 Hanting Yang1,2†
Hanting Yang1,2† Baimei Liu1,2†
Baimei Liu1,2† Haiyan Ding1
Haiyan Ding1 Zhaoyu Liu2,3
Zhaoyu Liu2,3 Xiaodong Li4Tianxing He1
Xiaodong Li4Tianxing He1 Ya Wu1Yuxuan Zhang1Can Wang1,2*
Ya Wu1Yuxuan Zhang1Can Wang1,2* Liang Leng1,2*Shilin Chen1,2*
Liang Leng1,2*Shilin Chen1,2* Chi Song1,2*
Chi Song1,2*- 1School of Pharmacy/School of Modern Chinese Medicine Industry, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 2Institute of Herbgenomics, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 3School of Chinese Materia Medica, Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 4Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan, China
Introduction: Cepharanthine (CEP), a bisbenzylisoquinoline alkaloid (bisBIA) extracted from Stephania japonica, has received significant attention for its anti-coronavirus properties. While ethylene response factors (ERFs) have been reported to regulate the biosynthesis of various alkaloids, their role in regulating CEP biosynthesis remains unexplored.
Methods: Genome-wide analysis of the ERF genes was performed with bioinformatics technology, and the expression patterns of different tissues, were analyzed by transcriptome sequencing analysis and real-time quantitative PCR verification. The nuclear-localized ERF gene cluster was shown to directly bind to the promoters of several CEP-associated genes, as demonstrated by yeast one-hybrid assays and subcellular localization assays.
Results: In this work, 59 SjERF genes were identified in the S. japonica genome and further categorized into ten subfamilies. Notably, a SjERF gene cluster containing three SjERF genes was found on chromosome 2. Yeast one-hybrid assays confirmed that the SjERF gene cluster can directly bind to the promoters of several CEP-associated genes, suggesting their crucial role in CEP metabolism. The SjERFs cluster-YFP fusion proteins were observed exclusively in the nuclei of Nicotiana benthamiana leaves. Tissue expression profiling revealed that 13 SjERFs exhibit high expression levels in the root, and the qRT-PCR results of six SjERFs were consistent with the RNA-Seq data. Furthermore, a co-expression network analysis demonstrated that 24 SjERFs were highly positively correlated with the contents of various alkaloids and expression levels of CEP biosynthetic genes.
Conclusion: This study provides the first systematic identification and analysis of ERF transcription factors in the S.japonica genome, laying the foundation for the future functional research of SjERFs transcription factors.
1 Introduction
The COVID-19 outbreak in 2019 had a severe global impact, prompting scientists worldwide to collaborate in the search for effective drugs (Li et al., 2022; Yang et al., 2024a). Cepharanthine (CEP) has demonstrated the ability to inhibit the entry of SARS-CoV-2 into cells by blocking the virus’s attachment to its intended target cells (Kumar et al., 2022). This characteristic makes CEP a promising therapeutic agent for potential anti-COVID-19 treatments (Kumar et al., 2022; Fan et al., 2020). CEP, a bisBIA isolated from Stephania japonica, with the biological activities of antioxidant (Chen et al., 2019), antitumor (Zhang et al., 2021), and immunomodulatory (Xu et al., 2021). CEP predominantly accumulates in the roots of S. japonica, followed by the leaves and stems (Leng et al., 2024). S. japonica (Thunb.) Miers, a tangled deciduous woody vine belonging to the Menispermaceae family and Stephania genus (Al-Amin et al., 2022), is commonly used in traditional Chinese folk medicine for its heat-clearing, detoxifying, and “wind and blockage” dispelling properties in the human body (Xiao et al., 2019). Given the increasing clinical demand for CEP, it is crucial to investigate its biosynthesis and transcriptional regulation.
The biosynthesis of CEP primarily initiates with dopamine and 4-hydroxyphenyl acetaldehyde catalyzed by norclaurane synthesis (NCS) (Minami et al., 2007), norclaurane 6-O-methyltransferase (6OMT) (Li et al., 2020), coclaurine N-methyltransferase (CNMT) (Zhao et al., 2020), and undergoes multi-step reaction catalyzed by OMT/CYP80A (Stadler et al., 1988; Carina and Till, 2019). The oxidase CYP80A1 selectively couples two N-methylcocoyl units in their benzylic portion, forming the simplest bisBIA (Kraus and Kutchan, 1995). The biosynthesis of guattegaumerine and berbamunine of bisBIA s has been elucidated (Payne et al., 2021). In S. japonica, SjNCS2 and SjNCS4 possessed NCS functionality and exhibited superior enzymatic activities compared with the Coptis chinensis NCS (Leng et al., 2024). However, the downstream biosynthetic pathways of CEP remain unclear (Supplementary Figure S1).
ERF transcription factors are significant regulators in various plant biological processes, including alkaloid biosynthesis (Feng et al., 2020; Yamada and Sato, 2021). For instance, clustered ORCA transcription factors (ORCA2-6) regulate the expression of different monoterpene indole alkaloid biosynthetic genes in Catharanthus roseus (Paul et al., 2020; Singh et al., 2020). In Nicotiana benthamiana, NtERF189 acts as a master regulator of nicotine biosynthesis by recognizing GCC-box-like elements in the promoter of nicotine biosynthetic genes (Shoji et al., 2010; Shoji and Hashimoto, 2012). OpERF2 positively regulates the anti-cancer camptothecin biosynthesis in Ophiorrhiza pumila (Udomsom et al., 2016). In Eschscholzia californica, a luciferase reporter assay indicated that four Group IX AP2/ERF TFs, known as EcERF2, EcERF3, EcERF4, and EcERF12, can trans-activate Ec6OMT and EcCYP719A5, which are involved in BIA biosynthesis (Yamada et al., 2020). Transiently overexpressing PhERF1 in petunia leaves has an impact on the production of petuniolides and petuniaserones (Shoji et al., 2023). Overexpression of ScAPD1-like significantly increased the metabolites of the phenylpropanoid pathway by directly regulating the abundance of ScPAL and ScC4H transcripts (Li et al., 2023). The ERF transcription factor WAX INDUCER1 (WIN1) promotes the accumulation of total polyphenols in Nicotiana tabacum, including chlorogenic acid (He et al., 2024). However, a study on the ERF family in S. japonica that regulates the biosynthesis of bisBIA has yet to be reported.
An increasing number of medicinal plant genomes have been published, including Artemisia argyi, Mentha suaveolens, and C. roseus, which will provide a foundation for the identification of ERF families and functional genomics research (Chen et al., 2023; Yang et al., 2024b; Sun et al., 2023; Pei et al., 2024). ERF protein identification and characterization have been studied in various plant species, including Arabidopsis thaliana (Nakano et al., 2006), barley (Taketa et al., 2008), Fagopyum Tataricum (Liu et al., 2019), grape (Zhuang et al., 2009; Zhu et al., 2019), apple (Girardi et al., 2013), and ginger (Xing et al., 2021). The number of ERF TFs family in many plants are as follows: 136 (Oryza sativa), 122 (A. thaliana), 96 (Citrus junos), 92 (Camptotheca acuminata), 80 (Vitis vinifera), and 60 (E. californica). Genome-wide identification of ERF transcription factor and its significance in CEP biosynthesis have not been elucidated in Stephania plants. This study proved the systematic identification and analysis of 59 SjERFs in the S. japonica genome using a set of bioinformatics tools. Meanwhile, tissue expression profiling and co-expression analysis of SjERF, CEP biosynthetic genes, and BIAs metabolites were also conducted. Yeast one-hybrid assays indicated that the SjERFs cluster recognizes GCC boxes in the promoters of several CEP-associated genes. This work provides valuable insight into the roles of ERF transcription factors in CEP biosynthesis and enhances our understanding of the ERF gene family in plants.
2 Materials and methods
2.1 Plant materials
The S. japonica plants were cultivated and harvested in Wuhan, Hubei Province, China. Different tissues of S. japonica including stems, leaves, roots, and shoots, were collected for transcriptome sequencing and quantitative real-time polymerase chain reaction (qRT-PCR) experiments. Three biological replicates were conducted for each experiment.
2.2 Identification of SjERF genes in the S. japonica genome
Our research group has acquired the genome data of S. japonica., and has been archived under the China National GeneBank DataBase (CNGBdb) accession number CNP0003595 (https://db.cngb.org/search/?q=CNP0003595) (Leng et al., 2024). The AtERFs protein sequences of A. thaliana were downloaded from the Arabidopsis Information Resource (TAIR) database (http://www.arabidopsis.org/). The hidden Markov model (HMM, PF00847) was used to search for ERF candidate genes in the S. japonica genome, with a threshold of 0.01. Furthermore, to ensure the comprehensive identification of SjERF genes, 121 AtERF proteins were used to BLAST the S. japonica protein database for ERF-containing sequences (Supplementary Table S1), minimizing the risk of missing any SjERF genes. Then, candidate proteins with only one AP2 domain were manually screened (Sakuma et al., 2002; Riechmann and Meyerowitz, 1998). The Molecular weight (MW) and pI of SjERF proteins were analyzed using the Expasy website (https://prosite.expasy.org/). Finally, the subcellular localization of SjERFs was predicted using WoLF PSORT and CELLO online software (Horton et al., 2007).
2.3 Classification, gene structure, and protein motif analysis of SjERF genes in S. japonica
To explore different biological characteristics and evolutionary relationships of SjERF proteins in S. japonica, an unrooted phylogenetic tree of ERFs protein sequences (59 SjERFs and 121 AtERFs) from S. japonica and A. thaliana was constructed by MEGA11 with 1,000 bootstrap replicates (Tamura et al., 2021). Then, an evolutionary tree was beautified and decorated using Evolview (Zhang et al., 2012). The conserved motifs of SjERFs protein were identified using the MEME website (parameters: number of motifs: 10, wide: 10-50, others are default values) (Bailey et al., 2009). Gene structure and protein motif of SjERFs were visualized using TBtools (Chen et al., 2020).
2.4 Analysis of cis-elements, microsynteny, and evolutionary patterns of SjERF genes
The promoter sequences of the 59 SjERFs (-2,000 to -1 bp) were extracted using TBtools. Subsequently, cis-acting regulatory elements of SjERFs gene promoters have been predicted and identified by PlantCARE (Lescot et al., 2002). The chromosomal positions of SjERF genes were retrieved from the S. japonica genome database and graphically represented using TBtools software. The duplication events of the SjERFs were analyzed using MCScanX and BLASTP (Wang et al., 2012). The synonymous relationship between SjERFs and AtERFs, OsERFs, CrERFs, and NtERFs was analyzed and visualized by TBtools software. The genome data of O. sativa, C. roseus, and N. tabacum were retrieved from the National Center for Biotechnology Information (NCBI: https://www.ncbi.nlm.nih.gov/), respectively.
2.5 Chromosome structure prediction and cluster prediction
The Topologically Associated Domains (TADs) were identified based on previous reports (Sun et al., 2020). Initially, the Hi-C read pairs were aligned to the S. japonica genome, and contact matrixes were generated using HiC-Pro (Servant et al., 2015). Subsequently, the Hi-C contact matrixes were imported into HiCExplorer (Wolff et al., 2018) and converted using the built-in function (hicConvertFormat). Then, the contact matrixes were normalized using hicNormalize with the KR correction method and corrected using hicCorrectMatrix with a filter threshold of -1.5 to 5. Next, the hicFindTADs algorithm was applied to identify TADs at various resolutions. The specific parameters used for this analysis were a minimum depth of 5, maximum depth of 10, step size of 2, and a threshold for comparisons set at 0.01.
2.6 Yeast one-hybrid assays
Yeast one-hybrid assays were performed to determine whether SjERF9-11 could bind to the GCC motif. The functional protein sequences of CEP biosynthetic genes with known functions were retrieved from the NCBI database, including NCS, 6OMT, and CNMT (Supplementary Table S2). The candidate genes involved in CEP biosynthesis were predicted using BLASTP (option: e-value 1e−10). Subsequently, the functional proteins and candidate genes were used to construct phylogenetic trees with 1,000 bootstrap replicates. Additionally, the ERF binding elements in CPE biosynthetic gene promoters were predicted using PlantCARE software. The open reading frame (ORF) fragment of SjERF9-11 was individually cloned into the effector plasmid pB42AD. Additionally, the triple tandem copy of the GCC motif (GCCGCC) or the ERF binding element from CEP-biosynthetic gene promoters was inserted into the reporter plasmid pLacZ. The effector and reporter plasmids were jointly transformed into the yeast strain EGY48 and grown on SD/-Ura/-Trp medium. Subsequently, the co-transformed cells were assayed on SD/-Ura/-Trp medium containing 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) for 24 hours, as previously described (Wang C. et al., 2022). Empty plasmids (pB42AD and pLacZ) were used as a negative control for the transformation. All primers utilized in this study are provided in Supplementary Table S3.
2.7 Subcellular localization
To analyze the subcellular localization of three SjERFs, the SjERF9-11 ORF fragments were amplified and individually integrated into the modified plant expression vector pHB-YFP. The plasmids pHB-SjERFs-YFP and the empty vector pHB-YFP (serving as the negative control) were introduced into the Agrobacterium tumefaciens strain GV3101 and transiently infected the epidermal cells of 5-week-old N. benthamiana leaves, as previously described (Wang C. et al., 2021; Hao et al., 2023). YFP signals were analyzed 48 h post-infection using an LSM880 confocal laser microscope (Carl Zeiss, Germany). Nuclei were stained with, 4’6-diamidino-2-phenylindole (DAPI, Sigma, Code No. D9542). Three biological replicates were performed as reported previously, to ensure the reliability of the results.
2.8 RNA-seq and qRT-PCR
For RNA-seq analysis, qualified RNA samples underwent testing for database establishment. The quality of the constructed library was assessed using an Agilent 2100 Bioanalyzer, while sequencing was performed using DNBSEQ technology. All raw sequencing data have been deposited under the National Center for Biotechnology Information (NCBI) GenBank accession number PRJNA888087. The expression pattern of SjERFs in different tissues was analyzed by TBtools according to the FPKM values. Total RNA was extracted from the roots, stems, leaves, and shoots of S. japonica using a plant total RNA Extraction Kit (Foregene Biotech, Chengdu, China, Code No. RE-05011). Subsequently, reverse transcription was carried out according to the instructions provided with the gDNA Eraser reagent Kit (Foregene Biotech, Chengdu, China, Code No. RT-01032) for qRT-PCR analysis. The qRT-PCR was carried out according to previous reports, and three biological replicates were conducted for each experiment. For qRT-PCR normalization, SjGAPDH, a housekeeping gene in S. japonica, was employed as an internal control of all samples (Yang et al., 2023; Jain et al., 2018; Barber et al., 2005). The specific primers used for the analysis are detailed in Supplementary Table S3. The relative expression levels of SjERFs across various tissues were determined using the 2−ΔΔCt method.
2.9 Co-expression network of SjERFs involved in CEP biosynthesis pathway
46 SjERFs and 9 CEP biosynthetic genes all exhibiting FPKM values exceeding 1, underwent co-expression analysis using Pearson’s correlation test. We employed untargeted metabolomics to profile BIAs across various tissues of the S. japonica (Leng et al., 2024). According to the expression patterns of SjERFs, two BIA precursors, alongside 23 BIA-type structures in roots, stems, and leaves of S. japonica, the partial correlation coefficient (PCC) method was used to calculate the Pearson correlation coefficient. The co-expression network of SjERFs, CEP biosynthetic genes, and BIAs metabolites was exhibited using Cytoscape, with the following parameters: absolute value of correlation coefficient > 0.9 and p-value < 0.05 (Shannon et al., 2003). The correlations between SjERFs, CEP biosynthetic genes, and BIAs metabolites were displayed in cluster heatmap using TBtools software.
3 Results
3.1 Genome-wide identification of 59 SjERF TFs in S. japonica genome
59 non-redundant SjERF genes have been identified in the S. japonica genome using HMMER and BLAST (Table 1). All identified ERF genes in S. japonica were named SjERF1- SjERF59 according to their chromosome distribution (Huang et al., 2020). All SjERFs were then manually confirmed by CDD’s online software and a Simple Modular Architecture Analysis Tool (SMART) for the presence of a core domain (Supplementary Figure S2). The CDS sequence length of SjERF genes was between 605 bp (SjERF21) and 1305 bp (SjERF29), encoding 202–434 amino acids (Supplementary Table S4). The molecular weight (Mw) of SjERFs ranged from 22.58 kDa (SjERF21) to 47.55 kDa (SjERF29), with theoretical pI values ranging from 4.53 (SjERF19) to 10.00 (SjERF37). Almost all SjERFs were predicted to be located in the nucleus, only SjERF57 was located in the cytoplasmic (Table 1).
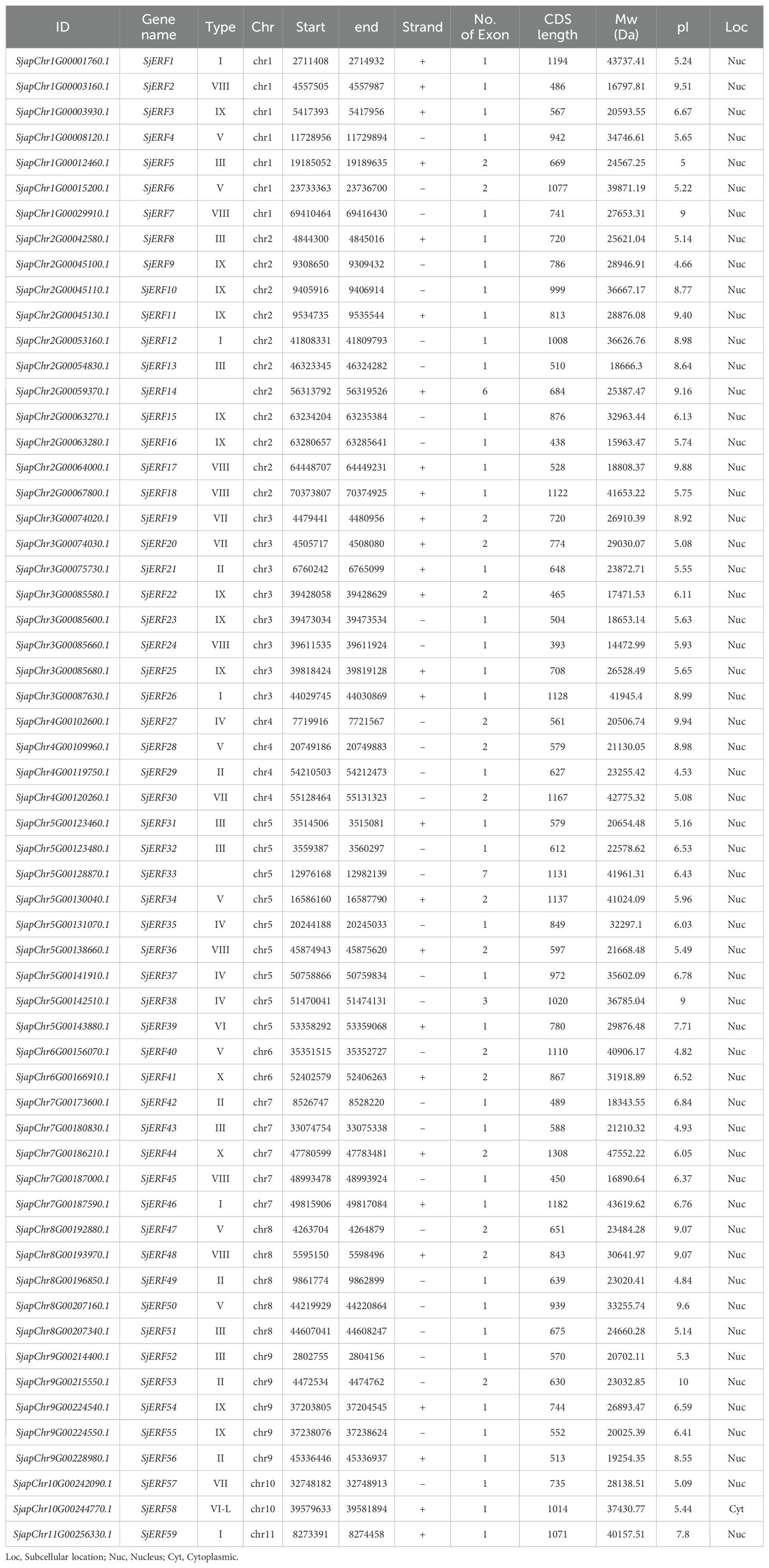
Table 1. Sequence characteristics of 59 SjERFs.
3.2 Phylogenetic relationship of SjERF genes
The unrooted phylogenetic tree of 59 SjERFs and 121 AtERFs has been constructed to explore the evolutionary relationship. 59 SjERFs have been divided into 10 subgroups, namely, groups I to X. A previous study has further divided the ERF family into ERF and CBF/DREB subfamily, and the ERF subfamily always classified into six groups (B1 to B6) (Zhang et al., 2015). In this analysis, group I to IV belong to the DREB subfamily, and group IV to X, and VI-L belong to the ERF subfamily, and there is no SjERF in group Xb-L. SjERF1, 12, 26, 46, and 59 were branched into group I, SjERF21, 29, 42, 49, 53, and 56 were branched into group II, group IX was the largest group with 11 members (SjERF3, 9, 10, 11, 15, 16, 22, 23, 25, 54, 55). As shown in Figure 1, group VI was the smallest with SjERF39, SjERF58 belongs to group VI-L, and SjERF14 and SjERF33 don’t belong to any subfamily.
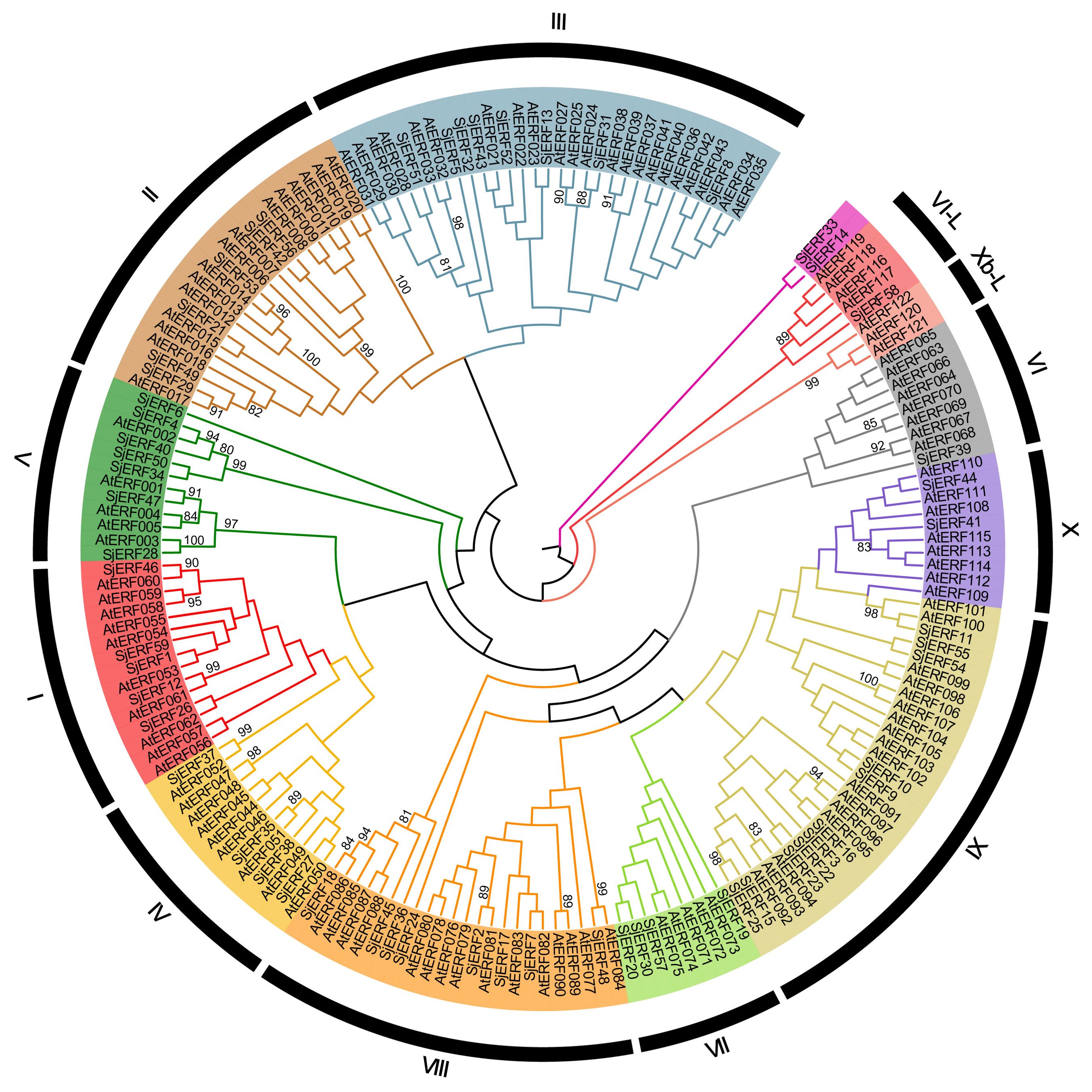
Figure 1. Phylogenetic tree of 59 SjERFs and 121 AtERFs. The ERF protein sequences of S. japonica and A. thaliana were used to construct the phylogenetic tree using the Neighbor-Joining (NJ) method, with 1,000 bootstrap replicates.
3.3 Gene structure and motif analysis of SjERFs in S. japonica genome
To better understand the evolution and structural diversity of the S. japonica ERF family, the MEME (Multiple Em for Motif Elicitation) was used to analyze the conserved sequence of the 59 SjERFs protein. The basic information (width and best possible match sequence) of the consensus sequences of these motifs are shown in Supplementary Table S5. The frequent motifs of SjERFs are motif1 (RVWLGTFDTAEEAARAYDEAAFKLRG), motif2 (YRGVRQRPWGKWVAEIRDP), and motif3 (SKAKLNFPEE). The results showed that each motif contained 10-29 kinds of amino acids, and each SjERF contained motif1. Almost all SjERFs contain motif2, only SjERF14 and SjERF33 don’t belong to any subfamily that does not contain motif2, while they only contain one conserved motif (motif1), and SjERF33 had two conserved motifs (motif1, motif3). SjERF44, SjERF18, SjERF37, SjERF31, SjERF59, and SjERF7 had six conserved motifs. 59 SjERFs contained ERF conservative domain (Figure 2B; Supplementary Figure S3). Additionally, these different motif patterns show their degree of deviation among different groups. For example, motif 5 is the representative of group IX. Motif 7 is only found in group III. Motif 6 is unique to group II and VII (Figures 2A, B).
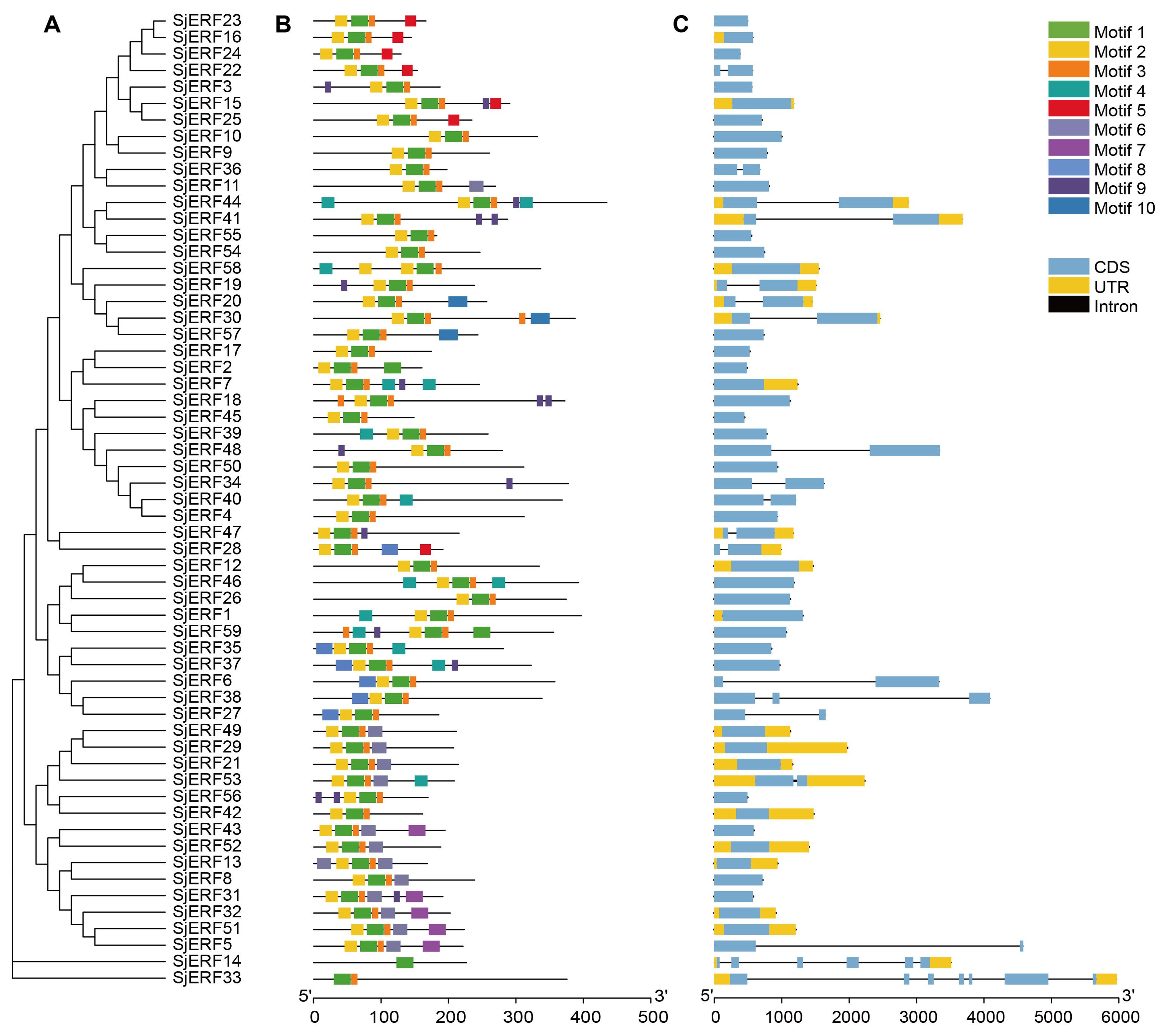
Figure 2. Schematic diagram of phylogenetic analysis, exon/intron distribution, and motifs analysis of SjERF TFs. (A) Phylogenetic tree of 59 SjERF proteins. (B) Motif distribution of 59 SjERF proteins. (C) The exon-intron structure of 59 SjERF genes. Yellow rectangle: UTR; black line: intron; blue rectangle: CDS.
In this study, gene structures of 19 SjERFs (SjERF5, 6, 14, 19, 20, 22, 27, 28, 30, 33, 34, 36, 38, 40, 41, 44, 47, 48, 53) have one intron, and SjERF38, SjERF33, and SjERF14 contained two or more introns. The other 40 SjERFs contain only one exon and no intron, accounting for 67.8% of the total SjERFs in S. japonica. Most SjERF genes contained only one exon in group I, II, III, and IX, except for SjERF53, SjERF5, and SjERF22. The unnamed subfamily genes (SjERF14, SjERF33) have 6 and 7 exons (Figure 2C).
3.4 Analysis of cis-acting elements in the SjERF genes promoter
The study identified various cis-acting elements located in the promoter regions of SjERFs, with the majority participating in hormone responses, and abiotic and biotic stress. In plant growth and development, 31 CAT-boxes were implicated in meristem expression across 26 SjERFs promoter regions, while 7 A-boxes participated in meristem expression in 7 SjERFs promoter regions (Figure 3; Supplementary Table S6). Furthermore, 18 GCN4_motif, 4 HD-Zip, 36 O2-site, 16 circadian control elements, and 7 seed-specific regulation elements were identified in the promoter regions of SjERFs (Supplementary Figure S4). In hormone responses, various cis-acting regulatory elements were identified, including 164 ABRE, 33 TGA-element, 7 AuxRR-core, 4 TGA-box, 13 GARE-motif, 10 TATC-box, 22 P-box, 123 CGTCA and TGACG-motif. However, the abiotic and biotic stress cis-acting elements were not found in the promoter regions of SjERF2, 7, 32, and 50.
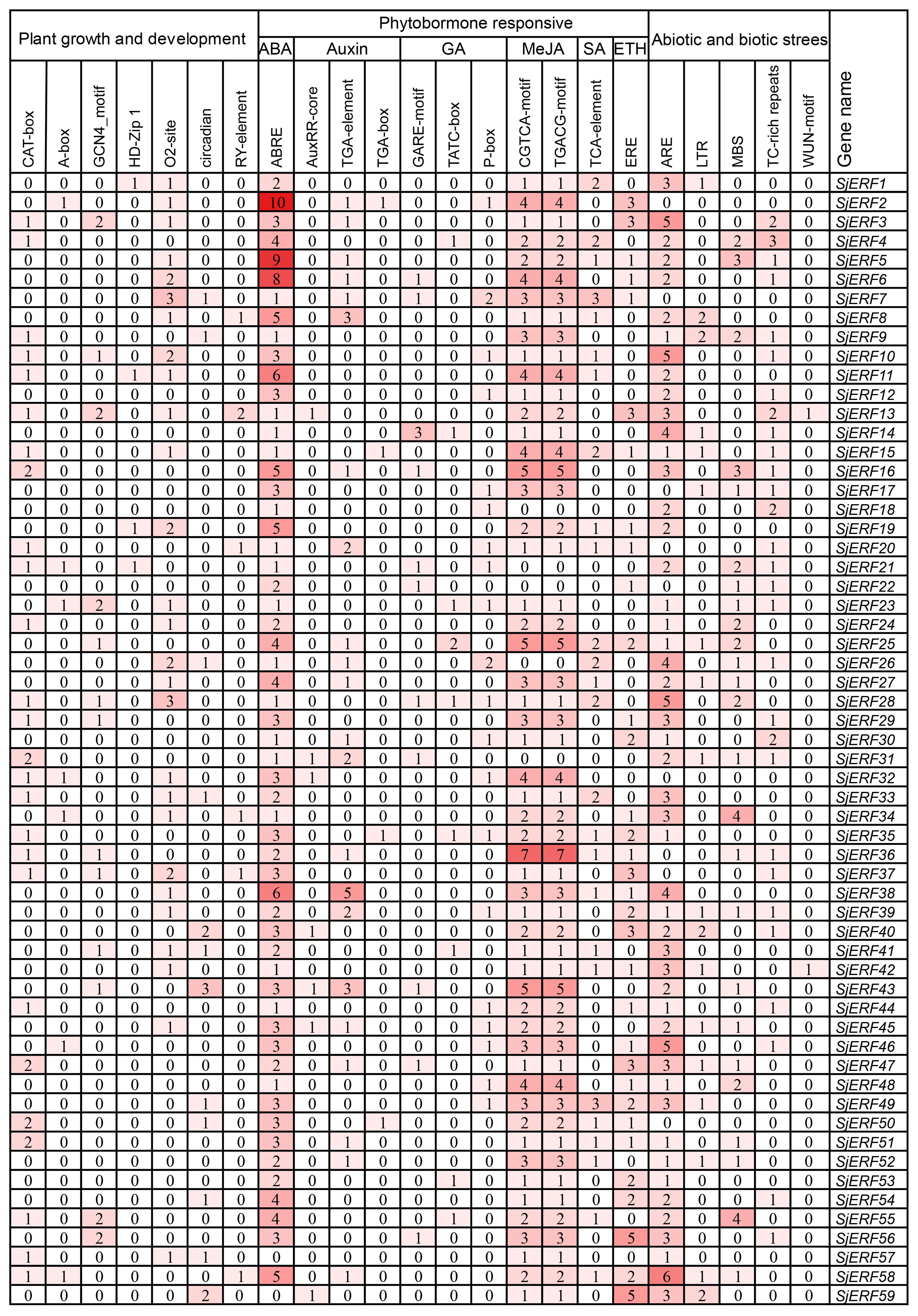
Figure 3. Pivotal cis−elements in the promoter of SjERF TFs.
3.5 Chromosome distribution and synteny analysis of SjERFs
Chromosome localization analysis found that 59 SjERFs were disproportionately distributed on eleven S. japonica chromosomes (Figure 4A). Seven SjERFs were distributed on Chr1 and Chr3, eleven SjERFs on Chr2, four SjERFs distributed on Chr4, nine SjERFs distributed on Chr5, two SjERFs distributed on Chr6 and Chr10, five SjERFs distributed on Chr7, Chr8 and Chr9, and only one SjERF distributed on Chr11. Interestingly, three SjERF genes containing SjERF9, SjERF10, and SjERF11 were distributed on S. japonica chromosome 2 (9.30 - 9.54 Mb) and formed an ERF gene cluster. Similar results have also been found in C. roseus and N. tabacum, such as the ORCA gene cluster and NICOTINE2 (NIC2) ERF cluster (Yuan, 2020; Shoji et al., 2010; Shoji and Yuan, 2021). The phylogenetic tree showed that SjERF9, SjERF10, and SjERF11 and functional ERF cluster were converging into one branch, and belonging to the IX subfamily (Figure 4B). Additionally, three SjERFs and four other genes were located in the same topologically associating domains (TADs) region (Figure 4C). Overall, the SjERF gene cluster found in S. japonica genome may play an important role in the biosynthesis of secondary metabolism.
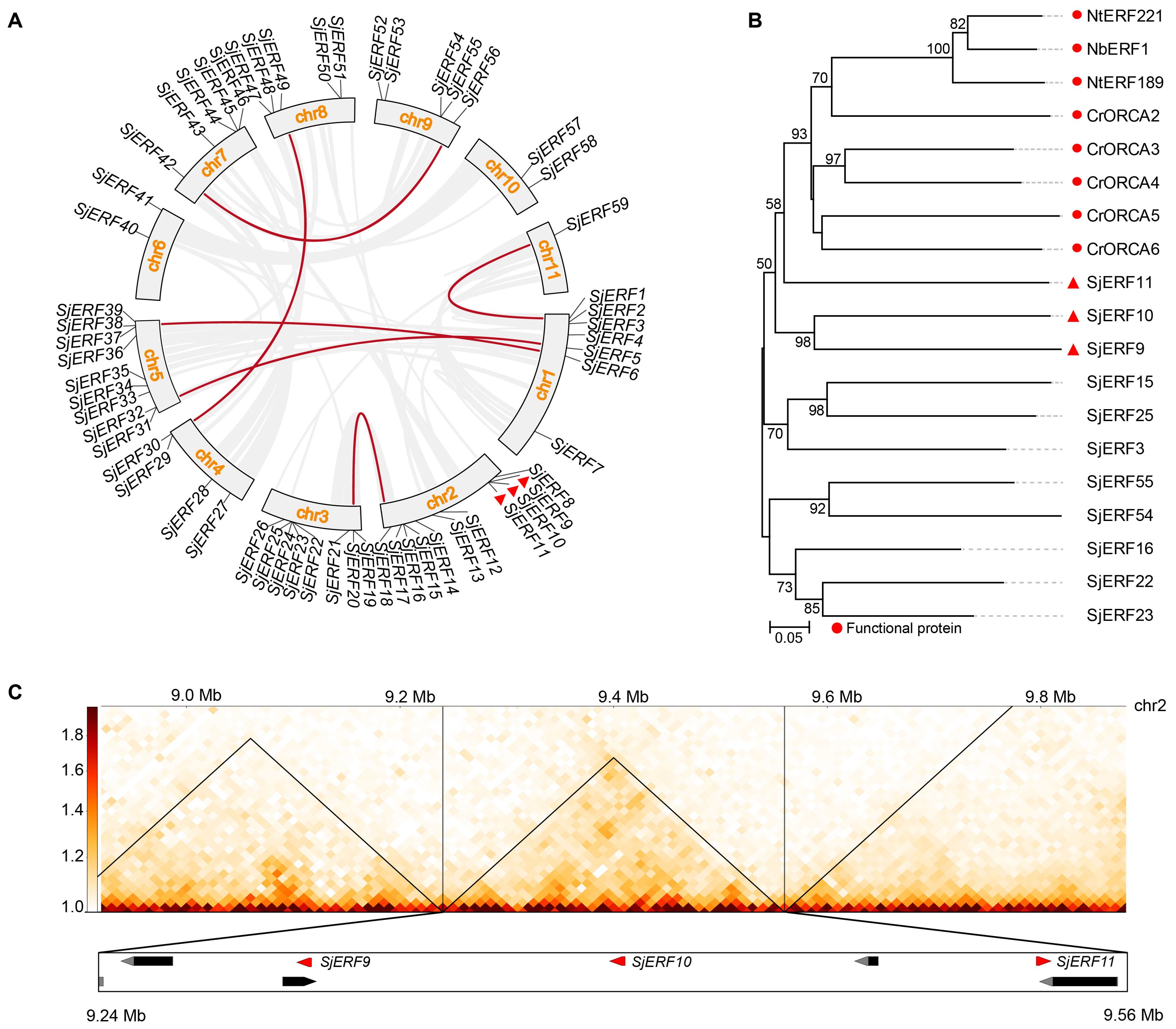
Figure 4. The chromosome distribution and synteny analysis of SjERFs. (A) Chromosomal locations and their synteny of SjERFs. The connecting lines indicate duplicated gene pairs in 59 SjERFs. (B) The phylogenetic tree of SjERFs and functional ERF cluster. (C) The topologically associating domains (TADs) region of three SjERFs.
Through collinear analysis, the potential relationship and gene duplication type between SjERF genes in the S. japonica genome were explored (Figure 4A). A total of seven SjERF genes were found in three segmental duplication events, such as Chr1(SjERF1)/Chr11(SjERF59), Chr1(SjERF5)/Chr5(SjERF31), Chr1(SjERF6)/Chr5(SjERF38), Chr7(SjERF42)/Chr9(SjERF56), Chr4(SjERF29)/Chr8(SjERF49). To further infer the evolutionary mechanism of the ERF family in S. japonica, we constructed a synteny diagram of S. japonica with C. roseus, N. tabacum, A. thaliana, and O. sativa (Supplementary Figure S5). Between S. japonica and A. thaliana, O. sativa, C. roseus, and N. tabacum, 56, 46, 50, and 32 syntenic SjERF gene pairs were identified, respectively (Supplementary Table S7). Some SjERF genes had multiple orthologous gene pairs (one SjERF associated with multiple AtERFs). For instance, three synteny events occurred in three SjERFs, such as SjERF9, SjERF30, and SjERF41. Interestingly, the SjERF1, SjERF8, SjERF9, SjERF11, SjERF26, SjERF29, SjERF30, SjERF38, SjERF44, SjERF46, SjERF49, and SjERF51 genes exhibited a conserved homologous relationship across all four species, suggesting that they might play a significant role in plant function.
3.6 SjERFs cluster specifically bind to the GCC-boxes in the promoters of CEP-associated genes in vitro
To predict the SjERFs involved in the CEP biosynthesis pathway, NCS, 6-OMT, and CNMT genes were identified in the S. japonica genome using the BLASTP approach with p-value < 1e-10 (Supplementary Table S2). Subsequently, the well-supported subfamily containing the functional protein sequence was defined as candidate functional genes in the CEP biosynthesis pathway using a phylogenetic tree. Finally, five NCS, three 6-OMT, and five CNMT genes were identified as candidate functional genes in S. japonica genome (Figure 5). Meanwhile, the majority of CEP-biosynthetic genes have high transcriptional levels in one or more tissues of S. japonica, except for SjNCS1, SjCNMT3 and SjCNMT5 (Figure 5; Supplementary Table S8). For instance, Sj6OMT1, SjNCS3-5, and SjCNMT4 have the highest expression level in S. japonica root (FPKM >30), while Sj6OMT3, SjNCS2, and SjCNMT1,2 exhibited preferential expression patterns in S. japonica shoots.
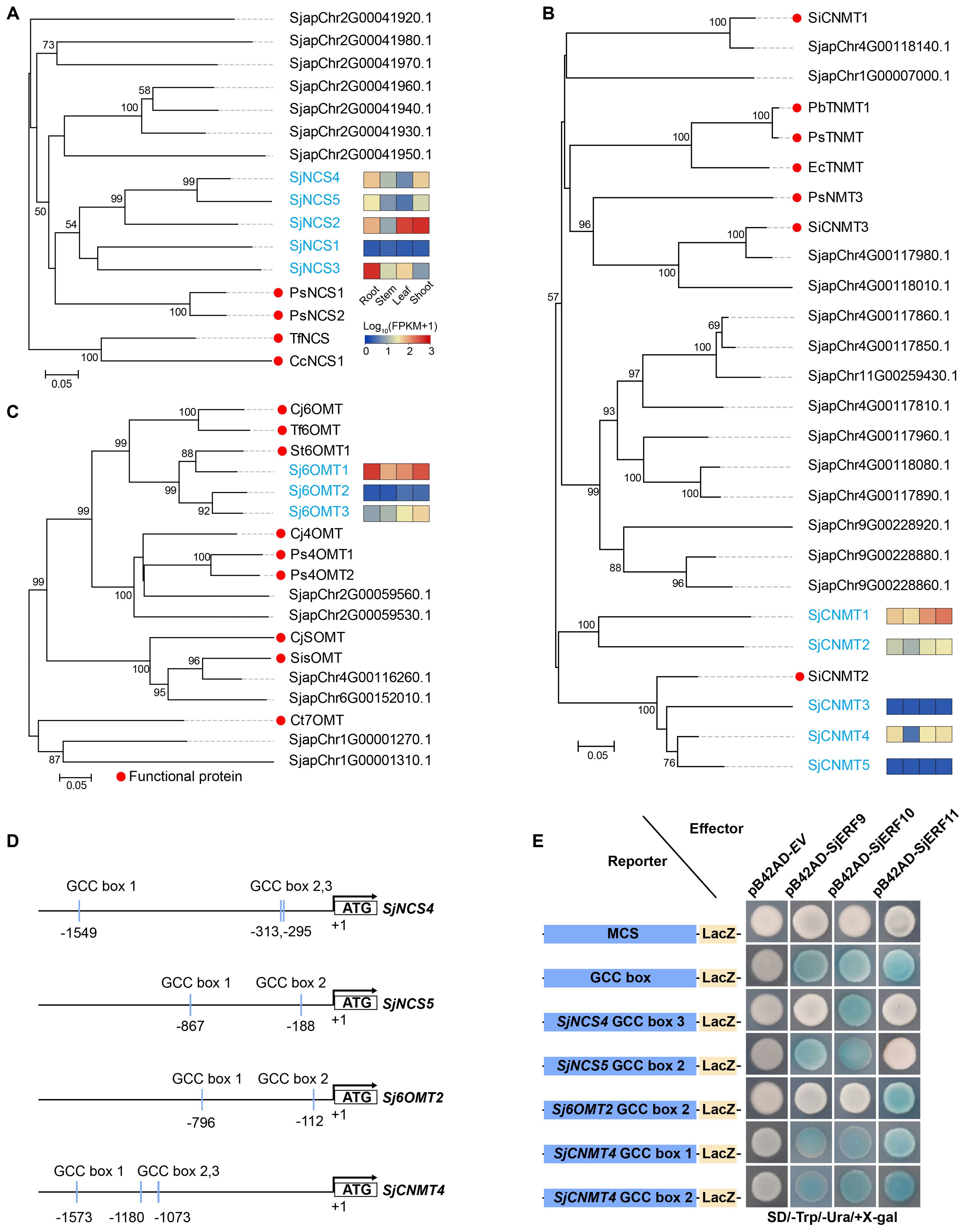
Figure 5. Members of the SjERFs cluster specifically bind to the GCC boxes in the promoters of CEP-associated genes in vitro. (A–C) Phylogenetic tree of CEP biosynthetic genes using MEGA11 with 1000 bootstrap replicates by Neighbor-joining (NJ) method. (D) Schematic diagrams of the SjNCS4, SjNCS5, Sj6OMT2, and SjCNMT4 promoters. The positions of potential GCC boxes are shown as blue Rectangles. (E) Yeast one-hybrid (Y1H) assay indicates that the SjERFs cluster binds to the GCC box in the promoters of CEP-associated genes, including SjNCS4, SjNCS5, Sj6OMT2 and SjCNMT4. Yeast cells transformed with different combinations of constructs were grown on SD/−Ura/−Trp/+X-gal medium. Photographs were taken after 3 d of incubation at 30°C. Y1H assays were repeated three times.
An increasing amount of data suggests that the ERF gene clusters play a crucial role in secondary metabolism (Paul et al., 2020; Shoji and Yuan, 2021). The cis-acting elements of CEP biosynthetic gene promoters were analyzed. Seven of the thirteen promoters (SjNCS1, SjNCS3, SjNCS4, SjNCS5, Sj6OMT1, Sj6OMT2, and SjCNMT4) contained either a predicted GCC motif or a GCC-like element (Figure 5D). Among them, three GCC-boxes were found in the promoter region of SjNCS4 and SjCNMT4, whereas two GCC-boxes were identified in the SjNCS5 and Sj6OMT2 promoter. To further identify the SjERFs gene cluster involved in CEP biosynthesis, Y1H assays were carried out in this study. As depicted in Figure 5E, binding of the AD-SjERF9/10/11 (GAL4 AD-prey protein) fusion protein, but not AD-EV (GAL4 AD empty vector) alone, to three tandem repeats of the GCC-box, strongly activated the expression of the LacZ reporter gene in the Y1H system. Moreover, the SjERF9 transcription factor regulates the expression of SjNCS5 by directly binding the GCC-box2 of the SjNCS5 promoter. SjERF10 could directly bind to the SjNCS5 promoter, while SjERF11 recognizes GCC-box2 of the Sj6OMT2 promoter. Interestingly, the SjERFs gene cluster was observed to bind to the GCC-box1, 2 in the promoter of SjCNMT4, indicating their potential crucial role in CEP metabolism. Additionally, the SjERFs gene cluster-YFP fusion proteins were observed exclusively in the nuclei, which is consistent with their putative role as transcription factors in the nucleus (Figure 6). In conclusion, our findings suggest that the SjERFs gene cluster regulates CEP biosynthesis by directly binding to the GCC-boxes in the promoters of CEP-associated genes.
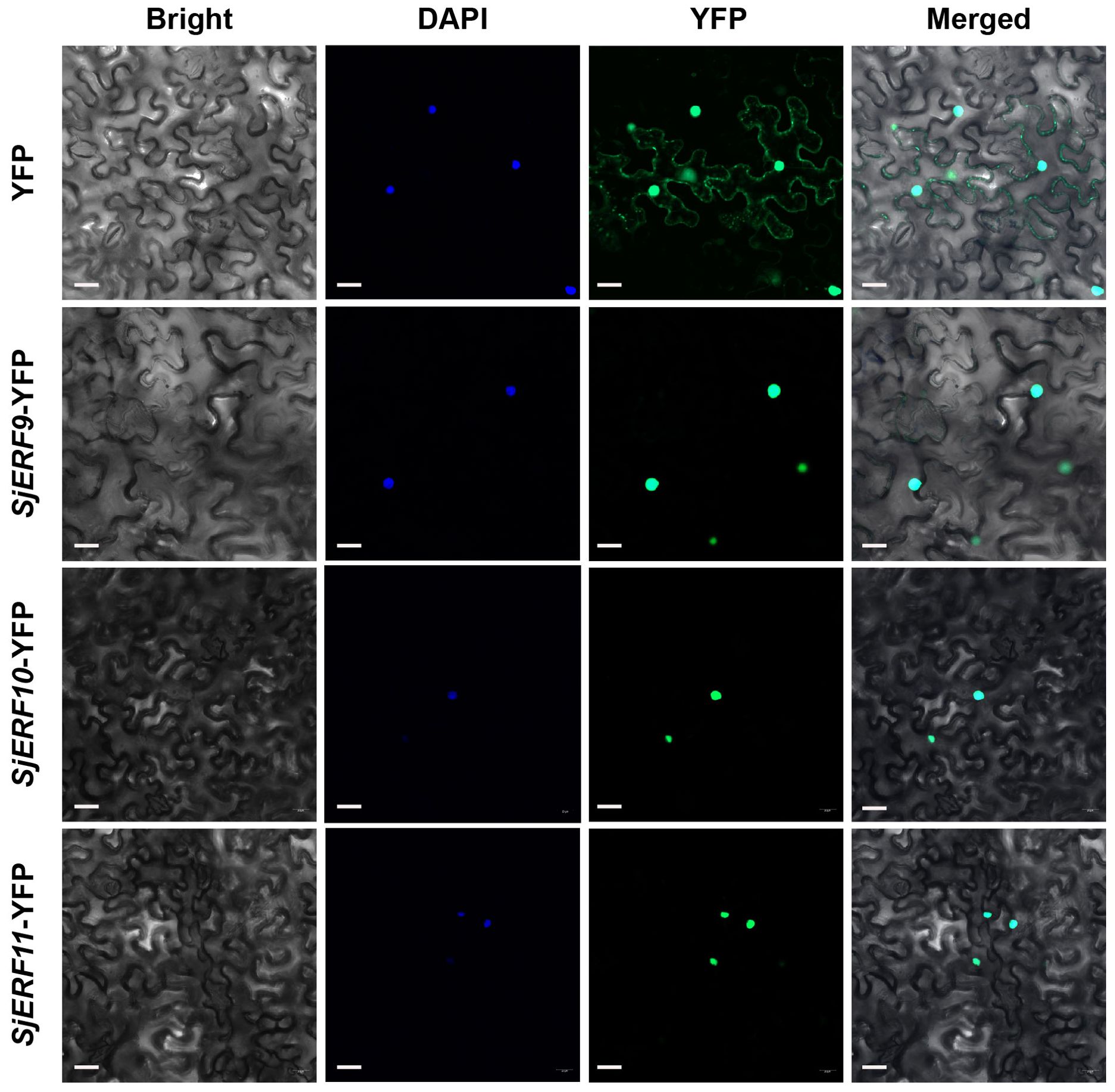
Figure 6. SjERFs protein fused to a yellow fluorescent protein (YFP) transiently expressed in N. benthamiana. Scale bar: 20 μm.
3.7 Tissue expression profiling of SjERF TFs
We analyzed the expression level of 59 SjERFs in roots, stems, leaves, and shoots of S. japonica from the available transcriptome data. Heat-map analysis showed that eleven SjERF genes were highly expressed in roots, stems, leaves, and shoots of S. japonica (FPKM > 50), including SjERF5, 9, 17, 20, 29, 43, 45, 49, 51, 55, and 57 (Figure 7A). Among them, SjERF20, 29, 43, 45, and 57 showed the highest expression level in S. japonica roots, and stems, respectively. However, thirteen genes had nearly no expression in the roots, stems, leaves, and shoots of S. japonica (FPKM < 1). Furthermore, some SjERF genes with tissue-specific or preferential expression patterns were observed in vegetative tissues of S. japonica. For example, SjERF5 and SjERF49 with the highest expressions were observed in S. japonica leaves. 13 SjERFs were observed with higher expression in root tissues of S. japonica. To validate the accuracy of RNA-seq, real-time qPCR was performed on six SjERFs, which exhibited significantly higher expression levels in the root of S. japonica. Overall, the results indicated that these SjERFs exhibited higher expression levels in the roots and lower expression in the leaves of S. japonica (Figure 7B). The qRT-PCR results of six SjERFs were consistent with the RNA-Seq data, indicating strong reliability of the RNA-Seq data.
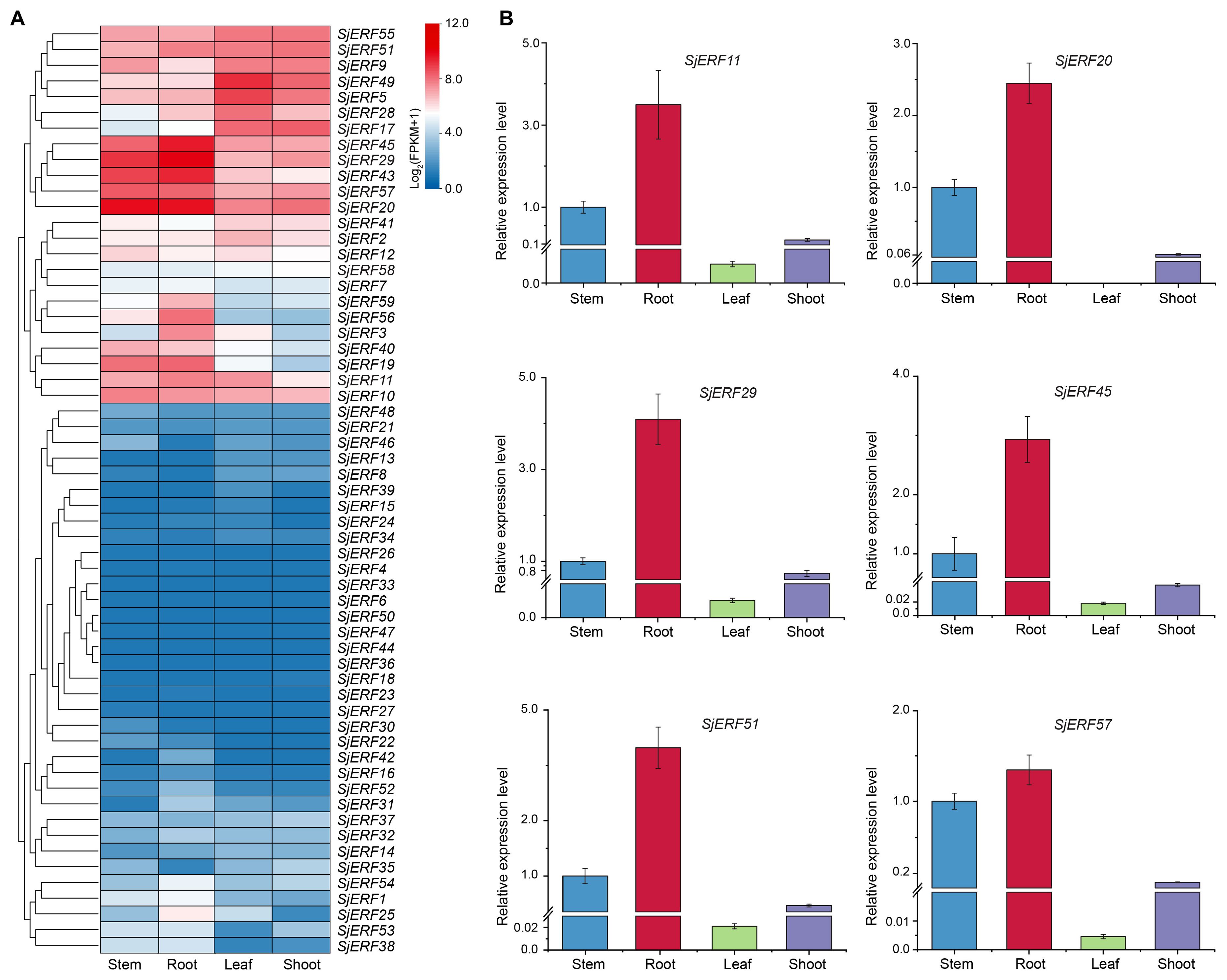
Figure 7. Expression patterns of 59 SjERFs in different tissues of S. japonica. (A) Hierarchical clustering of the expression level of SjERFs with RNA-Seq. (B) The expression profiles of six SjERFs in different tissues with the qRT-PCR method.
3.8 Co-expression analyses of SjERFs involved in CEP biosynthesis
Co-expression analysis of SjERFs, CEP biosynthetic genes, and BIAs metabolites was visualized using the Cytoscape tool. The co-expression network analysis revealed a strong correlation between the expression levels of 35 SjERFs and CEP biosynthetic genes in S. japonica (Pearson correlation coefficient r > 0.9 and p-value < 0.05). It is worth noting that SjERF17 and SjERF58 were strongly positively correlated with the three CEP biosynthetic genes, respectively (Figure 8; Supplementary Table S9). The expression profile of SjERF10 was correlated strongly with SjNCS2 and SjCNMT2 genes. Additionally, SjERF54 was highly positively correlated with ten BIAs, while SjERF35 was highly negatively correlated with these metabolites, including (S)-Norcoclaurine, N-Methylcoclaurine, 3-Hydroxy-N-methylcoclaurine, Magnoflorine, Coptisine, (S)-Tetrahydrocolumbamine, Guattegaumerine, Daurisoline, Fangchinoline, and Cepharanthine. SjERF42 and SjERF52 were highly positively correlated with seven BIAs. In summary, these findings suggest that these SjERFs may be involved in the biosynthesis of CEP and its precursors.
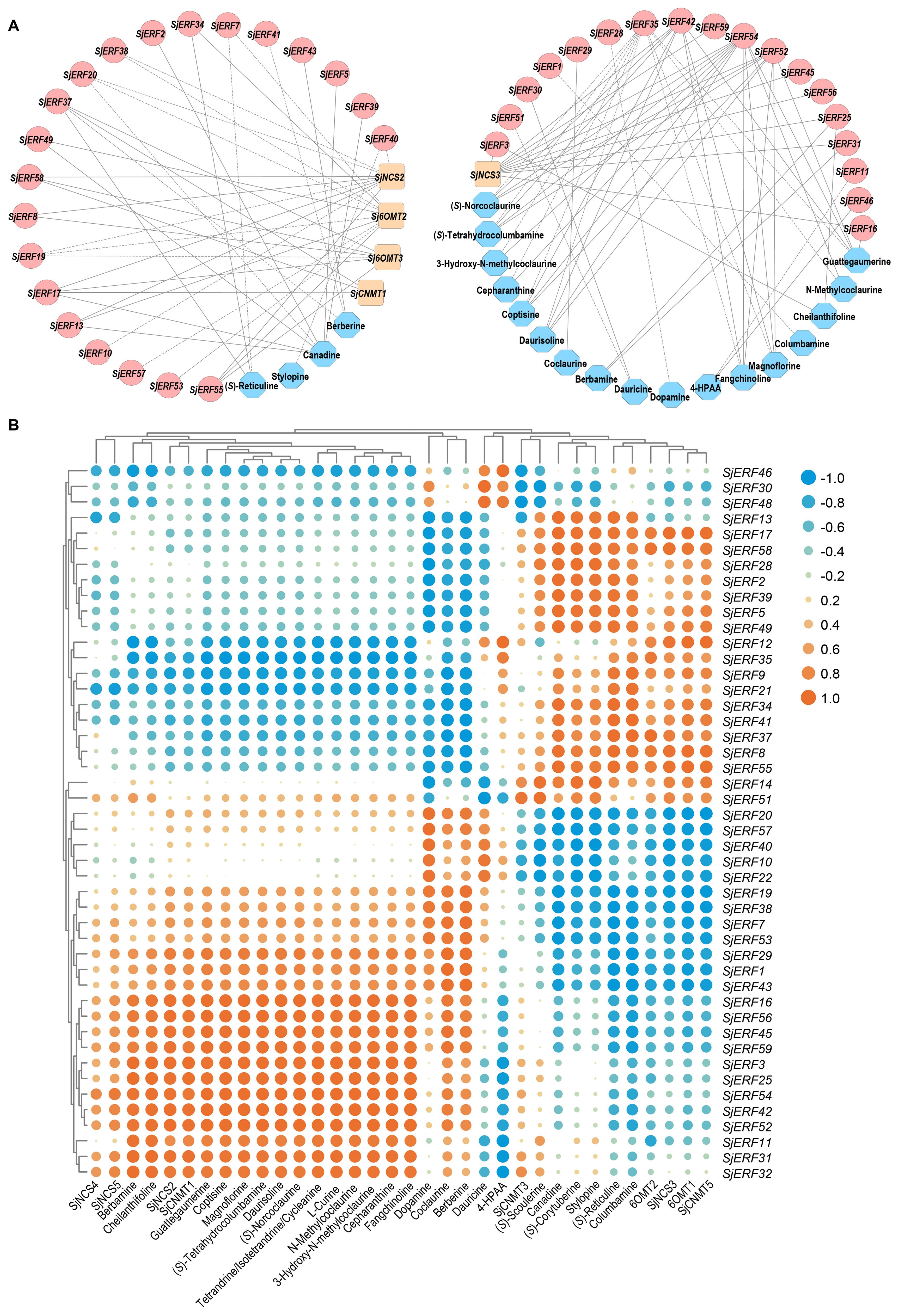
Figure 8. Analysis of correlation between SjERFs, CEP-biosynthetic genes, and BIAs metabolites. (A) Co-expression network of SjERF genes, CEP biosynthetic genes, and BIAs metabolites with |r| > 0.9 and p-value < 0.05. Orange squares: CEP biosynthetic genes, red circles: SjERF genes. Blue octagons: BIAs metabolites. (B) The cluster heatmap shows the expression correlations between five CEP biosynthetic genes, and two BIA precursors, alongside 23 BIA-type structures, and forty-six SjERFs.
4 Discussion
The AP2/ERF gene family is a plant-specific group of transcription factors, characterized by an AP2 domain for DNA binding (Licausi et al., 2013). Typically, members of this family usually contain one to two highly conserved AP2 domains. The AP2 subfamily members consist of members with two repeated AP2 domains, while the ERF subfamily contains members with a single AP2 domain (Sakuma et al., 2002). ERF transcription factors have significant effects in regulating the biosynthesis of the main pharmaceutical active components in medicinal plants (Gu et al., 2017; Wang M. et al., 2021). Extensive research on the ERF family has been conducted in various plants, including soybean, tomato, apple, corn, barley, and common wheat (Gu et al., 2017; Feng et al., 2020). However, genome-wide identification of ERF protein in BIA-producing plants remains limited.
In this study, 59 SjERFs have been identified in the S. japonica genome (Table 1; Figure 1), which is similar to the 60 EcERF genes in E. californica (Yamada et al., 2020), 59 in Cannabis sativa (Tian et al., 2020), and 65 in Spirodela polyrhiza (Tian et al., 2020). Each of these ERF genes is characterized by a single conserved AP2 domain. Notably, the number of SjERF gene members in the S. japonica genome was less than Oryza sativa (139 genes), Zea mays (136), A. thaliana (122), Glycine max (122), and Triticum aestivum (99) (Nakano et al., 2006; Feng et al., 2020). Collinear analysis was performed to explore the potential relationships between the SjERF genes in the S. japonica genome. The analysis revealed that a total of twelve SjERF genes were involved in six segmental duplication events (Figure 4). These segmental duplication events have likely contributed to the expansion of the ERF family in S. japonica.
Two commonly used classification systems were established in A. thaliana (Riechmann and Meyerowitz, 1998; Nakano et al., 2006). Riechmann et al. classified 144 AP2/ERF transcription factors into three classes (Riechmann and Meyerowitz, 1998); Sakuma et al. divided 145 AP2/ERF transcription factors into five classes and further divided the DREB subfamily into six subgroups (A1 to A6), and the ERF subfamily into six subgroups (B1 to B6) (Sakuma et al., 2002). In contrast, Nakano et al. classified ERF subfamily transcription factors into ten groups, which were named groups I to X, instead of the two major subfamilies (DREB and ERF) (Nakano et al., 2006). A phylogenetic tree of this work showed that 59 SjERFs were further categorized into ten subfamilies based on 121 AtERFs (Figure 1). This classification was in harmony with the evolutionary analyses of Nakano et al (Nakano et al., 2006). Generally, ERFs within the same group exhibit evolutionary conservation and share similar gene structures (Cao et al., 2020). Gene structure analysis revealed that 67.8% of SjERFs, including those from group II and III, contained only one exon, indicating a conserved gene structure for most SjERFs (Figure 2C). These findings were consistent with pineapple, where 66.22% of AcERFs displayed a similar gene structure (Huang et al., 2020). Furthermore, cis-element analysis of the promoter regions demonstrated that the majority of SjERF genes were involved in light-responsive processes (119), phytohormone responses (ABA, MeJA) (253), as well as abiotic and biotic stress responses (565) (Figure 3). Specifically, 164 abscisic acid responsiveness cis-acting regulatory elements (ABREs) were detected in the promoter regions of 57 SjERFs, excluding SjERF57 and SjERF59. Additionally, MeJA-responsive elements were discovered in the promoter regions of 54 SjERF genes, including SjERF gene clusters. Previous studies demonstrated that JAs are key signaling molecules involved in alkaloid biosynthesis (Wang M. et al., 2021). Many ERF transcription factors respond to jasmonic acid and activate the expression of alkaloid-associated genes, such as CrORCA and NbERF189 (van der Fits and Memelink, 2000; Shoji et al., 2010). Thus, these findings indicated that SjERFs can be regulated by various cis-acting elements in their promoters during growth and stress responses.
ERF TFs not only affect plant growth and development but also play a crucial role in secondary metabolisms, such as terpenoids, phenylpropanoids, and alkaloids (Zhou and Memelink, 2016; Shoji and Yuan, 2021; Godbole et al., 2022). The majority of ERFs shown to participate in secondary metabolites biosynthesis are members of group IX. Several group IX AP2/ERFs form physically linked gene clusters and have been characterized in a limited number of plant species, including Nicotiana tabacum (Kajikawa et al., 2017), potato (Cárdenas et al., 2016), and C. roseus (Paul et al., 2017). For instance, AaORA positively regulates artemisinin biosynthesis in Artemisia annua and activates the expression of AaADS, AaCYP71AV1, and AaDBR2 (Lu et al., 2013). In C. roseus, the ORCA cluster, consisting of ORCA3, ORCA4, and ORCA5, is a crucial regulator in alkaloid biosynthesis (van der Fits and Memelink, 2000; Singh et al., 2020). It is also proved that ERF189, ERF221, and the NIC2-locus clustered ERFs in N. benthamiana activate the nicotine biosynthetic pathway by affecting several nicotine biosynthetic genes (Shoji et al., 2010). To date, only the genome-wide identification and systematic analysis of ERF transcription factors in E. californica, which produces BIA, have been completed. It has been found that four Group IX ERFs can activate the expression of key enzyme genes involved in BIA biosynthesis (Yamada et al., 2020). In Coptis chinensis, cis-acting elements of BIA biosynthetic gene promoters were conducted and showed the involvement of GCC-box and ERF transcription factors in the regulation of berberine biosynthesis (Yamada et al., 2016). Nonetheless, the role of the ERF subfamily in CEP biosynthesis remains unexplored. In the present study, the co-expression network between SjERFs, CEP-associated genes, and BIAs metabolites showed that SjERF17 and SjERF58 have a strong correlation with the expression levels of CEP biosynthetic enzyme genes. SjERF42 and SjERF52 show positive correlations with the content of seven BIA metabolites; These results suggested that they might be involved in regulating the biosynthesis of CEP and its precursors (Figure 8). Notably, an ERF cluster (SjERF9/10/11) has also been identified in the S. japonica genome and is localized to the nucleus, respectively. Yeast one-hybrid assays proved that three SjERFs could directly bind to several CEP biosynthetic genes, including SjCNMT4 (Figure 5). In summary, the findings of this study indicate that SjERF cluster may act as a direct regulator of CEP metabolism by regulating the expression of CEP-associated genes. This study provides a foundation for analyzing the underlying molecular mechanism of CEP biosynthesis and further investigating the functional genomics of candidate SjERF genes.
5 Conclusions
This is the first study that 59 SjERFs were identified and categorized into ten subfamilies in S. japonica genome. Through a series of bioinformatics analyses of 59 SjERFs, it was found that the gene structure of SjERF32, and SjERF54 in same group was highly similar. Through collinear analysis, we identified twelve SjERF genes from the ERF genome data of S. japonica that were involved in six segmental duplication events. One gene cluster containing three SjERF genes was found on chromosome 2, which is close to the evolution of functional ORCA genes in C. roseus. Furthermore, the SjERFs cluster was observed to bind to the CEP-associated gene promoters, suggesting that the SjERFs cluster may act as a direct regulator of CEP metabolism. The tissue expression profile revealed that most SjERF genes were highly expressed in S. japonica root. Furthermore, we constructed a co-expression network between SjERFs, CEP biosynthetic genes, and BIAs metabolites, and several SjERFs were highly positively correlated with the contents of diverse BIAs of S. japonica. These results provide a basis for further characterizing the biological function of the SjERF gene and analyzing its molecular mechanism of regulating CEP biosynthesis.
Data availability statement
The raw data of Genome and RNA-seq data sets for the transcriptome analysis are available in NCBI, under BioProject PRJNA888087.
Author contributions
HY: Writing – original draft, Data curation, Investigation, Validation, Visualization. BL: Formal analysis, Visualization, Writing – original draft. HD: Visualization, Writing – original draft. ZL: Visualization, Writing – original draft. XL: Resources, Writing – review & editing. TH: Writing – original draft. YW: Writing – original draft. YZ: Writing – original draft. CW: Funding acquisition, Methodology, Writing – review & editing. LL: Methodology, Resources, Writing – review & editing. SC: Funding acquisition, Writing – review & editing. CS: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by introduces the talented person scientific research start funds subsidization project of Chengdu University of Traditional Chinese Medicine (030040015, 030040017), Sichuan Province Innovative Talent Funding Project for Postdoctoral Fellows (BX202206), China Postdoctoral Science Foundation (2023M730383), and Hubei science and technology planning project (2020BCB038).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1433015/full#supplementary-material
References
Al-Amin, M. Y., Lahiry, A., Ferdous, R., Hasan, M. K., Kader, M. A., Alam, A. K., et al. (2022). Stephania japonica ameliorates scopolamine-induced memory impairment in mice through inhibition of acetylcholinesterase and oxidative stress. Adv. Pharmacol. Pharm. Sci. 2022, 8305271. doi: 10.1155/2022/8305271
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME Suite: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Barber, R. D., Harmer, D. W., Coleman, R. A., Clark, B. J. (2005). GAPDH as a housekeeping gene: analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol. Genomics 21, 389–395. doi: 10.1152/physiolgenomics.00025.2005
Cao, S., Wang, Y., Li, X., Gao, F., Feng, J., Zhou, Y. (2020). Characterization of the AP2/ERF Transcription Factor Family and Expression Profiling of DREB Subfamily under Cold and Osmotic Stresses in Ammopiptanthus nanus. Plants. 9, 455. doi: 10.3390/plants9040455
Cárdenas, P. D., Sonawane, P. D., Pollier, J., Vanden Bossche, R., Dewangan, V., Weithorn, E., et al. (2016). GAME9 regulates the biosynthesis of steroidal alkaloids and upstream isoprenoids in the plant mevalonate pathway. Nat. Commun. 7, 10654. doi: 10.1038/ncomms10654
Carina, W., Till, O. (2019). Bisbenzylisoquinoline alkaloids. Alkaloids Chem. Biol. 81, 1–114. doi: 10.1016/bs.alkal.2018.07.001
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, M. L., Gou, J. M., Meng, X. L., Chen, C. L., Liu, X. N. (2019). Cepharanthine, a bisbenzylisoquinoline alkaloid, inhibits lipopolysaccharide-induced microglial activation. Pharmazie. 74, 606–610. doi: 10.1691/ph.2019.9562
Chen, H., Guo, M., Dong, S., Wu, X., Zhang, G., He, L., et al. (2023). A chromosome-scale genome assembly of Artemisia argyi reveals unbiased subgenome evolution and key contributions of gene duplication to volatile terpenoid diversity. Plant Commun. 4, 100516. doi: 10.1016/j.xplc.2023.100516
Fan, H.-H., Wang, L. Q., Liu, W. L., An, X. P., Liu, Z. D., He, X. Q., et al. (2020). Repurposing of clinically approved drugs for the treatment of coronavirus disease 2019 in a 2019-novel coronavirus-related coronavirus model. Chin. Med. J. (Engl). 133, 1051–1056. doi: 10.1097/CM9.0000000000000797
Feng, K., Hou, X. L., Xing, G. M., Liu, J. X., Duan, A. Q., Xu, Z. S., et al. (2020). Advances in AP2/ERF super-family transcription factors in plant. Crit. Rev. Biotechnol. 40, 750–776. doi: 10.1080/07388551.2020.1768509
Girardi, C. L., Rombaldi, C. V., Dal Cero, J., Nobile, P. M., Laurens, F., Bouzayen, M., et al. (2013). Genome-wide analysis of the AP2/ERF superfamily in apple and transcriptional evidence of ERF involvement in scab pathogenesis. Sci. Hortic. 151, 112–121. doi: 10.1016/j.scienta.2012.12.017
Godbole, R. C., Pable, A. A., Singh, S., Barvkar, V. T. (2022). The interplay of transcription factors orchestrates the biosynthesis of plant alkaloids. 3 Biotech. 12, 250. doi: 10.1007/s13205-022-03316-x
Gu, C., Guo, Z. H., Hao, P. P., Wang, G. M., Jin, Z. M., Zhang, S. L. (2017). Multiple regulatory roles of AP2/ERF transcription factor in angiosperm. Bot. Stud. 58, 6. doi: 10.1186/s40529-016-0159-1
Hao, X., Wang, C., Zhou, W., Ruan, Q., Xie, C., Yang, Y., et al. (2023). OpNAC1 transcription factor regulates the biosynthesis of the anticancer drug camptothecin by targeting loganic acid O-methyltransferase in Ophiorrhiza pumila. J. Integr. Plant Biol. 65, 133–149. doi: 10.1111/jipb.13377
He, S., Gao, J., Li, B., Luo, Z., Liu, P., Xu, X., et al. (2024). NtWIN1 regulates the biosynthesis of scopoletin and chlorogenic acid by targeting NtF6'H1 and NtCCoAMT genes in Nicotiana tabacum. Plant Physiol. Biochem. 214, 108937. doi: 10.1016/j.plaphy.2024.108937
Horton, P., Park, K. J., Obayashi, T., Fujita, N., Harada, H., Adams-Collier, C. J., et al. (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587. doi: 10.1093/nar/gkm259
Huang, Y., Liu, Y., Zhang, M., Chai, M., He, Q., Jakada, B. H., et al. (2020). Genome-wide identification and expression analysis of the ERF transcription factor family in pineapple (Ananas comosus (L.) Merr.). PeerJ. 8, e10014. doi: 10.7717/peerj.10014
Jain, N., Vergish, S., Khurana, J. P. (2018). Validation of house-keeping genes for normalization of gene expression data during diurnal/circadian studies in rice by RT-qPCR. Sci. Rep. 8, 3203. doi: 10.1038/s41598-018-21374-1
Kajikawa, M., Sierro, N., Kawaguchi, H., Bakaher, N., Ivanov, N. V., Hashimoto, T., et al. (2017). Genomic insights into the evolution of the nicotine biosynthesis pathway in Tobacco. Plant Physiol. 174, 999–1011. doi: 10.1104/pp.17.00070
Kraus, P. F., Kutchan, T. M. (1995). Molecular cloning and heterologous expression of a cDNA encoding berbamunine synthase, a C–O phenol-coupling cytochrome P450 from the higher plant berberis stolonifera. Proc. Natl. Acad. Sci. U S A. 92, 2071–2075. doi: 10.1073/pnas.92.6.2071
Kumar, P., Mathayan, M., Smieszek, S. P., Przychodzen, B. P., Koprivica, V., Birznieks, G., et al. (2022). Identification of potential COVID-19 treatment compounds which inhibit SARS Cov2 prototypic, Delta and Omicron variant infection. Virology. 572, 64–71. doi: 10.1016/j.virol.2022.05.004
Leng, L., Xu, Z., Hong, B., Zhao, B., Tian, Y., Wang, C., et al. (2024). Cepharanthine analogs mining and genomes of Stephania accelerate anti-coronavirus drug discovery. Nat. Commun. 15, 1537. doi: 10.1038/s41467-024-45690-5
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Li, Q., Bu, J., Ma, Y., Yang, J., Hu, Z., Lai, C., et al. (2020). Characterization of O-methyltransferases involved in the biosynthesis of tetrandrine in Stephania tetrandra. J. Plant Physiol. 250, 153181. doi: 10.1016/j.jplph.2020.153181
Li, J., Chen, G., Meng, Z., Wu, Z., Gan, H., Zhu, X., et al. (2022). Bioavailability enhancement of Cepharanthine via Pulmonary administration in Rats and Its therapeutic potential for Pulmonary Fibrosis associated with COVID-19 infection. Molecules. 27, 2745. doi: 10.3390/molecules27092745
Li, X., Yang, R., Liang, Y., Gao, B., Li, S., Bai, W., et al. (2023). The ScAPD1-like gene from the desert moss Syntrichia caninervis enhances resistance to Verticillium dahliae via phenylpropanoid gene regulation. Plant J. 113, 75–91. doi: 10.1111/tpj.16035
Licausi, F., Ohme-Takagi, M., Perata, P. (2013). APETALA2/Ethylene Responsive Factor (AP2/ERF) transcription factors: mediators of stress responses and developmental programs. New Phytol. 199, 639–649. doi: 10.1111/nph.12291
Liu, M., Sun, W., Ma, Z., Zheng, T., Huang, L., Wu, Q., et al. (2019). Genome-wide investigation of the AP2/ERF gene family in tartary buckwheat (Fagopyum Tataricum). BMC Plant Biol. 19, 84. doi: 10.1186/s12870-019-1681-6
Lu, X., Zhang, L., Zhang, F., Jiang, W., Shen, Q., Zhang, L., et al. (2013). AaORA, a trichome-specific AP2/ERF transcription factor of Artemisia annua, is a positive regulator in the artemisinin biosynthetic pathway and in disease resistance to Botrytis cinerea. New Phytol. 198, 1191–1202. doi: 10.1111/nph.12207
Minami, H., Dubouzet, E., Iwasa, K., Sato, F. (2007). Functional analysis of Norcoclaurine Synthase in Coptis japonica. J. Biol. Chem. 282, 6274–6282. doi: 10.1074/jbc.M608933200
Nakano, T., Suzuki, K., Fujimura, T., Shinshi, H. (2006). Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol. 140, 411–432. doi: 10.1104/pp.105.073783
Paul, P., Singh, S. K., Patra, B., Liu, X., Pattanaik, S., Yuan, L. (2020). Mutually regulated AP2/ERF gene clusters modulate biosynthesis of specialized metabolites in Plants. Plant Physiol. 182, 840–856. doi: 10.1104/pp.19.00772
Paul, P., Singh, S. K., Patra, B., Sui, X., Pattanaik, S., Yuan, L. (2017). A differentially regulated AP2/ERF transcription factor gene cluster acts downstream of a MAP kinase cascade to modulate terpenoid indole alkaloid biosynthesis in Catharanthus roseus. New Phytol. 213, 1107–1123. doi: 10.1111/nph.14252
Payne, J. T., Valentic, T. R., Smolke, C. D. (2021). Complete biosynthesis of the bisbenzylisoquinoline alkaloids guattegaumerine and berbamunine in yeast. Proc. Natl. Acad. Sci. U S A. 118, e2112520118. doi: 10.1073/pnas.2112520118
Pei, Y., Leng, L., Sun, W., Liu, B., Feng, X., Li, X., et al. (2024). Whole-genome sequencing in medicinal plants: current progress and prospect. Sci. China Life Sci. 67, 258–273. doi: 10.1007/s11427-022-2375-y
Riechmann, J. L., Meyerowitz, E. M. (1998). The AP2/EREBP family of plant transcription factors. Biol. Chem. 379, 633–646. doi: 10.1515/bchm.1998.379.6.633
Sakuma, Y., Liu, Q., Dubouzet, J. G., Abe, H., Shinozaki, K., Yamaguchi-Shinozaki, K. (2002). DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration- and cold-inducible gene expression. Biochem. Biophys. Res. Commun. 290, 998–1009. doi: 10.1006/bbrc.2001.6299
Servant, N., Varoquaux, N., Lajoie, B. R., Viara, E., Chen, C.-J., Vert, J.-P., et al. (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259. doi: 10.1186/s13059-015-0831-x
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shoji, T., Hashimoto, T. (2012). DNA-binding and transcriptional activation properties of tobacco NIC2-locus ERF189 and related transcription factors. Plant Biotechnol. 29, 35–42. doi: 10.5511/plantbiotechnology.11.1216a
Shoji, T., Kajikawa, M., Hashimoto, T. (2010). Clustered transcription factor genes regulate nicotine biosynthesis in tobacco. Plant Cell. 22, 3390–3409. doi: 10.1105/tpc.110.078543
Shoji, T., Yuan, L. (2021). ERF Gene Clusters: working together to regulate metabolism. Trends Plant Sci. 26, 23–32. doi: 10.1016/j.tplants.2020.07.015
Shoji, T., Sugawara, S., Mori, T., Kobayashi, M., Kusano, M., Saito, K. (2023). Induced production of specialized steroids by transcriptional reprogramming in Petunia hybrida. PNAS Nexus 2, pgad326. doi: 10.1093/pnasnexus/pgad326
Singh, S. K., Patra, B., Paul, P., Liu, Y., Pattanaik, S., Yuan, L. (2020). Revisiting the ORCA gene cluster that regulates terpenoid indole alkaloid biosynthesis in Catharanthus roseus. Plant Sci. 293, 110408. doi: 10.1016/j.plantsci.2020.110408
Stadler, R., Loeffler, S., Cassels, B. K., Zenk, M. H. (1988). Bisbenzylisoquinoline biosynthesis in Berberis stolonifera cell cultures. Phytochemistry. 27, 2557–2565. doi: 10.1016/0031-9422(88)87027-4
Sun, L., Jing, Y., Liu, X., Li, Q., Xue, Z., Cheng, Z., et al. (2020). Heat stress-induced transposon activation correlates with 3D chromatin organization rearrangement in Arabidopsis. Nat. Commun. 11, 1886. doi: 10.1038/s41467-020-15809-5
Sun, S., Shen, X., Li, Y., Li, Y., Wang, S., Li, R., et al. (2023). Single-cell RNA sequencing provides a high-resolution roadmap for understanding the multicellular compartmentation of specialized metabolism. Nat. Plants. 9, 179–190. doi: 10.1038/s41477-022-01291-y
Taketa, S., Amano, S., Tsujino, Y., Sato, T., Saisho, D., Kakeda, K., et al. (2008). Barley grain with adhering hulls is controlled by an ERF family transcription factor gene regulating a lipid biosynthesis pathway. Proc. Natl. Acad. Sci. U S A. 105, 4062–4067. doi: 10.1073/pnas.0711034105
Tamura, K., Stecher, G., Kumar, S.. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tian, F., Yang, D. C., Meng, Y. Q., Jin, J., Gao, G. (2020). PlantRegMap: charting functional regulatory maps in plants. Nucleic Acids Res. 48, D1104–D1113. doi: 10.1093/nar/gkz1020
Udomsom, N., Rai, A., Suzuki, H., Okuyama, J., Imai, R., Mori, T., et al. (2016). Function of AP2/ERF transcription factors involved in the regulation of specialized metabolism in Ophiorrhiza pumila revealed by transcriptomics and metabolomics. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01861
van der Fits, L., Memelink, J. (2000). ORCA3, a jasmonate-responsive transcriptional regulator of plant primary and secondary metabolism. Science. 289, 295–297. doi: 10.1126/science.289.5477.295
Wang, C., Hao, X., Wang, Y., Maoz, I., Zhou, W., Zhou, Z., et al. (2022). Identification of WRKY transcription factors involved in regulating the biosynthesis of the anti-cancer drug camptothecin in Ophiorrhiza pumila. Hortic. Res. 9, uhac099. doi: 10.1093/hr/uhac099
Wang, C., Hao, X., Wang, Y., Shi, M., Zhou, Z. G., Kai, G. (2021). Genome-Wide identification and comparative analysis of the teosinte branched 1/cycloidea/proliferating cell factors 1/2 transcription factors related to Anti-cancer drug camptothecin biosynthesis in Ophiorrhiza pumila. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.746648
Wang, M., Qiu, X., Pan, X., Li, C. (2021). Transcriptional factor-mediated regulation of active Component biosynthesis in medicinal plants. Curr. Pharm. Biotechnol. 22, 848–866. doi: 10.2174/1389201021666200622121809
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. doi: 10.1093/nar/gkr1293
Wolff, J., Bhardwaj, V., Nothjunge, S., Richard, G., Renschler, G., Gilsbach, R., et al. (2018). Galaxy HiCExplorer: a web server for reproducible Hi-C data analysis, quality control and visualization. Nucleic Acids Res. 46, W11–W16. doi: 10.1093/nar/gky504
Xiao, J., Hao, T., Chen, G., Song, J., Lin, B., Li, W., et al. (2019). Natural neuroprotective alkaloids from Stephania japonica (Thunb.) Miers. Bioorg Chem. 91, 103175. doi: 10.1016/j.bioorg.2019.103175
Xing, H., Jiang, Y., Zou, Y., Long, X., Wu, X., Ren, Y., et al. (2021). Genome-wide investigation of the AP2/ERF gene family in ginger: evolution and expression profiling during development and abiotic stresses. BMC Plant Biol. 21, 561. doi: 10.1186/s12870-021-03329-3
Xu, W., Chen, S., Wang, X., Tanaka, S., Onda, K., Sugiyama, K., et al. (2021). Molecular mechanisms and therapeutic implications of tetrandrine and Cepharanthine in T cell acute lymphoblastic leukemia and autoimmune diseases. Pharmacol. Ther. 217, 107659. doi: 10.1016/j.pharmthera.2020.107659
Yamada, Y., Nishida, S., Shitan, N., Sato, F. (2020). Genome-wide identification of AP2/ERF transcription factor-encoding genes in California poppy (Eschscholzia californica) and their expression profiles in response to methyl jasmonate. Sci. Rep. 10, 18066. doi: 10.1038/s41598-020-75069-7
Yamada, Y., Sato, F. (2021). Transcription factors in alkaloid engineering. Biomolecules. 11, 1719. doi: 10.3390/biom11111719
Yamada, Y., Yoshimoto, T., Yoshida, S. T., Sato, F. (2016). Characterization of the promoter region of biosynthetic enzyme genes involved in Berberine biosynthesis in Coptis japonica. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01352
Yang, H., Liu, Z., Yu, C., Song, C., Wang, C. (2023). Expression relationship between microRNA and transcription factors in Stephania japonica. Medicinal Plant Biol. 2, 7. doi: 10.48130/MPB-2023-0007
Yang, H., Wang, Y., Liu, W., He, T., Liao, J., Qian, Z., et al. (2024a). Genome-wide pan-GPCR cell libraries accelerate drug discovery. Acta Pharm. Sin. B, 2211–3835. doi: 10.1016/j.apsb.2024.06.023
Yang, H., Wang, C., Zhou, G., Zhang, Y., He, T., Yang, L., et al. (2024b). A haplotype-resolved gap-free genome assembly provides novel insight into monoterpenoid diversification in Mentha suaveolens ‘Variegata.’. Hortic. Res. 11, uhae022. doi: 10.1093/hr/uhae022
Yuan, L. (2020). Clustered ERF Transcription Factors: not all created equal. Plant Cell Physiol. 61, 1025–1027. doi: 10.1093/pcp/pcaa067
Zhang, H., Gao, S., Lercher, M. J., Hu, S., Chen, W. H. (2012). EvolView, an online tool for visualizing, annotating and managing phylogenetic trees. Nucleic Acids Res. 40, W569–W572. doi: 10.1093/nar/gks576
Zhang, M., Li, S., Nie, L., Chen, Q., Xu, X., Yu, L., et al. (2015). Two jasmonate-responsive factors, TcERF12 and TcERF15, respectively act as repressor and activator of tasy gene of taxol biosynthesis in Taxus chinensis. Plant Mol. Biol. 89, 463–473. doi: 10.1007/s11103-015-0382-2
Zhang, X., Zhang, G., Zhao, Z., Xiu, R., Jia, J., Chen, P., et al. (2021). Cepharanthine, a novel selective ANO1 inhibitor with potential for lung adenocarcinoma therapy. Biochim. Biophys. Acta Mol. Cell Res. 1868, 119132. doi: 10.1016/j.bbamcr.2021.119132
Zhao, W., Shen, C., Zhu, J., Ou, C., Liu, M., Dai, W., et al. (2020). Identification and characterization of methyltransferases involved in benzylisoquinoline alkaloids biosynthesis from Stephania intermedia. Biotechnol. Lett. 42, 461–469. doi: 10.1007/s10529-019-02785-0
Zhou, M., Memelink, J. (2016). Jasmonate-responsive transcription factors regulating plant secondary metabolism. Biotechnol. Adv. 34, 441–449. doi: 10.1016/j.bioteChadv.2016.02.004
Zhu, Y., Li, Y., Zhang, S., Zhang, X., Yao, J., Luo, Q., et al. (2019). Genome-wide identification and expression analysis reveal the potential function of ethylene responsive factor gene family in response to Botrytis cinerea infection and ovule development in grapes (Vitis vinifera L.). Plant Biol. (Stuttg). 21, 571–584. doi: 10.1111/plb.12943
Keywords: ERF, Stephania japonica, Cepharanthine biosynthesis, expression patterns, genome-wide analysis
Citation: Yang H, Liu B, Ding H, Liu Z, Li X, He T, Wu Y, Zhang Y, Wang C, Leng L, Chen S and Song C (2024) Genome-wide analysis of the ERF Family in Stephania japonica provides insights into the regulatory role in Cepharanthine biosynthesis. Front. Plant Sci. 15:1433015. doi: 10.3389/fpls.2024.1433015
Received: 15 May 2024; Accepted: 15 August 2024;
Published: 04 September 2024.
Edited by:
Weizhen Liu, Wuhan University of Technology, ChinaReviewed by:
Pawan Kumar, Agricultural Research Organization (ARO), IsraelXiaoxu Li, Chinese Academy of Agricultural Sciences (CAAS), China
Copyright © 2024 Yang, Liu, Ding, Liu, Li, He, Wu, Zhang, Wang, Leng, Chen and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Can Wang, d2FuZ2NhbkBjZHV0Y20uZWR1LmNu; Liang Leng, bGxpbmdAY2R1dGNtLmVkdS5jbg==; Shilin Chen, c2xjaGVuQGNkdXRjbS5lZHUuY24=; Chi Song, c29uZ2NoaUBjZHV0Y20uZWR1LmNu
†These authors have contributed equally to this work