 Zahid Khorshid Abbas1
Zahid Khorshid Abbas1 Arwa Abdulkreem Al-Huqail2*Aesha H. Abdel Kawy3Rabab A. Abdulhai4Doha A. Albalawi1,5Manal Abdullah AlShaqhaa6Moodi Saham Alsubeie7Doaa Bahaa Eldin Darwish1Ahmed Ali Abdelhameed8Fathia A. Soudy9Rania M. Makki10Maha Aljabri11
Arwa Abdulkreem Al-Huqail2*Aesha H. Abdel Kawy3Rabab A. Abdulhai4Doha A. Albalawi1,5Manal Abdullah AlShaqhaa6Moodi Saham Alsubeie7Doaa Bahaa Eldin Darwish1Ahmed Ali Abdelhameed8Fathia A. Soudy9Rania M. Makki10Maha Aljabri11 Nadiah Al-Sulami10
Nadiah Al-Sulami10 Mohammed Ali12
Mohammed Ali12 Muhammad Zayed13*
Muhammad Zayed13*- 1Department of Biology, Faculty of Sciences, University of Tabuk, Tabuk, Saudi Arabia
- 2Department of Biology, College of Science, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia
- 3Plant Ecophysiology Unit, Plant Ecology and Range Management Department, Desert Research Center, Cairo, Egypt
- 4Botany Department, Faculty of Women, Ain Shams University, Cairo, Egypt
- 5Biodiversity Genomics Unit, Faculty of Science, University of Tabuk, Tabuk, Saudi Arabia
- 6Department of Biology, College of Science, King Khalid University, Abha, Saudi Arabia
- 7Biology Department, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh, Saudi Arabia
- 8Agricultural Botany Department (Genetics), Faculty of Agriculture, Al-Azhar University, Assuit, Egypt
- 9Genetics and Genetic Engineering Department, Faculty of Agriculture, Benha University, Moshtohor, Egypt
- 10Department of Biological Sciences, Faculty of Science, King Abdulaziz University (KAU), Jeddah, Saudi Arabia
- 11Department of Biology, Faculty of Science, Umm Al-Qura University, Makkah, Saudi Arabia
- 12Maryout Research Station, Genetic Resources Department, Desert Research Center, Cairo, Egypt
- 13Department of Botany and Microbiology, Faculty of Science, Menoufia University, Shebin El-Kom, Egypt
Introduction: Carbohydrate compounds serve multifaceted roles, from energy sources to stress protectants, found across diverse organisms including bacteria, fungi, and plants. Despite this broad importance, the molecular genetic framework underlying carbohydrate biosynthesis pathways, such as starch, sucrose, and glycolysis/gluconeogenesis in Salvia guaranitica, remains largely unexplored.
Methods: In this study, the Illumina-HiSeq 2500 platform was used to sequence the transcripts of S. guaranitica leaves, generating approximately 8.2 Gb of raw data. After filtering and removing adapter sequences, 38 million reads comprising 210 million high-quality nucleotide bases were obtained. De novo assembly resulted in 75,100 unigenes, which were annotated to establish a comprehensive database for investigating starch, sucrose, and glycolysis biosynthesis. Functional analyses of glucose-6-phosphate isomerase (SgGPI), trehalose-6-phosphate synthase/phosphatase (SgT6PS), and sucrose synthase (SgSUS) were performed using transgenic Arabidopsis thaliana.
Results: Among the unigenes, 410 were identified as putatively involved in these metabolic pathways, including 175 related to glycolysis/gluconeogenesis and 235 to starch and sucrose biosynthesis. Overexpression of SgGPI, SgT6PS, and SgSUS in transgenic A. thaliana enhanced leaf area, accelerated flower formation, and promoted overall growth compared to wild-type plants.
Discussion: These findings lay a foundation for understanding the roles of starch, sucrose, and glycolysis biosynthesis genes in S. guaranitica, offering insights into future metabolic engineering strategies for enhancing the production of valuable carbohydrate compounds in S. guaranitica or other plants.
1 Introduction
Salvia guaranitica belongs to the Lamiaceae family. It is a valuable medicinal and fragrant plant that is native to South America and is widely cultivated in moderate and tropical climates across the globe, particularly in Uruguay, Paraguay, Brazil, Egypt, China, West Asia, and East Asia (Abd El-Wahab et al., 2015; Ali et al., 2018; Santa Cruz et al., 2021; Ali, 2023). The genus Salvia comprises numerous species that are marketed in traditional medicine for their healing properties, including S. officinalis, S. aegyptiaca, S. japonica, S. acerifolia, S. santolinifolia, S. acuminata, S. hydrangea, S. aethiopis, S. tomentosa, S. africana, S. tuxtlensis, S. africana, S. miltiorrhiza, S. arrabidae, S. chloroleuca, S. amplifrons, S. nipponica, S. algeriensis, S. fruticosa, S. aureus, S. amplifrons, S. przewalskii, S. argentea, S. epidermindis, S. isensis, S. arabica, S. arizonica, S.aethiopis, S. aequidens, and S. arenaria, as listed in the Plant List database of the World Flora Online (WFO) http://www.theplantlist.org/1.1/browse/A/Lamiaceae/Salvia/. Most sage species possess a significant concentration of essential oils, notably monoterpenes and sesquiterpenes (Ali et al., 2017, 2018, 2022c, 2022a, 2022b; El-ramah et al., 2022; Khater, 2022).
In plants, carbohydrates such as glucose, sucrose, trehalose, and starch are mono-, di-, and polysaccharides that are created during photosynthesis (Trouvelot et al., 2014). These carbohydrates and other essential chemicals are produced by numerous metabolic processes; including glycolysis/gluconeogenesis (KEGG: map00010), citrate cycle (KEGG: map00020), pentose phosphate pathway (KEGG: map00030), pentose and glucuronate interconversions (KEGG: map00040), fructose and mannose metabolism (KEGG: map00051), ascorbate and aldarate metabolism (KEGG: map00053), starch and sucrose metabolism (KEGG: map00500), amino sugar and nucleotide sugar metabolism (KEGG: map00520), pyruvate metabolism (KEGG: map00620), glyoxylate and dicarboxylate metabolism (KEGG: map00630), propanoate metabolism (KEGG: map 00640), butanoate metabolism (KEGG: map00650), C5-branched dibasic acid metabolism (KEGG: map 00660) and inositol phosphate metabolism (KEGG: map00562) (accessed on 5 Mar 2024) (Kanehisa, 2000, 2019; Kanehisa et al., 2023). Carbohydrate compounds are crucial for various biological processes in plants, including photosynthesis, plant development, signaling, growth, defense mechanisms, biochemical processes within plant cells, regulation of defense genes, plant immunity, symbiotic interactions in legume plants, and coordination of metabolism in response to biotic and abiotic stresses caused by environmental changes. Additionally, carbohydrates also serve as antioxidants and help regulate the generation of reactive oxygen species (ROS) (Koch, 1996, 2004; Sheen et al., 1999; Rolland et al., 2002, 2006; Smeekens et al., 2010; Walters et al., 2013; Keunen et al., 2013; Delaunois et al., 2014; Ali et al., 2022a, 2022b, 2022c). Consequently, breeders and biotechnology experts are primarily focused on augmenting the levels of diverse carbohydrate molecules in plants and crops (Kanehisa et al., 2023).
Currently, RNA-Sequencing (RNA-Seq), a Next-Generation Sequencing (NGS) technology, has emerged as a powerful tool for gene identification. It serves as a significant approach for discovering new genes and elucidating their associations with specific metabolic pathways (Li et al., 2018, 2023; Badawi et al., 2019; Safavi-Rizi et al., 2020; Zayed and Badawi, 2020; Mehmood et al., 2021; Bairakdar et al., 2023; Kanehisa et al., 2023). For example, the starch metabolism in barley grain (Collins et al., 2021), starch accumulation and biosynthesis in sorghum seeds (Ke et al., 2022; Xiao et al., 2022), starch metabolism in Triticum aestivum, and Fagopyrum tataricum (Gu et al., 2021; Huang et al., 2022), starch metabolism in Castanea henryi seeds (Liu et al., 2020), regulator of starch synthesis in Oryza sativa (Liu et al., 2022), starch biosynthesis in Zea mays (Zhang et al., 2019). Trehalose metabolism and biosynthesis in T. eastivum, Manihot esculenta, Medicago truncatula, Zea mays (Macovei et al., 2019; Luo et al., 2021; Lyra et al., 2021; Sukko et al., 2023), and sucrose metabolism in Z. mays, Ipomoea batatas, Vitis vinifera, and Arachis hypogaea (Zhu et al., 2017; Li et al., 2021; Jiang et al., 2023).
Recently, there have been several research on plant genes implicated in glycolysis/gluconeogenesis, starch and sucrose production and regulation. Furthermore, glycolysis/gluconeogenesis, starch, and sucrose regulatory genes from several plant species were discovered, cloned, and characterized, and then employed for engineering the metabolism of many plant species (Huang et al., 2022). For examples, cloning genes encoding the starch biosynthetic enzymes from O. sativa, F. tataricum and Z. mays (Jeon et al., 2010; Huang et al., 2021, 2022), sucrose-phosphate synthase (SPS), and sucrose synthase (SuSy) from Z. mays, Solanum lycopersicum, and Saccharum spp. hybrids (Nguyen‐Quoc and Foyer, 2001; Chandra et al., 2015; Xiao et al., 2024), and trehalose-6-phosphate synthase besides trehalose-6-phosphate phosphatase from A. thaliana, Rosa hybrida, Citrus sinensis (Singh et al., 2011; Liu and Zhou, 2022; Fan et al., 2023). Given that the genes for glycolysis/gluconeogenesis, starch and sucrose biosynthesis and regulation are unknown in S. guaranitica, this study focuses on identifying the candidate genes that are associated with glycolysis/gluconeogenesis, starch, and sucrose biosynthesis from S. guaranitica. Therefore, the identification and functional characterization of three enzymes-encoding genes from S. guaranitica which are glucose-6-phosphate isomerase (SgGPI), trehalose 6-phosphate synthase/phosphatase (SgT6PS) and sucrose synthase (SgSUS) is the focal point to elucidate glycolysis/gluconeogenesis, starch, and sucrose biosynthesis from S. guaranitica. Also, our work resolves their biological roles when transformed into the model plant, A. thaliana. These results illustrate the functioning of the regulatory network governing glycolysis/gluconeogenesis, starch, and sucrose metabolism in S. guaranitica. Moreover, the results provide valuable genetic resources for enhancing plant traits.
2 Materials and methods
2.1 Plant sampling, RNA library preparation, and sequencing
To study the transcriptome profiles, three biological replicates were collected using three separate S. guaranitica plants. Each replicate included a pooled sample of both young and mature leaves. The plants utilized in this study were two years old. Moreover, three biological replicates were gathered from mature leaves, tender leaves, flowering parts, flower buds, stems, and roots for the qRT-PCR assays. The samples were swiftly stored in liquid nitrogen and thereafter held onto -80°C till needed (Mehmood et al., 2021). TRIzol™ Reagent (Invitrogen, CA, US) was employed to obtain the total RNA from different samples as per the guidelines provided by the manufacturer. After treating the extracted RNA from various samples with DnaseI (Takara, China), its overall quality was assessed by subjecting it to electrophoresis on a 1.25% agarose-formaldehyde gel, followed by visualization with ethidium bromide. Additionally, the NanoDrop™ 2000/2000c Spectrophotometers (MA, USA) was utilized to calculate the RNA quality and concentration from different samples. Ten µg of RNA was used for cDNA synthesis via the reverse transcription kit (M-MLV, China) (Huang et al., 2012; Rastogi et al., 2014; Zhu et al., 2019; Santa Cruz et al., 2021). High-quality RNAs extracted from different samples were utilized to construct cDNA libraries (Ali et al., 2017, 2018). Sequencing was achieved on the high-quality libraries using an Illumina HiSeq 2500 platform, generating paired-end reads. Clean reads were acquired by filtering out adapters, poly-N sequences, and low-quality reads. Subsequently, the assembly was performed by the Trinity platform https://github.com/trinityrnaseq/trinityrnaseq/wiki with the parameters “min_kmer_cov set to 2”. Subsequently, the values of Q20, Q30, GC content, and sequence duplication level were estimated (Yang et al., 2017).
2.2 In silico differential gene expression and protein domain analysis
To examine the putative transcription levels of SgGPI, SgT6PS, and SgSUS across various tissues, the A. thaliana eFPbrowsers (http://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi) was employed. Moreover, the predicted subcellular localizations of their orthologs from A. thaliana were retrieved. Subsequently, the image envisioned their cellular localizations was built, and then the putative domains were estimated through the InterPro database (https://www.ebi.ac.uk/interpro/) (El-ramah et al., 2022; Elsherbeny et al., 2022; Makhadmeh et al., 2022a, 2022b; Abdelhameed et al., 2024b).
2.3.Validation and relative expression analysis of glycolysis/gluconeogenesis, starch and sucrose metabolism genes
Towards examining the activity levels of glycolysis/gluconeogenesis, starch, and sucrose biosynthesis genes in S. guaranitica at different tissues, twenty candidate genes were chosen. The expression profiles for these selected genes were compared within various tissue samples to disclose their ‘transcriptional control’, offering insights into the epistatic relationship regarding mRNA copies, and the products and the end-products. The expression profiles of our chosen candidates: SgGPI, SgT6PS, SgSUS, SgPFK9, SgALDH, SgALDO, SgPYK, SgFBP, SgACS, SgPCKA, SgGlGA, SgGlGC, SgBMY, SgGBE1, SgAGL, SgBGL, SgHK, SgPYG, SgUGDH and SgINV, across different tissues were investigated.
2.4 Cloning of full-length starch, sucrose and glycolysis synthase cDNAs
The SgGPI, SgT6PS, and SgSUS full-length cDNAs were amplified using gene-specific primers designed from the Illuimina sequencing data of S. guaranitica leaves (Supplementary Table S1). The initial PCR was conducted with the short primers, KOD-Plus-DNA polymerase (Toyobo, Japan), and leaf cDNA using the program: 96°C for 4 min, 98°C for 12 s, 60°C for 4 s, 68°C for 2.5 min, and 34 cycles followed by 68°C for 15 min. Regarding the following PCR, the products from the initial PCR were used as templates with the long primers under the same PCR conditions. The products were then cleaned and then transferred to the Gateway entry vector (pDONR221), then subsequently sub-cloned into the destination vector (pB2GW7). The vectors were incorporated into A. thaliana flowers via Agrobacterium tumefaciens strain GV3101 by electroporation. The cloning steps were verified using Sanger sequencing (Rehman et al., 2018; Ali et al., 2022a, 2022c, 2022b).
2.5 Functional characterization of SgGPI, SgT6PS and SgSUS in transgenic A. thaliana leaves
The genes SgGPI, SgT6PS and SgSUS were chosen to be characterized and expressed in A. thaliana utilizing the Agrobacterium-mediated floral dip technique. The transformation was performed using A. tumefaciens GV3101 harboring pB2GW7-SgGPI, pB2GW7-SgT6PS, and pB2GW7-SgSUS plasmids driven by the 35S promoters. Using the procedure outlined by (Ali et al., 2018, 2022a, 2022c, 2022b; Ali, 2023). Briefly, the A. thaliana seeds were germinated in advance and the plants were made ready for transformation after two months. The secondary inflorescences were immersed in a solution containing Agrobacterium carrying the pB2GW7-vector, specifically targeting the gynoecium of the flower. The plants were cultivated until the siliques reached a brown and dried state. Subsequently, the seeds were collected, cultivated again, and subjected to BASTA treatment – a herbicide containing glufosinate-ammonium – to select the desired transgenic seedlings carrying the resistance gene against BASTA. Moreover, the presence of target genes in positive transgenic lines was verified through semi-quantitative RT-PCR (semi-qRT-PCR). The physiological and biochemical parameters of different transgenic lines were evaluated. A total of twelve 45-day-old plants, including putative transgenic and wild-type plants, were chosen for the purpose of harvesting mature leaves. These leaves were then subjected to semi-qRT-PCR for measuring the activity levels of the Salvia-derived genes (SgGPI, SgT6PS and SgSUS) into transgenic A. thaliana plants.
2.6 Determination of relevant physiological and biochemical indices
Soluble sugars, including sucrose, glucose, and fructose, were analyzed as described by (Sonnewald et al., 1991; Ortiz-Marchena et al., 2014). Briefly, the quantification involved extraction from both wild-type and genetically modified A. thaliana plant leaves using 80% ethanol in 10 mM HEPES-KOH (pH 7.7) at 80°C for 2 hours. The supernatant was utilized to measure glucose, fructose, and sucrose concentrations through the sequential addition of specific enzymes—5 units each of glucose-6-phosphate dehydrogenase and hexokinase, 2 units of glucose-6-phosphate isomerase, and 20 units of invertase—followed by the monitoring of NAD+ reduction at 340 nm absorbance at various intervals.
Each parameter was tested with three biological replicates. Furthermore, the quantities of chlorophyll a, b, and total (a+b), were measured following (El-Mahdy et al., 2024). Concisely, about 20-30 mg of fresh leaf samples were weighted from each treatment, then each sample was transferred to a centrifuge tube with 4 mL dimethylformamide (DMF) and maintained away from light to preserve chlorophyll integrity. The level of chlorophyll a, b, and total (a+b) in the extracts were accomplished following (El-Mahdy et al., 2024). The absorbance readings of the chlorophyll samples were taken at 664 and 647 nm through JENWAY 6505 UV/Vis spectrophotometer.
2.7 Statistical analysis
The results were analyzed using SPSS (IBM Corp., 2020), incorporating three biological replicates. Significance levels were indicated as (*) for P-values less than 0.05, (**) for P < 0.01, (***) for P < 0.001, and (****) for P < 0.0001, demonstrating the highest degree of significance.
3 Results
3.1 Illumina-based sequencing, de novo assembly, and functional annotation
Lately, the Illumina sequencing technology has emerged as a robust technique for genome analysis and discovery in non-model plants. In this research, transcriptome sequences were obtained from pooled leaves of Salvia guaranitica using the Illumina HiSeq 2500 platform. This process yielded approximately 8.2 Gb of raw data from the S. guaranitica leaves. Post-filtering and removal of adapter sequences, 38,521,658 reads (38.52 million) were obtained, containing 210,521,170 high-quality nucleotide bases. The quality metrics indicated that 94.95% of the bases had a quality score of Q20, 90.54% had a quality score of Q30, and the GC content was 48.58%. Our findings were consistent with previously obtained results of many other studies which utilized transcriptome tools to detect and identify key genes associated with various biomolecules in several species, including Vicia sativa, Dendrobium nobile, S. officinalis, Ocimum sanctum, Ocimum basilicum, and Cunninghamia lanceolata (Rastogi et al., 2014; Ali et al., 2017; Zhu et al., 2019; Huang et al., 2021; Zhang et al., 2023).
3.2 De novo assembly and transcriptome analysis
With respect to de novo assembly and transcriptome study, high-quality and pure reads were assembled using Trinity program (Ali et al., 2017, 2018; Mehmood et al., 2021). The assembly results yielded 200,298 RNA variants, the N50 length was 1,850 bp, the N90 length was 520 bp, and the mean length was 1,125 bp. Additionally, 75,100 unigenes were identified, with N50 equals to 1,524 bp, N90 is 320 bp, and a mean length of 965 bp. The assembled lengths were 200 to approximately 2,000 bp. The majority of transcripts (83,387 transcripts, 41.652%) were between 200 and 500 bp, then 48,252 transcripts (24.102%) between 1,000 and 2,000 bp, and 41,197 transcripts (20.578%) between 500 and 1,000 bp. Conversely, the fewest transcripts (27,462 transcripts, 13.731%) were longer than 2,000 bp. Similarly, the lengths of the unigene assemblies ranged from 200 to over 2,000 bp, with the majority (39,145 unigenes, 52.125%) between 200 and 500 bp, followed by 16,866 unigenes (22.458%) between 500 and 1,000 bp, and 13,837 unigenes (18.425%) between 1,000 and 2,000 bp. The fewest unigenes (5,251 unigenes, 6.992%) were longer than 2,000 bp. The length profile of the transcripts and unigenes is presented in Supplementary Table S2. Our findings correspond with results observed in other species, such as, Boehmeria nivea, Curcuma longa, M. sativa, S. officinalis, Centella asiatica, and Apium graveolens, where transcript and unigene lengths predominantly fell within 75 to 500 bp (Srividya et al., 2015; Zhu et al., 2019; Xiao et al., 2024).
3.3 Unigene annotation and classification
Nearly 75,100 unigenes served as search queries across NR (http://www.ncbi.nlm.nih.gov/), NT, KO, Swiss-Prot (http://www.ebi.ac.uk/uniprot/), PFAM, GO (http://www.geneontology.org/), and KEGG (https://www.kegg.jp/kegg/kegg2.html) Supplementary Table S3. The BLAST2GO program facilitated the sorting and ranking of the functions of all annotated unigenes, with 29,695 unigenes (39.54% of the assembled unigenes) assigned with at least one GO term. Inferred from the homology results, the unigenes were sorted into 56 functional groups across three main categories: 63,008 assigned to biological processes (BP), 48,517 to cellular components (CC), and 20,546 to molecular functions (MF). In the CC section, the most enriched GO terms were “cell part” (9,357) and “cell” (9,281). In the MF section, “binding” (14,258) and “catalytic activity” (12,754) were predominant. Within the BP section, “metabolic process” (15,326) and “cellular process” (14,820) were highly enriched (Figure 1). These findings are consistent with previous studies on the RNA profiles of S. miltiorrhiza, S. officinalis, O. sanctum, and O. basilicum, that also reported high percentages of these GO terms (Ali et al., 2017, 2018; Mehmood et al., 2021).
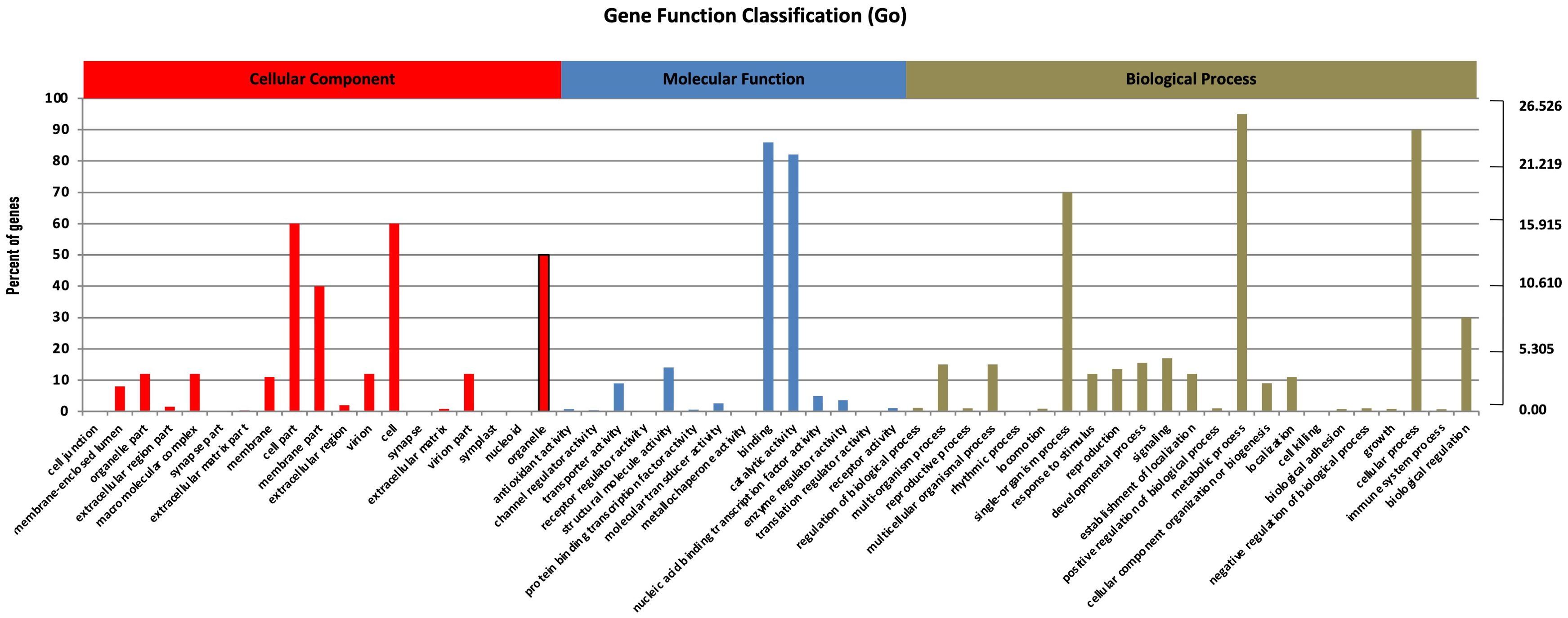
Figure 1. Unigenes annotations in S. guaranitica. The three main categories are BP, CC, and MF. In order to ascertain the biofunctions of the S. guaranitica transcriptome, KEGG pathways of the 75,100 unigenes were determined, with 11,746 unigenes (15.64%) ascribed to 270 pathways. Primarily, five major pathways were identified: (A) Cellular Processes, (B) Environmental Information Processing, (C) Genetic Information Processing, (D) Metabolism, and (E) Organismal Systems (Figure 2).
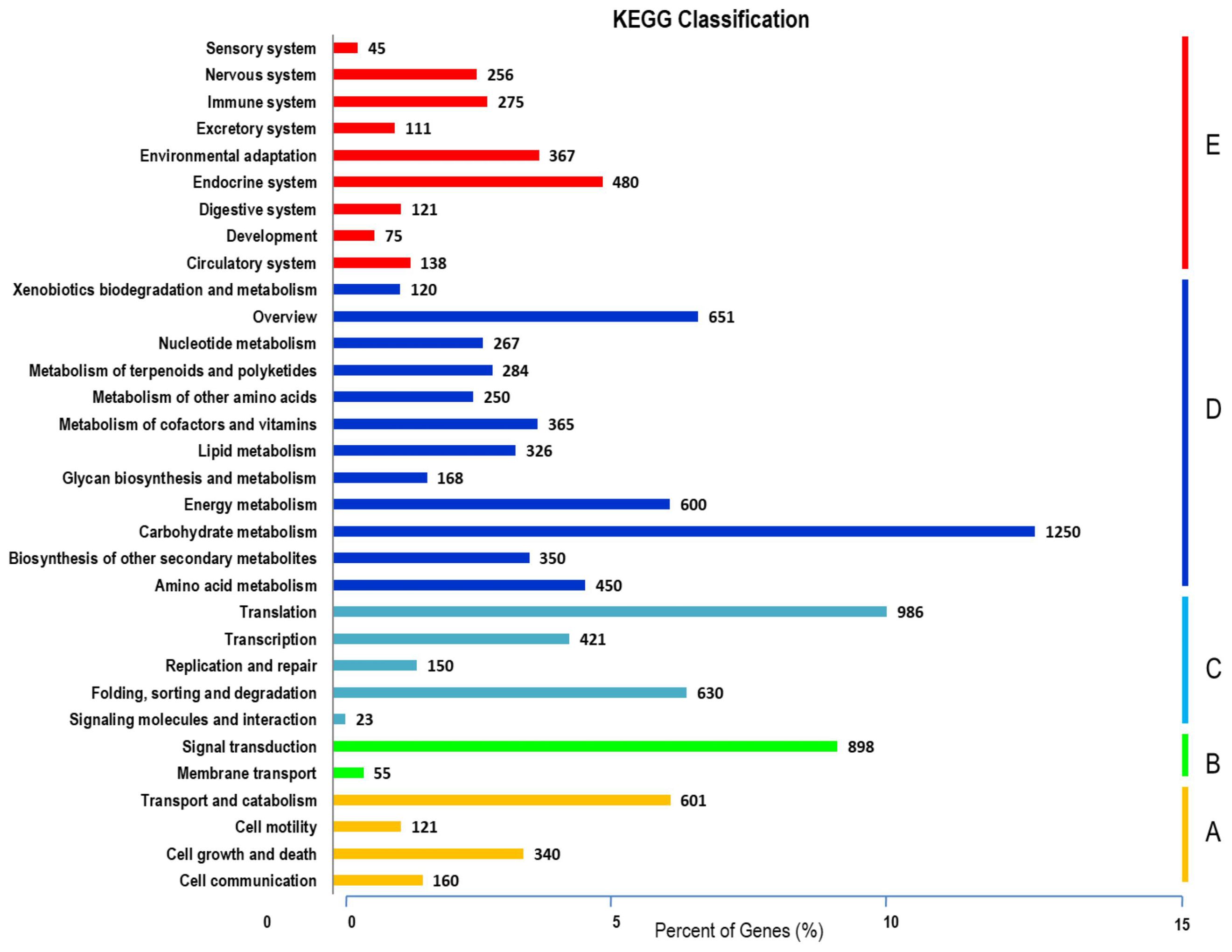
Figure 2. KEGG cellular processes pathways were classified into five largest categories; (A), environmental information processing (B), genetic information processing (C), metabolism (D) and organismal systems (E).
Most transcripts belonged to the Metabolism section (5,081), subsequently, Genetic Information Processing (2,187), Organismal Systems (1,868), Cellular Processes (1,222), and Environmental Information Processing (976). From the data analysis, 1,250 transcripts were related to carbohydrate metabolism, with 410 putatively linked to starch, sucrose, and glycolysis metabolism. This included 175 genes involved in glycolysis/gluconeogenesis biosynthesis and 235 genes related to starch and sucrose biosynthesis. The levels of gene expressions were estimated through the UniProt database for annotation against the transcriptome libraries. Normalization and calculation were performed using the DESeq package (1.10.1), represented as fragments per kilobase of transcripts per million mapped fragments (FPKM) as shown in Figure 3 and presented in Supplementary Tables S4 and S5.
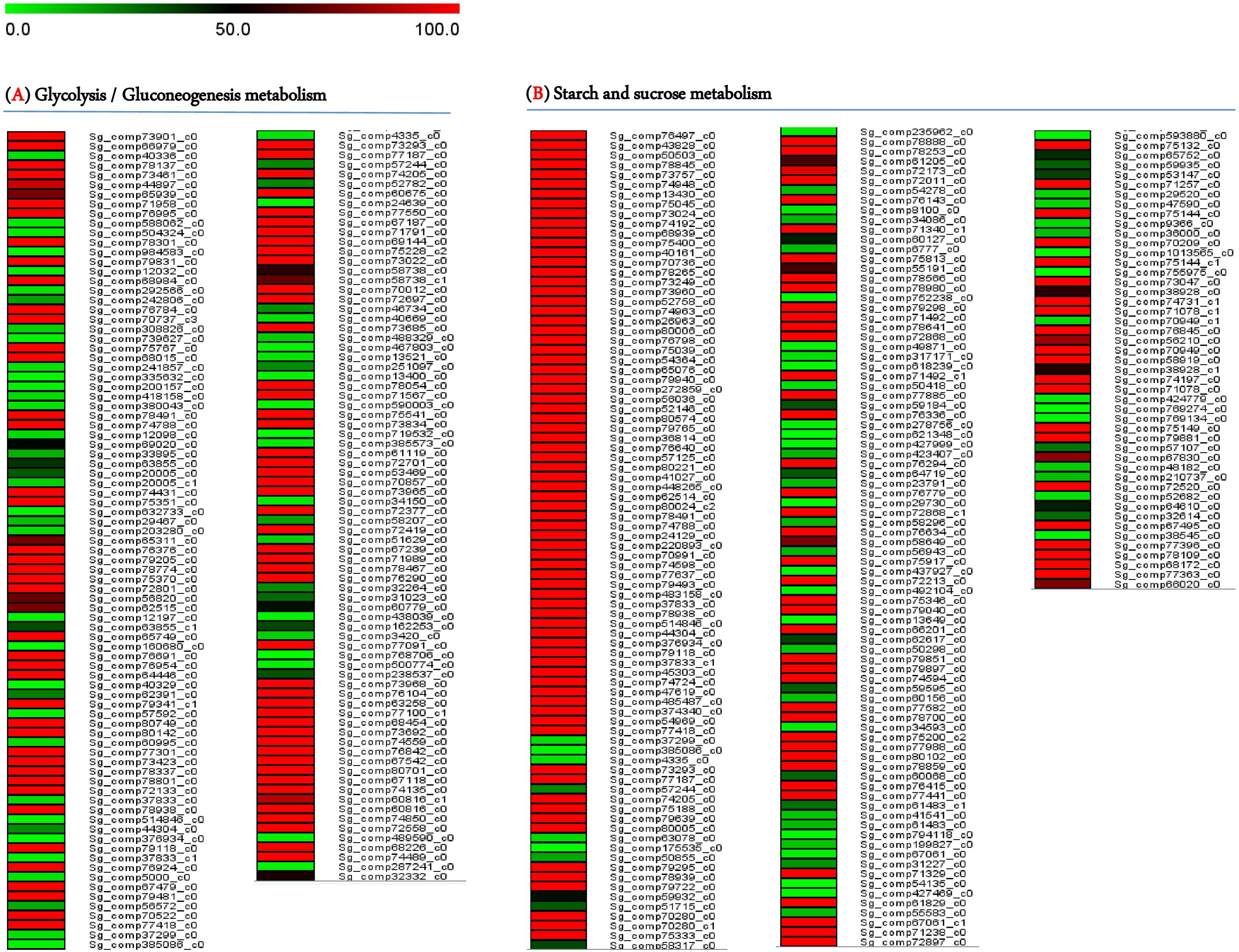
Figure 3. A heatmap illustrating the transcript levels of genes regulating (A) Glycolysis/Gluconeogenesis pathways and (B) Starch and Sucrose pathways.
3.4 Patterns of tissue expressions, subcellular localizations and protein domain analysis
To identify the physiological roles of the SgGPI, SgT6PS, and SgSUS genes, we investigated their accumulation patterns across forty-seven tissues. The analysis was facilitated by the high similarity between SgGPI, SgT6PS, SgSUS, and the AT4G24620, AT1G06410, and AT5G37180 from A. thaliana, respectively. The relative expressions of SgGPI, SgT6PS, and SgSUS were observed in various tested tissues (Figures 4A–C). These results align with results reported (Ali et al., 2017, 2018, 2022a) who used similar tools in the BAR database to predict the putative expression patterns for several genes, such as, SoNEOD, SoHUMS, SoFLDH, SoLINS2, GmTPS21, SgTPSV, SgGERIS, and SgFARD, from S. officinalis, Glycine max, and S. guaranitica, respectively, showing heightened manifestation in leaves, roots, and seeds. Additionally, the localizations of SgGPI, SgT6PS, and SgSUS revealed that they are present in various cell organelles. For example, the SgGPI is predominantly located in the plastid, cytosol, extracellular space, and mitochondria. However, SgT6PS is mainly found in the mitochondrion, cytosol, plasma membrane, Golgi apparatus, nucleus, peroxisome, and vacuole. Meanwhile, SgSUS is primarily present in the cytosol, mitochondrion, plastid, nucleus, and plasma membrane (Figures 4D–F).
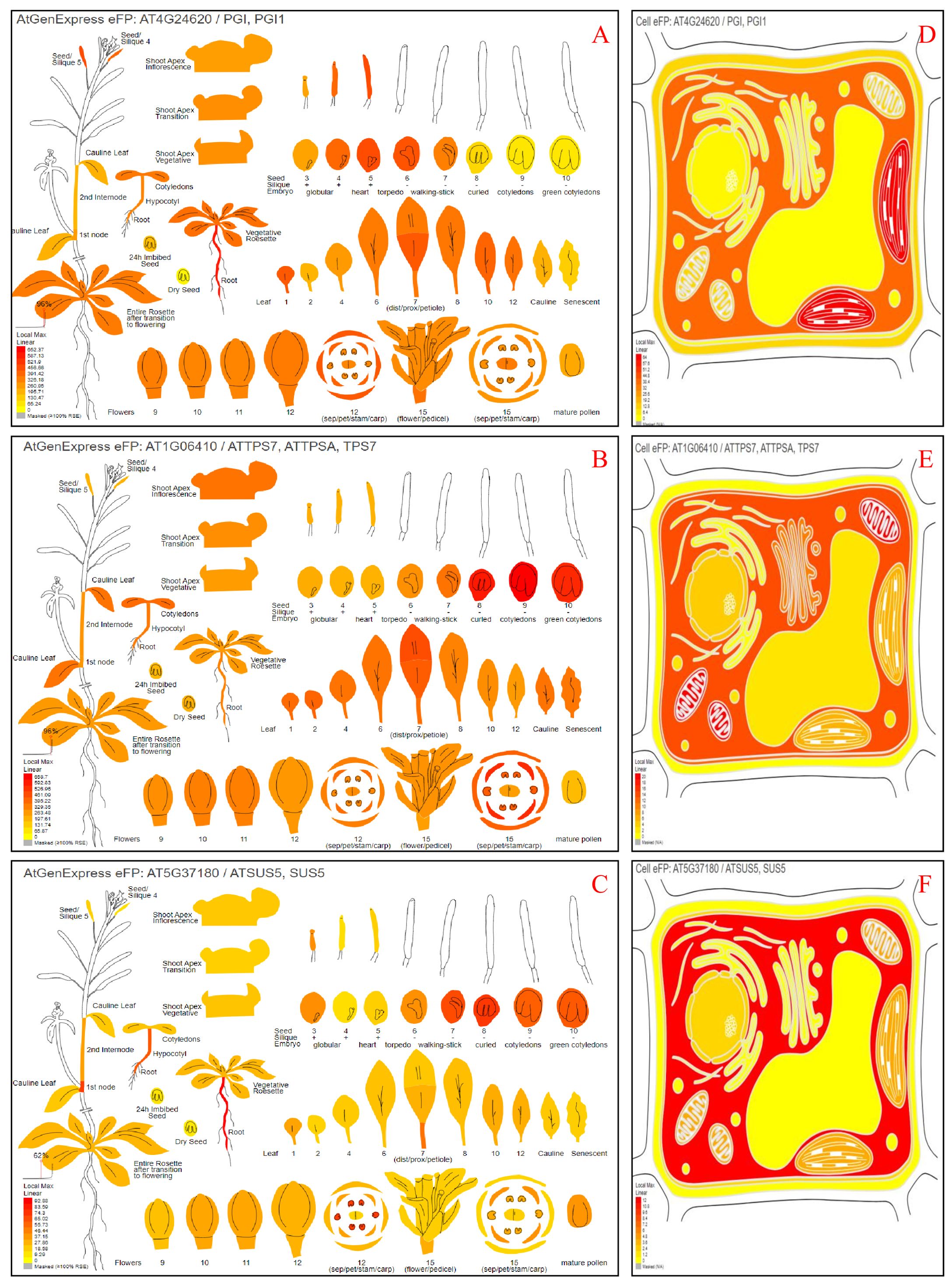
Figure 4. Depicting the potential an ‘electronic fluorescent pictograph’ website for investigating A. thaliana orthologous genes’ potential tissue expression and their proteins’ cellular localizations as retrieved from the eFPbrowsers (http://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi). Panels (A, D) demonstrate where cells may express SgGPI (AT4G24620). Panels (B, E) indicate where cells may express SgT6PS (AT1G06410). Panels (C, F) demonstrate where cells may express SgSUS (AT5G37180). The color box shows expression scale (greater red indicates more gene expression).
3.5 Quantitative RT-PCR analysis
To elucidate the differential gene expression of glycolysis/gluconeogenesis, starch and sucrose biosynthesis genes (Supplementary Table S6; Figure 5) across different treatments, we employed the Bio-Rad Nucleic Acid Amplification and Detection systems (CFX384) with SYBR Green fluorescence and ROX as a passive reference dye (Newbio Industry, China), following protocols outlined in previous studies (Rastogi et al., 2014; Zhu et al., 2019; Huang et al., 2021; Santa Cruz et al., 2021). Primers were designed using the IDTdna tool (https://eu.idtdna.com/scitools/Applications/RealTimePCR/), as listed in Supplementary Tables S6 and S2. The cycle threshold (CT) of the target genes was calculated using SgB-ACTIN as a reference gene to normalize gene expression levels. Relative gene expression levels were then determined through delta-delta Ct method (Supplementary Tables S6; Figure 5). The relative expression levels of several genes, including SgGPI, SgT6PS, SgSUS, SgPFK9, SgALDH, SgALDO, SgPYK, SgFBP, SgACS, SgPCKA, SgGlGA, SgGlGC, SgBMY, SgGBE1, SgAGL, SgBGL, SgHK, SgPYG, SgUGDH, and SgINV, were detected. For instance, SgBMY, SgGlGA, SgSUS, SgALDO, SgGlGC, SgPFK9, SgGPI, and SgAGL genes exhibited the top expression levels in mature leaves. In contrast, SgUGDH and SgACS genes showed the peak expression levels in immature leaves. Additionally, the SgT6PS gene demonstrated the maximum expression levels in flowers, whereas the SgALDH gene had the highest expression levels in flower buds. Moreover, SgBGL, SgINV, SgHK, SgPYK, and SgFBP genes demonstrated the peak levels in stems (Figure 5). Finally, SgPCKA, SgPYG, and SgGlGC genes were found with the greatest levels in roots. Interestingly, the qPCR analyses of these genes were in line with their expressions as measured by Illumina HiSeq 2500 (Hussain et al., 2017; Darwish et al., 2022; Ali et al., 2023b).
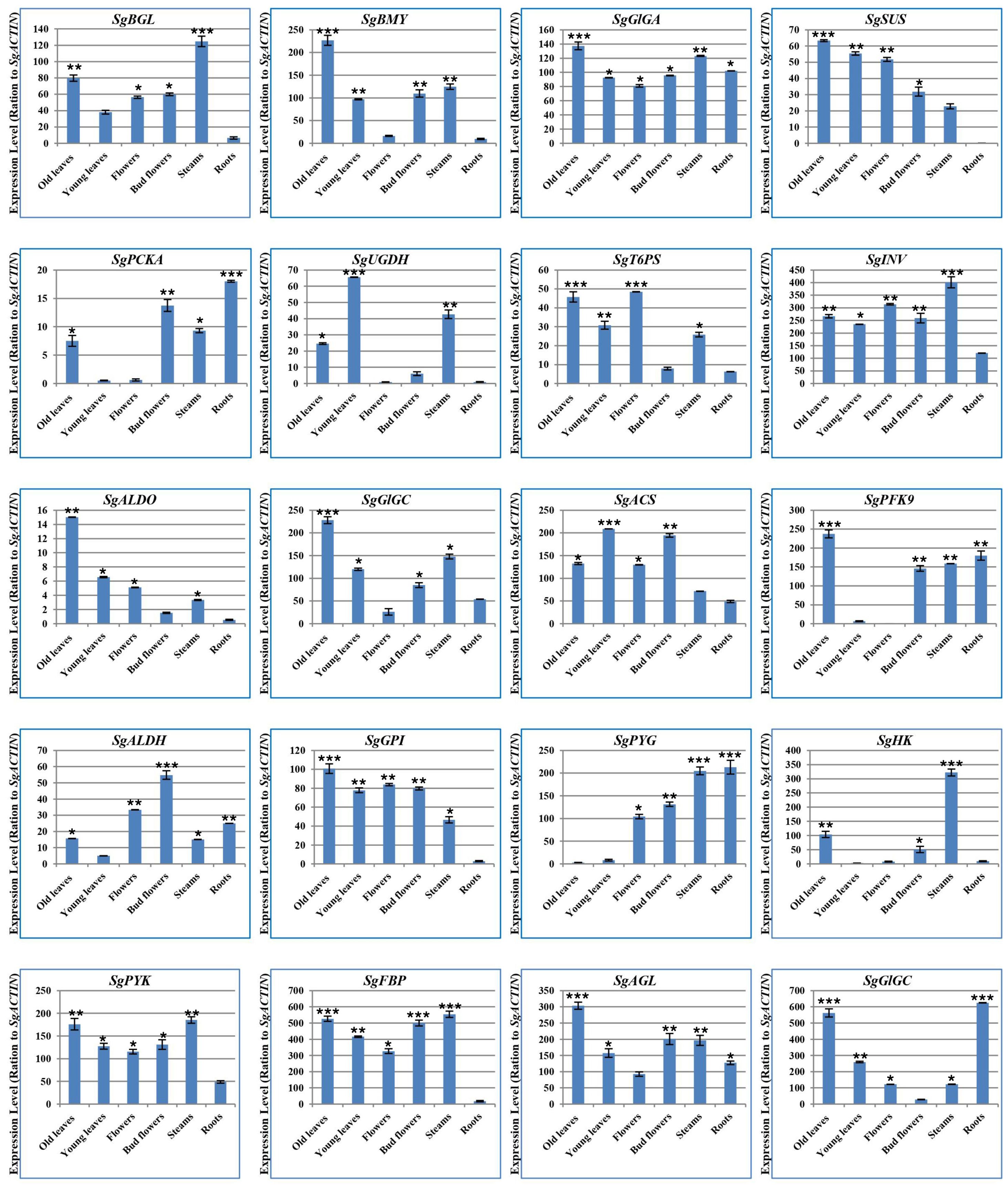
Figure 5. Quantitative RT-PCR of glycolysis/gluconeogenesis, starch and sucrose metabolism genes. The relative expressions of SgGPI, SgT6PS, SgSUS, SgPFK9, SgALDH, SgALDO, SgPYK, SgFBP, SgACS, SgPCKA, SgGlGA, SgGlGC, SgBMY, SgGBE1, SgAGL, SgBGL, SgHK, SgPYG, SgUGDH, and SgINV were calculated. The values are means ± SE of three biological replicates. Significance levels were indicated as (*) for P-values less than 5%, (**) for P < 1%, and (***) for P < 0.1%.
3.6 Phenotypic and functional characterization of SgGPI, SgT6PS, and SgSUS in A. thaliana
To further investigate the biological function of SgGPI, SgT6PS, and SgSUS, we transferred the overexpression vector pB2GW7-SgGPI, pB2GW7-SgT6PS and pB2GW7-SgSUS directed by 35S promoter into A. thaliana. We obtained twelve homozygous transgenic lines from each transformed gene and the successful transformed lines was confirmed by PCR and semi-qRT-PCR analysis (Figures 6A, B). Compared to the WT, a significant difference in leaf development and plant growth were observed in all lines overexpressing these genes (Figure 6A). Interestingly, the flowering process of transgenic A. thaliana was accelerated earlier than that of W.T plants (Figure 6A).

Figure 6. Overexpression of the SgGPI, SgT6PS and SgSUS genes from S. guaranitica in transgenic A. thaliana. (A) Comparison of the phenotypes of the transgenic A.. thaliana and wild type A. thaliana. (B) Semi-qRT-PCR of the genes regulating starch, sucrose, and glycolysis biosynthesis.
3.7 Overexpression of SgGPI, SgT6PS and SgSUS alters various physiological and biochemical attributes in transgenic plants
Sugars serve as the primary carbon source for synthesizing various pathways involved in both primary and secondary metabolites. The levels of soluble sugars were assessed in transgenic A. thaliana plants overexpressing SgGPI, SgT6PS, and SgSUS, revealing significant increases compared to the wild-type (WT). Furthermore, starch, total sugar, glucose, and fructose contents were quantified in these transgenic plants. Results indicated higher levels of these components in all transgenic lines compared to WT (Figure 7). Specifically, the average starch content increased by approximately 5.033-fold (10.033/5.0) for A. thaliana plants overexpressing SgGPI, 6.566-fold (11.866/5.3) for A. thaliana plants overexpressing SgT6PS, and 7.133-fold (11.700/4.6) for A. thaliana plants overexpressing SgSUS compared to WT. Similarly, average sugar contents visually increased by 2.2-fold (5.2/3) for A. thaliana plants overexpressing SgGPI, 3.766-fold (6.566/2.8) for A. thaliana plants overexpressing SgT6PS, and 6.233-fold (8.733/2.5) for A. thaliana plants overexpressing SgSUS. Glucose levels showed respective increases of 0.87-fold (1.97/1.1), 1.2-fold (2.4/1.2), and 1.04-fold (2.25/1.21), while fructose levels increased by 1.0-fold (2.2/1.2), 0.94-fold (2.24/1.3), and 1.52-fold (2.733/1.25) for A. thaliana plants overexpressing SgGPI, A. thaliana plants overexpressing SgT6PS, and A. thaliana plants overexpressing SgSUS compared to WT.
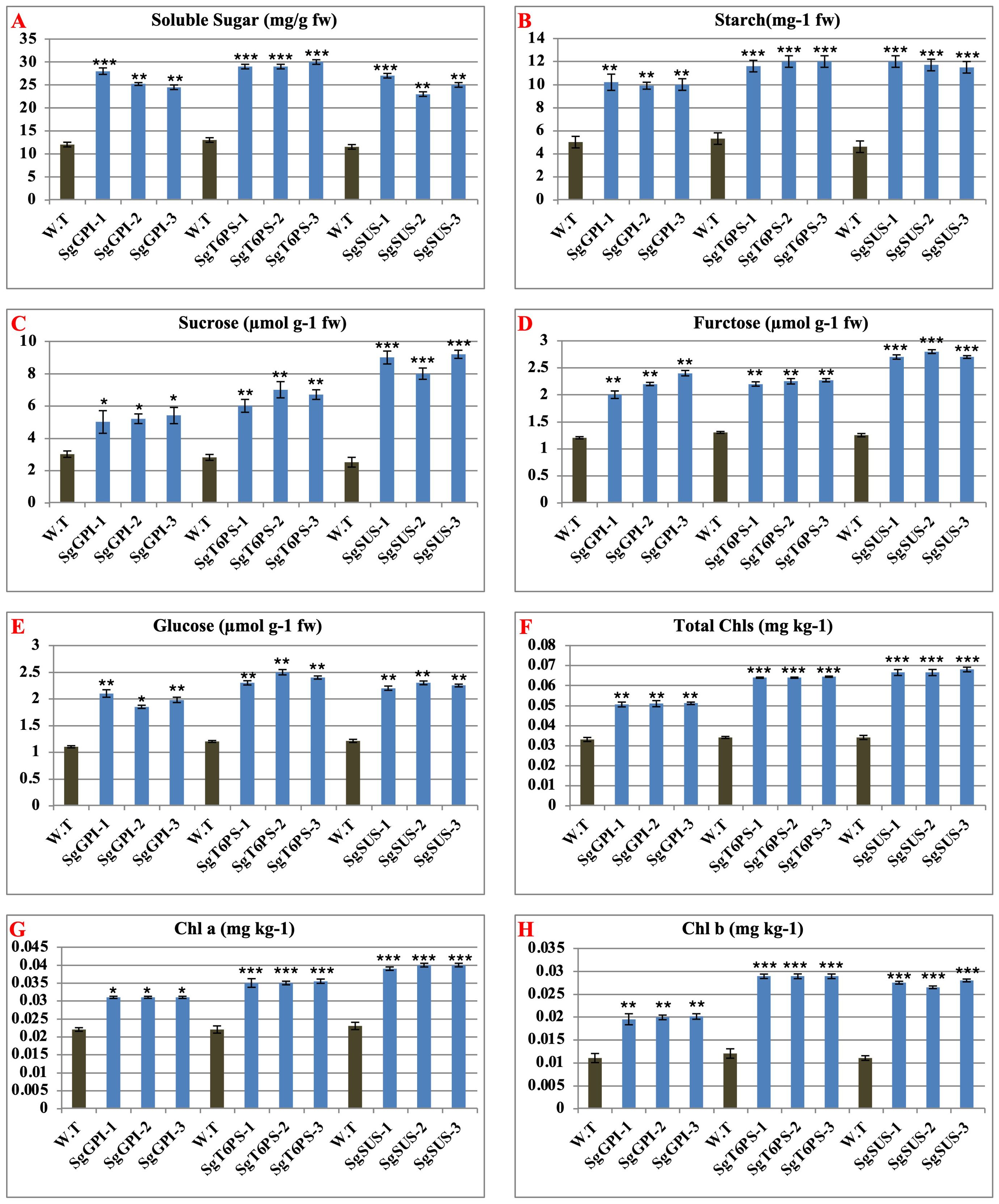
Figure 7. Analysis of physiological and biochemical parameters from wild and transgenic A thaliana under the effects of overexpression of SgGPI, SgT6PS and SgSUS separately. (A) soluble sugar; (B) starch; (C) sugar; (D) fructose; (E) glucose; (F) total chlorophyll; (G) chlorophyll a; (H) chlorophyll (b) Significance levels were indicated as (*) for P-values less than 0.05, (**) for P < 0.01, and (***) for P < 0.001.
Additionally, the total chlorophyll, chlorophyll a, and chlorophyll b contents were evaluated in these transgenic plants. Results indicated higher levels of these chlorophyll components in all transgenic lines compared to WT (Figure 7). For instance, the average total chlorophyll content increased by 0.017-fold (0.0508/0.033) for A. thaliana plants overexpressing SgGPI, 0.030-fold (0.0640/0.034) for A. thaliana plants overexpressing SgT6PS, and 0.033-fold (0.067/0.034) for A. thaliana plants overexpressing SgSUS compared to WT. Similarly, average chlorophyll a level increased by 0.009-fold (0.031/0.022) for A. thaliana plants overexpressing SgGPI, 0.0131-fold (0.0351/0.022) for A. thaliana plants overexpressing SgT6PS, and 0.016-fold (0.039/0.023) for A. thaliana plants overexpressing SgSUS, while chlorophyll b levels increased by 0.068-fold (0.0795/0.011) for A. thaliana plants overexpressing SgGPI, 0.0747-fold (0.0867/0.012) for A. thaliana plants overexpressing SgT6PS, and 0.071-fold (0.082/0.011) for A. thaliana plants overexpressing SgSUS compared to WT.
4 Discussion
Carbohydrate biosynthesis is the primary driver of plant growth and development. Carbohydrates are highly susceptible to biotic and abiotic stresses, which influence yield parameters. Therefore, identifying genes that regulate carbohydrate production in S. guaranitica will facilitate the development of targeted breeding techniques to enhance desirable traits such as growth, stress resilience, and medicinal properties. S. guaranitica is renowned for its rich array of primary and secondary metabolites, including disaccharides, starch, terpenoids, and flavonoids. Disaccharides, found across various organisms, play diverse roles in plant growth, development, and resistance to environmental stresses through pathways like glycolysis/gluconeogenesis and starch/sucrose metabolism, which share common enzymes and substrates, illustrating their interdependence. This study employed RNA-Seq technology to identify key enzymes in these pathways from S. guaranitica leaf transcriptomes, revealing over 75,100 unigenes encompassing all metabolic pathways. Gene ontology analysis highlighted 410 genes potentially involved in starch, sucrose, and glycolysis metabolism, including 175 related to glycolysis/gluconeogenesis and 235 to starch/sucrose biosynthesis, as illustrated Figure 3 and detailed in Supplementary Tables S4 and S5. These findings underscore RNA-Seq’s efficacy in elucidating mechanisms underlying metabolite synthesis (Liu et al., 2020; De Vega et al., 2021; Yang et al., 2023).
Functional characterization of SgGPI, SgT6PS, and SgSUS through putative tissue expression patterns, subcellular localization, and protein domain analysis using tools like eFP browsers, Cell-eFP browsers, and InterPro database enhances our understanding of their roles and expression levels see Figure 4. In silico analyses validate their predicted functions, excluding irrelevant pathways, and affirm their significance in plant biology. Furthermore, these previous tools were used in various studies such as; (Ali et al., 2022a, 2022c, 2022b, 2023b, 2023a; Abdelhameed et al., 2024b, 2024a).
In general, the expression levels of multiple genes which encoded glycolysis/gluconeogenesis, starch and sucrose enzymes (e.g. SgGPI, SgT6PS, SgSUS, SgPFK9, SgALDH, SgALDO, SgPYK, SgFBP, SgACS, SgPCKA, SgGlGA, SgGlGC, SgBMY, SgGBE1, SgAGL, SgBGL, SgHK, SgPYG, SgUGDH and SgINV) were detected at different tissues especially in leaves, and these previous genes may effectively enhance the entire pathways Figure 5. Moreover, any changes in the expression profiles of the aforementioned genes are often bound to the accumulation levels of biochemical contents from soluble sugar, starch, sugar, fructose, glucose, total chlorophyll, chlorophyll a and chlorophyll b. Also, these changes in the genes expression levels and their relationship to the accumulation of previous compounds explain the extent of the plant’s response to growth and the increase in the plant growth and plant biomass (Sung et al., 1988; Stein and Granot, 2019; Li et al., 2020; Taujale et al., 2021).
In comparison to control plants (non-transgenic), overexpression of SgGPI, SgT6PS and SgSUS controlled under 35S promoter caused manifest difference in phenotypic traits such as; plant biomass, leaves area, leaves number and early flowering (Figure 6). The results describe that transformation of previous genes under 35S promoter is expressed in whole transgenic A. thaliana plants as we confirmed by semi-qRT-PCR data, and this expression is related with vegetative growth and early flowering formation as well. The current findings and results are in close agreement with previous investigations by (Zhang et al., 2016; Ahmed et al., 2020; Liu et al., 2020; De Vega et al., 2021) which study the roles of various glycolysis, starch and sucrose genes from different plants (e.g., Allium cepa L., Sorghum, Taro Corm, Miscanthus hybrids and Castanea henryi) in plant growth and increase of biomass yield. Through our previous results, we can infer that SgGPI, SgT6PS and SgSUS encode essential enzymes involved in S. guaranitica development, as the activity of these enzymes are correlated with plant growth and increase of biomass yield.
Furthermore, this study evaluated changes in physiological and biochemical traits (soluble sugars, starch, sugars, fructose, glucose, total chlorophyll, chlorophyll a, and chlorophyll b) in wild-type and transgenic A. thaliana plants overexpressing SgGPI, SgT6PS, and SgSUS, as shown in Figure 7. These traits directly influence physiological and morphological variations that impact plant growth, consistent with earlier findings by (Dian et al., 2003, 2005; Akihiro et al., 2005; Wang and Ruan, 2013; Ahmed et al., 2018, 2020).
In conclusion, our study contributes valuable information to the limited transcriptome resources of Salvia, one of the largest genera in the Lamiaceae families, known for its diverse specialized secondary and primary metabolites. The S. guaranitica transcriptome data we provide here should be valuable for metabolic engineering, fundamental biological research, and plant improvement programs. Transgenic A. thaliana lines with high phenotypic traits and biochemical contents generated in this study should serve as a useful adjunct in enhancing the maximize the benefit of these genes in improving plants and increasing production.
5 Conclusions
In this study, we employed NGS technology to generate high-quality transcriptome data from S. guaranitica leaves. Utilizing de novo sequencing and analysis tools, we assembled data obtained from the Illumina HiSeq 2500 system to characterize and identify genes associated with starch, sucrose, and glycolysis biosynthesis pathways. The transcriptome analysis revealed numerous genes encoding enzymes pivotal to these metabolic pathways in S. guaranitica. Specifically, we conducted cloning and bioinformatics analyses of S. guaranitica. SgGPI, SgT6PS, and SgSUS. Our findings confirm that these genes encode prototypical GPI, T6PS, and SUS proteins. Overexpression of SgGPI, SgT6PS, and SgSUS in A. thaliana resulted in enhanced leaf growth, plant development, and accelerated flowering. Furthermore, these genes significantly increased soluble sugar, starch, sugars (glucose and fructose), total chlorophyll, chlorophyll a, and chlorophyll b contents in transgenic tobacco lines compared to wild-type (WT). Overall, our study provides insights into the regulatory mechanisms of SgGPI, SgT6PS, and SgSUS and their roles in starch, sucrose, and glycolysis metabolism pathways. These findings open new avenues for biotechnological applications utilizing these genes.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: NCBI, PRJNA1163088.
Author contributions
ZA: Formal analysis, Methodology, Validation, Writing – review & editing, Software. AA-H: Validation, Writing – review & editing, Data curation, Investigation, Supervision, Visualization, Writing – original draft. AK: Formal analysis, Methodology, Validation, Writing – review & editing, Data curation. RA: Formal analysis, Validation, Writing – review & editing, Data curation, Software, Visualization. DA: Formal analysis, Validation, Writing – review & editing, Data curation, Investigation, Visualization. MAA: Formal analysis, Validation, Writing – review & editing, Data curation, Software. MSA: Formal analysis, Validation, Writing – review & editing, Data curation, Software. DD: Formal analysis, Validation, Writing – review & editing, Data curation, Software. AA: Formal analysis, Methodology, Validation, Writing – review & editing, Data curation, Investigation, Software, Visualization. FS: Formal analysis, Methodology, Validation, Writing – review & editing, Software, Visualization. RM: Formal Analysis, Methodology, Validation, Writing – review & editing, Data curation, Software. MaA: Methodology, Validation, Writing – review & editing, Data curation, Investigation, Formal analysis. NA-S: Formal analysis, Methodology, Validation, Writing – review & editing, Data curation, Investigation. MoA: Conceptualization, Formal analysis, Methodology, Software, Writing – original draft, Writing – review & editing, Data curation, Investigation, Validation, Visualization. MZ: Investigation, Methodology, Supervision, Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Software, Validation, Visualization.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
All authors’ thanks and extend their appreciation to University and Research Institutes which they belong for supporting this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer OH declared a past co-authorship with the author MA, and declared a shared affiliation with the author MZ to the handling editor at the time of the review.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1467432/full#supplementary-material
References
Abdelhameed, A. A., Ali, M., Darwish, D. B. E., AlShaqhaa, M. A., Selim, D. A.-F. H., Nagah, A., et al. (2024a). Induced genetic diversity through mutagenesis in wheat gene pool and significant use of SCoT markers to underpin key agronomic traits. BMC Plant Biol. 24, 673. doi: 10.1186/s12870-024-05345-5
Abdelhameed, A. A., Eissa, M. A., El-kholy, R. I., Darwish, D. B. E., Abeed, A. H. A., Soudy, F. A., et al. (2024b). Molecular cloning and expression analysis of geranyllinalool synthase gene (SgGES) from salvia guaranitica plants. Horticulturae 10, 668. doi: 10.3390/horticulturae10070668
Abd El-Wahab, M., Toaima, W., Hamed, E. (2015). Effect of different planting locations in Egypt on salvia fruticosa mill. Plants. Egypt. J. Desert Res. 65, 291–307. doi: 10.21608/ejdr.2015.5955
Ahmed, M., Iqbal, A., Latif, A., Din, S., Sarwar, M. B., Wang, X., et al. (2020). Overexpression of a sucrose synthase gene indirectly improves cotton fiber quality through sucrose cleavage. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.476251
Ahmed, M., Shahid, A. A., Akhtar, S., Latif, A., Din, S., Fanglu, M., et al. (2018). Sucrose synthase genes: a way forward for cotton fiber improvement. Biol. (Bratisl). 73, 703–713. doi: 10.2478/s11756-018-0078-6
Akihiro, T., Mizuno, K., Fujimura, T. (2005). Gene expression of ADP-glucose pyrophosphorylase and starch contents in rice cultured cells are cooperatively regulated by sucrose and ABA. Plant Cell Physiol. 46, 937–946. doi: 10.1093/pcp/pci101
Ali, M. (2023). Cloning, molecular characterization and functional analysis of the cis-muuroladiene synthase (Sgcms) gene from leaves of salvia guaranitica plant. Egypt. J. Desert Res. 73, 239–263. doi: 10.21608/ejdr.2023.209304.1142
Ali, M., Abdelkawy, A. M., Darwish, D. B. E., Alatawi, H. A., Alshehri, D., Al-Amrah, H., et al. (2023a). Changes in Metabolite Profiling and Expression Levels of Key Genes Involved in the Terpenoid Biosynthesis Pathway in Garden Sage (Salvia officinalis) under the Effect of Hydrazine Hydrate. Metabolites 13, 807. doi: 10.3390/metabo13070807
Ali, M., Alshehri, D., Alkhaibari, A. M., Elhalem, N. A., Darwish, D. B. E. (2022a). Cloning and characterization of 1,8-cineole synthase (SgCINS) gene from the leaves of salvia guaranitica plant. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.869432
Ali, M., El-ramah, F. A., Elsherbeny, E. A., Suliman, M. N. S., Rizk, M. S. (2023b). Cloning and Characterization of Terpene synthase 3 (SoTPS3) Gene from Leaves of Garden Sage (Salvia officinalis). Jordan J. Biol. Sci. 16, 673–685. doi: 10.54319/jjbs/160413
Ali, M., Hussain, R. M., Rehman, N. U., She, G., Li, P., Wan, X., et al. (2018). De novo transcriptome sequencing and metabolite profiling analyses reveal the complex metabolic genes involved in the terpenoid biosynthesis in Blue Anise Sage (Salvia guaranitica L.). DNA Res. 25, 597–617. doi: 10.1093/dnares/dsy028
Ali, M., Li, P., She, G., Chen, D., Wan, X., Zhao, J. (2017). Transcriptome and metabolite analyses reveal the complex metabolic genes involved in volatile terpenoid biosynthesis in garden sage (Salvia officinalis). Sci. Rep. 7, 16074. doi: 10.1038/s41598-017-15478-3
Ali, M., Miao, L., Soudy, F. A., Darwish, D. B. E., Alrdahe, S. S., Alshehri, D., et al. (2022b). Overexpression of terpenoid biosynthesis genes modifies root growth and nodulation in soybean (Glycine max). Cells 11, 2622. doi: 10.3390/cells11172622
Ali, M., Nishawy, E., Ramadan, W. A., Ewas, M., Rizk, M. S., Sief-Eldein, A. G. M., et al. (2022c). Molecular characterization of a Novel NAD+-dependent farnesol dehydrogenase SoFLDH gene involved in sesquiterpenoid synthases from Salvia officinalis. PloS One 17, e0269045. doi: 10.1371/journal.pone.0269045
Badawi, M. A., Agharbaoui, Z., Zayed, M., Li, Q., Byrns, B., Zou, J., et al. (2019). Genome-wide identification and characterization of the wheat remorin (TaREM) family during cold acclimation. Plant Genome 12. doi: 10.3835/plantgenome2018.06.0040
Bairakdar, M. D., Tewari, A., Truttmann, M. C. (2023). A meta-analysis of RNA-Seq studies to identify novel genes that regulate aging. Exp. Gerontol. 173, 112107. doi: 10.1016/j.exger.2023.112107
Chandra, A., Verma, P. K., Islam, M. N., Grisham, M. P., Jain, R., Sharma, A., et al. (2015). Expression analysis of genes associated with sucrose accumulation in sugarcane (Saccharum spp. hybrids) varieties differing in content and time of peak sucrose storage. Plant Biol. 17, 608–617. doi: 10.1111/plb.12276
Collins, H. M., Betts, N. S., Dockter, C., Berkowitz, O., Braumann, I., Cuesta-Seijo, J. A., et al. (2021). Genes that mediate starch metabolism in developing and germinated barley grain. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.641325
Darwish, D. B. E., Ali, M., Abdelkawy, A. M., Zayed, M., Alatawy, M., Nagah, A. (2022). Constitutive overexpression of GsIMaT2 gene from wild soybean enhances rhizobia interaction and increase nodulation in soybean (Glycine max). BMC Plant Biol. 22, 431. doi: 10.1186/s12870-022-03811-6
Delaunois, B., Farace, G., Jeandet, P., Clément, C., Baillieul, F., Dorey, S., et al. (2014). Elicitors as alternative strategy to pesticides in grapevine? Current knowledge on their mode of action from controlled conditions to vineyard. Environ. Sci. pollut. Res. 21, 4837–4846. doi: 10.1007/s11356-013-1841-4
De Vega, J. J., Peel, N., Purdy, S. J., Hawkins, S., Donnison, L., Dyer, S., et al. (2021). Differential expression of starch and sucrose metabolic genes linked to varying biomass yield in Miscanthus hybrids. Biotechnol. Biofuels 14, 98. doi: 10.1186/s13068-021-01948-4
Dian, W., Jiang, H., Chen, Q., Liu, F., Wu, P. (2003). Cloning and characterization of the granule-bound starch synthase II gene in rice: gene expression is regulated by the nitrogen level, sugar and circadian rhythm. Planta 218, 261–268. doi: 10.1007/s00425-003-1101-9
Dian, W., Jiang, H., Wu, P. (2005). Evolution and expression analysis of starch synthase III and IV in rice. J. Exp. Bot. 56, 623–632. doi: 10.1093/jxb/eri065
El-Mahdy, M. T., Ali, M., Pisam, W. M. M., Abeed, A. H. A. (2024). Physiological and molecular analysis of pitaya (Hylocereus polyrhizus) reveal up-regulation of secondary metabolites, nitric oxide, antioxidant defense system, and expression of responsive genes under low-temperature stress by the pre-treatment of hydroge. Plant Physiol. Biochem. 213, 108840. doi: 10.1016/j.plaphy.2024.108840
El-ramah, F., Ali, M., Elsherbeny, E., Ahmed, M. (2022). Molecular cloning and characterization of beta-amyrin synthase (soamys) gene from salvia officinalis plant. Egypt. J. Desert Res. 72, 27–45. doi: 10.21608/ejdr.2022.122501.1099
Elsherbeny, E. A., ALI, M., El-Ramah, F. A., Ahmed, M. K. (2022). Molecular cloning and characterization of terpene synthase 4 (sgtps4) gene from salvia guaranitica plant. Egypt. J. Genet. Cytol. 51, 1–20. Available at: https://journal.esg.net.eg/index.php/EJGC/article/view/352.
Fan, Y., Gao, P., Zhou, T., Pang, S., Zhang, J., Yang, T., et al. (2023). Genome-Wide Identification and Expression Analysis of the Trehalose-6-phosphate Synthase and Trehalose-6-phosphate Phosphatase Gene Families in Rose (Rosa hybrida cv ‘Carola’) under Different Light Conditions. Plants 13, 114. doi: 10.3390/plants13010114
Gu, Y., Han, S., Chen, L., Mu, J., Duan, L., Li, Y., et al. (2021). Expression and regulation of genes involved in the reserve starch biosynthesis pathway in hexaploid wheat (Triticum aestivum L.). Crop J. 9, 440–455. doi: 10.1016/j.cj.2020.08.002
Huang, H.-H., Xu, L.-L., Tong, Z.-K., Lin, E.-P., Liu, Q.-P., Cheng, L.-J., et al. (2012). De novo characterization of the Chinese fir (Cunninghamia lanceolata) transcriptome and analysis of candidate genes involved in cellulose and lignin biosynthesis. BMC Genomics 13, 648. doi: 10.1186/1471-2164-13-648
Huang, J., Tang, B., Ren, R., Wu, M., Liu, F., Lv, Y., et al. (2022). Understanding the potential gene regulatory network of starch biosynthesis in tartary buckwheat by RNA-seq. Int. J. Mol. Sci. 23, 15774. doi: 10.3390/ijms232415774
Huang, L., Tan, H., Zhang, C., Li, Q., Liu, Q. (2021). Starch biosynthesis in cereal endosperms: An updated review over the last decade. Plant Commun. 2, 100237. doi: 10.1016/j.xplc.2021.100237
Hussain, R. M., Ali, M., Feng, X., Li, X. (2017). The essence of NAC gene family to the cultivation of drought-resistant soybean (Glycine max L. Merr.) cultivars. BMC Plant Biol. 17, 55. doi: 10.1186/s12870-017-1001-y
Jeon, J.-S., Ryoo, N., Hahn, T.-R., Walia, H., Nakamura, Y. (2010). Starch biosynthesis in cereal endosperm. Plant Physiol. Biochem. 48, 383–392. doi: 10.1016/j.plaphy.2010.03.006
Jiang, Z., Zhang, H., Gao, S., Zhai, H., He, S., Zhao, N., et al. (2023). Genome-wide identification and expression analysis of the sucrose synthase gene family in sweet potato and its two diploid relatives. Int. J. Mol. Sci. 24, 12493. doi: 10.3390/ijms241512493
Kanehisa, M. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kanehisa, M. (2019). Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. doi: 10.1002/pro.3715
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M., Ishiguro-Watanabe, M. (2023). KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. doi: 10.1093/nar/gkac963
Ke, F., Zhang, K., Li, Z., Wang, J., Zhang, F., Wu, H., et al. (2022). Transcriptomic analysis of starch accumulation patterns in different glutinous sorghum seeds. Sci. Rep. 12, 11133. doi: 10.1038/s41598-022-15394-1
Keunen, E., Peshev, D., Vangronsveld, J., Van Den Ende, W., And Cuypers, A. (2013). Plant sugars are crucial players in the oxidative challenge during abiotic stress: extending the traditional concept. Plant Cell Environ. 36, 1242–1255. doi: 10.1111/pce.12061
Khater, R. (2022). Evaluating the productivity of salvia officinalis, l. Plants using of fertilizers and spraying with vitamins. Egypt. J. Desert Res. 72, 47–71. doi: 10.21608/ejdr.2022.115098.1097
Koch, K. E. (1996). Carbohydrate-modulated gene expression in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47, 509–540. doi: 10.1146/annurev.arplant.47.1.509
Koch, K. (2004). Sucrose metabolism: regulatory mechanisms and pivotal roles in sugar sensing and plant development. Curr. Opin. Plant Biol. 7, 235–246. doi: 10.1016/j.pbi.2004.03.014
Li, J., Jiang, S., Yang, G., Xu, Y., Li, L., Yang, F. (2023). RNA-sequencing analysis reveals novel genes involved in the different peel color formation in eggplant. Hortic. Res. 10. doi: 10.1093/hr/uhad181
Li, Q., Byrns, B., Badawi, M. A., Diallo, A. B., Danyluk, J., Sarhan, F., et al. (2018). Transcriptomic insights into phenological development and cold tolerance of wheat grown in the field. Plant Physiol. 176, 2376–2394. doi: 10.1104/pp.17.01311
Li, W., Huang, L., Liu, N., Pandey, M. K., Chen, Y., Cheng, L., et al. (2021). Key regulators of sucrose metabolism identified through comprehensive comparative transcriptome analysis in peanuts. Int. J. Mol. Sci. 22, 7266. doi: 10.3390/ijms22147266
Li, Y., Yao, Y., Yang, G., Tang, J., Ayala, G. J., Li, X., et al. (2020). Co-crystal structure of thermosynechococcus elongatus sucrose phosphate synthase with UDP and sucrose-6-phosphate provides insight into its mechanism of action involving an oxocarbenium ion and the glycosidic bond. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.01050
Liu, B., Lin, R., Jiang, Y., Jiang, S., Xiong, Y., Lian, H., et al. (2020). Transcriptome analysis and identification of genes associated with starch metabolism in castanea henryi seed (Fagaceae). Int. J. Mol. Sci. 21, 1431. doi: 10.3390/ijms21041431
Liu, K., Zhou, Y. (2022). Genome-wide identification of the trehalose-6-phosphate synthase gene family in sweet orange ( Citrus sinensis ) and expression analysis in response to phytohormones and abiotic stresses. PeerJ 10, e13934. doi: 10.7717/peerj.13934
Liu, Z., Jiang, S., Jiang, L., Li, W., Tang, Y., He, W., et al. (2022). Transcription factor OsSGL is a regulator of starch synthesis and grain quality in rice. J. Exp. Bot. 73, 3417–3430. doi: 10.1093/jxb/erac068
Luo, Y., Xie, Y., Li, W., Wei, M., Dai, T., Li, Z., et al. (2021). Physiological and transcriptomic analyses reveal exogenous trehalose is involved in the responses of wheat roots to high temperature stress. Plants 10, 2644. doi: 10.3390/plants10122644
Lyra, D. H., Griffiths, C. A., Watson, A., Joynson, R., Molero, G., Igna, A., et al. (2021). Gene-based mapping of trehalose biosynthetic pathway genes reveals association with source- and sink-related yield traits in a spring wheat panel. Food Energy Secur. 10. doi: 10.1002/fes3.292
Macovei, A., Pagano, A., Cappuccio, M., Gallotti, L., Dondi, D., De Sousa Araujo, S., et al. (2019). A snapshot of the trehalose pathway during seed imbibition in medicago truncatula reveals temporal- and stress-dependent shifts in gene expression patterns associated with metabolite changes. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.01590
Makhadmeh, I., Albalasmeh, A. A., Ali, M., Thabet, S. G., Darabseh, W. A., Jaradat, S., et al. (2022a). Molecular Characterization of Tomato (Solanum lycopersicum L.) Accessions under Drought Stress. Horticulturae 8, 600. doi: 10.3390/horticulturae8070600
Makhadmeh, I. M., Thabet, S. G., Ali, M., Alabbadi, B., Albalasmeh, A., Alqudah, A. M. (2022b). Exploring genetic variation among Jordanian Solanum lycopersicon L. landraces and their performance under salt stress using SSR markers. J. Genet. Eng. Biotechnol. 20, 45. doi: 10.1186/s43141-022-00327-2
Mehmood, N., Yuan, Y., Ali, M., Ali, M., Iftikhar, J., Cheng, C., et al. (2021). Early transcriptional response of terpenoid metabolism to Colletotrichum gloeosporioides in a resistant wild strawberry Fragaria nilgerrensis. Phytochemistry 181, 112590. doi: 10.1016/j.phytochem.2020.112590
Nguyen-Quoc, B., Foyer, C. H. (2001). A role for ‘futile cycles’ involving invertase and sucrose synthase in sucrose metabolism of tomato fruit. J. Exp. Bot. 52, 881–889. doi: 10.1093/jexbot/52.358.881
Ortiz-Marchena, M. I., Albi, T., Lucas-Reina, E., Said, F. E., Romero-Campero, F. J., Cano, B., et al. (2014). Photoperiodic control of carbon distribution during the floral transition in arabidopsis. Plant Cell 26, 565–584. doi: 10.1105/tpc.114.122721
Rastogi, S., Meena, S., Bhattacharya, A., Ghosh, S., Shukla, R. K., Sangwan, N. S., et al. (2014). De novo sequencing and comparative analysis of holy and sweet basil transcriptomes. BMC Genomics 15, 588. doi: 10.1186/1471-2164-15-588
Rehman, N., Ali, M., Ahmad, M. Z., Liang, G., Zhao, J. (2018). Strigolactones promote rhizobia interaction and increase nodulation in soybean (Glycine max). Microb. Pathog. 114, 420–430. doi: 10.1016/j.micpath.2017.11.049
Rolland, F., Baena-Gonzalez, E., Sheen, J. (2006). SUGAR SENSING AND SIGNALING IN PLANTS: conserved and novel mechanisms. Annu. Rev. Plant Biol. 57, 675–709. doi: 10.1146/annurev.arplant.57.032905.105441
Rolland, F., Moore, B., Sheen, J. (2002). Sugar sensing and signaling in plants. Plant Cell 14, S185–S205. doi: 10.1105/tpc.010455
Safavi-Rizi, V., Herde, M., Stöhr, C. (2020). RNA-Seq reveals novel genes and pathways associated with hypoxia duration and tolerance in tomato root. Sci. Rep. 10, 1692. doi: 10.1038/s41598-020-57884-0
Santa Cruz, J., González, D., Valdebenito, S., Peñaloza, P. (2021). Salvia guaranitica A. St.-Hil. ex Benth. (Lamiaceae): a new record for the alien flora of Chile. Gayana. Botánica 78, 95–98. doi: 10.4067/S0717-66432021000100095
Sheen, J., Zhou, L., Jang, J.-C. (1999). Sugars as signaling molecules. Curr. Opin. Plant Biol. 2, 410–418. doi: 10.1016/S1369-5266(99)00014-X
Singh, V., Louis, J., Ayre, B. G., Reese, J. C., Shah, J. (2011). TREHALOSE PHOSPHATE SYNTHASE11 -dependent trehalose metabolism promotes Arabidopsis thaliana defense against the phloem-feeding insect Myzus persicae. Plant J. 67, 94–104. doi: 10.1111/j.1365-313X.2011.04583.x
Smeekens, S., Ma, J., Hanson, J., Rolland, F. (2010). Sugar signals and molecular networks controlling plant growth. Curr. Opin. Plant Biol. 13, 273–278. doi: 10.1016/j.pbi.2009.12.002
Sonnewald, U., Brauer, M., von Schaewen, A., Stitt, M., Willmitzer, L. (1991). Transgenic tobacco plants expressing yeast-derived invertase in either the cytosol, vacuole or apoplast: a powerful tool for studying sucrose metabolism and sink/source interactions. Plant J. 1, 95–106. doi: 10.1111/j.1365-313X.1991.00095.x
Srividya, N., Davis, E. M., Croteau, R. B., Lange, B. M. (2015). Functional analysis of (4 S )-limonene synthase mutants reveals determinants of catalytic outcome in a model monoterpene synthase. Proc. Natl. Acad. Sci. 112, 3332–3337. doi: 10.1073/pnas.1501203112
Stein, O., Granot, D. (2019). An overview of sucrose synthases in plants. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00095
Sukko, N., Kalapanulak, S., Saithong, T. (2023). Trehalose metabolism coordinates transcriptional regulatory control and metabolic requirements to trigger the onset of cassava storage root initiation. Sci. Rep. 13, 19973. doi: 10.1038/s41598-023-47095-8
Sung, S. S., Xu, D., Galloway, C. M., Black, C. C. (1988). A reassessment of glycolysis and gluconeogenesis in higher plants. Physiol. Plant 72, 650–654. doi: 10.1111/j.1399-3054.1988.tb09177.x
Taujale, R., Zhou, Z., Yeung, W., Moremen, K. W., Li, S., Kannan, N. (2021). Mapping the glycosyltransferase fold landscape using interpretable deep learning. Nat. Commun. 12, 5656. doi: 10.1038/s41467-021-25975-9
Trouvelot, S., Héloir, M.-C., Poinssot, B., Gauthier, A., Paris, F., Guillier, C., et al. (2014). Carbohydrates in plant immunity and plant protection: roles and potential application as foliar sprays. Front. Plant Sci. 5. doi: 10.3389/fpls.2014.00592
Walters, D. R., Ratsep, J., Havis, N. D. (2013). Controlling crop diseases using induced resistance: challenges for the future. J. Exp. Bot. 64, 1263–1280. doi: 10.1093/jxb/ert026
Wang, L., Ruan, Y.-L. (2013). Regulation of cell division and expansion by sugar and auxin signaling. Front. Plant Sci. 4. doi: 10.3389/fpls.2013.00163
Xiao, N., Ma, H., Wang, W., Sun, Z., Li, P., Xia, T. (2024). Overexpression of ZmSUS1 increased drought resistance of maize (Zea mays L.) by regulating sucrose metabolism and soluble sugar content. Planta 259, 43. doi: 10.1007/s00425-024-04336-y
Xiao, Q., Huang, T., Cao, W., Ma, K., Liu, T., Xing, F., et al. (2022). Profiling of transcriptional regulators associated with starch biosynthesis in sorghum (Sorghum bicolor L.). Front. Plant Sci. 13. doi: 10.3389/fpls.2022.999747
Yang, H., Sun, M., Lin, S., Guo, Y., Yang, Y., Zhang, T., et al. (2017). Transcriptome analysis of Crossostephium chinensis provides insight into the molecular basis of salinity stress responses. PloS One 12, e0187124. doi: 10.1371/journal.pone.0187124
Yang, H., Wei, Z., Wu, Y., Zhang, C., Lyu, L., Wu, W., et al. (2023). Transcriptomic and metabolomic profiling reveals the variations in carbohydrate metabolism between two blueberry cultivars. Int. J. Mol. Sci. 25, 293. doi: 10.3390/ijms25010293
Zayed, M., Badawi, M. A. (2020). In-silico evaluation of a new gene from wheat reveals the divergent evolution of the CAP160 homologous genes Into monocots. J. Mol. Evol. 88, 151–163. doi: 10.1007/s00239-019-09920-5
Zhang, C., Zhang, H., Zhan, Z., Liu, B., Chen, Z., Liang, Y. (2016). Transcriptome Analysis of Sucrose Metabolism during Bulb Swelling and Development in Onion (Allium cepa L.). Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01425
Zhang, X., Xie, S., Han, J., Zhou, Y., Liu, C., Zhou, Z., et al. (2019). Integrated transcriptome, small RNA, and degradome analysis reveals the complex network regulating starch biosynthesis in maize. BMC Genomics 20, 574. doi: 10.1186/s12864-019-5945-1
Zhang, Y.-W., Shi, Y.-C., Zhang, S.-B. (2023). Metabolic and transcriptomic analyses elucidate a novel insight into the network for biosynthesis of carbohydrate and secondary metabolites in the stems of a medicinal orchid Dendrobium nobile. Plant Divers. 45, 326–336. doi: 10.1016/j.pld.2022.10.004
Zhu, X., Wang, M., Li, X., Jiu, S., Wang, C., Fang, J. (2017). Genome-wide analysis of the sucrose synthase gene family in grape (Vitis vinifera): structure, evolution, and expression profiles. Genes (Basel). 8, 111. doi: 10.3390/genes8040111
Keywords: Salvia guaranitica, transcriptome, glycolysis, gluconeogenesis, starch, sucrose, transgenic Arabidopsis thaliana, functional characterization
Citation: Abbas ZK, Al-Huqail AA, Abdel Kawy AH, Abdulhai RA, Albalawi DA, AlShaqhaa MA, Alsubeie MS, Darwish DBE, Abdelhameed AA, Soudy FA, Makki RM, Aljabri M, Al-Sulami N, Ali M and Zayed M (2024) Harnessing de novo transcriptome sequencing to identify and characterize genes regulating carbohydrate biosynthesis pathways in Salvia guaranitica L.. Front. Plant Sci. 15:1467432. doi: 10.3389/fpls.2024.1467432
Received: 19 July 2024; Accepted: 22 August 2024;
Published: 26 September 2024.
Edited by:
Ahmed Sallam, Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), GermanyReviewed by:
Omar A. Hewedy, Menoufia University, EgyptMuna Ahmed, Huazhong Agricultural University, China
Ihteram Ullah, Gomal University, Pakistan
Copyright © 2024 Abbas, Al-Huqail, Abdel Kawy, Abdulhai, Albalawi, AlShaqhaa, Alsubeie, Darwish, Abdelhameed, Soudy, Makki, Aljabri, Al-Sulami, Ali and Zayed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arwa Abdulkreem Al-Huqail, YWFhbGh1cWFpbEBwbnUuZWR1LnNh; Muhammad Zayed, bWhkYXRlZnpheWVkQGdtYWlsLmNvbQ==