 Tian-jiang Liao1,2
Tian-jiang Liao1,2 Hui-yan Xiong
Hui-yan Xiong Rui-jun Duan
Rui-jun Duan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 27 June 2024
Sec. Plant Bioinformatics
Volume 15 - 2024 | https://doi.org/10.3389/fpls.2024.1421702
This article is part of the Research TopicGenetically Deciphering the Adaptive Growth and Development of Photosynthetic Organisms: A Holistic Omics ApproachView all 7 articles
Three-amino-loop-extension (TALE) family belongs to the homeobox gene superfamily and occurs widely in plants, playing a crucial role in regulating their growth and development. Currently, genome-wide analysis of the TALE family has been completed in many plants. However, the systematic identification and hormone response analysis of the TALE gene family in barley are still lacking. In this study, 21 TALE candidate genes were identified in barley, which can be divided into KNOX and BELL subfamilies. Barley TALE members in the same subfamily of the phylogenetic tree have analogically conserved motifs and gene structures, and segmental duplications are largely responsible for the expansion of the HvTALE family. Analysis of TALE orthologous and homologous gene pairs indicated that the HvTALE family has mainly undergone purifying selective pressure. Through spatial structure simulation, HvKNOX5–HvKNOX6 and HvKNOX5–HvBELL11 complexes are all formed through hydrogen bonding sites on both the KNOX2 and homeodomain (HD) domains of HvKNOX5, which may be essential for protein interactions among the HvTALE family members. Expression pattern analyses reveal the potential involvement of most HvTALE genes in responses to exogenous hormones. These results will lay the foundation for regulation and function analyses of the barley TALE gene family in plant growth and development by hormone regulation.
Homeobox genes encode a prodigious superfamily of transcription factors (TFs), which occur widely in animals, plants, and other eukaryotes. A typical homeobox domain comprises a triple helix structure of 60 amino acids, forming a ring structure in the first and second helix regions and forming a helix-turn-helix structure in the second and third helix regions (Billeter et al., 1993). The first plant homeobox gene, maize Knotted1, was reported by Vollbrecht et al. (1991). Three-amino-loop-extension (TALE) TFs with a non-classical homeobox domain comprising 63 amino acids, and three further residues (P-Y-P) inserted between the first and second helices, are referred to as the TALE gene family. Based on protein sequences and evolution, this family is divided into a knotted-like homeodomain (KNOX) subfamily, and a BEL1-like homeodomain (BELL) subfamily, which interacts between specific individuals (Hake et al., 2004; Hamant and Pautot, 2010). Selective targeting of KNOX–BELL heterodimer to the nucleus may be the purpose of interaction between BELL and KNOX proteins (Bhatt et al., 2004; Kim et al., 2013). KNOX proteins usually have four domains (KNOX1, KNOX2, ELK, and homeodomain). According to gene structural characteristics, the KNOX subfamily has been divided into three classes: KNOX I, II, and III. The KNOX III class lacks homeodomains and was only reported in dicotyledons (Hake et al., 2004). BELL subfamily proteins, which contain a MID (also known as POX) composed of BELL and SKY domains and a homeodomain (HD), were less well studied (Müller et al., 2001; Cole et al., 2006). BEL1-like proteins have not been systematically classified to date.
The TALE family members play a vital role in regulating plant growth and development (Lin et al., 2013) and maintaining organ morphology (Belles-Boix et al., 2006), signal transduction (Cnops et al., 2006), hormone regulation (Shani et al., 2006), tuber formation (Kondhare et al., 2019), and resistance to abiotic stress (Tao et al., 2018). The KNOX gene subfamily has been intensively studied. KNOX I genes are primarily expressed in meristems and have distinct roles (Hake et al., 2004; Hay and Tsiantis, 2010; Qi and Zheng, 2013). For instance, Arabidopsis KNAT2 is expressed in the apical meristems, influences AGAMOUS (AG) ectopic expression in the carpel and ovule center, and causes the nucellar structure to homomorphically convert to a carpel-like structure (Pautot et al., 2001). The KNAT1 mutation causes a decrease in PIN2 expression levels in the root tip, enhances auxin accumulation in roots, and downregulates auxin transport in basal leaves. According to these findings, KNAT1 may adversely affect root tilt by regulating auxin transport (Qi and Zheng, 2013). KNOX II genes were expressed in a variety of tissues. Among these genes, KNAT7 is important for secondary cell wall (SCW) biosynthesis and cell elongation in Arabidopsis (AtKNAT7), rice (OsKNAT7), cotton (GhKNAT7-A03), and poplar (PoptrKNAT7) (Li et al., 2012; Ma et al., 2019; Yu, 2019). To prevent more rhizobia infection and nodule development, KNAT3/4/5-like genes may activate the EFD/RR4 pathway, partially inhibiting cytokinin signaling, thereby regulating the nodular organ boundary and shape in Medicago truncatula (Vernié et al., 2008). KNOX III gene in Arabidopsis contains just one KNATM, which also affects leaf development and leaf polarity (Magnani and Hake, 2008). The functions of ATH1, BEL1, BLH2, BLH4, PNF/BLH8, and PNY/BLH9 in the Arabidopsis BELL subfamily have also been confirmed, but the roles of the other BELL-like genes remain unknown. For example, ATH1 is a flowering inhibitor that controls the expression level of FLOWERING LOCUS C (FLC) gene (Proveniers et al., 2007). BLH9 stimulates flowering when it interacts with BLH8, while it inhibits flowering when it interacts with ATH1 (Rutjens et al., 2009). One or more KNOX genes were regulated by BLH2 and BLH4 to prevent the growth of specific leaf subdomains (Kumar et al., 2007).
At present, genome-wide analysis of the TALE family has been completed in many plants such as Arabidopsis thaliana (Hamant and Pautot, 2010), Populus trichocarpa (Zhao et al., 2019), Glycine max (Wang et al., 2021), and Triticum aestivum (Han et al., 2022). Meanwhile, considering the importance of this family in the development of flowers and leaves in the Poaceae family, it is urgent and necessary to conduct research on the barley TALE gene family. However, the systematic identification and hormone response analysis of the TALE gene family in barley (Hordeum vulgare) are still lacking. Here, a whole genome-wide analysis of the TALE family in barley was fulfilled and identified. We also fulfilled a phylogenetic analysis and confirmed chromosome location, gene structure, and homology analysis, protein interactions, expression patterns of barley TALE genes (HvTALEs). This study will help us to better understand the roles of TALE family members in the growth and development of hormone regulation in barley. These studies depicted the characterization and diversity of HvTALE genes, revealing their roles in the growth and development of barley in the future.
Barley protein sequences were downloaded from the Ensembl Plants database (http://plants.ensembl.org/index.Html) (Bolser et al., 2017). A Pfam domain (PF00046) was used to find members of the barley TALE family in the HMMER3 program (http://hmmer.org) using the hidden Markov model (HMM) searching method. To exclude genes lacking the conserved domain, the candidate barley TALE family members were submitted to the following databases for validation: Pfam (http://www.ebi.ac.uk/interpro/), SMART (https://smart.embl.de), and National Center for Biotechnology Information (NCBI) protein Batch CD-search (http://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi) (Yang et al., 2020; Mistry et al., 2021). Genes with HD and KNOX1 or KNOX2 domains were selected as KNOX subfamily members, and those with HD and POX domains were selected as BELL subfamily members, so barley TALE family members were identified in the barley genome. Then, amino acid (aa) numbers, isoelectric point (pI), and molecular weight (MW) of identified barley TALE proteins were predicted in ExPASy (https://web.expasy.org/compute_pi/) online tools (Artimo et al., 2012).
Conserved protein sequences were extracted for multi-species phylogenetic analysis. Required barley TALE CDs and genomic DNA sequences were downloaded from the Ensembl Plants database. TALE protein sequences of T. aestivum, G. max, and P. trichocarpa were obtained from published papers (Zhao et al., 2019; Wang et al., 2021; Han et al., 2022). Arabidopsis protein sequences were obtained from TAIR (https://www.arabidopsis.org/), while TALE protein sequences of Oryza sativa and Zea mays were obtained from PlantTFDB v4.0 (http://planttfdb.gao-lab.org/) (Jin et al., 2017). Using the neighbor-joining (NJ) method, phylogenetic trees were constructed in MEGA7 software (https://www.megasoftware.net/) (Kumar et al., 2016) and EvolView 10 (https://evolgenius.info//evolview-v2/#login). Using Mapchart software, the location information of barley TALE superfamily members comes from the Ensembl Plants database (Voorrips, 2002), and its members were mapped to seven barley chromosomes.
Using the MCScanX program, segmental and tandem duplications in the barley TALE genes were found (Gu et al., 2002; Kong et al., 2013; Wang et al., 2013). Duplicated gene pairs in the barley H. vulgare Morex v3 genome were prepared using Circos software (Krzywinski et al., 2009). Syntenic relationships between TALE superfamily members in barley and other species were identified using TBtools (Chen et al., 2020). Required barley TALE CDS and protein sequences were downloaded from the barley pan-genome, such as Akashinriki, Golden_Promise, and B1K-04–12 (Jayakodi et al., 2020), and the wild barley OUH602 genome (Liu et al., 2020). TALE orthologous gene pairs in barley were determined among Morex and four barley accessions by an alignment of full-length aa sequences. TBtools software was used to calculate non-synonymous/synonymous (Ks/Ka) ratios, and the Ka/Ks values were defined by three criteria: Ka/Ks < 1 (purifying selection), Ka/Ks = 1 (neutral selection), and Ka/Ks > 1 (positive selection) (Anisimova et al., 2001).
GSDS (Gene Structure Display Server 2.0) (http://gsds.gao-lab.org/Gsds_help.php) was used to show gene structures (Hu et al., 2015). Conserved motifs in predicted barley TALE proteins were examined using the Multiple Expectation Maximization for Motif Elicitation (MEME) program (http://meme-suite.org/tools/meme) (Bailey et al., 2009) and were visualized by TBtools.
The 2,000-bp region upstream of the start codon of barley TALE family members was used as the promoter sequence (Zhao et al., 2016). The promoter sequence was submitted to the PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) to identify cis-elements (Lescot et al., 2002).
The Orthovenn2 site (https://www.genengnews.com/best-of-the-web/orthovenn2/) was used to map barley genes with Arabidopsis gene homology. Eight Arabidopsis TALE proteins representing eight barley TALE proteins were submitted to the STRING online server (https://string-db.org/) to predict protein–protein interactions. The barley TALE protein interaction network was visualized using Cytoscape (https://cytoscape.org/).
We use the FlyFish program with AlphaFold to realize automatic 3D structure modeling (Jumper et al., 2021). FlyFish is bioinformatics software that enables data analysis, task management, database operations, and more. We sought to write a 3D structure modeling analysis and data processing program in FlyFish, then run this program to automatically perform batch analysis in AlphaFold, and export results and 3D structures data. We selected reported interacting proteins as positive controls to identify the possibility of interaction between the two proteins of interest by MEGADOCK 4.0 docking software (Ohue et al., 2014).
Transcriptional profiles were provided by the Leibniz Institute of Plant Genetics and Crop Plant Research (IPK) barley BARLEX server (https://pgrc.ipk-gatersleben.de/projects/barley/) in 14 tissues and at different development stages (Monat et al., 2019). The fragments per kilobase of transcript per million fragment mapped reads (FPKM) were used to quantify the levels of gene expression. A heatmap of gene expression data was constructed in TBtools software using log2-transformed mean FPKM values.
To explore expression patterns of barley TALE genes under hormone treatments, publicly available 57 RNA-seq samples were downloaded [nine RNA-seq samples of ABA and SA treatments from the Sequence Read Archive (SRA) database using transcripts per million (TPM) treats and 48 RNA-seq samples of ABA and MeJA treatments from Gene Expression Omnibus (GEO) BioProject in NCBI]. Accession numbers and sample information of RNA-seq are listed in Supplementary Table 1. Agilent Feature Extraction Software (v 9.1) was used for background subtraction and LOWESS normalization of GEO sample data. Two heatmaps for gene expression data were constructed in TBtools software using log2 (TPM+1) of SRA data and downloaded GEO sample data. All reads from these datasets were mapped to the Morex v3 genome (Mascher et al., 2021).
Barley variety ‘Morex’ was used for hormone treatments. After sterilization with 10% bleach for 5 min and rinsing thrice with deionized water, the seeds were put into Petri dishes to germinate in the dark for 48 h at 22°C. In growth chambers, sprouted seeds were grown under carefully regulated circumstances, including 55% relative humidity, 22°C, and a 16-h/8-h light/dark photoperiod with 20,000 lx of light intensity. Barley seedlings were exposed to 100 μM GA3, 100 μM ABA, and 100 μM 6-BA for hormone treatments at 12 d after germination. At 0 h, 3 h, 6 h, 9 h, 12 h, and 24 h, the leaves of barley seedlings were taken from all treatments and controls. TRIzol was used to extract RNA from barley leaves, and a FastKing RT Kit (TIANGEN, Beijing, China) was used to synthesize cDNA. Real-time quantitative PCR was carried out using PerfectStart™ GREEN qPCR SuperMix (SYBR Green I) (Transgen, Beijing, China). For every qRT-PCR analysis, the barley β-actin gene served as an internal reference. The 2−ΔΔCT value was utilized for the analysis of gene expression data. The qRT-PCR-specific primers are listed in Supplementary Table 2.
We identified 21 HvTALE genes including nine KNOX genes (HvKNOX1 to HvKNOX9) and 12 BELL genes (HvBELL1 to HvBELL12) from H. vulgare Morex v3 genome, named according to their gene coordinate on the seven barley chromosomes (Han et al., 2022) (Table 1). These 21 HvTALE genes were unevenly distributed on the seven barley chromosomes (Supplementary Figure 1). Notably, Chr4H enclosed the most HvTALE genes (10), whereas Chr2, Chr3, and Chr6H had a single gene only.
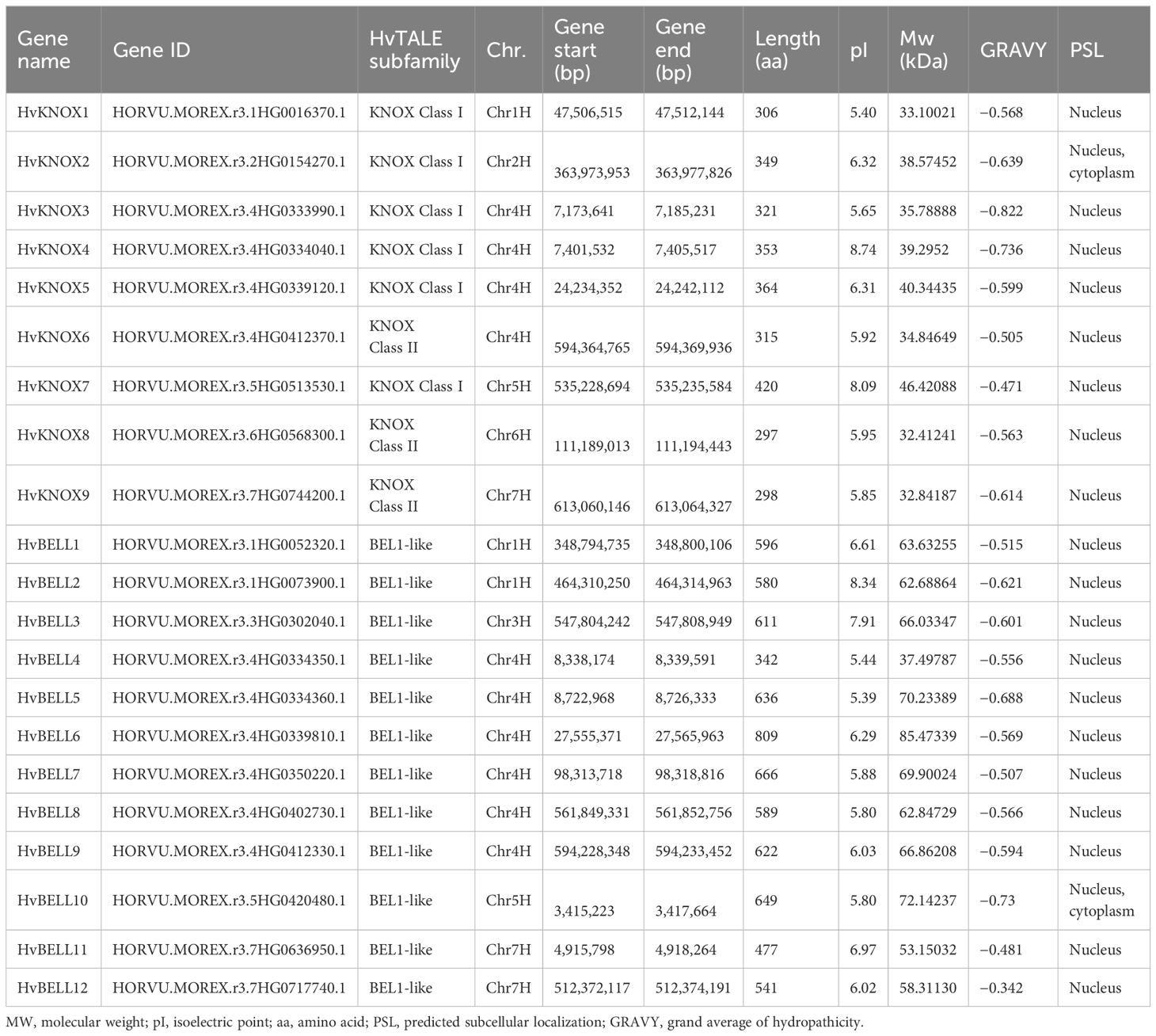
Table 1 Physical and biochemical properties of TALE genes identified in barley.
Fundamental physical and biochemical characteristics of HvTALE family members were explored (Table 1), including protein length, isoelectric point, MW, grand average of hydropathicity, and subcellular localization. Protein sizes of HvTALE members varied from 297 aa (HvKNOX8) to 809 aa (HvBELL6), and corresponding MW ranged from 32.41241 to 85.47339 kDa (Table 1); pI values ranged from 5.40 (HvKNOX1) to 8.74 (HvKNOX4). Subcellular location predictions indicated that HvTALE proteins (19 members) occurred mainly in the nuclear region. Two HvTALE members (HvKNOX2 and HvBELL10) may occur in nuclear and cytoplasm regions. Gene CDS and protein sequences are presented in Supplementary Table 3, and HD sequences of barley HvTALE members are presented in Supplementary Table 4.
A phylogenetic tree was built using 167 TALE protein sequences from five species (A. thaliana, H. vulgare, O. sativa, Z. mays, and T. aestivum) to investigate the phylogeny and taxonomic relationships of TALE superfamily genes (Figure 1). Additionally, an unrooted evolutionary tree with just barley TALE proteins was also built (Supplementary Figure 2). Based on structural characteristics and the classification in Arabidopsis (Supplementary Table 5), the 21 HvTALE family members were classified into KNOX and BEL1-like subfamilies (Table 1). Nine members of the KNOX subfamily were further divided into six members of class KNOX I and three members of class KNOX II.
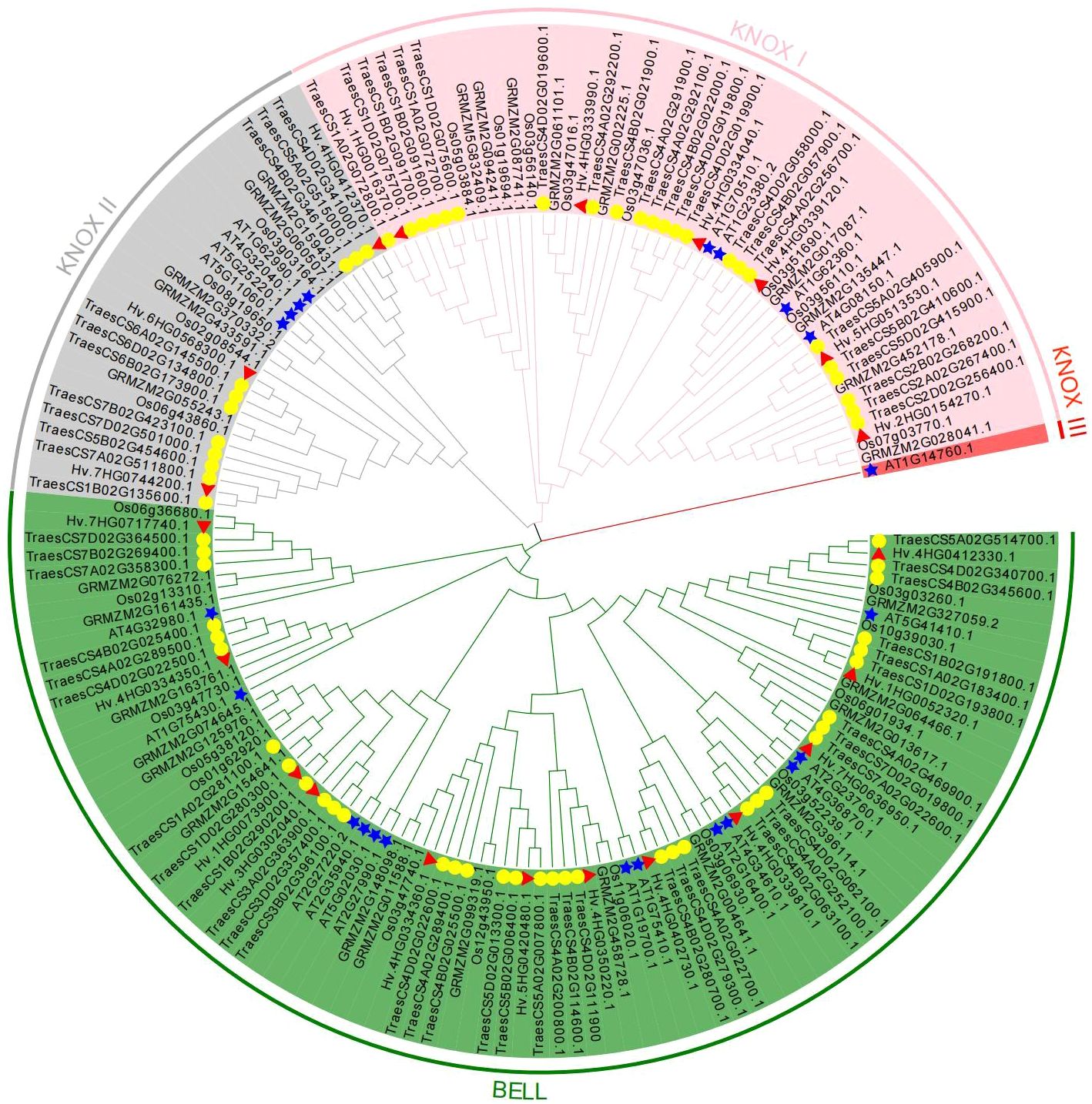
Figure 1 Phylogenetic analysis of HvTALE proteins. In total, 167 KNOX/BELL protein sequences from five species, including four monocotyledons (Hordeum vulgare, Oryza sativa, Zea mays, and Triticum aestivum) and one dicotyledon (Arabidopsis thaliana), were used to construct the unrooted neighbor-joining (NJ) tree. Different subclass genes are distinguished by color: KNOX I (pink), KNOX II (gray), KNOX III (red), and BELL (green). All barley TALE proteins are denoted by red triangles, Arabidopsis TALE proteins are represented by blue pentacles, and wheat TALE proteins are represented by yellow circles.
We investigated exon–intron patterns and conserved domain compositions of discovered HvTALE genes to gain a better understanding of possible relationships between the structure and function of HvTALE genes (Figure 2A). HvTALE genes displayed two to five exons (one HvTALE gene contains two exons, one HvTALE gene contains three exons, 10 HvTALE genes contain four exons, and nine HvTALE genes contain five exons). HvTALE genes in the same subfamily showed similar gene structures. Notably, except for HvBELL4 and HvBELL7, the BEL1-like subfamily genes had four exons, but the KNOX subfamily genes had five exons. Conserved domains of HvTALE members are depicted in Figure 2A. All HvTALE members contained HD domains, the BEL1-like subfamily members harbored POX domains, and KNOX1, KNOX2, and ELK domains occurred only in the KNOX subfamily. Significantly, the MEINOX domain, which mediates the formation of heterodimers between KNOX and BEL1-like proteins, is composed of the KNOX1 and KNOX2 domains (Bhatt et al., 2004; Kim et al., 2013).
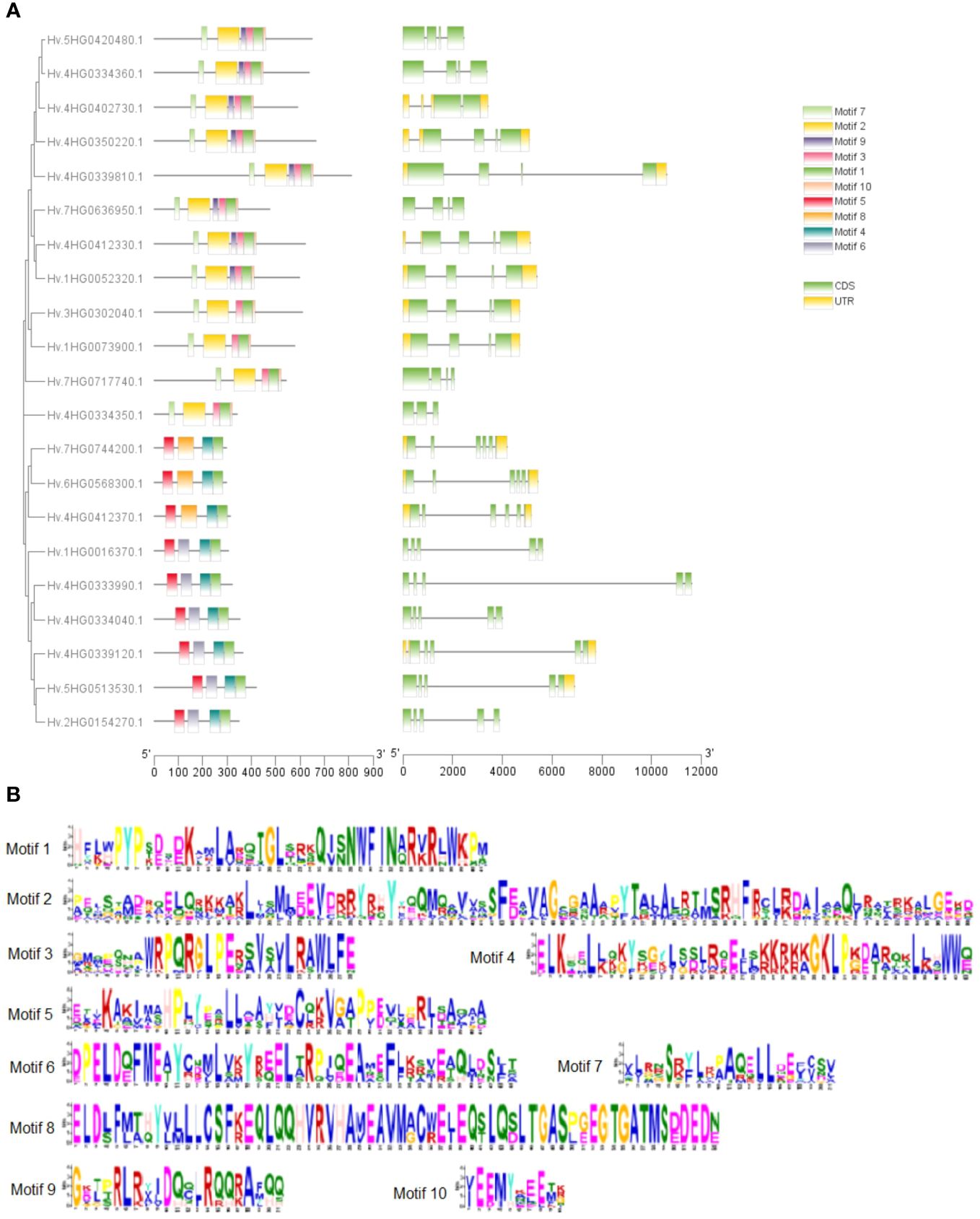
Figure 2 Gene structures and motif patterns of HvTALE members. (A) Gene structures and motif patterns of HvTALE genes. Colors: green boxes, exons; gray lines, introns; yellow boxes, untranslated 5′ and 3′ regions. Ten motifs are set by Multiple Expectation Maximization for Motif Elicitation (MEME) software. (B) Seq Logos of HvTALE members; 10 motifs are set by MEME software.
To better understand conserved domain patterns, we used MEME online software to scan HvTALE gene motifs and set the motif quantity at 10. A drawing was constructed using the 10 scanned MEME-motifs in Figure 2A, and the MEME-motifs’ Sequence Logos are shown in Figure 2B. HvTALE members of the same subfamily also display similar motif compositions. Motifs 1–3 and 7–10 were present in most BEL1-like subfamily members, while Motifs 1, 4–6, and 8 were present in KNOX subfamily members. The KNOX subfamily showed different motif components across different clades. Class KNOX II members were linked to Motifs 1, 4, 5, and 8, while class KNOX I members contained Motifs 1 and 4–6. The BELL subfamily exhibited two types of motif components: four HvBELL members contained Motifs 1, 2, 5, 7, and 10, and others were associated with Motifs 1, 2, 5, 7, 9, and 10.
Plant gene family expansions are thought to be primarily driven by tandem and segmental duplication events (Cannon et al., 2004). Tandem duplication events are defined as 200-kb chromosomal regions containing two or more genes (Han et al., 2022). We detected a tandem duplication event linked to two HvTALE genes (HvBELL4/HvBELL5) on Chr4H (Supplementary Figure 1). In contrast, segmental duplications result in a significant number of duplicated chromosomal blocks within genomes and frequently happen during chromosome rearrangement-related polyploidization events (Yu et al., 2005). In the barley genome, we found three segmental duplication events (Supplementary Table 6A, Figure 3). Compared with tandem duplications, the segmental duplications mainly drive the expansion of the HvTALE superfamily.
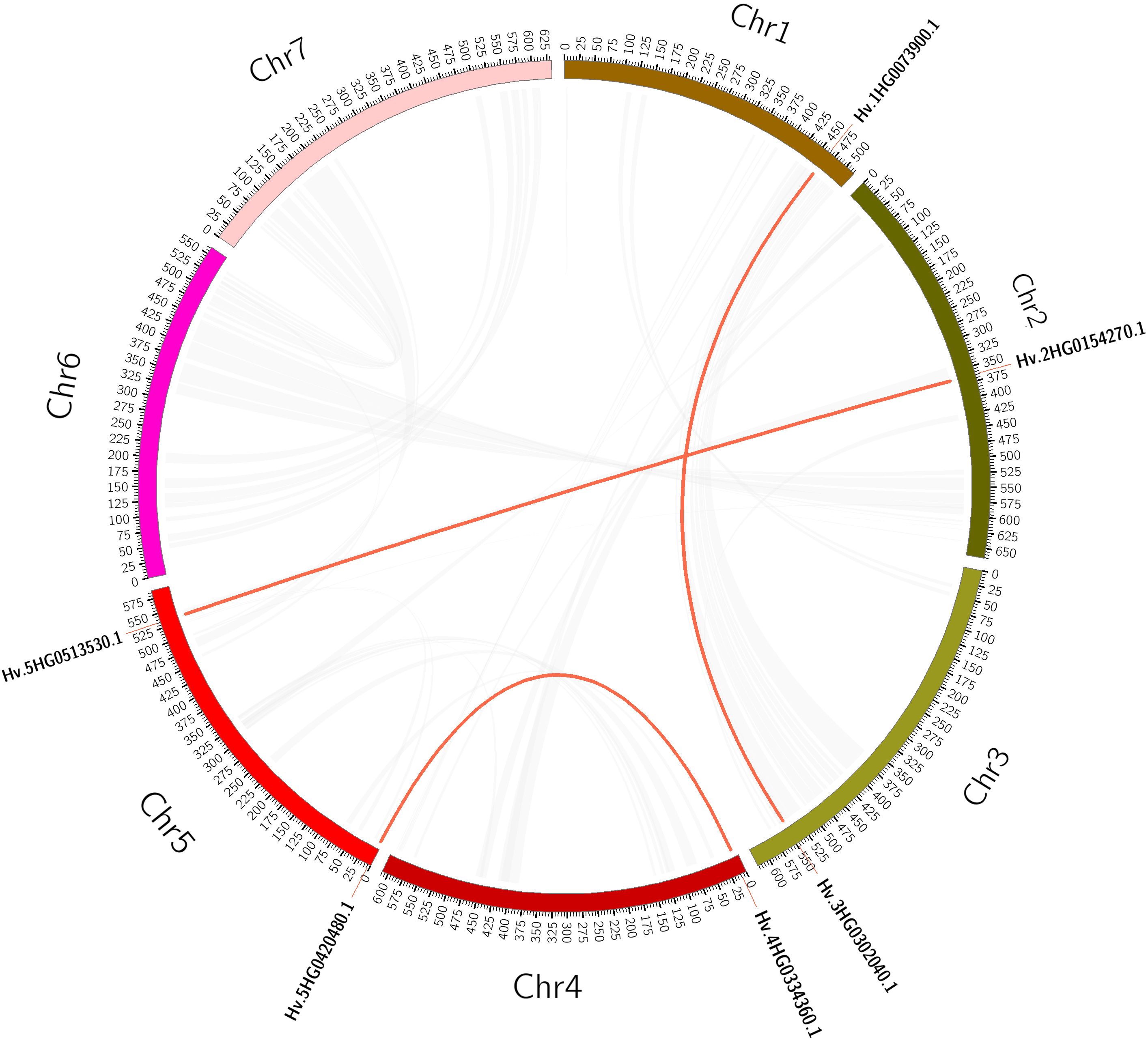
Figure 3 A circular figure showing the HvTALE genes’ collinearity. Duplicate HvTALE gene pairs are connected by red curves, and genome-wide collinear blocks are used as the background (gray).
To explore evolutionary clues of TALE members among barley (H. vulgare) and other species, three dicots (A. thaliana, G. max, and P. trichocarpa) and three monocots (O. sativa, Z. mays, and T. aestivum) were used for syntenic analyses (Supplementary Table 6B). In total, 3, 2, 5, 15, 56, and 14 HvTALE orthologous genes were identified for these species (Supplementary Figure 3). Output results were integrated into the comparative syntenic schematics in Figure 4. Two barley TALE members (HvBELL2 and HvBELL3) have collinear pairs with five species. Eight barley TALE members have collinear pairs with three monocot species. Thus, barley has more collinear pairs with the monocots O. sativa, Z. mays, and T. aestivum.
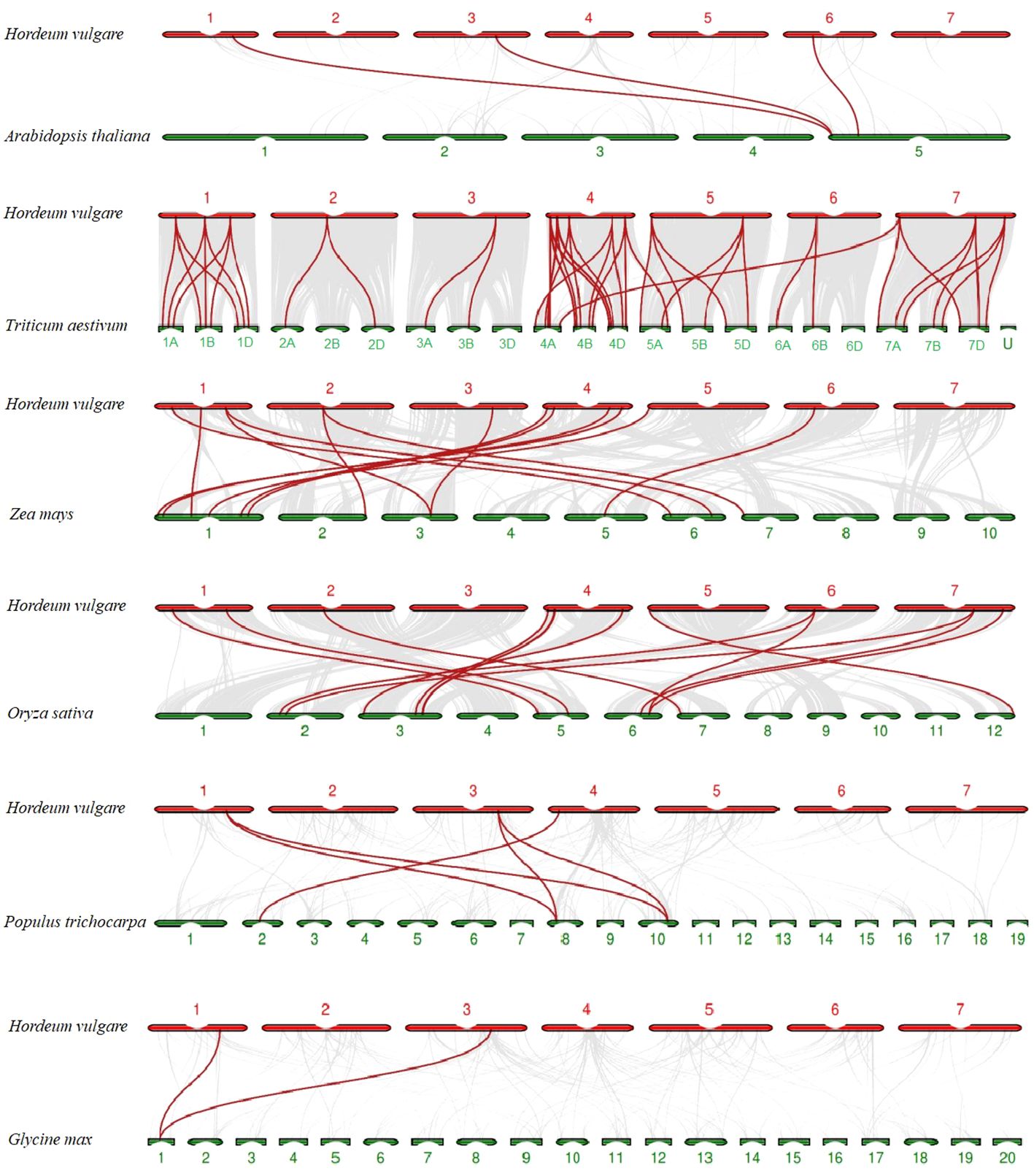
Figure 4 Syntenic relations of TALE members among barley and six plant species; 3, 2, 5, 15, 56, and 14 HvTALE orthologous genes were identified between barley and Arabidopsis thaliana, Glycine max, Populus trichocarpa, Oryza sativa, Zea mays, and Triticum aestivum, respectively. Gray lines (background) indicate collinear blocks within barley and other plant genomes; red lines highlight syntenic HvTALE gene pairs.
The Ka/Ks ratios of TALE orthologous gene pairs between barley and the six species were also calculated to uncover the evolutionary constraints on the TALE superfamily (Supplementary Table 6B). All orthologous TALE gene pairs show Ka/Ks < 1, indicating that the barley TALE superfamily has undergone purifying selective pressure in monocotyledons (Han et al., 2022). Obtained Ka/Ks ratio values among barley and three monocot species are shown in Figure 5A.
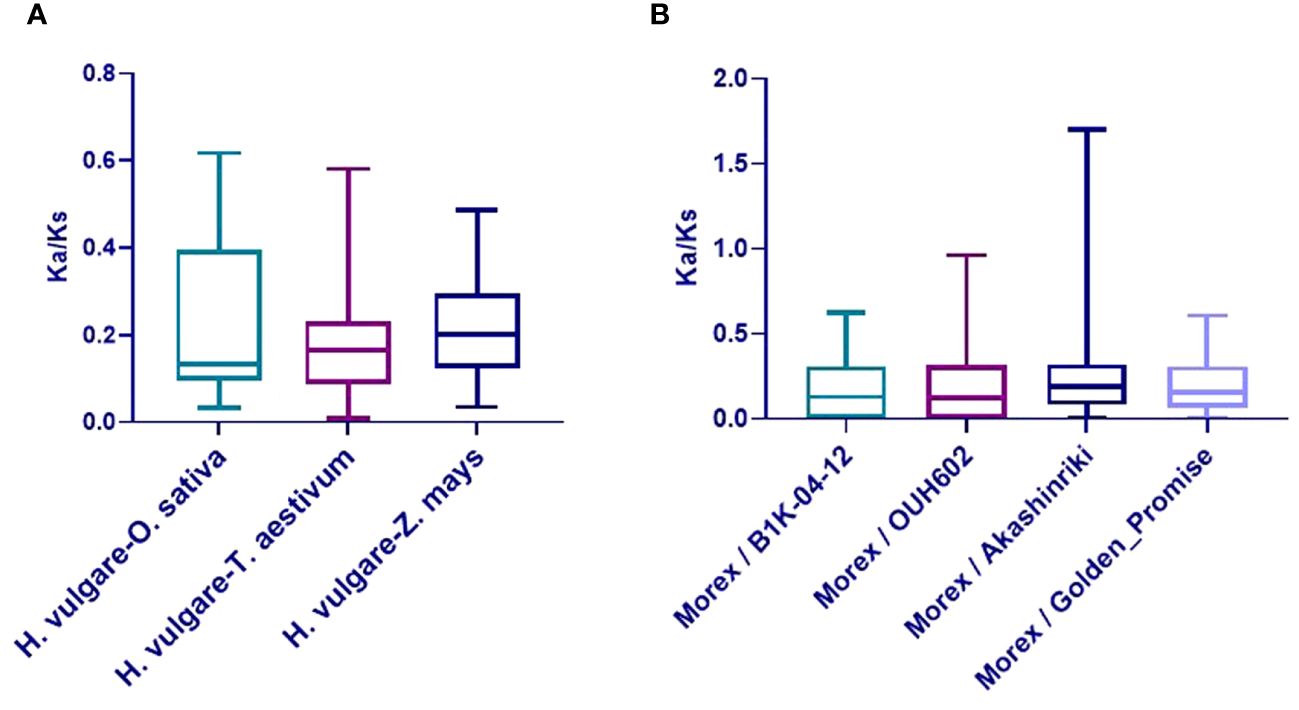
Figure 5 Boxplot of non-synonymous/synonymous substitutions (Ka/Ks) ratios in orthologous gene pairs. (A) Ka/Ks values of TALE orthologous gene pairs among barley (Hordeum vulgare) and three species (Oryza sativa, Zea mays, and Triticum aestivum). (B) Ka/Ks values of barley TALE homologous gene pairs among Morex and four barley accessions (Akashinriki, Golden_Promise, B1K-04–12, and OUH602).
Variations in TALE genes among five barley accessions (Morex, Akashinriki, Golden Promise, B1K-04–12, and OUH602) were investigated. Approximately 105 TALE family genes were identified from three cultivated and two wild accessions, with 21 genes in each (Supplementary Table 3). Ka/Ks values of barley TALE homologous gene pairs among Morex and four barley accessions were calculated to study selection pressure on the TALE superfamily during barley domestication (Supplementary Figure 3, Supplementary Table 7). Excepting BELL4 (Ka/Ks = 1.70) between Morex and Akashinriki, the Ka/Ks values were all <1, indicating that TALE superfamily genes are continuously evolving through purification selection. It is possible that BELL4 in Akashinriki (cultivated barley) had great artificial variation. Ka/Ks values are depicted in Figure 5B.
Protein–protein interactions are central mediators in biological processes. Clarifying interaction relationships among HvTALE proteins is important to characterize their potential functions and regulatory pathways. A protein interaction network among HvTALE proteins prepared using STRING software predicted eight HvTALE family members to interact with other proteins (Supplementary Table 8, Figure 6). Excepting HvBELL7, seven HvTALE proteins could interact with other HvTALE members. HvKNOX5 was central to the interaction network and interacted strongly with five HvTALE members, including one KNOX subfamily member (HvKNOX6) and four BELL subfamily members (HvBELL3, HvBELL5, HvBELL11, and HvBELL12). Additionally, the BELL subfamily member (HvBELL5) may interact with KNOX subfamily member HvKNOX6, and HvBELL3 may interact with HvKNOX1. This is consistent with previous studies to form KNOX–BELL heterodimer proteins.
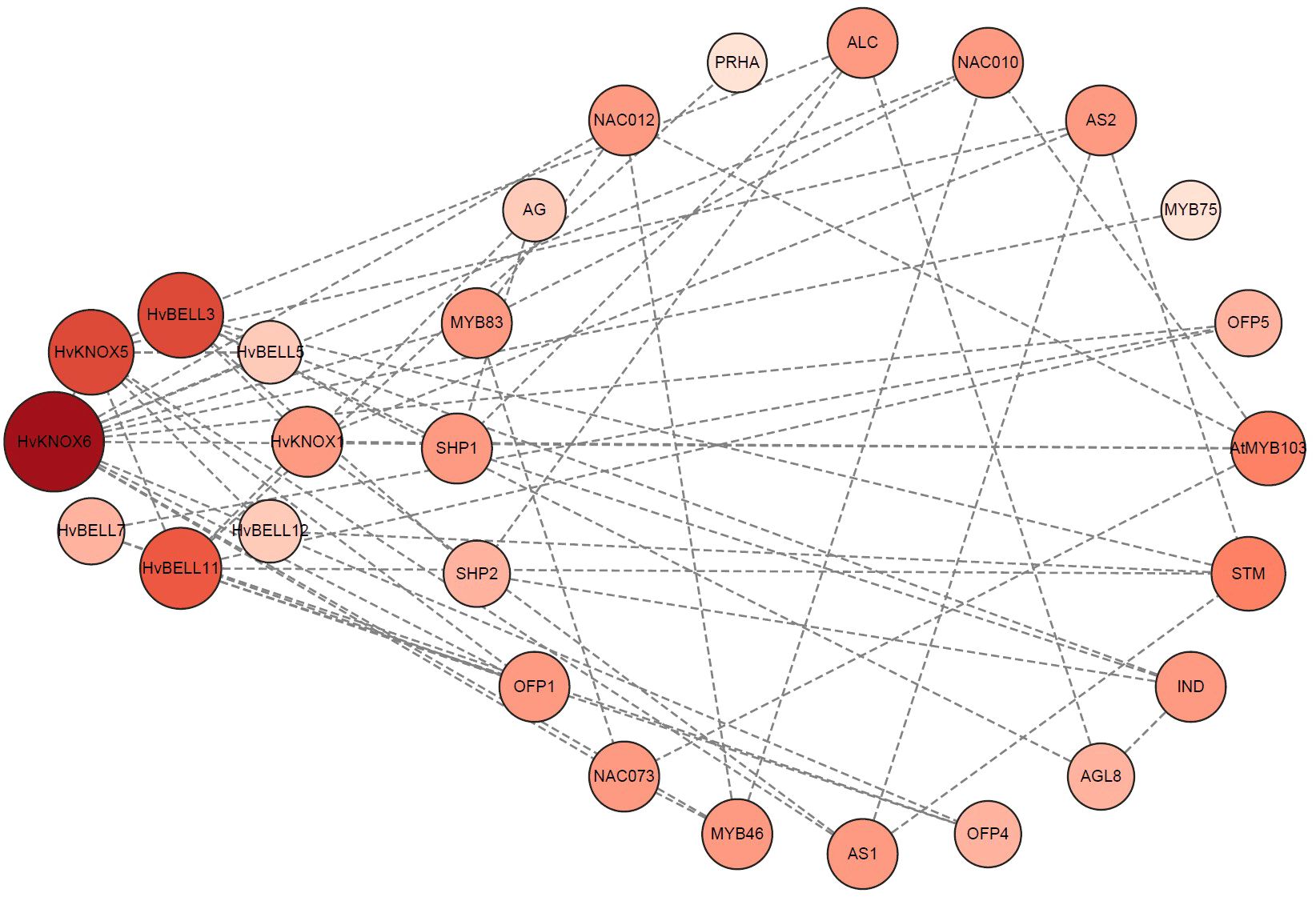
Figure 6 Predicted functional interaction networks of barley TALE proteins. The eight barley genes identified and marked in red are orthologs mapped using Arabidopsis genes. Medium confidence (0.400); 20 interactors.
On the basis of the HvTALE family interaction network, we selected HvKNOX6–HvKNOX5 and HvBELL11–HvKNOX5 protein interaction and simulated the spatial structure of their protein complexes. According to 3D structure modeling (Figure 7), HvKNOX5 and HvKNOX6 interact with each other through 11 hydrogen bonds and two salt bridges (Supplementary Figure 5A). The 3D modeling of the HvKNOX5–HvKNOX6 complex has a confidence score of 0.9775 (Supplementary Table 9). In the HvKNOX5–HvKNOX6 complex, the KNOX2 domain of the HvKNOX5 protein has one bonding site (Tyr174), and the C-terminal side of the HD domain has three consecutive hydrogen-bonding sites (Lys324, Arg325, and His326). HvKNOX5 and HvBELL11 interact with each other through 15 hydrogen bonds and one salt bridge (Supplementary Figure 5B). The 3D modeling of the HvKNOX5–HvBELL11 complex has a confidence score of 0.9763 (Supplementary Table 9). In the HvKNOX5–HvBELL11 complex, in addition to one hydrogen bonding site (Leu189) on the KNOX2 domain, there is another (Lys324) on the C-terminal side of the HD domain; the HvKNOX5 protein has two hydrogen bonding sites (Glu265 and Lys269) on the ELK domain and two (Ala303 and Glu306) on HD domains. HvKNOX5–HvKNOX6 and HvKNOX5–HvBELL11 complexes are formed through a hydrogen bonding site on the KNOX2 domain and a hydrogen bonding site on the C-terminal of the HD domain, which may be essential for the interaction between HvKNOX5 and other TALE superfamily members.
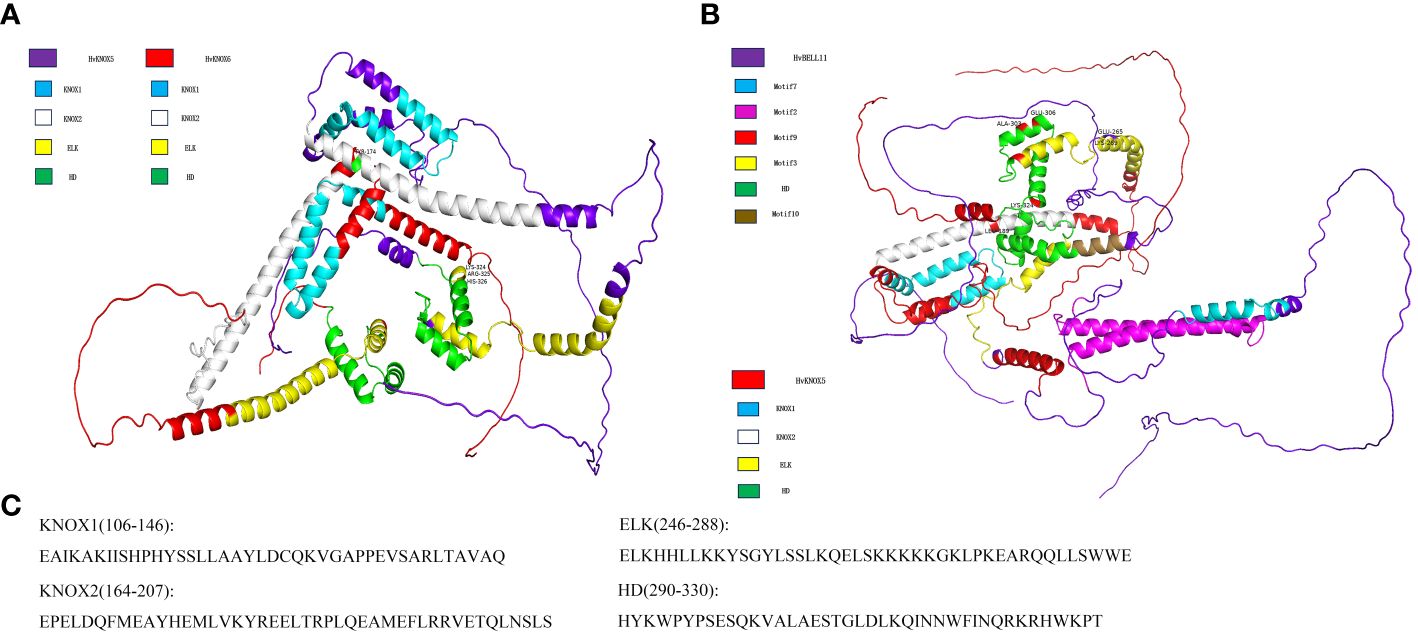
Figure 7 The 3D structure modeling of HvKNOX5–HvKNOX6 and HvKNOX5–HvBELL11 complexes. (A) HvKNOX5–HvKNOX6 complex with a hydrogen bonding site (Tyr174) on the KNOX2 domain of the HvKNOX5 protein and three consecutive bonding sites (Lys324, Arg325, and His326) on the C-terminal side of the HD domain of the HvKNOX5 protein. (B) HvKNOX5–HvBELL11 complex with a hydrogen bonding site (Leu189) on the KNOX2 domain of the HvKNOX5 protein, three hydrogen bonding sites (Ala303, Glu306, and Lys324) on the HD domain of the HvKNOX5 protein and two hydrogen bonding sites (Glu265 and Lys269) on the ELK domain of the HvKNOX5 protein. (C) KNOX1, KNOX2, ELK, and HD domain sequences of HvKNOX5.
cis-Elements are crucial for transcriptional control of gene expression (Wang et al., 2017). For cis-element studies, we extracted the HvTALE genes’ promoter region sequences, namely, the 2,000-bp upstream sequences from gene initiation codons (Supplementary Table 10). cis-Elements are proportionally displayed in Supplementary Figure 6. Interestingly, cis-elements with a wide distribution in gene promoter regions included light, hormone, defense, and stress responsiveness and anaerobic induction (Supplementary Table 11). The results also manifest that barley TALE members are probably linked to responses to plant hormones, such as abscisic acid (53), jasmonic acid methyl ester (76), salicylic acid (17), auxin (18), and gibberellin (23) (Figure 8). This means that the potential functions of HvTALE genes include responses to abiotic stressors and plant hormones and participation in various biological processes.
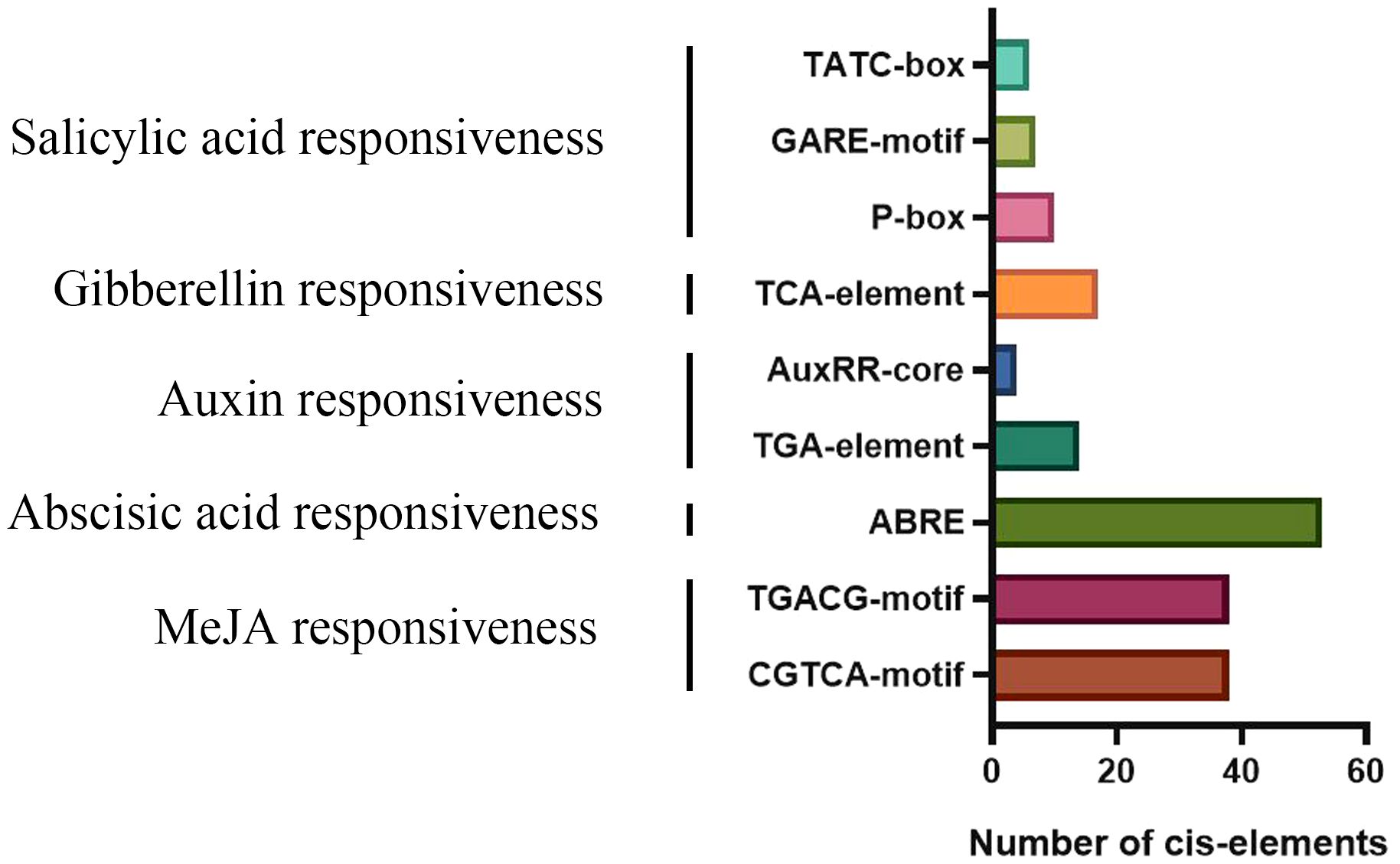
Figure 8 Number of plant hormone cis-elements detected in HvTALE genes.
Expression patterns of barley TALE genes were analyzed by generating a tissue-specific expression heatmap (Supplementary Table 1E, Figure 9). Four TALE genes show very low or no expression, with log2(FPKM+1) < 1 for all developmental stages; others were expressed with log2(FPKM+1) > 1 in at least one organ. Of them, transcription levels of HvKNOX8 (HORVU.MOREX.r3.6HG0568300.1), HvKNOX9 (HORVU.MOREX.r3.7HG0744200.1), HvBELL5 (HORVU.MOREX.r3.4HG0334360.1), and HvBELL10 (HORVU.MOREX.r3.5HG0420480.1) are high at all stages. Most HvTALE genes are expressed more strongly in the third internode of tillers than in the other organs. The expression of HvKNOX6 (HORVU.MOREX.r3.4HG0412370.1) was abundant in the third internode of tillers and lemma.
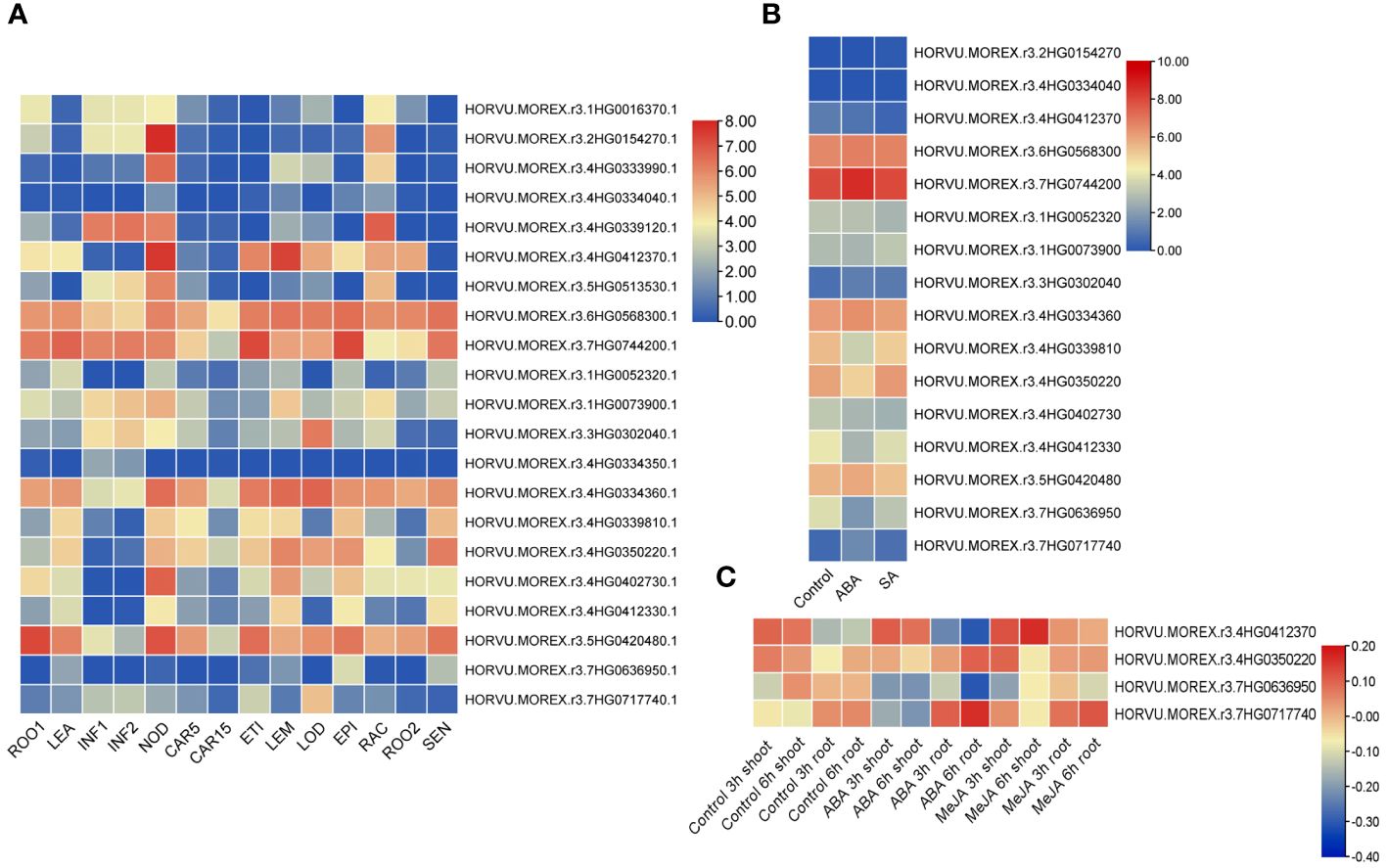
Figure 9 Expression heatmaps of HvTALE genes in different tissues and different responses to exogenous hormones. Expression heatmaps of HvTALE genes in (A) diverse tissues at different developmental stages [using profiles provided by the Leibniz Institute of Plant Genetics and Crop Plant Research (IPK)], (B) responses to ABA and SA [using Sequence Read Archive (SRA) samples], and (C) different responses to ABA and MeJA [using Gene Expression Omnibus (GEO) BioProject samples].
We also analyzed expression profiles of barley HvTALE genes responding to plant hormones based on public RNA-seq datasets. Based on mapping all reads from SRA and GEO datasets to the Morex v3 genome, we found that there were 16 reads in SRA datasets (ABA and SA treats) (Supplementary Table 1A) while only four reads in the GEO datasets (ABA and MeJA treats) (Supplementary Table 1C) mapping to TALE genes from the Morex v3 genome. From the overall trend, under ABA and SA stress, KNOX family members were basically unchanged compared with the control in 14-d leaves after seedling, and most BELL family members [e.g., HvBELL6 (HORVU.MOREX.r3.4HG0339810), HvBELL9 (HORVU.MOREX.r3.4HG0412330), and HvBELL11 (HORVU.MOREX.r3.7HG0636950)] were slightly downregulated (Figure 9). GEO sample data reveal that four genes in the development of shoots and roots exhibit expression changes under MeJA treatments (Figure 9), of which HvBELL12 (HORVU.MOREX.r3.7HG0717740), HvBELL7 (HORVU.MOREX.r3.4HG0350220), and HvKNOX6 (HORVU.MOREX.r3.4HG0412370) were upregulated compared with the control and HvBELL11 (HORVU.MOREX.r3.7HG0636950) was downregulated. Under ABA stress, HvKNOX6 (HORVU.MOREX.r3.4HG0412370) was upregulated in the shoot compared with the control, and three BELL family members [HvBELL12 (HORVU.MOREX.r3.7HG0717740), HvBELL7 (HORVU.MOREX.r3.4HG0350220), and HvBELL11 (HORVU.MOREX.r3.7HG0636950)] were downregulated. Additionally, HvBELL12 (HORVU.MOREX.r3.7HG0717740) and HvBELL7 (HORVU.MOREX.r3.4HG0350220) increased significantly in the roots in ABA treatments compared with the control, and HvKNOX6 (HORVU.MOREX.r3.4HG0412370) and HvBELL11 (HORVU.MOREX.r3.7HG0636950) genes showed much lower expressions.
Based on previous studies (Zhang et al., 2021; Han et al., 2022), we used qRT-PCR to detect expression levels under ABA, GA3, and 6-BA stress for four TALE genes that were relatively highly expressed in each tissue (Figure 10; Supplementary Table 12). When treated with ABA, the expression levels of HvKNOX6 and HvNOX8 decreased more significantly than those of their controls (the 0-h sample point). Compared with that of the control groups, the expression level of HvKNOX9 was downregulated at 3-h, 6-h, 9-h, and 24-h sample points but upregulated at 12 h. The expression level of HvBELL10 did not differ significantly from controls at 3 h, 6 h, 12 h, and 24 h but was significantly decreased at 9 h. For GA3 treatments, the expression levels of HvKNOX6 and HvNOX9 trended downward compared with controls, while those of HvBELL10 trended upward at all sample points. At 6 h, 9 h, and 12 h, the expression levels of HvNOX8 increased more significantly than control values. When treated with 6-BA, the expression level of HvKNOX6 also decreased more significantly than controls. The expression levels of HvNOX8 increased more significantly compared with controls at 12 h. The expression levels of HvNOX9 did not differ significantly from controls at 3 h, 9 h, and 24 h but increased significantly compared with its controls at 6 h and 12 h. The expression levels of HvBELL10 at 3 h, 9 h, and 24 h were significantly higher than for controls. For treatments of exogenous ABA, GA3, and 6-BA, three KNOX II genes showed relatively complicated expression patterns at 3 h, 6 h, 9 h, 12 h, and 24 h, possibly indicating that each gene had a different regulating function in barley (Matsumoto et al., 2014; Hartmann et al., 2022).
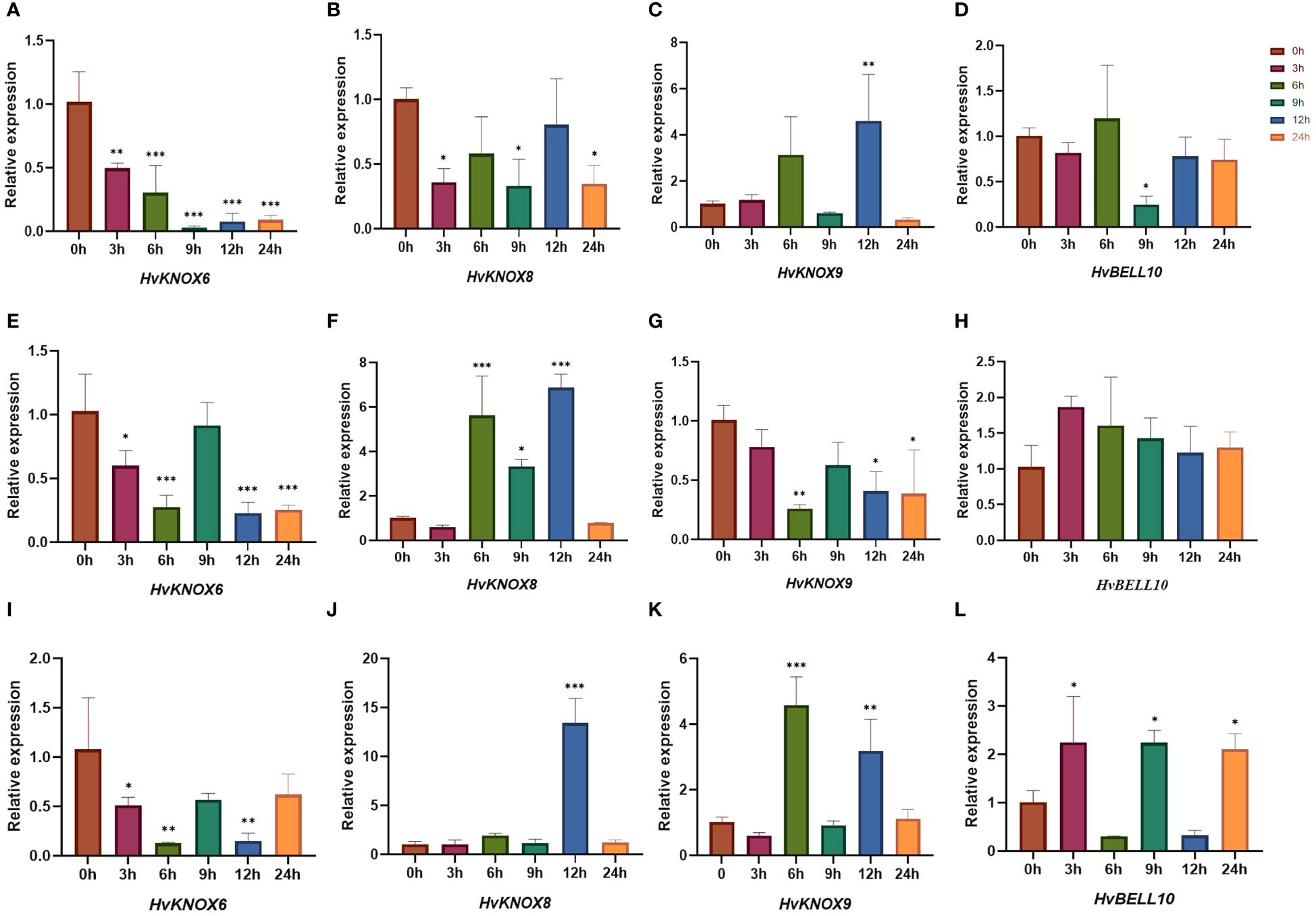
Figure 10 HvTALE gene expression patterns responding to exogenous hormones in seedling leaves. The expression patterns of (A) HvKNOX6, (B) HvKNOX8, (C) HvKNOX9, and (D) HvBELL10 in response to the ABA treatment. The expression patterns of (E) HvKNOX6, (F) HvKNOX8, (G) HvKNOX9, and (H) HvBELL10 in response to the GA3 treatment. The expression patterns of (I) HvKNOX6, (J) HvKNOX8, (K) HvKNOX9, and (L) HvBELL10 in response to the 6-BA treatment. Asterisks indicate significant differences from controls (0-h sample points) and others (3 h, 6 h, 9 h, 12 h, and 24 h): *p < 0.05, **p < 0.01, ***p < 0.001, Student’s t-test.
Members of the TALE superfamily are essential for regulating plant development, growth, and hormone responses. We comprehensively investigated the HvTALE gene family and provided perspectives into their biological functions. The number of TALE gene family members varies between taxa: Arabidopsis (22), poplar (35), soybean (68), and wheat (70) (Hamant and Pautot, 2010; Zhao et al., 2019; Wang et al., 2021; Han et al., 2022). In barley, the total number of TALE members identified (21) is close to that in diploid Arabidopsis but below that in polyploid plants (triploid poplar, tetraploid soybean, and hexaploid wheat). Therefore, we speculate that numbers of TALE superfamily genes are associated with species ploidy levels.
To explore the phylogeny and evolutionary relationships among TALE family genes, a phylogenetic tree comprising five species (four dicotyledons and one monocotyledon) was constructed. With reference to the classification in Arabidopsis, we divided barley TALE gene family members into KNOX and BELL-like subfamilies. In barley, KNOX subfamily members are further divided into class I KNOX and class II KNOX. The BELL-like subfamily has not been previously systematically classified. In terms of the conserved domains of HvTALE members, all HvTALE members contained HD domains. The BELL-like subfamily members harbored POX domains, but the KNOX subfamily had only KNOX1, KNOX2, and ELK domains (Müller et al., 2001; Cole et al., 2006). Our classification results are supported by the specific domains or motif combinations found in each HvTALE subfamily and hereditary class. Introns are essential for both evolution and the production of new gene family members (Roy and Penny, 2007). Investigating the evolution of HvTALE genes may be aided by understanding the intron distribution patterns of these genes. HvTALE gene structures are distinct in different subfamilies. All genes in the KNOX subfamily contain five exons, and most BEL1-like subfamily members contain four exons (except HvBELL4 and HvBELL7). Members of HvTALE may exhibit distinctive divergences and consistencies that indicate their functional differences and comparability, and members of the same TALE branch may perform comparable biological functions.
Segmental and tandem gene replication plays a driving role in the evolutionary expansion of plant gene families, and low-tandem, high segmental duplication classes are involved in various enzymatic functions (Cannon et al., 2004; Zhang et al., 2018). Gene duplication analysis revealed that most HvTALE genes had originated from segmental duplications, further identifying the crucial role that these segmental duplications play in barley TALE gene family expansion. Syntenic gene pairs among species may be useful for evolutionary research on the gene family (Han et al., 2022). To further investigate evolutionary clues from HvTALE genes, three dicotyledons and three monocotyledons were recruited for syntenic analysis (Figure 4). More HvTALE orthologous genes were detected in monocotyledons than in dicotyledons, and barley and wheat had the most syntenic pairs. This indicates that syntenies among TALE genes may parallel the evolutionary divergence of species. Important homologous pairs are often highly conserved, and they may exist before taxonomic differentiation (Xie et al., 2018; Wang et al., 2020). We report HvBELL2 and HvBELL3 to show homologous pairing in dicotyledonous and monocotyledonous, indicating that this conserved pairing may have existed before monocotyledons separated from dicotyledons.
Selective pressure at the protein level is usually measured by the non-synonymous/synonymous rate ratio (Ka/Ks), with Ka/Ks < 1, Ka/Ks = 1, and Ka/Ks > 1 indicating purifying (or negative) selection, neutral evolution, and diversifying (or positive) selection, respectively (Anisimova et al., 2001). Except for the BELL4 Ka/Ks value (1.70) between the Morex and Akashinriki varieties of barley, Ka/Ks values were all <1, indicating that the barley TALE superfamily member had undergone a strong purifying selection. The Ka/Ks value of BELL4 between the Morex and Akashinriki varieties was >1, possibly because BELL4 of Akashinriki had been subject to abnormal artificial mutation.
The protein interactions between members of the TALE gene family are widespread in plants. For example, the interaction between the KNAT3 protein in Arabidopsis and the BLH1 protein can influence how plants respond to ABA and may also indirectly influence how resilient plants are to adversity (Bhatt et al., 2004; Kim et al., 2013). Certain GhBEL1-like proteins in cotton interact with GhKNAT7 homologs to influence the network responsible for the formation of fiber SCWs (Ma et al., 2019). In protein interactions, some domains or patterns are crucial (Liu et al., 1999). The KNOX1 and KNOX2 domains comprise the MEINOX region. The KNOX1 domain is crucial for reducing the expression of the target gene, while the functionally essential KNOX2 domain is thought to be necessary for dimerization (Nagasaki et al., 2001); even the KNOX2 domain alone can interact with the BELL protein (Liu et al., 2014). In Arabidopsis, the MEINOX domain in KNOX proteins mediated BEL1 and particular KNOX protein interactions to form heterodimers (Bellaoui et al., 2001). KNATM proteins that have lost their HD domains also use the MEINOX domain to selectively bind with Arabidopsis BELL proteins (Magnani and Hake, 2008). The HD domain of MdKNOX15 in apple can also interact with MdBLH1 through yeast double hybridization (Jia et al., 2021a).
How KNOX–KNOX and KNOX–BELL interact in the TALE gene family may vary from species to species. Compared with KNOX proteins, there has been less research performed on BELL proteins, which contain an HD and MEINOX interacting domain composed of POX domains (Müller et al., 2001; Cole et al., 2006). In this study, through interaction network analysis, we report that barley HvKNOX5 has strong interactions with five HvTALE members (HvKNX6, HvBELL3, HvBELL5, HvBELL11, and HvBELL12). Accordingly, we chose HvKNOX5–HvKNOX6 and HvKNOX5–HvBELL11 complexes for protein interaction studies. In the HvKNOX5–HvKNOX6 complex, the KNOX2 domain of the HvKNOX5 protein has one hydrogen bonding site (Tyr174), and the HD domain has three consecutive hydrogen bonding sites (Lys324, Arg325, and His326). In the HvKNOX5–HvBELL11 complex, HvKNOX5 has two hydrogen bonding sites (Glu265 and Lys269) on the ELK domain, one hydrogen bonding site (Leu189) on the KNOX2 domain, and three hydrogen bonding sites (Ala303, Glu306, and Lys324) on the HD domain. The C-terminal of the MEINOX domain of KNOX3 that is the former name of HvKNOX5 in barley is necessary for KNOX–KNOX and KNOX–BELL complex interactions (Müller et al., 2001). We report the barley HvKNOX5 protein to have a hydrogen binding site on the KNOX2 domain, regardless of whether it binds to HvKNOX6 or HvBELL11.
Excepting in the third internode of tillers and developing grains 5 d and 15 d after anthesis, the expression level of KNOX I genes was relatively low in different barley developmental stages, consistent with results reporting KNOX I member expression in meristems (Hake et al., 2004; Hay and Tsiantis, 2010; Qi and Zheng, 2013). Additionally, KNOX genes control the metabolic processes and signaling pathways linked to several hormones, such as cytokinin, auxin, and gibberellin (Tsuda and Hake, 2015). Exogenous ethylene treatment could promote the downregulation of Bkn3, which has a 305-bp insertion sequence in the fourth intron of HvKNOX5 and change the phenotype of the barley hooded mutation (Osnato et al., 2010). In apple, MdKNOX19 (a class II KNOX gene) was significantly upregulated when ABA was applied to its seeds, leaves, and fruits (Jia et al., 2021b). We report many cis-elements in the 2,000-bp promoter sequence to respond to hormones, indicating that most TALE superfamily members may be inducible by exogenous phytohormones. Excepting HvKNOX9 at 6 h and 12 h, we report ABA to inhibit the expression of class II KNOX genes in barley leaves. From these SRA sample data, the expression of KNOX II family members under ABA stress is unchanged compared with control values. According to GEO sample data, the expression of the KNOX II family member HvKNOX6 differs in shoots and roots under ABA stress. Accordingly, we conclude that barley KNOX II subfamily members may have different expressions in different growth periods, tissues, and organs in ABA treatments. Class KNOX I genes are TFs that help preserve meristem identity by inhibiting GA production and stimulating cytokinin synthesis (Hay and Tsiantis, 2010). Compared with controls, we report the expression of HvKNOX6 and HvKNOX9 to trend downward and for HvKNOX8 to trend upward with GA3 treatment. Therefore, the expression of class II KNOX genes responds differently to GA3. In Arabidopsis, class KNOX I genes can be mediated by cytokinin to promote cell division and maintain meristem (Jasinski et al., 2005). Although we found no CK-responsive cis-elements in the promoter sequence of the barley KNOX family, qRT-PCR results indicate that cytokinin (6-BA) also has a regulatory effect on class II KNOX genes in barley. Therefore, we assume that CK regulates the expression of KNOX II genes by regulating other genes.
Few studies have examined interactions between BELL genes and hormones. We report HvBELL11 to be downregulated compared with control values under ABA, SA, and MeJA stress. From the interaction network among HvTALE proteins, HvBELL11 may interact with HvKNOX5. Given that HvKNOX5 plays an important regulatory role in the formation of barley awns (Osnato et al., 2010), the regulatory relationship between them warrants study in specific barley tissues (such as awns).
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
T-JL: Conceptualization, Data curation, Investigation, Writing – original draft, Writing – review & editing. TH: Formal analysis, Investigation, Software, Writing – original draft. H-YX: Funding acquisition, Writing – original draft, Writing – review & editing. J-CD: Data curation, Investigation, Writing – original draft. J-ZM: Data curation, Investigation, Writing – original draft. M-YD: Data curation, Investigation, Writing – original draft. R-JD: Conceptualization, Funding acquisition, Investigation, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The National Natural Science Foundation of China provided funding for this research (32260087) and the Fundamental Project for “Kunlun Talent—Top Innovation and Entrepreneurship Talents” of Qinghai Province funded this study.
We appreciate the help of Ruiyuan Biotechnology Co., Ltd. (Nanjing, China) in protein spatial structure simulation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1421702/full#supplementary-material
Anisimova, M., Bielawski, J. P., Yang, Z. (2001). Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol. Biol. Evol. 18, 1585–1592. doi: 10.1093/oxfordjournals.molbev.a003945
Artimo, P., Jonnalagedda, M., Arnold, K., Baratin, D., Csardi, G., de Castro, E., et al. (2012). ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 40, W597–W603. doi: 10.1093/nar/gks400
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME Suite: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Bellaoui, M., Pidkowich, M. S., Samach, A., Kushalappa, K., Kohalmi, S. E., Modrusan, Z., et al. (2001). The Arabidopsis BELL1 and KNOX TALE homeodomain proteins interact through a domain conserved between plants and animals. Plant Cell 13, 2455–2470. doi: 10.1105/tpc.010161
Belles-Boix, E., Hamant, O., Witiak, S. M., Morin, H., Traas, J., Pautot, V. (2006). KNAT6: an Arabidopsis homeobox gene involved in meristem activity and organ separation. Plant Cell 18, 1900–1907. doi: 10.1105/tpc.106.041988
Bhatt, A. M., Etchells, J. P., Canales, C., Lagodienko, A., Dickinson, H. (2004). VAAMANA—a BEL1-like homeodomain protein, interacts with KNOX proteins BP and STM and regulates inflorescence stem growth in Arabidopsis. Gene 328, 103–111. doi: 10.1016/j.gene.2003.12.033
Billeter, M., Qian, Y. Q., Otting, G., Müller, M., Gehring, W., Wüthrich, K. (1993). Determination of the nuclear magnetic resonance solution structure of an Antennapedia homeodomain-DNA complex. J. Mol. Biol. 234, 1084–1097. doi: 10.1006/jmbi.1993.1661
Bolser, D. M., Staines, D. M., Perry, E., Kersey, P. J. (2017). “Ensembl plants: integrating tools for visualizing, mining, and analyzing plant genomic data,” in Plant Genomics Databases: Methods and Protocols. Ed. van Dijk, A. D. J. (Springer New York, New York, NY), 1–31.
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D., May, G. (2004). The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 4, 10. doi: 10.1186/1471-2229-4-10
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cnops, G., Neyt, P., Raes, J., Petrarulo, M., Nelissen, H., Malenica, N., et al. (2006). The TORNADO1 and TORNADO2 genes function in several patterning processes during early leaf development in Arabidopsis thaliana. Plant Cell 18, 852–866. doi: 10.1105/tpc.105.040568
Cole, M., Nolte, C., Werr, W. (2006). Nuclear import of the transcription factor SHOOT MERISTEMLESS depends on heterodimerization with BLH proteins expressed in discrete sub-domains of the shoot apical meristem of Arabidopsis thaliana. Nucleic Acids Res. 34, 1281–1292. doi: 10.1093/nar/gkl016
Gu, Z., Cavalcanti, A., Chen, F. C., Bouman, P., Li, W. H. (2002). Extent of gene duplication in the genomes of Drosophila, nematode, and yeast. Mol. Biol. Evol. 19, 256–262. doi: 10.1093/oxfordjournals.molbev.a004079
Hake, S., Smith, H. M., Holtan, H., Magnani, E., Mele, G., Ramirez, J. (2004). The role of knox genes in plant development. Annu. Rev. Cell Dev. Biol. 20, 125–151. doi: 10.1146/annurev.cellbio.20.031803.093824
Hamant, O., Pautot, V. (2010). Plant development: a TALE story. C R Biol. 333, 371–381. doi: 10.1016/j.crvi.2010.01.015
Han, Y., Zhang, L., Yan, L., Xiong, X., Wang, W., Zhang, X. H., et al. (2022). Genome-wide analysis of TALE superfamily in Triticum aestivum reveals TaKNOX11-A is involved in abiotic stress response. BMC Genomics 23, 89. doi: 10.1186/s12864-022-08324-y
Hartmann, A., Berkowitz, O., Whelan, J., Narsai, R. (2022). Cross-species transcriptomic analyses reveals common and opposite responses in Arabidopsis, rice and barley following oxidative stress and hormone treatment. BMC Plant Biol. 22, 62. doi: 10.1186/s12870-021-03406-7
Hay, A., Tsiantis, M. (2010). KNOX genes: versatile regulators of plant development and diversity. Development 137, 3153–3165. doi: 10.1242/dev.030049
Hu, B., Jin, J., Guo, A. Y., Zhang, H., Luo, J., Gao, G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–1297. doi: 10.1093/bioinformatics/btu817
Jasinski, S., Piazza, P., Craft, J., Hay, A., Woolley, L., Rieu, I., et al. (2005). KNOX action in Arabidopsis is mediated by coordinate regulation of cytokinin and gibberellin activities. Curr. Biol. 15, 1560–1565. doi: 10.1016/j.cub.2005.07.023
Jayakodi, M., Padmarasu, S., Haberer, G., Bonthala, V. S., Gundlach, H., Monat, C., et al. (2020). The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 588, 284–289. doi: 10.1038/s41586-020-2947-8
Jia, P., Xing, L., Zhang, C., Chen, H., Li, Y., Zhang, D., et al. (2021a). MdKNOX15, a class I knotted-like transcription factor of apple, controls flowering and plant height by regulating GA levels through promoting the MdGA2ox7 transcription. Environ. Exp. Bot. 185, 104411. doi: 10.1016/j.envexpbot.2021.104411
Jia, P., Xing, L., Zhang, C., Zhang, D., Ma, J., Zhao, C., et al. (2021b). MdKNOX19, a class II knotted-like transcription factor of apple, plays roles in ABA signalling/sensitivity by targeting ABI5 during organ development. Plant Sci. 302, 110701. doi: 10.1016/j.plantsci.2020.110701
Jin, J., Tian, F., Yang, D. C., Meng, Y. Q., Kong, L., Luo, J., et al. (2017). PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, D1040–d1045. doi: 10.1093/nar/gkw982
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi: 10.1038/s41586-021-03819-2
Kim, D., Cho, Y. H., Ryu, H., Kim, Y., Kim, T. H., Hwang, I. (2013). BLH1 and KNAT3 modulate ABA responses during germination and early seedling development in Arabidopsis. Plant J. 75, 755–766. doi: 10.1111/tpj.12236
Kondhare, K. R., Vetal, P. V., Kalsi, H. S., Banerjee, A. K. (2019). BEL1-like protein (StBEL5) regulates CYCLING DOF FACTOR1 (StCDF1) through tandem TGAC core motifs in potato. J. Plant Physiol. 241, 153014. doi: 10.1016/j.jplph.2019.153014
Kong, X., Lv, W., Zhang, D., Jiang, S., Zhang, S., Li, D. (2013). Genome-wide identification and analysis of expression profiles of maize mitogen-activated protein kinase kinase kinase. PloS One 8, e57714. doi: 10.1371/journal.pone.0057714
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, R., Kushalappa, K., Godt, D., Pidkowich, M. S., Pastorelli, S., Hepworth, S. R., et al. (2007). The Arabidopsis BEL1-LIKE HOMEODOMAIN proteins SAW1 and SAW2 act redundantly to regulate KNOX expression spatially in leaf margins. Plant Cell 19, 2719–2735. doi: 10.1105/tpc.106.048769
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Li, E., Bhargava, A., Qiang, W., Friedmann, M. C., Forneris, N., Savidge, R. A., et al. (2012). The Class II KNOX gene KNAT7 negatively regulates secondary wall formation in Arabidopsis and is functionally conserved in Populus. New Phytol. 194, 102–115. doi: 10.1111/j.1469-8137.2011.04016.x
Lin, T., Sharma, P., Gonzalez, D. H., Viola, I. L., Hannapel, D. J. (2013). The impact of the long-distance transport of a BEL1-like messenger RNA on development. Plant Physiol. 161, 760–772. doi: 10.1104/pp.112.209429
Liu, M., Li, Y., Ma, Y., Zhao, Q., Stiller, J., Feng, Q., et al. (2020). The draft genome of a wild barley genotype reveals its enrichment in genes related to biotic and abiotic stresses compared to cultivated barley. Plant Biotechnol. J. 18, 443–456. doi: 10.1111/pbi.13210
Liu, L., White, M. J., MacRae, T. H. (1999). Transcription factors and their genes in higher plants functional domains, evolution and regulation. Eur. J. Biochem. 262, 247–257. doi: 10.1046/j.1432-1327.1999.00349.x
Liu, Y., You, S., Taylor-Teeples, M., Li, W. L., Schuetz, M., Brady, S. M., et al. (2014). BEL1-LIKE HOMEODOMAIN6 and KNOTTED ARABIDOPSIS THALIANA7 interact and regulate secondary cell wall formation via repression of REVOLUTA. Plant Cell 26, 4843–4861. doi: 10.1105/tpc.114.128322
Ma, Q., Wang, N., Hao, P., Sun, H., Wang, C., Ma, L., et al. (2019). Genome-wide identification and characterization of TALE superfamily genes in cotton reveals their functions in regulating secondary cell wall biosynthesis. BMC Plant Biol. 19, 432. doi: 10.1186/s12870-019-2026-1
Magnani, E., Hake, S. (2008). KNOX lost the OX: the Arabidopsis KNATM gene defines a novel class of KNOX transcriptional regulators missing the homeodomain. Plant Cell 20, 875–887. doi: 10.1105/tpc.108.058495
Mascher, M., Wicker, T., Jenkins, J., Plott, C., Lux, T., Koh, C. S., et al. (2021). Long-read sequence assembly: a technical evaluation in barley. Plant Cell 33, 1888–1906. doi: 10.1093/plcell/koab077
Matsumoto, T., Morishige, H., Tanaka, T., Kanamori, H., Komatsuda, T., Sato, K., et al. (2014). Transcriptome analysis of barley identifies heat shock and HD-Zip I transcription factors up-regulated in response to multiple abiotic stresses. Mol. Breed. 34, 761–768. doi: 10.1007/s11032-014-0048-9
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–d419. doi: 10.1093/nar/gkaa913
Monat, C., Padmarasu, S., Lux, T., Wicker, T., Gundlach, H., Himmelbach, A., et al. (2019). TRITEX: chromosome-scale sequence assembly of Triticeae genomes with open-source tools. Genome Biol. 20, 284. doi: 10.1186/s13059-019-1899-5
Müller, J., Wang, Y., Franzen, R., Santi, L., Salamini, F., Rohde, W. (2001). In vitro interactions between barley TALE homeodomain proteins suggest a role for protein-protein associations in the regulation of Knox gene function. Plant J. 27, 13–23. doi: 10.1046/j.1365-313x.2001.01064.x
Nagasaki, H., Sakamoto, T., Sato, Y., Matsuoka, M. (2001). Functional analysis of the conserved domains of a rice KNOX homeodomain protein, OSH15. Plant Cell 13, 2085–2098. doi: 10.1105/TPC.010113
Ohue, M., Shimoda, T., Suzuki, S., Matsuzaki, Y., Ishida, T., Akiyama, Y. (2014). MEGADOCK 4.0: an ultra–high-performance protein–protein docking software for heterogeneous supercomputers. Bioinformatics 30, 3281–3283. doi: 10.1093/bioinformatics/btu532
Osnato, M., Stile, M. R., Wang, Y., Meynard, D., Curiale, S., Guiderdoni, E., et al. (2010). Cross talk between the KNOX and ethylene pathways is mediated by intron-binding transcription factors in barley. Plant Physiol. 154, 1616–1632. doi: 10.1104/pp.110.161984
Pautot, V., Dockx, J., Hamant, O., Kronenberger, J., Grandjean, O., Jublot, D., et al. (2001). KNAT2: evidence for a link between knotted-like genes and carpel development. Plant Cell 13, 1719–1734. doi: 10.1105/TPC.010184
Proveniers, M., Rutjens, B., Brand, M., Smeekens, S. (2007). The Arabidopsis TALE homeobox gene ATH1 controls floral competency through positive regulation of FLC. Plant J. 52, 899–913. doi: 10.1111/j.1365-313X.2007.03285.x
Qi, B., Zheng, H. (2013). Modulation of root-skewing responses by KNAT1 in Arabidopsis thaliana. Plant J. 76, 380–392. doi: 10.1111/tpj.12295
Roy, S. W., Penny, D. (2007). A very high fraction of unique intron positions in the intron-rich diatom Thalassiosira pseudonana indicates widespread intron gain. Mol. Biol. Evol. 24, 1447–1457. doi: 10.1093/molbev/msm048
Rutjens, B., Bao, D., van Eck-Stouten, E., Brand, M., Smeekens, S., Proveniers, M. (2009). Shoot apical meristem function in Arabidopsis requires the combined activities of three BEL1-like homeodomain proteins. Plant J. 58, 641–654. doi: 10.1111/j.1365-313X.2009.03809.x
Shani, E., Yanai, O., Ori, N. (2006). The role of hormones in shoot apical meristem function. Curr. Opin. Plant Biol. 9, 484–489. doi: 10.1016/j.pbi.2006.07.008
Tao, Y., Chen, M., Shu, Y., Zhu, Y., Wang, S., Huang, L., et al. (2018). Identification and functional characterization of a novel BEL1-LIKE homeobox transcription factor GmBLH4 in soybean. Plant Cell Tissue Organ Culture (PCTOC) 134, 331–344. doi: 10.1007/s11240-018-1419-4
Tsuda, K., Hake, S. (2015). Diverse functions of KNOX transcription factors in the diploid body plan of plants. Curr. Opin. Plant Biol. 27, 91–96. doi: 10.1016/j.pbi.2015.06.015
Vernié, T., Moreau, S., de Billy, F., Plet, J., Combier, J. P., Rogers, C., et al. (2008). EFD Is an ERF transcription factor involved in the control of nodule number and differentiation in Medicago truncatula. Plant Cell 20, 2696–2713. doi: 10.1105/tpc.108.059857
Vollbrecht, E., Veit, B., Sinha, N., Hake, S. (1991). The developmental gene Knotted-1 is a member of a maize homeobox gene family. Nature 350, 241–243. doi: 10.1038/350241a0
Voorrips, R. E. (2002). MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered 93, 77–78. doi: 10.1093/jhered/93.1.77
Wang, L., Ding, X., Gao, Y., Yang, S. (2020). Genome-wide identification and characterization of GRAS genes in soybean (Glycine max). BMC Plant Biol. 20, 415. doi: 10.1186/s12870-020-02636-5
Wang, W., Jiang, W., Liu, J., Li, Y., Gai, J., Li, Y. (2017). Genome-wide characterization of the aldehyde dehydrogenase gene superfamily in soybean and its potential role in drought stress response. BMC Genomics 18, 518. doi: 10.1186/s12864-017-3908-y
Wang, Y., Li, J., Paterson, A. H. (2013). MCScanX-transposed: detecting transposed gene duplications based on multiple colinearity scans. Bioinformatics 29, 1458–1460. doi: 10.1093/bioinformatics/btt150
Wang, L., Yang, X., Gao, Y., Yang, S. (2021). Genome-wide identification and characterization of TALE superfamily genes in soybean (Glycine max L.). Int. J. Mol. Sci. 22, 4117. doi: 10.3390/ijms22084117
Xie, T., Chen, C., Li, C., Liu, J., Liu, C., He, Y. (2018). Genome-wide investigation of WRKY gene family in pineapple: evolution and expression profiles during development and stress. BMC Genomics 19, 490. doi: 10.1186/s12864-018-4880-x
Yang, M., Derbyshire, M. K., Yamashita, R. A., Marchler-Bauer, A. (2020). NCBI’s conserved domain database and tools for protein domain analysis. Curr. Protoc. Bioinf. 69, e90. doi: 10.1002/cpbi.90
Yu, Y. (2019). OsKNAT7 bridges secondary cell wall formation and cell growth regulation. Plant Physiol. 181, 385–386. doi: 10.1104/pp.19.01018
Yu, J., Wang, J., Lin, W., Li, S., Li, H., Zhou, J., et al. (2005). The Genomes of Oryza sativa: a history of duplications. PloS Biol. 3, e38. doi: 10.1371/journal.pbio.0030038
Zhang, B., Liu, J., Yang, Z. E., Chen, E. Y., Zhang, C. J., Zhang, X. Y., et al. (2018). Genome-wide analysis of GRAS transcription factor gene family in Gossypium hirsutum L. BMC Genomics 19, 348. doi: 10.1186/s12864-018-4722-x
Zhang, S., Pan, Y., Zhi, C., Zheng, Y., Wang, X., Li, X., et al. (2021). Genome-wide identification and characterization of KNOTTED-like homeobox (KNOX) homologs in garlic (Allium sativum L.) and their expression profilings responding to exogenous cytokinin and gibberellin. Int. J. Mol. Sci. 22, 9237. 10.3390/ijms22179237
Zhao, W., Liu, Y.-W., Zhou, J.-M., Zhao, S.-P., Zhang, X.-H., Min, D.-H. (2016). Genome-wide analysis of the lectin receptor-like kinase family in foxtail millet (Setaria italica L.). Plant Cell Tissue Organ Culture (PCTOC) 127, 335–346. doi: 10.1007/s11240-016-1053-y
Keywords: barley, TALE, gene family, KNOX, BELL, hormones treatment, growth and development
Citation: Liao T-j, Huang T, Xiong H-y, Duo J-c, Ma J-z, Du M-y and Duan R-j (2024) Genome-wide identification, characterization, and evolutionary analysis of the barley TALE gene family and its expression profiles in response to exogenous hormones. Front. Plant Sci. 15:1421702. doi: 10.3389/fpls.2024.1421702
Received: 22 April 2024; Accepted: 07 June 2024;
Published: 27 June 2024.
Edited by:
Maoteng Li, Huazhong University of Science and Technology, ChinaCopyright © 2024 Liao, Huang, Xiong, Duo, Ma, Du and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rui-jun Duan, cnVpanVuZHVhbkAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.