 Lei Wang1,2*†
Lei Wang1,2*† Fuxing Li1†Kexin Zhao1†
Fuxing Li1†Kexin Zhao1† Jie Yang3Haonan Sun4Xingyong Cui2
Jie Yang3Haonan Sun4Xingyong Cui2 Wenpan Dong2
Wenpan Dong2 Enze Li2*
Enze Li2* Ning Wang1*
Ning Wang1*- 1College of Horticulture and Plant Protection, Henan University of Science and Technology, Luoyang, China
- 2Laboratory of Systematic Evolution and Biogeography of Woody Plants, College of Ecology and Nature Conservation, Beijing Forestry University, Beijing, China
- 3College of Biology and Food, Shangqiu Normal University, Shangqiu, China
- 4College of Landscape and Tourism, Hebei Agricultural University, Baoding, China
Weigela Thunb. is a genus in the family Caprifoliaceae. All species in this genus have high ornamental and medicinal value. However, the genetic divergence between species and the phylogeny within Weigela is still unclear. Therefore, we sequenced and analyzed four plastomes from four different Weigela species to reveal the genetic divergence among species of this genus, and the phylogeny within Weigela. The four plastomes from Weigela ranged from 156,909 bp to 157,739 bp in size, and presented a typical circular quadripartite structure. Each complete plastome contained a pair of inverted repeat regions (23,592~24,957 bp), a larger single-copy (LSC) region (89,922~90,229 bp), and a small single-copy (SSC) region (17,668~20,429 bp). We identified three types of repeats, corresponding to 268 forward repeats, 128 palindromic repeats, and 867 tandem repeats, for a total of 1,263 long repeats. A total of 352 SSRs were identified from the four plastomes, and most of them were concentrated in the LSC region and the noncoding regions. Mononucleotide repeat units were the most frequently detected types of repeats, of which A/T repeat units were the most abundant. Three mutational hotspots (trnH-psbA, trnR-ndhF, and trnN-ndhF) were identified as candidate barcodes for Weigela species. Weigela belongs to Diervilloideae located at an early diverging position in the Caprifoliaceae. Within Weigela, W. japonica and W. floribunda were sister with W. subsessilis and W. florida. This study revealed the plastome structure and variation of four well-known Weigela species, and found three candidate barcodes for further study of four well-known Weigela species. In addition, the phylogenetic location of Weigela within the Caprifoliaceae was identified.
Introduction
Given that the plastome is primarily characterized by maternal inheritance, it has been extensively employed in phylogenetic studies at different levels (even the population level) (Dong et al., 2021; Wang et al., 2022a, b; Zhang et al., 2007). Due to their stable structure, GC content, and non-recombination during the process of heredity, plastome sequences can often be used for species identification (Liu et al., 2023). Despite the highly conserved structure of the plastome, evolutionary events including gene or intron loss can also occur during the course of species evolution (Claude et al., 2022).
Weigela Thunb. belongs to Caprifoliaceae, and all species in this genus have high ornamental and medicinal value (Liang et al., 2013; Yamada and Maki, 2012). The genus contains about 11~12 species, having a discontinuous distribution pattern in America and Asia (Kim and Kim, 1999). The phylogeny based on the ITS showed that Weigela could be divided into three major clades, one core group and two single species clades (Kim and Kim, 1999). The flowers are large with bell-shaped or funnel-shaped corollas, solitary or in cymes of two to six flowers in the axils or tips of lateral branches, with white or pink-to-crimson petals (Figure 1). Because of their unique flower shape and multifarious colors, species of Weigela are a unique ornamental resource for urban greening. However, to date, there have been few studies on the phylogeny and genetic diversity of Weigela. Meeler combined morphological evidence with nuclear ITS and chloroplast gene (trnS-G) sequences to reconstruct the phylogeny of Diervilla, showing that Diervilla and Weigela are clustered in the Diervilloideae (Meeler, 2018), and that, because of the sister-group relationship between Diervilla and W. middendorffiana (Carrière) K.Koch, Weigela is not a monophyletic group (Kim and Kim, 1999; Meeler, 2018). In addition, studies on the phylogeny of the entire family (Caprifoliaceae) have rarely involved Weigela, despite the phylogeny within Caprifoliaceae having been studied extensively based on morphological characters (Donoghue et al., 2003; Sun et al., 2022; Donoghue et al., 2001) or molecular evidence (Jacobs et al., 2009; Wang et al., 2021, 2020; Li et al., 2024; Bell et al., 2001). So far, there has been no clear conclusion on the phylogenetic relationships of Weigela, which has seriously restricted the development and utilization of this genus, especially in the breeding of new, high-quality ornamental varieties.
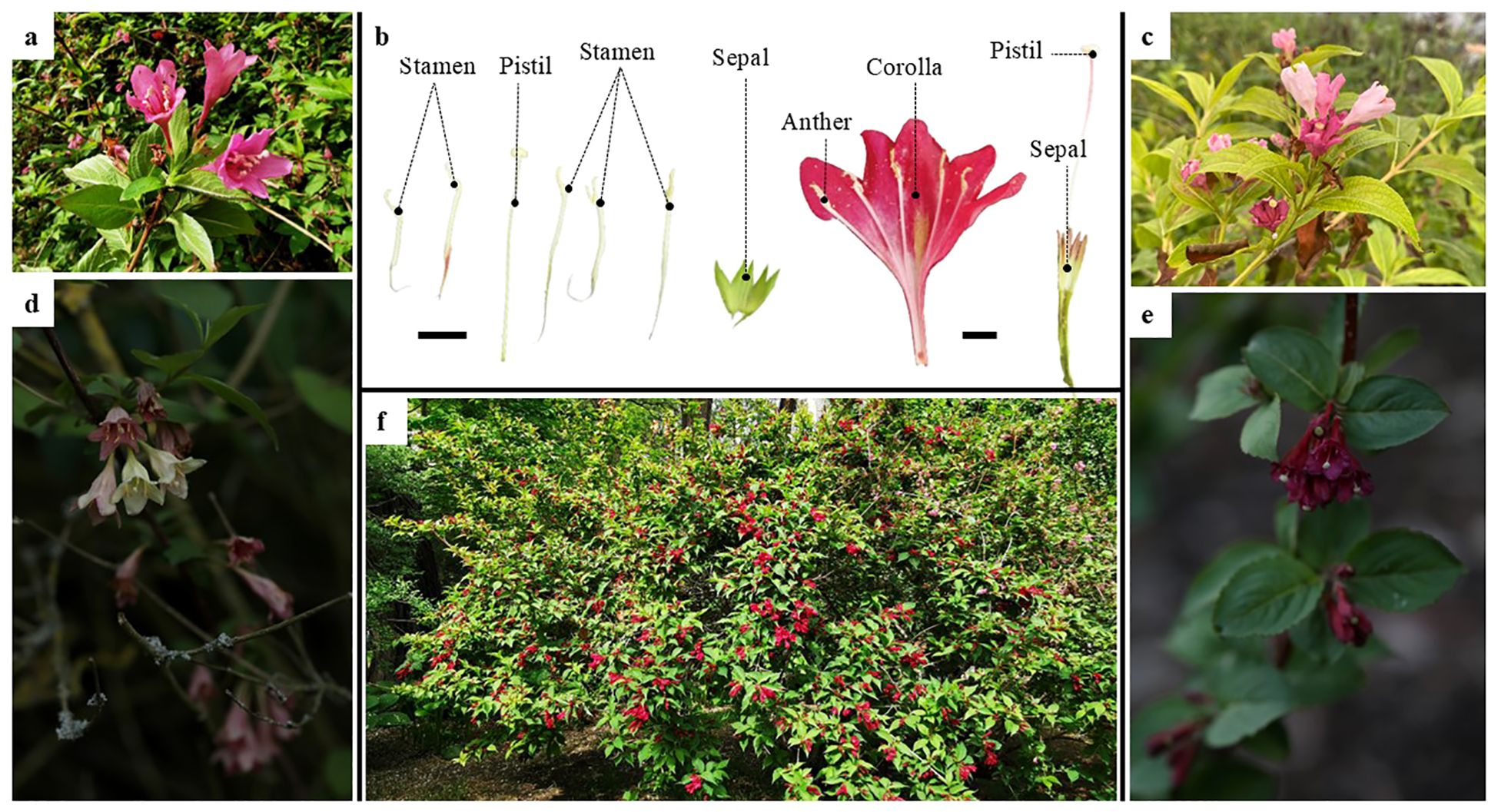
Figure 1. Floral anatomy of Weigela florida and its habitat. (A) Pink flowers of Weigela florida; (B) Floral anatomy of Weigela florida; (C) Pink flowers of Weigela japonica; (D) Flowers of Weigela subsessilis; (E) Flowers of Weigela floribunda; (F) Morphology and habitat of Weigela florida; Scale bars: 1.0 cm.
Based on the abovementioned state of the research and the urgent problems still to be solved, our study aims to reveal the genetic diversity of Weigela using whole plastome sequences. Furthermore, in order to clarify the phylogenetic status of Weigela, this study aims to reconstruct the phylogeny of Caprifoliaceae using plastomic data. The main objectives of this study include the following three aspects: (1) To comprehensively compare and analyze the structural characteristics of plastomes between four Weigela species, (2) to reveal the genetic divergence within Weigela, (3) and to clarify the phylogeny of Weigela and verify the phylogenetic position of Weigela within the Caprifoliaceae.
Materials and methods
Plant material, DNA extraction, and genome sequencing
In this study, four species found mainly in northeast Asia of the core clade in Weigela were selected based on previous studies (Kim and Kim, 1999; Meeler, 2018). The tissue of four Weigela species were obtained from the National Botanical garden in Beijing, China. These samples were identified by Dr. Wenpan Dong from Beijing Forestry University, and voucher specimens of these samples were deposited in the Herbarium at the College of Horticulture and Plant Protection, Henan University of Science and Technology, Henan province, China. The fresh leaves were preserved in silica gel, and the detailed information of these samples is given in Supplementary Table S1. Total DNA was isolated from each sample using a modified CTAB method (Li et al., 2013) and detected through electrophoresis on 0.8% agarose gels. The library preparation and Illumina-based Nova PE150 sequencing were carried out at Novogene Biotechnology Co., Ltd. in Tianjin, China, resulting in a total of 6 Gb of raw data.
Plastome assembly and annotation
The sequencing raw data were firstly filtered, and those low quality data located in the joints or ends were removed with Trimmomatic v0.39 (Bolger et al., 2014) using the default parameters. GetOrganelle v1.7.6.1 (Jin et al., 2020) was used to assemble the plastome with the default parameters. The plastomes were annotated by PGA (Plastid Genome Annotator) (Qu et al., 2019) with the reference of W. florida (Bunge) A. DC. (MN524626), and the annotation results were manually checked through Geneious Prime v2021 (Kearse et al., 2012) to ensure their accuracy, especially for those genes with high sequence differentiation and introns. Plastome maps of Weigela were drawn and visualized using OGDRAW v1.3.1 (Greiner et al., 2019). The annotated sequences of these complete plastomes were deposited in GenBank, with the accession numbers listed in Supplementary Table S1.
Comparison of plastome structures
The comparative analyses of plastome structures and the sequence similarity between the four Weigela species were conducted by the program mVISTA (Mayor et al., 2000). The annotated plastome of W. floribunda (Siebold & Zucc.) K.Koch was used as a reference. The online program IRscope (Amiryousefi et al., 2018) was used to compare the variation in the junction regions between LSC, IRb, SSC, and IRa.
The REPuter program (Kurtz et al., 2001) and the Perl script MISA (Beier et al., 2017) were used to analyse the repeat sequences of these plastomes. Various types of long repeats, including forward, reverse, palindromic, and complementary were identified across the four Weigela plastomes by the REPuter online program, with a Hamming distance of 3 and a minimum repeat size of 30 bp. The Perl script MISA was used to identify sequence repeats, including six types of SSRs (mono-, di-, tri-, tetra-, penta-, and hexanucleotides). The minimum thresholds for each SSR type were set at 10, 5, 4, 3, 3, and 3, respectively.
Divergence analyses
Firstly, we used MAFFT v7 (Katoh and Standley, 2013) to align the plastomes for identifing the mutational hotspot regions and genes. Subsequently, the nucleotide diversity (Pi) of the four plastomes were analysed by DnaSP v6.12.03 (Zhou et al., 2018), and the hotspot regions with high mutation rates were identified using a sliding window method. The step size was prescribed at 20 bp, and the window length was 100 bp.
Phylogenetic inference
In addition to the four newly assembled plastomes of Weigela, we also collected all the published plastome sequences of Caprifoliaceae from NCBI (Benson et al., 2018). A total of 84 species were used for the phylogenetic analysis including four species of the subfamily Diervilloideae, 34 of the subfamily Caprifolioideae, five of the subfamily Dipsacoideae, eight of the subfamily Valerianoideae, three of the subfamily Morinoideae, 17 of the subfamily Linnaeoideae, nine of Zabelia, and four species of Viburnum and Sambucus as the outgroups. All the voucher information and GenBank accessions of each taxon are provided in Supplementary Table S1. A total of 83 genes, consisting of 79 protein-coding genes and four rRNA genes, were extracted from each plastome for phylogenetic analyses. All extracted sequences for each sample were aligned through MAFFT v7 (Katoh and Standley, 2013).
The phylogeny of the family Caprifoliaceae was inferred through ML (Maximum Likelihood) method and BI (Bayesian Inference) method. The ML tree of Caprifoliaceae was reconstructed by RAxML-NG (Kozlov et al., 2019) with the model of GTR+G, and the node support was evaluated by 1,000 bootstrap replicates. The BI tree was reconstructed using MrBayes v3.2 (Ronquist et al., 2012), and four Markov chains were run for 5,000,000 generations, and each 1,000 generations were used for tree sampling. The convergence was analyzed in Tracer v1.6 (Rambaut et al., 2018) for stationary distribution along with the effective size for each parameter. To ensure the stable state of each chain, the initial 25% of the trees were removed as burn-in, and the PP (posterior probabilities) were calculated. The remaining trees were utilized for building the BI tree with posterior probabilities.
Results
The general features of Weigela plastomes
With the Illumina sequencing platform, we produced an average of about 6Gb raw data for each sample. The coverage depth of these plastomes were estimated by mapping at more than 40×. The complete plastomes were 156,909~157,739 bp in length and presented a typical circular quadripartite structure. Each complete plastome comprised a pair of inverted repeat (IR) regions (23,592~24,957 bp), a larger single-copy (LSC) region (89,922~90,229 bp), and a small single-copy (SSC) region (17,668~20,429 bp). The circular maps of the four Weigela plastomes are shown in Supplementary Figure S1.
In total, 113 unique genes were annotated for each Weigela plastome, consisting of 79 protein-coding genes, 30 tRNAs, and four rRNAs. In addition, 14 genes including three protein-coding genes, four rRNAs, and seven tRNAs were duplicated within the IR regions. On account of the expansion of the IR region in some species of Weigela (W. japonica Thunb., W. florida, and W. floribunda), the ycf1 within the IRb region evolved into a pseudogene (Supplementary Figure S1). Among these genes, 60 genes are related to self-replication, and 45 genes are involved in photosynthesis (Supplementary Table S2). Of all these genes identified, only ycf3 contained two introns, whereas 10 protein-coding genes and six tRNA genes only contained one intron (Supplementary Table S4). The longest intron (2,558 bp), including the matK gene, are located in the trnK-UUU gene. The rps12 gene is a trans-spliced gene, with its 3’ end situated in the IR region and its 5’ end in the LSC region. The total GC content of these complete plastomes was 37.9%~38.0%, and the GC content of the IR region (42.8%~43.3%) exceeded that of the LSC region (36.3%~36.4%) and the SSC region (32.7%~33.1%).
Repeat sequences and SSR analysis
The repeat sequences of these four complete plastomes were investigated. The long repeat sequences greater than 30 bp were selected for analysis. From the four plastomes, we identified three types of repeats. There were 268 forward repeats, 128 palindromic repeats, and 867 tandem repeats, with a total of 1,263 long repeats. The number of tandem repeats was the highest among these, corresponding to 219 (W. floribunda), 220 (W. florida), and 214 (W. japonica var. sinica and W. subsessilis (Nakai) L.H.Bailey), while the number of palindromic repeats was the lowest, at 34 (W. floribunda), 33 (W. florida), 40 (W. japonica var. sinica), and 21 (W. subsessilis). Reverse and complementary repeats were not detected (Figure 2). Most of the long repeat sequences were mainly located in the noncoding areas, covering intergenic regions and intron regions. Several of the long repeat sequences were present in the shared genes, for instance accD, trnN, and ycf2 (Supplementary Table S3).
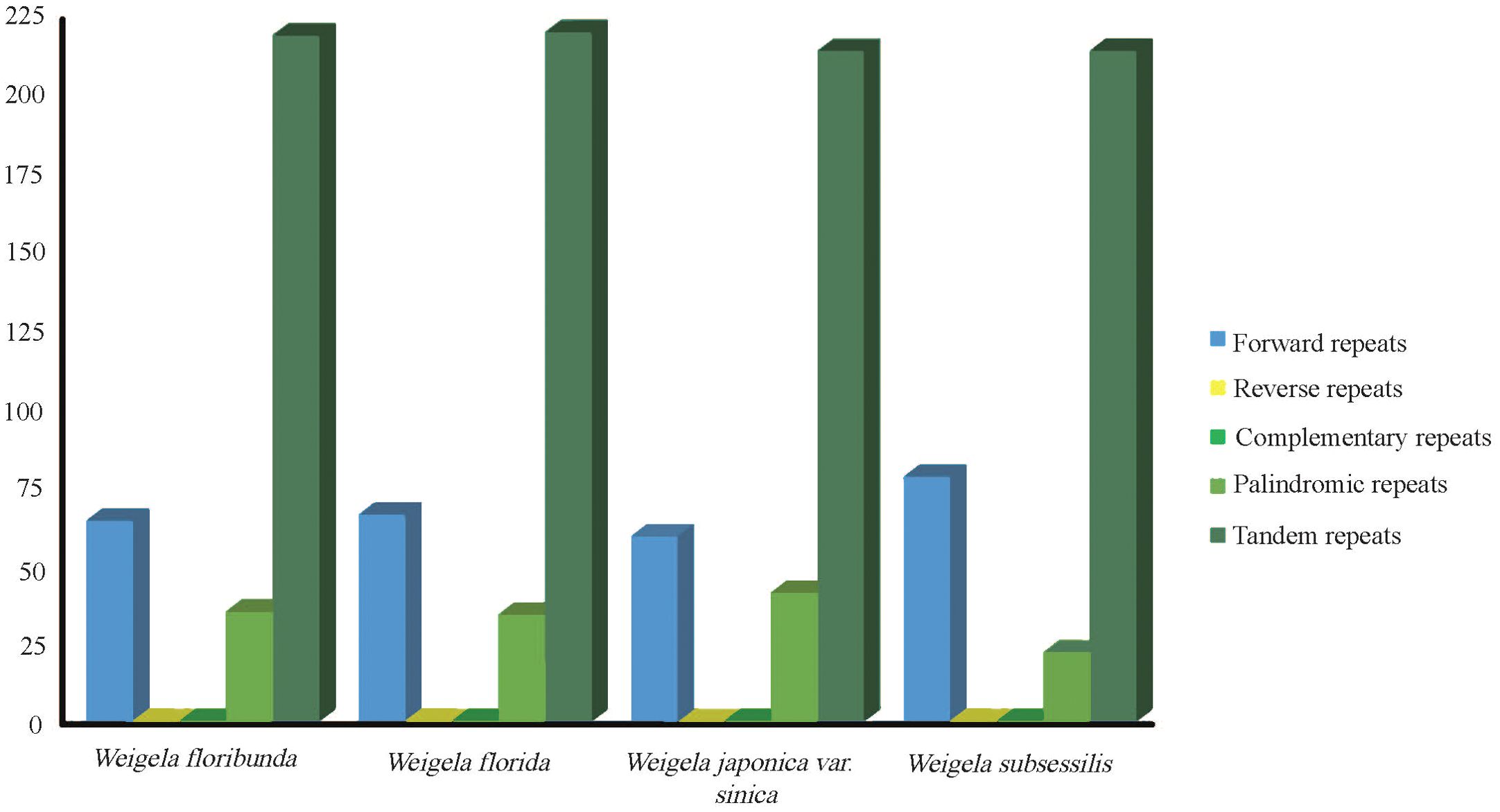
Figure 2. Analysis of repeated sequences in the plastomes of four Weigela species. Types and numbers of interspersed repeats within the four plastomes.
A total of 352 SSRs were found in these four plastomes, with the number of SSRs per species being 89 (W. subsessilis and W. florida), 88 (W. floribunda), and 86 (W. japonica var. sinica). Most of the SSRs were concentrated in the LSC region, accounting for 75% of the total, while the number of SSRs in the IR and SSC regions was relatively small, accounting for 16% and 9% of the total, respectively (Figures 3A, C). The most common types of repeats detected were mononucleotide repeat units, of which the A/T repeat units were the most common. Among the dinucleotide and tetranucleotide repeat units, AT/TA and AAAG/CTTT were the most frequent units respectively. Pentanucleotide repeats were absent from these four plastomes of Weigela, whereas a small number of repeats were detected in the hexanucleotide repeats, except for W. japonica var. sinica (Figures 3B, D). We also found several SSRs within the protein-coding gene regions, including rpoC1, trnG, and atpF (Supplementary Table S4).
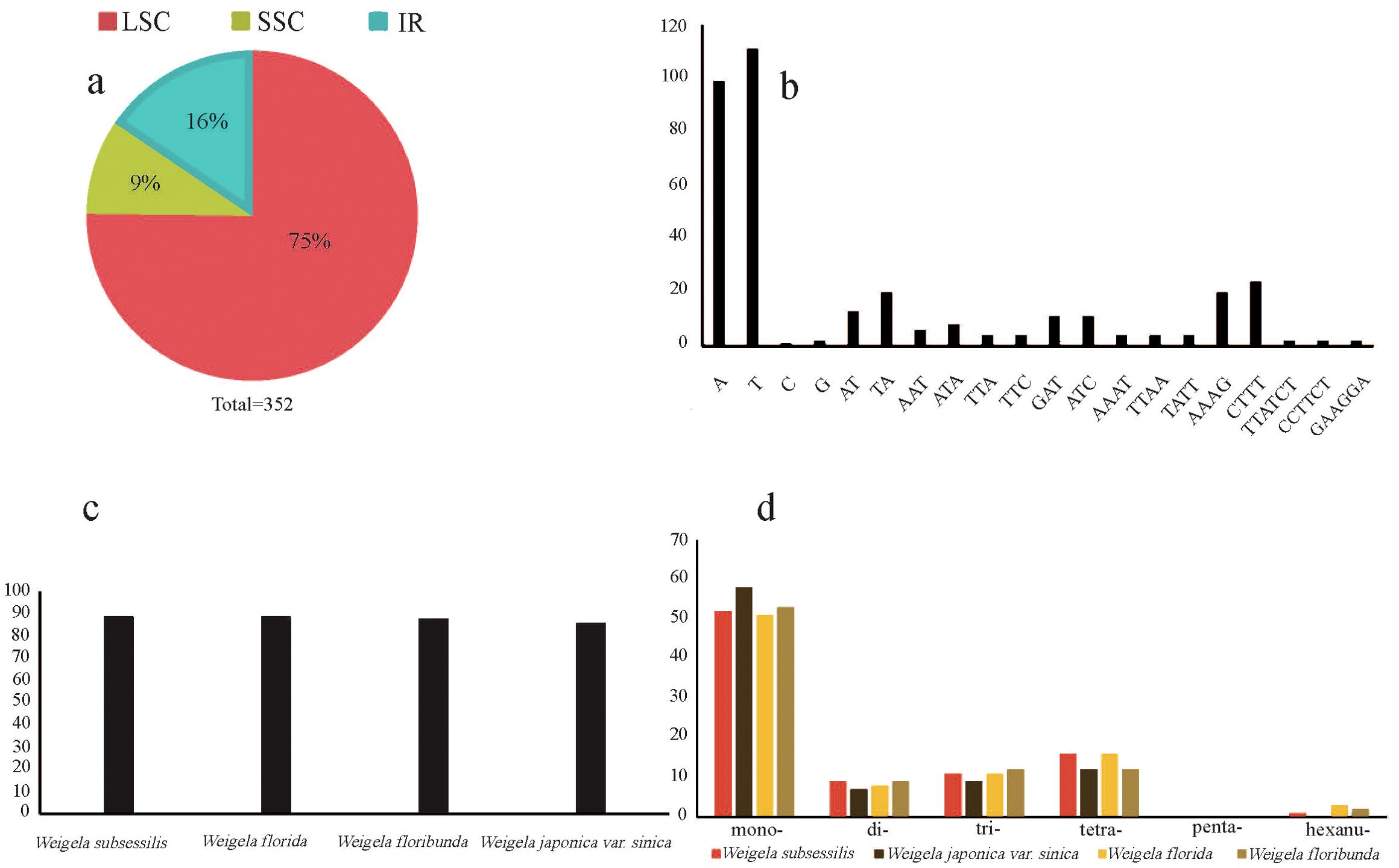
Figure 3. Frequency of the simple sequence repeat (SSR) sequences in the four Weigela plastomes. (A) The proportion of SSRs within the LSC, SSC, and IR regions; (B) Frequency of SSRs with the nucleotide motifs from di- to penta-; (C) The number of SSRs identified in the four Weigela species; (D) Number and type of SSRs.
Analysis of the plastome structure
Our comparative analysis of the Weigela plastomes showed that all four species have consistent gene content and very similar genome structures (Table 1, Supplementary Table S2, Supplementary Figure S1). All four plastomes showed a typical circular quadripartite structure, and the total genome size ranged from 156,909 bp (Weigela japonica var. sinica) to 157,739 bp (Weigela florida). The size of the LSC ranged from 89,922bp to 90,229 bp, and the size of the SSC was 17,668~20,429 bp. The four Weigela plastomes have consistent gene content and very similar GC content (37.9~38.0%) but differed by region. The GC content within the IR regions (42.8~43.3%) exceeded that of the LSC (36.3~36.4%) and SSC (32.7~33.1%) regions.
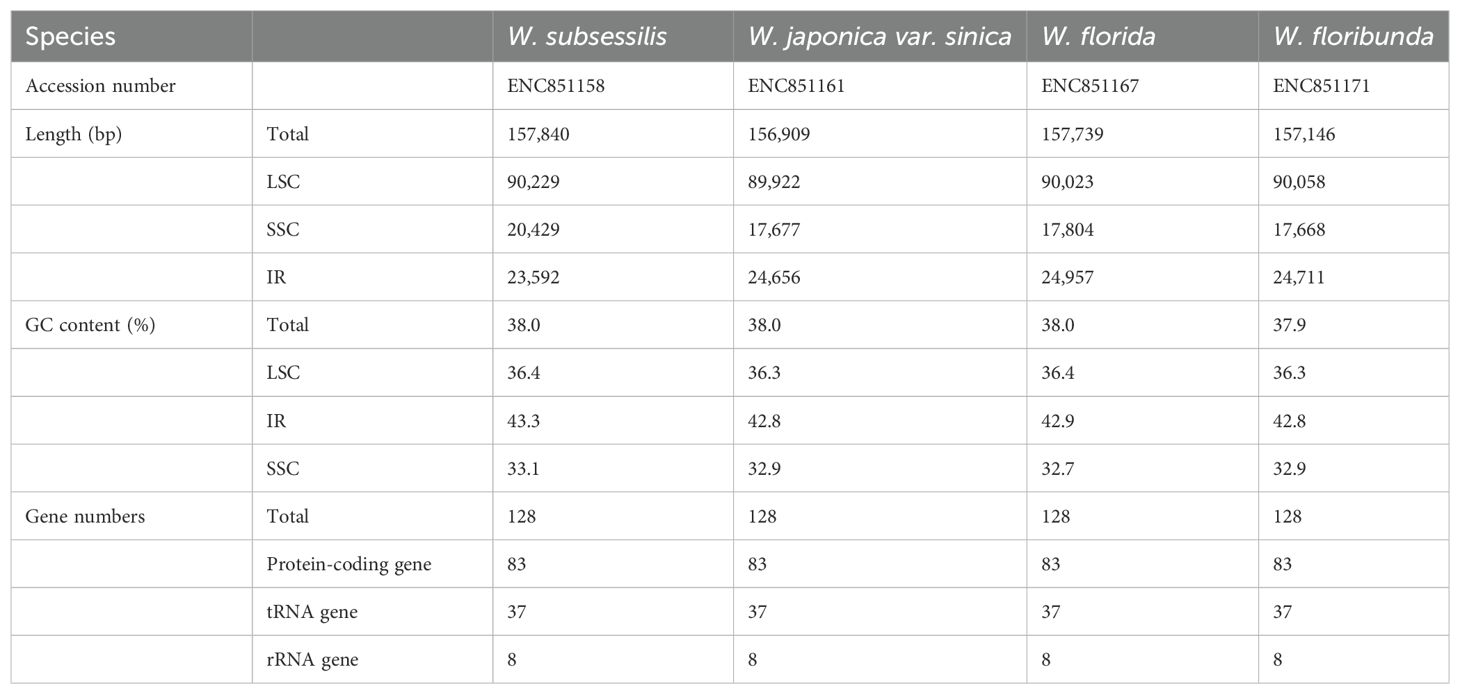
Table 1. Basic features of the plastome from the four species of Weigela.
Multiple sequence alignments indicated that there were no significant genomic rearrangements or large inversions in these four Weigela plastomes (Figure 4). These four plastomes of Weigela showed high conservation not only in gene order but also in basic features. As expected, the coding regions and inverted repeat (IR) regions were more conserved than the noncoding regions and single-copy (SC) regions. The intergenic spacer regions exhibited greater variability, such as trnH-psbA, trnR-ndhF, and trnN-ndhF. Moreover, only ycf1 displayed a high level of variation across all the coding regions.
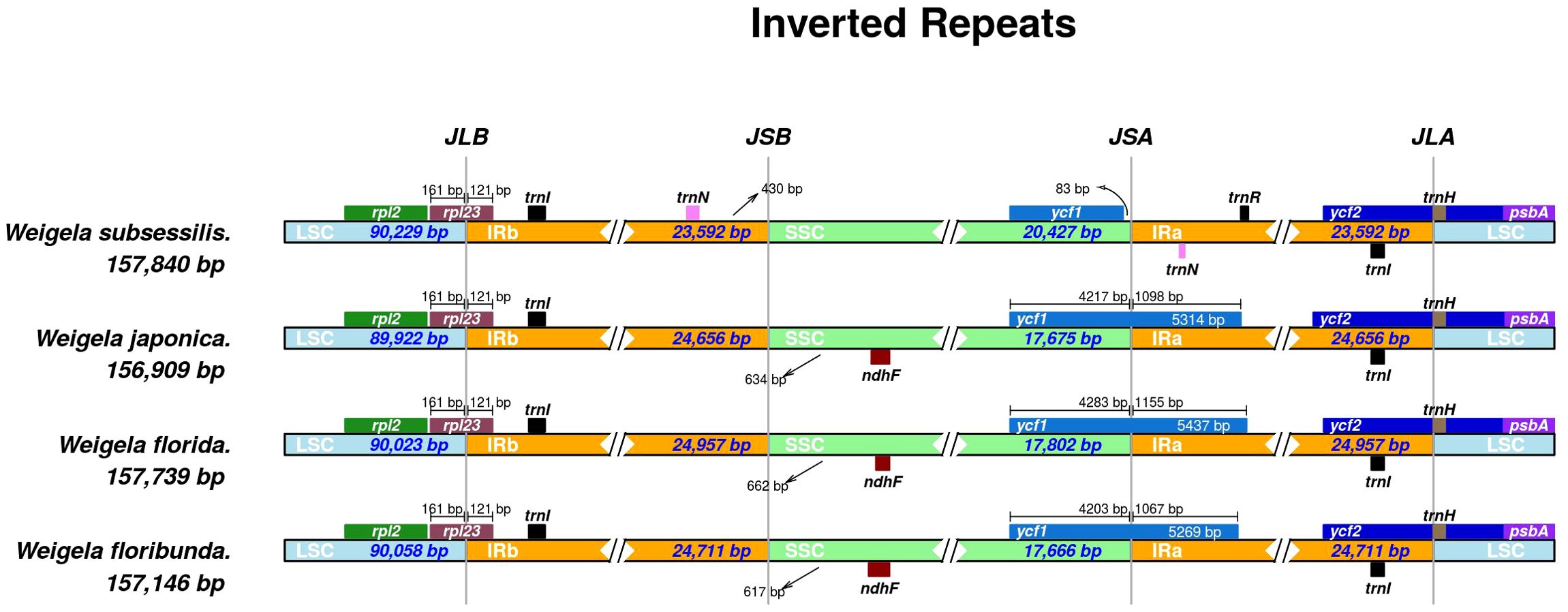
Figure 4. Comparison of the boundaries between LSC, SSC, and IR regions across the four Weigela species. The distance in the figure is not to scale.
Multiple sequence alignments from mVISTA indicated that the IR regions of the four Weigela plastomes were rather conserved (Table 2), and structural variation was only observed in the boundaries between SSC and IR regions (Figure 4). Among these four plastomes, there were two types of SSC/IR boundary identified. W. subsessilis belonged to a type in which ycf1 was located only in the SSC region, and 83 base pairs were identified between the ycf1 and IRa boundary in this species. The other three species displayed a different type of structure with the ycf1 gene spanning the boundaries between SSC and IRa regions. This result showed that the expansion of the IR regions caused a duplication of ycf1 in the plastomes of W. japonica, W. florida, and W. floribunda, by 1,089, 1,155, and 1,067 bp, respectively. Moreover, an intergenic region of 430 bp was found between the trnN and the boundaries the LSC/IRb in W. subsessilis. Hundreds of base pairs were identified between the ndhF and IRb boundary in three species, namely, W. japonica, W. florida, and W. floribunda, by 634, 662, and 617 bp, respectively. These results indicated that there were dynamic variations in the boundaries between the SSC/IRa and the IRb/SSC among these four species. The rpl23 gene crossed the LSC/IRb boundary in all the four plastomes with the length of 121 bp in the IRb region. The trnl and trnH genes were situated at the LSC/IRa boundary.
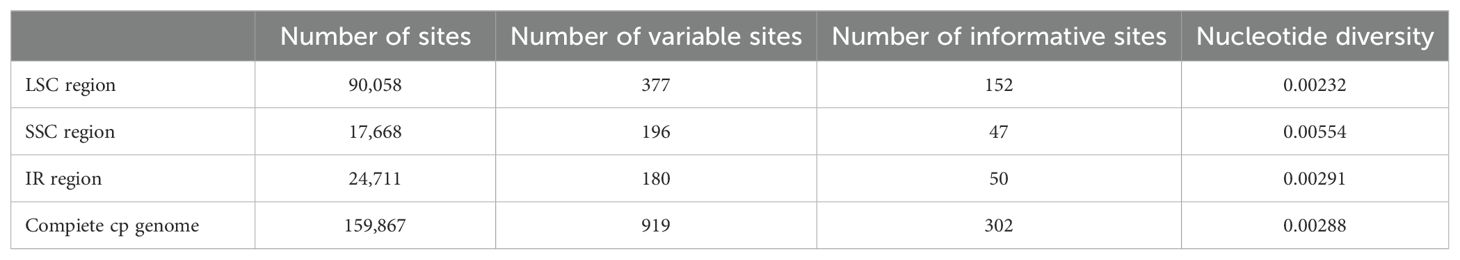
Table 2. Analyses of variable sites in plastomes from Weigela.
Plastome sequence divergence and barcode identification
To reveal the differences between the four Weigela species, we conducted a comprehensive analysis of the plastome sequences (Figure 5). We found that these plastomes of Weigela were highly similar, and no rearrangement had occurred. However, we detected some differences within the intergenic and intragenic regions among the plastomes in Weigela, especially in the LSC regions and the SSC regions (Figure 5). In view of this, there are a number of intergenic and intragenic areas for which DNA barcodes could be developed to differentiate Weigela species.
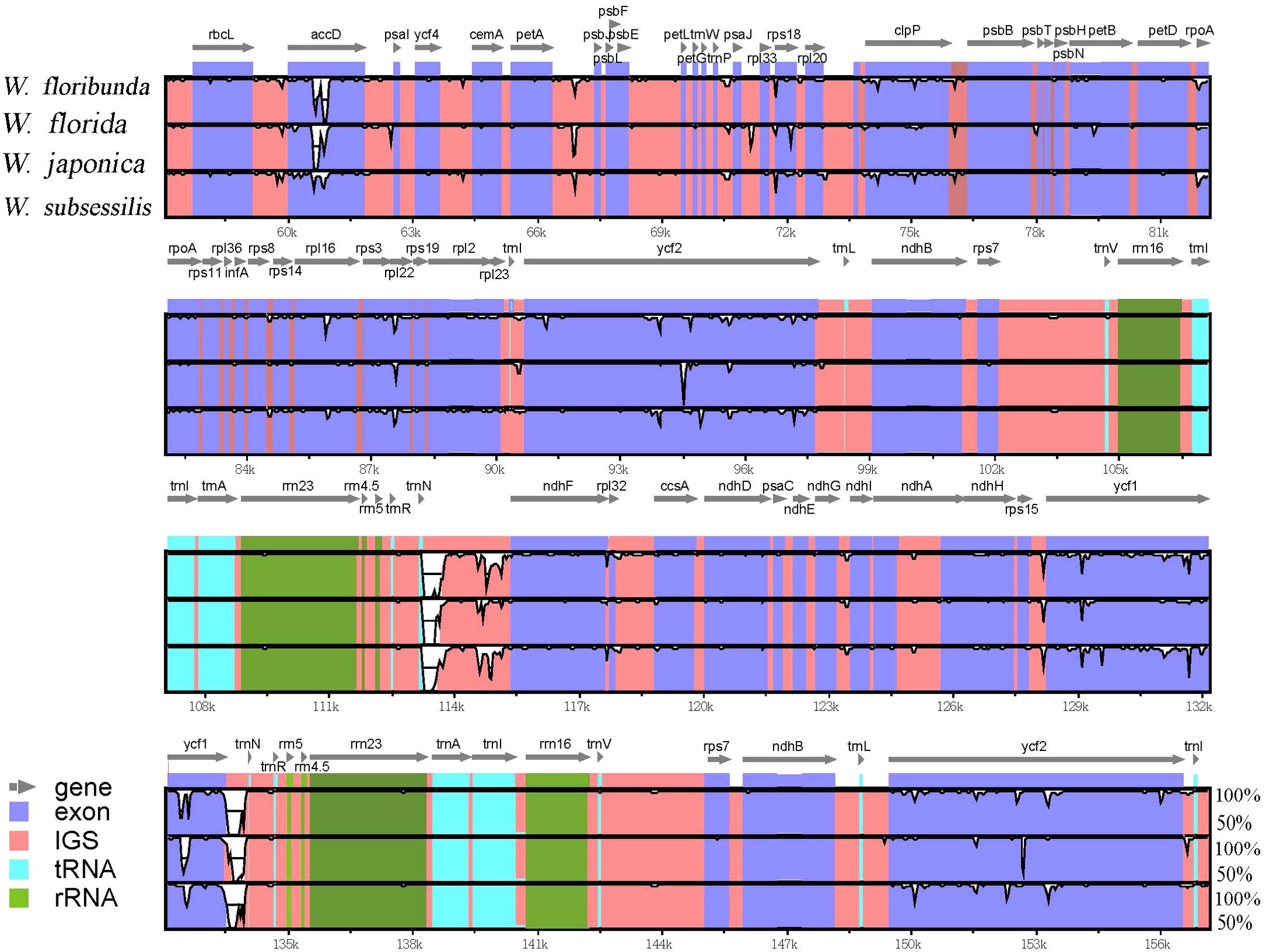
Figure 5. Sequence similarity analyses across the four Weigela plastomes generated through mVISTA. The vertical axis denotes the percentage of sequence identity, ranging from 50% to 100%. The horizontal axis corresponds to the coordinates within the chloroplast genome. Genome regions are color-coded to indicate protein-coding (exon), tRNAs or rRNAs, and intergenic regions. Genes are depicted by gray arrows.
To facilitate a deeper analysis of the mutational hotspots within the plastomes of Weigela, DnaSP v6.12.03 was employed to analyze the nucleotide diversity (Pi) across the alignment of the four plastomes (Figure 6, Table 3). The observed Pi values ranged from 0 to 0.12333. At the threshold of Pi > 0.02, three mutational hotspots (trnH-psbA, trnR-ndhF, and trnN-ndhF) were suitable as candidate barcodes for Weigela species. The analysis of the nucleotide diversity in protein-coding genes showed that at the threshold of Pi > 0.02, only ycf1 exhibited high nucleotide diversity and was deemed suitable for phylogenetic analysis.
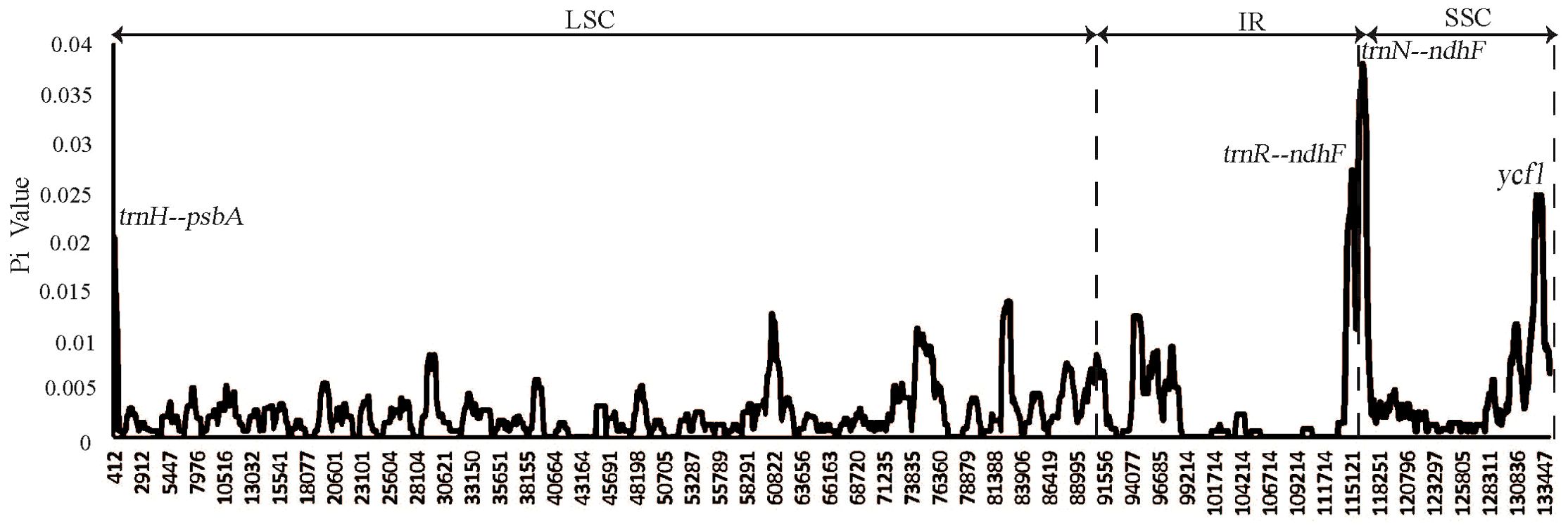
Figure 6. Nucleotide diversity (Pi) of plastomes among the four Weigela species. Four mutation hotspot areas (Pi > 0.002) were annotated. Pi values were calculated using an 800 bp sliding windows with 100 bp steps.
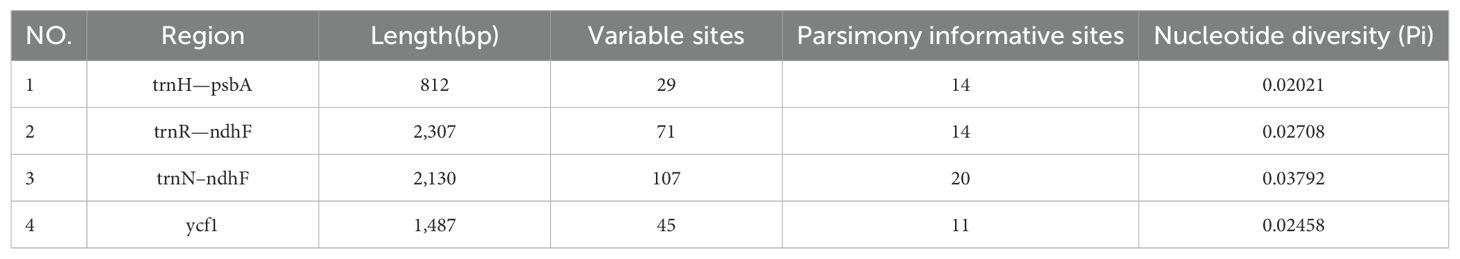
Table 3. Four hypervariable regions of Weigala.
Phylogenetic inference of the family Caprifoliaceae
The phylogenetic topologies of the Caprifoliaceae inferred through ML and BI methods based on 83 gene sequences were consistent (Figure 7). All species of Caprifoliaceae constituted a strongly supported monophyletic group. In addition, the Caprifoliaceae family could be divided into seven clades: Diervilloideae, Caprifolioideae, Dipsacoideae, Valerianoideae, Morinoideae, Zabelia, and Linnaeoideae. Diervilloideae was located at the base of the whole family, and was retrieved as sister to the rest of the members with robust support (bootstrap (BS) value = 100% for the ML tree and posterior probability (PP) = 1.00 for the BI tree). Caprifolioideae was retrieved as sister to the other five clades with strong support. Moreover, Zabelia and Morinoideae formed a clade and were sister to Linnaeoideae. Dipsacoideae was sister to Valerianoideae, and the two were sister to Morinoideae + Zabelia + Linnaeoideae. The phylogeny within Weigela showed that W. japonica and W. floribunda were sister and W. subsessilis and W. florida were sister, with all Weigela species forming a monophyletic group.
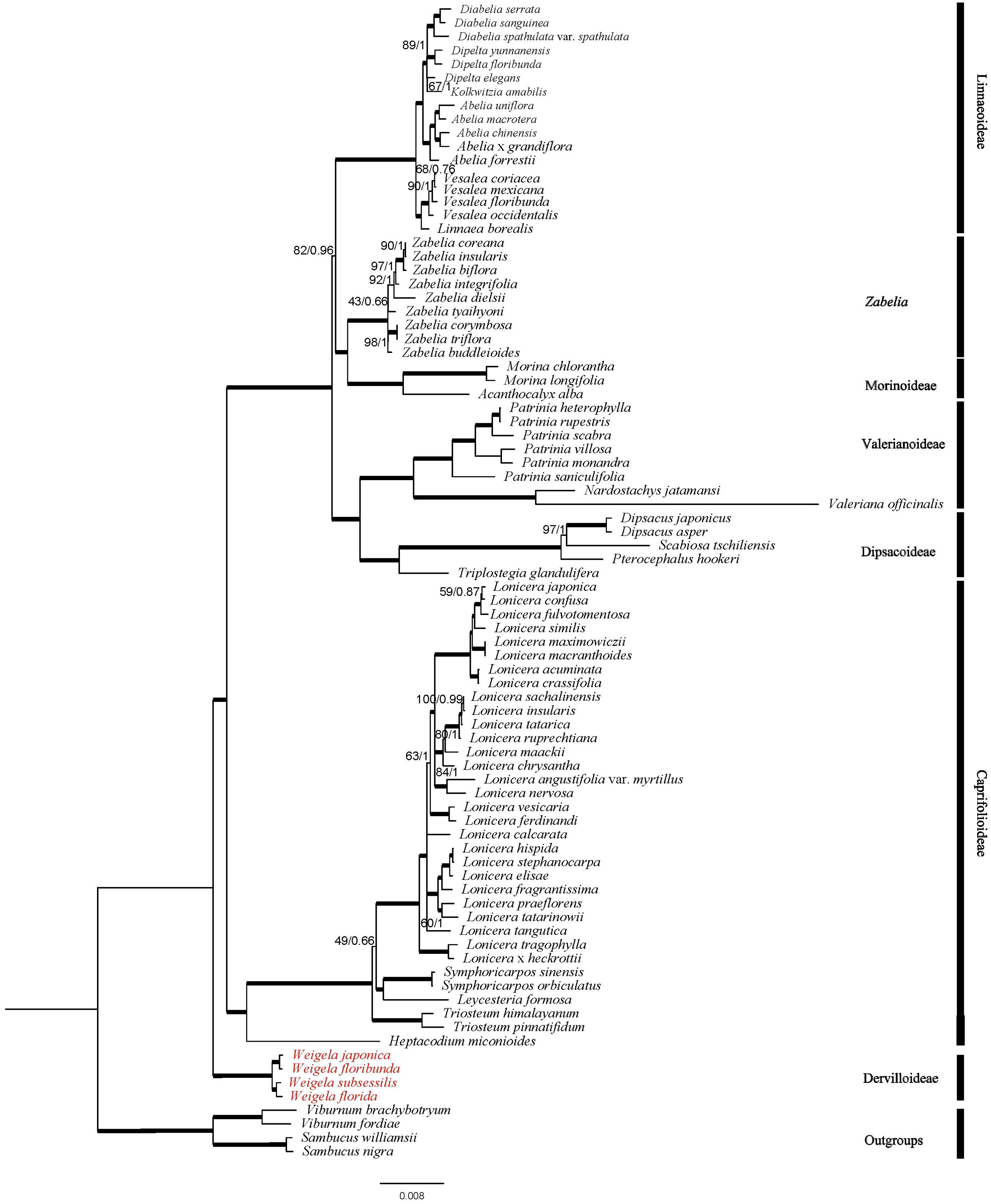
Figure 7. The phylogeny constructed by ML and BI methods based on 84 plastome genes. The numbers above the branches represent ML bootstrap support (BS) values and Bayesian posterior probabilities (PP) (100 BS/1.0 PP). Roots are thickened to indicate BS = 100 and PP = 1.
Discussion
In this study, we assembled four complete plastomes from W. japonica, W. floribunda, W. subsessilis, W. florida, and compared and analyzed the structural characteristics of these genomes. The four plastomes exhibited a high degree of conservation, with a typical quadripartite structure comprising one LSC, one SSC, and two IR regions. This result was consistent with the previous reports on plastomes of Caprifoliaceae (Fan et al., 2018; Peng et al., 2019; Park et al., 2021). The plastome size of Weigela species was 157,790~157,870 bp, which is not much different from other species of Caprifoliaceae (Fan et al., 2018; Peng et al., 2019).
The contractions or expansions at the boundaries of the IR regions are widely recognized as the primary processes leading to genome size variation (Chen et al., 2023; Zhang et al., 2022; Jia et al., 2024). Our results showed that the IR region of W. subsessilis was much shorter than that of the other three species, indicating that the expansion of IR regions caused a duplication of ycf1 in the plastomes of W. japonica, W. florida, and W. floribunda. There were two types of SSC/IR boundary in the four species (Figure 4). W. subsessilis had a genomic structure in which ycf1 was located only in the SSC region, and 83 bp were identified between the ycf1 and IRa boundary in this species. The other three species had a different genomic structure, with ycf1 located at the boundaries of the SSC and IRa regions. We also evaluated the variation in IRa and LSC junction regions, finding that the distributions and locations of genes within these regions displayed a high degree of variability. In consequence, changes in the SC/IR boundary may be the primary catalyst for the plastome size variation within Weigela species, especially in the IR region.
In a similar manner to the plastomes commonly found in angiosperms (Wicke et al., 2011; Daniell et al., 2016; Zhao et al., 2021), the plastomes of Weigela species exhibited a high similarity with other members of Caprifoliaceae (Yao et al., 2019; Wang et al., 2020). Nevertheless, some regions within these plastomes displayed a notable level of sequence variation. The mVISTA results showed that the sequence divergence in the IR region was lower compared to the LSC and the SSC regions (Figure 5). This result was attributed to the correction of sequence during gene replication and transcription of the two copies (Luo et al., 2021). Another reason contributing to the conservation of the IR regions is their crucial role in ensuring the structural stability of the plastome (Xiong et al., 2021; Wen et al., 2021; Park et al., 2021). From the results of the mVISTA analysis, we identified notable sequence variations in several intergenic regions and genes, including trnH-psbA, trnR-ndhF, trnN-ndhF, and ycf1 (Figure 6). These regions with high sequence divergence have previously been reported in some other closely related lineages within Caprifoliaceae. For instance, Fan et al. (2018). compared eight plastomes of Caprifoliaceae species and identified 23 variant hotspot regions (containing genes and intergenic spacers) as candidate DNA barcodes that might be used as markers for phylogenetic analyses and species identification within the family Caprifoliaceae. These variant hotspot regions are potentially useful for phylogenetic analyses and interspecific divergence in the Caprifoliaceae. Our new findings also support the LSC region having higher sequence divergence than the IR and SSC regions (Wang et al., 2022a; Dong et al., 2022).
Repeat sequences play a pivotal role in the genomic rearrangement and structural stabilization of plastomes (Deng et al., 2018; Chen et al., 2023; Skuza et al., 2022). Such repeats are also important in understanding phylogenetic and biogeographic relationships, as well as population genetic characteristics, among species (Chen et al., 2023). In this study, a total of 1,263 long repeats (30-190 bp) without reverse and complementary repeats, and a total of 352 SSRs, were identified from the four Weigela plastomes (Figure 2). Among these long repeats, the number of tandem repeats was the highest, and the number of palindromic repeats was the lowest. In particular, most long repeat sequences were mainly located in noncoding areas, and a few long repeat sequences were found in shared genes. These repeats exhibited patterns similar to those previously reported, which are crucial components in the evolutionary dynamics of these plastomes (Sun et al., 2022; Wei et al., 2021).
Owing to their characteristics of abundance, maternal inheritance, and haploid nature, plastome SSRs (cpSSRs) have mainly been used for analysis of population genetic variation and gene flow (Xue et al., 2021; Ebert and Peakall, 2009), and are usually considered to be informative markers. The application and significance of cpSSR markers in other angiosperms have been frequently reported (Huang et al., 2018). In addition, we analyzed the numbers, types, and distribution of the cpSSRs in four Weigela plastomes. W. floribunda exhibited the highest count of cpSSRs (69), whereas W. subsessilis had the fewest (20). Consistent with the most recent results (Wang et al., 2022a; Zhou et al., 2018), we also determined that the mononucleotide-type SSRs is predominant in the plastome, with a bias toward A/T nucleotides, which corresponds with an A/T-rich plastome.
In recent years, studies increasingly demonstrate the unique advantages of plastome sequences in inferring phylogenetic relationships across various taxonomic levels (Guo et al., 2021; Xu and Wang, 2020). Based on the complete plastome sequences, researchers have successfully resolved numerous phylogenetic questions at deep-node levels of taxonomy, for example, in identifying the phylogenetic relationships among different angiosperms (Xiong et al., 2023) or among seven species of Papaveraceae (Wang et al., 2022a). This approach could lead to a better understanding of some complex evolutionary relationships in plant lineages. Meanwhile, the use of fewer plastome genes in analyses can only resolve shallow phylogenetic problems to a certain extent. Previously, Li et al. (1999). reconstructed the internal phylogenetic relationships of Hamamelidaceae through the chloroplast gene matK. The results indicated that the plastome gene resolved the phylogenetic relationships among subfamilies/families within Hamamelidaceae well, but failed to resolve the phylogeny at the species level. In this study, we constructed the phylogeny of the Caprifoliaceae based on whole plastomes. The results showed that Weigela belongs to Diervilloideae, which is an early diverging lineage in the family Caprifoliaceae, and was retrieved as sister to the rest of the members with strong support. Zabelia and Morinoideae formed a clade and were sister to Linnaeoideae. Dipsacoideae was sister to Valerianoideae, and the two were retrieved as sister groups to Morinoideae + Zabelia + Linnaeoideae. This is consistent with the results inferred from transcriptomes and plastomes (Li et al., 2024). Furthermore, this phylogeny also confirmed the phylogenetic relationships of the four studied species in Weigela, and the support value of each clade was significantly higher than the previous phylogenies based on ITS sequences and cpDNA regions (Kim and Kim, 1999; Meeler, 2018). In addition, the short branch length of the four Weigela species indicated that species distributed in the northeast Asia might have undergone a rapid radiation (Chang, 1997).
Conclusions
In conclusion, we newly sequenced and assembled four complete plastomes of Weigela, and conducted comparative analysis between them. On this basis, we also reconstructed the phylogeny of the whole Caprifoliaceae family with the help of 75 published plastomes from other Caprifoliaceae genera. Our comparative plastomes analysis has shown that the plastomes of Weigela are highly conserved. Moreover, only a few regions of high sequence divergence were detected throughout the whole plastome. The results of the phylogenetic analysis provide a stable phylogenetic framework for Caprifoliaceae and the phylogenetic position of Weigela. Furthermore, the phylogenetic relationships of the four species of Weigela were confirmed.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
LW: Conceptualization, Funding acquisition, Project administration, Writing – original draft, Writing – review & editing. FL: Formal analysis, Investigation, Methodology, Visualization, Writing – review & editing. KZ: Data curation, Formal analysis, Investigation, Visualization, Writing – review & editing. JY: Investigation, Methodology, Validation, Writing – review & editing. HS: Data curation, Investigation, Resources, Writing – review & editing. XC: Methodology, Software, Visualization, Writing – review & editing. WD: Supervision, Methodology, Project administration, Writing – review & editing. EL: Supervision, Conceptualization, Validation, Writing – review & editing. NW: Conceptualization, Project administration, Supervision, Writing – review & editing.
Ethics statement
The samples utilized in this research were gathered from an unprotected region and adhered to the regulations stipulated by the People’s Republic of China on the conservation of wild plants, the IUCN Policy Statement on conducting studies involving endangered species, and the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES).
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research received supports from the Science and Technology Research Project of Henan Province (202102110232) and Doctoral Research Foundation of Henan University of Science and Technology (13480079).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1487725/full#supplementary-material
Supplementary Figure S1 | Gene map of four Weigela species plastome. Genes located outside the circle are transcribed counterclockwise, while genes inside the circle are transcribed clockwise. The dark gray area and light gray area of the inner circle represent the ratio of GC content to AT content in the genome, respectively. Different colored blocks represent genes from different functional groups.
References
Amiryousefi, A., Hyvönen, J., Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bell, C. D., Edwards, E. J., Kim, S.-T., Donoghue, M. J. (2001). Dipsacales phylogeny based on chloroplast dna sequences. Harv. Pap. Bot. 6, 481–499. doi: 10.2307/41761758
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Ostell, J., Pruitt, K. D., et al. (2018). GenBank. Nucleic Acids Res. 46, D41–D47. doi: 10.1093/nar/gkx1094
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Chang, C.-S. (1997). Flavonoid chemistry of weigela (Caprifoliaceae) in korea. J. Plant Res. 110, 275–281. doi: 10.1007/BF02509316
Chen, J., Wang, F., Zhou, C., Ahmad, S., Zhou, Y., Li, M., et al. (2023). Comparative phylogenetic analysis for aerides (Aeridinae, orchidaceae) based on six complete plastid genomes. Int. J. Mol. Sci. 24, (15). doi: 10.3390/ijms241512473
Claude, S. J., Park, S., Park, S. (2022). Gene loss, genome rearrangement, and accelerated substitution rates in plastid genome of Hypericum ascyron (Hypericaceae). BMC Plant Biol. 22, 135. doi: 10.1186/s12870-022-03515-x
Daniell, H., Lin, C.-S., Yu, M., Chang, W.-J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 1–29. doi: 10.1186/s13059-016-1004-2
Deng, C.-Y., Xin, G.-L., Zhang, J.-Q., Zhao, D.-M. (2018). Characterization of the complete chloroplast genome of Dalbergia hainanensis (Leguminosae), a vulnerably endangered legume endemic to China. Conserv. Genet. Resour. 11, 105–108. doi: 10.1007/s12686-017-0967-y
Dong, W., Liu, Y., Li, E., Xu, C., Sun, J., Li, W., et al. (2022). Phylogenomics and biogeography of Catalpa (Bignoniaceae) reveal incomplete lineage sorting and three dispersal events. Mol. Phylogenet Evol. 166, 107330. doi: 10.1016/j.ympev.2021.107330
Dong, W., Liu, Y., Xu, C., Gao, Y., Yuan, Q., Suo, Z., et al. (2021). Chloroplast phylogenomic insights into the evolution of Distylium (Hamamelidaceae). BMC Genomics 22, 293. doi: 10.1186/s12864-021-07590-6
Donoghue, M. J., Bell, C. D., Winkworth, R. C. (2003). The evolution of reproductive characters in dipsacales. Int. J. Plant Sci. 164, S453–S464. doi: 10.1086/376874
Donoghue, M. J., Eriksson, T., Reeves, P. A., Olmstead, R. G. (2001). Phylogeny and phylogenetic taxonomy of dipsacales, with special reference to Sinadoxa, and Tetradoxa (Adoxaceae). Harv. Pap. Bot. 6, 459–479.
Ebert, D., Peakall, R. (2009). Chloroplast simple sequence repeats (cpSSRs): technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour 9, 673–690. doi: 10.1111/j.1755-0998.2008.02319.x
Fan, W. B., Wu, Y., Yang, J., Shahzad, K., Li, Z. H. (2018). Comparative chloroplast genomics of dipsacales species: insights into sequence variation, adaptive evolution, and phylogenetic relationships. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00689
Greiner, S., Lehwark, P., Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Guo, Y. Y., Yang, J. X., Bai, M. Z., Zhang, G. Q., Liu, Z. J. (2021). The chloroplast genome evolution of Venus slipper (Paphiopedilum): IR expansion, SSC contraction, and highly rearranged SSC regions. BMC Plant Biol. 21, 248. doi: 10.1186/s12870-021-03053-y
Huang, L. S., Sun, Y. Q., Jin, Y., Gao, Q., Hu, X. G., Gao, F. L., et al. (2018). Development of high transferability cpSSR markers for individual identification and genetic investigation in Cupressaceae species. Ecol. Evol. 8, 4967–4977. doi: 10.1002/ece3.4053
Jacobs, B., Lens, F., Smets, E. (2009). Evolution of fruit and seed characters in the Diervilla and Lonicera clades (Caprifoliaceae, Dipsacales). Ann. Bot. 104, 253–276. doi: 10.1093/aob/mcp131
Jia, M., Wang, J., Cao, D., Jiang, C., Li, W., Tembrock, L. R., et al. (2024). The pan-plastome of Hemerocallis citrina reveals new insights into the genetic diversity and cultivation history of an economically important food plant. BMC Plant Biol. 24, 44. doi: 10.1186/s12870-023-04668-z
Jin, J.-J., Yu, W.-B., Yang, J.-B., Song, Y., dePamphilis, C. W., Yi, T.-S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kim, Y.-D., Kim, S.-H. (1999). Phylogeny of weigea and diewilla (Caprifoliaceae) based on nuclear rDNA ITS sequences: biogeographic and taxonomic implications. J. Plant Res. 112, 331–341. doi: 10.1007/PL00013887
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Li, J., Bogle, A. L., Klein, A. S. (1999). Phylogenetic relationships in the Hamamelidaceae: Evidence from the nucleotide sequences of the plastid genematK. Plant Systematics Evol. 218, 205–219. doi: 10.1007/BF01089228
Li, J., Wang, S., Jing, Y., Wang, L., Zhou, S. (2013). A modified CTAB protocol for plant DNA extraction. Chin. Bull. Bot. 48, 72–78.
Li, Y., Sun, Z., Jin, Z., Li, J. (2024). Genome sequencing provides insights into Caprifoliaceae genome evolution and the mechanism underlying second blooming phenomenon in Heptacodium miconioides. Hortic. Plant J. 10, 897–910. doi: 10.1016/j.hpj.2022.11.008
Liang, X.-Q., Li, Y., Kvaček, Z., Wilde, V., Li, C.-S. (2013). Seeds of Weigela (Caprifoliaceae) from the Early Miocene of Weichang, China and the biogeographical history of the genus. Taxon 62, 1009–1018. doi: 10.12705/625.20
Liu, X., Bai, Y., Wang, Y., Chen, Y., Dong, W., Zhang, Z. (2023). Complete chloroplast genome of hypericum perforatum and dynamic evolution in hypericum (Hypericaceae). Int. J. Mol. Sci. 24, (22). doi: 10.3390/ijms242216130
Luo, Y., He, J., Lyu, R., Xiao, J., Li, W., Yao, M., et al. (2021). Comparative analysis of complete chloroplast genomes of 13 species in epilobium, circaea, and chamaenerion and insights into phylogenetic relationships of onagraceae. Front. Genet. 12. doi: 10.3389/fgene.2021.730495
Mayor, C., Brudno, M., Schwartz, J. R., Poliakov, A., Rubin, E. M., Frazer, K. A., et al. (2000). VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16, 1046–1047. doi: 10.1093/bioinformatics/16.11.1046
Meeler, H. E. (2018). Taxonomy and phylogeny of the flowering plant genus Diervilla (Diervillaceae). North Carolina, Western Carolina University.
Park, S., Jun, M., Park, S., Park, S. (2021). Lineage-Specific Variation in IR Boundary Shift Events, Inversions, and Substitution Rates among Caprifoliaceae s.l. (Dipsacales) Plastomes. Int. J. Mol. Sci. 22, (19). doi: 10.3390/ijms221910485
Peng, F.-F., Chen, L., Yang, Q., Tian, B., Liu, Z.-L. (2019). The complete chloroplast genome of Dipelta yunnanensis (Caprifoliaceae), a vulnerable plant in China. Mitochondrial DNA Part B 4, 515–516. doi: 10.1080/23802359.2018.1551084
Qu, X.-J., Moore, M. J., Li, D.-Z., Yi, T.-S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15, 50. doi: 10.1186/s13007-019-0435-7
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., Suchard, M. A. (2018). Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Skuza, L., Gastineau, R., Sielska, A. (2022). The complete chloroplast genome of Secale sylvestre (Poaceae: Triticeae). J. Appl. Genet. 63, 115–117. doi: 10.1007/s13353-021-00656-x
Sun, Q., Morales-Briones, D., Wang, H., Landis, J., Wen, J., Wang, H.-F. (2022). Phylogenomic analyses of the East Asian endemic Abelia (Caprifoliaceae) shed insights into the temporal and spatial diversification history with widespread hybridization. Ann. Bot. 129, 201–216. doi: 10.1101/2021.04.13.439739
Wang, L., Li, F., Wang, N., Gao, Y., Liu, K., Zhang, G., et al. (2022a). Characterization of the Dicranostigma leptopodum chloroplast genome and comparative analysis within subfamily Papaveroideae. BMC Genomics 23, 794. doi: 10.1186/s12864-022-09049-8
Wang, H. X., Liu, H., Moore, M. J., Landrein, S., Liu, B., Zhu, Z. X., et al. (2020). Plastid phylogenomic insights into the evolution of the Caprifoliaceae s.l. (Dipsacales). Mol. Phylogenet Evol. 142, 106641. doi: 10.1016/j.ympev.2019.106641
Wang, H. X., Morales-Briones, D. F., Moore, M. J., Wen, J., Wang, H. F. (2021). A phylogenomic perspective on gene tree conflict and character evolution in Caprifoliaceae using target enrichment data, with Zabelioideae recognized as a new subfamily. J. Systematics Evol. 59, 897–914. doi: 10.1111/jse.12745
Wang, L., Yang, W., Zhang, X., Zhang, D., Zhang, G. (2022b). Insights into the evolutionary history and taxonomic status of Sinopteris (Pteridaceae). Mol. Phylogenet Evol. 180, 107672. doi: 10.1016/j.ympev.2022.107672
Wei, R., Yang, J., He, L.-J., Liu, H.-M., Hu, J.-Y., Liang, S.-Q., et al. (2021). Plastid phylogenomics provides novel insights into the infrafamilial relationship of Polypodiaceae. Cladistics. 37, 717–727. doi: 10.1111/cla.12461
Wen, F., Wu, X., Li, T., Jia, M., Liu, X., Liao, L. (2021). The complete chloroplast genome of Stauntonia chinensis and compared analysis revealed adaptive evolution of subfamily Lardizabaloideae species in China. BMC Genomics 22, 161. doi: 10.1186/s12864-021-07484-7
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Muller, K. F., Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Xiong, Q., Hu, Y., Lv, W., Wang, Q., Liu, G., Hu, Z. (2021). Chloroplast genomes of five Oedogonium species: genome structure, phylogenetic analysis and adaptive evolution. BMC Genomics 22, 707. doi: 10.1186/s12864-021-08006-1
Xiong, C., Huang, Y., Li, Z., Wu, L., Liu, Z., Zhu, W., et al. (2023). Comparative chloroplast genomics reveals the phylogeny and the adaptive evolution of Begonia in China. BMC Genomics 24, 648. doi: 10.1186/s12864-023-09563-3
Xu, X., Wang, D. (2020). Comparative chloroplast genomics of corydalis species (Papaveraceae): evolutionary perspectives on their unusual large scale rearrangements. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.600354
Xue, C., Geng, F. D., Li, J. J., Zhang, D. Q., Gao, F., Huang, L., et al. (2021). Divergence in the Aquilegia ecalcarata complex is correlated with geography and climate oscillations: Evidence from plastid genome data. Mol. Ecol. 30, 5796–5813. doi: 10.1111/mec.16151
Yamada, T., Maki, M. (2012). Impact of geographical isolation on genetic differentiation in insular and mainland populations of Weigela coraeensis (Caprifoliaceae) on Honshu and the Izu Islands. J. Biogeography 39, 901–917. doi: 10.1111/j.1365-2699.2011.02634.x
Yao, G., Jin, J. J., Li, H. T., Yang, J. B., Mandala, V. S., Croley, M., et al. (2019). Plastid phylogenomic insights into the evolution of Caryophyllales. Mol. Phylogenet Evol. 134, 74–86. doi: 10.1016/j.ympev.2018.12.023
Zhang, G., Zhang, X., Chen, Z., Liu, H., Yang, W. (2007). First insights in the phylogeny of Asian cheilanthoid ferns based on sequences of two chloroplast markers. TAXON 56, 369–378. doi: 10.1002/tax.562009
Zhang, H. J., Zhang, X., Landis, J. B., Sun, Y. X., Sun, J., Kuang, T. H., et al. (2022). Phylogenomic and comparative analyses of Rheum (Polygonaceae, Polygonoideae). J. Systematics Evol. 60, 1229–1240. doi: 10.1111/jse.12814
Zhao, C., Xu, W., Huang, Y., Sun, Q., Wang, B., Chen, C., et al. (2021). Chloroplast genome characteristics and phylogenetic analysis of the medicinal plant Blumea balsamifera (L.) DC. Genet. Mol. Biol. 44, e20210095. doi: 10.1590/1678-4685-GMB-2021-0095
Keywords: plastome phylogenetics, phylogeny, Weigela, Caprifoliaceae, maternal inheritance
Citation: Wang L, Li F, Zhao K, Yang J, Sun H, Cui X, Dong W, Li E and Wang N (2024) Comparative plastomes sheds light on phylogeny of Weigela. Front. Plant Sci. 15:1487725. doi: 10.3389/fpls.2024.1487725
Received: 28 August 2024; Accepted: 04 October 2024;
Published: 29 October 2024.
Edited by:
Zhiqiang Wu, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Yan-Yan Liu, Henan Agricultural University, ChinaShenglong Kan, Weihai, China
Luke R. Tembrock, Colorado State University, United States
Copyright © 2024 Wang, Li, Zhao, Yang, Sun, Cui, Dong, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Wang, d2xoYXVzdEAxMjYuY29t; Enze Li, bGllbnplQGJqZnUuZWR1LmNu; Ning Wang, NDc1NjYyNjI4QHFxLmNvbQ==
†These authors share first authorship