 Ian Tsang1,2*†
Ian Tsang1,2*† Pauline Thomelin1
Pauline Thomelin1 Eric S. Ober1†Stephen Rawsthorne3
Eric S. Ober1†Stephen Rawsthorne3 Jonathan A. Atkinson2†
Jonathan A. Atkinson2† Darren M. Wells2†
Darren M. Wells2† Lawrence Percival-Alwyn1†
Lawrence Percival-Alwyn1† Fiona J. Leigh1†
Fiona J. Leigh1† James Cockram1*†
James Cockram1*†- 1Plant Genetics Department, NIAB, Cambridge, United Kingdom
- 2Department of Plant Science, University of Nottingham, Nottingham, United Kingdom
- 3The Morley Agricultural Foundation, Wymondham, United Kingdom
Background: Root hairs are single-celled projections on root surfaces, critical for water and nutrient uptake. Here, we describe the first short root hair mutant in wheat (Triticum aestivum L.), identified in a mutagenized population and termed here short root hair 1 (srh1).
Results: While the srh1 mutant can initiate root hair bulges, lack of subsequent extension results in very short root hairs. Due to its semi-dominant nature, heterozygous lines displayed intermediate root hair lengths compared to wild-type. Bulked segregant analysis in a BC1F3 segregating population genotyped via exome capture sequencing localized the genetic control of this mutant to a region on the long arm of chromosome 3A. Via RNA sequencing and bioinformatic analysis, we identified two promising candidate genes. The first was a respiratory burst oxidase homolog (RBOH) encoding gene TaNOX3-A, orthologous to RBOH genes controlling root hair elongation in rice (OsNOX3) and maize (ZmRTH5), that carries a missense mutation in a conserved region of the predicted protein. RBOHs are membrane bound proteins that produce reactive oxygen species (ROS) which trigger cell wall extensibility, allowing subsequent root hair elongation. Notably, reduced ROS levels were observed in srh1 root hair bulges compared to wild-type. The second candidate was the calreticulin-3 encoding gene TaCRT3-A, located within the wider srh1 interval and whose expression was significantly downregulated in srh1 root tissues.
Conclusions: The identification of a major effect gene controlling wheat root hair morphology provides an entry point for future optimization of root hair architecture best suited to future agricultural environments.
Highlights
● We describe the wheat root hair mutant, short root hair 1 (srh1). Mapped to chromosome 3A, srh1 reduces reactive oxygen species production and affects root hair elongation, but not initiation.
Introduction
Hexaploid bread wheat (Triticum aestivum L.) is one of the world’s most important crops, and accounts for 20% of the global human calorie intake (FAO, 2022). Whilst not under direct selection by breeders, there is indication that across wheat breeding history, root hairs have been decreasing in size. With the impacts of climate change and focus on reduced input agriculture, current wheat root hair morphology and function may be sub-optimal for future agricultural environments. Thus, root hairs serve as a targetable trait for plant breeding with which to help optimize wheat yields and resilience, as well as reducing the environmental footprint of wheat cultivation (Tsang et al., 2024).
Root hair development in plants can be broadly split into four major stages (Grierson et al., 2014). The first - cell fate determination - regulates the fate of epidermal cells on the emerging primary root (radicle), where cells can differentiate into either a hair cell (trichoblast) or a non-hair cell (atrichoblast). The second stage involves bulge initiation, where the cell wall forms a bulge at the base of an emerging root hair (Grierson et al., 2014). Elongation of the bulge occurs in the third stage via a process known as tip growth, until the root hair reaches full length at maturity during the fourth and final stage (Grierson et al., 2014).
In the model plant Arabidopsis (Arabidopsis thaliana L.), the four stages of root hair development are each regulated by a complex network of genes and transcription factors (Balcerowicz et al., 2015). Across the world’s major cereal species, some of these genes have been characterized (recently reviewed by Tsang et al., 2024). For example, in rice (Oryza sativa L.), OsRHL1 encodes a basic helix-loop-helix (bHLH) transcription factor regulating root hair length (Ding et al., 2009). OsEXPA8 and OsEXPB5 encode expansins that facilitate cell wall extension to promote subsequent root hair elongation (Ma et al., 2013). OsRBOH5 encodes a reactive oxygen species (ROS) producing enzyme, catalyzing production of free oxygen radicals to aid cell wall extensibility and root hair bulge formation (Wang et al., 2018). Examples of characterized maize (Zea mays L.) root hair genes include ZmRTH3, which encodes a COBRA-like protein that functions as a Glycosylphosphatidylinositol (GPI) anchor, organizing synthesized cellulose bundles from cellulose synthases for root hair extension (Hochholdinger et al., 2008), and ZmRTH6, which encodes a cellulose synthase (Li et al., 2016). Despite the global importance of wheat, and the central role root hairs play in water and nutrient acquisition, current information on the genetic control of wheat root hair development is very limited (Han et al., 2016, Han et al., 2017; Zeng et al., 2024). While no wheat genes controlling root hair development have been positionally cloned to date, putative genes acting within the pathway have begun to be identified. Some of the first identified were the wheat Class II bHLH transcription factors TaRSL2 and TaRSL4, which share sequence homology with the Arabidopsis root hair genes ROOT HAIR DEFECTIVE-SIX LIKE 2 (AtRSL2) and AtRSL4 (Ma et al., 2013, Han et al., 2016, Han et al., 2017). Based on the observation that increase in root hair length in wheat species with increased ploidy is associated with higher TaRSL2 and TaRSL4 expression (Han et al., 2016), and that ectopic expression of TaRSL2 in Arabidopsis increased root hair length, these wheat genes have been hypothesized to function in a similar manner to their Arabidopsis homologs in root hair development. Historically, the size and complexity of the 17Gb hexaploid wheat genome has hindered progress in understanding the genetic control of wheat traits, including root hair development. However, recent advances including the availability of genome assemblies (The International Wheat Genome Sequencing Consortium (IWGSC) et al., 2018; Walkowiak et al., 2020) and resources such as Ethyl methanesulfonate (EMS) mutagenized Targeting Induced Local Lesions IN Genomes (TILLING) populations (Krasileva et al., 2017), have begun lowering the barriers to genetic locus and gene identification in wheat. These resources have been recently used to identify a candidate gene (stumpy) on chromosome 7B encoding a putative transmembrane protein associated with calcium ion (Ca2+) transport, mutation in which is thought to lead to increased root hair length when grown in high Ca2+ soils (Zeng et al., 2024).
In this paper, we report the discovery of a semi-dominant wheat mutant controlling root hair extension, which we term short root hair 1 (srh1). We identified a candidate gene (TaNOX3-A) that is located close to the peak of the srh1 genetic interval, is orthologous to genes known to control root hair length in maize and rice, and which contains a missense mutation in a conserved region in the predicted protein. Additionally, within the wider srh1 genetic interval, we identified a wheat Calreticulin-3 gene that was highly down-regulated in srh1 roots compared to wild-type, indicating a likely role of calreticulin in root hair development.
Materials and methods
Identification of the srh1 mutant
The wheat srh1 mutant was identified by phenotypic screening of a wheat cv. Cadenza TILLING population (Krasileva et al., 2017) using M4 generation seed sourced from the SeedStor, United Kingdom (UK) (https://www.seedstor.ac.uk). The mutation was identified visually in Cadenza TILLING line 1714, and M4 seedlings with the srh1 phenotype were then selfed to the M5 generation. M5 seed was phenotyped to check for phenotypic segregation, and two short root hair M5 lines derived from a single M4 plant were back-crossed to wild-type Cadenza to form the backcross-1 first filial (BC1F1) generation, and then selfed to yield BC1F2 and subsequent BC1F3 generations. Selected BC1F2 individuals shown phenotypically to be homogyzous for the short root hair phenotype were crossed to wild-type to generate a BC2F1, and selfed to generate BC2F2 and BC2F3 generations.
Root phenotyping
For root hair phenotypic measurements, BC1F2 seedlings were surface sterilized with 5% (v/v) bleach (sodium hypochlorite) and cold-treated at 4°C for 5 days in the dark. Seedlings were sown on agar (1%, pH7) plates which were sealed with micropore tape and left to germinate at room temperature for 3 days in the dark. Three-day old seedlings were imaged under a stereo-microscope (Leica MZ10F, Leica, Germany) with a Leica DFC 295 Camera (Leica, Germany). Measurement of root hair length (RHL), seminal root length (SRL), primary root length (PRL) and total root length (TRL) using the images obtained was performed using the software, FIJI (Schindelin et al., 2012). To capture images for root hair quantification, a Raspberry Pi HQ camera (Pi Hut, UK) with an attached macro lens (Pimoroni, UK) was used. Representative root hairs that grew flat on the agar surface were measured for 10 individuals of each genotype using FIJI (Schindelin et al., 2012)
To asses root hair phenotype in soil-grown plants, BC2F4 srh1 and wild-type segregant lines were grown in 12cm x 12cm petri dishes filled with moist soil: silty clay loam in the Hornbeam series (chromic endostagnic Luvisol) with pH 6.1, P index 1(12.4mg L-1 plant available Olsen P), K index 1 (94mg L-1 available K) and Mg index 2 (66mg L-1 available Mg). Six seedlings of each line were grown for seven days in a controlled environment chamber (18hr light at 20°C, 6hr dark at 16°C), after which roots were imaged using a Leica DM500 microscope and visually assessed for the presence/absence of root hairs.
Cryo-SEM
Wild-type Cadenza and BC1F2 srh1 mutants were sown on 1.5% agar medium for 5-7 days. Using a sterile scalpel, 1cm root segments from the root hair differentiation zone were excised and mounted on a stub covered with a glue/graphite mixture. Samples were immersed in liquid nitrogen and transferred to a vacuum chamber to be coated in platinum. Two sets of samples were produced: for batch one, water was removed via sublimation and shadowed with 7nm platinum; for batch two, samples were fractured and shadowed with 7nm platinum. Cryo-scanning electron microscopy (Cryo-SEM) imaging was carried out using a Zeiss EVO HD15 (Zeiss, Oberkochen, Germany). To calculate root hair density, measurements were taken from images captured under Cryo-SEM. Ten independent, randomly assigned field of views were each used to score root hair density in wild-type and srh1 root samples.
Bulked segregant analysis and exome capture
Library prep
Three-day old seedlings from the BC1F3 generation were phenotyped as homozygous mutant (short root hair) or homozygous wild-type (wild-type root hair length). The wild-type bulk contained 55 seedlings while the srh1 bulk contained 63 seedlings. Seedling root DNA from each bulk was extracted using a QIAGEN DNeasy Plant Mini Kit, following the manufacturer’s instructions. Exome capture was carried out via subcontract to Earlham Institute (UK): the sequencing library was constructed using a KAPA HTP kit (Roche) and were hybridized with exome capture baits (Gardiner et al., 2019). Fragments were sequenced using a 300 SP flow cell on a NovaSeq 6000 sequencing system (Illumina), generating 150bp paired-end reads.
Exome capture sequence alignment and bulked frequency ratio
Quality control of the reads before and after adapter removal was carried out using FastQC (Babraham Bioinformatics). Illumina adapter sequences were trimmed using Trim Galore (Babraham Bioinformatics). Reads with a Phred quality score of <25 and length <100bp were discarded. Trimmed reads were aligned to the indexed wheat reference genome of cv. Chinese Spring (RefSeq v1.1; IWGSC, 2018) using Burrows Wheeler Aligner (BWA) (Li and Durbin, 2009). Alignment files were sorted using Samtools (Danecek et al., 2021) prior to marking polymerase chain reaction (PCR) duplicates for removal using Picard MarkDuplicates (http://broadinstitute.github.io/picard). Alignments were then filtered using Samtools (Danecek et al., 2021) to retain properly paired reads while removing non-primary alignments and PCR duplicates. Variant calling was carried out using BCFtools mpileup (Danecek et al., 2021), discarding alignments with a minimum mapping quality < 30, and a coefficient for downgrading mapping quality of 50 was used, as recommended for BWA alignments.
To assess sequencing depths at gene regions, Samtools depth was used to construct a table of depths across genic regions +/- 50% of gene lengths for each bulk (Supplementary Figure S2).
Bulked allele frequencies were calculated as follows (as previously described by Martinez et al., 2020):
where BAF = bulked allele frequency, AA = alternate allele read depth, RD = total read depth. Bulked frequency ratio (BFR) was then calculated as:
Bulked frequency ratios (BFRs) between the wild-type and srh1 bulk were calculated from the variant calls after applying the following filters in VCFtools (Danecek et al., 2011): a minimum depth of 5, a maximum depth of 200 and exclusion of InDels and missing genotypes. BFR values were then computed using a bespoke python script across a 27.5Mb jumping window with a 1.5Mb jump. The outputted table was then visualized using CIRCOS (Krzywinski et al., 2009).
Variant effects
Variant annotation was carried out using SnpEff and Ensembl’s Variant Effect Predictor (VEP) (Cingolani et al., 2012; McLaren et al., 2016). Filtering of variants was carried out to discard single nucleotide polymorphisms (SNPs) with a quality score of ≤ 50 and a combined sample depth of ≤ 20. Non-exome variants were removed, and the genes containing variants were then filtered for those showing expression in root tissues using publicly available expression databases (expVIP, Borrill et al., 2016). Variant Sorting Intolerant From Tolerant (SIFT) scores were then predicted using VEP, retaining only non-synonymous variants with a score of ≤ 0.05. The remaining variants and the flanking amino acid regions surrounding the variant of interest were aligned against plant ‘orthologs’ as identified in Ensembl Plants (https://plants.ensembl.org/index.html), and filtered based on sequence homology (Supplementary Figure S3). Variants with an amino acid conservation of ≥ 70% across all plant orthologs were retained.
Reactive oxygen species quantification
For quantification of ROS levels in root hairs, seedlings from the BC2F3 generation were germinated on filter paper in the dark for 3 days at room temperature. 2′, 7′-Dichlorodihydrofluorescein diacetate (H2DCF-DA) incubation solution was prepared fresh as follows: 1mg of H2DCF-DA was dissolved in 1ml of DMSO, and the mixture diluted to 200ml with dH2O (Nestler et al., 2014). The seedlings were submerged in the H2DCF-DA solution for 20 minutes and rinsed twice in Phosphate Buffered Saline (PBS) buffer (Sigma Aldrich, P4417), then individually placed in cover glass bottomed chambers (Nunc LabTek II 1 Well Cover Glass, Thermo Fisher Ltd.). Roots were sandwiched between the glass and nutrient agar to force roots to lay flat. The chambers were imaged under a Leica SP8 Confocal Microscope (Leica, Wetzlar, Germany). Excitation was performed using an argon laser at 488nm and captured between 500-540nm. Z-projections were captured across the visible epidermal cell layers and maximal Z-projections were constructed in FIJI (Schindelin et al., 2012).
To quantify fluorescence intensity at root hair tips, a region of interest was first created around a 20µm section of the root hair tip in FIJI (Schindelin et al., 2012). Pixel thresholding was performed in the region of interest containing the root hair tip across all Z-stacks containing the same root hair. Thresholding boundaries for minimum and maximum pixel intensity were set at 10 and 255, respectively. The pixel value for each root hair was calculated as a mean across Z-stacks.
Sanger sequencing of TaNOX3-A
To confirm the presence of the candidate SNP in TaNOX3-A, mutant and wild-type individuals from the BC2F4 generation were grown in soil in a controlled growth environment under 16hr day; 20°C, 6hr night; 18°C conditions. First leaf tissue was harvested from mutant and wild-type plants after 2 weeks, and DNA was extracted from the leaves following the Tanksley DNA extraction protocol (Fulton et al., 1995). Phusion HF DNA Polymerase (Thermo Fisher Ltd) was used to PCR amplify the DNA fragment containing the target SNP with the following cycle (98°C 30s; 40 cycles of 98°C 10s, 66.5°C 30s, 72°C 15s; 72°C 10min) using homoeologue-specific primers (F-GAGGTGCGGCAGTAATGATAT, R-AGTTGCCTGAACTGACCTTC). The PCR products were size checked using electrophoresis across a 1% agarose gel and were subsequently excised from the gel under UV illumination, and extracted using the Thermo Fisher GeneJet Gel Extraction Kit (Thermo Fisher Ltd.) following manufacturer guidelines. DNA Sanger sequencing was outsourced to Source Bioscience, UK, and the sequence chromatograms were visualized using Geneious (https://www.geneious.com/).
RNA-seq
Library Prep
RNA extracted from 3-day old wild-type and srh1 seedlings from the BC2F3 generation were used to construct libraries for RNA sequencing (RNA-seq). The seedlings were germinated on filter paper at room temperature and grouped based on root hair phenotype. RNA was extracted using a QIAGEN RNeasy Plant Mini Kit from the seminal roots of 30 wild-type and 30 srh1 individual seedlings across three biological replicates per genotype. For each root, RNA was extracted from two regions: the elongation zone (∼ 1cm from root tip) and the mature zone (∼ 2-3cm above the root tip). Illumina libraries were prepared and sequenced via subcontract to Novogene (Cambridge, UK) using an Illumina NovaSeq 6000 S4 flowcell generating 150bp paired-end reads.
Alignment and read counts
FastQC (Babraham Bioinformatics) was used to assess read quality before and after adapter trimming. Adapter trimming was performed using Trim Galore (Babraham Bioinformatics). RNA-seq reads were aligned to the indexed wheat reference genome (RefSeq v1.0; IWGSC, 2018) using Hisat2 (Kim et al., 2019). Aligned reads were sorted using Samtools (Danecek et al., 2021). FeatureCounts (Liao et al., 2014) was used to output a counts matrix from the BAM files and gene models (RefSeq v1.1, IWGSC, 2018).
Differential gene expression
The RNA-seq data was used to determine differentially expressed genes (DEGs) between the following treatments: overall root tissue between srh1 and wild-type; mature root tissue between srh1 and wild-type, tip tissue between srh1 and wild-type; and mature tissue versus tip tissue for srh1 and wild-type. Differential gene expression analysis was conducted in RStudio (Version 4.3.2, RStudio Team, 2020) using r/DESeq2 (Love et al., 2014). Quality control, count normalization and clustering were performed in DESeq2, while shrinkage of Log2Fold Change (Log2FC) was carried out using lfcShrink() (Zhu et al., 2019). To yield manageable lists of DEGs, significance and Log2FC thresholds were adjusted according to each comparison. For differential gene expression analysis in overall root tissues, genes were deemed significantly differentially expressed with a false discovery rate (FDR) ≤ 0.01 and Log2FC ≥ abs(1.5). Across mature root tissues, thresholds for FDR and Log2FC were ≤ 0.001 and ≥ abs(2), respectively. For root tip tissues, thresholds for FDR and Log2FC were 0.01 and abs(1.5), respectively. Volcano plots were created using r/EnhancedVolcano (Bilghe et al., 2023).
Functional annotation of differentially expressed genes
To identify functional annotation terms associated with candidate genes, the IWGSC gene model IDs were searched against the gene functional annotations (IWGSC, 2018). Traits putatively associated with DEGs were predicted using KnetMiner (Hassani-Pak et al., 2021).
Homology and protein modeling
Wheat homologues of genes originally characterized in other plant species were identified using coding regions (CDS) as queries for BLASTn analysis of the wheat reference genome (RefSeq v1.0) in Ensembl Plants (Yates et al., 2022). For analysis of the predicted proteins encoded by homologous genes across many plant species, gene/protein lists were exported from Ensembl Plants using the “orthologues” function. Plots of amino acid conservation were generated using WebLogo (Crooks et al., 2004). To clarify comparison of homologous and/or orthologous genes between species, we prefix gene names with genus and species initials.
Protein modeling was carried out using SWISS-MODEL (https://swissmodel.expasy.org/). Protein sequences (in FASTA format) were downloaded from Ensembl Plants (https://plants.ensembl.org/index.html), protein models were constructed using the ‘Build Model’ option.
Results
The semi-dominant short root hair 1 mutant controls root hair elongation
The disrupted root hair phenotype of the srh1 mutant was initially identified visually at the seedling stage in the Cadenza TILLING line 1714 during a screen of the population. Examination of segregating BC1F2 lines under a microscope showed that very short root hairs were found on the root surface of both primary and seminal roots in the mutant (e.g. Figure 1A). High resolution cryo-SEM imaging revealed that root hairs successfully initiate, but fail to extend in mutant lines compared to wild-type (Figures 1D, E, G, H). In soil, the srh1 mutant also displays a vestigial root hair phenotype, while the wild-type segregant produces normal root hairs (Supplementary Figure S7). Root hair density was not significantly different in the mutant compared to wild-type (Figure 1K). No visual differences in trichoblast lengths were observed between srh1 and wild-type. Cross-section cryo-SEM images (Figures 1F, I) did not reveal any major root tissue structural differences between genotypes. Together, these data indicate that while root hair initiation occurs in the mutant, root hair tip growth is impeded. Accordingly, we named the mutant short root hair 1 (srh1).
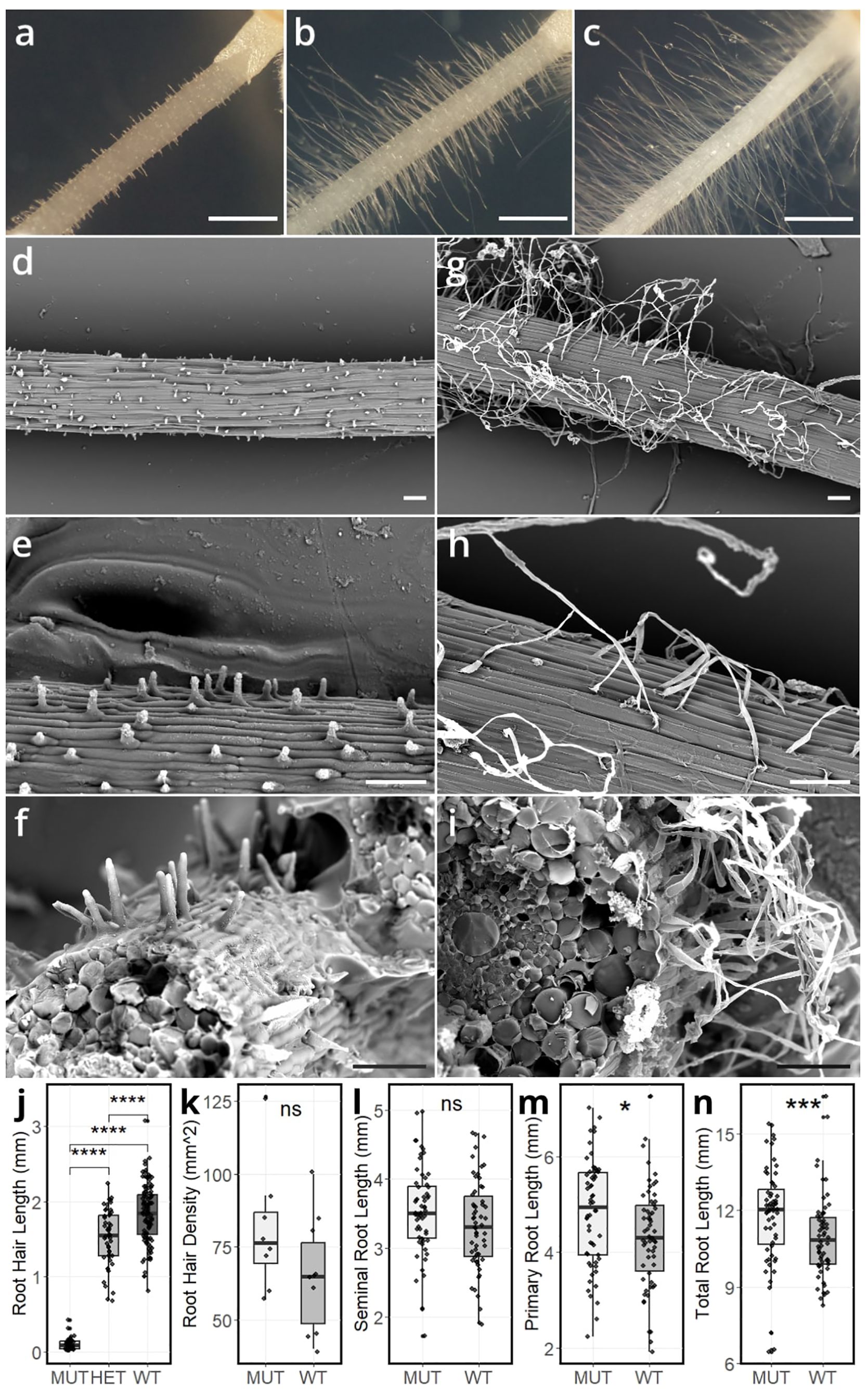
Figure 1. Seminal roots depicting root hair morphology in 3-day old BC1F2 seedlings segregating for srh1 and wild-type phenotype: (A): srh1 mutant (B), heterozygous (C) wild-type. Cryo-SEM images of the srh1 mutant (D–F) and wild-type (G–I) across increasing magnifications. Scale bars: (A–C) 1mm, (D–I) 100µm. (J–N): Root morphological differences between srh1, heterozygous and wild-type. (J): Root hair length on seminal roots. (K): Root hair density. (L): Seminal root length. (M): Primary root length, (N): Total root length (primary root + seminal roots). Significance as determined by T-test: * p <0.05, *** p <0.001, **** p <0.0001, Ns – not significant.
Analysis of 266 BC1F2 seedlings (three days old) found root hair length to segregate into three visually distinct (Figure 1J) phenotypic categories: 62 wild-type (long root hairs), 141 heterozygous (medium length root hairs) and 63 srh1 (no visible root hairs) (Figures 1A-C), consistent with a 1:2:1 segregation ratio, indicating srh1 is controlled in a Mendelian fashion via a single semi-dominant genetic locus (χ2 = 0.518, p<0.05). Primary root length at seedling stage in the srh1 mutant was significantly longer (4.82 ± 0.143 cm, p<0.05) than wild-type (4.31 ± 0.14 cm), as was srh1 total root length (11.8 ± 0.24 cm, p<0.001) when compared to wild-type (11 ± 0.24 cm). No significant differences in seminal root length, number or root hair density were observed between mutant and wild-type (Figures 1J-N).
ROS production is impaired in srh1
ROS production is involved in cell wall polymer cleaving - a process essential for subsequent root hair elongation. To determine whether srh1 affected ROS levels in root hairs, we used the fluorescent dye H2DCF-DA to detect fluorescence in root hair tips of srh1 and the wild-type seedlings. Mean pixel intensity in 20µm segments of root hair tips showed a significant (p<0.001) decrease between the mutant and wildtype (Figures 2A, C, D). A high level of fluorescence was maintained in elongating root hair tips in wild-type (Figures 2B, F), while low fluorescence was consistent across srh1 root hair bulges (Figure 2E).
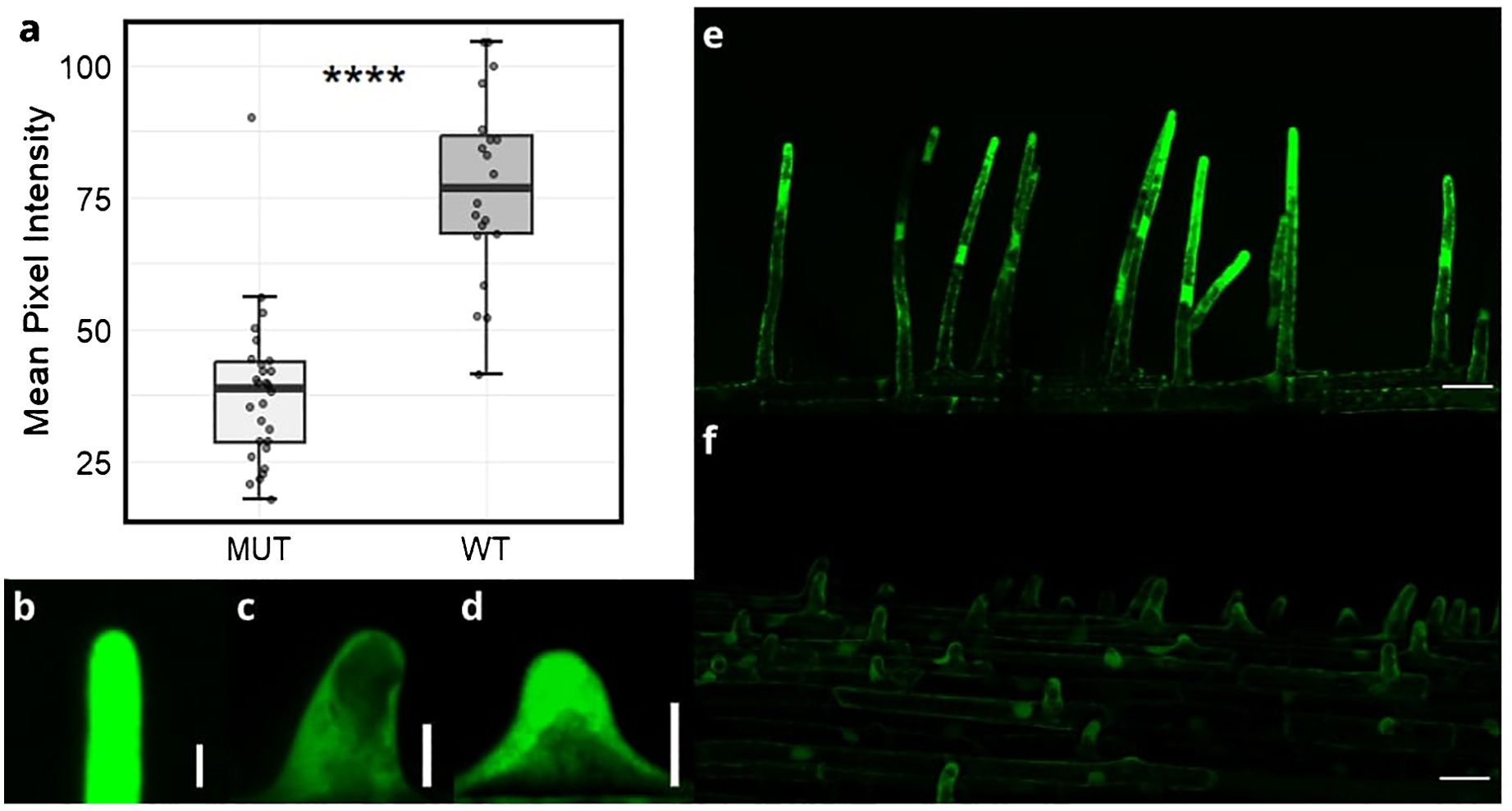
Figure 2. Reactive oxygen species (ROS) levels in emerging/short root hairs of the wheat srh1 mutant (MUT) versus wild-type (WT). (A): Difference in pixel intensity between root hair tips in srh1 and wild-type BC2F3 individuals (WT: n=20, srh1: n=28). (B–D): 20µm sections of root hair tips in (B): root hairs in wild-type, (C): srh1 tips, d: emerging wild-type tips. Fluorescence levels are much higher in (E): wild-type hairs compared to (F): srh1. Scale bars: (B–D): 10µm, (E, F): 50µm. Significance as determined by T-test: **** p <0.0001.
The short root hair 1 mutant phenotype is controlled by a genetic locus on chromosome 3A
To identify the genetic locus controlling srh1, bulked segregant analysis was performed via exome capture of genomic DNA extracted from bulks of 55 wild-type and 63 srh1 seedlings. Differences between allelic ratios were calculated following variant calling on the exome capture sequence alignments. At the genetic loci responsible for the srh1 phenotype, we expect differences in the ratio of alleles between the mutant and wild-type bulks due to high linkage disequilibrium with the underlying genetic mutation(s).
Bulked segregant analysis located srh1 to between 584-647Mb (defined as BFR ≥ 0.35) on the long arm of chromosome 3A (Figure 3). Within this interval, the highest difference in allelic ratios spanned a 22Mb interval between 595Mb - 617Mb (defined as BFR ≥ 0.5), with a maximal BFR value at ∼ 606Mb (BFR 0.66). Within this peak region, 204 of the 210 high confidence (HC) gene models in the reference genome contained associated variants assayed via exome capture. After discarding non-exome variants, we retained 39 SNPs across 33 genes. These variants were filtered for SIFT score and known expression in root tissue in public gene expression databases to identify a preliminary candidate gene list (Table 1). Seven SNPs across seven genes remained after filtering. Of these genes, four contained SNPs typically induced by EMS-mutagenesis (C to T, G to A), including: a predicted Glutamyl-tRNA amidotransferase (TraesCS3A02G347600), a predicted respiratory burst oxidase homolog (RBOH) (TraesCS3A02G354200), a pumilio-like protein (TraesCS3A02G361700) and a BEL-1 like homeodomain protein (TraesCS3A02G363900) (Table 1). The remaining three genes harbored atypical EMS-induced nucleotide substitutions, and were predicted to encode: a putative protein kinase (TraesCS3A02G351500), a Rop guanine nucleotide exchange factor (TraesCS3A02G364400) and a pentatricopeptide repeat containing protein (TraesCS3A02G367700).
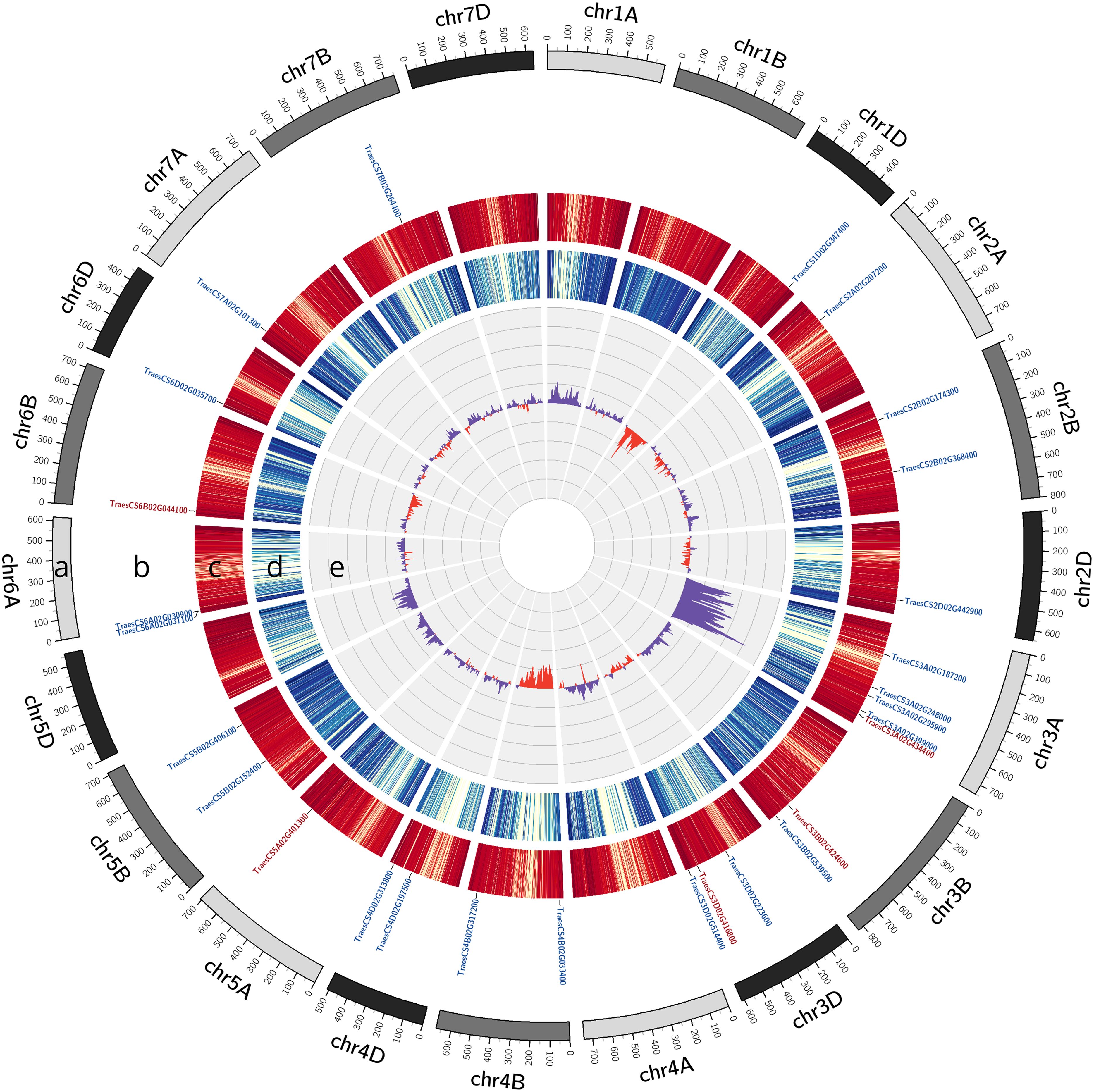
Figure 3. Genetic mapping of srh1 to the long arm of chromosome 3A via bulk segregant analysis (BSA). DNA variants were identified by exome capture sequencing of BC1F3 individuals, and mapped via analysis of bulked frequency ratios (BFR) between srh1 and wild-type bulks, aligned to the wheat reference genome. (A): Wheat karyotype. (B): Differentially expressed genes between srh1 and wild-type. Down-regulated genes in red, up-regulated genes in blue (Threshold FDR ≤ 0.01 and Log2FC ≥ abs(1.5)). (C): High confidence (HC) gene density, (D): Filtered SNP density, (E): BFR calculated from BFsrh1 - BFWT.
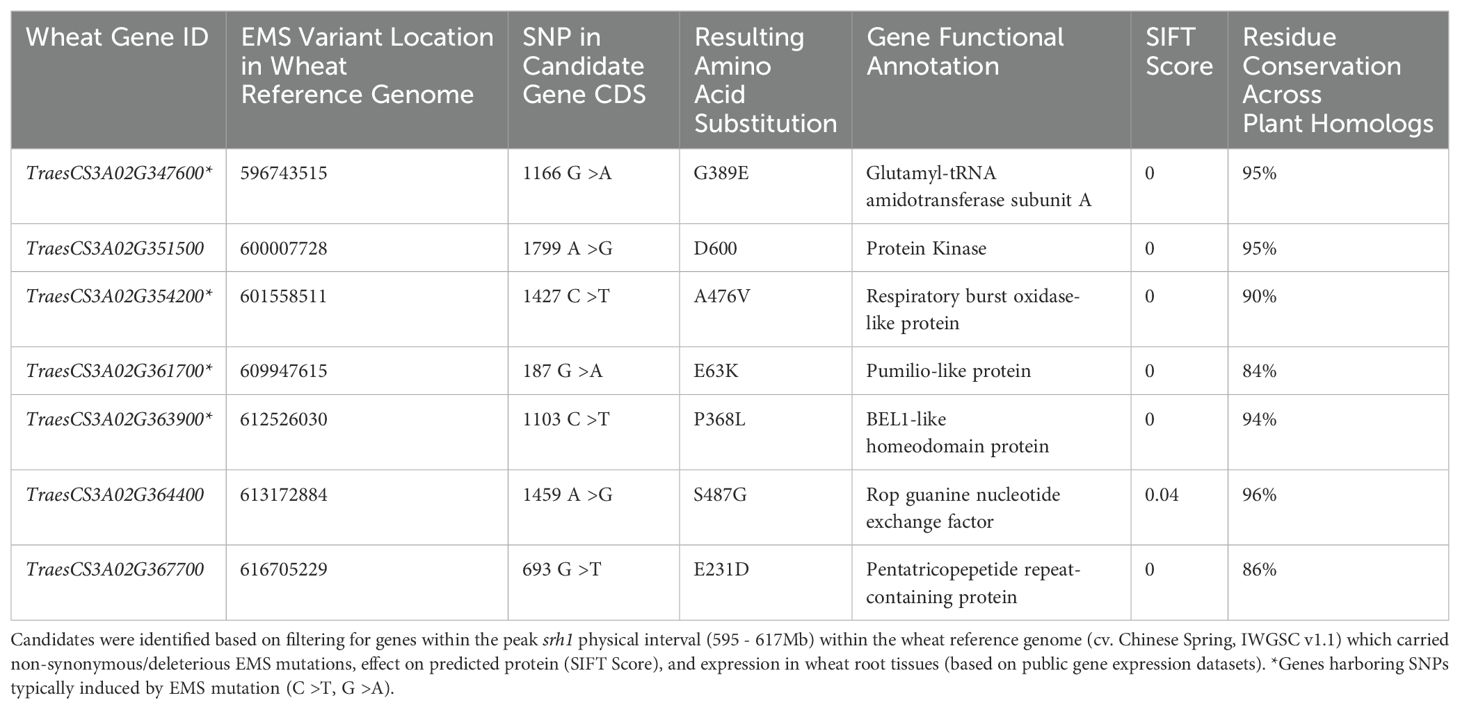
Table 1. srh1 candidate genes identified through bulked segregant analysis and exome capture.
The EMS induced SNPs across these seven genes all resulted in missense amino acid substitutions spanning highly conserved predicted protein domains (Supplementary Figure S3). The G to A DNA substitution at position 1166bp in the genomic sequence of the glutamyl-tRNA amidotransferase subunit A gene TraesCS3A02G347600 (g.1166G>A) resulted in a glycine (G) to glutamic acid (E) amino acid substitution at residue 389 in a predicted amidase signature domain within the predicted protein (a.G389E), with the G residue having 95% conservation across 152 plant homologs. The g.1799A>G substitution in the protein kinase TraesCS3A02G351500 resulted in an aspartic acid (D) to G amino acid substitution (a.D600Q) in a predicted protein kinase domain, with the D residue having 95% conservation across 159 plant homologs. In the RBOH gene TraesCS3A02G354200, the g.1427C>T mutation (Supplementary Figure S6A) resulted in an Alanine (A) to Valine (V) amino acid substitution (a.A476V) within a predicted ferric reductase transmembrane domain, with the A residue having 90% conservation across 157 homologs (Supplementary Figure S6B). The g.187G>A mutation in the pumilio-like gene TraesCS3A02G361700 resulted in a E to Lysine (K) substitution (a.E63K). Although not located in a predicted protein domain, the E residue at position 63 had 84% conservation across 244 plant homologs. In the BEL-1 like homeodomain gene TraesCS3A02G363900, the g.1103C>T mutation resulted in a Proline (P) to Leucine (L) substitution (a.P368L) at a residue with 94% conservation across 185 homologs within a predicted homeobox protein domain. The g.1459A>G mutation in TraesCS3A02G364400, a Rop guanine nucleotide exchange factor, resulted in a serine (S) to G amino acid substitution (a.S487G). While not predicted to lie within an annotated protein domain, the S residue was 96% conserved across 24 plant homologs. The g.693G>T mutation in the pentatricopeptide repeat containing gene TraesCS3A02G367700 resulted in an E to D substitution (a.E231D) within a pentatricopeptide repeat domain, with the E residue conserved at 86% across 119 plant homologs (Supplementary Figure S3). Additionally, within the wider srh1 genetic interval, we identified a g.1077G>A substitution at in the coding sequence of TraesCS3A02G39900 (TaCRT3-A), which resulted in an amino acid substitution of tryptophan (W) >STOP (a.W359STOP) (SIFT score = 0) resulting in the truncation of an entire alpha helix towards the C-terminus of the predicted protein (Supplementary Figure S1).
Differentially expressed genes
To help inform candidate gene analysis, as well as identify additional genes whose expression may have been altered in the srh1 mutant background, we analyzed RNA-seq data from root tissues extracted from two zones of primary roots (root tip and mature zone) of BC1F3 lines with either mutant srh1 or wild-type phenotype. Across srh1 and wild-type tissues from both root zones (i.e. considering tip and mature zone together), we identified a total of 67651 genes.
Overall primary root tissues
Using false discovery rate (FDR) ≤ 0.01 and a Log2 fold change in gene expression (Log2FC) ≥ abs(1.5) (equating to a 2.83 fold change in gene expression), 29 genes were significantly differentially regulated in “combined” (i.e tip + mature zone) srh1 root tissues relative to wild-type (Table 2, Figure 4A, Supplementary Figure S4. See also Figure 3B). Of these, 24 genes were down-regulated (Log2F C ≤ -1.5) and 5 were up-regulated (Log2FC ≥ 1.5).
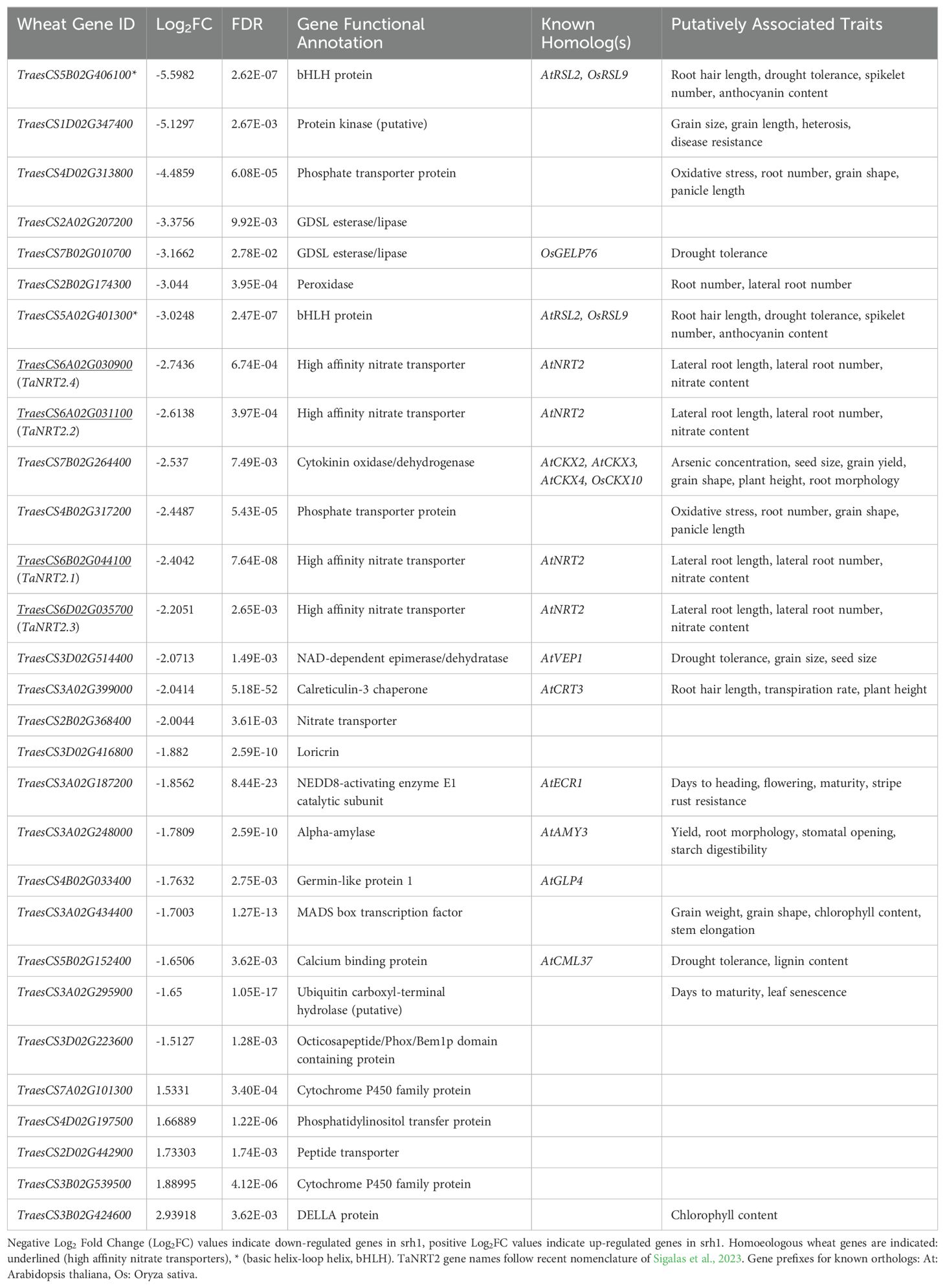
Table 2. Differentially expressed genes in “combined” root tissues between srh1 and wild-type.
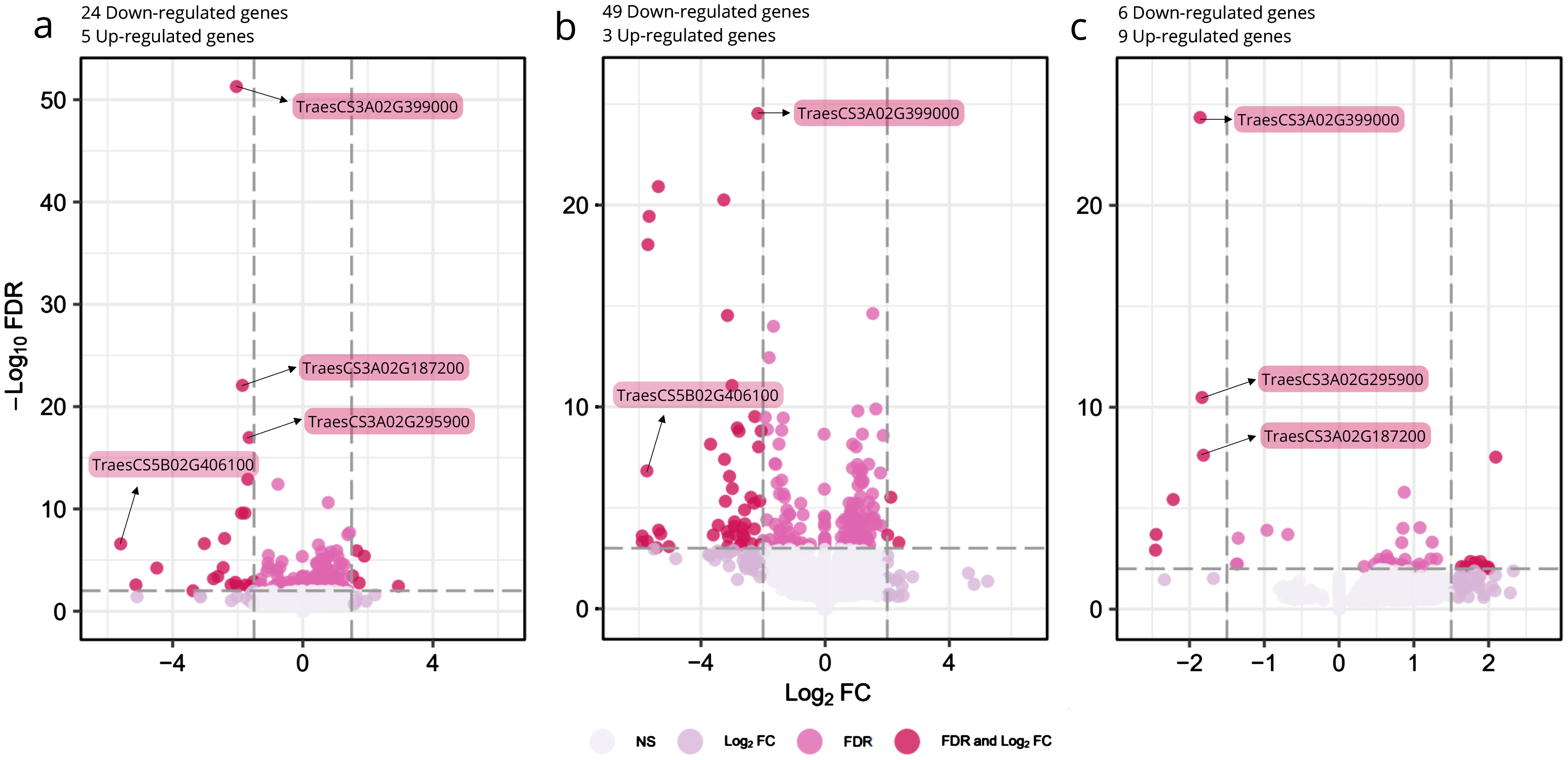
Figure 4. Differentially expressed genes between srh1 and wild-type, based on RNA-seq of root tissues from BC2F3 germplasm. (A): Overall primary root tissue, (B): Mature primary root tissue, (C): Tip primary root tissue. For (A–C): the x-axis shows Log2 fold change (Log2FC) in gene expression, with negative and positive values indicating down-regulation and up-regulation respectively. The y-axis shows the significance, -Log10 False Discovery Rate (FDR). Dark red points illustrate individual genes satisfying the Log2FC and FDR significance thresholds. Highlighted genes are those with either highly significant FDR or large fold changes. Dashed lines illustrate Log2FC and FDR cutoffs.
Across “combined” root tissues, the four most significantly down-regulated genes were all located on chromosome 3A (Log2FC ≥ -1.6, FDR ≤ 1.27E-13). Of these, TaCRT3-A (TraesCS3A02G399000, located at 646.1Mb) was the most significantly down-regulated gene between the “combined” root tissues (Log2FC -2.04, FDR 5.18E-52) in srh1 versus wild-type. This pattern was repeated for TaCRT3-A in mature and root tip tissues (Figures 4B, C). Additional down-regulated genes located on the long arm of chromosome 3A and down-regulated was a putative Ubiquitin carboxyl-terminal hydrolase (TraesCS3A02G295900, at 530.2Mb) and a MADS-box transcription factor (TraesCS3A02G434400, at 676.2Mb) orthologous to rice gene OsMADS65. The most strongly down-regulated (FDR ≤2.62E-03) DEGs were the bHLH gene TraesCS5B02G406100, a homolog of the Arabidopsis root hair gene AtRSL2; a leucine rich protein kinase (TraesCS1D02G347400); a phosphate transporter protein (TraesC S4D02G313800), and a GDSL esterase/lipase (TraesCS2A02G207200) (Log2FC -5.60, -5.13, -4.49 and -3.38 respectively).
Five significantly up-regulated genes were identified in srh1 root tissues, of which four were predominantly upregulated in the srh1 mature zone (Supplementary Figure S4). These include TraesCS3B02G424600, predicted to encode a DELLA protein; TraesCS3B02G539500 and TraesCS7A02G101300, predicted to encode Cytochrome P450 oxidoreductases; TraesCS2D02G442900, predicted to encode a peptide transporter, and TraesCS4D02G197500, predicted to encode a Phosphatidylinositol transfer protein (Log2FC 2.94, 1.89, 1.53, 1.73, 1.67 respectively, all with FDR ≤ 1.73E -03, Table 2).
We identified 10 DEGs located on Chromosome 3A, and while none of these colocalized with the peak region of the srh1 locus (596 - 617Mb, BFR ≥ 0.5), the calreticulin-3 chaperone gene TaCRT3-A was located within the wider srh1 interval (584 - 647Mb).
Discussion
Root Hair mutants as an entry point for trait improvement
In cereal crop breeding, while much emphasis has been placed on targeting above ground traits for increased crop yield and performance, less focus has been placed historically on root traits. With increasing interest in cereal root phenotypes (Ober et al., 2021), optimization of root hair morphology is likely to play an ever increasing role in ensuring adequate crop nutrition and resilience under future agricultural environments characterized by reduced inputs and the challenges of climate change. To date, the only previously characterized wheat root hair mutant is the Stumpy locus on chromosome 7B, which results in long root hairs and very short roots, but only when grown in soil with high levels of (Ca2+ (Zeng et al., 2024). In this study, we provide detailed characterization of the first known mutant to display short root hairs in wheat, termed here srh1.
srh1 is a root hair elongation mutant with altered root growth
Unlike other crop species, little is known about the genetics, genes and gene pathways regulating wheat root hair development. Here, the srh1 mutant was identified in a mutagenized TILLING population constructed in a spring wheat (cv. Cadenza) background.
While numerous genes controlling the first two stages of root hair development have been described in Arabidopsis (epidermal cell fate determination and bulge initiation), to date, none have been identified in the world’s most important cereal crops - rice, maize or wheat (Tsang et al., 2024). In the wheat srh1 mutant, while most root hairs fail to elongate via tip growth after bulge initiation, some hairs still undergo partial root hair elongation, which is more evident on seminal roots (Figure 1A). Notably, the root hair phenotype caused by early termination of tip growth in srh1 is also found in root hair mutants across different cereal species, including the barley (Hordeum vulgare L.) cellulose synthase mutant hvsclc1 (Gajek et al., 2021) and rice expansin mutant osexpa17 (ZhiMing et al., 2011). Wheat srh1 also had longer primary and overall root length compared to wild-type at the seedling stage (Figures 1M, N). As we did not observe any differences in trichoblast length between the two genotypes (data not shown), the longer roots in srh1 are unlikely to be a result of differential cell elongation, or a negative compensatory correlation between root hair and trichoblast length.
srh1 candidate genes
Through exome capture and BSA, we localized srh1 to a chromosomal region of interest (584 - 647Mb), within which a peak region was located between 595 - 617Mb. Within this peak region, seven candidate genes were identified based on filtering for genes with non-synonymous/premature stop mutations, SIFT score and expression in root tissue (Table 1). All seven of these genes were found to be expressed in our RNA-seq data, though none were found to be differentially expressed based on the FDR and Log2FC criteria used. The amino acid residues of the mutations present in these genes in srh1 were all conserved by at least 70% across other plant homologs. Below we consider these seven candidate genes with reference to gene expression data in the wheat gene expression atlas (Ramírez-González et al., 2018, Supplementary Figure S5) and the wheat single-cell RNA-seq atlas (Zhang et al., 2023).
Analysis of publicly available single-cell RNA-seq data (Zhang et al., 2023) showed that three of the seven genes are not expressed in root hair cells (protein kinase, TraesCS3A02G351500; pumilio-like, TraesCS3A02G361700; BEL-1 like homoeodomain, TraesCS3A02G363900), and are less likely to encode SRH1. Of the remaining four candidates, TaNOX3-A (TraesCS3A02G354200) is notable in that it is predicted to encode a respiratory burst oxidase homolog (RBOH) orthologous to known root hair genes in both maize (ZmRTH5, Nestler et al., 2014) and rice (OsNOX3, Wang et al., 2018). TaNOX 3-A is highly expressed in wheat root hair cells relative to all other root tissues (Zhang et al., 2023). Missense mutations in RBOH encoding genes in other cereal species (Supplementary Figure S1) result in arrested root hair phenotype. In Arabidopsis, ROS production is key for regulating cell growth and root hair development (Foreman et al., 2003; Carol, 2006). Close Arabidopsis homologs of TaNOX3-A include RBOHH and RBOHJ (as identified via the ‘Orthologues’ function in Ensembl). RBOHH and RBOHJ are primarily expressed in mature pollen grains and regulate pollen tube growth - a process identical to how root hairs elongate (Zhou et al., 2020). Mutations in both RBOHH and RBOHJ result in defective pollen tube growth, as well as a defective root hair phenotype (Zhou et al., 2020). In ZmRTH5, a missense mutation resulting in an a.C821Y substitution in the predicted protein encodes the loss-of-function allele, whereby zmrth5 root hairs fail to elongate and are less dense than wild-type maize (Nestler et al., 2014). In the rice osnox3 mutant, a missense mutation leading to an a.S676N substitution in the predicted protein results in reduced root hair length and density (Wang et al., 2018). RBOHs are membrane bound proteins that produce ROS in root hair tips, which is critical for cell wall extensibility and subsequent root hair initiation and elongation (Nestler et al., 2014). Both amino acid substitutions in zmrth5 and osnox3 span NAD binding domains towards the C-termini of their predicted proteins, while the a.A476V substitution in TaNOX3-A spans a ferric reductase transmembrane domain further towards the C-terminus. The TaNOX3-A alanine amino acid residue at position 476 is 100% conserved across wheat homoeologues present in 8 other cultivars with sequenced genomes (International Wheat Genome Sequencing Consortium et al., 2018; Walkowiak et al., 2020) (Supplementary Figure S6b), and highly conserved across all cereal orthologues, including the predicted proteins encoded by OsNOX3, ZmRTH5, barley (HORVU.MOREX.r3.3HG0303410.1) and Brachypodium distachyon (BRADI_2g54240v3). While the TaNOX3-A a.A476V substitution is not predicted to result in a major conformation change (Supplementary Figure S1), it’s location in a well conserved ferric reductase transmembrane domain (Figure 5) could alter the stability of membrane binding, the ability of the protein to interact with downstream signaling factors, or its ability to produce ROS. Our results indicated ROS production in root hair tips was much lower in srh1 than wild-type, supporting TaNXO3-A as a candidate for srh1.
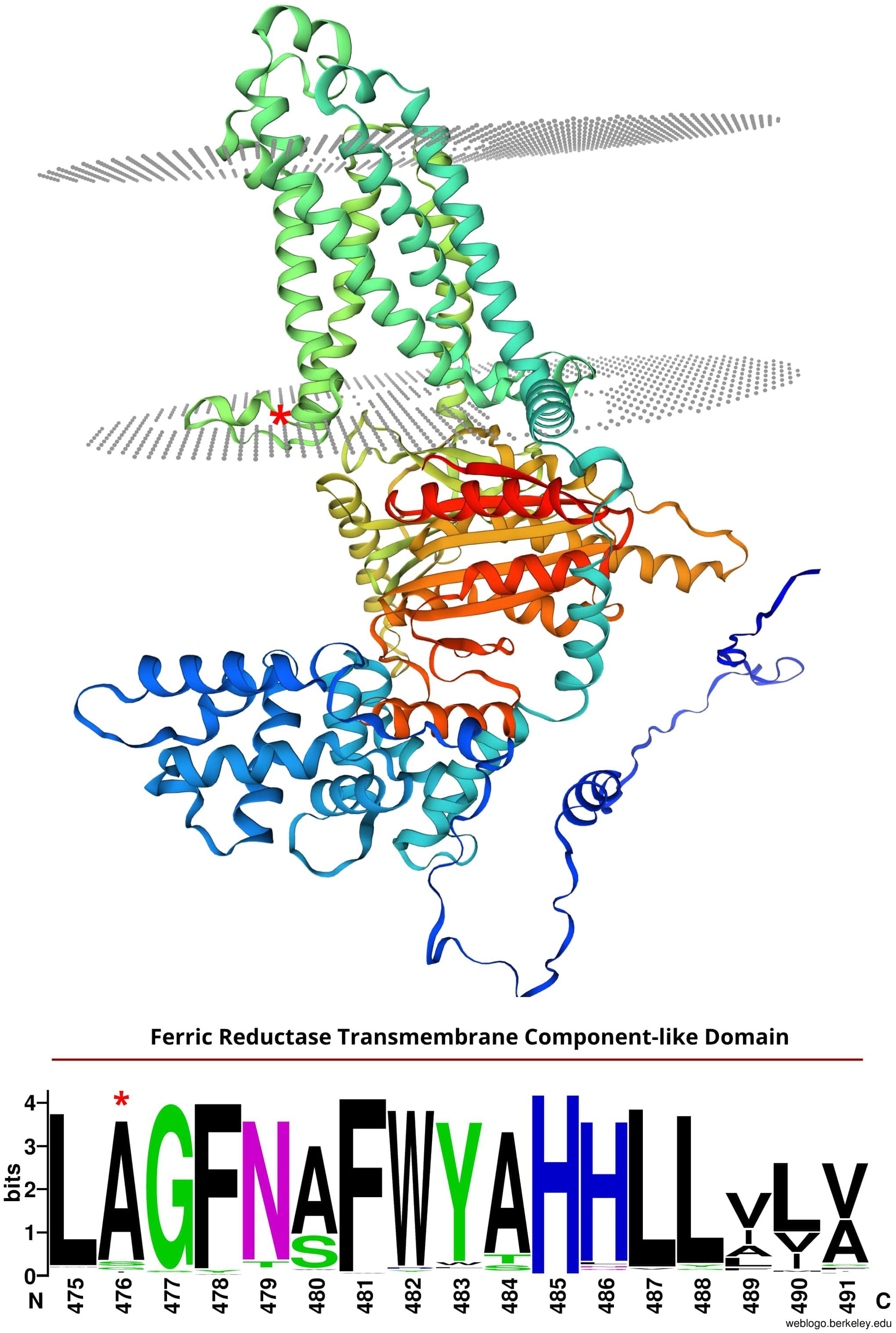
Figure 5. Top: Predicted structure of the srh1 candidate protein, TaNOX3-A, predicted to encode a respiratory burst oxidase homolog (RBOH). Grey dots illustrate membrane layers. Model predicted using SWISS-MODEL (https://swissmodel.expasy.org/). Bottom: Conservation of amino acid resides 475-491 in TaNOX3-A along with 157 other plant homologs. Letter size illustrates degree of conservation. *TaNOX3-A missense substitution in srh1.
Insights into the gene pathways disrupted in the srh1 mutant
Through RNA-seq of primary root tissues of srh1 and wild-type, and using relatively stringent significance (FDR ≤ 0.01) and gene expression fold change (Log2FC ≥ abs(1.5)) criteria, we identified 29 DEGs. Of these, five were notable for their exclusive and high gene expression in the wheat seedling root tip in the gene expression atlas (Zhang et al., 2023), including down-regulated homoeologous bHLH genes in srh1 tissues (TraesCS5A02G401300 and TraesCS5B02G406100) which show sequence conservation with RSL genes controlling root hair elongation in Arabidopsis (e.g. Vijayakumar et al., 2016). Previously, increased expression of the wheat RSL -like genes TaRSL2-A and TaRSL2-B on the group 4 chromosomes (most homologous to Arabidopsis AtRSL4) have been correlated with longer root hairs that are linked to increased wheat ploidy (Han et al., 2017). Indeed, RSL DEG TraesCS5B02G406100 was the most down-regulated gene identified in the srh1 mutant. Thus, our results further support the role of the members of the wheat RSL gene family in root hair differentiation (Supplementary Figure S5). As may be expected in a loss-of-function mutant, the majority of DEGs (>80%) were down-regulated (24 out of 29), of which, seven were predicted to encode nitrate or phosphate transporters. Plant root and root hair development is known to be controlled by the interplay between genetic factors and external nutrient concentrations (reviewed by Liu et al., 2020), and previous studies have demonstrated that nitrate transporters are progressively upregulated in root hairs over developmental time (Shanks et al., 2024). Analysis of the wheat gene expression atlas shows that three of the high-affinity nitrate transport genes we identified as DEGs are most highly expressed in the root tissues at the tillering stage, while the fourth is most highly expressed during anthesis (Supplementary Figure S5). As such, downregulation of genes involved in these pathways fits with the disrupted srh1 root hair phenotype.
While it is possible that the hypothetical loss or alteration-of function in TaNOX3-A affects the downstream expression of the down-regulated genes (such as the aforementioned nitrate and phosphate transporters), it may be more likely that the downregulation is primarily associated with the root hair phenotype itself. With shorter root hairs in the srh1 mutant, less physical space is available on the surface of the root hair membrane for the transporters to be inserted, and thus, lower expression of these genes may be expected.
Of the five DEGs that were upregulated in srh1, TraesCS3B02G424600 (encoding a GRAS-domain DELLA protein) had the highest increase in expression in the mutant compared to wild-type (Log2FC 2.94, Table 2). Members of this class of DELLA protein are known to act as repressors of signaling by the plant phytohormone gibberellin, and can affect root elongation and radial patterning (Hirsch and Oldroyd, 2009). These GRAS encoding genes also promote beneficial root-fungal associations, including in the cereal species rice and barley, highlighting further possible mechanisms for the interaction between root structure and function (Li et al., 2022).
Of all DEGs identified in srh1, TaCRT3-A was most significantly down-regulated in the mutant, in both mature root and root tip root tissues (Supplementary Text 1). Located at 646Mb on chromosome 3A, while TaCRT3-A is not within the peak of the srh1 locus as identified via BSA (595 - 617Mb), it is within the wider srh1 interval (584 - 647Mb). TaCRT3-A is predicted to encode a wheat calreticulin - an endoplasmic reticulum bound chaperone involved in calcium signaling. Calreticulin functions in plants are associated with tip growth (Suwińska et al., 2017), drought stress (Jia et al., 2008) and plant height (Jin et al., 2009). Importantly, calcium signaling is known to be a crucial process in controlling plant root hair development. For example, the calcium protein kinase ZmCPK9 in maize is responsible for root hair elongation by maintaining a high Ca2+ concentration at the root hair apex (Zhang et al., 2020). The formation of intracellular calcium gradients in a root hair also aids the establishment of cell polarity (Brost et al., 2019). Indeed, this class of gene has also been shown to affect root hair development in Arabidopsis (Vu et al., 2017). The recent finding of a wheat root hair mutant that only exhibits the long root hair phenotype under high soil Ca2+ levels (Zeng et al., 2024), further highlights the role of calcium signaling in the control of wheat root hair growth.
Collectively, our DEG analysis indicates that disrupting root hair development through srh1 impacts expression of many genes with a range of putative functions, several of which are linked to root hair development in Arabidopsis and other plant species through either direct or indirect mechanisms. In the near future, we aim to explore how the srh1 mutant performs in the field in contrast to the wild-type, and subsequently, quantify the effect of root hairs on wheat field performance.
Conclusions
Wheat root hairs are involved in numerous root/soil interactions. Here, we describe the first mutant repressing root hair elongation in wheat, providing an entry point into the genes and gene networks controlling wheat root hair development and function. The ability to understand the genetic control of their development, structure, and function, and how this interacts with the environment, will aid the design and development of new, better performing, resilient wheat varieties to support food production under future agricultural scenarios.
Data availability statement
The data presented in the study are deposited in Sequence Read Archive (SRA) under the project number PRJNA1170241.
Author contributions
IT: Data curation, Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. PT: Writing – review & editing, Data curation. LP-A: Supervision, Writing – review & editing. EO: Writing – review & editing. DW: Supervision, Writing – review & editing. JA: Supervision, Writing – review & editing. SR: Supervision, Writing – review & editing. FL: Supervision, Writing – review & editing. JC: Supervision, Conceptualization, Project administration, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. IT was funded by the Biotechnology and Biological Sciences Research Council (BBSRC) as part of the Collaborative Training Program for Sustainable Agricultural Innovation (CTP-SAI) (grant BB/W009439/1), in partnership with The Morley Agricultural Foundation (TMAF). PT, EO, and JC were supported by the International Wheat Yield Project 'Rooty' (IWYP122), funded in part by the BBSRC (grant BB/S012826/1). FL was funded by BBSRC through the Cross-Institute Strategic Programme ‘Designing Future Wheat’ (grant BB/P016855/1).
Acknowledgments
The authors would like to thank James Hutton Institute (JHI) for the operation and maintenance of the UK Crop Diversity HPC (BBSRC grant BB/S019669/1), which has been instrumental for the analysis of data described in this paper.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1490502/full#supplementary-material
Supplementary Figure 1 | Impact of TILLING mutations on the predicted proteins of srh1 candidate genes. Predicted protein alignment of the missense substitutions between wheat TaNOX3-A, rice OsNOX3 and maize ZmRTH5 and other plant homologs. (A): srh1 A >V, (B): osnox3 S >N, (C): zmrth5 C >Y. Residues in red indicate mutation position. (D): SWISS-MODEL (https://swissmodel.expasy.org/) protein model of TaCRT3-A, highlighting the TILLING mutation in the srh1 donor line Cadenza1714 that results in a stop codon at amino acid residue 359 (colored red in the magnified section, and highlighting the disruption of numerous hydrogen bond, dashed lines, with adjacent amino acid residues) and the resulting truncated region (highlighted in green).
Supplementary Figure 2 | Exome capture sequencing gene (including flanks of +/- 50% of gene lengths up and downstream of each gene) depths in bulked populations of (A) Cadenza wild-type and (B) srh1 mutant BC1F3 lines. To remove noise seen by spuriously mapped reads, we introduced a lower depth cutoff of 3 and an upper limit of 200. Turquoise lines represent 10th and 90th depth percentiles, respectively. Orange and red lines illustrate median and mode depths, respectively, following the removal of depths<3 and >200. For bulked segregant analysis (BSA), lower and upper depth cutoffs were established at 5 and 200, respectively.
Supplementary Figure 3 | Amino acid conservation in the predicted proteins of the seven candidate genes identified at the srh1 genetic locus.
Supplementary Figure 4 | Heatmap of differentially expressed genes (DEGs) in seminal root tissues of three day old wild-type versus srh1 mutant BC2F3 segregants, identified in this study via RNA-seq. WM = Wild-type Mature Tissues, WT = Wild-type Tip Tissues, MM = Mutant Mature Tissues, MT = Mutant Tip Tissues. Expression in each of the three biological replicates assayed per tissue type are shown.
Supplementary Figure 5 | Heatmap illustrating expression of differentially expressed genes (DEGs) identified by RNA-seq in srh1 versus wild-type. Expression is shown in key tissues across varying growth stages, normalized across each row via a Z-score. Blues indicate higher relative expression of a gene in a particular tissue; reds indicate lower relative expression across the same tissue. Tissues color coded based on growth stage, root tissues underlined. RNAseq expression data from cv. Azhurnaya spring wheat (Wheat eFP browser, Ramírez-González et al., 2018).
Supplementary Figure 6 | TaNOX3-A sequences. (A): Sanger sequencing of the mutation in TaNOX3-A. 1st and 3rd traces illustrate the genomic sequence of the mutant, 2nd and 4th traces illustrate the genomic sequence of the wild-type. The C >T substitution is clearly visible at sequence position 734 (marked by a red asterisk). (B): Alignment of TaNOX3-A homoeologues from 8 other sequenced wheat genomes (International Wheat Genome Sequencing Consortium et al., 2018; Walkowiak et al., 2020) and orthologues (in rice and maize) within the region spanning the srh1 mutation. Homoeologues from the wheat reference genome of cv. Chinese Spring are outlined via a black box. The position of the A > V amino acid substitution is marked by a red asterisk. Region shown via Sanger sequencing in (A) is correspondingly annotated on (B).
Supplementary Figure 7 | Seven day old BC2F4 srh1 mutant and wild-type seedling root hair phenotype in soil. (A-C): Wild-type, (D-F): Mutant. Root hairs are clearly visible in the wild-type, and are either absent or vestigial in the mutant.
References
Balcerowicz, D., Schoenaers, Sébastjen, Vissenberg, K. (2015). Cell fate determination and the switch from diffuse growth to planar polarity in Arabidopsis root epidermal cells. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.01163
Bilghe, K., Rana, S., Lewis, M. (2023). EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. Available at: https://github.com/kevinblighe/EnhancedVolcano.
Borrill, P., Ramirez-Gonzalez, R., Uauy, C. (2016). expVIP: a Customizable RNA-seq Data Analysis and Visualization Platform. Plant Physiol. 170.4, 2172–2186. doi: 10.1104/pp.15.01667. 0032-0889, 1532-2548.
Brost, C., Studtrucker, T., Reimann, R., Denninger, P., Czekalla, J., Krebs, M., et al. (2019). Multiple cyclic nucleotide-gated channels coordinate calcium oscillations and polar growth of root hairs. Plant J. 99, 910-923. doi: 10.1111/tpj.14371. 0960-7412, 1365-313.
Carol, R. J. (2006). The role of reactive oxygen species in cell growth: lessons from root hairs. J. Exp. Bot. 57.8, 1829–1834. doi: 10.1093/jxb/erj201
Cingolani, P., Platts, A., Wang, Le L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6.2, 80–92. doi: 10.4161/fly.19695
Crooks, G. E., Hon, G., Chandonia, J.-M., Brenner, S. E. (2004). WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Danecek, P., Bonfield, J. K., Liddle, J., Marshal, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. GigaScience 10, giab008. doi: 10.1093/gigascience/giab008
Ding, W., Yu, Z., Tong, Y., Huang, W., Chen, H., Wu, P. (2009). A transcription factor with a bHLH domain regulates root hair development in rice. Cell Res. 19, 1309–1311. doi: 10.1038/cr.2009.109
FAO. (2022). FAO Cereal Supply and Demand Brief. Available at: https://www.fao.org/worldfoodsituation/csdb/en/.
Foreman, J., Demidchik, V., Bothwell, J. H.F., Mylona, P., Miedema, H., Torres, M. A., et al. (2003). Reactive oxygen species produced by NADPH oxidase regulate plant cell growth. Nature 452, 587–600. doi: 10.1038/nature01485
Fulton, T. M., Chunwongse, J., Tanksley, S. D. (1995). Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol. Biol. Rep. 13, 207–209. doi: 10.1007/BF02670897
Gajek, K., Janiak, A., Korotko, U., Chmielewska, B., Marzec, M., Szarejko, I. (2021). Whole Exome Sequencing-Based Identification of a Novel Gene Involved in Root Hair Development in Barley (Hordeum vulgare L.). Int. J. Mol. Sci. 22.24, 13411. doi: 10.3390/ijms222413411
Gardiner, L.-J., Brabbs, T., Akhunov, A., Jordan, K., Budak, H., Richmond, T., et al. (2019). Integrating genomic resources to present full gene and putative promoter capture probe sets for bread wheat. GigaScience 8.4. doi: 10.1093/gigascience/giz018
Grierson, C., Nielsen, E., Ketelaarc, T., Schiefelbein, J. (2014). Root hairs. Arabidopsis Book 12, e0172. doi: 10.1199/tab.0172
Han, H., Wang, H., Han, Y., Hu, Z., Xin, M., Peng, H., et al. (2017). Altered expression of the TaRSL2 gene contributed to variation in root hair length during allopolyploid wheat evolution. Planta 246, 1019–1028. doi: 10.1007/s00425-017-2735-3
Han, Y., Xin, M., Huang, Ke, Xu, Y., Liu, Z., Hu, Z., et al. (2016). Altered expression of TaRSL4 gene by genome interplay shapes root hair length in allopolyploid wheat. New Phytol. 209, 721–732. doi: 10.1111/nph.13615
Hassani-Pak, K., Singh, A., Brandizi, M., Hearnshaw, J., Parsons, J. D., Amberkar, S., et al. (2021). KnetMiner: a comprehensive approach for supporting evidence-based gene discovery and complex trait analysis across species. Plant Biotechnol. J. 19.8, 1670–1678. doi: 10.1111/pbi.13583
Hirsch, S., Oldroyd, G. E. D. (2009). GRAS-domain transcription factors that regulate plant development. Plant Signaling Behav. 4.8, 698–700. doi: 10.4161/psb.4.8.9176
Hochholdinger, F., Wen, T.-J., Zimmermann, R., ChimotMarolle, P., Silva, O. da C. e, Bruce, W., et al. (2008). The maize (Zea mays L.) roothairless3 gene encodes a putative GPI-anchored, monocot-specific, COBRAlike protein that significantly affects grain yield. Plant J. 54.5, 888–898. doi: 10.1111/j.1365-313X.2008.03459.x
International Wheat Genome Sequencing Consortium, Alaux, M., Rogers, J., Flores, R., Alfama, F., Pommier, C., et al. (2018). Linking the International Wheat Genome Sequencing Consortium bread wheat reference genome sequence to wheat genetic and phenomic data. Genome Biol. 19.1, 111. doi: 10.1186/s13059-018-1491-4
Jia, X.-Y., Xu, C.-Y., Jing, R.-L., Li, R.-Z., Mao, X.-G., Wang, J.-P., et al. (2008). Molecular cloning and characterization of wheat calreticulin (CRT) gene involved in drought-stressed responses. J. Exp. Bot. 59.4, 739–751. doi: 10.1093/jxb/erm369
Jin, H., Hong, Z., Su, W., Li, J. (2009). A plant-specific calreticulin is a key retention factor for a defective brassinosteroid receptor in the endoplasmic reticulum. Proc. Natl. Acad. Sci. 106.32, 13612–13617. doi: 10.1073/pnas.0906144106
Kim, D., Paggi, J. M., Park, C., Bennett, C., Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37.8, 907–915. doi: 10.1038/s41587-019-0201-4
Krasileva, K. V., Vasquez-Gross, H. A., Howell, T., Bailey, P., Paraiso, F., Clissold, L., et al. (2017). Uncovering hidden variation in polyploid wheat. Proc. Natl. Acad. Sci. 114.6, 1091–6490. doi: 10.1073/pnas.1619268114
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Res. 19.9, 1639–1645. doi: 10.1101/gr.092759.109
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25.14, 1754–1760. Available at: https://academic.oup.com/bioinformatics/article/25/14/1754/225615.
Li, L., Hey, S., Liu, S., Liu, Q., McNish, C., Hu, H.-C., et al. (2016). Characterization of maize roothairless6 which encodes a D-type cellulose synthase and controls the switch from bulge formation to tip growth. Sci. Rep. 6.1, 34395. doi: 10.1038/srep34395
Li, X.-R., Sun, J., Albinsky, D., Zarrabian, D., Hull, R., Lee, T., et al. (2022). Nutrient regulation of lipochitooligosaccharide recognition in plants via NSP1 and NSP2. Nat. Commun. 13.1, 6421. doi: 10.1038/s41467-022-33908-3
Liao, Y., Smyth, G. K., Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Liu, B., Wu, J., Yang, S., Schiefelbein, J., Gan, Y. (2020). Nitrate regulation of lateral root and root hair development in plants. J. Exp. Bot. 71.15, 4405–4414. doi: 10.1093/jxb/erz536
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15.12, 550. doi: 10.1186/s13059-014-0550-8
Ma, N., Wang, Y., Qiu, S., Kang, Z., Che, S., Wang, G., et al. (2013). Overexpression of OsEXPA8, a root-specific gene, improves rice growth and root system architecture by facilitating cell extension. PloS One 8, e75997. doi: 10.1371/journal.pone.0075997
Martinez, S. A., Shorinola, O., Conselman, S., See, D., Skinner, D. Z., Uauy, C., et al. (2020). Exome sequencing of bulked segregants identified a novel TaMKK3-A allele linked to the wheat ERA8 ABA-hypersensitive germination phenotype. Theor. Appl. Genet. 133.3, 719–736. doi: 10.1007/s00122-019-03503-0
McLaren, W., Gil, L., Sarah, E., Riat, H. S., Ritchie, G. R.S., et al. (2016). The ensembl variant effect predictor. Genome Biol. 17.1, 122. doi: 10.1186/s13059-016-0974-4
Nestler, J., Liu, S., Wen, T.-J., Paschold, A., Marcon, C., Tang, H. M., et al. (2014). Roothairless5, which functions in maize (Zea mays L.) root hair initiation and elongation encodes a monocot-specific NADPH oxidase. Plant J. 79.5, 729–740. doi: 10.1111/tpj.12578
Ober, E. S., Alahmad, S., Cockram, J., Forestan, C., Hickey, L. T., Kant, J., et al. (2021). Wheat root systems as a breeding target for climate resilience. Theor. Appl. Genet. 134.6, 1645–1662. doi: 10.1007/s00122-021-03819-w
Ramírez-González, R. H., Borrill, P., Lang, D., Harrington, S. A., Brinton, J., Venturini, L., et al. (2018). The transcriptional landscape of polyploid wheat. Science 361.6403, eaar6089. doi: 10.1126/science.aar6089
RStudio Team (2020). RStudio: integrated development environment for R. RStudio (Boston, MA: PBC). Available at: http://www.rstudio.com/.
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9.7, 676–682. doi: 10.1038/nmeth.2019
Shanks, C. M., Rothkegel, K., Brooks, M. D., Cheng, C.-Y., Alvarez, JoséM., Ruffel, S., et al. (2024). Nitrogen sensing and regulatory networks: it’s about time and space. Plant Cell 36, 1482–1503. doi: 10.1093/plcell/koae038
Sigalas, P. P., Buchner, P., Kröper, A., Hawkesford, M. J. (2023). The functional diversity of the high-affinity nitrate transporter gene family in hexaploid wheat: insights from distinct expression profiles. Int. J. Mol. Sci. 25.1, 509. doi: 10.3390/ijms25010509
Suwińska, A., Wasąg, P., Zakrzewski, Przemysław, Lenartowska, M., Lenartowski, R. (2017). Calreticulin is required for calcium homeostasis and proper pollen tube tip growth in Petunia. Planta 245.5, 909–926. doi: 10.1007/s00425-017-2649-0
The International Wheat Genome Sequencing Consortium (IWGSC), Appels, R., Eversole, K., Stein, N., Feuillet, C., Keller, B., et al. (2018a). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361, eaar7191. doi: 10.1126/science.aar7191
Tsang, I., Atkinson, J. A., Rawsthorne, S., Cockram, J., Leigh, F. (2024). Root hairs: an underexplored target for sustainable cereal crop production. J. Exp. Bot. 75, 5484–5500. doi: 10.1093/jxb/erae275
Vijayakumar, P., Datta, S., Dolan, L. (2016). ROOT HAIR DEFECTIVE SIX LIKE 4 (RSL4) promotes root hair elongation by transcriptionally regulating the expression of genes required for cell growth. New Phytologist 212, 944–953. doi: 10.1111/nph.14095
Vu, K. V., Nguyen, N. T., Jeong, C. Y., Lee, Y.-H., Lee, H., Hong, S.-W. (2017). Systematic deletion of the ER lectin chaperone genes reveals their roles in vegetative growth and male gametophyte development in Arabidopsis. Plant J. 89, 972–983. doi: 10.1111/tpj.13435
Walkowiak, S., Gao, L., Monat, C., Haberer, G., Kassa, M. T., Brinton, J., et al. (2020). Multiple wheat genomes reveal global variation in modern breeding. Nature 588, 277–283. doi: 10.1038/s41586-020-2961-x
Wang, S. S., Zhu, X. N., Lin, J. X., Zheng, W. J., Zhang, B. T., Zhou, J. Q., et al. (2018). OsNOX3, encoding a NADPH oxidase, regulates root hair initiation and elongation in rice. Biol. Plantarum. doi: 10.1007/s10535-018-0814-3
Yates, A. D., Allen, J., Amode, R. M., Azov, A. G., Barba, M., Becerra, A., et al. (2022). Ensembl Genomes 2022: an expanding genome resource for non-vertebrates. Nucleic Acids Res. 50, D996–D1003. doi: 10.1093/nar/gkab1007
Zeng, D., Ford, B., Doležel, J., Karafiátová, M., Hayden, M. J., Rathjen, T. M., et al. (2024). A conditional mutation in a wheat (Triticum aestivum L.) gene regulating root morphology. Theor. Appl. Genet. 137, 48. doi: 10.1007/s00122-024-04555-7
Zhang, L., He, C., Lai, Y., Wang, Y., Kang, L., Liu, A., et al. (2023). Asymmetric gene expression and cell-type-specific regulatory networks in the root of bread wheat revealed by single-cell multiomics analysis. Genome Biol. 24, 65. doi: 10.1186/s13059-023-02908-x
Zhang, X., Mi, Y., Mao, H., Liu, S., Chen, L., Qin, F. (2020). Genetic variation in ZmTIP1 contributes to root hair elongation and drought tolerance in maize. Plant Biotechnol. J. 18, 1271–1283. doi: 10.1111/pbi.13290
ZhiMing, Yu, Bo, K., XiaoWei, He, ShaoLei, Lv, YouHuang, B., WoNa, D., et al. (2011). Root hair-specific expansins modulate root hair elongation in rice: Rice root hair elongation requires expansins. Plant J. 66, 725–734. doi: 10.1111/j.1365-313X.2011.04533.x
Zhou, X., Xiang, Yu, Li, C., Yu, G. (2020). Modulatory role of reactive oxygen species in root development in model plant of Arabidopsis thaliana. Front. Plant Sci. 11, 485932. doi: 10.3389/fpls.2020.485932
Keywords: temperate cereal species, sustainable crop production, root system architecture, exome capture, RNA sequencing (RNA-Seq), differential gene expression (DEG)
Citation: Tsang I, Thomelin P, Ober ES, Rawsthorne S, Atkinson JA, Wells DM, Percival-Alwyn L, Leigh FJ and Cockram J (2024) A novel root hair mutant, srh1, affects root hair elongation and reactive oxygen species levels in wheat. Front. Plant Sci. 15:1490502. doi: 10.3389/fpls.2024.1490502
Received: 03 September 2024; Accepted: 04 October 2024;
Published: 30 October 2024.
Edited by:
John Hancock, University of the West of England, United KingdomReviewed by:
Gerasimos Daras, Agricultural University of Athens, GreeceJuli Jing, Cornell University, United States
Copyright © 2024 Tsang, Thomelin, Ober, Rawsthorne, Atkinson, Wells, Percival-Alwyn, Leigh and Cockram. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ian Tsang, aWFuLnRzYW5nQG5pYWIuY29t; James Cockram, amFtZXMuY29ja3JhbUBuaWFiLmNvbQ==
†ORCID: Ian Tsang, orcid.org/0009-0006-1999-3251
Eric S. Ober, orcid.org/0000-0002-4832-5897
Jonathan A. Atkinson, orcid.org/0000-0003-2815-0812
Darren M. Wells, orcid.org/0000-0002-4246-4909
Lawrence Percival-Alwyn, orcid.org/0000-0001-7725-9203
Fiona J. Leigh, orcid.org/0000-0002-3221-5327
James Cockram, orcid.org/0000-0002-1014-6463