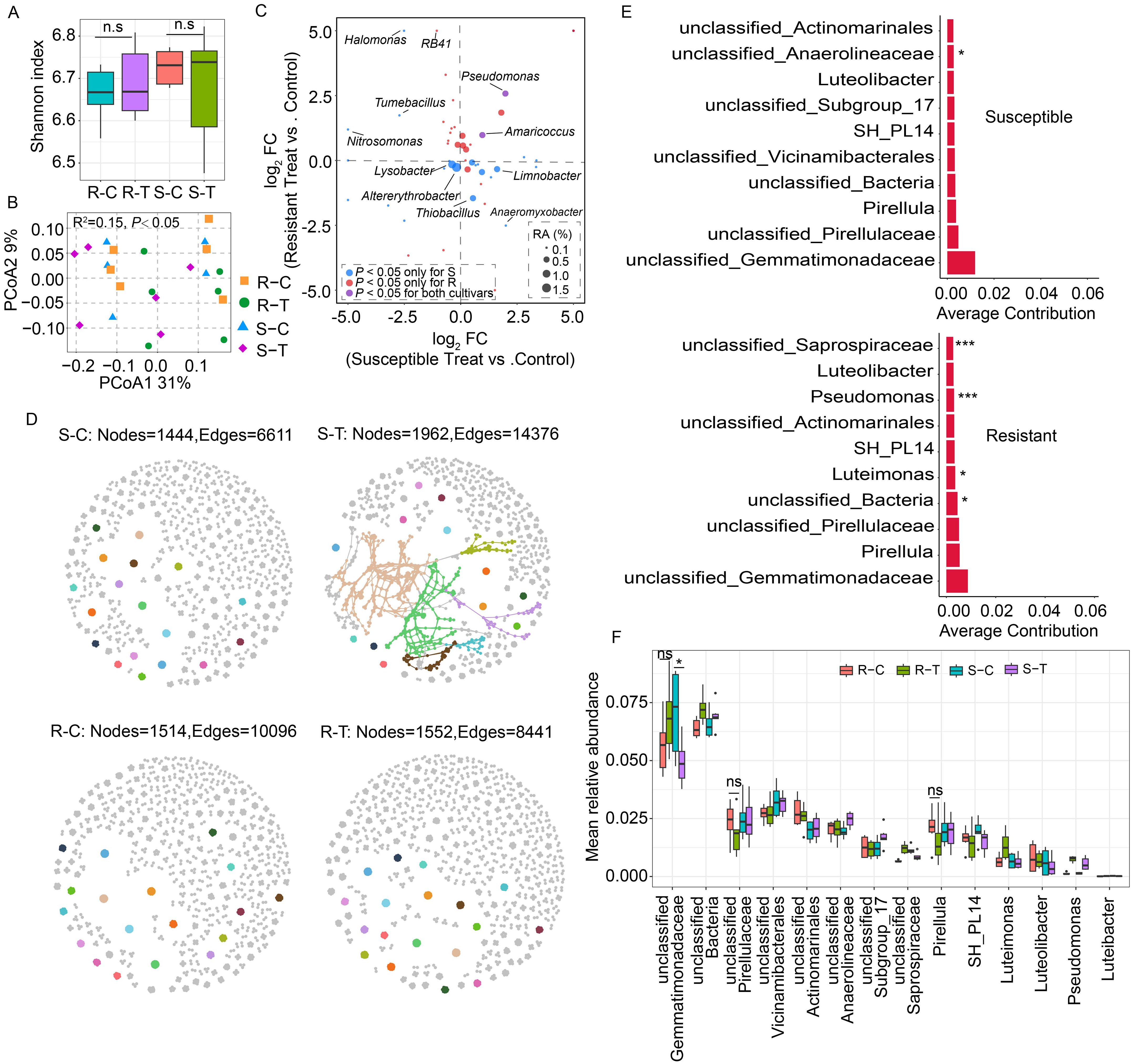
Figure 2. Comparison of rhizosphere bacterial community diversity between clubroot-susceptible and resistant oilseed rape cultivars. (A) Rhizosphere bacterial α-diversity (Shannon index) in clubroot susceptible cultivars treated with water (C36 and H62, S-C) and P. brassicae (S-T); clubroot-resistant cultivars treated with water (H62R and Menhir, R-C) and P. brassicae (R-T). “n.s.” means no significant difference between two samples (P > 0.05, n = 3, Student's t-test). (B) PCoA analysis of rhizosphere bacterial community structure between susceptible and resistant cultivars. (C) Comparison analysis of bacterial genera that underwent significant changes in the susceptible and resistant cultivars. FC means fold change in relative abundance level between the treated and control samples. RA means the relative abundance of bacterial genus in each sample. The blue dot means bacterial genera in relative abundance levels that only showed significant changes in susceptible cultivars. The red dot means bacterial genera in relative abundance levels that only showed significant changes in resistant cultivars. The purple dot indicates bacterial genera in relative abundance level that showed significant changes in both susceptible and resistant cultivars. (D) Rhizosphere bacterial co-occurrence networks between clubroot-susceptible and resistant oilseed rape cultivars treated with or without P. brassicae. (E) SIMPER analysis of the top ten bacterial genera that contribute to differences in the bacterial co-occurrence network in the susceptible and resistant cultivars between the control and P. brassicae-treated groups. *, *** indicate significant differences among the samples (P<0.05, P<0.005, n=6, Permutation test). (F) Mean relative abundance of the top 10 contributing bacterial genera in the susceptible and resistant cultivar samples. “ns” means no significant difference between two samples (P > 0.05, n = 6, Student's t-test). “*” indicate significant differences among the samples (P < 0.05, n = 6, Student’s t-test).
Moreover, a rhizosphere bacterial co-occurrence network was generated to evaluate the interaction of rhizosphere bacterial communities of oilseed rape with or without P. brassicae infection. Compared to the control group (susceptible control, S-C), increased connectedness and robustness were exhibited in the rhizosphere bacterial community in susceptible cultivars infected with P. brassicae (susceptible treat, S-T) (Figure 2D). Consistently, the number of nodes and edges in the bacterial co-occurrence network in the S-T treatment was also higher than those in the S-C treatment group (S-C: nodes = 1,444, edges = 6,611, S-T: nodes = 1,962, edges = 14,376). On the contrary, similar changes were not observed in clubroot-resistant cultivars, while the connectedness and robustness of the bacterial co-occurrence network was slightly affected by P. brassicae infections (Figure 2D). The number of nodes and edges in the bacterial co-occurrence network showed a similar level between R-T and R-C treatments (R-C: nodes = 1,514, edges = 10,096; R-T: nodes = 1,552, edges = 8,441), while it was lower than those in the S-T treatment (Figure 2D). The above-mentioned results implied that P. brassicae infection had a significant impact on the interaction of the rhizosphere bacterial community in susceptible cultivars. The SIMPER analysis identified the top 10 bacterial genera responsible for differences in microbial co-occurrence network between susceptible and resistant cultivars (Figure 2E). Meanwhile, seven bacterial genera coexist in both susceptible and resistant cultivars, with the top three contributing bacterial genera being unclassified_Gemmatimonadaceae, unclassified_Pirellulaceae, and Pirellula. Compared to the controls, the relative abundance of unclassified_Gemmatimonadaceae was upregulated in resistant cultivars when subjected to P. brassicae infestation, while it was significantly downregulated in susceptible cultivars (P < 0.05) (Figure 2F). However, the relative abundance of unclassified_Pirellulaceae and Pirellula, respectively, were downregulated in resistant cultivars, while it was upregulated in susceptible cultivars (P > 0.05).
Metagenomic analysis was used to investigate functional differences in the rhizosphere microbial community between clubroot-susceptible and clubroot-resistant cultivars when exposed to P. brassicae infection. An analysis of functional diversity annotated based on GO and KEGG databases showed that there was a significant difference between susceptible and resistant cultivars with or without P. brassicae infestation. Compared to the controls, rhizosphere microbial functional α- and β-diversity based on GO and KEGG databases showed significant changes in susceptible cultivars exposed to P. brassicae infection, while there was no significant change in resistant cultivars (Figures 3A, B). In contrast to the two types previously mentioned, rhizosphere microbial functional α- and β-diversity based on CAZy database showed no significant changes in both of the two cultivars exposed to P. brassicae infection compared with the controls (Figure 3C).
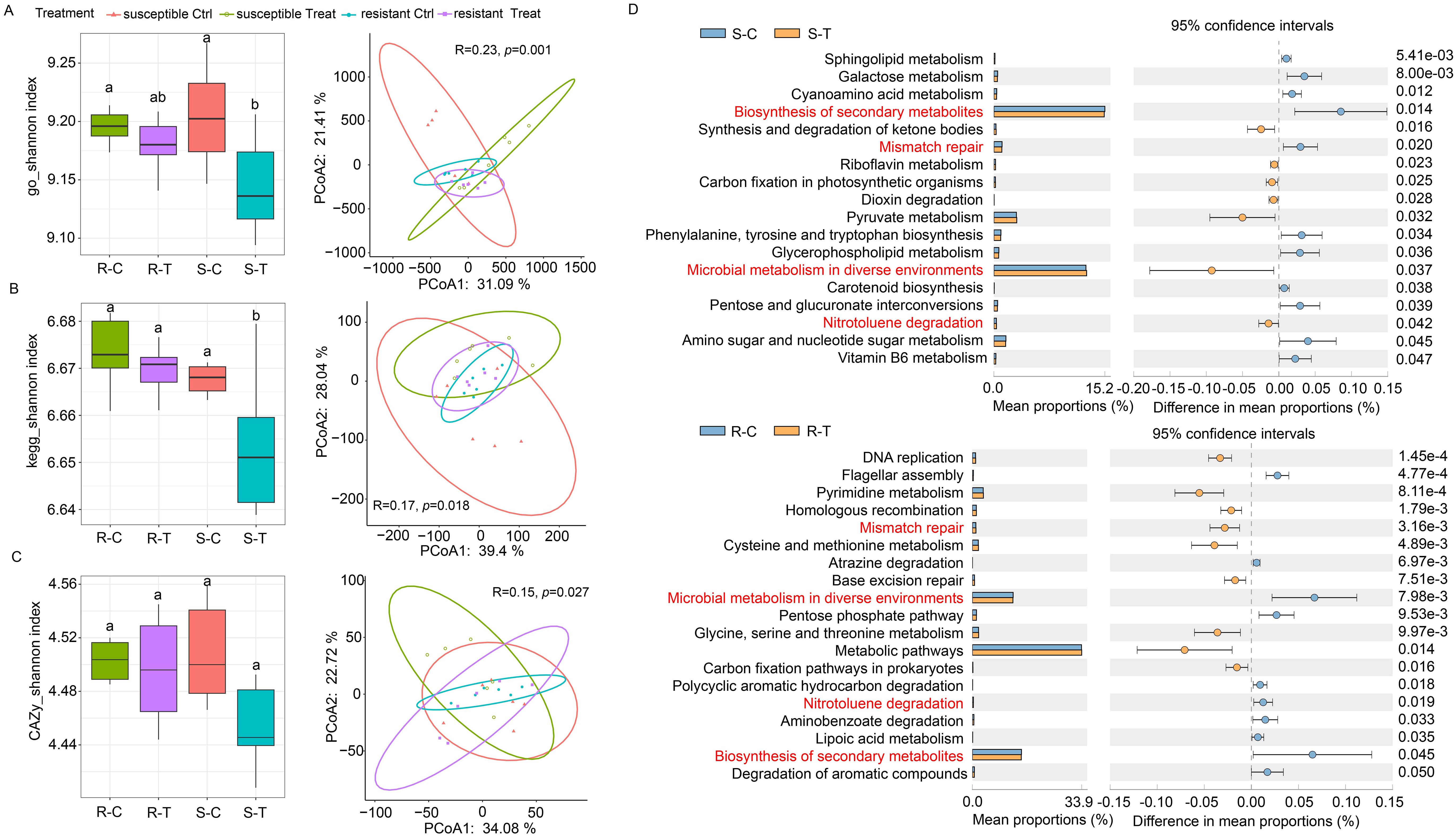
Figure 3. Functional diversity of rhizosphere microbiomes between clubroot-susceptible and resistant oilseed rape cultivars when exposed to P. brassicae infestation. (A–C) Functional diversity (Shannon index and PCoA) of rhizosphere microbiomes based on GO, KEGG, and CAZy databases between susceptible and resistant cultivars. Different letters represent significant differences among the treatments (P < 0.05, n = 6, Tukey test). (D) Differential KEGG pathways of rhizosphere microbiomes in the susceptible and resistant cultivars between the control and P. brassicae-treated groups.
An analysis of KEGG pathways showed that the relative abundance of multiple pathways was significantly changed in susceptible and resistant cultivars under P. brassicae infection compared with the controls. In susceptible cultivars, the relative abundance of 11 pathways were significantly downregulated when exposed to P. brassicae infection compared with the controls, while the other seven pathways were significantly upregulated (Figure 3D). In the resistant cultivars, the relative abundance of 10 pathways was significantly downregulated when exposed to P. brassicae infection compared with the controls, while the other nine pathways were significantly upregulated (Figure 3D). Among them, most of the pathways belonged to microbial metabolism, and three pathways (biosynthesis of secondary metabolites, microbial metabolism in diverse environments, and nitrotoluene degradation) showed significant changes in both susceptible and resistant cultivars. However, microbial metabolism in diverse environments and nitrotoluene degradation pathways showed opposite changes between susceptible and resistant cultivars. Both of the two pathways were significantly upregulated in the susceptible cultivars under P. brassicae infection compared with the controls, while they were significantly downregulated in resistant cultivars.
Subsequently, we focused on the variation of genes and pathways in the nitrogen (N) cycle between susceptible and resistant cultivars with or without P. brassicae infection. A total of 351 unigene protein sequences (identity ≥90%) were matched to 23 genes involved in the N cycle pathway, including nitrification (amoB, hao, and nxrB; number of unigenes = 3), denitrification (nirS, nirK, norB, and nosZ; number of unigenes = 249), nitrogen fixation (nirH; number of unigenes = 4), assimilatory nitrate reduction (nasA, narB, and nirA; number of unigenes = 8), dissimilatory nitrate reduction (narG, narH, napA, and nrfA; number of unigenes = 14), and organic nitrogen metabolism (nmo, gdh_K00261, gdh_K00262, gdh_K15371, glsA, glnA, ureA, and ureC; number of unigenes = 73) (Figure 4A). Among them, 16 genes were further analyzed. Compared to the controls, multiple genes showed different changes between susceptible and resistant cultivars when exposed to P. brassicae infection. In susceptible cultivars, we observed a higher abundance of genes associated to nitrification (amoB and nxrB), dissimilatory nitrate reduction (narH and nrfA), and denitrification (nirS, nirK, norB, and nosZ) when exposed to P. brassicae infection compared with the controls (Figure 4B). A variation in the nxrB gene abundance in the nitrification pathway (NO2-→NO3-) indicated an accelerated process in NO3- synthesis, which was consistent with the results above (Figure 2). However, in the resistant cultivars, a number of genes associated with assimilatory nitrate reduction (nasA, narB, and nirA), denitrification (nirS and nosZ), nitrogen fixation (nirH), and nitrification (amoB and hao) were upregulated when infected with P. brassicae compared with the controls, while the other genes associated with denitrification (nirK) and organic nitrogen metabolism (nmo) were downregulated. Additionally, a variation in the abundance of nasA, narB, and nirA genes in the assimilatory nitrate reduction pathway indicated an accelerated process in NO3- assimilation.
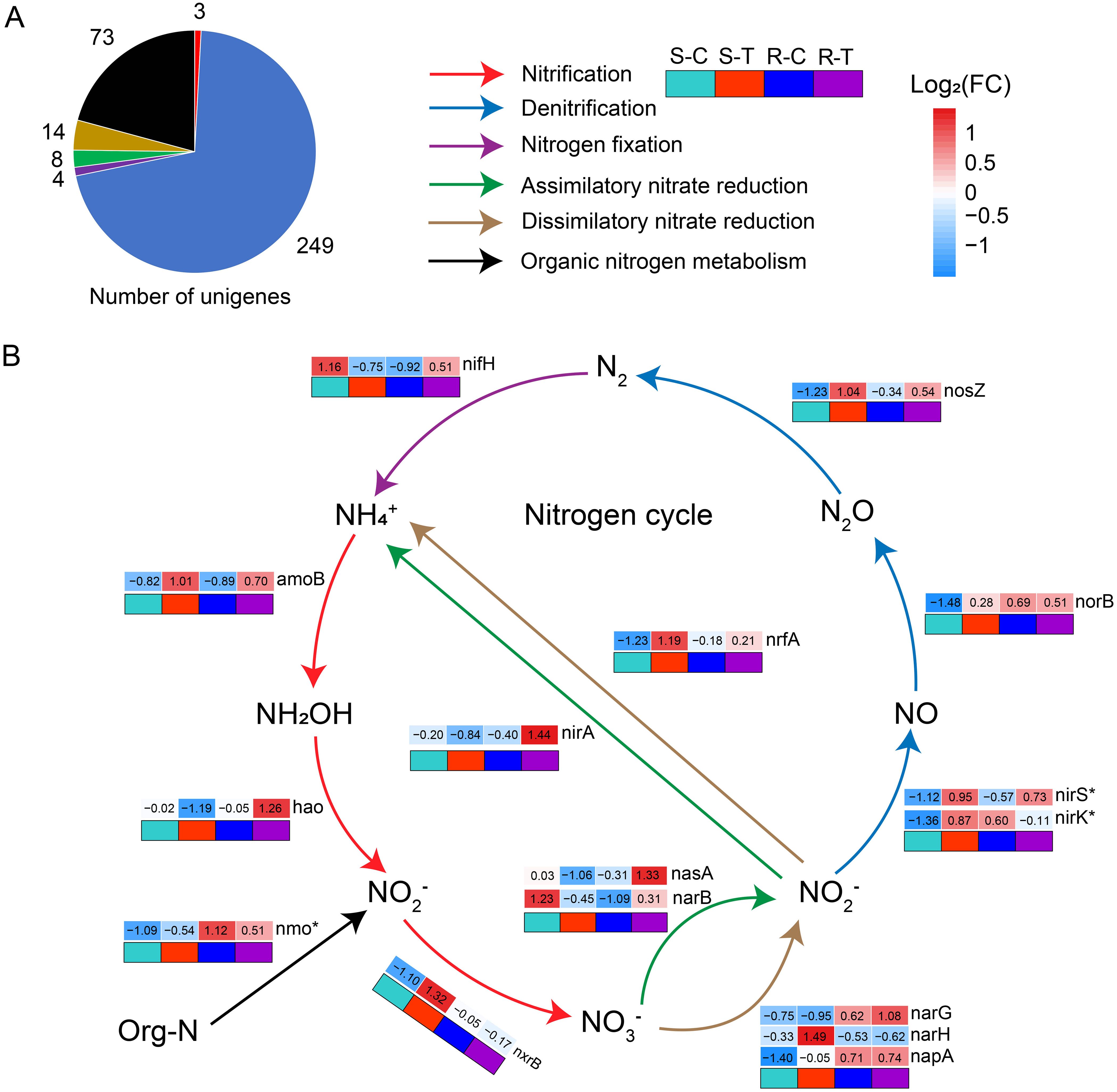
Figure 4. Metagenomic analysis of nitrogen cycle pathways of rhizosphere microbiomes in susceptible and resistant cultivars. (A) Number of unigenes annotated to genes related to the nitrogen cycle pathway. The color of the cake is consistent with that shown in (B), indicating the type of nitrogen cycle pathway. (B) The red arrow indicates the nitrification pathway (amoB, hao, and nxrB) of the nitrogen cycle. The blue arrow indicates the denitrification pathway (nirS, nirK, norB, and nosZ) of the nitrogen cycle. The purple arrow indicates the nitrogen fixation pathway (nifH) of the nitrogen cycle. The green arrow represents the assimilatory nitrate reduction pathway (nasA, narB, and nirA) of the nitrogen cycle. The brown arrow indicates the dissimilatory nitrate reduction pathway (narG, narH, napA, and nrfA) of the nitrogen cycle. The black arrow represents the organic nitrogen metabolism pathway (nmo) of N cycle. “*” indicates significant differences among the samples (P < 0.05, n = 6, Student's t-test).
Alterations in the rhizosphere microbiome always occur in plants after being infected by soil-borne pathogens; this phenomenon has been reported in several crop/pathogen disease systems, including clubroot (Kwak et al., 2018; Hu et al., 2020; Ni et al., 2022). However, studies on the differences in response of rhizosphere bacterial community to P. brassicas infection between clubroot-susceptible and clubroot-resistant cultivars are limited. Herein we investigated the variation in rhizosphere bacterial community in oilseed rape cultivars with different susceptibility to P. brassicae infection. Our results revealed a distinct shift in rhizosphere bacterial communities in the four cultivars when infected with P. brassicae compared with the control, and this also varied among the cultivars (Figure 1). The results of this study indicated that the response of rhizosphere bacterial community to P. brassicae infection was different among the four cultivars, and the cultivar type was the main driving factor that led to variations in the rhizosphere bacterial community (Figure 1). Our results are consistent with those of previous studies, indicating that the host genotype has a significant impact on shaping the plant rhizosphere microbial community, which has also been extensively confirmed in other crops (Zhang et al., 2019)—for example, the Indica variety of rice showed a higher nitrogen use efficiency than that of the Japonica variety through recruiting more microbial taxa with nitrogen metabolism functions, which is determined by a nitrate transporter and sensor named NRT1.1B. Moreover, this distinct shift in rhizosphere bacterial community is a result of the interaction between the host plant and P. brassicae. Previous studies have also proven that a distinct transcriptome landscape existed in the roots of clubroot-susceptible and clubroot-resistant Chinese cabbage lines after P. brassicae infection, indicating an obvious difference in clubroot resistance mechanisms and root exudations (Jia et al., 2017). A variation in the root transcription landscape could also lead to alterations in root metabolites, which, in turn, may affect the rhizosphere bacterial community (Pedras et al., 2008; Li et al., 2022). Moreover, shaping plant rhizosphere microbiomes can be achieved through the secretion of root exudates (Hu et al., 2018). In cereal crops such as wheat and maize, plant root would release benzoxazinoids to alter root-associated fungal and bacterial communities under a pathogen’s infestation.
Although four cultivars showed significant differences in their rhizosphere bacterial community when infected with P. brassicae, clubroot occurrence performance and commonality analysis of variation in bacterial genera also confirmed that cultivars C36 and H62, H62R, and Menhir, could be classified into clubroot-susceptible and clubroot-resistant types (Table 2; Figure 1). A comparative analysis further revealed differences in rhizosphere bacterial communities between clubroot-susceptible and clubroot-resistant cultivars in response to P. brassicae infection (Figure 2). Compared to the controls, some bacterial genera, such as Limnobacter, Thiobacillus, Anaeromyxobacter Nitrosomonas, Tumebacillus, and Halomonas, showed significant changes in relative abundance level between susceptible and resistant cultivars when exposed to P. brassicae infection. Meanwhile, Limnobacter, Thiobacillus, and Anaeromyxobacter were reported to be associated with BNF (biological N2 fixation) and were significantly upregulated in susceptible cultivars infected with P. brassicae and were downregulated in resistant cultivars. Additionally, Thiobacillus and Anaeromyxobacter were reported to be associated with arsenite oxidation-dependent biological nitrogen fixation, and Limnobacter was reported to be associated with nitrification-anammox (PN/A) processes (Li et al., 2023; Wang P. et al., 2024). Our results suggested that variations in the relative abundance of those three bacterial genera may lead to an accumulation of NO3- and SO42- in the soil and a reduction in pH value, which may be conducive to P. brassicae infection by reducing the soil’s pH value (Wang et al., 2023). Moreover, Nitrosomonas, Tumebacillus, and Halomonas were significantly downregulated in susceptible cultivars but were upregulated in resistant cultivars. Nitrosomonas, Tumebacillus, and Halomonas were reported to be associated with nitrosation and aerobic denitrification; variations in the relative abundance of those bacterial genera have also been shown to lead to an accumulation of NO3- and SO42- in soil samples from susceptible cultivars (Arp et al., 2002; Zhang et al., 2014; González-Domenech et al., 2010).
A variation in bacterial taxa is also reflected in microbial interactions. The bacterial co-occurrence network also showed a distinct interaction intensity within rhizosphere bacterial communities in susceptible cultivars infected with P. brassicae compared with the controls. Meanwhile, increased connectedness and robustness of the bacterial co-occurrence network were observed in susceptible cultivars exposed to P. brassicae infestation compared with the controls, while there were slight changes in resistant cultivars. The SIMPER analysis revealed that unclassified_Gemmatimonadaceae, unclassified_Pirellulaceae, and Pirellula were the three most common bacterial genera that contributed to the differences in bacterial co-occurrence network between the P. brassicae-treated and control samples in susceptible and resistant cultivars (Figure 2E). Among them, the relative abundance of unclassified_Gemmatimonadaceae was upregulated in resistant cultivars exposed to P. brassicae, while it was significantly downregulated in susceptible cultivars (Figure 2F). Gemmatimonadaceae was reported to be associated with plant root metabolites, which had a negative correlation with organic acid and a positive correlation with ketone content (Wang M. et al., 2024). Flavonoids, a type of ketone, have been reported to be associated with clubroot disease resistance in Arabidopsis thaliana (Päsold et al., 2010). The relative abundance of unclassified_Pirellulaceae and Pirellula was downregulated in the resistant cultivars when exposed to P. brassicae, while it showed slight changes in susceptible cultivars. Pirellula belonged to Planctomycetes and has been shown to be associated with anaerobic ammonia oxidation (Huang et al., 2014). In this study, a decrease in the relative abundance of Pirellula in resistant cultivars may also lead to the degradation of NO3- in the soil.
Variations in bacterial taxa and interaction within rhizosphere bacterial communities may also be reflected in microbial metabolisms. Functional diversity analysis based on metagenomic sequencing data further confirmed that changes in bacterial communities resulted in alterations in microbial metabolism. Our results showed that multiple pathways associated with microbial metabolism were significantly different in susceptible and resistant cultivars when exposed to P. brassicae infection compared with the controls (Figure 3). Among them, pathways associated with the nitrogen cycle exhibited distinct differences between susceptible and resistant cultivars (Figure 3D). Our results also demonstrated that the synthesis (nitrification) and assimilation (assimilatory nitrate reduction) processes of NO3- content were promoted in susceptible and resistant cultivars, respectively. Meanwhile, the abundance of nxrB gene related to nitrification (NO2-″NO3-) was upregulated in susceptible cultivars exposed to P. brassicae infection compared with the control, while it only changed slightly in resistant cultivars. Moreover, the expression of nasA, narB, and nirA genes, which are related to assimilatory nitrate reduction, was upregulated in resistant cultivars when exposed to P. brassicae infections compared with the controls, while it was downregulated in susceptible cultivars (Figure 4). In this study, the differences in nitrification and assimilatory nitrate reduction pathways between susceptible and resistant cultivars were due to rhizosphere microbiomes in response to B. napus/P. brassicae interaction. Our results suggested that NO3- may be one of the critical factors that affect B. napus/P. brassicae interaction and could reduce the incidence of clubroot.
Nitrogen, which is widely considered as a central element in soil ecosystems, has a huge impact on plant/pathogen interactions (Cui et al., 2014). Meanwhile, nitrogen supply could enhance the development of biotrophic pathogens, while the opposite effect is observed for necrotrophic pathogens (Solomon et al., 2003; Mur et al., 2016). The different forms of nitrogen supply (ammonium NH4+ or nitrate NO3-) can have various effects on the occurrence of plant disease due to differences in assimilation and metabolism pathways (Mur et al., 2016). NO3- feeding can strengthen host hypersensitive response (HR)-mediated resistance through enhancing the production of polyamines, while NH4+ nutrition can attenuate host defense. Regarding clubroot, although some studies suggest that the occurrence of clubroot is reduced with the application of high-nitrogen fertilizers, this may be attributed to the fact that oilseed rape requires a relatively large amount of nitrogen fertilizer during its entire growth period for growth and disease resistance (Gossen et al., 2014; Rathke et al., 2005). However, recent studies have shown that a high nitrogen supply could promote the occurrence of clubroot in susceptible B. napus cultivars by regulating the transcriptomic profile of P. brassicae, including pathogenicity-related genes (NUDIX and NEP-proteins) and genes associated to obligate biotrophic functions (glutamine synthetase, associated with nitrogen metabolism), whereas the effect differs in resistant cultivars (Gazengel et al., 2021). The above-mentioned results suggest that nitrogen supply may be beneficial for P. brassicae infection. Germination of P. brassicae’s resting spores is crucial for the occurrence of clubroot disease. Recent studies have proven that a diverse bacterial community, rather than root exudates, is necessary to stimulate the germination of the resting spores of P. brassicae (Wang et al., 2023). Meanwhile, the relative abundance of Sphingobacteriia, Flavobacteriia, and Bacteroidetes was significantly enriched in the “high”-germination-rate group, while Proteobacteria dominated in the “low”-germination-rate group. Moreover, the addition of NO3-, not NH4+, was conducive for the induction of the microbial community, leading to the germination of resting spores. The NO3- supply may be utilized as nutrients by certain microorganisms and could enhance nitrogen cycle pathways within microbial communities. The results of this study are consistent with the conclusions of previous studies. The NO3- synthesis pathways in the rhizosphere microbiomes were promoted in susceptible cultivars when exposed to P. brassicae infection compared with the controls, while the NO3- assimilation pathways in the rhizosphere microbiomes were promoted in resistant cultivars. Furthermore, in this study, variations in the NO3- assimilation and synthesis pathways in rhizosphere microbiomes were a result of the occurrence of clubroot, suggesting that changes in rhizosphere microbial community were directed by the B. napus/P. brassicae interaction. We considered that this rhizosphere microbial ecology environment associated with NO3- accumulation in the susceptible cultivar may be conductive to the further development of clubroot. In the clubroot-susceptible cultivar, once the host’s own defense system was breached, P. brassicae may control the rhizosphere microbial community to facilitate further infection by regulating the host’s metabolism. However, in clubroot-resistant varieties, the rhizosphere microbial community may be continuously manipulated by the host to jointly resist the P. brassicae infection. The differences between susceptible and resistant cultivars may be determined by their distinct resistance mechanisms. Multiple studies have also shown that Brassica crop roots showed different changes in genes, transcription, metabolomics, and proteome perspectives after P. brassicae infection, while the situation varied between susceptible and resistant cultivars due to the CR gene (Chen et al., 2015; Zhang et al., 2016; Pedras et al., 2008; Cao et al., 2008; Li et al., 2022). In this study, although we revealed the differences in NO3- assimilation and synthesis pathways in the rhizosphere microbiomes between susceptible and resistant cultivars after infection with P. brassicae, it is still unclear that soil NO3- participated in the interaction between hosts, P. brassicae, and rhizosphere microbiomes. In this study, the results of microbial diversity were conducted based on relative abundance indicators, and we only focused on limited microbial taxa that were differentially distributed among different samples. Hence, we might ignore those microbial taxa that showed significant changes in absolute abundance level across different samples while having similar relative abundance levels, and these microbes might be crucial for regulating microbial ecological functions. Moreover, all the conclusions obtained in this study were completed in the greenhouse condition, so several environmental impact factors, such as soil property, fertilizer regime, and cultivation pattern, were overlooked compared to field experiments. Our further study will validate the effect of NO3- pathway of microbial community on clubroot occurrence and clarify the relationship between soil NO3- content and microbial community function. We will also investigate the succession pattern of soil nitrogen cycling during the occurrence of the clubroot disease process, which is conducive to clarify the occurrence mechanism of clubroot disease and lay a theoretical foundation for clubroot disease control by using soil microorganisms.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA1084241.
YD: Data curation, Formal analysis, Investigation, Resources, Software, Writing – original draft, Writing – review & editing. WW: Conceptualization, Investigation, Resources, Visualization, Writing – review & editing. XH: Conceptualization, Funding acquisition, Supervision, Validation, Visualization, Writing – review & editing. XY: Conceptualization, Methodology, Supervision, Writing – review & editing. YY: Investigation, Supervision, Validation, Writing – review & editing. ZZ: Conceptualization, Investigation, Supervision, Validation, Writing – review & editing. ZH: Software, Supervision, Validation, Writing – review & editing. XZ: Investigation, Writing – review & editing. KZ: Conceptualization, Formal Analysis, Software, Supervision, Validation, Visualization, Writing – review & editing. YL: Conceptualization, Data curation, Funding acquisition, Investigation, Validation, Visualization, Writing – review & editing. LZ: Conceptualization, Data curation, Funding acquisition, Project administration, Supervision, Visualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported financially by the Natural Science Foundation of Sichuan Province (2022NSFSC0168 and 2023NSFSC0150). This work was also supported financially by the Independent Innovation Project (2022ZZCX023), the Science and Technology Project (1 + 9KJGG006), and the Talent Research Fund (NKYRCZX2024018) of Sichuan Academy of Agricultural Sciences.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Amato, K. R., Yeoman, C. J., Kent, A., Righini, N., Carbonero, F., Estrada, A., et al. (2013). Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 7, 1344–1353. doi: 10.1038/ismej.2013.16
Arp, D. J., Sayavedra-Soto, L. A., Hommes, N. G. (2002). Molecular biology and biochemistry of ammonia oxidation by Nitrosomonas europaea. Arch. Microbiol. 178, 250–255. doi: 10.1007/s00203-002-0452-0
Bardgett, R. D., van der Putten, W. H. (2014). Belowground biodiversity and ecosystem functioning. Nature. 515, 505–511. doi: 10.1038/nature13855
Berendsen, R. L., Pieterse, C. M. J., Bakker, P. A. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/S2095-3119(21)63817-0
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Oxford, England. Bioinformatics. 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Buchfink, B., Xie, C., Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Cao, T., Luo, Y., Shi, M., Tian, X., Kuzyakov, Y. (2024). Microbial interactions for nutrient acquisition in soil: Miners, scavengers, and carriers. Soil Biol. Biochem. 188, 109215. doi: 10.1016/j.soilbio.2023.109215
Cao, T., Srivastava, S., Rahman, M. H., Kav, N. N. V., Hotte, N., Deyholos, M. K., et al. (2008). Proteome-level changes in the roots of Brassica napus as a result of Plasmodiophora brassicae infection. Plant Sci. 174, 97–115. doi: 10.1016/j.plantsci.2007.10.002
Chai, A. L., Xie, X. W., Shi, Y. X., Li, B. J. (2014). Special issue: research status of clubroot (Plasmodiophora brassicae) on cruciferous crops in China. Can. J. Plant Pathol. 36, 142–153. doi: 10.1080/07060661.2013.868829
Chen, J., Pang, W., Chen, B., Zhang, C., Piao, Z. (2015). Transcriptome analysis of Brassica rapa near-isogenic lines carrying clubroot-resistant and -susceptible alleles in response to Plasmodiophora brassicae during early infection. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.01183
Compant, S., Samad, A., Faist, H., Sessitsch, A. (2019). A review on the plant microbiome: Ecology, functions, and emerging trends in microbial application. J. Adv. Res. 19, 29–37. doi: 10.1016/j.jare.2019.03.004
Cordero-Elvia, J., Galindo-González, L., Fredua-Agyeman, R., Hwang, S.-F., Strelkov, S. E. (2024). Clubroot-induced changes in the root and rhizosphere microbiome of susceptible and resistant canola. Plants. 13, 1880. doi: 10.3390/plants13131880
Cui, X., Yan, Q. W., Sun, J. L., Xiao, S., Xie, F. C., Chen, Y. J. (2014). Research progress on nitrogen use and plant growth. J. Northeast Agric. Univ. 21, 68–74. doi: 10.1016/S1006-8104(14)60036-2
Daval, S., Gazengel, K., Belcour, A., Linglin, J., Guillerm-Erckelboudt, A. Y., Sarniguet, A., et al. (2020). Soil microbiota influences clubroot disease by modulating Plasmodiophora brassicae and Brassica napus transcriptomes. Microb. Biotechnol. 13, 1648–1672. doi: 10.1111/1751-7915.13634
Diederichsen, E., Frauen, M., Linders, E. G. A., Hatakeyama, K., Hirai, M. (2009). Status and perspectives of clubroot resistance breeding in crucifer crops. J. Plant Growth Regul. 28, 265–281. doi: 10.1007/s00344-009-9100-0
Dixon, G. R. (2009). The occurrence and economic impact of Plasmodiophora brassicae and clubroot disease. J. Plant Growth Regul. 28, 194–202. doi: 10.1007/s00344-009-9090-y
Donald, E. C., Porter, I. J. (2014). Clubroot in Australia: The history and impact of Plasmodiophora brassicae in Brassica crops and research efforts directed towards its control. Can. J. Plant Pathol. 36, 66–84. doi: 10.1080/07060661.2013.873482
Edwards, J., Johnson, C., Santos-Medellín, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U.S.A. 112, E911–E920. doi: 10.1073/pnas.1414592112
Gao, Y. Y., Zhang, G. X., Jiang, S. Y., Liu, Y. X. (2024). Wekemo Bioincloud: A user-friendly platform for meta-omics data analyses. IMeta. 3, e175. doi: 10.1002/imt2.175
Gazengel, K., Aigu, Y., Lariagon, C., Humeau, M., Gravot, A., Manzanares-Dauleux, M. J., et al. (2021). Nitrogen supply and host-plant genotype modulate the transcriptomic profile of Plasmodiophora brassicae. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.701067
González-Domenech, C. M., Martínez-Checa, F., Béjar, V., Quesada, E. (2010). Denitrification as an important taxonomic marker within the genus Halomonas. Syst. Appl. Microbiol. 33, 85–93. doi: 10.1016/j.syapm.2009.12.001
Gossen, B. D., Deora, A., Peng, G., Hwang, S.-F., Mcdonald, M. R. (2014). Effect of environmental parameters on clubroot development and the risk of pathogen spread. Can. J. Plant Pathol. 36, 37–48. doi: 10.1080/07060661.2013.859635
Hasan, J., Megha, S., Rahman, H. (2021). Clubroot in Brassica: recent advances in genomics, breeding, and disease management. Genome. 64, 735–760. doi: 10.1139/gen-2020-0089
Hu, J., Wei, Z., Kowalchuk, G. A., Xu, Y., Shen, Q., Jousset, A. (2020). Rhizosphere microbiome functional diversity and pathogen invasion resistance build up during plant development. Environ. Microbiol. 22, 5005–5018. doi: 10.1111/1462-2920.15097
Hu, L., Robert, C. A. M., Cadot, S., Zhang, X., Ye, M., Li, B., et al. (2018). Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 9, 2738. doi: 10.1038/s41467-018-05122-7
Huang, P. B., Jiao, N. Z., Feng, J., Shu, Q. L. (2014). Research progress on Planctomycetes’ diversity and ecological function in marine environments. Microbiol. China. 41, 1891–1902. doi: 10.13344/j.microbiol.China.130874
Javed, M. A., Schwelm, A., Zamani-Noor, N., Salih, R., Silvestre Vañó, M., Wu, J., et al. (2023). The clubroot pathogen Plasmodiophora brassicae: A profile update. Mol. Plant Pathol. 24, 89–106. doi: 10.1111/mpp.13283
Jia, H., Wei, X., Yang, Y., Yuan, Y. X., Wei, F., Zhao, Y. Y., et al. (2017). Root RNA-seq analysis reveals a distinct transcriptome landscape between clubroot-susceptible and clubroot-resistant Chinese cabbage lines after P. brassicae infection. Plant Soil. 421, 93–105. doi: 10.1007/s11104-017-3432-5
Jiang, L. C., Huang, C., Pu, X. B., Zhang, J. F., Li, H. J., Jiang, J., et al. (2011). Breeding of new high-yield, extensive-adaptability and double-low NEA-cytoplasmic hybrid variety Chuanyou No. 36 in Brassica napus L. (in Chinese). Southwest China J. Agri Sci. 24, 1660–1664. doi: 10.16213/j.cnki.scjas.2011.05.032
Kageyama, K., Asano, T. (2009). Life cycle of Plasmodiophora brassicae. J. Plant Growth Regul. 28, 203–211. doi: 10.1007/s00344-009-9101-z
Kang, H. J., Chai, A. L., Lin, Z. L., Shi, Y. X., Xie, X. W., Li, L., et al. (2024). Deciphering differences in microbial community diversity between clubroot-diseased and healthy soils. Microorganisms. 12, 251–251. doi: 10.3390/microorganisms12020251
Kramer, J., Özkaya, Ö., Kümmerli, R. (2020). Bacterial siderophores in community and host interactions. Nat. Rev. Microbiol. 18, 152–163. doi: 10.1038/s41579-019-0284-4
Kuginuki, Y., Yoshikawa, H., Hirai, M. (1999). Variation in virulence of Plasmodiophora brassicae in Japan tested with clubroot-resistant cultivars of chinese cabbage (Brassica rapa L. ssp. pekinensis). Eur. J. Plant Pathol. 105, 327–332. doi: 10.1023/A:1008705413127
Kwak, M.-J., Kong, H. G., Choi, K., Kwon, S.-K., Song, J. Y., Lee, J., et al. (2018). Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 36, 1100–1109. doi: 10.1038/nbt.4232
Lebreton, L., Guillerm-Erckelboudt, A. Y., Gazengel, K., Linglin, J., Ourry, M., Glory, P., et al. (2019). Temporal dynamics of bacterial and fungal communities during the infection of Brassica rapa roots by the protist Plasmodiophora brassicae. PloS One 14, e0204195. doi: 10.1371/journal.pone.0204195
Li, J., Huang, T., Lu, J., Xu, X., Zhang, W. (2022). Metabonomic profiling of clubroot-susceptible and clubroot-resistant radish and the assessment of disease-resistant metabolites. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1037633
Li, M., Wei, Z., Wang, J., Jousset, A., Friman, V. P., Xu, Y., et al. (2019). Facilitation promotes invasions in plant-associated microbial communities. Ecol. Lett. 22, 149–158. doi: 10.1111/ele.13177
Li, Q., Nadil, S., Zhou, Y. W., Hou, Z. K., Gong, J. F., Liu, J., et al. (2021). Breeding of a novel clubroot disease-resistant Brassica napus variety Huayouza 62R. Acta Agronomica Sin. 47, 210–223. doi: 10.3724/SP.J.1006.2021.04086
Li, Y., Guo, L., Yang, R., Yang, Z., Zhang, H., Li, Q., et al. (2023). Thiobacillus spp. and Anaeromyxobacter spp. mediate arsenite oxidation-dependent biological nitrogen fixation in two contrasting types of arsenic-contaminated soils. J. Hazard Mater. 443, 130220. doi: 10.1016/j.jhazmat.2022.130220
Liu, Y., Lai, J., Sun, X., Huang, L., Sheng, Y., Zhang, Q., et al. (2024). Comparative metagenomic analysis reveals rhizosphere microbiome assembly and functional adaptation changes caused by clubroot disease in Chinese Cabbage. Microorganisms. 12, 1370. doi: 10.3390/microorganisms12071370
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Mori, H., Maruyama, F., Kato, H., Toyoda, A., Dozono, A., Ohtsubo, Y., et al. (2013). Design and experimental application of a novel non-degenerate universal primer set that amplifies prokaryotic 16S rRNA genes with a low possibility to amplify eukaryotic rRNA genes. DNA Res. 21, 217–227. doi: 10.1093/dnares/dst052
Mur, L. A. J., Simpson, C., Kumari, A., Gupta, A. K., Gupta, K. J. (2016). Moving nitrogen to the centre of plant defence against pathogens. Ann. Bot. 119, 703–709. doi: 10.1093/aob/mcw179
Nagaoka, T., Doullah, M. A. U., Matsumoto, S., Kawasaki, S., Ishikawa, T., Hori, H., et al. (2010). Identification of QTLs that control clubroot resistance in Brassica oleracea and comparative analysis of clubroot resistance genes between B. rapa and B. oleracea. Theor. Appl. Genet. 120, 1335–1346. doi: 10.1007/s00122-010-1259-z
Ni, H., Zong, R., Sun, J., Wu, Y., Yu, L., Liu, Y., et al. (2022). Response of bacterial community to the occurrence of clubroot disease in Chinese cabbage. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.922660
Pacheco, A. R., Moel, M., Segrè, D. (2019). Costless metabolic secretions as drivers of interspecies interactions in microbial ecosystems. Nat. Commun. 10, 103. doi: 10.1038/s41467-018-07946-9
Panke-Buisse, K., Poole, A. C., Goodrich, J. K., Ley, R. E., Kao-Kniffin, J. (2015). Selection on soil microbiomes reveals reproducible impacts on plant function. ISME J. 9, 980–989. doi: 10.1038/ismej.2014.196
Päsold, S., Siegel, I., Seidel, C., Ludwig-Müller, J. (2010). Flavonoid accumulation in Arabidopsis thaliana root galls caused by the obligate biotrophic pathogen Plasmodiophora brassicae. Mol. Plant Pathol. 11, 545–562. doi: 10.1111/j.1364-3703.2010.00628.x
Pedras, M. S. C., Zheng, Q. A., Strelkov, S. (2008). Metabolic changes in roots of the oilseed canola infected with the biotroph Plasmodiophora brassicae: phytoalexins and phytoanticipins. J. Agric. Food Chem. 56, 9949–9961. doi: 10.1021/jf802192f
Rahman, H., Peng, G., Yu, F., Falk, K. C., Kulkarni, M., Selvaraj, G. (2014). Genetics and breeding for clubroot resistance in canadian spring canola (Brassica napus L.). Can. J. Plant Pathol. 36, 122–134. doi: 10.1080/07060661.2013.862571
Rathke, G. W., Christen, O., Diepenbrock, W. (2005). Effects of nitrogen source and rate on productivity and quality of winter oilseed rape (Brassica napus L.) grown in different crop rotations. Field Crops Res. 94, 103–113. doi: 10.1016/j.fcr.2004.11.010
Saleem, M., Hu, J., Jousset, A. (2019). More than the sum of its parts: microbiome biodiversity as a driver of plant growth and soil health. Annu. Rev. Ecol. Evol. S. 50, 145–168. doi: 10.1146/annurev-ecolsys-110617-062605
Schuller, A., Ludwig-Müller, J. (2016). Histological methods to detect the clubroot pathogen Plasmodiophora brassicae during its complex life cycle. Plant Pathol. 65, 1223–1237. doi: 10.1111/ppa.12520
Shah, N., Sun, J., Yu, S., Yang, Z., Wang, Z., Huang, F., et al. (2019). Genetic variation analysis of field isolates of clubroot and their responses to Brassica napus lines containing resistant genes CRb and PbBa8.1 and their combination in homozygous and heterozygous state. Mol. Breeding. 39, 1–11. doi: 10.1007/s11032-019-1075-3
Solomon, P. S., Tan, K. C., Oliver, R. P. (2003). The nutrient supply of pathogenic fungi; a fertile field for study. Mol. Plant Pathol. 4, 203–210. doi: 10.1046/j.1364-3703.2003.00161.x
Strelkov, S. E., Tewari, J. P., Smith-Degenhardt, E. (2006). Characterization of Plasmodiophora brassicae populations from Alberta, Canada. Can. J. Plant Pathol. 28, 467–474. doi: 10.1080/07060660609507321
Sun, Q., Wu, H. L., Chen, F., Kang, J. H. (2020). Characteristics of soil nutrients and fungal community composition in crop rhizosphere under different rotation patterns. Environ. Sci. 41, 4682–4689. doi: 10.13227/j.hjkx.202001031
Sun, X., Xu, Z., Xie, J., Hesselberg-Thomsen, V., Tan, T., Zheng, D., et al. (2022). Bacillus velezensis stimulates resident rhizosphere Pseudomonas stutzeri for plant health through metabolic interactions. ISME J. 16, 774–787. doi: 10.1038/s41396-021-01125-3
Tu, Q., Lin, L., Cheng, L., Deng, Y., He, Z. (2019). NCycDB: a curated integrative database for fast and accurate metagenomic profiling of nitrogen cycling genes. Bioinformatics. 35, 1040–1048. doi: 10.1093/bioinformatics/bty741
Wang, M., Mu, C., Lin, X., Ma, W., Wu, H., Si, D., et al. (2024). Foliar application of nanoparticles reduced cadmium content in wheat (Triticum aestivum L.) grains via long-distance “leaf-root-microorganism” regulation. Environ. Sci. Technol. 58, 6900–6912. doi: 10.1021/acs.est.3c10506
Wang, P., Ou, R., Tan, J., Li, N., Zheng, M., Jin, Q., et al. (2024). Effect of sludge redistribution strategy on stability of partial nitrification-anammox process: Further exploration of the potential value of sludge. Chemosphere. 355, 141707. doi: 10.1016/j.chemosphere.2024.141707
Wang, Y., Zheng, X., Sarenqimuge, S., von Tiedemann, A. (2023). The soil bacterial community regulates germination of Plasmodiophora brassicae resting spores rather than root exudates. PloS Pathog. 19, e1011175. doi: 10.1371/journal.ppat.1011175
Williams, P. H. (1966). A system for the determination of races of Plasmodiophora brassicae that infect cabbage and rutabaga. Phytopathology. 56, 624–626.
Wu, W. X., Huang, X. Q., Zhang, L., Yang, X. X., Li, H. Z., Liu, Y. (2020). Crucifer clubroot disease changes the microbial community structure of rhizosphere soil. Acta Ecologica Sinica. 40, 1532–1541. doi: 10.5846/stxb201901190151
Zegeye, E. K., Brislawn, C. J., Farris, Y., Fansler, S. J., Hofmockel, K. S., Jansson, J. K., et al. (2019). Selection, succession, and stabilization of soil microbial consortia. mSystems. 4, e00055–e00019. doi: 10.1128/mSystems.00055-19
Zhan, Z. X., Jiang, Y. F., Zhu, Z. Y., Zhang, C. S., Yang, Q. Y., Li, Q., et al. (2015). Development of close linked marker to PbBa8.1 con-ferring canola resistance to Plasmodiophora brassicae. Chin. J. Oil Crop Sci. 37, 772–779. doi: 10.7505/j.issn.1007-9084.2015.06.005
Zhang, F. T., Huang, H. Q., Cui, Y., Sun, Q. G., Zhu, J., Liu, M., et al. (2014). Isolation and diversity of bacillus species from Jiaxi tropical rain forest soil. J. Microbiol. 34, 42–46. doi: 10.3969/j.issn.1005-7021.2014.04.008
Zhang, J., Ahmed, W., Dai, Z., Zhou, X., He, Z., Wei, L., et al. (2022). Microbial consortia: an engineering tool to suppress clubroot of chinese cabbage by changing the rhizosphere bacterial community composition. Biology. 11, 918. doi: 10.3390/biology11060918
Zhang, J., Liu, Y. X., Zhang, N., Hu, B., Jin, T., Xu, H., et al. (2019). NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 37, 676–684. doi: 10.1038/s41587-019-0104-4
Zhang, X., Liu, Y., Fang, Z., Li, Z., Yang, L., Zhuang, M., et al. (2016). Comparative transcriptome analysis between broccoli (Brassica oleracea var. italica) and wild cabbage (Brassica macrocarpa Guss.) in response to Plasmodiophora brassicae during different infection stages. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01929
Zhou, Q., Galindo-González, L., Manolii, V., Hwang, S. F., Strelkov, S. E. (2020). Comparative transcriptome analysis of rutabaga (Brassica napus) cultivars indicates activation of salicylic acid and ethylene-mediated defenses in response to Plasmodiophora brassicae. Int. J. Mol. Sci. 21, 8381. doi: 10.3390/ijms21218381
Keywords: rhizosphere microbiome, Plasmodiophora brassicae, susceptible cultivar, resistant cultivar, microbial metabolism, nitrogen cycle
Citation: Deng Y, Wu W, Huang X, Yang X, Yu Y, Zhang Z, Hu Z, Zhou X, Zhou K, Liu Y and Zhang L (2025) Characterization of rhizosphere bacterial communities in oilseed rape cultivars with different susceptibility to Plasmodiophora brassicae infection. Front. Plant Sci. 15:1496770. doi: 10.3389/fpls.2024.1496770
Received: 15 September 2024; Accepted: 02 December 2024;
Published: 06 January 2025.
Edited by:
Mahendar Thudi, Fort Valley State University, United StatesReviewed by:
Venkatesh Bollina, Agriculture and Agri-Food Canada (AAFC), CanadaCopyright © 2025 Deng, Wu, Huang, Yang, Yu, Zhang, Hu, Zhou, Zhou, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Zhang, emhhbmdsZWk5Mjk2QGFsaXl1bi5jb20=; Yong Liu, bGl1eW9uZ2RyQDE2My5jb20=; Kang Zhou, emhvdWthbmcxOTk1MDYwMUAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.