 Le Wang
Le Wang Xue Liu
Xue Liu Yongde Wang1*
Yongde Wang1*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 30 July 2024
Sec. Functional and Applied Plant Genomics
Volume 15 - 2024 | https://doi.org/10.3389/fpls.2024.1429545
The genus Dendrobium, part of the Orchidaceae family, encompasses species of significant medicinal, nutritional, and economic value. However, many Dendrobium species are threatened by environmental stresses, low seed germination rates, and overharvesting. Mitochondria generate the energy necessary for various plant life activities. Despite their importance, research on the mitochondrial genomes of Dendrobium species is currently limited. To address this gap, we performed a comprehensive genetic analysis of four Dendrobium species—D. flexicaule, D. nobile, D. officinale, and D. huoshanense—focusing on their mitochondrial and chloroplast genomes to elucidate their genetic architecture and support conservation efforts. We utilized advanced sequencing technologies, including Illumina for high-throughput sequencing and Nanopore for long-read sequencing capabilities. Our findings revealed the multichromosomal mitochondrial genome structures, with total lengths ranging from 596,506 bp to 772,523 bp. The mitochondrial genomes contained 265 functional genes, including 64-69 protein-coding genes, 23-28 tRNA genes, and 3 rRNA genes. We identified 647 simple sequence repeats (SSRs) and 352 tandem repeats, along with 440 instances of plastid-to-mitochondrial gene transfer. Additionally, we predicted 2,023 RNA editing sites within the mitochondrial protein-coding genes, predominantly characterized by cytosine-to-thymine transitions. Comparative analysis of mitochondrial DNA across the species highlighted 25 conserved genes, with evidence of positive selection in five genes: ccmFC, matR, mttB, rps2, and rps10. Phylogenetic assessments suggested a close sister relationship between D. nobile and D. huoshanense, and a similar proximity between D. officinale and D. flexicaule. This comprehensive genomic study provides a critical foundation for further exploration into the genetic mechanisms and biodiversity of Dendrobium species, contributing valuable insights for their conservation and sustainable utilization.
Plastids and mitochondria are essential organelles in plant cells, each containing a genome distinct from the nuclear genome. Plastids play a crucial role in photosynthesis and various biosynthetic processes, while mitochondria are the primary sites for oxidative phosphorylation and ATP synthesis, both vital for plant growth and development (Liu et al., 2023; Lian et al., 2024; Wang et al., 2024). The plastid genome is maternally inherited, rich in gene content, structurally conserved, and evolves relatively slowly. These characteristics make plastid genomes valuable for phylogenetic studies, biological research, and examining the degradation of photosynthetic genes in plants. In contrast, plant mitochondrial genomes (mtDNA) have traditionally garnered less attention than plastid or nuclear genomes due to their highly variable structures and repetitive DNA content, which present significant challenges for comprehensive genomic sequencing (Xiong et al., 2021; Han et al., 2022). Meanwhile, variations in mtDNA size are largely due to differences in the amount of non-coding content, which originates from diverse sources. These sources include repeats, large duplications (Maréchal and Brisson, 2010), intracellular gene transfers (IGT) from nuclear or plastid genomes (Gandini et al., 2019), and horizontal gene transfers (HGT). Notably, RNA editing events are considered a general feature of plant mtDNAs (Varré et al., 2019). Particularly, Cytosine (C)-to-Uracil (U) RNA editing occurs at approximately 200-800 mRNA sites (Mower, 2008; Sloan et al., 2010; Rice et al., 2013; Richardson et al., 2013). Additionally, 92% of RNA editing sites result in changes to amino acid sequences. For example, hydrophilic amino acids are often converted into hydrophobic ones, which can enhance protein folding and gene functionality (Giegé and Brennicke, 1999). The study of mitochondrial genomes in angiosperms not only elucidates phylogenetic relationships between species but also facilitates the investigation of intraspecific differentiation.
To date, it is known that the majority of plant mitochondrial genomes exhibit a single circular chromosome structure, but this is not universal (Qu et al., 2024). For example, the mitochondrial genomes of Lilium tsingtauense (Qu et al., 2024), Populus simonii (Bi et al., 2022), Amborella trichopoda, Gastrodia elata (Yuan et al., 2018), Populus deltoides (Qu et al., 2023), Cucumis sativus, wheat and rape exhibit complex multichromosomal structures (Alverson et al., 2011; Yu et al., 2022). Interestingly, the largest circular chromosome (1,556 kb) in the C. sativus mtDNA has greater protein-coding capability than the other two smaller chromosomes (45 and 84 kb). This suggests that the three mitochondrial chromosomes of C. sativus replicate independently (Alverson et al., 2011; Wu et al., 2022). Similarly, the mitochondrial genomes of Lophophytum mirabile and Silene noctiflora consist of 54 and 128 distinct circular chromosomes, respectively, highlighting the extreme complexity of plant mitochondrial structures (Bi et al., 2022). Currently, the D. officinale and D. huoshanense mtDNAs were found to have 22 and 19 chromosomes, respectively. This indicated that the differently length isoforms during rapid evolution of Dendrobium mtDNAs structure might be caused by repeat-mediated rearrangement events (Wang et al., 2023). Exploring mitochondrial genome differences among species or subspecies of endangered plants elucidates the role of mitochondria in biological evolution. This understanding can significantly enhance applications in genetic breeding, conservation, and research.
Dendrobium, the second-largest genus in the Orchidaceae family, encompasses approximately 80 species, many of which hold significant medicinal, nutritional, ornamental, and economic value (Xu et al., 2022). However, extreme ecological conditions and insufficient protective measures for wild Dendrobium resources, several species, notably D. huoshanense and D. flexicaule, are considered rare and endangered. Due to the significant morphological similarities among D. flexicaule, D. nobile, D. officinale, and D. huoshanense, we sequenced the mitochondrial and plastid genomes of four important medicinal Dendrobium species using both Illumina and Nanopore sequencing technologies in this study. Furthermore, we analyzed codon usage preference, sequence repeat, Ka/Ks ratio, RNA editing sites and collinearity in the four Dendrobium mtDNAs. Additionally, we also analyzed homologous fragments between mitochondria and chloroplasts and phylogenetic analysis. Our analyses provide the theoretical foundation for the identification, conservation, and the development of molecular markers and genetic breeding strategies of Dendrobium species.
Fresh and healthy leaves from four individual plants (D. flexicaule, D. nobile, D. officinale, D. huoshanense) were collected and cultivated in the germplasm resources herb garden at the Chongqing Academy of Chinese Materia Medica, Chongqing, China (Supplementary Table S2.1). The integrity and concentration of DNA were assessed using 0.75% agarose gel electrophoresis, a NanoDrop One spectrophotometer (Thermo Fisher Scientific), and a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). DNA samples were sheared using a Covaris ultrasonic disruptor and prepared into 300 bp insert size Illumina sequencing libraries using the Nextera DNA Flex Library Prep Kit (Illumina, San Diego, CA, USA). Sequencing was performed on the Illumina NovaSeq platform (Supplementary Figure S1). Raw reads were processed to remove adapters, unknown nucleotides (Ns), and low-quality bases using SOAPnuke v.2.1.4 (https://github.com/BGI-flexlab/SOAPnuke), retaining clean data for further analysis. For Oxford Nanopore sequencing, libraries were prepared using the SQK-LSK109 ligation kit and sequenced on a PromethION sequencer (Oxford Nanopore Technologies, Oxford, UK) for 48-hour runs. Base-calling was conducted using GUPPY software (Shen et al., 2022).
We combined next-generation sequencing (NGS) data from Illumina and third-generation sequencing (TGS) data from Nanopore to assemble the mitochondrial genomes of four Dendrobium species. Using GetOrganelle v.1.7.5 (Jin et al., 2020), we assembled the plant mitochondria with default parameters, incorporating both next-generation and third-generation DNA sequencing data to produce a graphical plant mitochondrial genome (Jin et al., 2020). The bandage tool was employed to visualize this mitochondrial genome and manually eliminate extended segments originating from chloroplast and nuclear genomes (Wick et al., 2015). Subsequently, bwa software was used to align the Nanopore data to the graphical mitochondrial genome fragment, which helped resolve repetitive regions within the plant mitochondrial genome (Li and Durbin, 2009). As a result, we obtained the mitochondrial genomes of the four Dendrobium species, each with multiple branches.
For annotating the protein-coding genes (PCGs) of the mitochondrial genome, we used reference genomes from the complete mitochondrial genomes of Dendrobium (LC704589.1-LC704614.1). The Geseq software program (Tillich et al., 2017) was employed for mitochondrial genome annotation. tRNA-encoding genes were annotated with tRNAscan-SE software (Lowe and Eddy, 1997), and rRNA-encoding genes were annotated using BLASTN software (Chen et al., 2015). Any annotation errors in the mitochondrial genome were manually corrected using Apollo software (Lewis et al., 2002).
The Phylosuite software program was used with default parameters to extract protein-coding sequences from the mitochondrial genomes of four Dendrobium species (Zhang et al., 2020). The MEGA v.7.0 software was employed to analyze the coding bias of mitogenomic protein-coding genes (PCGs) and to calculate the relative synonymous codon usage (RSCU) values (Kumar et al., 2016). The online tool CUSP (https://www.bioinformatics.nl/cgi-bin/emboss/cusp) was used to determine the overall GC content, as well as GC1, GC2, and GC3 content in the mtDNA of the four Dendrobium species. The CodonW v.1.4.2 software with default settings was used to compute the effective number of codons (ENC) (Sharp and Li, 1986), which measures the diversity of codon usage in a gene, with values typically ranging from 20 (indicating each amino acid uses only one codon) to 61 (indicating equal usage of all codons) (Wright, 1990). The GC content at different positions in the four Dendrobium species was visualized using TBtools software with default parameters (Chen C. et al., 2020). Additionally, a scatter plot of the average GC1 and GC2 (GC12) values against GC3 values was created using Microsoft Excel 2021 to analyze neutrality in the four Dendrobium species.
The online tools MISA (https://webblast.ipk-gatersleben.de/misa/), TRF (https://tandem.bu.edu/trf/trf.unix.help.html), and the REPuter network server were utilized for analyzing simple sequence repeats (SSRs), tandem repeats, and dispersed repeats, respectively. The parameters for MISA were set as 1-10, 2-5, 3-4, 4-3, 5-3, and 6-3. For TRF, the parameters were 2 7 7 80 10 50 2000 –f -d -m. REPuter was used with a hamming distance of 3, a maximum of 5,000 computed repeats, and a minimal repeat size of 30. Default parameters were applied for the analysis of dispersed repeats (Benson, 1999; Kurtz et al., 2001; Beier et al., 2017; Shen et al., 2024).
We assembled and annotated the chloroplast genome using Getorganelle software and CPGAVAS2 respectively (Shi et al., 2019; Jin et al., 2020). The Circos package and Excel 2021 were employed to visualize the resulting plots. Additionally, BLASTN software (parameters: e-value=1e−6; word size=7) was used for analyzing homologous fragments, and the Circos package was utilized to visualize intracellular gene transfers (IGT) (Zhang et al., 2013; Chen et al., 2015).
RNA editing sites were predicted using PREPACT3 (http://www.prepact.de/) with a cutoff value of 0.001 (Lenz et al., 2018). Homologous sequences longer than 500 bp were retained to construct multiple synteny plots as conserved collinear blocks and pairwise comparisons of individual mitochondrial genomes were conducted using BLASTN with the parameters set to e-value of ≤1e−10 and a matching rate of ≥80% (Qu et al., 2023). The MCscanX program was employed to generate multiple synteny plots for the four Dendrobium species (Wang et al., 2012).
23 species closely related to the four Dendrobium species were selected, and their mitochondrial genomes were downloaded from NCBI. Magnolia biondii and Liriodendron tulipifera were designated as outgroups. Twenty-one conserved protein-coding genes (atp1, atp4, atp6, ccmB, ccmC, ccmFC, ccmFN, cob, cox1, cox2, cox3, matR, nad1, nad2, nad3, nad4, nad5, nad6, nad7, nad9, and rps12) were aligned using the MAFFT software with default parameters (Katoh and Standley, 2013). A phylogenetic tree was constructed using IQ-TREE v.1.6.12 with the parameters “-m MFP -B 1000 –bnni -T AUTO “ and the model “GTR+F+I+I+R2”. The results were visualized using ITOL v.4.0 software (Nguyen et al., 2015; Bi et al., 2022).
Illumina sequencing generated a total of 47.7 Gb of raw data, with the following breakdown: D. flexicaule (11.3 Gb), D. nobile (13.2 Gb), D. huoshanense (11.5 Gb), and D. officinale (11.7 Gb). Nanopore sequencing contributed approximately 45.9 Gb of data across the four species: D. flexicaule (11.3 Gb), D. nobile (12.9 Gb), D. huoshanense (10.5 Gb), and D. officinale (11.2 Gb). The mitochondrial and plastidial genomes of these species are available in GenBank (https://www.ncbi.nlm.nih.gov), with corresponding accession numbers provided in Supplementary Tables S2.2–S2.5. The mitochondrial and plastidial genomes were analyzed to identify variations and conservations within the Dendrobium species. Using Bandage, we graphically represented the assembled mtDNAs based on Nanopore data, manually removing nodes from the nuclei and chloroplasts (Figure 1). These nodes are independent of each other and capable of self-organizing into circular structures, revealing that the mitochondrial genomes of these species exhibit a complex multibranched conformation with multiple single circular structures (Figure 2). Detailed analysis showed that D. flexicaule’s mitochondrial DNA comprises 20 circular chromosomes, D. nobile has 25, D. officinale has 21, and D. huoshanense has 20 circular chromosomes. The sizes of these mitochondrial genomes vary significantly: D. flexicaule measures 596,506 bp, D. nobile 772,523 bp, D. officinale 625,267 bp, and D. huoshanense 650,957 bp. The GC content was highly similar among the mitochondrial genomes of the four Dendrobium species (Table 1).

Figure 1 Sketch of the mitochondrial genome of Dendrobium nobile. Based on the Nanopore, sketch of the mitogenomes were assembled. Sketches of mitochondrial genomes of D. huoshanense, D. flexicaule and D. officinale were uploaded in Additional File 1 (Supplementary Figure S2).

Figure 2 Dendrobium nobile mitogenome map. Genes are classified into different categories based on different colors, with the gray area within the circles representing different GC contents. Mitogenome maps of D. huoshanense, D. flexicaule and D. offcinale were uploaded in Additional File 1 (Supplementary Figure S3).
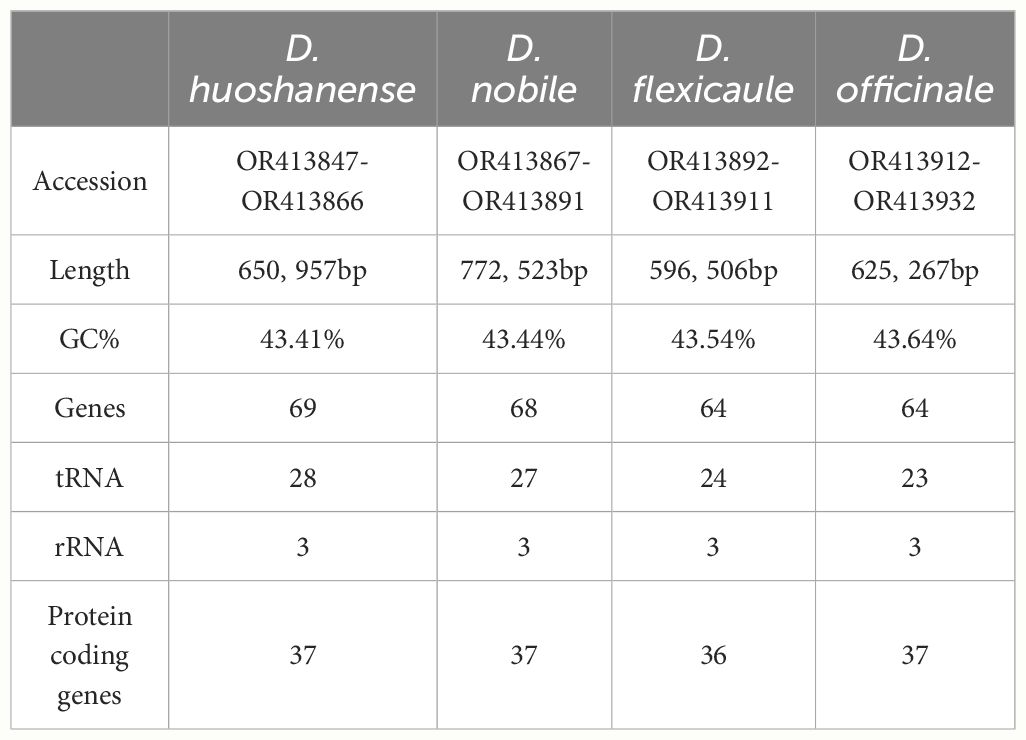
Table 1 Features of four Dendrobium mitogenomes.
Among the four Dendrobium species, D. huoshanense possesses the most genes with a total of 69, followed closely by D. nobile with 68, and both D. flexicaule and D. officinale with 64 genes each (Supplementary Tables S2.6–S2.9). All species showed significant conservation in the number and types of rRNA genes, though differences were noted in their tRNA-encoding genes and protein-coding genes (PCGs). Specifically, D. flexicaule was unique in the loss of the rps11 gene. Variations in the types and quantities of tRNA-encoding genes were observed among the species, particularly in trnM-CAU, trnG-GCC, trnN-GUU, and trnQ-UUG. Compared to D. flexicaule, D. huoshanense had three additional tRNA-encoding genes (trnL-CAA, trnR-ACG, and trnT-UGU) but lacked trnH-GUG. trnI-CAU gene was missing in D. nobile but additional genes trnR-ACG and trnT-UGU were present. D. officinale lacked trnH-GUG but had an additional trnT-UGU gene (Supplementary Table S2.10).
Chloroplast genomes of four Dendrobium species have also been sequenced and assembled. We found that the chloroplast genomes displayed smaller variations in size compared to their mitochondrial counterparts. The lengths of cpDNA were 150,602 bp for D. nobile, 150,529 bp for D. huoshanense, 152,588 bp for D. flexicaule, and 152,213 bp for D. officinale (Supplementary Figure S4). D. nobile and D. huoshanense shared similar GC content, as well as comparable tRNA, rRNA, and protein-coding regions (Table 2). The results of the mVISTA analysis demonstrated that the chloroplast genomes of the four species are highly similar, especially in the IR and coding regions, with lower differentiation observed in the SC and non-coding areas (Supplementary Figure S5). This pattern of conservation and variation provides valuable insights into the evolutionary dynamics and functional adaptations within the genus Dendrobium.

Table 2 Features of chloroplast genomes of the four Dendrobium species based on the Orchidaceae.
Codon usage bias is crucial for understanding gene expression and evolutionary patterns. In our analysis, we assessed the codon usage of 36 unique protein-coding genes (PCGs) from D. flexicaule, and 37 from each of D. officinale, D. nobile, and D. huoshanense. The results revealed a distinct amino acid bias, with Relative Synonymous Codon Usage (RSCU) values above 1 indicating preferences (Figure 3, Supplementary Tables S2.11–S2.14). The mitochondrial genomes showed a general codon bias; for instance, Ala favored GCU, with RSCU values around 1.62 in D. officinale and D. huoshanense, and 1.61 in both D. flexicaule and D. nobile. Gln showed a preference for CAA in D. huoshanense and D. officinale, while His in D. flexicaule and D. nobile had an RSCU of 1.51. Notably, Lys, Phe, and the stop codons exhibited values below 1.2, suggesting minimal codon bias for these amino acids.
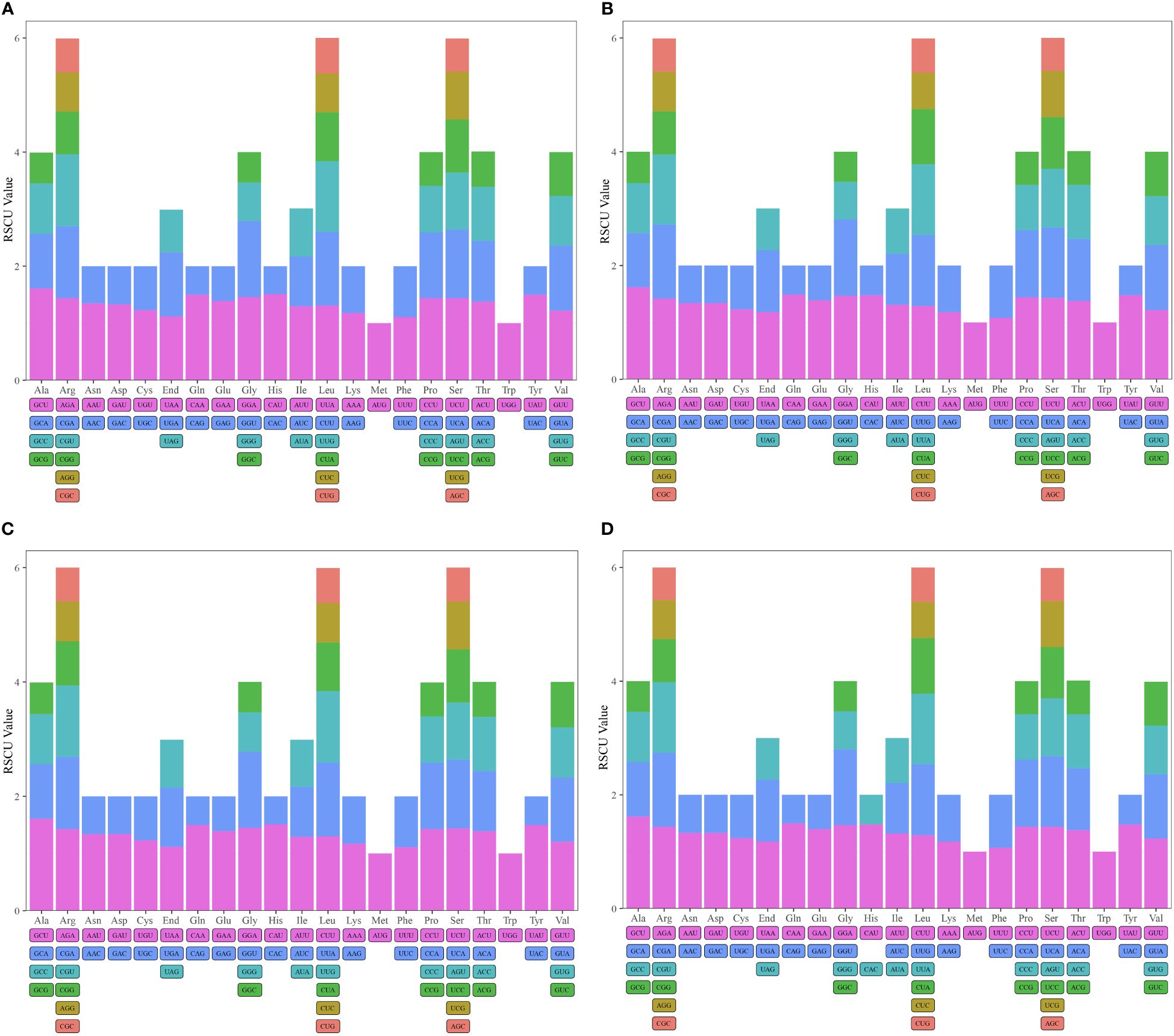
Figure 3 Relative synonumous codon usage (RSCU) in mitogenomes of D. huoshanense (A), D. nobile (B), D. flexicaule (C), D. officinale (D). Codon families are indicated on the x-axis. RSCU values are shown on the y-axis.
The mitochondrial genomes of D. huoshanense, D. nobile, D. flexicaule, and D. officinale comprise 17 coding sequences (CDs), including essential genes such as atp4, atp6, and cox1. Notably, D. nobile uniquely possesses the cox2 gene. The GC content across these species ranges from 35.64% to 51.34%, indicating minor disparities in genomic regions (Supplementary Figure S6). The average effective number of codons (ENC) exceeding 35 across the species suggests a weak codon usage bias. Neutrality plots illustrating the balance between GC content at different codon positions show minimal slope, indicating that mutation pressure may play a subdued role in shaping codon usage (Supplementary Figure S7).
We computed the non-synonymous to synonymous mutation ratio (Ka/Ks) for 25 common protein-coding genes (PCGs) across the Dendrobium species to identify evolutionary adaptations in response to environmental pressures (Supplementary Figure S8, Supplementary Table S2.15). Notably, genes such as ccmFC, matR, mttB, nad7, rps1, rps2, and rps10 exhibited higher Ka/Ks values, suggesting these genes may be under positive selection. In particular, the rps10 gene displayed a Ka/Ks ratio of 2.196, indicating significant evolutionary divergence (P<0.05). This analysis underscores the adaptive responses of mitochondrial genomes to environmental factors, highlighting the potential evolutionary dynamics within the Dendrobium genus.
Mitochondrial DNA (mtDNA) in flowering plants, including the Dendrobium genus, typically exhibits high complexity due to the substantial presence of repeated sequences. Our investigation into four Dendrobium species revealed varied repeat sequences within their mtDNA, detailed comprehensively in Table 3 and illustrated in Figure 4. Across these species, a total of 647 simple sequence repeats (SSRs) were cataloged. The distribution was as follows: D. officinale possessed 154 SSRs, D. flexicaule had 153, D. huoshanense recorded 149, and D. nobile had the highest with 191 SSRs. Tetranucleotide repeats were the predominant SSR type in all four species’ mtDNAs, with percentages of 29.87% in D. officinale, 30.07% in D. flexicaule, 28.19% in D. huoshanense, and 34.03% in D. nobile. Conversely, hexanucleotide repeats were significantly less common, with no occurrences noted in D. huoshanense’s mtDNA.

Table 3 Number of repeat sequences from the Dendrobium species.
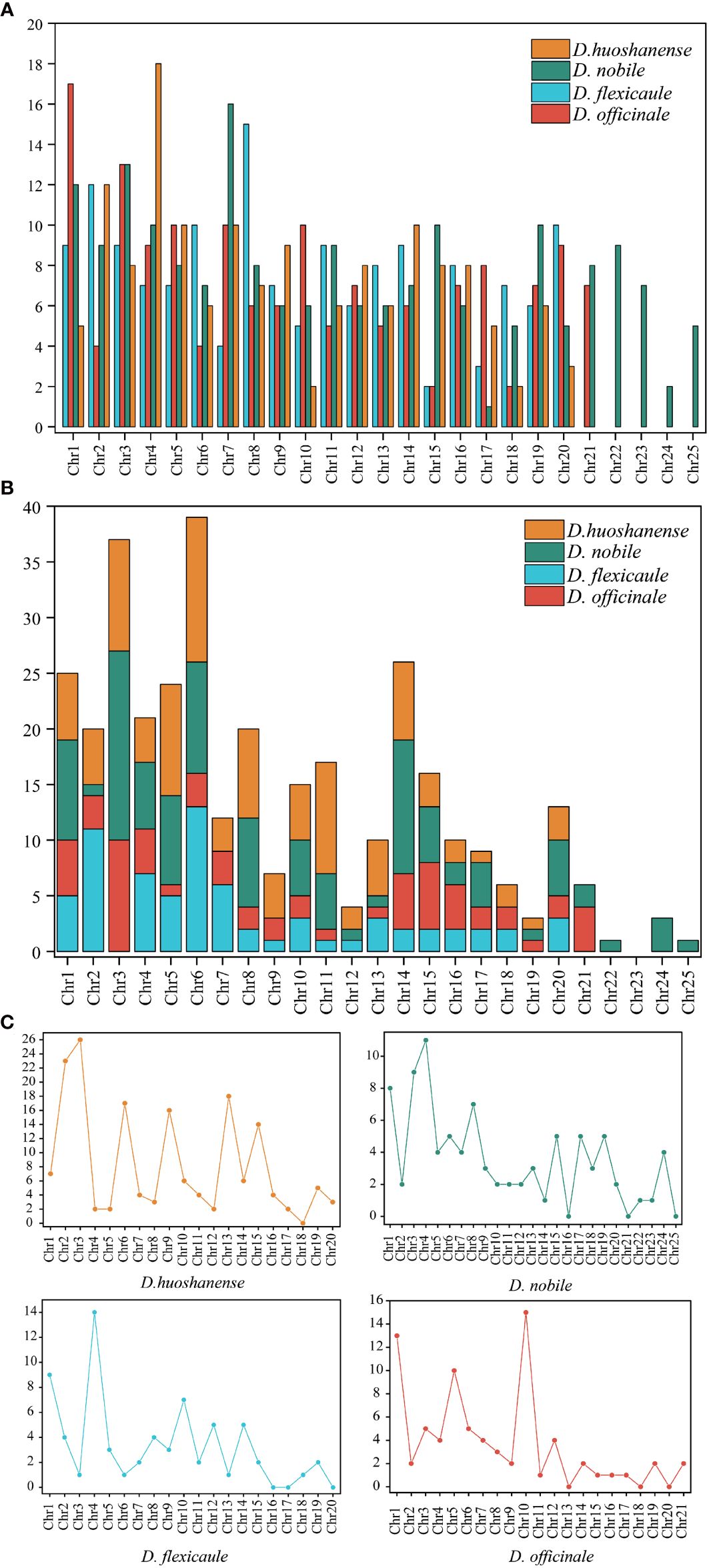
Figure 4 Analysis of repetitive sequences in mitogenomes of four Dendrobium species. (A) Simple sequence repeats types. (B) Tandem repeats types. (C)Dispersed repeats types (≥30 bp).
Furthermore, the mtDNA of these species contained a total of 345 tandem repeats, distributed as follows: 63 in D. officinale, 71 in D. flexicaule, 104 in D. huoshanense, and 107 in D. nobile. Additionally, 396 dispersed repeats were identified. Of these, forward repeats constituted 76.52% of all dispersed repeats across the mtDNA of the four species, while palindromic repeats accounted for 23.23%. D. huoshanense’s mtDNA featured a single 30-bp-long reverse repeat, representing just 0.25% of the total repeat content. These repeat elements were primarily located in the intergenic spacer regions between trnH and trnL.
Notably, certain chromosomes within the mitochondrial genomes of these species did not harbor any dispersed repeats, indicating unique structural characteristics within their genomic architecture. These chromosomes include D. officinale (chromosomes 13, 18, and 20), D. flexicaule (chromosomes 16, 17, and 20), D. huoshanense (chromosome 18), and D. nobile (chromosomes 16, 21, and 25).
Global alignment of the organelle genomes revealed heterogeneously distributed homologous regions within certain areas of the plastomes (Supplementary Table S2.16). Our analysis highlighted considerable variations in the lengths of sequences transferred from the plastomes to the mtDNAs across the four species: D. nobile (91,810 bp), D. flexicaule (93,329 bp), D. huoshanense (103,176 bp), and D. officinale (82,308 bp) (Figure 5). These segments, integrated due to active recombination and rearrangement, contributed to a total of 173 complete genes across the mitochondrial genomes, encompassing 115 protein-coding genes (PCGs) and 58 tRNA-encoding genes. The specific impacts on gene complements were as follows: 48 genes in D. nobile, 45 in D. flexicaule, 40 in D. huoshanense, and 40 in D. officinale (Table 4).

Figure 5 Intracellular genome transfer in Dendrobium huoshanense (A), Dendrobium nobile (B), Dendrobium flexicaule (C), Dendrobium officinale (D). Mitogenomes are shown in blue, chloroplast genomes are shown in green, and the purple arcs within the circular ring represent homologous segments between mitochondrial genome and chloroplast genome.
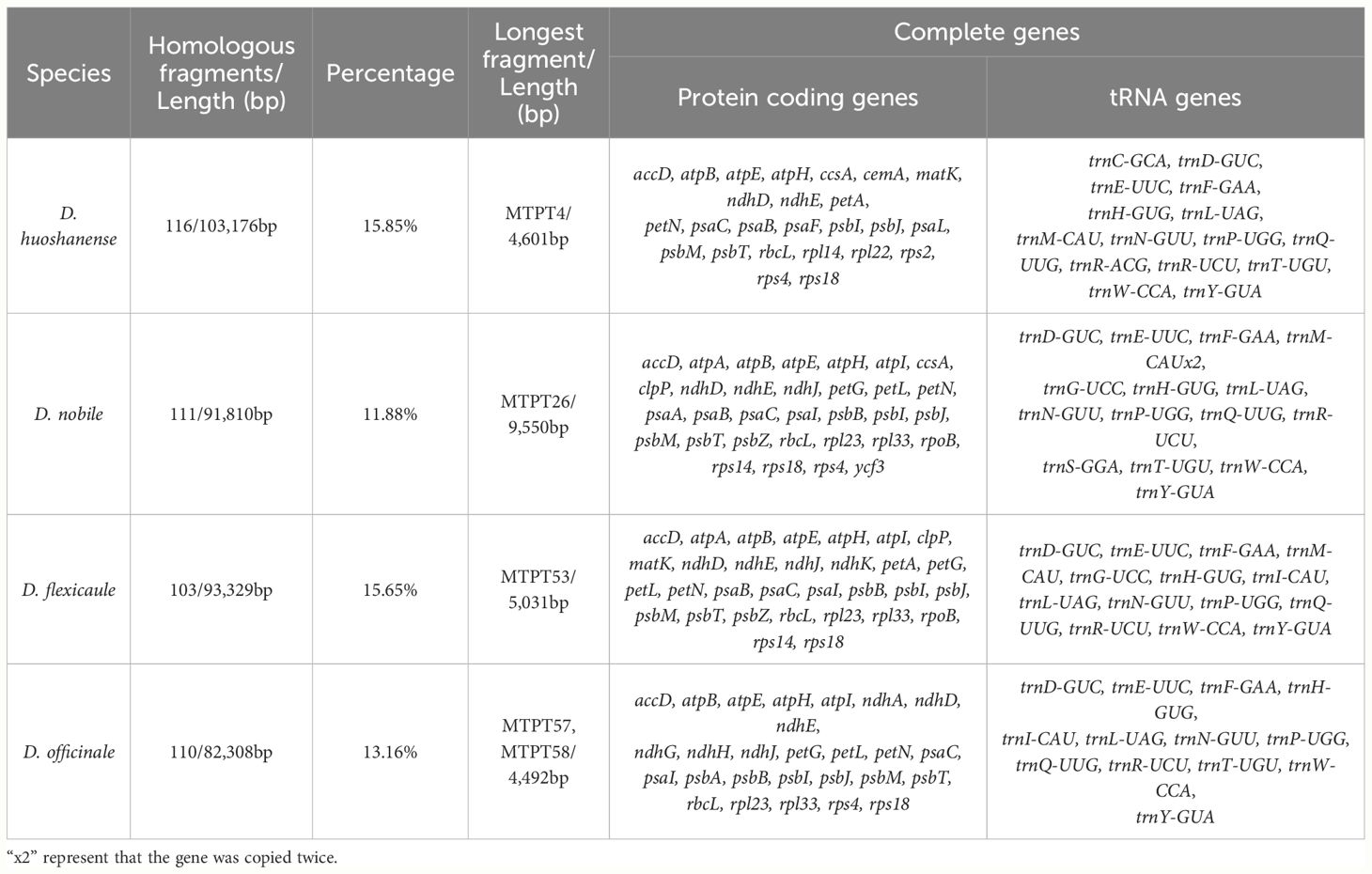
Table 4 Analysis of homologous fragments based on the four Dendrobium species .
In D. flexicaule, the transferred fragments, representing 15.65% of the total transferred DNA, included 45 complete genes (31 PCGs and 14 tRNAs). Notably, the longest homologous fragment identified was MTPT53, measuring 5,031 bp and containing several complete genes such as psbZ, trnG-UCC, trnM-CAU, rps14, psaB, and ndhK, along with the partial gene psaA. The shortest sequence, MTPT68, spanned merely 33 bp and constituted an intergenic spacer.
D. huoshanense exhibited the most extensive collection of homologous sequences among the species studied, totaling 116 mitochondrial-transferred plastid fragments (MTPTs), which accounted for 15.85% of its mitochondrial genome. The longest sequence in this collection was 4,601 bp, incorporating segments of the incomplete genes rpoC1 and rpoB. D. officinale and D. nobile followed closely with 110 and 111 homologous fragments, respectively. In D. officinale, the most substantial sequences were MTPT57 and MTPT58, which included parts of the incomplete genes ycf2 and trnI-CAU. For D. nobile, the longest sequence, MTPT26, spanned 9,550 bp and encompassed eight complete genes: psbZ, trnG-UCC, trnM-CAU, rps14, psaA, psaB, ycf3, and trnS-GGA.
Interestingly, the ycf3 gene was exclusive to the mitochondrial PCGs of D. nobile and absent in D. flexicaule, D. officinale, and D. huoshanense. The petL gene appeared in the PCGs of the homologous segments from D. flexicaule, D. nobile, and D. officinale, while the petA gene was specific to the PCGs of D. flexicaule and D. huoshanense. A comprehensive analysis of the homologous sequences across the four species revealed a significant presence of complete tRNA-encoding genes. Notably, the trnC-GCA gene, with an alignment length of 728 bp, was found exclusively in the homologous sequences of D. huoshanense’s mtDNA and was absent in the other three species. Additionally, the trnS-GGA gene was unique to D. nobile, highlighting the specialized function and distribution of these genes within the chloroplast genomes of D. huoshanense and D. nobile.
We identified several partial pseudogenes in the homologous fragments of the four Dendrobium species, including ψndhA, ψndhD, ψndhE, ψndhG, ψndhH, ψndhJ, and ψndhK (Table 5). The lengths of these pseudogenes range from 451 to 5,031 bp. An in-depth analysis revealed that these pseudogenes are predominantly distributed in the large single copy (LSC) and small single copy (SSC) regions of the chloroplast genome. Furthermore, no complete pseudogenes were found in the homologous fragments of D. nobile. In contrast, D. flexicaule and D. officinale exhibited the highest number of pseudogenes, while D. huoshanense had the lowest pseudogene count.

Table 5 Sequences transferred from the plastomes to the mitogenomes of four Dendrobium species.
An extensive analysis of the mitochondrial genomes across four Dendrobium species identified a significant number of RNA editing sites, totaling 2,023 across 147 protein-coding genes (PCGs). The distribution of PCGs undergoing RNA editing was fairly consistent, with 36 unique PCGs in D. flexicaule and 37 in D. huoshanense, D. nobile, and D. officinale, as detailed in Supplementary Figure S9. Notably, the ccmFN gene exhibited the highest number of RNA editing events across all four species, with a cumulative total of 168 sites. For D. officinale and D. huoshanense, the ccmB gene experienced significant RNA editing, with each species showing 35 editing events. Similarly, in D. nobile and D. flexicaule, both the ccmB and mttB genes demonstrated 35 RNA editing instances each.
An interesting observation was the minimal RNA editing activity in the atp8 gene, with only a single editing event predicted across all four species. This consistency suggests low variability in RNA editing for this particular gene. RNA editing events predominantly converted cytosine (C) to uridine (U), leading to changes in the amino acid properties of the encoded proteins. Analysis revealed that 23.93%-24.55% of the amino acids retained their hydrophobic characteristics post-editing, while 43.23%-44.12% of amino acids transitioned from hydrophilic to hydrophobic properties. Leucine was the most common amino acid resulting from RNA editing, affecting 178-183 positions, followed by phenylalanine alterations at 113-116 sites. Some RNA editing events even resulted in the transformation of regular amino acids into stop codons, specifically UAA and UGA.
The collinearity analysis of mitochondrial genomes within the four Dendrobium species used BLASTN to identify conserved homologous sequences. These sequences, considered conserved collinear blocks, were included in the analysis if it exceeded 500 bp and met a size threshold of 0.5 kb. The results indicated an increase in the number of homologous blocks across the four species (Figure 6), although these blocks were shorter compared to those in reference species. Notably, a total of 65 homologous collinear blocks ≥7,000 bp were found among the four Dendrobium species. The longest block (24.11 kb) was between chromosome 9 of D. nobile and chromosome 1 of D. flexicaule, while the shortest block (7.15 kb) was identified between chromosome 17 of D. nobile and chromosome 15 of D. flexicaule. D. flexicaule and D. nobile were grouped in the same clade in the phylogenetic tree. Additionally, the longest collinear block between D. huoshanense and D. nobile was found on chromosome 4 of D. huoshanense and chromosome 22 of D. nobile, measuring 19.93 kb, while the shortest was observed on chromosome 2 of D. huoshanense and chromosome 6 of D. nobile, measuring 7.15 kb. D. nobile and D. huoshanense are closely related species.

Figure 6 Comparison and analysis of collinearity in four Dendrobium species. Perform collinearity analysis between five species and four Dendrobium species on the left. Each contig in the figure represents a chromosome. The regions of red arcs suggest inversion areas. The gray regions suggest homologous areas.
The presence of blank areas within these plots highlighted notable variations and distinctions in mitochondrial genome homology when compared to other species. The results suggested a divergent order in the arrangement of collinear blocks among Asparagales, indicating significant gene rearrangements within the Dendrobium species. The shorter lengths of these collinear blocks across the mitochondrial genomes underscore a lower conservation of sequence order and frequent gene recombination among the mitochondrial DNA of these and other Asparagales species. This complex genetic architecture may reflect adaptive evolutionary processes in response to environmental stresses, influencing the mitogenomic stability and evolution in the Dendrobium genus.
Mitochondrial genomes from 27 species across five orders of angiosperms were constructed phylogenetic trees in this study (Figure 7). The phylogenetic tree was based on the mitogenomic sequences of 21 conserved protein-coding genes (PCGs), including atp1, atp4, atp6, ccmB, ccmC, ccmFC, ccmFN, cob, cox1, cox2, cox3, matR, nad1, nad2, nad3, nad4, nad5, nad6, nad7, nad9, and rps12. Two mitochondrial genomes from the Magnoliales were used as the outgroup. The resulting phylogenetic tree had a topological structure consistent with the classification of the angiosperm phylogeny group, indicating its alignment with the latest classification systems. Among the species analyzed, the four Dendrobium species belonged to the Asparagales order within the Orchidaceae family. Within this group, D. nobile and D. huoshanense were identified as closely related sister species, while D. officinale and D. flexicaule were also closely related.
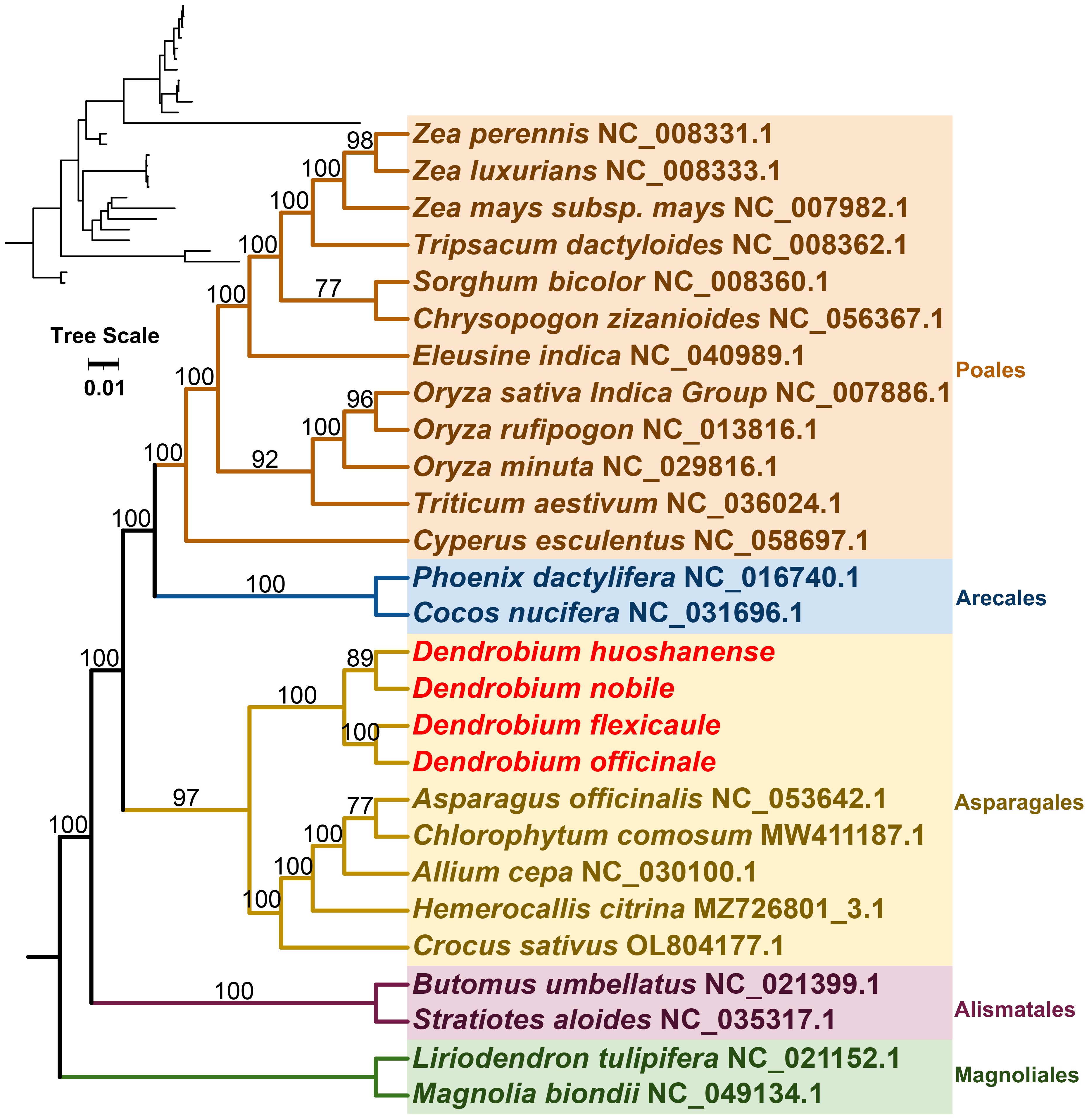
Figure 7 The 21 conserved DNA sequences of protein-coding genes (PCGs) in complete mitogenomes required to construct a phylogenetic tree by selecting 27 species of five orders (Polaes, Arecales, Asparagales, Alismatales, and Magnoliales) in angiosperms. Tree scale was 0.01. The number on each node is the bootstrap values. Magnolia biondii and Liriodendron tulipifera were selected as outgroups. The accession numbers of the sequences in each species are indicated in the map.
Plant mitochondrial genomes are known for their significant variability in size, ranging from several hundred kilobases to several megabases (Skippington et al., 2015; Jackman et al., 2020; Sullivan et al., 2020; Bi et al., 2023; Zhou et al., 2024). The elongation of mitochondrial genomes in seed plants primarily arises from non-coding content (Alverson et al., 2010; Dong et al., 2018; Bi et al., 2024), including duplicated regions, fragments from plastid genomes acquired through intracellular gene transfer (IGT), and foreign mitogenomic sequences obtained through horizontal gene transfer (HGT) (Gandini et al., 2019). These non-coding regions, such as duplicated sequences and intergenomic transfer fragments, are the main contributors to this size variation (Kubo et al., 2000). Additionally, the multi-chromosome structure of mitochondrial genomes further contributes to their complexity (Chen T-C. et al., 2020; Zhou et al., 2023). In this study, we successfully assembled the complete mitochondrial genomes of four Dendrobium species—D. flexicaule, D. nobile, D. huoshanense, and D. officinale—using a hybrid assembly approach that combined Illumina and Nanopore sequencing technologies. We uncovered a common multibranch conformation in the mitochondrial structures of these four Dendrobium species, a characteristic feature of the Orchidaceae family. The mitochondrial genome sizes varied substantially, ranging from 596,506 bp in D. flexicaule to 772,523 bp in D. nobile. Notably, the total length of the Cymbidium ensifolium mtDNA (19 chromosomes, 560,647 bp) also exhibited significant variation compared to D. flexicaule within the Orchidaceae family (Shen et al., 2024). Similarly, D. nobile (25 chromosomes) and Paphiopedilum micranthum (26 chromosomes) differ by only one chromosome in their mtDNAs, yet their mitochondrial genome lengths vary significantly, with a difference of 325,155 bp (Yang et al., 2023a). Meanwhile, the mitochondrial genome of Apostasia shenzhenica, (Ke et al., 2023) a closely related species to the four Dendrobium species, consists of only one circular molecule, highlighting the remarkable diversity in plant mitochondrial genome structure (Petersen et al., 2020). Interestingly, we also found that non-coding regions and gene numbers might cause variation of mitochondrial genome sizes in Orchidaceae through comparing D. flexicaule, D. nobile, D. huoshanense, D. officinale, A. shenzhenica and C. ensifolium. Despite this variation, the GC content across these genomes was incredibly stable, ranging from 43.41% to 43.64%, suggesting a conservation of GC content across angiosperms over evolutionary timescales (Fan et al., 2022; Niu et al., 2022). Unlike the more conserved and structurally compact animal mitochondria and plastomes, plant mitochondrial genomes are characterized by rapid structural evolution and a high degree of rearrangement.
In our study, the mitochondrial genomes of the four Dendrobium species exhibited a sparse distribution of genes amidst a plethora of non-coding sequences. Specifically, D. flexicaule and D. officinale contained 64 genes, D. nobile had 68, and D. huoshanense had 69. This pattern mirrors observations in other species (Fan et al., 2022; Li et al., 2022; Niu et al., 2022; Yu et al., 2022). For example, non-coding sequences occupied 91.96% of the total mtDNA of P. deltoides (Qu et al., 2023). Additionally, the four Dendrobium mtDNAs shared 56 unique genes, comprising 36 protein-coding genes (PCGs), three rRNA-encoding genes, and 17 tRNA-encoding genes. Notably, the rps11 gene and several tRNA-encoding genes were absent (Supplementary Table S2.10), a trend also seen in other species such as Broussonetia spp. (Lai et al., 2022) and Abelmoschus esculentus (Li et al., 2022). Meanwhile, we found that three (chromosome 11, chromosome 20 and chromosome 24) out of 25 chromosomes in the D. nobile mtDNA were found to contain no functional genes, which is similar with previous studies (Alverson et al., 2011; Bi et al., 2022).
RNA editing is a critical post-transcriptional mechanism in plant organelles that modifies RNA nucleotide sequences to ensure the production of functional proteins (Yan et al., 2018; Zhu et al., 2022). These events can influence protein stability and quantity (Jiang et al., 2022). Within mitochondrial genomes, RNA editing is vital for the accurate translation of mitochondrial genes and the proper functioning of mitochondrial proteins. Additionally, RNA editing plays a crucial role in various aspects of plant biology, including normal biosynthesis in mitochondria and chloroplasts, plant adaptability to environmental conditions, and signal transduction (Yan et al., 2018; Wang and Yang, 2020). This process is essential for the stability and activity of mitochondrial and chloroplast proteins. In the studied Dendrobium species, we identified 2,023 RNA editing sites in the mitochondrial genomes of four species, predominantly involving cytosine-to-uridine transitions, which are common in plant mitochondrial genomes (Verhage, 2020; Edera and Sanchez-Puerta, 2021). Notably, while the number of RNA editing sites in D. flexicaule (501) and D. nobile (502) mtDNAs was similar, there was a significant difference compared to D. officinale (510) and D. huoshanense (510). Further analysis suggested that the RNA editing events in atp6, ccmFN, cob, mttB, nad3, nad4, nad7, nad9, and rps19 genes were the main factors contributing to this variation. The highest number of RNA editing sites in the four Dendrobium species was found in NADH dehydrogenase and cytochrome c biogenesis genes, which is consistent with observations in C. ensifolium, Ilex metabaptista (Zhou et al., 2023), and Bupleurum chinense (Qiao et al., 2022).
The consistency of RNA editing events across the four Dendrobium species underscores their evolutionary conservation and functional necessity (Edera et al., 2018). This pattern emphasizes the importance of RNA editing in fine-tuning mitochondrial gene products, ensuring appropriate protein functions, and maintaining efficient energy production. Further analysis revealed that the ccmFN gene had the highest number of RNA editing sites (42 sites), suggesting extensive modifications crucial for its role in the cytochrome c maturation pathway, essential for mitochondrial electron transport and energy metabolism (Unseld et al., 1997; Giegé et al., 2008). Additionally, 35 RNA editing events were observed in the ccmB genes of D. officinale and D. huoshanense, as well as in the ccmB and mttB genes of D. nobile and D. flexicaule. These RNA editing events in specific genes might be functionally significant for their corresponding protein products or essential for their proper assembly into functional complexes involved in mitochondrial electron transport and energy production (Mower et al., 2012). In contrast, the atp8 gene, which encodes a subunit of the ATP synthase complex critical for ATP production during oxidative phosphorylation, had fewer RNA editing events. This suggests that the atp8 gene’s nucleotide sequence might be highly conserved and functionally well-adapted in these Dendrobium species. Analyzing the patterns of RNA editing events provides insights into the molecular mechanisms governing mitochondrial gene expression and function in these orchids. Comparing these patterns with other orchid species and across different plant lineages might also reveal the evolutionary dynamics of mitochondrial gene regulation and its importance in plant adaptation and diversification.
The mitochondrial genomes of flowering plants often exhibit a high level of complexity due to the prevalence of repetitive DNA sequences (Guo et al., 2017; Ma et al., 2022). These repeat sequences are divided into two types: tandem and dispersed repeats. Simple sequence repeats (SSRs), a type of tandem repeat, are commonly used as molecular markers for species identification and genetic diversity (Khera et al., 2015; Ma et al., 2017; Li and Ye, 2020; Liu et al., 2022; Tan et al., 2023; Yang et al., 2023b, c). In our analysis of four Dendrobium species, we identified a significant number of SSRs within their mitochondrial DNA (mtDNA). Specifically, D. officinale had 154 SSRs, D. flexicaule had 153 SSRs, D. huoshanense had 149 SSRs, and D. nobile had the highest number with 191 SSRs. Tetranucleotide repeats were the most abundant type of SSR across these species, consistent with findings in the mitochondrial genomes of C. ensifolium, A. shenzhenica, and L. tsingtauense (Ke et al., 2023; Qu et al., 2024; Shen et al., 2024). This highlights their potential as molecular markers for genetic diversity assessments and species differentiation within the Dendrobium genus (Qiu et al., 2010).
Further examination revealed 352 tandem repeats and 396 dispersed repeats across the mtDNA of these species, with forward repeats accounting for over 76% of all dispersed repeats. These repetitive elements are significant not only for their structural roles but also for facilitating frequent recombination events within the mitochondrial genomes. Such recombination can lead to genomic rearrangements that may influence gene function and plant adaptation (Cole et al., 2018; Dong et al., 2018; Sullivan et al., 2020).
The study of codon usage bias provides essential insights into the evolutionary adaptations and functional constraints of genomic sequences (Nair et al., 2013; Kannaujiya et al., 2014; Mazumdar et al., 2017). Various factors influence codon preferences, including natural selection, mutation, gene sequence base composition bias, tRNA abundance, GC content, gene length, protein hydrophobicity, and amino acid conservativeness (Bulmer, 1991; Novembre, 2002; Parvathy et al., 2022). Our analysis revealed that preferred codons typically end in A or U, indicating a conservation of nucleotide composition. This pattern aligns with the evolutionary trends observed in other plant mitochondrial genomes (Jin et al., 2019; Tang et al., 2020).
Our analysis of codon usage neutrality indicated a generally weak correlation across the species, supporting the theory that natural selection, rather than mutational pressure, predominantly shapes codon usage in these genomes. The mitochondrial gene rps3 in D. flexicaule had the lowest ENC values, while the atp6 gene’s ENC value was below 35 in all four species. Beyond these two genes, the ENC values of mitochondrial genes in the four Dendrobium species ranged from 36.61 to 61.00, with most coding genes exhibiting ENC values above 50.00%, indicating a weaker codon preference. Determining the optimal codons can further enhance gene expression efficiency, providing a foundation for future research on the expression regulation of functional genes in Dendrobium, as well as predictions of protein structure and function. This research also offers insights into the conservation of Dendrobium germplasm resources and artificial cultivation practices. Analyzing codon bias in plant mitochondrial genomes is crucial for studying genetic regulations, phylogenetic relationships, and the evolution of mtDNA.
The Ka/Ks ratio is often used to examine divergence in protein-coding genes (PCGs) under positive or purifying selection (Hurst, 2002). Our study further explored the non-synonymous/synonymous mutation ratios (Ka/Ks) to identify genes under positive selective pressure, indicative of adaptive evolutionary processes (Xie et al., 2019). Faced with environmental pressures and shifts, species may undergo positive selection on specific genes to alter their protein-coding sequences, enhancing their adaptability to new environments and stress responses (Pimpinelli and Piacentini, 2020). Genes with high Ka/Ks ratios might be involved in essential biological processes related to environmental adaptation (Zhang et al., 2016). For example, the Ka/Ks values for the ccmFC, matR, mttB, and rps2 genes in the four Dendrobium mtDNAs were all greater than one, suggesting that these genes could be closely tied to energy metabolism, redox reactions, or other adaptive processes. The notably high Ka/Ks ratio observed for the rps10 gene hints at its critical role in species’ adaptive evolution, possibly due to its involvement in key cellular processes or environmental responses. Elevated Ka/Ks ratios might also suggest that these genes help maintain genetic diversity among species (Zhong et al., 2023). By subjecting these genes to positive selection, species could bolster their adaptive capabilities and resilience, enhancing their resistance to various environmental pressures (Bijlsma and Loeschcke, 2012). This analysis highlights the importance of mitochondrial genomes in the adaptive evolution of Dendrobium species, facilitating their survival and diversification through modifications in key functional genes.
In plant cells, there is frequent and significant transfer of genetic material between chloroplast and mitochondrial genomes, often resulting in chloroplast DNA sequences comprising 1%-12% of the mitochondrial genome (Wendel et al., 2012). In our study of Dendrobium species—D. huoshanense (116), D. nobile (111), D. flexicaule (103), and D. officinale (110)—we discovered substantial DNA transfers from chloroplasts to mitochondria, totaling 82,308 bp, 91,810 bp, 93,329 bp, and 103,176 bp, respectively. These transfers constitute a notable proportion of the mitochondrial genome—11.88%, 15.65%, 13.16%, and 15.85%, respectively—highlighting significant variation in the extent of gene transfer among species (Adams et al., 2002). Although the length of these transfer fragments was extremely large compared to C. ensifolium (38,163 bp), the number of sequences transferred from the plastomes to the mtDNAs was similar (Shen et al., 2024). Additionally, the ratio of transfer fragments to whole mtDNA length was consistent with previous studies (Yang et al., 2023a).
This influx of genetic material is largely attributable to the integration of repetitive and homologous sequences, which are pivotal in the evolutionary expansion of mitochondrial genomes. Such integration events have occurred gradually over evolutionary timescales, incorporating not just coding regions but also often leading to the formation of non-functional pseudogenes due to recombination events (Richardson et al., 2013). Our analysis reveals that some chloroplast genes become pseudogenes upon integration into the mitochondrial genome. These pseudogenes accounted for 0.94%-16.77% of the mtDNAs in three Dendrobium species (D. huoshanense, D. flexicaule, and D. officinale) (Table 5) and included NADH dehydrogenase genes. In contrast, the sequence transferred from the plastome to the mtDNA on chromosome 8 of the D. flexicaule mtDNA was the largest in length, at 5,031 bp. This result indicates that these transfer fragments undergo multiple rounds of recombination (Yang et al., 2023). Interestingly, shareable ndhD, ndhG, ndhH, ndhJ, and ndhK genes have been lost in the three Dendrobium plastomes, but only a pseudo copy of the shareable ψndhD gene was identified in the three Dendrobium mtDNAs. Published studies have indicated that the loss of NADH genes in some plant plastomes results in nonfunctional pseudogenes (Li et al., 2021). However, tRNA genes tend to retain their functionality, suggesting a conservation of transport functions in mitochondrial genomes, a trait especially pronounced in higher plants.
The specific mechanisms and functional consequences of gene transfer between the mitochondrial and chloroplast genomes in Dendrobium species remain poorly understood. However, ongoing advancements in whole-genome sequencing are expected to shed light on these processes. This mitogenomic research enhances our understanding of mitochondrial evolution in flowering plants and sets the stage for further exploration of molecular markers and evolutionary relationships within the Orchidaceae family.
Comparative genomics has become indispensable in the study of medicinal plants, providing insights into gene differentiation and evolutionary trends (Daniell et al., 2016). By analyzing 21 conserved mitochondrial protein-coding genes (PCGs) across 27 angiosperm species, we constructed a phylogenetic tree that suggests close relationships within Dendrobium species. However, discrepancies in phylogenetic alignment between mitochondrial and chloroplast-derived trees highlight potential inconsistencies that warrant further investigation (Zhu et al., 2018).
In conclusion, this study successfully assembled the complete chloroplast and mitochondrial genomes of four Dendrobium species employing a hybrid assembly method. The mitochondrial genomes of these species are characterized by their multi-chromosomal structure, with chromosome size ranging from 20 to 25 and sizes varying between 596,506 bp and 772,523 bp. The observed mitochondrial genome rearrangements among the four Dendrobium species suggest the occurrence of homologous recombination within the mitochondrial genomes of this genus. Phylogenetic analyses reveal that D. nobile shares a close evolutionary relationship with D. huoshanense, whereas D. officinale and D. flexicaule are more closely aligned. Overall, our findings contribute significantly to the existing knowledge on the diversity, evolutionary dynamics, and potential molecular markers within Dendrobium mitochondrial genomes, thereby providing a robust foundation for further studies on the genetic and functional characteristics of these species.
The organelle sequences supporting the conclusions of this article are available in GenBank (https://www.ncbi.nlm.nih.gov/) with accession numbers: OR413847-OR413866 (Dendrobium huoshanense), OR413867-OR413891(Dendrobium nobile), OR413892-OR413911(Dendrobium flexicaule), OR413912-OR413932 (Dendrobium officinale) (mitochondrial genomes); OR387325 (Dendrobium huoshanense), OR387323 (Dendrobium nobile), OQ360111 (Dendrobium flexicaule), OR387324 (Dendrobium officinale) (chloroplast genomes). The raw data has been released through NCBI with the following accession numbers: (1) Dendrobium huoshanense: BioProject PRJNA1129804, BioSample SAMN42166854, SRA SRR29650379 (Illumina), SRA SRR29650378 (Nanopore); (2) Dendrobium nobile: BioProject PRJNA1129805, BioSample SAMN42166915, SRA SRR29654108 (Illumina), SRA SRR29654107 (Nanopore); (3) Dendrobium flexicaule: BioProject PRJNA1129490, BioSample SAMN42166852, SRA SRR29649768 (Illumina), SRA SRR29649767 (Nanopore); (4) Dendrobium officinale: BioProject PRJNA1129803, BioSample SAMN42166853, SRA SRR29649991 (Illumina), SRA SRR29649990 (Nanopore).
LW: Conceptualization, Data curation, Methodology, Software, Writing – original draft. XL: Writing – review & editing. YW: Investigation, Visualization, Writing – review & editing. XM: Writing – review & editing. JQ: Software, Validation, Writing – review & editing. YZ: Software, Validation, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was Supported by the National Key Research and Development Program of China (2022YFD1201600), Reproductive ecology and endangering factors of Dendrobium flexicaule (jbky20210006), Ecological and genetic basis of the unique growth characteristics of Dendrobium flexicaule (cstc2021jscx-dxwtBX0015) and Research on the medicinal plants of Yintiaoling.
We thank the Editor and the reviewers for their insightful comments and suggestions on the manuscript. The authors thank Wuhan Benagen Technology Co., Ltd. for help in genome sequencing and the analysis of RNA editing. We thank the assistance of Changbing Zheng from Chongqing Yintiaoling National Nature Reserve Management Affairs Center in the sampling work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1429545/full#supplementary-material
PCGs, Protein-coding genes; mtDNA, Mitochondrial genome; cpDNA, Chloroplast genome; Ka/Ks, Non-synonymous/synonymous mutation ratio; RSCU, Relative synonymous codon usage; MTPT, Mitochondrial plastid DNA sequence; tRNA, Transfer RNA; rRNA, Ribosomal RNA; SSRs, simple sequence repeats; ENC, Effective number of codons; IGT, intracellular gene transfer
Adams, K. L., Qiu, Y. L., Stoutemyer, M., Palmer, J. D. (2002). Punctuated evolution of mitochondrial gene content: high and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. U.S.A. 99, 9905–9912. doi: 10.1073/pnas.042694899
Alverson, A. J., Rice, D. W., Dickinson, S., Barry, K., Palmer, J. D. (2011). Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell. 23, 2499–2513. doi: 10.1105/tpc.111.087189
Alverson, A. J., Wei, X., Rice, D. W., Stern, D. B., Barry, K., Palmer, J. D. (2010). Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 27, 1436–1448. doi: 10.1093/molbev/msq029
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bi, C., Qu, Y., Hou, J., Wu, K., Ye, N., Yin, T. (2022). Deciphering the multi-chromosomal mitochondrial genome of populus simonii. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.914635
Bi, C., Shen, F., Han, F., Qu, Y., Hou, J., Xu, K., et al. (2024). PMAT: an efficient plant mitogenome assembly toolkit using low-coverage HiFi sequencing data. Hortic. Res. 11, uhae023. doi: 10.1093/hr/uhae023
Bi, C., Sun, N., Han, F., Xu, K., Yang, Y., Ferguson, D. K. (2023). The first mitogenome of Lauraceae (Cinnamomum chekiangense). Plant Divers. 46, 144–148. doi: 10.1016/j.pld.2023.11.001
Bijlsma, R., Loeschcke, V. (2012). Genetic erosion impedes adaptive responses to stressful environments. Evol. Appl. 5, 117–129. doi: 10.1111/j.1752-4571.2011.00214.x
Bulmer, M. (1991). The selection-mutation-drift theory of synonymous codon usage. Genetics 129, 897–907. doi: 10.1093/genetics/129.3.897
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, T.-C., Su, Y.-Y., Wu, C.-H., Liu, Y.-C., Huang, C.-H., Chang, C.-C. (2020). Analysis of mitochondrial genomics and transcriptomics reveal abundant RNA edits and differential editing status in moth orchid, Phalaenopsis aphrodite subsp. formosana. Sci. Hortic. 267, 109304. doi: 10.1016/j.scienta.2020.109304
Chen, Y., Ye, W., Zhang, Y., Xu, Y. (2015). High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res. 43, 7762–7768. doi: 10.1093/nar/gkv784
Cole, L. W., Guo, W., Mower, J. P., Palmer, J. D. (2018). High and variable rates of repeat-mediated mitochondrial genome rearrangement in a genus of plants. Mol. Biol. Evol. 35, 2773–2785. doi: 10.1093/molbev/msy176
Daniell, H., Lin, C. S., Yu, M., Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134. doi: 10.1186/s13059-016-1004-2
Dong, S., Zhao, C., Chen, F., Liu, Y., Zhang, S., Wu, H., et al. (2018). The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genomics 19, 614. doi: 10.1186/s12864-018-4991-4
Edera, A. A., Gandini, C. L., Sanchez-Puerta, M. V. (2018). Towards a comprehensive picture of C-to-U RNA editing sites in angiosperm mitochondria. Plant Mol. Biol. 97, 215–231. doi: 10.1007/s11103-018-0734-9
Edera, A. A., Sanchez-Puerta, M. V. (2021). Computational detection of plant RNA editing events. Methods Mol. Biol. 2181, 13–34. doi: 10.1007/978-1-0716-0787-9_2
Fan, W., Liu, F., Jia, Q., Du, H., Chen, W., Ruan, J., et al. (2022). Fragaria mitogenomes evolve rapidly in structure but slowly in sequence and incur frequent multinucleotide mutations mediated by microinversions. New Phytol. 236, 745–759. doi: 10.1111/nph.18334
Gandini, C. L., Garcia, L. E., Abbona, C. C., Sanchez-Puerta, M. V. (2019).The complete organelle genomes of Physochlaina orientalis: Insights into short sequence repeats across seed plant mitochondrial genomes. Mol. Phylogenet. Evol. 137, 274–284. doi: 10.1016/j.ympev.2019.05.012
Giegé, P., Brennicke, A. (1999). RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc. Natl. Acad. Sci. U.S.A. 96, 15324–15329. doi: 10.1073/pnas.96.26.15324
Giegé, P., Grienenberger, J. M., Bonnard, G. (2008). Cytochrome c biogenesis in mitochondria. Mitochondrion 8, 61–73. doi: 10.1016/j.mito.2007.10.001
Guo, W., Zhu, A., Fan, W., Mower, J. P. (2017). Complete mitochondrial genomes from the ferns Ophioglossum californicum and Psilotum nudum are highly repetitive with the largest organellar introns. New Phytol. 213, 391–403. doi: 10.1111/nph.14135
Han, F., Qu, Y., Chen, Y., Xu, L., Bi, C. (2022). Assembly and comparative analysis of the complete mitochondrial genome of Salix wilsonii using PacBio HiFi sequencing. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1031769
Hurst, L. D. (2002). The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 18, 486. doi: 10.1016/s0168-9525(02)02722-1
Jackman, S. D., Coombe, L., Warren, R. L., Kirk, H., Trinh, E., MacLeod, T., et al. (2020). Complete mitochondrial genome of a gymnosperm, sitka spruce (Picea sitchensis), indicates a complex physical structure. Genome Biol. Evol. 12, 1174–1179. doi: 10.1093/gbe/evaa108
Jiang, H., Lu, Q., Qiu, S., Yu, H., Wang, Z., Yu, Z., et al. (2022). Fujian cytoplasmic male sterility and the fertility restorer gene OsRf19 provide a promising breeding system for hybrid rice. Proc. Natl. Acad. Sci. U.S.A. 119, e2208759119. doi: 10.1073/pnas.2208759119
Jin, G., Wang, L., Long, L., Wu, F., Tang, Y., Qin, J., et al. (2019). Analysis of codon usage bias in the mitochondrial protein-coding genes of Oryza rufipogon. Plant Sci. J. 37, 188–197. doi: 10.11913/PSJ.2095-0837.2019.20188
Jin, J., Yu, W., Yang, J., Song, Y., dePamphilis, C., Yi, T., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Kannaujiya, V. K., Rastogi, R. P., Sinha, R. P. (2014). GC constituents and relative codon expressed amino acid composition in cyanobacterial phycobiliproteins. Gene 546, 162–171. doi: 10.1016/j.gene.2014.06.024
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Ke, S.-J., Liu, D.-K., Tu, X.-D., He, X., Zhang, M.-M., Zhu, M.-J., et al. (2023). Apostasia mitochondrial genome analysis and monocot mitochondria phylogenomics. Int. J. Mol. Sci. 24, 7837. doi: 10.3390/ijms24097837
Khera, P., Saxena, R., Sameerkumar, C. V., Saxena, K., Varshney, R. K. (2015). Mitochondrial SSRs and their utility in distinguishing wild species, CMS lines and maintainer lines in pigeonpea (Cajanus cajan L.). Euphytica 206, 737–746. doi: 10.1007/s10681-015-1504-2
Kubo, T., Nishizawa, S., Sugawara, A., Itchoda, N., Estiati, A., Mikami, T. (2000). The complete nucleotide sequence of the mitochondrial genome of sugar beet (Beta vulgaris L.) reveals a novel gene for tRNA(Cys)(GCA). Nucleic Acids Res. 28, 2571–2576. doi: 10.1093/nar/28.13.2571
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lai, C., Wang, J., Kan, S., Zhang, S., Li, P., Reeve, W. G., et al. (2022). Comparative analysis of mitochondrial genomes of Broussonetia spp. (Moraceae) reveals heterogeneity in structure, synteny, intercellular gene transfer, and RNA editing. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1052151
Lenz, H., Hein, A., Knoop, V. (2018). Plant organelle RNA editing and its specificity factors: enhancements of analyses and new database features in PREPACT 3.0. BMC Bioinf. 19, 255. doi: 10.1186/s12859-018-2244-9
Lewis, S. E., Searle, S. M., Harris, N., Gibson, M., Lyer, V., Richter, J., et al. (2002). Apollo: a sequence annotation editor. Genome Biol. 3, Research0082. doi: 10.1186/gb-2002-3-12-research0082
Li, J., Li, J., Ma, Y., Kou, L., Wei, J., Wang, W. (2022). The complete mitochondrial genome of okra (Abelmoschus esculentus): using nanopore long reads to investigate gene transfer from chloroplast genomes and rearrangements of mitochondrial DNA molecules. BMC Genomics. 23, 481. doi: 10.1186/s12864-022-08706-2
Li, X., Yang, J.-B., Wang, H., Song, Y., Corlett, R. T., Yao, X., et al. (2021). Plastid NDH pseudogenization and gene loss in a recently derived lineage from the largest hemiparasitic plant genus pedicularis (Orobanchaceae). Plant Cell Physiol. 62, 971–984. doi: 10.1093/pcp/pcab074
Li, J., Ye, C. (2020). Genome-wide analysis of microsatellite and sex-linked marker identification in Gleditsia sinensis. BMC Plant Biol. 20, 338. doi: 10.1186/s12870-020-02551-9
Lian, Q., Li, S., Kan, S., Liao, X., Huang, S., Sloan, D. B., et al. (2024). Association analysis provides insights into plant mitonuclear interactions. Mol. Biol. Evol. 41, msae028. doi: 10.1093/molbev/msae028
Liu, C., Chen, H.-H., Tang, L.-Z., Khine, P. K., Han, L.-H., Song, Y., et al. (2022). Plastid genome evolution of a monophyletic group in the subtribe Lauriineae (Laureae, Lauraceae). Plant Divers. 44, 377–388. doi: 10.1016/j.pld.2021.11.009
Liu, H., Hou, Z., Xu, L., Ma, Q., Wei, M., Tembrock, L. R., et al. (2023). Comparative analysis of organellar genomes between diploid and tetraploid Chrysanthemum indicum with its relatives. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1228551
Lowe, T. M., Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Ma, Q., Li, S., Bi, C., Hao, Z., Sun, C., Ye, N. (2017). Complete chloroplast genome sequence of a major economic species, Ziziphus jujuba (Rhamnaceae). Curr. Genet. 63, 117–129. doi: 10.1007/s00294-016-0612-4
Ma, Q., Wang, Y., Li, S., Wen, J., Zhu, L., Yan, K., et al. (2022). Assembly and comparative analysis of the first complete mitochondrial genome of Acer truncatum Bunge: a woody oil-tree species producing nervonic acid. BMC Plant Biol. 22, 29. doi: 10.1186/s12870-021-03416-5
Maréchal, A., Brisson, N. (2010). Recombination and the maintenance of plant organelle genome stability. New Phytol. 186, 299–317. doi: 10.1111/j.1469-8137.2010.03195.x
Mazumdar, P., Binti Othman, R., Mebus, K., Ramakrishnan, N., Ann Harikrishna, J. (2017). Codon usage and codon pair patterns in non-grass monocot genomes. Ann. Bot. 120, 893–909. doi: 10.1093/aob/mcx112
Mower, J. P. (2008). Modeling sites of RNA editing as a fifth nucleotide state reveals progressive loss of edited sites from angiosperm mitochondria. Mol. Biol. Evol. 25, 52–61. doi: 10.1093/molbev/msm226
Mower, J. P., Sloan, D. B., Alverson, A. J. (2012). “Plant mitochondrial genome diversity: the genomics revolution,” in Plant genome diversity volume 1. Eds. Wendel, J., Greilhuber, J., Dolezel, J., Leitch, I. (Vienna: Springer), 123–144.
Nair, R. R., Nandhini, M. B., Sethuraman, T., Doss, G. (2013). Mutational pressure dictates synonymous codon usage in freshwater unicellular α - cyanobacterial descendant Paulinella chromatophora and β - cyanobacterium Synechococcus elongatus PCC6301. Springerplus 2, 492. doi: 10.1186/2193-1801-2-492
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Niu, Y., Gao, C., Liu, J. (2022). Complete mitochondrial genomes of three Mangifera species, their genomic structure and gene transfer from chloroplast genomes. BMC Genomics 23, 147. doi: 10.1186/s12864-022-08383-1
Novembre, J. A. (2002). Accounting for background nucleotide composition when measuring codon usage bias. Mol. Biol. Evol. 19, 1390–1394. doi: 10.1093/oxfordjournals.molbev.a004201
Parvathy, S. T., Udayasuriyan, V., Bhadana, V. (2022). Codon usage bias. Mol. Biol. Rep. 49, 539–565. doi: 10.1007/s11033-021-06749-4
Petersen, G., Anderson, B., Braun, H.-P., Meyer, E. H., Møller, I. M. (2020). Mitochondria in parasitic plants. Mitochondrion 52, 173–182. doi: 10.1016/j.mito.2020.03.008
Pimpinelli, S., Piacentini, L. (2020). Environmental change and the evolution of genomes: Transposable elements as translators of phenotypic plasticity into genotypic variability. Funct. Ecol. 34, 428–441. doi: 10.1111/1365–2435.13497
Qiao, Y., Zhang, X., Li, Z., Song, Y., Sun, Z. (2022). Assembly and comparative analysis of the complete mitochondrial genome of Bupleurum chinense DC. BMC Genomics 23, 664. doi: 10.1186/s12864-022-08892-z
Qiu, L., Yang, C., Tian, B., Yang, J. B., Liu, A. (2010). Exploiting EST databases for the development and characterization of EST-SSR markers in castor bean (Ricinus communis L.). BMC Plant Biol. 10, 278. doi: 10.1186/1471-2229-10-278
Qu, K., Chen, Y., Liu, D., Guo, H., Xu, T., Jing, Q., et al. (2024). Comprehensive analysis of the complete mitochondrial genome of Lilium tsingtauense reveals a novel multichromosome structure. Plant Cell Rep. 43, 150. doi: 10.1007/s00299-024-03232-9
Qu, Y., Zhou, P., Tong, C., Bi, C., Xu, L. (2023). Assembly and analysis of the Populus deltoides mitochondrial genome: the first report of a multicircular mitochondrial conformation for the genus Populus. J. For. Res. 34, 717–733. doi: 10.1007/s11676-022-01511-3
Rice, D. W., Alverson, A. J., Richardson, A. O., Young, G. J., Sanchez-Puerta, M. V., Munzinger, J., et al. (2013). Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 342, 1468–1473. doi: 10.1126/science.1246275
Richardson, A. O., Rice, D. W., Young, G. J., Alverson, A. J., Palmer, J. D. (2013). The “fossilized” mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 11, 29. doi: 10.1186/1741-7007-11-29
Sharp, P. M., Li, W. H. (1986). Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’ codons. Nucleic Acids Res. 14, 7737–7749. doi: 10.1093/nar/14.19.7737
Shen, J., Li, X., Li, M., Cheng, H., Huang, X., Jin, S. (2022). Characterization, comparative phylogenetic, and gene transfer analyses of organelle genomes of Rhododendron × pulchrum. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.96976
Shen, B., Shen, A., Liu, L., Tan, Y., Li, S., Tan, Z. (2024). Assembly and comparative analysis of the complete multichromosomal mitochondrial genome of Cymbidium ensifolium, an orchid of high economic and ornamental value. BMC Plant Biol. 24, 255. doi: 10.1186/s12870-024-04962-4
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73. doi: 10.1093/nar/gkz345
Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. (2015). Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. U.S.A. 112, E3515–E3524. doi: 10.1073/pnas.1504491112
Sloan, D. B., MacQueen, A. H., Alverson, A. J., Palmer, J. D., Taylor, D. R. (2010). Extensive loss of RNA editing sites in rapidly evolving Silene mitochondrial genomes: selection vs. retroprocessing as the driving force. Genetics 185, 1369–1380. doi: 10.1534/genetics.110.118000
Sullivan, A. R., Eldfjell, Y., Schiffthaler, B., Delhomme, N., Asp, T., Hebelstrup, K. H., et al. (2020). The mitogenome of Norway spruce and a reappraisal of mitochondrial recombination in plants. Genome Biol. Evol. 12, 3586–3598. doi: 10.1093/gbe/evz263
Tan, C., Ferguson, D. K., Tang, Z., Yang, Y. (2023). Distribution and conservation of the lauraceae in China. Glob. Ecol. Conserv. 46, e02566. doi: 10.1016/j.gecco.2023.e02566
Tang, X., Yang, S., Chen, H., Sun, Z., Lai, Z., Zeng, W., et al. (2020). Comparative analysis on codon usage bias inmitogenome of two species in genus Glycine. Guihaia 40, 926–934. doi: 10.11931/guihaia.gxzw201911033
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–w11. doi: 10.1093/nar/gkx391
Unseld, M., Marienfeld, J. R., Brandt, P., Brennicke, A. (1997). The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet. 15, 57–61. doi: 10.1038/ng0197-57
Varré, J. S., D’Agostino, N., Touzet, P., Gallina, S., Tamburino, R., Cantarella, C., et al. (2019). Complete sequence, multichromosomal architecture and transcriptome analysis of the Solanum tuberosum mitochondrial genome. Int. J. Mol. Sci. 20, 4788. doi: 10.3390/ijms20194788
Verhage, L. (2020). Targeted editing of the Arabidopsis mitochondrial genome. Plant J. 104, 1457–1458. doi: 10.1111/tpj.15097
Wang, J., Kan, S., Liao, X., Zhou, J., Tembrock, L. R., Daniell, H., et al. (2024). Plant organellar genomes: much done, much more to do. Trends Plant Sci. 29, 754–769. doi: 10.1016/j.tplants.2023.12.014
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. doi: 10.1093/nar/gkr1293
Wang, H., Yang, J. (2020). Identification and analysis of RNA editing sites chloroplast genome in Nicotiana tabacum. Mol. Plant Breed. 18, 6649–6656. doi: 10.13271/j.mpb.018.006649
Wang, M., Yu, W., Yang, J., Hou, Z., Li, C., Niu, Z., et al. (2023). Mitochondrial genome comparison and phylogenetic analysis of Dendrobium (Orchidaceae) based on whole mitogenomes. BMC Plant Biol. 23, 586. doi: 10.1186/s12870-023-04618-9
Wendel, J. F., Greilhuber, J., Dolezel, J., Leitch, I. J. (2012). Plant genome diversity volume 1: Plant genomes, their residents, and their evolutionary dynamics (Vienna: Springer).
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1101/018333
Wright, F. (1990). The ‘effective number of codons’ used in a gene. Gene 87, 23–29. doi: 10.1016/0378-1119(90)90491-9
Wu, Z., Liao, X., Zhang, X., Ternbrock, L. R., Broz, A. (2022). Genomic architectural variation of plant mitochondria—A review of multichromosomal structuring. J. Syst. Evol. 60, 160–168. doi: 10.1111/jse.12655
Xie, D. F., Yu, H. X., Price, M., Xie, C., Deng, Y. Q., Chen, J. P., et al. (2019). Phylogeny of Chinese Allium species in section Daghestanica and adaptive evolution of Allium (Amaryllidaceae, Allioideae) species revealed by the chloroplast complete genome. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00460
Xiong, Y., Yu, Q., Xiong, Y., Zhao, J., Lei, X., Liu, L., et al. (2021). The complete mitogenome of Elymus sibiricus and insights into its evolutionary pattern based on simple repeat sequences of seed plant mitogenomes. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.802321
Xu, Q., Niu, S. C., Li, K. L., Zheng, P. J., Zhang, X. J., Jia, Y., et al. (2022). Chromosome-scale assembly of the Dendrobium nobile genome provides insights into the molecular mechanism of the biosynthesis of the medicinal active ingredient of Dendrobium. Front. Genet. 13. doi: 10.3389/fgene.2022.844622
Yan, J., Zhang, Q., Yin, P. (2018). RNA editing machinery in plant organelles. Sci. China Life Sci. 61, 162–169. doi: 10.1007/s11427-017-9170-3
Yang, J.-X., Dierckxsens, N., Bai, M.-Z., Guo, Y.-Y. (2023a). Multichromosomal mitochondrial genome of paphiopedilum micranthum: compact and fragmented genome, and rampant intracellular gene transfer. Int. J. Mol. Sci. 24, 3976. doi: 10.3390/ijms24043976
Yang, Z., Ferguson, D. K., Yang, Y. (2023b). New insights into the plastome evolution of Lauraceae using herbariomics. BMC Plant Biol. 23, 387. doi: 10.1186/s12870-023-04396-4
Yang, Z., Ferguson, D. K., Yang, Y. (2023c). Plastome phylogeny and taxonomy of cinnamomum guizhouense (Lauraceae). Forests 14, 310. doi: 10.3390/f14020310
Yu, R., Sun, C., Zhong, Y., Liu, Y., Sanchez-Puerta, M. V., Mower, J. P., et al. (2022). The minicircular and extremely heteroplasmic mitogenome of the holoparasitic plant Rhopalocnemis phalloides. Curr. Biol. 32, 470–479.e475. doi: 10.1016/j.cub.2021.11.053
Yuan, Y., Jin, X., Liu, J., Zhao, X., Zhou, J., Wang, X., et al. (2018). The Gastrodia elata genome provides insights into plant adaptation to heterotrophy. Nat. Commun. 9, 1615. doi: 10.1038/s41467-018-03423-5
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, Z. F., Li, Y. Y., Xiao, B. Z. (2016). Comparative transcriptome analysis highlights the crucial roles of photosynthetic system in drought stress adaptation in upland rice. Sci. Rep. 6, 19349. doi: 10.1038/srep19349
Zhang, H., Meltzer, P., Davis, S. (2013). RCircos: an R package for Circos 2D track plots. BMC Bioinf. 14, 1–5. doi: 10.1186/1471-2105-14-244
Zhong, F., Ke, W., Li, Y., Chen, X., Zhou, T., Xu, B., et al. (2023). Comprehensive analysis of the complete mitochondrial genomes of three Coptis species (C. chinensis, C. deltoidea and C. omeiensis): the important medicinal plants in China. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1166420
Zhou, J., Nie, L., Zhang, S., Mao, H., Arimura, S., Jin, S., et al. (2024). Mitochondrial genome editing of WA352 via mitoTALENs restore fertility in cytoplasmic male sterile rice. Plant Biotechnol. J. 22, 1960–1962. doi: 10.1111/pbi.14315
Zhou, P., Zhang, Q., Li, F., Huang, J., Zhang, M. (2023). Assembly and comparative analysis of the complete mitochondrial genome of Ilex metabaptista (Aquifoliaceae), a Chinese endemic species with a narrow distribution. BMC Plant Biol. 23, 393. doi: 10.1186/s12870-023-04377-7
Zhu, S., Niu, Z., Xue, Q., Wang, H., Xie, X., Ding, X. (2018). Accurate authentication of Dendrobium officinale and its closely related species by comparative analysis of complete plastomes. Acta Pharm. Sin. B. 8, 969–980. doi: 10.1016/j.apsb.2018.05.009
Keywords: Dendrobium species, mitochondrial genome, chloroplast genome, homologous sequence, RNA editing
Citation: Wang L, Liu X, Wang Y, Ming X, Qi J and Zhou Y (2024) Comparative analysis of the mitochondrial genomes of four Dendrobium species (Orchidaceae) reveals heterogeneity in structure, synteny, intercellular gene transfer, and RNA editing. Front. Plant Sci. 15:1429545. doi: 10.3389/fpls.2024.1429545
Received: 08 May 2024; Accepted: 16 July 2024;
Published: 30 July 2024.
Edited by:
Changwei Bi, Nanjing Forestry University, ChinaReviewed by:
Zhigang Hao, China Agricultural University, ChinaCopyright © 2024 Wang, Liu, Wang, Ming, Qi and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xue Liu, bGl1MDkwNnh1ZUAxNjMuY29t; Yongde Wang, d3lkMjAyMzA4MThAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.