 Shanbin Xu1†
Shanbin Xu1† Jie Zheng1†Haoqiang Du1†Xiaodong Du2Chong Li1Yuxuan Duan1Yanan Cai1Jingguo Wang1Hualong Liu1
Jie Zheng1†Haoqiang Du1†Xiaodong Du2Chong Li1Yuxuan Duan1Yanan Cai1Jingguo Wang1Hualong Liu1 Luomiao Yang1Wei Xin1Yan Jia1
Luomiao Yang1Wei Xin1Yan Jia1 Detang Zou1*
Detang Zou1* Hongliang Zheng1*
Hongliang Zheng1*- 1Key Laboratory of Germplasm Enhancement and Physiology & Ecology of Food Crop in Cold Region, Ministry of Education/College of Agriculture, Northeast Agricultural University, Harbin, China
- 2Rice Research Institute, Heilongjiang Academy of Agricultural Sciences, Jiamusi, China
Background: Soil salinization is one of the significant factors limiting high crop yields and expansion of arable land, seriously affecting global agricultural production. Rice is an essential food crop throughout the world, and its seedlings are particularly susceptible to salt stress, which can directly affect the growth and development of rice and its final yield. We used the natural population as the material for genome-wide association study (GWAS) and the recombinant inbred line (RIL) population from CD (salt sensitive)/WD20342 (salt tolerant) hybridization as the material for linkage analysis.
Results: The degree of salt tolerance was evaluated by using the relative root length (RRL), relative root number (RRN), relative root fresh weight (RRFW), and relative root dry weight (RRDW) as indicators. Fifteen and six major quantitative trait loci (QTLs) were identified by GWAS and linkage analysis, respectively. Meanwhile, the GWAS identified the lead SNP (Chr2_22340368), which was located within qRRL2 and qRRDW2 identified by linkage analysis. GWAS, combined with linkage analysis, selected a 196-kb overlapping region on chromosome 2, including 22 candidate genes. LOC_Os02g36880 was discovered as the candidate gene involved in salt tolerance by haplotype analysis, qRT-PCR, and sequence analysis. The score of salinity toxicity (SST) and seedling survival rate (SSR) were determined for CRISPR/Cas9 mutants (CR-1 and CR-15) and wild-type (ZH11), respectively.
Conclusion: The phenotypic validation indicated that LOC_Os02g36880 negatively regulated the salt tolerance at the seedling stage. This study provides resources for breeding Japonica rice to improve its response to salt stress.
1 Introduction
Rice is an essential crop globally, and with the growing global population, the demand for higher rice yields is increasingly urgent. However, various abiotic factors including drought, salinity, alkalinity, and cold, significant impact crop yields (Radha et al., 2023; Raj and Nadarajah, 2023; Sarma et al., 2023). Among these, salt damage is one of the most critical factors reducing rice productivity, as rice is highly sensitive to saline conditions (Ruan et al., 2011; Chen et al., 2022). Soil salinity affects a substantial portion of arable land, and the shrinking availability of agricultural land further constrains global agricultural productivity. High salt concentrations lead to ionic toxicity, hyperosmotic stress, and secondary stresses, which together disrupt essential rice processes throughout its life cycle, from germination to senescence (Hanin et al., 2016; Zhu, 2016). Rice is particularly vulnerable to the adverse effects of salt stress during the seedling stages. Thus, understanding the genetic mechanisms and identifying relevant QTLs or genes is crucial for developing salt-tolerant rice cultivars, which forms the theoretical and practical foundation for breeding varieties.
Salt tolerance in rice is a typical quantitative trait jointly controlled by multiple genes and the integrated expression of various mechanisms (Liu et al., 2022; Phosuwan et al., 2024). Significant progress has been made by employing forward genetics strategies, such as GWAS and linkage analysis, to identify numerous QTLs and genes associated with salt tolerance. For instance, Kumar et al. (2015) evaluated twelve salt-related agronomic traits using a natural population of 220 rice accessions. By combining these traits with a gene microarray containing 6,000 single nucleotide polymorphisms (SNPs), they identified sixty-four loci, which explained 5% to 18% of the phenotypic variance. Similarly, Al-Tamimi et al. (2016) conducted a GWAS on 553 rice varieties identified 12 efficient QTLs associated with salt tolerance based on indicators such as relative growth rate, transpiration rate, and transpiration efficiency. Batayeva et al. (2018) identified 26 QTLs linked to salt tolerance in 203 rice accessions, with eleven of these QTLs overlapping with previously known loci. Rohila et al. (2019) used a hydroponic system to subject 162 rice accessions to salt stress during the seedling stage, leading to the identification of nine genomic regions associated with salt tolerance by analyzing over three million SNPs. Additionally, they discovered six new loci and identified sixteen potential genes. Yuan et al. (2020) utilized 664 cultivated rice materials for GWAS, identifying 21 QTLs associated with salt tolerance.
Meanwhile, researchers have also found several QTLs using linkage analysis. De Leon et al. (2016) utilized a RIL population derived from a cross between the Bengal (salt-sensitive) and the Pokkali (salt-tolerant) to identify 85 additive QTLs for nine salt-related traits, including salt injury score, shoot length, root length. Nakhla et al. (2021) identified 23 and 46 QTLs controlling salt tolerance at the germination and seedling stages, respectively, through linkage mapping. Zeng et al. (2021) used the a BC1F2 population derived from a cross between WJZ and Nipponbare to identify the major salt tolerance QTL qGR6.2, during the germination stage and predicted 11 candidate genes. Geng et al. (2023) employed a RIL population derived from a cross between Milyang 23 (salt-tolerant) and Jileng 1 (salt-sensitive) to identify 12 QTLs through linkage mapping.
In this study, we explored the genetic foundation of salt tolerance in rice using GWAS and linkage analysis with RRL, RRN, RRFW, and RRDW during the rice seedling salt treatment. Both GWAS and linkage analysis are powerful tools for detecting QTLs associated with complex traits, and their combination can significantly enhance the accuracy of QTL detection. Our GWAS identified a lead SNP (Chr2_22340368), within the QTLs qRRL2 and qRRDW2, which were also identified through linkage analysis. A 196-kb region on chromosome 2 was selected for further examination based on the overlapping regions identified through both GWAS and linkage mapping. Knockout mutants were subsequently used for the functional verification, leading to the identification of LOC_Os02g36880 as a gene that contributes to salinity resistance.
2 Materials and methods
2.1 Plant materials
The natural population was conducted using the 295 Japonica rice materials gathered from Chinese provinces such as Heilongjiang, Ningxia, and Jilin, along with foreign varieties mainly from Japan, Russia, and Korea (Li et al., 2019). The linkage mapping population consisted of 189 RILs, a cross between the CD (salt-sensitive) and WD20342 (salt-tolerant) (Li et al., 2023). Supplementary Figure S1 shows CD and WD20342 salt tolerance phenotypes. In Heilongjiang Province, China, at the Northeast Agricultural University Experimental Station (45°52′N, 127°04′E), all materials were planted.
2.2 Phenotype data
All materials were baked for 48 hours at 55°C, after choosing and disinfecting the seeds for 20 minutes with a 3% NaClO solution. The seeds were cultured hydroponically at 30°C for 4 days. Using the Yoshida nutrient solution, sixty-four evenly germinated seedlings of each type were divided into salt-treated and control groups for hydroponic cultivation. The germinated seeds were incubated at 26 and 24°C, with a light and dark cycle of 14 and 10 hours, respectively. Once the seedlings reached two leaves and one heart, the control group received standard nutrient solution application; the treatment group received salt stress treatment. Pretreatment of the seedlings was 50 mM NaCl, which was applied for 3 days, and formally treated with 120 mM NaCl for 7 days. Five plants were randomly chosen for the measurement in each material, including the root length, the root number, the root fresh weight, and the root dry weight under control and salt treatment. Further calculations were performed to determine the RRL, RRN, RRFW, and RRDW: the relative values represent the ratio of phenotypic values during salt treatments to phenotypic values under control conditions. Three replications of the experiment were set up.
2.3 GWAS
Using Plink 2.0 software (Chang et al., 2015), 788,396 SNPs were identified with a minor allele frequency (MAF) of ≥5% and a missing rate of ≤20%. The 295 materials were analysed by GWAS using 788,396 SNPs. The calculated population structure (Q) and kinship between individuals were used to adjust the population structure of GWAS (Li et al., 2019). The mixed linear model (MLM) included in TASSEL 5.0 (Bradbury et al., 2007) was used for GWAS. The threshold for the identification of SNPs significantly associated with traits was set to –log10(P) > 5.26, determined by the genetic type 1 error calculator (GEC; http://statgenpro.psychiatry.hku.hk/gec/), which calculates the effective number of independent markers. Generate the Manhattan map using the R program ‘qqman’. To determine the leading SNPs with the lowest P-value, redundant SNPs were filtered using a least-distance interval. Utilizing the LDBlockShow software, the pairwise R2 value between any two SNPs within the interval of leading SNPs ± 2 Mb was determined. The LD attenuation interval of leading SNPs between 1.5 and 2.0 Mb was the average of the top 10% R2 values plus 0.2 (Dong et al., 2020).
2.4 Linkage analysis
Using 10 K Array targeted sequencing technology from MOLBREEDING Biotechnology Company, the genotypes of the RIL lines were determined. Following biparental polymorphism analysis and de-redundant analysis and by the IciMapping-Bin project, there was a grouping of specific SNP markers with the same genotype into one “BIN”. Using 189 RILs from CD and WD20342 and 978 bin markers, a genetic linkage mapping was produced. With an average distance of 2.52 cM between markers, the map contains 2465.33 cm of the genome (Supplementary Figure S2). The inclusive composite interval mapping method used in ICIMapping Ver.4.2 was utilized for linkage analysis, with LOD > 2.5 as the threshold value (Lei et al., 2021).
2.5 Haplotype analysis
Non-synonymous mutant SNPs in the promoter (1.5 kb before ATG) and exonic regions of genes were retrieved using the RiceSNPSeekDatabase website (https://snp-seek.irri.org/_snp.zul). Haplotype analysis was done by the DnaSP software application. The number of materials needs to be ten or more to be classified as different haplotypes.
2.6 qRT-PCR and sequence analysis
Using qRT-PCR analysis in both standard and salinized conditions, the expression levels of the candidate genes associated with both CD and WD20342 were confirmed. Roche LightCycler96 was used to carry out the qRT-PCR analysis. Supplementary Table S3 contains all of the primer sequences. The promoter and exonic regions of genes were amplified using PCR.
2.7 LOC_Os02g36880 mutant plants
By utilizing CRISPR/Cas9 technology, the mutants were created, and the T1 generation LOC_Os02g36880 knockout mutant seeds in the Zhonghua11 (ZH11) background were made by BIOGLE GENETECH company (http://www.biogle.cn/). PCR amplification was used to identify two homozygous mutants (CR-1, CR-15) in the T1 generation. These plants were then selfed to produce the T3 generation. The experiment was conducted with three replications using the wild-type (ZH11) and T3 generation homozygous mutants as the experimental materials. The experimental materials were separated into two groups. One group got a regular nutrient solution when they reached two leaves and one heart, while the other group received a nutrient solution with 120 mM NaCl. The therapy lasted 7 days, followed by a recovery period of 10 days. The treatment lasted for 7 days, and the recovery period lasted for 10 days. Salt tolerance was determined by measuring the SST and SSR.
3 Results
3.1 Phenotypes of GWAS and RIL population
Data from 295 Japonica rice materials and RIL lines were analyzed statistically to determine four salt tolerance phenotypes: RRL, RRN, RRFW, and RRDW. Under the salt stress during the seedling stage, the RRL, RRN, RRFW, and RRDW among the 295 materials varied in range from 0.39-0.88, 0.41-0.88, 0.38-0.87, and 0.39-0.87, respectively, with corresponding coefficients of variation of 13.65%, 12.79%, 14.74%, and 14.71% (Supplementary Table S1). The ranges of variation for RRL, RRN, RRFW, and RRDW in the RIL lines were 0.37-0.87, 0.44-0.88, 0.40-0.89, and 0.39-0.89, with the coefficients of variation of 17.05%, 13.89%, 17.22%, and 17.78%, respectively (Supplementary Table S1). The phenotypic data demonstrate a wide range of phenotypic diversity to Japonica rice seedling salt tolerance among the 295 materials and RIL lines. The phenotypic values of four traits significantly differed between WD20342 and CD, revealing that WD20342 was more salinity-tolerant than CD (Supplementary Table S1). The RRL, RRN, RRFW, and RRDW phenotypic values of the 295 materials and RIL lines were typically distributed and showed continuous phenotypic variation for each trait, suggesting that these traits are quantitative and affected by multiple factors (Figure 1).
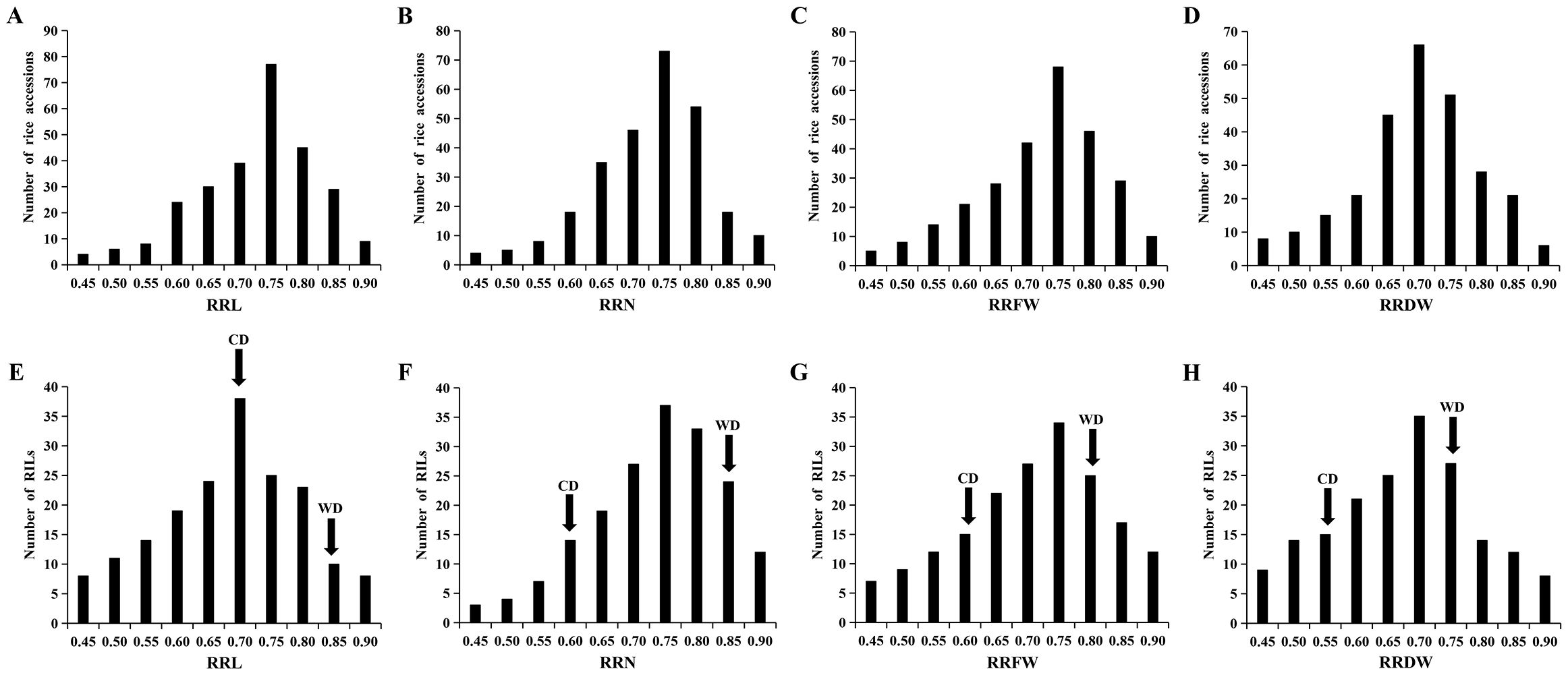
Figure 1. Phenotypic variation in the RRL, RRN, RRFW, and RRDW. (A-D) The RRL, RRN, RRFW, and RRDW of 295 materials. (E-H) The RRL, RRN, RRFW, and RRDW of RIL lines.
3.2 GWAS of salt-related traits in the seedling stage
GWAS was performed using the 788,396 SNPs acquired from previous research (Duan et al., 2022; Li et al., 2022). Fifteen lead SNPs were significantly related to RRL, RRN, RRFW, and RRDW by GWAS (Table 1; Figure 2). Four QTLs linked to RRL were identified on chromosomes 2, 3, 6, and 11, with R2 values varying between 9.52% and 11.30%. Three QTLs linked to RRN were identified on chromosomes 6, 10, and 11, with R2 values ranging from 8.35% to 11.12%. Five QTLs linked to RRFW were identified on chromosomes 4, 5, 6, 7, and 12, with R2 values ranging from 10.14% to 12.21%. Meanwhile, three QTLs linked to RRDW were identified on chromosomes 4, 6, and 9, with R2 values ranging from 8.42% to 10.27%.
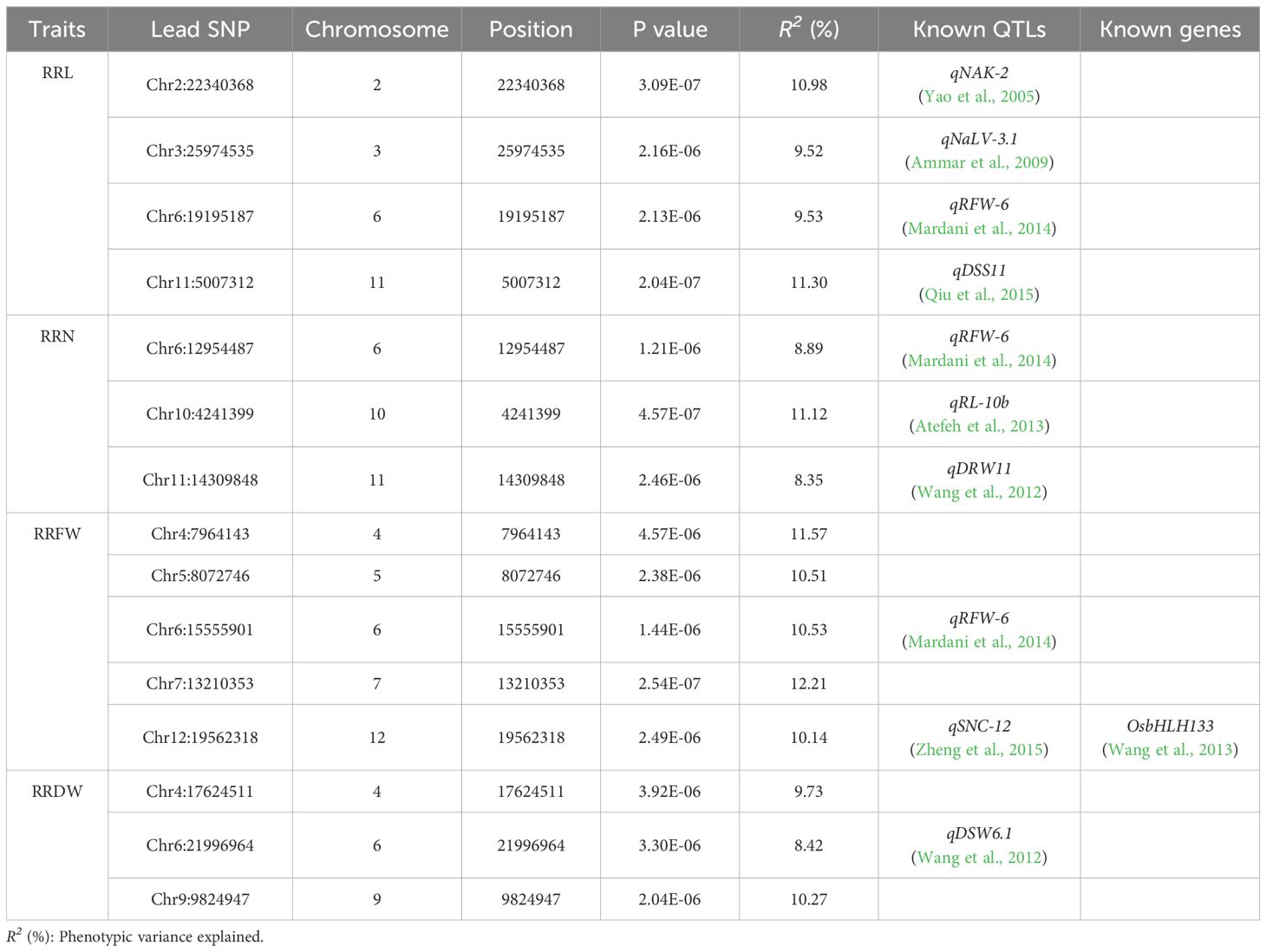
Table 1. Lead SNPs associated with RRL, RRN, RRFW, and RRDW were identified by GWAS.

Figure 2. Manhattan plots and Q-Q plots. (A-D) Manhattan plot for the RRL, RRN, RRFW, and RRDW. (E-H) Q-Q plot for the RRL, RRN, RRFW, and RRDW.
3.3 QTL mapping of RIL
Six major QTLs were found on chromosomes 1, 2, 6, 10, and 11 in the RIL lines. The explained phenotypic variance varied from 4.82% to 17.26%, while the LOD values ranged from 2.55 to 7.57. There was one QTL associated with RRL (qRRL2), one QTL associated with RRN (qRRN6), two QTLs associated with RRFW (qRRFW1 and qRRFW10), and two QTLs associated with RRDW (qRRDW2 and qRRDW11) (Table 2). Furthermore, qRRL2 and qRRDW2 were defined as the same QTL because of the same interval, located in the physical region between markers C2_21864234 and C2_24239570 and explaining 9.38-17.26% of the phenotypic variance.
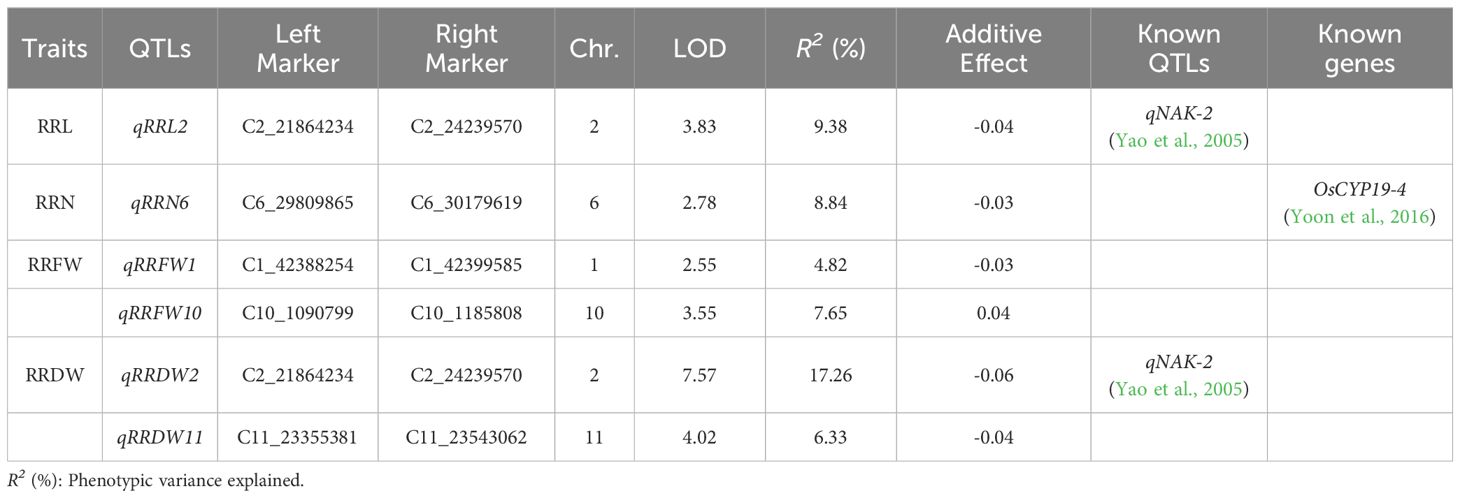
Table 2. QTLs for RRL, RRN, RRFW, and RRDW were identified by linkage analysis.
Significantly, the GWAS discovered the lead SNP related to RRL (Chr2_22340368), which was located within qRRL2 and qRRDW2. On chromosome 2, the LD block region was estimated to be 22.241-22.437 Mb (196 kb). Using GWAS and linkage analysis, a 196-kb overlap region was identified (Figure 3). Therefore, we identified the 196-kb overlap region as the significant QTL for candidate gene identification.
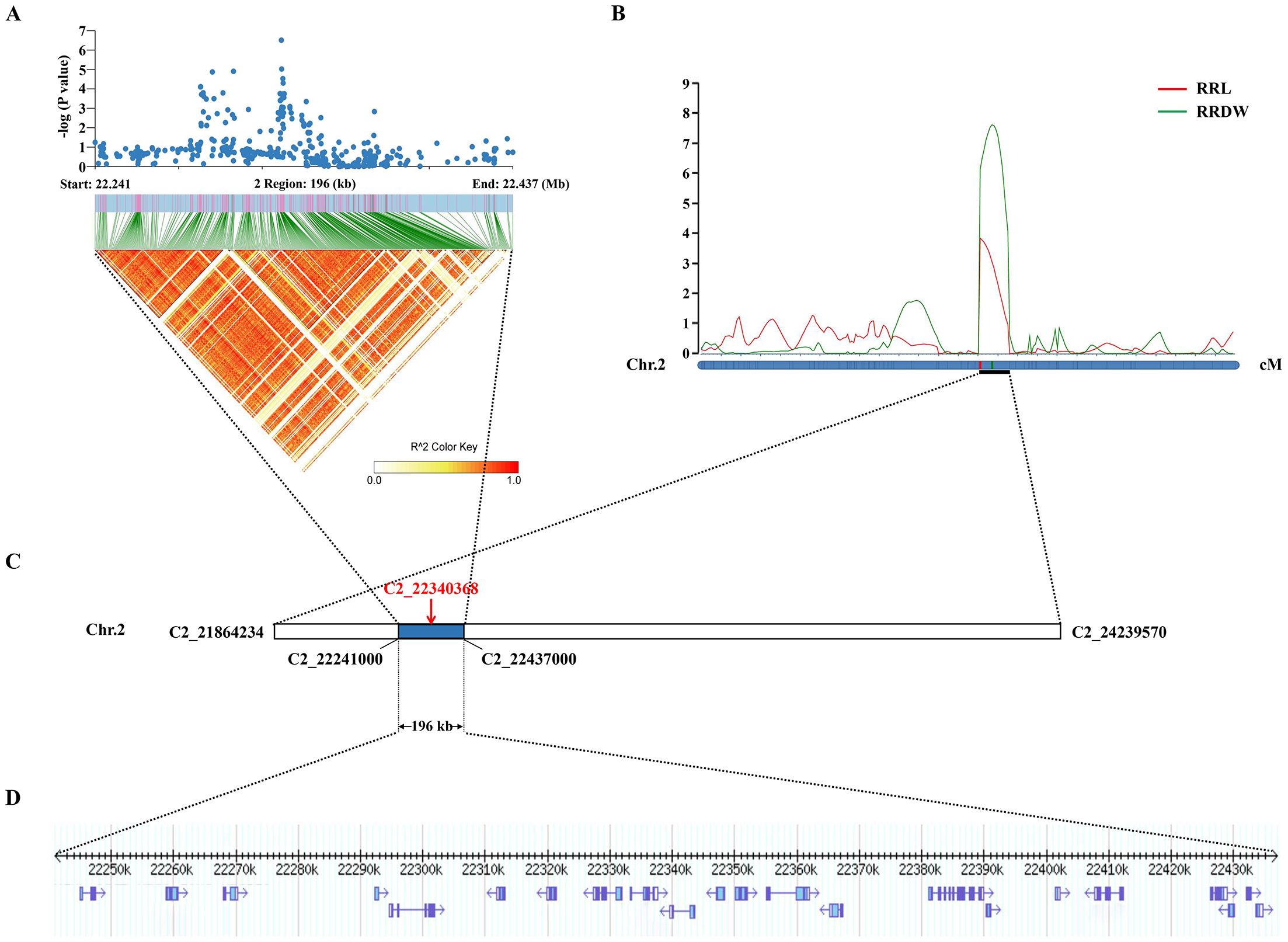
Figure 3. Co-location results of GWAS and linkage analysis. (A) The local Manhattan plot and LD heatmap surround the lead SNP. (B) Mapped to the interval between markers C2_21864234 and C2_24239570 by linkage analysis. (C) The lead SNP (C2_22340368) was identified by the GWAS. (D) The 196-kb region.
3.4 Haplotype analysis of candidate genes
The Rice Genome Annotation Project confirms that the 196-kb region contained 22 genes (Supplementary Table S2). We conducted haplotype analysis on 22 genes and identified 4 genes (LOC_Os02g36880, LOC_Os02g36950, LOC_Os02g37000, LOC_Os02g37080) in the overlapping region. LOC_Os02g36880 was classified into three haplotypes based on non-synonymous mutant SNPs in the exon region. LOC_Os02g36950, LOC_Os02g37000, and LOC_Os02g37080 were separated into two haplotypes due to non-synonymous mutation SNPs in the exon region (Figure 4).

Figure 4. Haplotype analysis of candidate genes. (A-D) The haplotype analysis of LOC_Os02g36880, LOC_Os02g36950, LOC_Os02g37000, and LOC_Os02g37080. (The * and ** suggest significance of ANOVA at P < 0.05 and P < 0.01, respectively).
3.5 Identification of candidate genes
Both parents, CD and WD20342, were treated with 120 mM NaCl for 0, 1, 3, 6, 12, and 24 h, respectively. Four genes (LOC_Os02g36880, LOC_Os02g36950, LOC_Os02g37000, LOC_Os02g37080) were determined by qRT-PCR analysis. LOC_Os02g36950 was no induction at all under salt stress, and expression of the other genes (LOC_Os02g36880, LOC_Os02g37000, and LOC_Os02g37080) were up-regulated under stress. Interestingly, only LOC_Os02g36880 showed significant upregulation of CD and WD20342 expression under salt stress, with significant differences in expression levels. In the 24h salt stress, the expression level of LOC_Os02g36880 is more than four times higher in CD than WD20342 (Figure 5). Meanwhile, qRT-PCR analysis of other functionally annotated genes within the 196-kb region showed that the eight genes expressed between the two parents were not differentially expressed (Supplementary Figure S3).
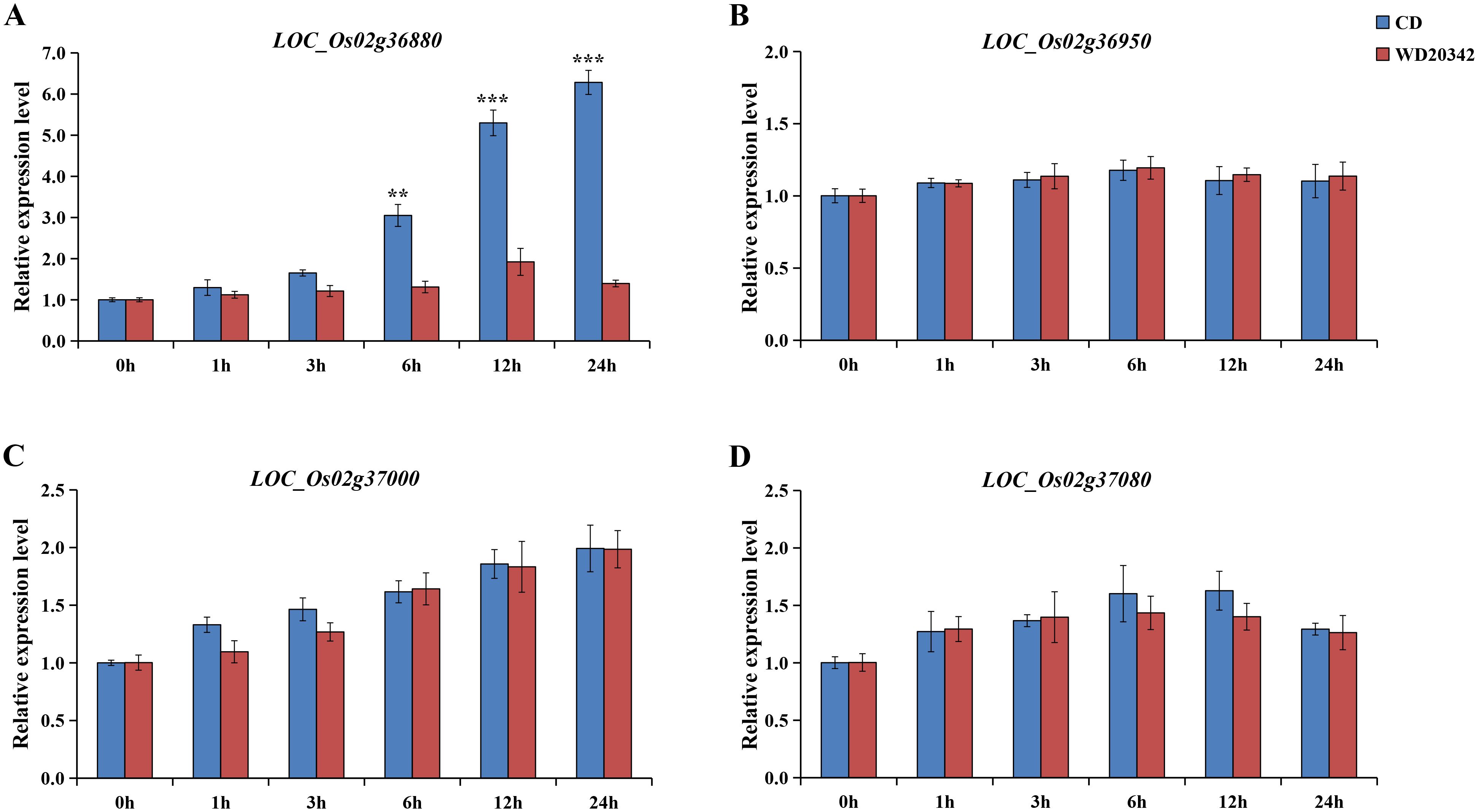
Figure 5. The candidate gene expression patterns were confirmed. (A-D) The expression of LOC_Os02g36880, LOC_Os02g36950, LOC_Os02g37000, and LOC_Os02g37080 genes under control conditions and salt stress was analyzed. (** P < 0.01, *** P < 0.001, Students’t-test).
The sequencing of promotor and exonic regions of the genes (LOC_Os02g36880, LOC_Os02g36950, LOC_Os02g37000, and LOC_Os02g37080) was conducted for the two parents, CD and WD20342, after analyzing the initial findings. LOC_Os02g36880 in the CD (salt-sensitive) matches the reference sequence from Nipponbare, as shown by the findings. Contrarily, LOC_Os02g36880 in WD20342 (salt-tolerant) had one SNP mutation in the promoter region, 6-bp insertion mutations in the first exon region, one SNP mutation in the second exon region, and two SNP mutations in the third exon region (Supplementary Figure S4). The three genes (LOC_Os02g36950, LOC_Os02g37000, and LOC_Os02g37080) in CD and WD20342 was not differ regarding mutations. Consequently, LOC_Os02g36880 was found to be the functional gene in salt tolerance, and its function was further verified.
3.6 Validation of the LOC_Os02g36880 mutant
We selected two homozygous CRISPR/Cas9 mutants, CR-1 with a one-base (A) insertion and CR-15 with a one-base (T) deletion (Figure 6) to investigate whether LOC_Os02g36880 was associated with salt tolerance. These plants were propagated to the T3 generation for further analysis. The analysis found no significant difference in wild-type and CRISPR/Cas9 mutant seedlings under normal conditions. While treated with salt, mutants CR-1 and CR-15 showed higher salt tolerance than wild-type, with average SST values of 3.4 and 4.2, and SSR values of 77.0% and 72.0%, respectively. In contrast, the wild-type was an average SST of 7.8 and an average SSR of 27.0% (Figure 7). The findings suggest that the knockout mutant of LOC_Os02g36880 enhances salt tolerance in seedlings.
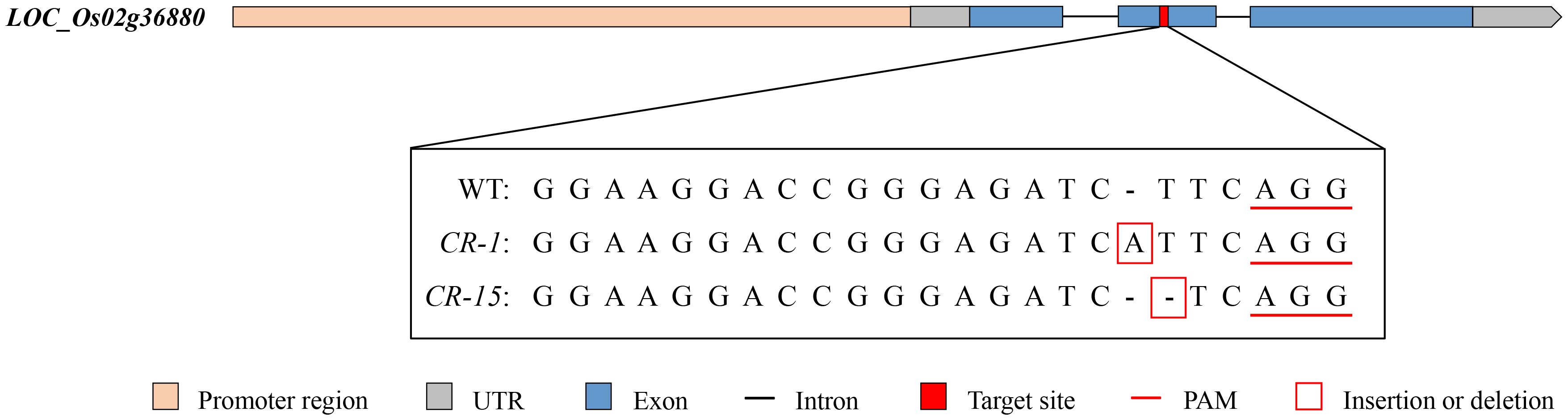
Figure 6. Target sequences of WT (ZH11) and knockout mutants (CR-1, CR-15).
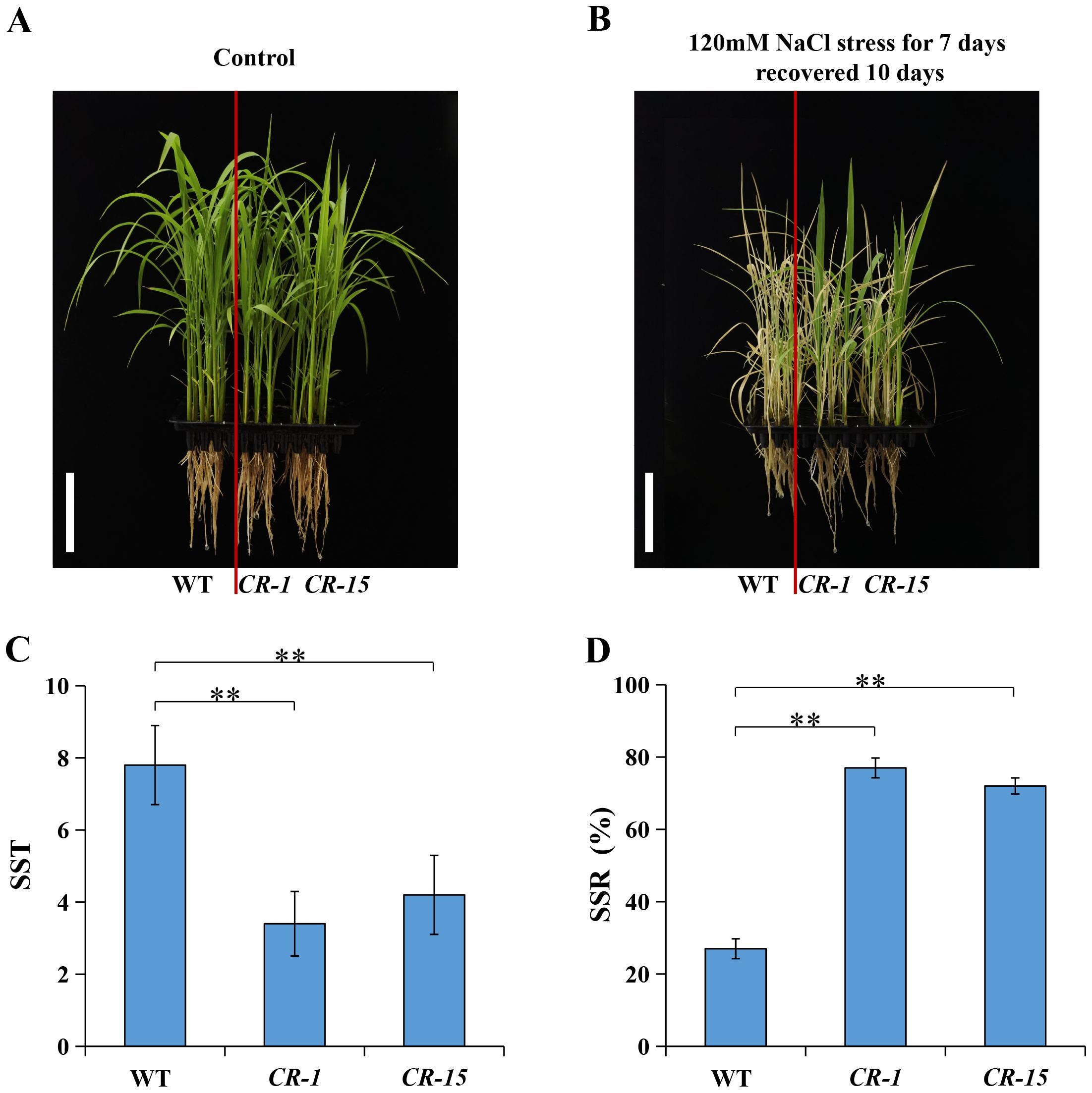
Figure 7. Salt tolerance phenotype of WT and knockout mutants. (A) Phenotype of control conditions; (B) Phenotype of the salt stress conditions; (C) The SST under salt stress; (D) The SSR under salt stress. (** P < 0.01; Students’ t-test).
4 Discussion
The combination of GWAS and linkage analysis to locate gene loci for traits can effectively improve the accuracy and precision of association loci and obtain reliable target intervals. In recent years, this method has been widely applied in genetic studies of rice traits, such as salt tolerance (Kong et al., 2021), alkalinity tolerance (Li et al., 2020), disease resistance (Xiao et al., 2019), grain size (Kang et al., 2020), flag leaf trait (Wang et al., 2022). In this study, we combined these two approaches to characterize seedling salt tolerance in 295 materials and 189 RIL lines and obtained fifteen and six major QTLs, respectively. Comparing these genes to the findings of earlier research, they were near or overlapped with the loci of several known QTLs/genes. Ammar et al. (2009) found a salt tolerance-associated QTL (qNaLV-3.1), and the lead SNP (Chr3_25974535), detected by GWAS, were located within qNaLV-3.1. A QTL (qRFW-6) associated with the radicle fresh weight under salt stress was discovered by Mardani et al. (2014) on chromosome 6, and it contained the lead SNPs Chr6_19195187, Chr6_12954487, and Chr6_15555901 that were identified by GWAS. Atefeh et al. (2013) found a QTL (qRL-10b) on chromosome 10 associated with root length under osmotic stress, where the lead SNP (Chr10_4241399) identified by GWAS was located. The lead SNP (Chr12_19562318) detected by GWAS was located within the QTL (qSNC-12) for the concentrations of Na+ in shoots under salt stress detected by Zheng et al. (2015) using linkage analysis, and was close to the gene regulating iron distribution, OsbHLH133 (Wang et al., 2013). The QTL qRRN6 contained known genes OsCYP19-4 (Yoon et al., 2016), which has peptidyl-prolyl cis-trans isomerase activity, and, OsCYP19-4 overexpression plants enhance cold tolerance. Meanwhile, the GWAS identified the lead SNP (Chr2_22340368), which is located within qRRL2 and qRRDW2 identified by linkage analysis, both within the QTL (qNAK-2) associated with salt tolerance identified by Yao et al. (2005).
We found that LOC_Os02g36880, a NAC transcription factor, contains an apical meristem structural domain. NAC transcription factors have a role in regulating abiotic stress tolerance, leaf senescence, lateral root growth, programmed cell death, etc (Huysmans et al., 2018; Xia et al., 2018; Zhang et al., 2018; Singh et al., 2021; Wang et al., 2021). For example, the OsNAC106 gene is a NAC transcription factor that negatively regulates salt tolerance, and the osnac106 deletion mutant is more salinity tolerant than normal plants (Sakuraba et al., 2015). Nakashima et al. (2007) found that transgenic overexpressing OsNAC6 had increased tolerance to high salt and drought stresses. Hong et al. (2016) identified ONAC022 as a transcriptional activator that positively regulates salt tolerance by modulating abscisic acid signaling, and overexpression of OsNAC022 transgenic plants were more salt-tolerant, accumulating less Na+ in roots and shoots than wild-type. It was shown that the NAC family of transcription factors is crucial in the study of salt tolerance.
In the previous study, Fang et al (Fang et al., 2014). found that the expression level of LOC_Os02g36880 was significantly reduced under drought and increased under high salt stress. Wang et al. (2023) combined metabolomic and transcriptomic profiles and found that LOC_Os02g36880 may be associated with low-nitrogen tolerance. The gene LOC_Os02g36880 was determined to be the most likely functional gene linked to salt tolerance. Seedlings of the CRISPR/Cas9 mutant of the LOC_Os02g36880 gene were substantially more saline-tolerant than wild-type, demonstrating that LOC_Os02g36880 negatively regulates salt tolerance. We combined with haplotype analysis and found that CD belongs to Hap1 (AC) and WD20342 belongs to Hap2 (GA), as WD20342 is more salt-tolerant than CD, indicating that Hap2 belongs to the dominant salt tolerant haplotype. This provides a theoretical basis for breeding new salt-tolerant rice varieties and is essential for improving the utilization efficiency of saline land.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
SX: Formal analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. JZ: Formal analysis, Investigation, Validation, Writing – original draft, Writing – review & editing. HD: Formal analysis, Investigation, Validation, Writing – original draft, Writing – review & editing. XD: Formal analysis, Writing – review & editing. CL: Validation, Writing – review & editing. YD: Validation, Writing – review & editing. YC: Writing – review & editing. JW: Writing – review & editing. HL: Writing – review & editing. LY: Writing – review & editing. WX: Writing – review & editing. YJ: Writing – review & editing. DZ: Conceptualization, Supervision, Writing – review & editing. HZ: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by Key Research and Development Projects of Heilongjiang Province (grant No. 2022ZX02B04), the National Natural Science Foundation of China (grant No. U20A2025), the “Academic Backbone” Project of Northeast Agricultural University (grant No. 20XG24), and the Mining of Salinity Tolerant Germplasm Resources and Variety Selection and Breeding of Coldland Early Japonica Rice (grant No. CZKYF2022-1-C005).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1462856/full#supplementary-material
Supplementary Figure 1 | Phenotypes of CD and WD20342 seedlings under control and salt stress. (A) Under control conditions. (B) Under salt stress. Bar = 5 cm.
Supplementary Figure 2 | Linkage analysis results of RIL lines derived from CD/WD20342.
Supplementary Figure 3 | Expression patterns of the other eight genes under normal growth and salt stress.
Supplementary Figure 4 | The gene structure and sequence difference analysis of LOC_Os02g36880. (A) The gene structure of LOC_Os02g36880. (B) The sequence difference analysis of LOC_Os02g36880 in WD20342, CD, and Nipponbare.
References
Al-Tamimi, N., Brien, C., Oakey, H., Berger, B., Saade, S., Ho, Y. S., et al. (2016). Salinity tolerance loci revealed in rice using high-throughput non-invasive phenotyping. Nat. Commun. 7, 11. doi: 10.1038/ncomms13342
Ammar, M. H. M., Pandit, A., Singh, R. K., Sameena, S., Chauhan, M. S., Singh, A. K., et al. (2009). Mapping of QTLs controlling Na+, K+ and Cl- ion concentrations in salt tolerant indica rice variety CSR27. J. Plant Biochem. Biotechnol. 18, 139–150. doi: 10.1007/bf03263312
Atefeh, Sabouri, Hossein, Sabouri, Marjorie, De, et al. (2013). Genetic analysis seedling 333 vigour under osmotic stress in rice by QTL mapping. Russ. Agric. Sci.
Batayeva, D., Labaco, B., Ye, C. R., Li, X. L., Usenbekov, B., Rysbekova, A., et al. (2018). genome-wide association study of seedling stage salinity tolerance in temperate japonica rice germplasm. BMC Genet. 19, 11. doi: 10.1186/s12863-017-0590-7
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., Buckler, E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Chang, C. C., Chow, C. C., Tellier, L., Vattikuti, S., Purcell, S. M., Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 16. doi: 10.1186/s13742-015-0047-8
Chen, G., Hu, K. M., Zhao, J. H., Guo, F. F., Shan, W. F., Jiang, Q. Q., et al. (2022). genome-wide association analysis for salt-induced phenotypic and physiologic responses in rice at seedling and reproductive stages. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.822618
De Leon, T. B., Linscombe, S., Subudhi, P. K. (2016). Molecular dissection of seedling salinity tolerance in rice (Oryza sativa L.) using a high-density GBS-based snp linkage map. Rice 9, 22. doi: 10.1186/s12284-016-0125-2
Dong, S. S., He, W. M., Ji, J. J., Zhang, C., Yang, T. L. (2020). LDBlockShow: a fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Briefings Bioinf. 22, 6. doi: 10.1101/2020.06.14.151332
Duan, Y. X., Zheng, H. L., Wen, H. R., Qu, D., Cui, J. N., Li, C., et al. (2022). Identification of candidate genes for salt tolerance at the germination stage in japonica rice by genome-wide association analysis. Agriculture-Basel 12, 15. doi: 10.3390/agriculture12101588
Fang, Y. J., Xie, K. B., Xiong, L. Z. (2014). Conserved miR164-targeted NAC genes negatively regulate drought resistance in rice. J. Exp. Bot. 65, 2119–2135. doi: 10.1093/jxb/eru072
Geng, L. Y., Zhang, W., Zou, T., Du, Q., Ma, X. D., Cui, D., et al. (2023). Integrating linkage mapping and comparative transcriptome analysis for discovering candidate genes associated with salt tolerance in rice. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1065334
Hanin, M., Ebel, C., Ngom, M., Laplaze, L., Masmoudi, K. (2016). New insights on plant salt tolerance mechanisms and their potential use for breeding. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01787
Hong, Y. B., Zhang, H. J., Huang, L., Li, D. Y., Song, F. M. (2016). Overexpression of a stress-responsive NAC transcription factor gene ONACO22 improves drought and salt tolerance in rice. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00004
Huysmans, M., Buono, R. A., Skorzinski, N., Radio, M. C., De Winter, F., Parizot, B., et al. (2018). NAC transcription factors ANAC087 and ANAC046 control distinct aspects of programmed cell death in the arabidopsis columella and lateral root cap. Plant Cell 30, 2197–2213. doi: 10.1105/tpc.18.00293
Kang, J. W., Kabange, N. R., Phyo, Z., Park, S. Y., Lee, S. M., Lee, J. Y., et al. (2020). Combined linkage mapping and genome-wide association study identified QTLs associated with grain shape and weight in rice (Oryza sativa L.). Agronomy-Basel 10, 22. doi: 10.3390/agronomy10101532
Kong, W. L., Zhang, C. H., Zhang, S. C., Qiang, Y. L., Zhang, Y., Zhong, H., et al. (2021). Uncovering the novel QTLs and candidate genes of salt tolerance in rice with linkage mapping, RTM-GWAS, and RNA-seq. Rice 14, 12. doi: 10.1186/s12284-021-00535-3
Kumar, V., Singh, A., Mithra, S. V. A., Krishnamurthy, S. L., Parida, S. K., Jain, S., et al. (2015). Genome-wide association mapping of salinity tolerance in rice (Oryza sativa). DNA Res. 22, 133–145. doi: 10.1093/dnares/dsu046
Lei, L., Han, Z. H., Cui, B. W., Yang, L. M., Liu, H. L., Wang, J. G., et al. (2021). Mapping of a major QTL for salinity tolerance at the bud burst stage in rice (Oryza sativa L) using a high-density genetic map. Euphytica 217, 8. doi: 10.1007/s10681-021-02901-0
Li, J., Xin, W., Wang, W. P., Zhao, S. J., Xu, L., Jiang, X. D., et al. (2022). Mapping of candidate genes in response to low nitrogen in rice seedlings. Rice 15, 16. doi: 10.1186/s12284-022-00597-x
Li, N., Zheng, H. L., Cui, J. N., Wang, J. G., Liu, H. L., Sun, J., et al. (2019). Genome-wide association study and candidate gene analysis of alkalinity tolerance in japonica rice germplasm at the seedling stage. Rice 12, 12. doi: 10.1186/s12284-019-0285-y
Li, S. S., Xu, S. B., Zheng, J., Du, H. Q., Li, C., Shen, S., et al. (2023). Joint QTL mapping and transcriptome sequencing analysis reveal candidate genes for salinity tolerance in oryza sativa L. ssp. japonica seedlings. Int. J. Mol. Sci. 24, 15. doi: 10.3390/ijms242417591
Li, X. W., Zheng, H. L., Wu, W. S., Liu, H. L., Wang, J. G., Jia, Y., et al. (2020). QTL mapping and candidate gene analysis for alkali tolerance in japonica rice at the bud stage based on linkage mapping and genome-wide association study. Rice 13, 11. doi: 10.1186/s12284-020-00412-5
Liu, C. T., Mao, B. G., Yuan, D. Y., Chu, C. C., Duan, M. J. (2022). Salt tolerance in rice: physiological responses and molecular mechanisms. Crop J. 10, 13–25. doi: 10.1016/j.cj.2021.02.010
Mardani, Z., Rabiei, B., Sabouri, H., Sabouri, A. (2014). Identification of molecular markers linked to salt-tolerant genes at germination stage of rice. Plant Breed. 133, 196–202. doi: 10.1111/pbr.12136
Nakashima, K., Tran, L. S. P., Nguyen, D. V., Fujita, M., Maruyama, K., Todaka, D., et al. (2007). Functional analysis of a NAC-type transcription factor OsNAC6 involved in abiotic and biotic stress-responsive gene expression in rice. Plant J. 51, 617–630. doi: 10.1111/j.1365-313X.2007.03168.x
Nakhla, W. R., Sun, W. Q., Fan, K., Yang, K., Zhang, C. P., Yu, S. B. (2021). Identification of QTLs for salt tolerance at the germination and seedling stages in rice. Plants-Basel 10, 12. doi: 10.3390/plants10030428
Phosuwan, S., Nounjan, N., Theerakulpisut, P., Siangliw, M., Charoensawan, V. (2024). Comparative quantitative trait loci analysis framework reveals relationships between salt stress responsive phenotypes and pathways. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1264909
Qiu, X. J., Yuan, Z. H., Liu, H., Xiang, X. J., Yang, L. W., He, W. J., et al. (2015). Identification of salt tolerance-improving quantitative trait loci alleles from a salt-susceptible rice breeding line by introgression breeding. Plant Breed. 134, 653–660. doi: 10.1111/pbr.12321
Radha, B., Sunitha, N. C., Sah, R. P., Azharudheen, M. T. P., Krishna, G. K., Umesh, D. K., et al. (2023). Physiological and molecular implications of multiple abiotic stresses on yield and quality of rice. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.996514
Raj, S. R. G., Nadarajah, K. (2023). QTL and candidate genes: techniques and advancement in abiotic stress resistance breeding of major cereals. Int. J. Mol. Sci. 24, 35. doi: 10.3390/ijms24010006
Rohila, J. S., Edwards, J. D., Tran, G. D., Jackson, A. K., McClung, A. M. (2019). Identification of superior alleles for seedling stage salt tolerance in the USDA rice mini-core collection. Plants-Basel 8, 23. doi: 10.3390/plants8110472
Ruan, S. L., Ma, H. S., Wang, S. H., Fu, Y. P., Xin, Y., Liu, W. Z., et al. (2011). Proteomic identification of OsCYP2, a rice cyclophilin that confers salt tolerance in rice (Oryza sativa L.) seedlings when overexpressed. BMC Plant Biol. 11, 15. doi: 10.1186/1471-2229-11-34
Sakuraba, Y., Piao, W., Lim, J. H., Han, S. H., Kim, Y. S., An, G., et al. (2015). Rice ONAC106 inhibits leaf senescence and increases salt tolerance and tiller angle. Plant Cell Physiol. 56, 2325–2339. doi: 10.1093/pcp/pcv144
Sarma, B., Kashtoh, H., Tamang, T. L., Bhattacharyya, P. N., Mohanta, Y. K., Baek, K. H., et al. (2023). Abiotic stress in rice: visiting the physiological response and its tolerance mechanisms. Plants-Basel 12, 41. doi: 10.3390/plants12233948
Singh, S., Koyama, H., Bhati, K. K., Alok, A. (2021). The biotechnological importance of the plant-specific NAC transcription factor family in crop improvement. J. Plant Res. 134, 475–495. doi: 10.1007/s10265-021-01270-y
Wang, Z. F., Cheng, J. P., Chen, Z. W., Huang, J., Bao, Y. M., Wang, J. F., et al. (2012). Identification of QTLs with main, epistatic and QTL x environment interaction effects for salt tolerance in rice seedlings under different salinity conditions. Theor. Appl. Genet. 125, 807–815. doi: 10.1007/s00122-012-1873-z
Wang, Y. R., Jiang, J., Qian, Y. K., Miao, S. Y., Wang, W. S., Xu, J. L., et al. (2023). Multi-Omics analysis reveals the regulatory and metabolic mechanisms underlying low-nitrogen tolerance at the flowering stage in rice. Agronomy-Basel 13, 16. doi: 10.3390/agronomy13020578
Wang, J. X., Wang, T., Wang, Q., Tang, X. D., Ren, Y., Zheng, H. Y., et al. (2022). QTL mapping and candidate gene mining of flag leaf size traits in japonica rice based on linkage mapping and genome-wide association study. Mol. Biol. Rep. 49, 63–71. doi: 10.1007/s11033-021-06842-8
Wang, L., Ying, Y. H., Narsai, R., Ye, L. X., Zheng, L. Q., Tian, J. L., et al. (2013). Identification of OsbHLH133 as a regulator of iron distribution between roots and shoots in oryza sativa. Plant Cell Environ. 36, 224–236. doi: 10.1111/j.1365-3040.2012.02569.x
Wang, H. L., Zhang, Y., Wang, T., Yang, Q., Yang, Y. L., Li, Z., et al. (2021). An alternative splicing variant of PtRD26 delays leaf senescence by regulating multiple NAC transcription factors in Populus. Plant Cell 33, 1594–1614. doi: 10.1093/plcell/koab046
Xia, J. N., Zhao, Y. J., Burks, P., Pauly, M., Brown, P. J. (2018). A sorghum NAC gene is associated with variation in biomass properties and yield potential. Plant Direct 2, 11. doi: 10.1002/pld3.70
Xiao, S. Z., Wang, B. X., Liu, Y. Q., Miao, T. H., Zhang, H. L., Wen, P. Z., et al. (2019). Genome-wide association study and linkage analysis on resistance to rice black-streaked dwarf virus disease. Mol. Breed. 39, 10. doi: 10.1007/s11032-019-0980-9
Yao, M. T., Wang, J. F., Chen, H. Y., Zhai, H. Q., Zhang, H. S. (2005). Inheritance and QTL mapping of salt tolerance in rice. Rice Sci. 12, 25–32.
Yoon, D. H., Lee, S. S., Park, H. J., Lyu, J. I., Chong, W. S., Liu, J. R., et al. (2016). Overexpression of OsCYP19-4 increases tolerance to cold stress and enhances grain yield in rice (Oryza sativa). J. Exp. Bot. 67, 69–82. doi: 10.1093/jxb/erv421
Yuan, J., Wang, X. Q., Zhao, Y., Khan, N. U., Zhao, Z. Q., Zhang, Y. H., et al. (2020). Genetic basis and identification of candidate genes for salt tolerance in rice by GWAS. Sci. Rep. 10, 9. doi: 10.1038/s41598-020-66604-7
Zeng, P., Zhu, P. W., Qian, L. F., Qian, X. M., Mi, Y. X., Lin, Z. F., et al. (2021). Identification and fine mapping of qGR6.2, a novel locus controlling rice seed germination under salt stress. BMC Plant Biol. 21, 14. doi: 10.1186/s12870-020-02820-7
Zhang, L., Yao, L., Zhang, N., Yang, J. W., Zhu, X., Tang, X., et al. (2018). Lateral root development in potato is mediated by stu-mi164 regulation of NAC transcription factor. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00383
Zheng, H. L., Zhao, H. W., Liu, H. L., Wang, J. G., Zou, D. T. (2015). QTL analysis of Na+ and K+ concentrations in shoots and roots under NaCl stress based on linkage and association analysis in japonica rice. Euphytica 201, 109–121. doi: 10.1007/s10681-014-1192-3
Keywords: Japonica rice seedling, salt tolerance, QTL, candidate gene, GWAS, linkage analysis
Citation: Xu S, Zheng J, Du H, Du X, Li C, Duan Y, Cai Y, Wang J, Liu H, Yang L, Xin W, Jia Y, Zou D and Zheng H (2024) GWAS combined with linkage analysis reveals major QTLs and candidate genes of salt tolerance in Japonica rice seedlings. Front. Plant Sci. 15:1462856. doi: 10.3389/fpls.2024.1462856
Received: 10 July 2024; Accepted: 25 September 2024;
Published: 01 November 2024.
Edited by:
Meng Jiang, Zhejiang University, ChinaReviewed by:
Biaolin Hu, Jiangxi Academy of Agricultural Sciences, ChinaXiaodeng Zhan, Chinese Academy of Agricultural Sciences, China
Copyright © 2024 Xu, Zheng, Du, Du, Li, Duan, Cai, Wang, Liu, Yang, Xin, Jia, Zou and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongliang Zheng, emhlbmdob25nbGlhbmcwMDhAMTI2LmNvbQ==; Detang Zou, em91ZHRuZWF1QDEyNi5jb20=
†These authors have contributed equally to this work