 Kun Liu
Kun Liu Yonglin Qin
Yonglin Qin Lan Wu
Lan Wu Liguo Jia
Liguo Jia Mingshou Fan
Mingshou Fan- College of Agronomy, Inner Mongolia Agricultural University, Hohhot, Inner Mongolia, China
The HAK/KUP/KT (High-affinity K+ transporters/K+ uptake permeases/K+ transporters) is the largest and most dominant potassium transporter family in plants, playing a crucial role in various biological processes. However, our understanding of HAK/KUP/KT gene family in potato (Solanum tuberosum L.) remains limited and unclear. In this study, 24 HAK/KUP/KT genes (StHAKs) were identified through a genome-wide analysis and were found to be unevenly distributed across ten chromosomes. Based on phylogenetic analysis, these StHAK gene family members were classified into four distinct clusters. All StHAK protein sequences contained the conserved motifs and domains. Promoter cis-acting elements analysis revealed that most StHAK gene family members in potatoes were associated with responses to light and hormones such as abscisic acid or methyl jasmonate, however, many motifs responsive to hormones and stress conditions have not been clearly studied or reported in plants. Synteny analysis suggested that 33, 19, 8, 1 StHAK genes were orthologous to those in soybean, cassava, Arabidopsis and rice, respectively. The previously published RNA-seq results, transcriptomic data and qRT-PCR experiments indicated that the expression profiles of these StHAK genes were tissue-specific and were influenced by multiple factors, including biotic and abiotic stress, hormone, potassium fertilizer. To provide a clear and convenient view of StHAK gene expression across different tissues in potato, we generated a cartoon heatmap to vividly illustrate the tissue-specific expression of StHAK genes, which is unprecedented in the gene family analysis of potato. At last, we identified genes such as StHAK8, StHAK14, and StHAK22 with high expression in potato tubers using qRT-PCR, suggesting their potential involvement in tuber growth and development. This can contribute to a deeper understanding of the mechanism of potassium absorption and transportation in potatoes. It has laid a solid theoretical foundation for the genetic regulation of potassium nutritional efficiency in potatoes and the breeding of potato varieties with high potassium efficiency.
1 Introduction
As one of the most abundant and essential mineral nutrient element in plant, potassium regulates numerous physiological and metabolic procedures, and most importantly, it determines the yield and quality of crop production (Sardans and Peñuelas, 2021). Due to its high mobility in plants, potassium is involved in regulating critical metabolic pathways and contributing to synthesis and transport of proteins and sugars (Fan et al., 2019). Additionally, potassium is engaged in stabilizing balance between anion and cation and in regulating cellular osmotic potential, strengthening the water content of protoplasm and enhancing the drought resistance of plant (Torabian et al., 2021). After wheat (Triticum aestivum), rice (Oryza sativa) and maize (Zea mays), potato (Solanum tuberosum L.) is the fourth most important crop in world and has a great demand for potassium, which is the predominant element in ash, accounting for 50-70% of the total ash content (Fan et al., 2019).To produce one ton of tubers, potatoes need to absorb 4.0-6.0 kg of K2O, 1.0-1.5 kg of P2O5, and 3.0-4.0 kg of N from the soil (Chen et al., 2024; Kong et al., 2024; Shi et al., 2019). A sufficient supply of potassium strengthens stems, making them more robust and resilient, thereby enhancing resistance to collapse, freezing, and diseases in potatoes. Additionally, it promotes tuber bulking and significantly improves potato yield (Shi et al., 2019). Some quality index, such as protein, starch and vitamins in tubers, can also be improved with adequate potassium supply, and the tuber hollow rate in potatoes is significantly reduced (Naumann et al., 2020). With insufficient potassium, potato stolons are significantly shortened, and root development and nutrient absorption are noticeably decreased (Fan et al., 2019).
In plants, K+ is primarily absorbed by the roots through dual-affinity mechanisms, which include high-affinity and low-affinity systems for potassium assimilation. When the external potassium concentration is below 0.2 mM, a high-affinity transport system (HATS) is activated, facilitating the movement of K+ from the extracellular to the intracellular environment via potassium transporters. Conversely, when the external potassium concentration exceeds 0.3 mM, a low-affinity transport system (LATS) via potassium channels is initiated (Li et al., 2018). For optimal plant function, the K+ concentration in various cellular compartments, including the nucleus, cytosol, chloroplast stroma, and mitochondrial matrix, should be maintained between 100 and 150 mM (Britto and Kronzucker, 2008). The minimum vacuolar and tissue concentration of K+ present in cell are approximately 10 mM and 5 mg K (g dry matter (DM))-1, respectively (White, 2013). In many cultivated soils, potassium application is insufficient to maintain healthy growth and development of crop, with K+ concentration in soil typically varying between 0.01 and 1 mM, representing only 0.1%~0.2% of the total soil K (White, 2013). Consequently, the acquisition of K+ from the surrounding soil by plant roots is primarily mediated through HATS, while the translocation of K+ from root to shoot and its distribution from source to sink within the plants is predominantly mediated through both HATS and LATS (Wang et al., 2021).
In higher plants, K+ transport occurs primarily through potassium transporter and ion channel protein families, with K+ channel proteins encoded by three main families: Shaker-like K+ channels, tandem-pore K+ (TPK) channels, and K+ inward rectifiers (Kir). and K+ transporters classified into four families: HAK/KUP/KT (High-affinity K+ transporters/K+ uptake permeases/K+ transporters), HKT (High-affinity K+ transporters), NHX (Na+/H+ exchangers) and CHX (Cation/H+ exchangers) (Johnson et al., 2022). Among these transporter protein families, the KUP/HAK/KT family (abbreviated as HAK) is the largest and plays a crucial role in mediating intracellular K+ accumulation for maintaining plant growth and development (Wang et al., 2021). HAK functions as a K+/H+ symporter, facilitating K+ uptake in plants by linking high-affinity K+ transport with H+ gradients (Santa-Maria et al., 2018; Sze and Chanroj, 2018).The plant HAK/KUP/KT transporters were first identified from barley (Hordeum vulgare) (Santa-María et al., 1997). Since then, they have been discovered in a wide range of plant species, including rice, wheat, maize, sugarcane (Saccharum spontaneum), Brassica napus, grape (Vitis vinifera), tomato (Lycopersicon esculentum), among others (Zhou et al., 2020; Luo et al., 2021; Lei et al., 2022; Li et al., 2022; Run et al., 2022; Che et al., 2024; Wang et al., 2024);
Under K+-deficient conditions, Arabidopsis HAK/KUP/KT transporter HAK5 is primarily expressed in the root and responsible for K+ absorbed from the rhizosphere soil (Jiménez-Estévez et al., 2024). Like AtHAK1, AtKUP3, AtHAK5, HvHAK1, OsHAK1, and CaHAK1, they also serve as highly efficient potassium transporters across various plant species (Feng et al., 2020). In addition to K+ uptake and transport, HAK/KUP/KT genes are also associated with stress response and tolerance. The expression levels of CeqHAK11 and CeqHAK6 shows a significant increase as the duration of salt treatment progressed, and overexpression of CeqHAK6 or CeqHAK11 in Arabidopsis shows higher germination, survival rates and longer root length than wild-type under salt stress (Wang et al., 2023). OsHAK16 is induced by K+ deficiency or salt stress, and its knockout mutant exhibits significantly increased salt sensitivity (Feng et al., 2019). ZmHAK4 can promote Na+ exclusion from shoots and improve salt tolerance, contributing to the development of salt-resistance maize varieties (Zhang et al., 2019). All KT/HAK/KUPs in Citrullus lanatus and Citrullus amarus exhibit specific expression responses to drought stress (Cheng et al., 2024). RT-qPCR data shows that all six MsHAK genes are highly induced by salt and drought treatment, and all RT-qPCR for six MsHAK genes are positively correlated with transcriptome data (Li et al., 2022).
Besides their roles in K+ acquisition, translocation and stress tolerance, members of HAK/KUP/KT genes family are also involved in regulating root morphogenesis and development. In the presence of 10 μM K+, roots of F130S-1 and F130S-2 lines (Both strains are mutant lines of the AtHAK5 gene) shows significantly larger roots than those of WT and akt1 ones (63 and 66% larger with respect to akt1, respectively) (Jiménez-Estévez et al., 2024). The addition of K+ significantly stimulated adventitious root development, with roots beginning to emerge at 8 days and showing a marked increase in number at 16 days compared to the K+-free control. Notably, several MdHAKs demonstrated strong root-specific expression in stem cuttings of B9 apple rootstock, with their expression levels sharply upregulated during the initiation and emergence phases of adventitious root formation under K+ treatment (Tahir et al., 2023). TINY ROOT HAIR 1 (TRH1), a HAK/KUP/KT transporter in Arabidopsis root cells, regulates intracellular ionic gradients and polar auxin transport, driving root epidermal differentiation and gravitropic responses (Templalexis et al., 2022). AtKUP9, highly expressed in roots and specifically expressed in quiescent center cells in root tips, controls primary root growth in Arabidopsis, and kup9 displayed a short-root phenotype that resulted from reduced numbers of root cells under low-K+ conditions (Zhang et al., 2020). Although the crucial functions of HAK genes have been characterized in different plant species, the regulatory mechanisms of HAK in potato remain poorly understood, and a systematic study of the HAK gene family in potato is still lacking.
Currently, the HAK gene family has been reported in many plants (Guo et al., 2024). The HAK family genes are widely expressed in various tissues, including roots, stems, leaves, and tubers, enabling plants to efficiently absorb potassium in low-potassium environments, maintaining cellular ion balance and osmotic pressure, and regulating stress responses and metabolic processes, but the relationship between evolution and functional characterization of HAK family genes remains unclear (Jin et al., 2021; Johnson et al., 2022). Research in this field is particularly lacking in potato. In this study, a genome-wide analysis of HAK genes family in potato was conducted and 24 HAK gene family members were identified. The study also explored phylogenetic relationships, conserved motifs and domains, cis-regulatory elements, collinearity and syntenic relationships, as well as expression profiles across various tissues and potassium gradients to elucidate their potential biological functions in potato. Notably, our findings indicated that the StHAK genes exhibited distinct expression patterns across different tissues, and their transcription was induced by a range of biotic and abiotic stresses, including potassium availability. These studies will enhance our understanding of the functions of the HAK/KUP/KT gene family and have important implications for improving potassium utilization efficiency, as well as for advancing agricultural practices and production in potatoes.
2 Materials and methods
2.1 Identification of StHAK gene family in potato
In this study, the candidate HAK genes in potato were identified. The coding sequences (CDS), transcript sequences (cDNA), protein sequences and locus (genomic) sequences were download from Phytozome 13.0 (Goodstein et al., 2012) and Potato Genomic Resource (http://spuddb.uga.edu/). A profile hidden Markov model (pHMM) file for the ‘K_trans’ (Pfam: PF02705) domain was obtained from the Pfam protein family database (El-Gebali et al., 2018). The K_trans.hmm file was used to search against protein sequences from various plant genomic databases using the Hmmsearch program (HMMer package version 3.1b1). An E-value threshold of 1e-10 was set to identify plausible domains, and multiple sequence alignment was performed using Cluster W (V2.1). In consideration of the different predicted isoforms of genes loci, the primary isoforms were selected based on the annotation. If no annotation was available, the longest protein sequence was chosen for follow-up studies. To confirm the ‘K_trans’ domain in the predicted protein, sequences were upload to SMART (http://smart.emblheidelberg.de/), NCBI CDD (https://www.ncbi.nlm.nih.gov/cdd/) and HMMER (http://www.hmmer.org/) for online analysis (Finn et al., 2011; Letunic and Bork, 2018; Lu et al., 2020). Only genes with an intact ‘K_trans’ domain were retained for further analysis and research.
The isoelectric point (pI) and Molecular weight (Mw) of StHAKs were predicted by the ExPASy server (https://web.expasy.org/compute_pi/). Transmembrane domains (TMS) in the StHAK proteins were predicted by TMHHM Server v.2.0 (http://www.cbs.dtu.dk/services/TMHMM/). Conserved domains of the StHAK proteins were identified using the NCBI Conserved Domain Database (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi). Additionally, the subcellular localization of all StHAK proteins were predicted using WOLF PSORT (https://www.genscript.com/wolf-psort.html) (Horton et al., 2007).
2.2 Phylogenetic analysis of StHAK proteins
The full-length amino acid sequences of HAK genes identified from S. tuberosum, A. thaliana, O. sativa and Z. mays were aligned using ClusterW with default parameters (Larkin et al., 2007). A phylogenetic tree of the HAK gene family was then constructed using the neighbor-joining (NJ) method with 1000 bootstrap replicates in MEGA7.0 software. In addition, the phylogenetic relationships of HAKs were also analyzed using IQ-TREE with the maximum likelihood (ML) method. The resulting phylogenetic trees were decorated and visualized using the online tool iTOL (Letunic and Bork, 2007).
2.3 Gene structure and conserved motif analysis of StHAKs
The exon/intron organization of StHAKs was analyzed by comparing cDNA sequences with their corresponding genomic DNA sequences and visualized using the Gene Structure Display Server 2.0 (GSDS; http://gsds.cbi.pku.edu.cn/) (Hu et al., 2015). The conserved motifs in StHAKs were identified through Multiple Ems for Motif Elicitation (MEME) online program (http://meme-suite.org/ ) with the default parameters and the maximum number of conserved motifs was set to 10. The schematic paragraph of the phylogenetic tree, gene structure and conserved motifs were re-edited in PDF and assembled using TBtools software (Chen et al., 2023a).
2.4 Chromosomal distribution, gene duplication, collinearity and synteny analysis of StHAKs
The chromosomal distribution, gene duplication events, and collinearity analysis of the StHAK gene family were carried out using Advanced Cricos tool in TBtools (Chen et al., 2023a). The synteny relationships of HAK genes between potato and other plants were analyzed via Multiple Collinearity Scan toolkit (MCScanX) (http://chibba.pgml.uga.edu/mcscan2/) (Wang et al., 2012). The plant species included in the analysis were Arabidopsis thaliana, Solanum lycopersicum, Glycine max, Manihot esculenta, Oryza sativa, Sorghum bicolor, Zea mays, Vitis vinifera.
2.5 Analysis of cis-acting elements in StHAK genes promoter region
The 2000 bp of 5’ upstream StHAK CDS sequences were retrieved from the Spud DB online database (http://solanaceae.plantbiology.msu.edu/). These sequences were then submitted to the PlantCARE online program (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) (Lescot et al., 2002). We predicted cis-regulatory elements related to plant growth and development, phytohormone responsiveness, as well as stress response in the promoter regions of the HAK genes. Some cis-acting elements that were unrelated or lacked explicit annotations were excluded. The identified cis-regulatory elements were visualized using the Simple Biosequence Viewer program in TBtools.
2.6 Gene expression level analysis of StHAK genes based on microarray data
According to the Spud DB database (https://spuddb.uga.edu), expression profiles of StHAK genes were retrieved based on published RNA-seq data from 15 types of tissues, 3 types of abiotic stresses, 3 types of biotic elicitors and 4 types of hormonal treatments (Xu et al., 2011). The genomic locus information for S. tuberosum Group Phureja DM 1–3 v1.6 was used to analyze the expression levels of HAK genes. The gene expression values (Transcripts per million, TPM) from 219 potato Microarray libraries in the SRA were retrieved from the database. The tissues examined included sepals, leaves, roots, shoots, callus, tubers, stolons, petioles, petals, stamens, carpels, flowers, mature fruits, immature fruits, mesocarp & endocarp. Abiotic stresses contained NaCl (150 mM) for salinity treatment, mannitol (260 μM) for dehydration treatment, and 35°C for heat treatment. Biotic stresses included BABA (β-aminobutyric acid), BTH (benzothiadiazole), and pathogen, with leaves treated for 72 hours. The whole potato plants were incubated for 24 hours under four hormonal condition: 6-benzylaminopurine (BAP, 10 μM), indole-3-acetic acid (IAA, 10 μM), abscisic acid (ABA, 50 μM), and gibberellic acid (GA3, 50 μM). The expression matrix data for StHAK genes were used to generate a heatmap with the heatmap program in TBtools. For statistical convenience, each TPM value was log2-transformed (TPM+1).
2.7 Plant materials and different treatment
The potato research was conducted under field conditions between 2021 and 2022 in Siziwang County (41°46′N, 111°46′E) and County Chayouzhong (41°30′N, 112°64′E), Inner Mongolia Autonomous Region, China. The experimental sites are located in the Inner Mongolia plateau region, with an average altitude of approximately 1700 meters and an average annual temperature of 1.3 °C. The frost-free period in this region is about 100 d, and the annual evaporation exceeds 2000 mm (Cui et al., 2020). Precipitation at the trial sites during the growing season was approximately 250 mm in 2021 and 350 mm in 2022. The soil at the experimental sites is calcareous with a sandy texture. The basic physical and chemical properties of the 0~20 cm upper soil are detailed in Supplementary Table 1.
The experiments were preformed based on a randomized block design with three replicates per treatment. Each plot measured 72 m² (8×9 meters), with potato plants (cultivar Zhongjia-2) spaced 24–26 cm apart within rows, and rows separated by 90 cm. During the growing period, a drip irrigation system was extensively applied (Shi et al., 2019). Three potassium levels were tested in the field experiment: K0 (0 kg/hm2), K1 (300 kg/hm2), K2 (600 kg/hm2). In the controlled field experiment, nitrogen fertilizer was applied at 300 kg/hm2 as coated urea (N 46%), phosphorus fertilizer was applied at 180 kg/hm2 as triple superphosphate (P2O5 50%), and potassium sulfate was used as the potassium source (K2O 50%). Nitrogen, phosphorus and potassium fertilizers were all broadcasted at sowing for each treatment. Potatoes were sown on May 12, 2021, and May 8, 2022, and harvested on September 3, 2021, and September 13, 2022, respectively.
Before the potassium fertilization treatments, the soil in the experimental plots was homogenized to avoid localized potassium nutrient imbalances. During the potassium treatment, the total irrigation amount during the potato entire growing season was 120 mm/667 m², and other cultivation and field management practices followed the same methods as local farmers. Sampling was collected at 45 days after potato emergence (during the tuber bulking stage). The sampling time was chosen in the morning (8:00-10:00), preferably on clear, windless, and rain-free days, in order to avoid the effects of midday sun exposure, which could lead to photorespiration or photoinhibition in potatoes. The samples were divided into five types of tissue: roots, stems, leaves, tubers, and flowers. Each tissue was rapidly frozen in liquid nitrogen. Roots were cleaned with distilled water to remove any soil before freezing.
2.8 Transcriptome sequencing analysis of StHAK genes expression levels
Under field experimental conditions, samples from tissues (roots, leaves, tubers) and potassium fertilizer treatments (0, 300, 600 kg/hm2) were collected at 50 days after potato emergence (tuber expansion stage). Total RNA was extracted from these samples using the RNAprep Pure Plant Kit (Tiangen, Beijing, China) according to the instructions provided by the manufacturer. RNA concentration and purity was assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). RNA integrity was evaluated with the RNA Nano 6000 Assay Kit on the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). A total of 1 μg RNA per sample was used as input material for RNA sample preparation. Sequencing libraries were generated using Hieff NGS Ultima Dual-mode mRNA Library Prep Kit for Illumina (Yeasen Biotechnology (Shanghai) Co., Ltd.) following manufacturer’s recommendations and index codes were added to attribute sequences to each sample. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. First-strand cDNA synthesis was followed by second-strand cDNA synthesis. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3’ ends of DNA fragments, NEBNext Adaptor with hairpin loop structure were ligated to prepare for hybridization. The library fragments were purified with AMPure XP system (Beckman Coulter, Beverly, USA). Then 3 μl USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37°C for 15 min followed by 5 min at 95°C before PCR. Then PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers and Index (X) Primer. At last, PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system. The libraries were sequenced on an Illumina NovaSeq platform to generate 150 bp paired-end reads. The raw reads were further processed with a bioinformatic pipelinetool, BMKCloud (www.biocloud.net) online platform. The gene sequences of DMv6.1 from spud was selected as the reference genome (Xu et al., 2011). Quantification of gene expression levels were estimated by fragments per kilobase of transcript per million fragments mapped (FPKM) (Trapnell et al., 2010). The selection criteria for DEGs were as follows: genes with a Fold Change (FC) ≥1.5 and p-value ≤ 0.01 were considered significantly differentially expressed. After standardizing and quality-controlling the raw data, differential analysis was conducted using edgeR_DESeq2. All these analysis were performed using BMKCloud (www.biocloud.net).
2.9 qRT-PCR and expression pattern assay of StHAKs
For tissue samples, 45-day-old potato plants after emergence were randomly sampled with three replicates. The samples were divided into five different tissues including roots, stems, leaves, flowers and tubers. Tissue samples were clipped with DEPC water-socked scissors, and roots and tubers were carefully washed with distilled water to clean the soil. All tissues were immediately stored into liquid nitrogen. For the potassium fertilizer treatment experiment, three randomly selected potato leaves (the fourth from the top) were collected at each potassium level and rapidly frozen into liquid nitrogen.
Real-time PCR assays were conducted according to our previous studies with minor modifications (Liu et al., 2019). Total RNA of each samples was extracted via a total RNA extraction kit (Tiangen, China). Quality evaluation of RNA was done by agarose gel electrophoresis and Quawell micro volume spectrophotometer (Q5000, USA), with the concentrations of total RNA ranging from 200 to 400 ng/μL and A260/280 ratios between 2.0~2.1. Then, 1μg total RNA was digested by DNase I and reverse-transcribed into cDNA based on TransScript gDNA Removal and cDNA Synthesis SuperMix Kit (TransGen, Beijing, China, Cat# AT311). The qRT-PCR was carried out through the QuantiNova SYBR Green PCR kit (QIAGEN, Germany) on a LightCycler 480 system (Roche, Basel, Switzerland). The thermal cycling parameters was set as follows: 95°C for 30 s, followed by 40 cycles of 95°C for 5 s, 60°C for 30 s and 72°C for 15 s. After 40 cycles, the temperature continuous boost from 60 to 90°C and fluorescence signal was continuous collected. At this period, the melting curve was analyzed to access the purity of amplified product. The qRT-PCR experiment for different tissues and potassium treatments were preformed with three technical replicates. The expression level of StHAK genes were calculated through 2-ΔΔCt method and StActin was chosen as internal reference gene in real time PCR assay (Livak and Schmittgen, 2001). All primers used in this study are listed in (Supplementary Table 2).
3 Results
3.1 Identification of the StHAKs in potato
Based on the characteristics of the ‘K-trans’ domain (PF02705), which is specific to the HAK/KUP/KT potassium transporter family, a total of 24 StHAK genes were identified in potato (S. tuberosum) (Table 1). After comparing with Phytozome and Spud databases, redundant candidates with low visibility and incomplete domains were excluded. The result showed that all HAK genes were unevenly distributed on ten chromosomes. Chromosome 1, 3, 8 and 10 each contained only one gene, chromosome 5 and 6 contained two genes, chromosome 2 and 9 contained three genes, chromosome 4 and 12 contained five genes. Notably, no StHAK genes were localized on chromosome 7 and 11.
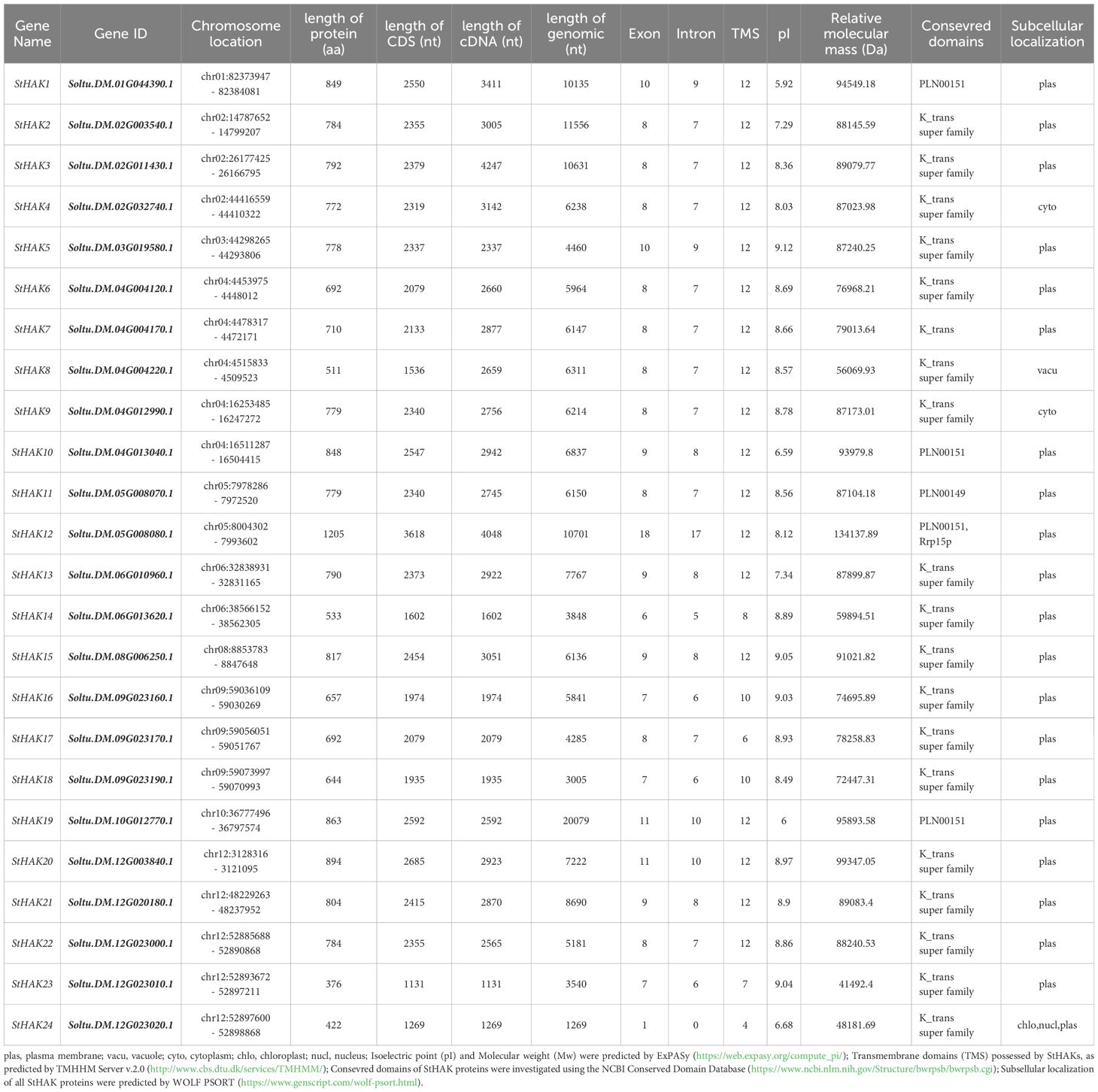
Table 1. Overview and identification of StHAKs in Solanum tuberosum.
All genes were named based on their order of location on chromosomes, and all 24 StHAKs possess the typical “K-trans” domain. The amino acid lengths of these genes ranged from 422 to 1205, with an average length of 734. StHAK12 had the longest amino acids sequences, while StHAK24 had the shortest. The theoretical isoelectric point (pI) and relative molecular mass (Da) of these genes varied from 5.92 to 9.12 (with an average of 8.20) and 41.49 to 134.14 kDa (with an average of 82.79 kDa), respectively. The number of transmembrane domains (TMS) ranges from 4 to 12, with most HAK proteins containing 12 transmembrane helices. Except for StHAK24, all genes contained multiple exons and introns. The subcellular location of StHAKs predicted by WoLF PSORT indicated that most protein were located in the plasma membrane, which were consistent with their role in maintaining K+ homeostasis in potato. However, StHAK4 and StHAK9 were predicated to be localized in the cytoplasm, and StHAK8 was located in vacuole (Table 1).
3.2 Classification and phylogenetic analysis of StHAKs in potato
To explore the evolutionary relationships of StHAKs in potato, the full-length protein sequences of 13 KUPs in Arabidopsis (MaüSer et al., 2001), 27 HAKs in rice (Gupta et al., 2008), 27 HAKs in maize (Zhang et al., 2012), 24 HAKs in potato were aligned with Cluster W. A phylogenetic tree was then constructed via the neighbor-joining method and maximum likelihood method (Figure 1 and Supplementary Data sheet 1). The trees showed that the StHAKs could be categorized into four distinct clusters based on the classification criteria of HAK transporters in maize, rice and Arabidopsis. Besides, these clusters were further subdivided into sub-cluster A and B (Figure 1 and Supplementary Data sheet 1).
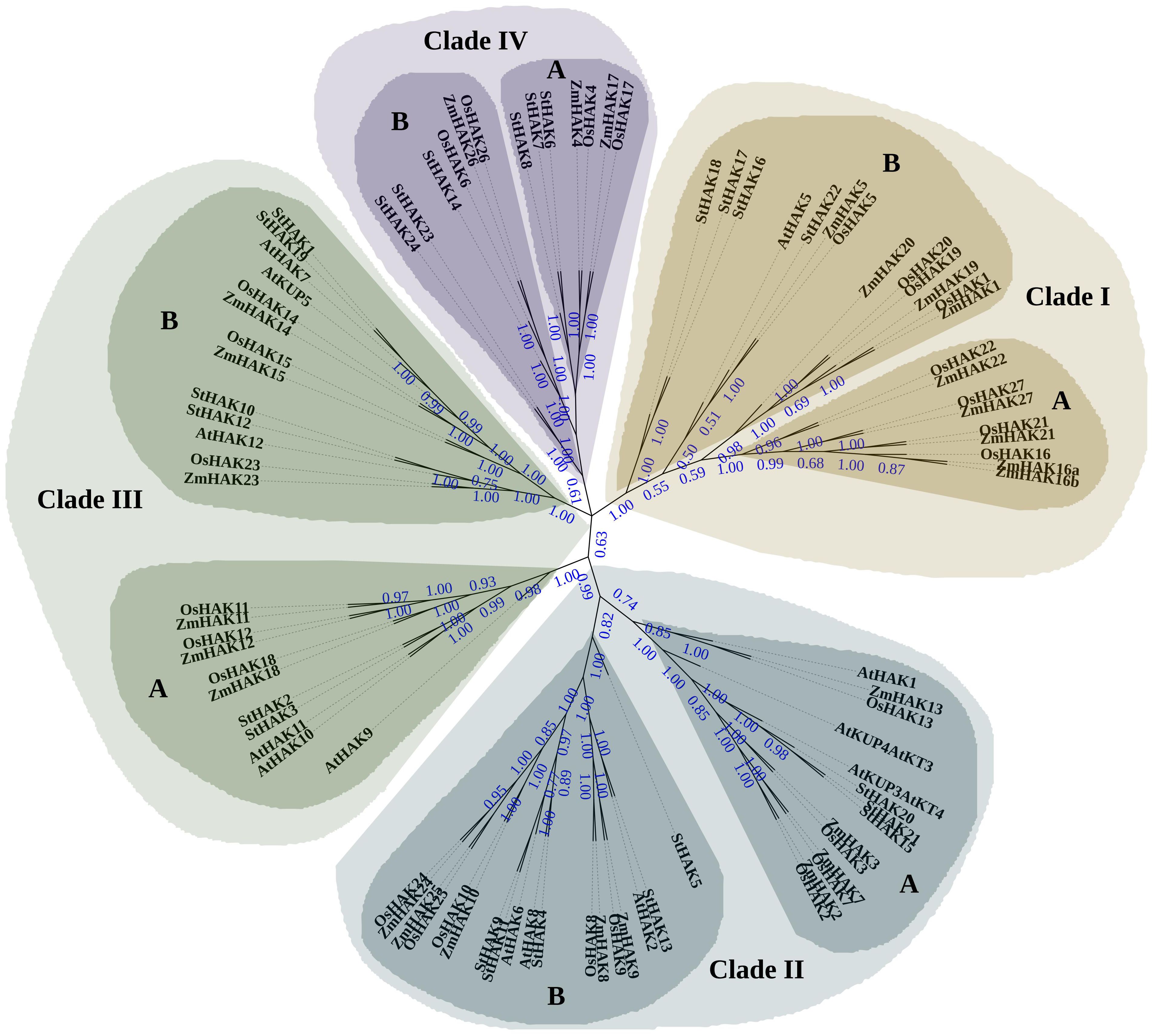
Figure 1. Phylogenetic analysis of HAK/KUP/KT family proteins from potato, rice, maize and Arabidopsis. The purple dots on the branches represent bootstrap values. Higher bootstrap values indicated a relatively higher confidence level for the corresponding branch. The full-length protein sequences of 13 KUPs from Arabidopsis, 27 HAKs from rice, 27 HAKs from maize, and 24 HAKs from potato were aligned using ClustalW. A phylogenetic tree was subsequently constructed using the neighbor-joining method (NJ) with MEGA7.0. The StHAKs could be grouped into four distinct clusters and were indicated with different colors, consistent with the classification criteria for HAK transporters in maize, rice, and Arabidopsis. Each cluster was further divided into two sub-clusters, designated as A and B. St, Solanum tuberosum; Os, Oryza sativa; Zm, Zea mays; At, Arabidopsis thaliana.
StHAKs from closely related species tended to clustered together. Clade I contained 22 transporters, including 9 from maize, 8 from rice, 1 from Arabidopsis (AtHAK5) and 4 from potato (StHAK16, StHAK17, StHAK18, StHAK22). Clade II consisted of 32 transporters, with 9 each from maize and rice, 6 from Arabidopsis and 8 from potato (StHAK4, StHAK5, StHAK9, StHAK11, StHAK13, StHAK15, StHAK20, StHAK21). 24 transporters were categorized into Clade III, with 6 from maize, rice and Arabidopsis, and potato (StHAK1, StHAK2, StHAK3, StHAK10, StHAK12, StHAK19), respectively. Clade IV had the fewest StHAKs, with 3 from maize, 4 from rice and 6 from potato (StHAK6, StHAK7, StHAK8, StHAK14, StHAK23, StHAK24). No StHAKs were belonged in Clade IA.
3.3 Gene structures, conserved motifs and domains analysis in StHAKs
To investigate the gene structures, conserved motifs and domains of StHAKs, detailed information on the domains of the 24 StHAK protein sequences was assayed using SMART, HMMER, and CDD database, as shown in Table 1. The results predicted by SMART and HMMER were largely consistent, while those from CDD database showed some discrepancies. Most StHAK proteins contained only a single ‘K_trans’ domain, but two ‘K_trans’ domain were identified in StHAK12, StHAK14, StHAK17 and StHAK18 via the SMART and HMMER tools. In contrast, the CDD database predominantly identified a single ‘K_trans superfamily’ domain in most StHAK proteins, except for StHAK12, which contained both ‘K_trans superfamily’ and ‘Rrp15p’ domains. The presence of similar domains suggested that the HAK family members with the same domain may belong to the same subfamily, for instance, StHAK16, StHAK17 and StHAK18, all of which belonged to Clade IB, indicating that the protein structure was conserved within this specific subfamily.
In this study, 10 distinct conserved motifs were identified in the StHAKs via the MEME online program, and they were labeled as motifs 1 to 10. The sequence and logos for these conserved motifs and their distribution in each StHAK protein were illustrated in Figure 2 and Supplementary Tables 2, 3. Generally, motif 1, 2, 4 and 6 were found in nearly all StHAKs, with the exception of StHAK24. StHAK24, which belonged to Clade IB, had the fewest motifs, containing only 4 motifs (Figures 2A, B). Similarly, StHAK23, another member of Clade I, exhibited a motif distribution similar to StHAK24, possessing only 5 motifs. The fewer conserved motifs in StHAK23 and StHAK24 may be related to their shorter lengths of protein (Table 1). In contrast, the number of conserved motifs in Clade II and Clade III were consistent, with all 10 conserved motifs were present in these subfamilies, suggesting potential functional similarities among these StHAKs. Within Clade IV, StHAK6, StHAK8 and StHAK14 contained 9, 7 and 8 conserved motifs, respectively, while all motifs were present in StHAK7 (Figure 2B). The variation in motif categories among different subfamilies may reflect the conservation and unique functions of the HAK protein family. The presence and arrangement of certain motifs within the same subfamily indicated that the structure of StHAKs is both conserved and distinctive within specific subfamilies.
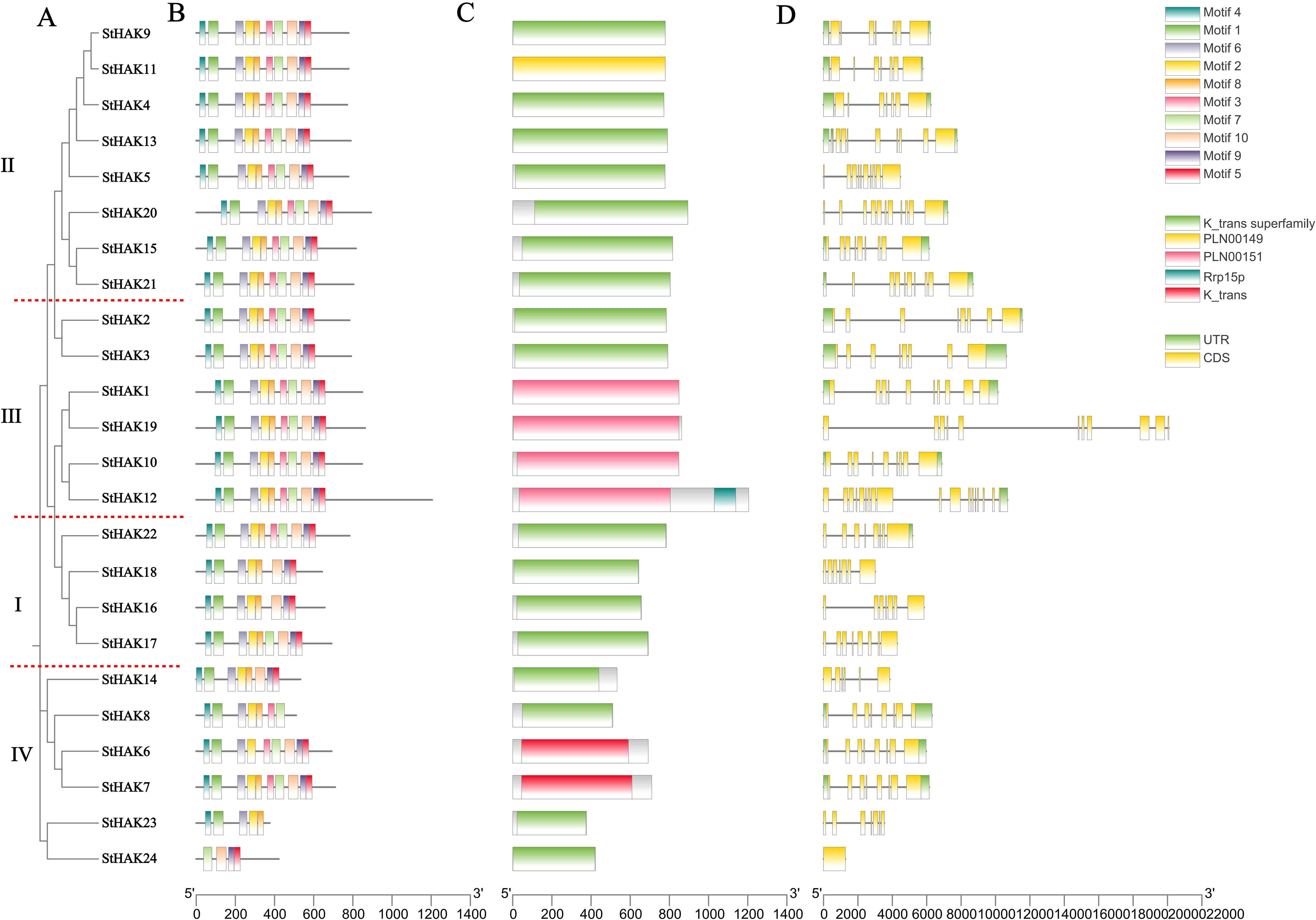
Figure 2. Phylogenetic relationships, conserved motifs and gene structures of the StHAKs. (A) The phylogenetic tree was constructed according to the full-length sequences of 24 potato HAK/KUP/KT family proteins. The different Clades were separated by red dashed lines. (B) The conserved motifs of StHAK proteins. The gray horizontal lines represented the amino acid lengths of the sequences, and the different colors on the sequences denoted various motif types. (C) The domain of StHAK proteins. The gray regions indicated areas without specific domains, while the colored sections marked the locations of the K_trans domain. (D) The Exon-intron structures of StHAK genes. UTR, untranslated region. the gray horizontal lines represented intron regions, green corresponded to UTRs, and yellow corresponded to CDS regions. At the bottom of the figures, “5′ “ and “3′ “ indicated direction of the sequences, while the numbers represented the length of the sequences in amino acids. The legend comprised three parts: the top section used different colors to represent various motif types, the middle section indicated different domain types, and the bottom section showed the positions of the UTR and CDS regions.
Conserved domain prediction results revealed that all StHAKs contain the typical ‘K_trans’ domains within their sequences, confirming their classification as members of the HAK/KUP/KT gene family (Figure 2C). To investigate the structural characteristics of the StHAKs, we analyzed the gene structure of the 24 StHAKs using GSDS2.0, mapping the exon/intron organization alongside conserved motifs and a phylogenetic tree (Figure 2D). The number of exon and intron varied across the four Clades, with Clade III exhibiting a higher exon/intron count compared to the other three Clades (Figure 2D; Supplementary Table 4). Notably, StHAK12, a member of Clade III, had the most exons, totaling 18. The exon number in Clade IV ranged from 1 to 8, with an average of 6.3, which is lower than in the other Clades. Interestingly, StHAK24, a Clade IV member, harbored only one exon and no intron, which with the fewest exon in the entire HAK gene family (Supplementary Table 4). These findings suggested that a reconstruction of gene structure occurred during the evolutionary divergence of altered HAKs subfamilies in potato.
3.4 Chromosomal location, collinearity and synteny analysis of StHAKs in potato
The StHAKs were mapped onto chromosomes based on the potato reference genome (Potato Genomics Resource). The StHAK genes were generally distributed across the genome, while their distribution across the chromosomes is uneven (Figure 3). Five StHAK genes (20.83%) were located on both chromosomes 4 and 12, which is the highest concentration among the ten chromosomes. In contrast, only one gene (4.17%) was found on chromosomes 1, 3, 8 and 10 were distributed with two (8.33%) on chromosomes 5 and 6, and three (12.5%) on chromosomes 2 and 9. However, no HAK genes were distributed on chromosomes 7 and 11.
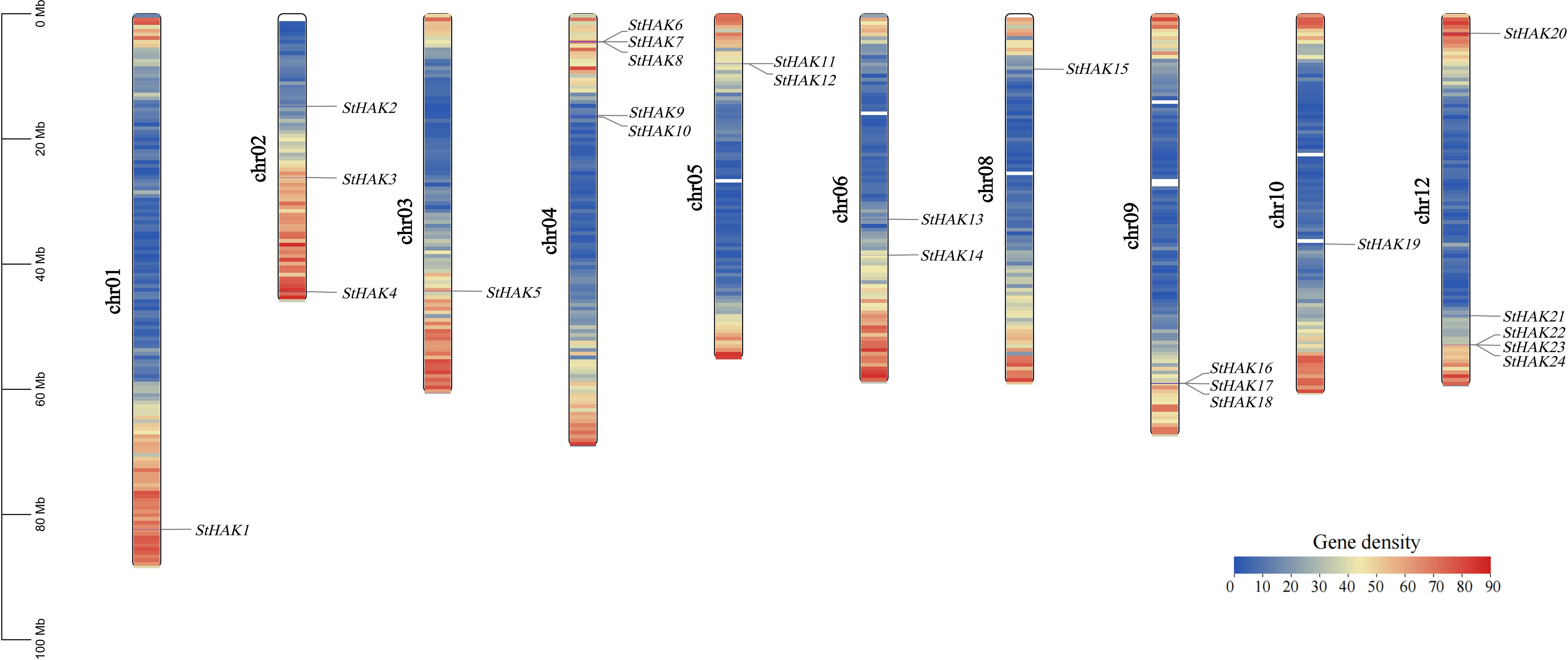
Figure 3. The Chromosomal localization of the StHAK genes. The black lines indicated the position of each StHAK genes. Different colors within the chromosomes represented the gene density. The legend on the left indicated the size of the chromosomes. From left to right, the chromosomes were arranged in order from 1 to 12, except for chromosomes 7 and 11. The white regions within the chromosomes represented areas without corresponding gene information. In the gene density color key, the numbers indicated the number of genes.
Gene density varied across different regions of genomes. In the StHAK genes family, gene density profiles were assayed via Gene Destiny Profile of TBtools. Numerous StHAK genes were unevenly distributed on chromosomes, with a higher destiny observed at the ends of the long and short arms and a lower destiny in the mid-chromosomal regions (Figure 3). The majority of StHAK genes family members were located in the terminal regions of the chromosomes. The color shift from blue to red on the chromosomes maps indicated an accumulation of genes, whereas blank areas represent regions with lower gene density.
Collinear gene pairs within the potato genome were analyzed to elucidate the relationship among the HAK genes and other potential gene duplication events (Figure 4). The results of collinearity analysis revealed that six genes pairs (StHAK4/11, StHAK9/11, StHAK10/12, StHAK15/21, StHAK15/20,StHAK20/21) were found in potato genome, and were distributed across different chromosomes. No significant tandem duplication or segmental duplication gene pairs were identified within the potato genome. To further investigate the syntenic relationship of HAK gene families between potato and other species, syntenic analyses were conducted with eight species, including three monocots (O.Sativa and S. bicolor, Z.mays) and five dicots (A.thaliana, V.vinifera, G.max, M.esculenta, S. lycopersicum) (Figure 5; Supplementary Table 5). A total of 91 orthologous gene pairs were identified. The analysis showed that 33 gene pairs were found between G.max and S.tuberosum, followed by M.esculenta (19), S. lycopersicum (17), V.vinifera (10), A.thaliana (8), S. bicolor (2), O.Sativa (1), Z.mays (1). In comparison with dicotyledonous plants, fewer collinear relationships were detected between S.tuberosum and monocotyledonous plants like O.Sativa and Z.mays. The fewest collinear pairs were found on chromosomes 8 and 12, while the highest number were observed on chromosomes 2 and 6, and the distribution of these collinear pairs on chromosome 6 were rather equally across species. Interestingly, the collinear relationships of monocots were all identified on chromosome 6, suggesting that these orthologous pairs may have existed prior to the ancestral divergence.
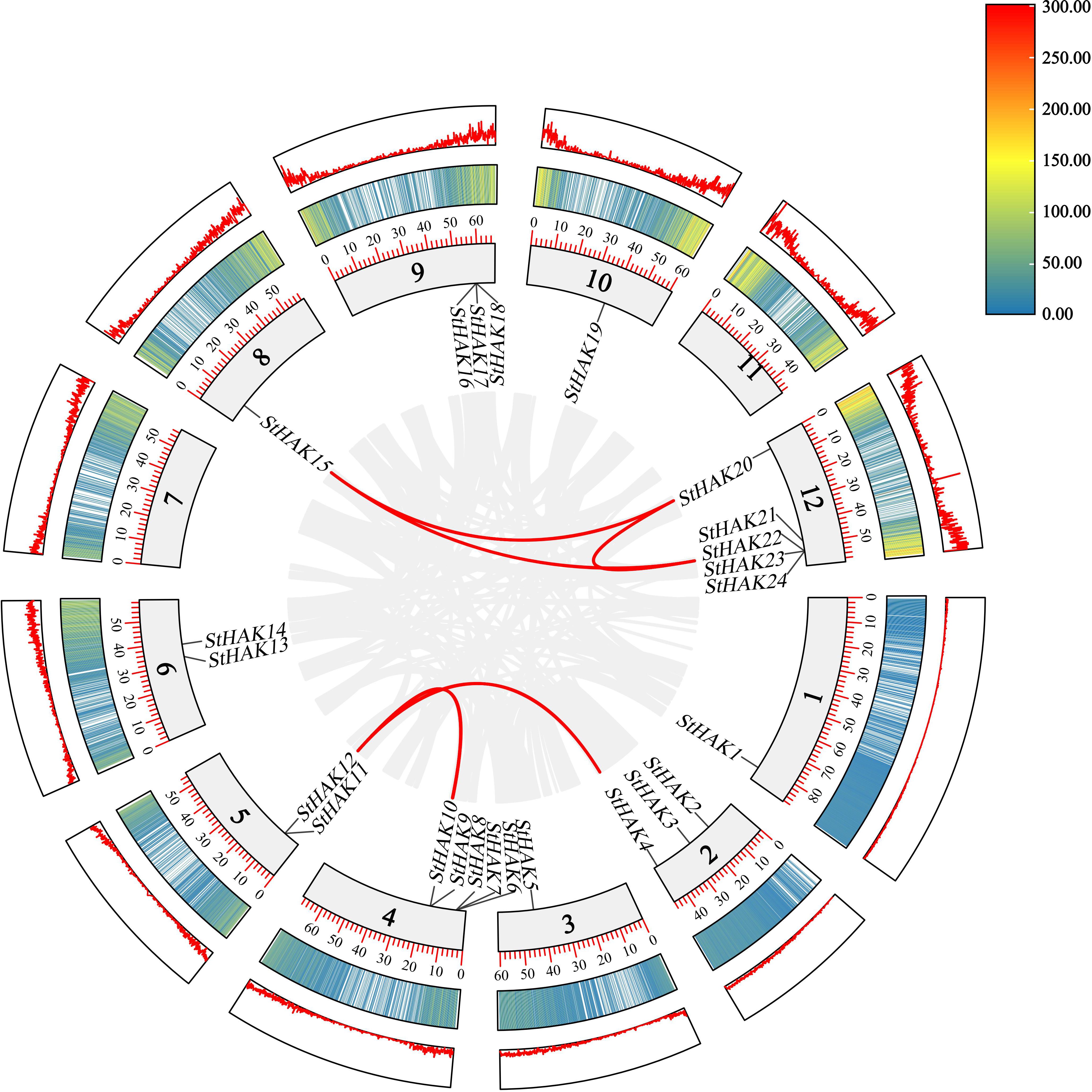
Figure 4. Collinearity analysis of HAK genes in potato. Collinearity refers to the conserved order of genes along a chromosome. It emphasizes the sequential arrangement of homologous genes, often focusing on their relative positions and orientation across chromosomes within or between species. It is often used to analyze evolutionary events like genome duplications or rearrangements. The grey lines on the inside of the ring represented all collinear blocks within the potato genome. The red lines on the inside of the ring indicated the duplicated HAK gene pairs in potato. The two rings on the outside of the chromosome ring represented gene density, with different colors indicating different gene densities. The numbers in the color key at the top right represented the number of genes.
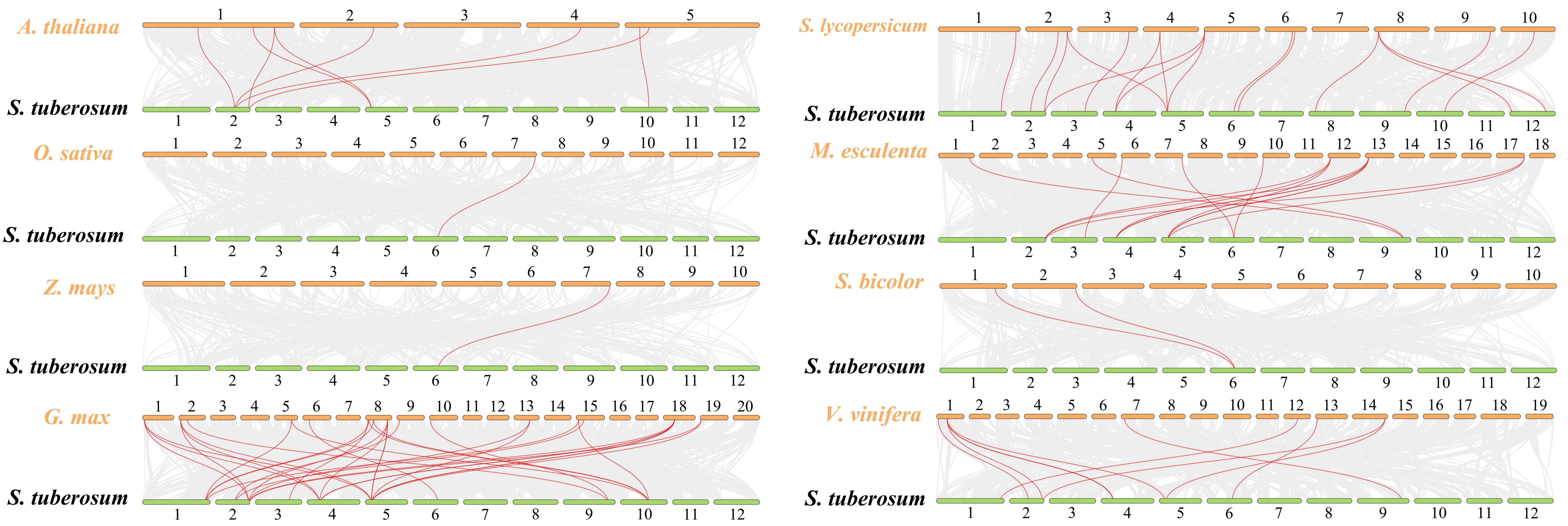
Figure 5. Synteny analyses between HAK genes of potato and other eight species (Arabidopsis thaliana, Oryza sativa, Zea mays, Glycine max, Solanum lycopersicum, Manihot esculenta, Sorghum bicolor, Vitis vinifera). Synteny refers to the co-localization of genes on the same chromosome, without necessarily preserving their order or orientation. Synteny simply indicates that a set of genes appears together on the same chromosome in different species or genomes, regardless of the sequence. Small green rectangles represented potato chromosomes, while small orange rectangles represented chromosomes from eight species. Grey lines indicated synteny blocks between potato and other plant genomes, while red lines highlighted syntenic HAK gene pairs. The number of syntenic gene pairs between potato and other species are as follows: Arabidopsis thaliana (8), rice (1), maize (1), soybean (33), tomato (17), cassava (19), sorghum (2), and grape (10).
3.5 Analysis of promoter cis-acting elements of StHAKs in potato
The cis-acting elements of the StHAK genes were analyzed within the 2000-bp upstream regulatory region from the start codon ATG. A total of 533 cis-elements were identified in the promoter regions of the StHAK genes and mainly four groups were categorized: hormone responsive (118, 22.1%), stress (80, 15.0%), light (295, 55.4%) and development/tissue specificity (40, 7.5%) (Figure 6; Supplementary Table 6). Plenty of phytohormone-responsive elements were present in the promoter regions of the StHAK genes, including abscisic acid responsive (ABA) (36, 30.5%), auxin-responsive (IAA) (7, 5.9%), gibberellin-responsive (GA) (24, 20.3%), methyl jasmonate-responsive (MeJA) (40, 33.9%), salicylic acid responsive (SA) (11, 9.3%) (Figure 6A; Supplementary Table 6). Stress related cis-regulatory elements, such as anaerobic induction (44, 55%), anoxic specific inducibility (3. 3.7%), defense and stress responsive (7, 8.7%), drought-inducibility (14. 17.5%), low-temperature responsive (10. 12.5%) and wound-responsive element (2, 2.5%), were also detected in the StHAKs promoter regions (Figure 6A; Supplementary Table 6). The number of light-responsive elements in the StHAKs promoter regions was the highest (295), accounting for nearly 56% of the total, and these elements were distributed almost all StHAKs gene promoter regions (Figure 6B). Elements related to plant development and tissue specificity, such as meristem expression (22, 55%), zein metabolism regulation (7, 17.5%), endosperm expression (4, 10%), seed specific (1, 2.5%) and palisade mesophyll cells differentiation (1, 2.5%), were also found in the promoter regions of the StHAK genes. What’s more, elements involved in cell cycle regulation (1, 2.5%) and circadian control (3, 7.5%) were categorized as development/tissue specificity elements. Overall, these results suggested that multiple cis-acting elements may involve in regulation of StHAK genes in response to growth, hormone induction, and abiotic stress.
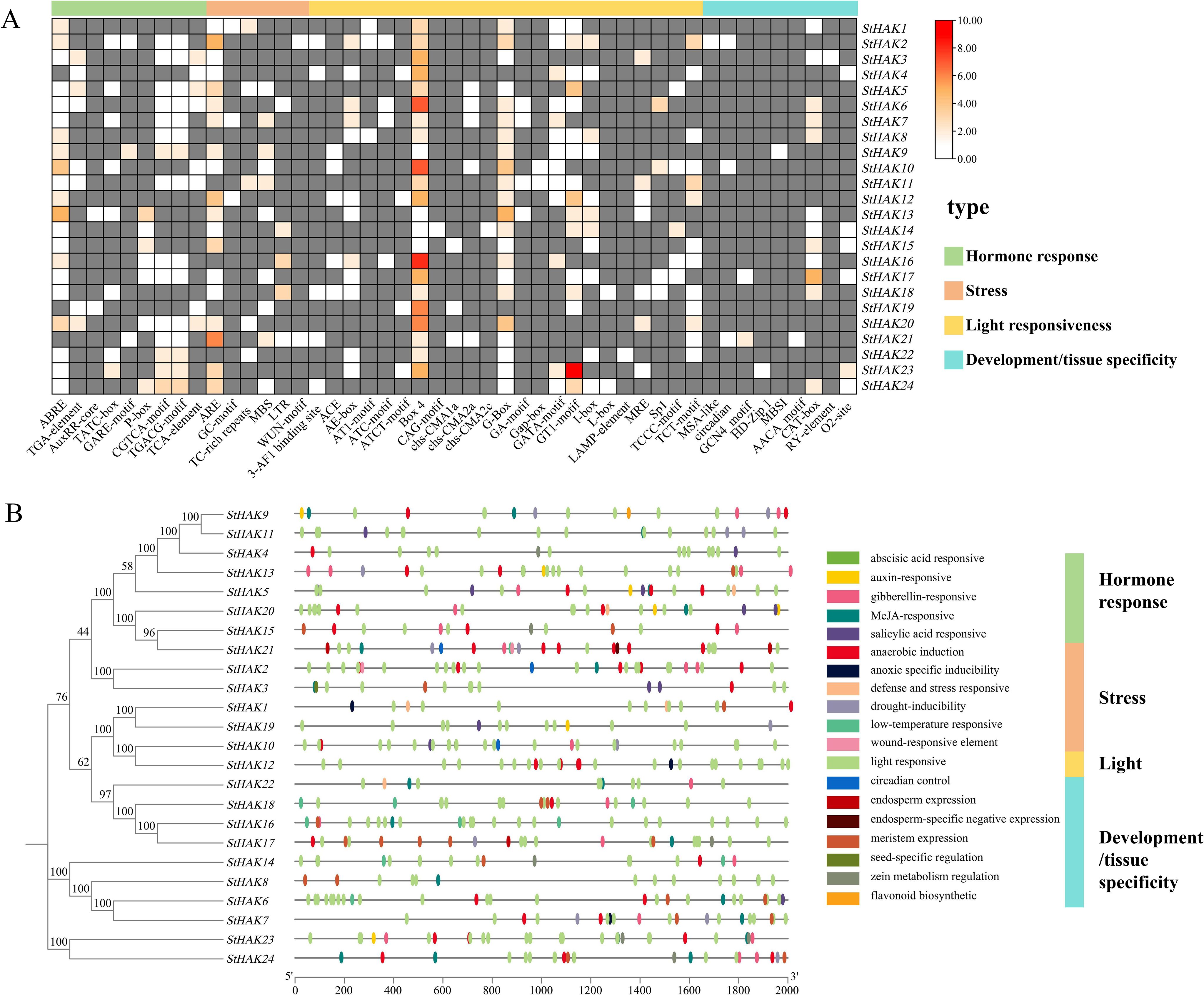
Figure 6. Analysis of cis-acting elements in StHAKs. Analysis of cis-acting elements was conducted on 2 kb sequences upstream of coding sequences of StHAKs. (A) Classification and number of cis-acting elements on the promoter region of StHAKs. The promoter regions of the StHAKs genes were primarily divided into four major categories of elements: light green represents hormone response-related elements, light pink represents stress response-related elements, yellow represents light response-related elements, and blue represents development and tissue-specific elements. The small boxes within the large rectangles indicated the number of these elements in the StHAKs genes, with a gradient from white to red signifying an increasing number of elements. Gray indicated the absence of these elements in the StHAKs genes. The color key on the top right referred to the number of cis-elements. (B) Distribution of cis-acting elements in promoter region of StHAKs. The phylogenetic tree was constructed according to the full-length sequences of 24 potato HAK/KUP/KT family proteins, which were presented in left. On the right side, the selected promoter lengths (2000 bp) were displayed, along with the cis-acting elements located at different positions within the promoters, represented by small ovals in different colors.
3.6 Array of expression profiles of StHAKs based on microarray data
To investigate the expression level of StHAKs in potatoes, their expression patterns were analyzed across various tissues, stress and hormone-induced treatments by means of Microarray data downloaded from Potato Genomics Resource. According to the RNA-Seq data, the expression levels of all 24 StHAKs were further examined and interpreted (Figure 7; Supplementary Table 7). The expression of StHAKs in 15 different tissues were analyzed, including sepals, leaves, roots, shoots, callus, tubers, stolons, petioles, petals, stamens, carpels, flowers, mature fruits, immature fruits, mesocarp & endocarp. The results indicated that the expression of the StHAKs were significant difference in these tissues (Figure 7A). Notably, the expression levels of StHAKs of Clade I and Clade IV were generally lower than other Clades in different tissues, such as StHAK22, StHAK23, StHAK24 from Clade I and StHAK6, StHAK7, StHAK8 from Clade IV. While StHAK18, a member of Clade I, was highly expressed in stamen, carpel and flower and almost no expression in other tissues, indicating apparent tissue specificity (Figure 7A). Similarly, StHAK20, a Clade II member, displayed the same expression pattern as StHAK18. Members of Clade III generally showed higher mRNA transcript levels in different tissues, especially StHAK2 and StHAK10. The expression profiles of StHAK1, StHAK3, StHAK12 and StHAK19, all belonging to Clade III, were somewhat lower in shoot tissues such as leaves and petioles, but were more highly expressed in carpels, stamens, roots, mature fruits, mesocarp & endocarp (Figure 7A). The expression pattern of StHAK21 (Clade II member) was similar to that of StHAK2, which was also highly expressed in sepals, callus and petals.
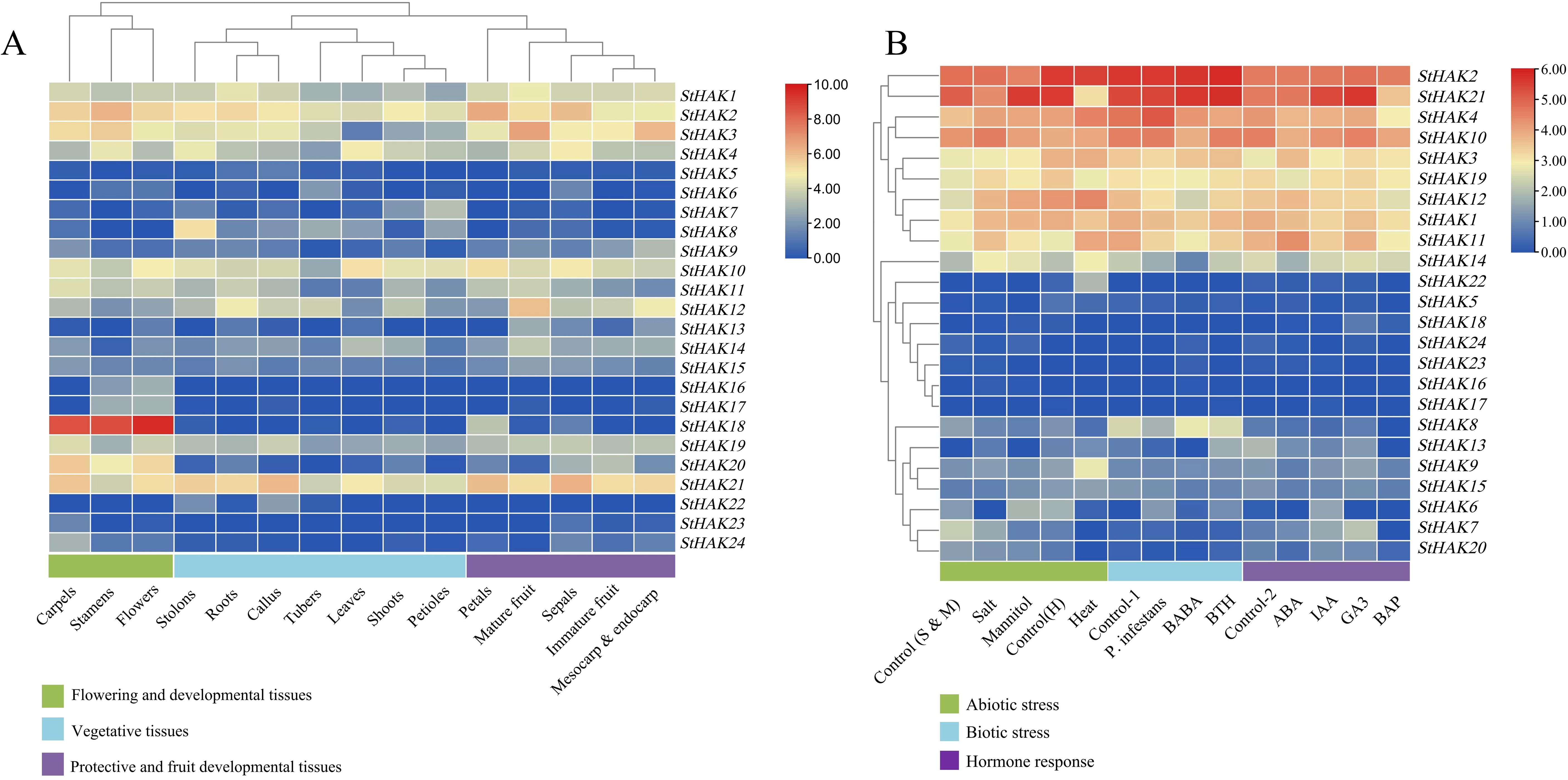
Figure 7. Expression profiling of StHAKs based on Microarray data. (A) The mircoarray data was download from Potato Genomics Resource (Spud) and the expression level of StHAKs in 15 various tissues were analyzed. We performed clustering on tissues with similar StHAKs expression levels, categorizing the 15 tissues into three major groups. Green boxes represented flowering and developmental tissues (including carpels, stamens and flowers), blue boxes represented vegetative tissues (including stolons, roots, callus, tubers, leaves, shoots and petioles), and purple boxes represented protective and fruit developmental tissues (petals, mature fruits, sepals, immature fruits, mesocarp & endocarp). The color key indicated the gene expression levels (TPM values), with colors ranging from blue to red, where blue represented lower expression and red represented higher expression. (B) Expression levels of StHAKs under three types of abiotic stresses, three types of biotic stresses and four types of phytohormone treatments. Control (S & M) referred to the control treatment for salt stress and mannitol stress. Control (H) referred to the control treatment for heat stress. Control-1 referred to the control for biotic stress, and control-2 referred to the control for different hormone treatments. The color key on the top right indicated the gene expression levels (TPM values).The TPM values were normalized by log2 (TPM) transformation.
The RNA transcript levels of StHAKs under abiotic and biotic stress were also examined (Figure 7B). In comparison with control, the expression of StHAK1, StHAK5, StHAK11, StHAK12, StHAK14, and StHAK19 genes was significantly upregulated under salt stress, with some genes showing more than a 2-fold increase. Conversely, the expression level of StHAK6 was markedly reduced under salt stress. To further explored whether the transcript levels of StHAK genes are affected by biotic stress, the expression profiles of StHAKs were analyzed under three biotic elicitors: β-aminobutyric acid (BABA), benzothiadiazole (BTH) and pathogen treatment (Figure 7B). Under pathogen treatment, the expression levels of most StHAKs were reduced, except for StHAK4, although the reduction was not significant. Similarly, under BABA treatment, the expression pattern of StHAKs resembled that observed after pathogen infection, with most StHAKs exhibiting a downward trend in expression. In contrast, under BTH treatment, the expression levels of StHAK9, StHAK13, StHAK20, and StHAK21 were significantly upregulated (Figure 7B).
Phytohormones play a crucial role in plant development and growth, ionic homeostasis and stress response. Expression data for StHAKs were also obtained from Spud for several hormonal treatments. StHAK3 and StHAK5 were highly expressed under ABA and GA3 treatment, whereas their expression was lower under IAA treatment (Figure 7B). Conversely, StHAK20 and StHAK21 were highly expressed under IAA and GA3 treatments, but StHAK20 expression was reduced by approximately half under ABA treatment. The expression of StHAK21 remained relatively unchanged after ABA treatment. StHAK7 was induced by ABA, IAA and GA3, but inhibited by BAP treatment. StHAK6 was particularly sensitive to IAA, with expression levels increasing over 20-fold, but it did not respond to other hormonal treatments. With the exception of StHAK3 and StHAK18, the RNA transcript levels of other genes were significantly lower under BAP treatment (Figure 7B), indicating that most StHAKs were insensitive to BAP treatment.
3.7 Array of expression profiles of StHAKs based on transcriptome sequencing data and qRT-PCR
To investigate the expression profiles of StHAKs in different tissues of potato, three tissues-leaves, roots and tubers-were selected for transcriptome sequencing (Figure 8A). The results revealed significant differences in the expression of StHAKs in these tissues (Figure 8B; Supplementary Table 8). In general, the mRNA transcript levels of StHAK4, StHAK5, StHAK9 in Clade II, StHAK24 in Clade I and StHAK14 in Clade IV were higher in leaves compared to roots and tubers. Among these, StHAK4 showed the most pronounced variation, which expressed 53.65 times higher in leaves than in tubers, followed by StHAK9 and StHAK24, which expressed 13.22 and 13.14 times higher than in tubers, respectively (Figure 8C). Besides, StHAK5 was 5.32 times more abundant in leaves than in roots. Members of Clade IV were predominantly expressed in roots, with expression levels generally 7-fold higher than in tubers, and StHAK6 exhibiting the highest increase at 9.57-fold. The mRNA transcript levels of StHAK4, StHAK9, StHAK11, and StHAK13 in Clade II showed a similar trend, with the expression of StHAK9 and StHAK4 in roots were 48.57-fold and 21.20-fold higher than that in tubers, respectively. Notably, StHAK24 (Clade I) exhibited the most significant increase, with expression in roots 134.43 times higher than in tubers (Figure 8C). Overall, the expression of StHAKs was lower in tubers compared to leaves and roots, however, StHAK12 was expressed 1.28 times more in tubers than in leaves, and StHAK20 was expressed 1.41 times more in tubers than in roots (Figure 8C).
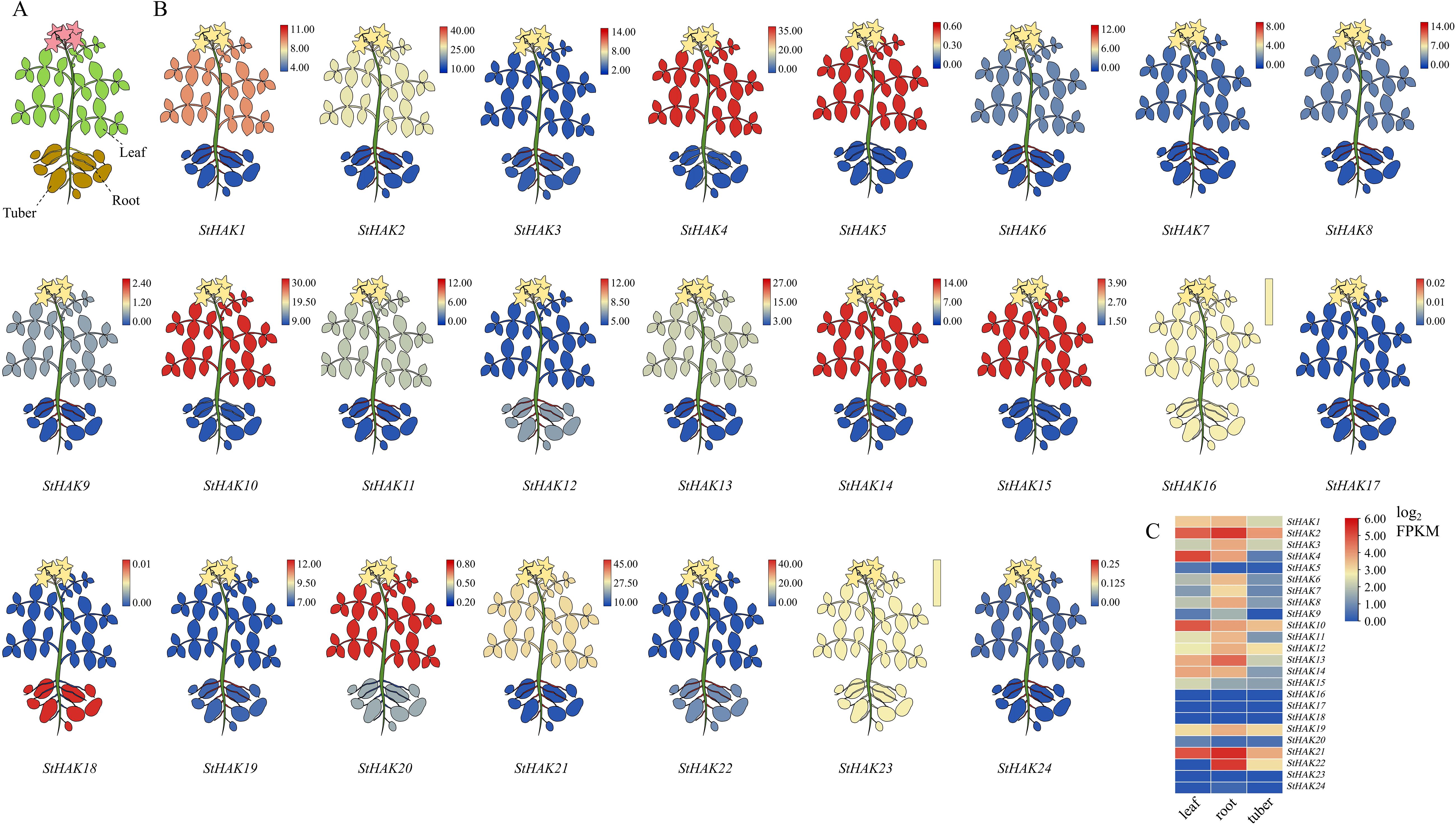
Figure 8. Expression profiling of StHAKs based on transcriptomic data. (A) Tissues for transcriptome sequencing. The tissues used for transcriptome sequencing mainly included leaf, root and tuber. (B) Cartoon plot of StHAKs expression in leaf, root and tuber. Different colors represented variations in gene expression levels, ranging from blue to red, with expression levels gradually increasing. The color key indicated the FPKM values of expression levels. The expression levels of StHAK16 and StHAK23 were consistently very low across different tissues. (C) Heatmap of StHAKs expression in leaf, root and tuber. Different colors represented variations in gene expression levels, ranging from blue to red, with expression levels gradually increasing. The FPKM values were normalized by log2 (FPKM) transformation.
To explore the response of StHAKs to varying potassium concentrations, we established three potassium gradients in the field and sequenced the transcriptomes of roots, stems, and leaves of potato during the tuber expansion stage. The results showed significant differences in the response of StHAKs to potassium in different tissues (Figure 9; Supplementary Table 9). In leaves, as potassium concentration increased, StHAK2 was upregulated, while the expression of StHAK3, StHAK4 and StHAK12 was initially increased followed by decreased. In contrast, StHAK5, StHAK9 and StHAK10 were downregulated. In addition, the mRNA transcript levels of several genes such as StHAK16, StHAK17, StHAK18, StHAK22, StHAK23, StHAK24 were extremely low under different doses of potassium (Figure 9A). In roots, the expression levels of ten StHAKs (StHAK4, StHAK6, StHAK7, StHAK8, StHAK9, StHAK11, StHAK13, StHAK14, StHAK15, StHAK19) increased and then decreased with rising potassium concentration, suggesting that a moderate potassium level (300 kg/hm2) promotes StHAKs expression, while excessive potassium (600 kg/hm2) suppressed it. The mRNA transcript levels of StHAK21, StHAK22, and StHAK24 gradually declined with increasing potassium concentration, whereas the expression level of StHAK20 gradually increased. In contrast, StHAK16, StHAK17, StHAK18 and StHAK23 were almost not expressed under varying potassium concentration treatments (Figure 9B). In tubers, the expression patterns of StHAKs were comparable to those in roots, with eleven genes showing an initial increase followed by a decrease in response to rising potassium levels. Meanwhile, three genes (StHAK7, StHAK13, StHAK14) displayed a decreasing trend, while StHAK5 and StHAK12 showed an increasing trend (Figure 9C).
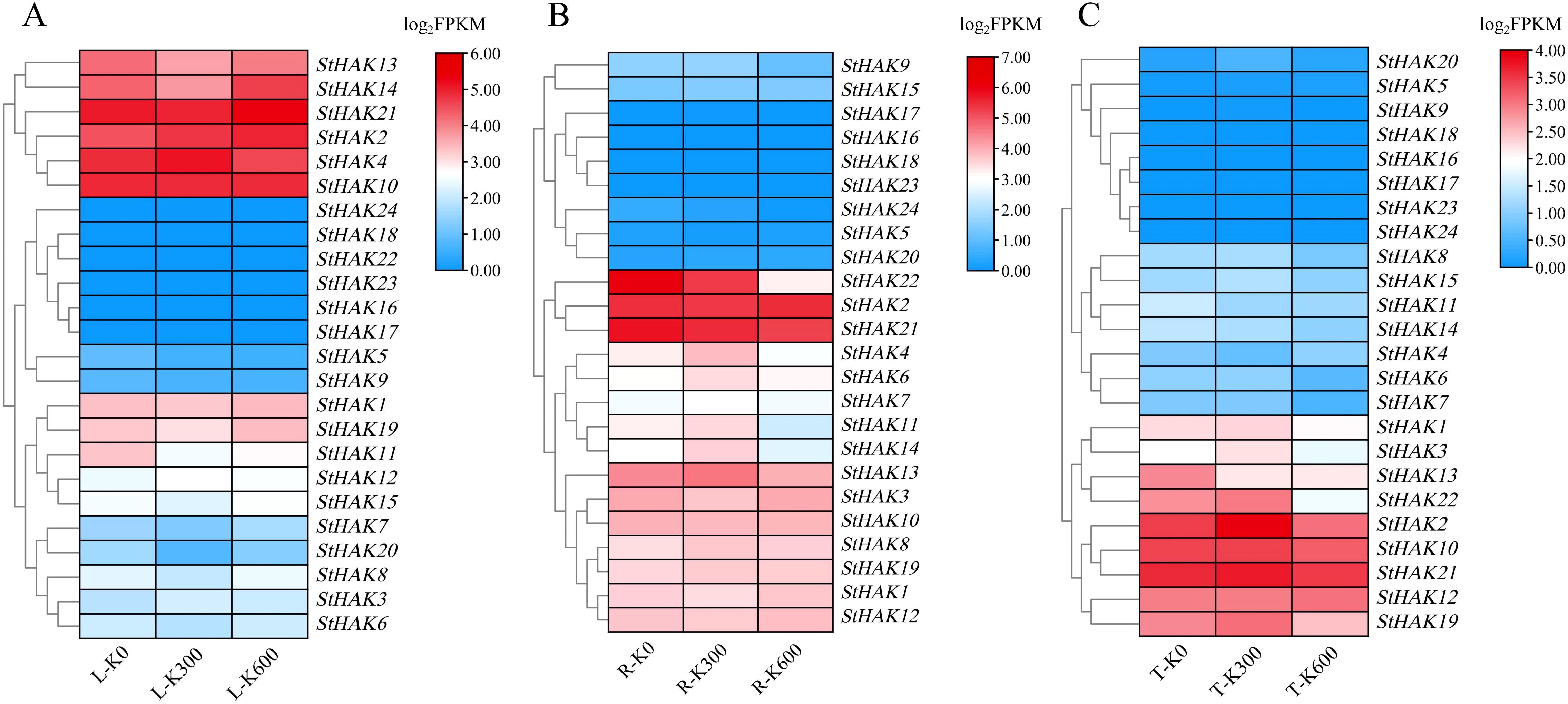
Figure 9. The transcript levels of StHAKs in leaf, root and tuber of potato under potassium fertilizer treatment based on transcriptomic data. (A) Expression levels of StHAKs in leaves. (B) Expression levels of StHAKs in roots. (C) Expression levels of StHAKs in tubers. R, roots; L, leaves; T, tubers; K0, 0 kg/hm2; K300, 300 kg/hm2; K600, 600 kg/hm2; Genes with similar expression levels were clustered together. Different colors represented variations in gene expression levels, ranging from blue to red, with expression levels gradually increasing. FPKM values were normalized by log2 (FPKM) transformation.
To better understand the expression patterns of StHAKs in various potato tissues, we selected all genes from each Clade for qRT-PCR analysis. The tissues examined included roots, stems, leaves, tubers and flowers (Figure 10A). In short, most of the selected StHAKs were expressed at relatively low levels in the roots (Figures 10B, C). StHAK8, StHAK10, StHAK14, StHAK19, StHAK22 were highly expressed in stems and tubers, but were poorly expressed in other tissues. Among them, the expression of StHAK14 in stems and tubers was 560 and 661 times higher than in root, and 320 and 388 times higher than in flowers, respectively (Figures 10B, C). Many genes showed higher expression in tubers compared to other tissues. In addition to StHAK4, there were nine genes, which primarily belonged to Clade IV and Clade III, might play an important role in the growth and development of potato tubers. The transcript levels of StHAK4 and StHAK21, which were all belonged to Clade II, were higher in leaves compared to other tissues, while the expression of StHAK1, StHAK3, StHAK8, StHAK10, StHAK14, StHAK17, StHAK18, and StHAK22 in leaves was significantly lower than in stems and tubers. These results showed that StHAK4 and StHAK21 might involved in potato leaf development or in pathways related to the photosynthetic response in leaves. The mRNA levels of StHAK2, StHAK11 and StHAK18 were higher in flowers, whereas StHAK1, StHAK5, StHAK19, StHAK20 and StHAK23 were expressed at somewhat lower levels in flowers (Figures 10B, C).
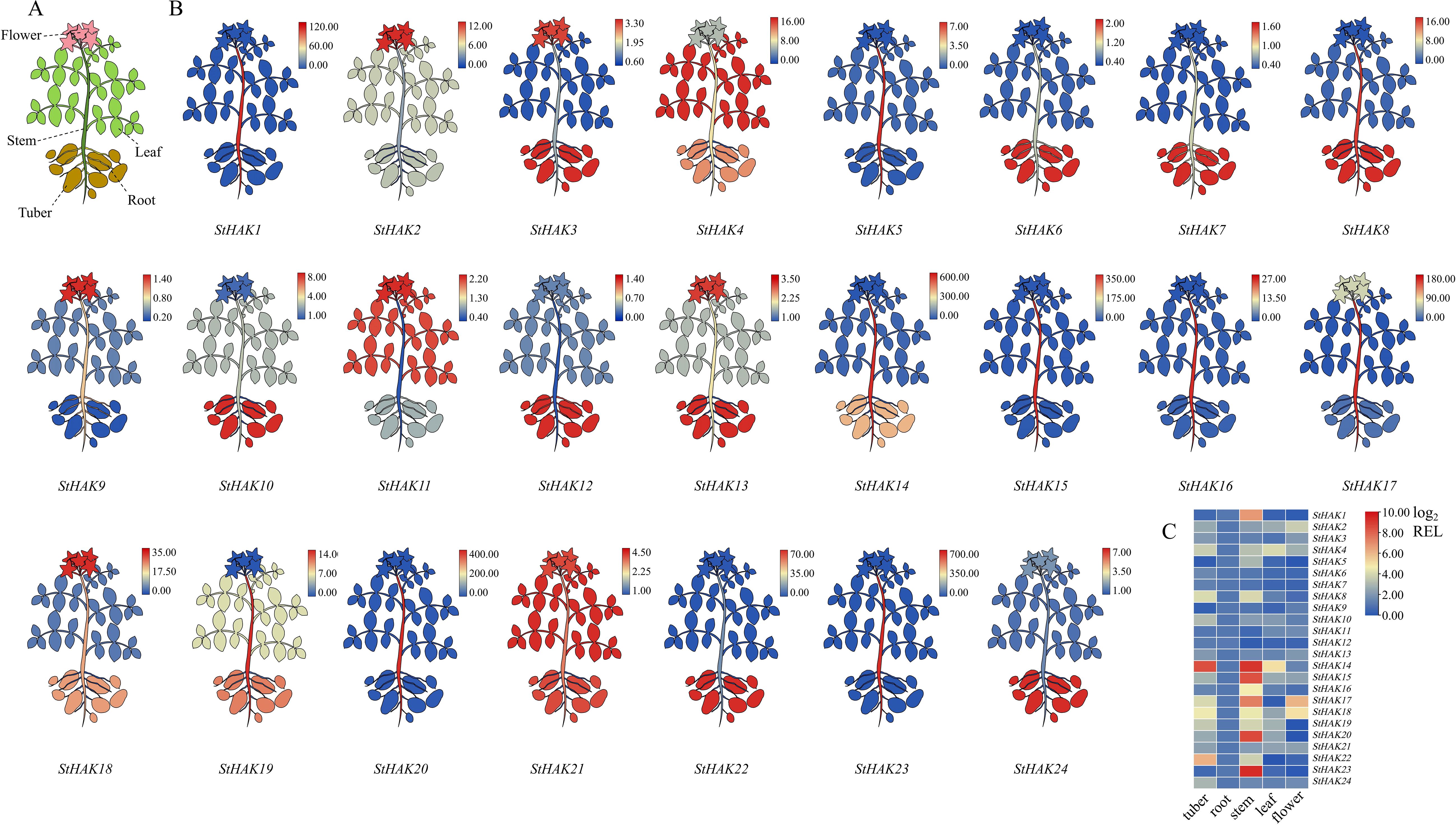
Figure 10. The quantitative real-time-PCR (qRT-PCR) analysis of expression patterns of StHAKs at different organs. (A) Five tissues for qRT-PCR. The tissues used for qRT-PCR mainly included root, stem, leaf, tuber and flower. (B) 24 StHAKs were selected for expression level analysis. Different colors represented variations in gene expression levels, ranging from blue to red, with expression levels gradually increasing. The color key indicated the Relative genes expression values of expression levels. (C) Heatmap of StHAK genes expression in tubers, roots, stems, leaves, flower. The color key indicated the relative genes expression values of expression levels. Relative genes expression values were normalized by log2 (REL) transformation.
The relative abundance levels of StHAKs exhibited varying trends under different potassium fertilizer treatments (Figure 11). Compared to the K0 treatment, nearly all StHAK genes showed a significant decrease in expression under K2 and K3 treatments, which was related to HAK-mediated K+ uptake; the expression of StHAK genes was significantly upregulated only under very low external potassium concentrations. Interestingly, StHAK24 displayed a contrasting pattern, with a notable increase in expression under K2 treatment and a decrease under K3 treatment.
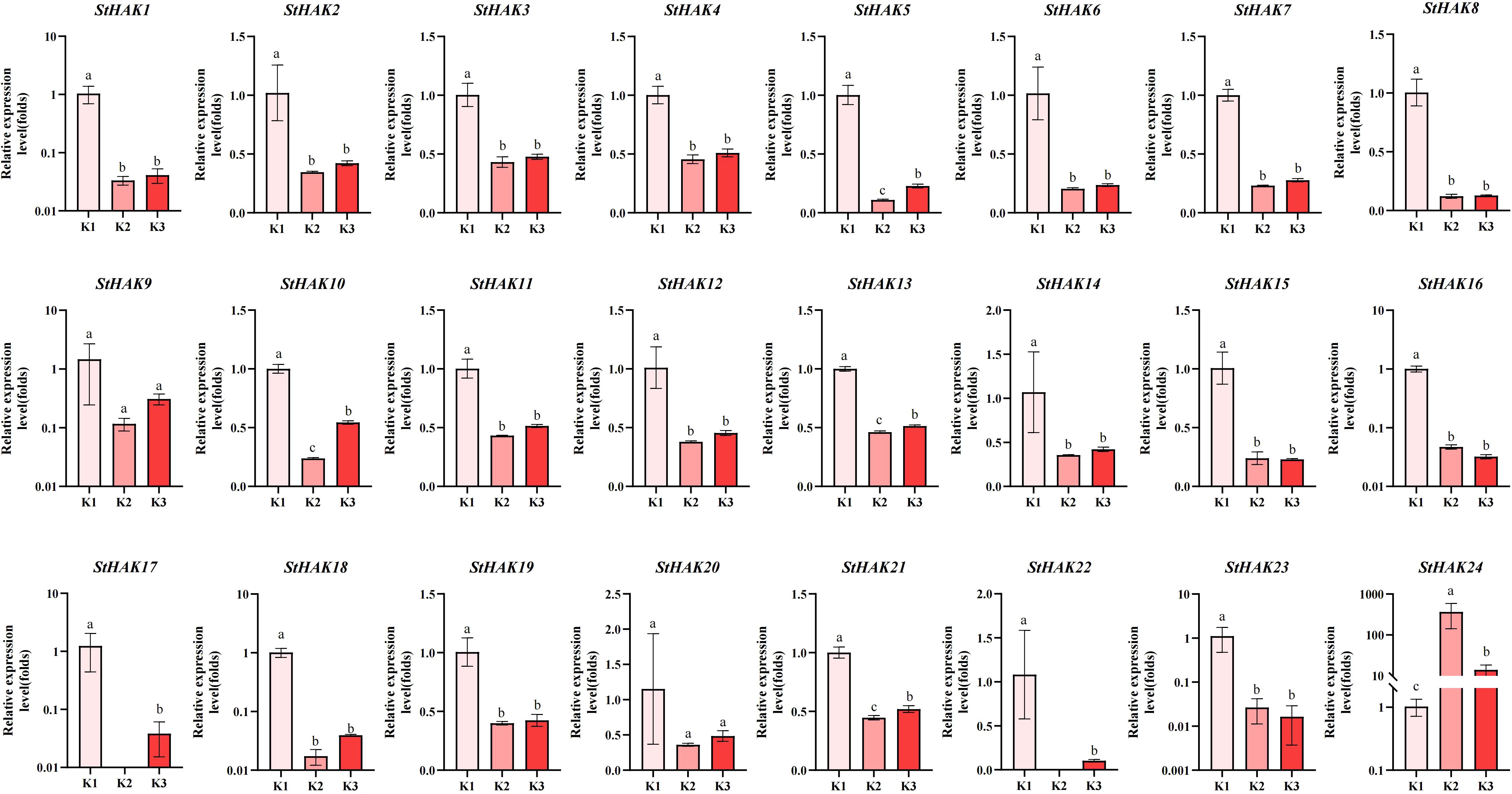
Figure 11. The qRT-PCR analysis of expression patterns of StHAKs under different potassium fertilizers. K0, 0 kg/hm2; K1, 300 kg/hm2; K2, 600 kg/hm2; The horizontal axis represented potassium fertilizer treatments, and the vertical axis represented relative gene expression levels. K1 was selected as the control group, while K2 and K3 were used as treatment groups. Vertical bars indicate ± SD (N = 3). Different letters indicated significant differences between treatments (p < 0.05).
4 Discussion
Members of the HAK gene family have been identified in a broad range of plants, including Arabidopsis, rice, wheat, barley, moso bamboo, cassava (Gierth et al., 2005; Gupta et al., 2008; Cheng et al., 2018; Cai et al., 2021; Guo et al., 2024; Luo et al., 2024). These genes are involved in the uptake and transport of potassium, as well as in various processes related to plant growth and development (Wang and Wu, 2013). In this study, we identified a total of 24 StHAKs, constructed a phylogenetic tree, and analyzed their evolutionary relationships and potential biological functions. What’ more, we examined the gene structure, conserved motifs, chromosomal localization, collinearity, syntenic relationships, and cis-acting elements in the promoter regions of these 24 StHAKs to gain a comprehensive understanding of their roles. Finally, we analyzed the transcript levels of StHAKs in different tissues of potato and under various potassium fertilizer treatments.
4.1 Phylogenetic analysis of StHAK genes
The StHAKs were divided into four clades according to phylogenetic analysis (Figure 1), consistent with the HAK gene families of rice, maize and barely (Gupta et al., 2008; Zhang et al., 2012; Cai et al., 2021). The number of OsHAKs, ZmHAKs, and HvHAKs is 27 for each, with minor differences in the distribution among clades, particularly in clade II and clade III, where the number of family members is identical (Gupta et al., 2008; Zhang et al., 2012; Cai et al., 2021). The total number of PeHAKs is 41, with significantly more members in clade I and clade II compared to OsHAKs, ZmHAKs, HvHAKs, and StHAKs (Guo et al., 2024). This disparity in the number of PeHAKs raises the question of whether it is due to differences in genome sizes. For instance, the genome sizes of mosaic bamboo, maize, and barley are 2051 Mb, 2066 Mb, and 3800-4500 Mb, respectively (Jayakodi et al., 2020). In comparison, the genome sizes of moso bamboo is more than three times larger than that of the potato (DM8.1) genome (773.36 Mb), yet the number of PeHAK gene families is less than twice as large (Yang et al., 2023). Hence, this suggested that species genome size is not a dominant determinant for number of gene family members. The main reason, probably, is intra-genomic covariance and gene duplication events, which lead to the expansion of the HAK gene family members in moso bamboo (Guo et al., 2024). In tuber crops, the number of StHAK family genes (24) is slightly higher than that in MeHAK (21) and IbHAK (22) (Jin et al., 2021). Unlike the four clades of HAK gene family in other species, IbHAKs are divided into five clades (clade I to clade V), with clade II further subdivided into A, B and C groups. This is inconsistent with other crops like Arabidopsis, rice, and cassava, which have only A and B groups in clade II. Meanwhile, OsHAK3 and OsHAK7, which were affiliated with clade IIA, were not classified in the phylogenetic analysis of IbHAK. This discrepancy may be due to the methods used in construction of phylogenetic tree, which showed a significance of difference between the Maximum likelihood method (ML) and Neighbor joining method (NJ) (Jin et al., 2021).
After aligning protein sequences of HAKs from different species using ClusterW, some blank regions appeared in the 5’ and 3’end of the StHAK23 and StHAK24 sequences. To ensure accuracy in our phylogenetic analysis of HAK family proteins from various species, we removed these blank regions before constructing the phylogenetic tree. The result showed that both StHAK23 and StHAK24 belonged to Clade IVB (Figure 1). Interestingly, AtHAK5, classified as clade I, but was neither fall into IA nor IB, and previous studies has classified it as a separate group, named IC (Gupta et al., 2008). In cassava, AtHAK5 is grouped into clade IB, while it is placed in clade IA in moso bamboo (Ou et al., 2018; Guo et al., 2024). We used ClusterW to align the amino acid sequence of AtHAK5 with OsHAKs and ZmHAKs, and the results suggested that it clusters with StHAK22, ZmHAK5, and OsHAK5, all of which belong to clade IB (Figure 1). Interestingly, similar to StHAK23 and StHAK24, AtHAK5 also presented blank regions at the 5’ and 3’ ends of the CDS sequences. After truncating these blank regions and constructing a phylogenetic tree, we found that AtHAK5 clustered with StHAK23 and StHAK24 in clade IVB. Although these results were inconsistent with most previous reports, the reliability and confidence of the constructed tree were still high.
4.2 Gene structures and conserved domains analysis of StHAKs
The gene structures of barley HvHAKs consist of 3-10 exons and 2-9 introns. While the gene structures of PeHAKs exhibits significant variability, ranging from 1-10 exons and 2-10 introns. The majority of MeKUP genes harbored 6 to 10 exons. Similarly, the gene structures of the 22 StHAKs comprise 6-11 exons, of which 10 genes containing 8 exons and 4 genes containing 9 exons. Distinctly, StHAK24 contains only 1 exon, while StHAK12 has 18 exons. The majority of StHAKs owned 6-10 introns, with the exceptions of StHAK24, which has no introns, and StHAK14, which has only 5 introns. The differences in exon/intron organization among HAK genes may provide insight into the functional diversity and evolutionary adaptation of these genes. In plants, exon/intron structure can influence gene expression regulation, mRNA splicing efficiency, and stability under various environmental conditions. Genes with more exons, like StHAK12, may undergo alternative splicing, potentially producing multiple protein isoforms that allow plants to flexibly respond to different stresses, including nutrient availability and abiotic stress (Laloum et al., 2018). Moreover, the introns may also play a crucial role in mediating regulation of gene expression (Rose, 2008). Tan have preformed a large-scale of bioinformatics analysis of GT47 gene family, and identified two Clades of intron-poor genes with putative functions in drought stress responses and seed development in maize (Tan et al., 2018). These findings suggest that exon/intron organization can influence gene responsiveness under different environmental conditions, which may be particularly crucial for plant adaptation and stress responses.
To predict the conserved structural domains of StHAKs, three databases (SMART, HMMER, CDD) were chosen (Table 1 and Figure 2). The results predicted by SMART and HMMER databases were similar, as all StHAK proteins contained ‘K_trans’ domain, indicating that these genes belong to the HAK/KUP/KT superfamily. In NCBI’ s CDD database, there are seven types of K_trans superfamily (cl15781) structural domains, including pfam02705, TIGR00794, PLN00149, PRK10745, PLN00148, PLN00150, PLN00151. Most StHAK proteins contained the ‘K_trans superfamily’ domain, and StHAK1, StHAK10, StHAK12, StHAK19 specifically contained the ‘PLN00151’ domain. In addition to the ‘PLN00151’ domain, StHAK12 also contained a domain belonging to the cl06777 superfamily, which corresponds to Rrp15p, however, detailed information about this specific protein superfamily was not available in NCBI’ s CDD database. Furthermore, similar to StHAK6, StHAK7 and StHAK11, which contained the ‘K_trans (pfam02705)’ and’PLN00149’domain respectively, the HvHAK and CqHAK proteins also possessed the ‘K_trans superfamily’, ‘PLN00151’ and ‘K_trans (pfam02705) ‘ domains (Cai et al., 2021; Chen et al., 2023b). Finally, the number of motifs predicts in different species is variable, with 10 motifs in potato, 3 in both rice and maize, 7 in quinoa, 10 in barley and mosaic bamboo, and 16 in cassava. This variation in the number of motifs may be due to differences in screening criteria (E values) or the limitations on the maximum number of motifs.
4.3 Analysis of collinearity, synteny, and cis-acting elements of StHAK genes
To further understand the evolutionary patterns of HAK genes in potato, the collinearity and synteny analysis were conducted on StHAKs both within the potato genome and between potato and other crops. We identified a total of six co-linear gene pairs of the HAK gene family within the potato genome, which located primarily on chromosomes 5, 8 and 12 (Figure 4). Synteny analysis between species revealed that StHAKs shared more covariant gene pairs with GmHAKs, MeHAKs and SlHAKs, while fewer covariant gene pairs were found with monocotyledon like ZmHAKs, OsHAKs and SbHAKs (Figure 5). These findings suggested that the genomes of potato, soybean, cassava and tomato shared many homologous gene pairs within the HAK gene family, indicating a closer genetic relationship among these species. In dicotyledons, StHAK4 and StHAK11, located on chromosomes 2 and 5 respectively, appeared more frequently in covariant gene pairs and formed more gene pairs, whereas StHAK13 on chromosome 6 appeared more frequently in several monocotyledons. This suggested that these homologous genes may have originated from a common ancestor and have remained relatively conserved over the span of the species evolution. The study of the synteny and evolutionary relationships of the HAK gene family members in potatoes not only deepens our understanding of the genetic mechanisms of potatoes but also provides precise strategies for breeding programs, including gene mapping, parental selection, and marker-assisted selection. Additionally, it lays a solid foundation for optimizing biotechnological methods such as gene editing, transgenic operations, and gene expression regulation, significantly promoting the genetic improvement of potato traits and their agricultural applications.
In this study, the elements in the promoter regions of StHAKs were categorized into four types based on their functions. The most numerous were light-responsive element (55.4%), which is similar to HvHAK (42.3%) but opposite to OsHAK and ZmHAK. Among the light-responsive elements, Box4 was the most prevalent (Figure 6) (Gupta et al., 2008; Zhang et al., 2012; Cai et al., 2021). The proportion of stress response elements was higher in CqHAK and PeHAK (45.3% and 14%, respectively), but lower in potato and barley (15% and 9.6%) (Cai et al., 2021; Chen et al., 2023b; Guo et al., 2024). The number of promoter/enhancer elements (CAAT box and TATA box) in StHAKs was not analyzed, whereas it was 13.5% and 52% in HvHAKs and PeHAKs, respectively (Cai et al., 2021; Guo et al., 2024). Interestingly, the Ca2+ response elements was numerically and widely distributed in the promoter regions of OsHAKs and ZmHAKs, but were absent in the promoter regions of StHAK and HvHAK (Gupta et al., 2008; Zhang et al., 2012; Cai et al., 2021). In the promoter regions of StHAKs, the most relevant cis-acting elements to stress response are ARE (Anaerobic Responsive Element), MBS (MYB Binding Site) and LTR (Low-Temperature Responsive Element), and the most relevant cis-acting elements to hormone regulation are ABRE (Abscisic Acid Responsive Element), TGA-element (Auxin Responsive Element), TATC-box (Cytokinin Responsive Element), P-box (Gibberellin Responsive Element), CGTCA-motif (Methyl Jasmonate Responsive Element), TGACG-motif (Methyl Jasmonate Responsive Element), TCA-element (Salicylic Acid Responsive Element). As reported, that MYB77 directly bind to MYB factor binding motifs MYBcore (CNGTTR) and MRE(AACC), which located in the HAK5 gene promoter region to involve in potassium uptake (Feng et al., 2021). Four transcription factors dwarf and delayed flowering 2 (DDF2), jagged lateral organs (JLO), basic helix-loop-helix 121 (bHLH121) and transcription initiation factor II_A gamma chain (TFII_A) induced the expression of HAK5 via binding to the HAK5 promoter when plants were grown under low K conditions, while the specific cis-acting elements they bind to were still not clearly defined (Hong et al., 2013; Mulet et al., 2023). Although various transcription factors (TFs) that respond to abiotic stress signals have been reported to induce the expression of HAK genes, specific motifs within the HAK gene promoter regions targeted by these TFs remain largely unidentified. In terms of hormone signaling, low potassium levels also trigger responses involving phytohormones such as cytokinin, auxin, jasmonic acid (JA), and ethylene (ET) (Johnson et al., 2022). HAK5 expression, for instance, is upregulated by reactive oxygen species (ROS) and ethylene signals under potassium-deficient conditions (Schachtman, 2015). Another K+ transporter, OsCHX14, which is preferentially expressed in flowers, is regulated by JA (Chen et al., 2016). Although similar findings exist, these studies have yet to clarify the specific upstream and downstream regulatory interactions, especially regarding the functional role of specific motifs within the HAK promoter region, such as the CGTCA-motif, TGACG-motif, and P-box. Understanding how these motifs are involved in hormone or stress-induced regulation of HAK gene expression remains limited (Hong et al., 2013; Armengaud et al., 2004; Johnson et al., 2022). Therefore, research focused on hormone- or stress-induced motifs that regulate HAK gene expression could provide significant insights.
4.4 Expression of StHAK genes across various tissues and their responses to different stresses
A well-understanding of the expression properties of StHAKs is crucial for elucidating their functions. In the study, the transcript levels of StHAKs in diverse tissues of potato and under different potassium fertilizer treatments were assayed through publicly available RNA-Seq data, transcriptome data and qRT-PCR. Microarray data revealed that most StHAKs in clade I and IV were expressed at relatively low levels across all organs, while those in clade II and III showed higher expression levels in all test tissues (Figure 7). These results are consistent with studies on other species such as Arabidopsis, barley, and quinoa (MaüSer et al., 2001; Cai et al., 2021; Chen et al., 2023b). Notably, StHAK18 and StHAK20 were exclusively expressed in carpels, stamens and mature flowers of potato, indicating a specific role in regulating the reproductive growth. The high mRNA levels of StHAK8 in stolons, compared to other tissues, indicated its potential involvement in potato tuber formation. Transcriptomic data revealed that StHAK4, StHAK9, and StHAK24 were more abundant in leaves and roots than in tubers, suggesting their involvement in K+ uptake by roots, photosynthesis in leaves, and photosynthate allocation in potatoes. This requires further experimental validation. Chen et al. (2017) showed that OsHAK1 is highly expressed in rice root tips but weakly in aerial parts, indicating its key role in root K+ uptake. According to qRT-PCR results, StHAK14 was expressed significantly in tubers, suggesting that it may be involved in the development of potato tubers (Figure 10B). This finding was somewhat not the same with RNA-seq or transcriptome data, which did not show a apparent expression of StHAK14. StHAK14 shared 48% similarity with AtKUP8, which maintains K+ homeostasis and responds to heavy metal stress (e.g., Cd, Cr and Cu) in Arabidopsis (Sanz-Fernandez et al., 2021). AtKUP8 is also involving in promoting cellular osmotic adaptation by maintaining K+ homeostasis under drought conditions, while the exact function of StHAK14 in tubers remains unclear, which needs to further investigate (Osakabe et al., 2013). StHAK4 was abundant in potato leaves, consistent with both qRT-PCR and transcriptomic data (Figures 8, 10). StHAK8 was enriched in stolons in RNA-seq, and also expressed obviously in stems and tubers according to qRT-PCR results (Figures 7, 10). Additionally, qRT-PCR results showed that StHAK18 and StHAK21 were apparently expressed in flowers, which consistent with Microarray findings (Figures 7, 10).
Different paralogs of StHAKs show significant expression in various tissues such as roots, stems, leaves, stolon, tubers and flowers (including carpels, stamens, sepals, petals), as indicated in the results. As previously reported, AtHAK5 is chiefly expressed in roots and regulates K+ uptake under low potassium conditions, while AtHAK4 and AtHAK9 are primarily expressed in stems and leaves, suggesting their involvement in K+ transport and distribution (Ahn et al., 2004). Similarly, in rice, genes such as OsHAK1 and OsHAK2 are expressed in roots to promote K+ absorption, while OsHAK5 and OsHAK16 are expressed in leaves, likely contributing to K+ redistribution and leaf function. OsHAK10 and OsHAK15 are expressed specifically in stamens, suggesting they play key roles in pollination or fertilization (Gupta et al., 2008). In cassava, five genes including MeKUP3 and MeKUP4, are broadly expressed in stems, leaves and store roots, while MeKUP1, MeKUP19, and MeKUP20 are expressed significantly only in leaves, indicating their involvement in K+ redistribution (Ou et al., 2018). This suggest that: (1) The HAKs gene family is widely involved in critical physiological processes, such as K+ absorption and distribution, and the development of vegetative organs (e.g., tubers) and reproductive organs (e.g., flowers); (2) Different HAKs paralogs perform specific biological roles in distinct tissues, optimizing K+ uptake and utilization under diverse environmental conditions. Therefore, investigating the expression profiles of HAKs paralogs can provide insights into how these genes regulate K+ homeostasis through gene differentiation and contribute to plant adaptation to environmental changes. This work also provides valuable information for a deeper understanding of HAKs gene functions.
Moreover, StHAKs are crucial in responding to abiotic stresses. As we can see, StHAK1, StHAK6, StHAK11, and StHAK22 responded to at least two abiotic stresses, while StAHK14 was able to respond to three (salt, mannitol, and heat) (Figure 7B). As reported previously, the expression levels of AtHAK2, AtHAK6 and AtHAK11 were significantly raised under high salt stress (Maathuis, 2006). Likewise, the relative abundance levels of three and eight TaHAKs are dramatically up-regulated under salt and drought stress, respectively (Cheng et al., 2018). Most HAK gene family members in moso bamboo are capable of responding to at least two abiotic stresses (Guo et al., 2024). In addition to salt and mannitol stress, StHAK6 was also induced by IAA (Figure 7B). StHAK20 and StHAK21 were co-induced by IAA and GA3, however, StHAK20 was suppressed by ABA (Figure 7B). Like StHAK20, some PeHAKs are involved in the cross-pathways of multiple hormone signals, such as ABA and IAA, or NAA and GA (Guo et al., 2024). Moreover, certain StHAKs, such as StHAK4 and StHAK9, were also somewhat capable of responding to biotic stresses (Figure 7B).
K+ is a crucial ion in regulating stomatal movement in plant (Zhang et al., 2020). Stomatal closure is triggered by the efflux of K+ from guard cells, leading to water loss from the cells, which causes them to deflate and closes the stomata. This process is critical during drought conditions to reduce water loss through transpiration. As we all known, potassium transporters facilitate this movement of K+. HAKs (High-affinity potassium transporters) play a significant role in potassium uptake, particularly under low potassium conditions, which can be exacerbated during drought stress. Studies on Arabidopsis and wheat have shown that specific members of the HAK family are involved in transporting K+ from roots to shoots, ensuring that adequate K+ levels are maintained in guard cells to modulate stomatal movements under stress. The similar mechanisms may be expected in potato, although specific research on StHAK members like StHAK5 remains limited. Potatoes, like many plants, rely on K+ for maintaining turgor pressure in guard cells to regulate stomatal aperture. Under drought conditions, potassium transporters such as HAKs and KUPs likely play a critical role in maintaining this function, thus reducing water loss. Further experimental evidence, such as gene expression studies under drought conditions, could provide more specific insights into which K+ transporters are most active during drought stress in potato. The future studies would be focused on elucidating the specific roles of potato HAK/KUP transporters in stomatal regulation during drought stress. Some experiments such as gene expression analysis, knockout studies, and physiological assays under drought conditions need to conduct, which could provide clearer insights into these transporters’ functional roles. In summary, potassium transporters are integral to stomatal regulation, and evidence from other species suggests that potato HAK transporters may perform similar functions. Of course, this hypothesis requires further validation.
Based on our in-depth analysis of the HAK gene family in potatoes, we believe these genes have significant potential in regulating potassium ion balance and enhancing drought and cold resistance, as well as tolerance to high light intensity in crops. In the future, gene-editing techniques (like CRISPR/Cas9) could be used to optimize the expression of these key genes, thus enabling the development of potato or other crops with improved stress resilience. Additionally, these findings could serve as targets for improving stress tolerance in other crops, providing new approaches to address global climate change challenges and enhance the stability of agricultural production.
5 Conclusion
In this study, a total of 24 StHAK genes were identified and divided into four subfamilies with potentially similar functions. The expression levels of these StHAKs varied significantly across different tissues and displayed a obvious difference under various abiotic stress, hormone treatments and potassium fertilizer conditions. This suggests that these genes were expressed tissue-specificity as well as are involved in both abiotic stress response and potassium uptake. To clearly display HAK gene expression across potato tissues, we created a cartoon heatmap illustrating their tissue-specific patterns, an unprecedented approach in potato gene family analysis. Finally, StHAK genes like StHAK8, StHAK14, and StHAK22 as highly expressed in tubers, suggesting their role in tuber growth and development. Consequently, these findings would contribute to a deeper understanding of the biological functions of HAK/KUP/KT genes, and provide a solid basis for further functional analysis of these genes in potato growth and potassium responses.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author contributions
KL: Writing – original draft. YQ: Writing – original draft, Writing – review & editing. LW: Writing – review & editing. RY: Writing – review & editing. XuS: Writing – original draft, Writing – review & editing. JY: Writing – review & editing. XoS: Writing – review & editing. WD: Writing – review & editing. LJ: Writing – original draft, Writing – review & editing. MF: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work was funded by Inner Mongolia Autonomous Region Natural Science Foundation of China (grant no. 2022QN03013), Basic research funds directly under the university of Inner Mongolia Autonomous Region (grant no. BR220502), The Innovation Team of Inner Mongolia Agricultural University (grant no. BR-22-13-01).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1487794/full#supplementary-material
References
Armengaud, P., Breitling, R., Amtmann, A. (2004). The potassium-dependent transcriptome of Arabidopsis reveals a prominent role of jasmonic acid in nutrient signaling. Plant Physiol. 136, 2556–2576. doi: 10.1104/pp.104.046482
Ahn, S. J., Shin, R., Schachtman, D. P. (2004). Expression of KT/KUP genes in Arabidopsis and the role of root hairs in K+ uptake. Plant Physiol. 134, 1135–1145. doi: 10.1104/pp.103.034660
Britto, D. T., Kronzucker, H. J. (2008). Cellular mechanisms of potassium transport in plants. Physiol. Plant 133, 637–650. doi: 10.1111/j.1399-3054.2008.01067.x
Cai, K., Zeng, F., Wang, J., Zhang, G. (2021). Identification and characterization of HAK/KUP/KT potassium transporter gene family in barley and their expression under abiotic stress. BMC Genomics 22, 317. doi: 10.1186/s12864-021-07633-y
Che, J., Yamaji, N., Wang, S. F., Xia, Y., Yang, S. Y., Su, Y. H., et al. (2024). OsHAK4 functions in retrieving sodium from the phloem at the reproductive stage of rice. Plant J. 120, 76–90. doi: 10.1111/tpj.16971
Chen, C., Wu, Y., Li, J., Wang, X., Zeng, Z., Xu, J., et al. (2023a). TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 16, 1733–1742. doi: 10.1016/j.molp.2023.09.010
Chen, G., Liu, C., Gao, Z., Zhang, Y., Jiang, H., Zhu, L., et al. (2017). OsHAK1, a high-affinity potassium transporter, positively regulates responses to drought stress in rice. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01885
Chen, Y., Lin, Y., Zhang, S., Lin, Z., Chen, S., Wang, Z. (2023b). Genome-Wide Identification and Characterization of the HAK Gene Family in Quinoa (Chenopodium quinoa Willd.) and Their Expression Profiles under Saline and Alkaline Conditions. Plants. 12, 3747. doi: 10.3390/plants12213747
Chen, Y., Ma, J., Miller, A. J., Luo, B., Wang, M., Zhu, Z., et al. (2016). OsCHX14 is involved in the K+ homeostasis in rice (Oryza sativa) flowers. Plant and Cell Physiol. 57, 1530–1543. doi: 10.1093/pcp/pcw088
Chen, Y., Shi, X., Chen, Y., Yu, J., Qin, Y., Jia, L., et al. (2024). Meeting the demand for different nitrogen forms in potato plants without the use of nitrification inhibitors. Plants 13, 3177. doi: 10.3390/plants13223177
Cheng, X., Liu, X., Mao, W., Zhang, X., Chen, S., Zhan, K., et al. (2018). Genome-wide identification and analysis of HAK/KUP/KT potassium transporters gene family in wheat (Triticum aestivum L.). Int. J. Mol. Sci. 19, 3969. doi: 10.3390/ijms19123969
Cheng, R., Zhao, Z., Tang, Y., Gu, Y., Chen, G., Sun, Y., et al. (2024). Genome-wide survey of KT/HAK/KUP genes in the genus Citrullus and analysis of their involvement in K+-deficiency and drought stress responses in between C. lanatus and C. amarus. BMC Genomics 25, 836. doi: 10.1186/s12864-024-10712-5
Cui, S., Qin, Y., Yu, J., Shi, X., Jia, L., Fan, M. (2020). Improving tuber yield and phosphorus use efficiency using split phosphorus application to potatoes in Inner Mongolia. Am. J. Potato Res. 97, 318–324. doi: 10.1007/s12230-020-09783-3
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2018). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. doi: 10.1093/nar/gky995%J
Fan, M., Xing, H., Yu, J., Suyala, Q., Chen, Y., Qin, Y., et al. (2019). Physiology of mineral nutrition and nutrient management in potato (Beijing: China Agricultural Press).
Feng, C., Luo, Y., Wang, P., Gilliham, M., Long, Y. (2021). MYB77 regulates high-affinity potassium uptake by promoting expression of HAK5. New Phytol. 232, 176–189. doi: 10.1111/nph.17589
Feng, H., Tang, Q., Cai, J., Xu, B., Xu, G., Yu, L. (2019). Rice OsHAK16 functions in potassium uptake and translocation in shoot, maintaining potassium homeostasis and salt tolerance. Planta. 250, 549–561. doi: 10.1007/s00425-019-03194-3
Feng, X., Wang, Y., Zhang, N., Wu, Z., Zeng, Q., Wu, J., et al. (2020). Genome-wide systematic characterization of the HAK/KUP/KT gene family and its expression profile during plant growth and in response to low-K+ stress in Saccharum. BMC Plant Biol. 20, 20. doi: 10.1186/s12870-019-2227-7
Finn, R. D., Clements, J., Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Gierth, M., Maser, P., Schroeder, J. I. (2005). The potassium transporter AtHAK5 functions in K+ deprivation-induced high-affinity K+ uptake and AKT1 K+ channel contribution to K+ uptake kinetics in Arabidopsis roots. Plant Physiol. 137, 1105–1114. doi: 10.1104/pp.104.057216
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Guo, H., Tan, J., Jiao, Y., Huang, B., Ma, R., Ramakrishnan, M., et al. (2024). Genome-wide identification and expression analysis of the HAK/KUP/KT gene family in Moso bamboo. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1331710
Gupta, M., Qiu, X., Wang, L., Xie, W., Zhang, C., Xiong, L., et al. (2008). KT/HAK/KUP potassium transporters gene family and their whole-life cycle expression profile in rice (Oryza sativa). Mol. Genet. Genomics 280, 437–452. doi: 10.1007/s00438-008-0377-7
Hong, J., Takeshi, Y., Kondou, Y., Schachtman, D., Matsui, M., Shin, R. (2013). Identification and characterization of transcription factors regulating Arabidopsis HAK5. Plant Cell Physiol. 54, 1478–1490. doi: 10.1093/pcp/pct094
Horton, P., Park, K. J., Obayashi, T., Fujita, N., Harada, H., Adams-Collier, C. J., et al. (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587. doi: 10.1093/nar/gkm259
Hu, B., Jin, J., Guo, A. Y., Zhang, H., Luo, J., Gao, G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics. 31, 1296–1297. doi: 10.1093/bioinformatics/btu817
Jayakodi, M., Padmarasu, S., Haberer, G., Bonthala, V. S., Gundlach, H., Monat, C., et al. (2020). The barley pan-genome reveals the hidden legacy of mutation breeding. Nature. 588, 284–289. doi: 10.1038/s41586-020-2947-8
Jiménez-Estévez, E., Martínez-Martínez, A., Amo, J., Yáñez, A., Miñarro, P., Martínez, V., et al. (2024). Increased tolerance to low K+, and to cationic stress of Arabidopsis plants by expressing the F130S mutant version of the K+ transporter AtHAK5. Plant Physiol. Biochem. 212, 108768. doi: 10.1016/j.plaphy.2024.108768
Jin, R., Jiang, W., Yan, M., Zhang, A., Liu, M., Zhao, P., et al. (2021). Genome-wide characterization and expression analysis of HAK K+ transport family in Ipomoea. 3 Biotech. 11, 3. doi: 10.1007/s13205-020-02552-3
Johnson, R., Vishwakarma, K., Hossen, M. S., Kumar, V., Shackira, A. M., Jos, T., et al. (2022). Potassium in plants: Growth regulation, signaling, and environmental stress tolerance. Plant Physiol. Biochem. 172, 56–69. doi: 10.1016/j.plaphy.2022.01.001
Kong, S., Qin, Y., Shi, X., Yu, J., Jia, L., Chen, Y., et al. (2024). Establishing a critical phosphorus dilution curve for potato in semi-arid regions based on a Bayesian analysis. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1458741
Laloum, T., Martín, G., Duque, P. (2018). Alternative splicing control of abiotic stress responses. Trends Plant Sci. 23, 140–150. doi: 10.1016%2Fj.tplants.2017.09.019
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., Mcgettigan, P. A., Mcwilliam, H., et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics. 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Lei, H., Li, J., Song, Z. (2022). Genome-wide identification and expression profiling of the shaker K+ Channel and HAK/KUP/KT transporter gene families in grape (Vitis vinifera L.). Phyton. 91, p2519. doi: 10.32604/phyton.2022.021268
Lescot, M., Dehais, P., Thijs, G., Marchal, K., Moreau, Y., Van De Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Letunic, I., Bork, P. (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 23, 127–128. doi: 10.1093/bioinformatics/btl529
Letunic, I., Bork, P. (2018). 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496. doi: 10.1093/nar/gkx922
Li, Q., Du, W., Tian, X., Jiang, W., Zhang, B., Wang, Y., et al. (2022). Genome-wide characterization and expression analysis of the HAK gene family in response to abiotic stresses in Medicago. BMC Genomics 23, 791. doi: 10.1186/s12864-022-09009-2
Li, W., Xu, G., Alli, A., Yu, L. (2018). Plant HAK/KUP/KT K+ transporters: Function and regulation. Semin. Cell Dev. Biol. 74, 133–141. doi: 10.1016/j.semcdb.2017.07.009
Liu, K., Yang, Q., Yang, T., Wu, Y., Wang, G., Yang, F., et al. (2019). Development of Agrobacterium-mediated transient expression system in Caragana intermedia and characterization of CiDREB1C in stress response. BMC Plant Biol. 19, 237. doi: 10.1186/s12870-019-1800-4
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48, D265–D268. doi: 10.1093/nar/gkz991
Luo, M., Chu, J., Wang, Y., Chang, J., Zhou, Y., Jiang, X. (2024). Positive Regulatory Roles of Manihot esculenta HAK5 under K+ Deficiency or High Salt Stress. Plants. 13, 849. doi: 10.3390/plants13060849
Luo, H. B., Huang, C. M., Wei, Y. W., Zhu, H. M., Cao, H. Q., Jiang, S. L., et al. (2021). Uncovering expression and functional analysis of newly discovered high-affinity K+ transporter family members from sugarcane. J. Plant Biochem. Biotechnol. 31, 826–836. doi: 10.1007/s13562-021-00726-5
MaüSer, P., Thomine, S. B., Schroeder, J. I., Ward, J. M., Hirschi, K., Sze, H., et al. (2001). Phylogenetic relationships within cation transporter families of arabidopsis. Plant Physiol. 126, 1646–1667. doi: 10.1104/pp.126.4.1646%JPlantPhysiology
Maathuis, F. J. (2006). The role of monovalent cation transporters in plant responses to salinity. J. Exp. Bot. 57, 1137–1147. doi: 10.1093/jxb/erj001
Mulet, J., Porcel, R., Yenush, L. (2023). Modulation of potassium transport to increase abiotic stress tolerance in plants. J. Exp. Bot. 74, 5989–6005. doi: 10.1093/jxb/erad333
Naumann, M., Koch, M., Thiel, H., Gransee, A., Pawelzik, E. (2020). The importance of nutrient management for potato production part II: Plant nutrition and tuber quality. Potato Res. 63, 121–137. doi: 10.1007/s11540-019-09430-3
Osakabe, Y., Arinaga, N., Umezawa, T., Katsura, S., Nagamachi, K., Tanaka, H., et al. (2013). Osmotic stress responses and plant growth controlled by potassium transporters in Arabidopsis. Plant Cell. 25, 609–624. doi: 10.1105/tpc.112.105700
Ou, W., Mao, X., Huang, C., Tie, W., Yan, Y., Ding, Z., et al. (2018). Genome-Wide Identification and Expression Analysis of the KUP Family under Abiotic Stress in Cassava (Manihot esculenta Crantz). Front. Physiol. 9. doi: 10.3389/fphys.2018.00017
Rose, A. B. (2008). Intron-mediated regulation of gene expression. Curr. Top. Microbiol. Immunol. 326, 277–290. doi: 10.1007/978-3-540-76776-3_15
Run, Y., Cheng, X., Dou, W., Dong, Y., Zhang, Y., Li, B., et al. (2022). Wheat potassium transporter TaHAK13 mediates K+ absorption and maintains potassium homeostasis under low potassium stress. Front. Plant Sci. 13, 1103235. doi: 10.3389/fpls.2022.1103235
Santa-Maria, G. E., Oliferuk, S., Moriconi, J. I. (2018). KT-HAK-KUP transporters in major terrestrial photosynthetic organisms: A twenty years tale. J. Plant Physiol. 226, 77–90. doi: 10.1016/j.jplph.2018.04.008
Santa-María, G. E., Rubio, F., Dubcovsky, J., Rodríguez-Navarro, A. (1997). The HAK1 gene of barley is a member of a large gene family and encodes a high-affinity potassium transporter. Plant Cell. 9, 2281–2289. doi: 10.1105/tpc.9.12.2281%J
Sanz-Fernandez, M., Rodriguez-Gonzalez, A., Sandalio, L. M., Romero-Puertas, M. C. (2021). Role of potassium transporter KUP8 in plant responses to heavy metals. Physiol. Plant 173, 180–190. doi: 10.1111/ppl.13345
Sardans, J., Peñuelas, J. (2021). Potassium control of plant functions: ecological and agricultural implications. Plants 10, 419. doi: 10.3390/plants10020419
Schachtman, D. P. (2015). The role of ethylene in plant responses to K+ deficiency. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.01153
Shi, X., Zhang, X., Kang, W., Chen, Y., Fan, M. (2019). Possibility of recommending potassium application rates based on a rapid detection of the potato petiole K status with a portable K ion meter. Am. J. Potato Res. 96, 48–54. doi: 10.1007/s12230-018-9687-4
Sze, H., Chanroj, S. (2018). Plant endomembrane dynamics: studies of K+/H+ Antiporters provide insights on the effects of pH and ion homeostasis. Plant Physiol. 177, 875–895. doi: 10.1104/pp.18.00142%JPlantPhysiology
Tahir, M. M., Tong, L., Xie, L., Wu, T., Ghani, M. I., Zhang, X., et al. (2023). Identification of the HAK gene family reveals their critical response to potassium regulation during adventitious root formation in apple rootstock. Hortic. Plant J. 9, 45–59. doi: 10.1016/j.hpj.2022.11.001
Tan, J., Miao, Z., Ren, C., Yuan, R., Tang, Y., Zhang, X., et al. (2018). Evolution of intron-poor clades and expression patterns of the glycosyltransferase family 47. Planta. 247, 745–760. doi: 10.1007/s00425-017-2821-6
Templalexis, D., Tsitsekian, D., Liu, C., Daras, G., Šimura, J., Moschou, P., et al. (2022). Potassium transporter TRH1/KUP4 contributes to distinct auxin-mediated root system architecture responses. Plant Physiol. 188, 1043–1060. doi: 10.1093/plphys/kiab472
Torabian, S., Farhangi-Abriz, S., Qin, R., Noulas, C., Sathuvalli, V., Charlton, B., et al. (2021). Potassium: A vital macronutrient in potato production—A review. Agronomy 11, 543. doi: 10.3390/agronomy11030543
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., Van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Wang, Y., Wu, W. H. (2013). Potassium transport and signaling in higher plants. Annu. Rev. Plant Biol. 64, 451–476. doi: 10.1146/annurev-arplant-050312-120153
Wang, Y., Chen, Y. F., Wu, W. H. (2021). Potassium and phosphorus transport and signaling in plants. J. Integr. Plant Biol. 63, 34–52. doi: 10.1111/jipb.13053
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. doi: 10.1093/nar/gkr1293
Wang, L., Wang, Y., Yin, P., Jiang, C., Zhang, M. (2024). ZmHAK17 encodes a Na+-selective transporter that promotes maize seed germination under salt conditions. New Crops 1, 100024. doi: 10.1016/j.ncrops.2024.100024
Wang, Y., Zhang, Y., Wei, Y., Meng, J., Zhong, C., Fan, C. (2023). Characterization of HAK protein family in Casuarina equisetifolia and the positive regulatory role of CeqHAK6 and CeqHAK11 genes in response to salt tolerance. Front. Plant Sci. 13. doi: 10.3389/fpls
White, P. J. (2013). Improving potassium acquisition and utilisation by crop plants. J. Plant Nutr. Soil Sc. 176, 305–316. doi: 10.1002/jpln.201200121
Xu, X., Pan, P., Cheng, S., Zhang, B., Mu, D., Ni, P., et al. (2011). Genome sequence and analysis of the tuber crop potato. Nature. 475, 189–195. doi: 10.1038/nature10158
Yang, X., Zhang, L., Guo, X., Xu, J., Zhang, K., Yang, Y., et al. (2023). The gap-free potato genome assembly reveals large tandem gene clusters of agronomical importance in highly repeated genomic regions. Mol. Plant 16, 314–317. doi: 10.1016/j.molp.2022.12.010
Zhang, M. L., Huang, P. P., Ji, Y., Wang, S., Wang, S. S., Li, Z., et al. (2020). KUP9 maintains root meristem activity by regulating K+ and auxin homeostasis in response to low K. EMBO Rep. 21, e50164. doi: 10.15252/embr.202050164
Zhang, L., Li, D., Yao, Y., Zhang, S. (2020). H2O2, Ca2+, and K+ in subsidiary cells of maize leaves are involved in regulatory signaling of stomatal movement. Plant Physiol. Bioch 152, 243–251. doi: 10.1016/j.plaphy.2020.04.045
Zhang, M., Liang, X., Wang, L., Cao, Y., Song, W., Shi, J., et al. (2019). A HAK family Na+ transporter confers natural variation of salt tolerance in maize. Nat. Plants. 5, 1297–1308. doi: 10.1038/s41477-019-0565-y
Zhang, Z., Zhang, J., Chen, Y., Li, R., Wang, H., Wei, J. (2012). Genome-wide analysis and identification of HAK potassium transporter gene family in maize (Zea mays L.). Mol. Biol. Rep. 39, 8465–8473. doi: 10.1007/s11033-012-1700-2
Keywords: potato, HAK/KUP/KT, potassium ion transport, gene expression, potassium response
Citation: Liu K, Qin Y, Wu L, Yi R, Shi X, Yu J, Shi X, Dong W, Jia L and Fan M (2025) Genome-wide identification, characterization and expression pattern analysis of HAK/KUP/KT potassium transporter gene family in potato. Front. Plant Sci. 15:1487794. doi: 10.3389/fpls.2024.1487794
Received: 28 August 2024; Accepted: 18 December 2024;
Published: 16 January 2025.
Edited by:
Fenglin Deng, Yangtze University, ChinaReviewed by:
Jose M. Mulet, Universitat Politècnica de València, SpainAyan Sadhukhan, Indian Institute of Technology Jodhpur, India
Wei Jiang, Nanjing Agricultural University, China
Copyright © 2025 Liu, Qin, Wu, Yi, Shi, Yu, Shi, Dong, Jia and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liguo Jia, bm5kamlhbGlndW9AMTYzLmNvbQ==; Mingshou Fan, Zm1zd2hAMTI2LmNvbQ==