 Xionghui Bai
Xionghui Bai Pengfei Qiao
Pengfei Qiao Hanxiao Liu
Hanxiao Liu Jie Guo
Jie Guo Keli Dai
Keli Dai- College of Agronomy, Key Laboratory of Sustainable Dryland Agriculture (Co-construction by Ministry and Province), Ministry of Agriculture and Rural Affairs, Shanxi Agricultural University, Jinzhong, China
Introduction: Wheat (Triticum aestivum L.) is among themost important crop worldwide. Given a growing population and changing climate, enhancing wheat yield is of great importance. Yield is closely associated with flower and spike development, and E-class genes play important roles in the flower and kernel development of plants. Currently, the absence of systematic analysis on the E gene family hinders our comprehension of their roles in plant growth and development.
Methods: Identify E-class genes based on homologous sequence searches. Analyze the identified E-class genes through a series of gene family analyses. Determine the expression levels of wheat E-class genes by searching public databases. Validate the functions of these genes by transforming them into Arabidopsis. Finally, determine the interactions between the genes through yeast two-hybrid experiments.
Results: Fifteen E-class genes (TaEs) were identified in common wheat. Nine E-class genes were detected in five ancestral/closely related species, including one in Aegilops tauschii (AtE), one in T. Urartu (TuEs), two in T. turgidum (TtEs), two in T. dicoccoides (TdEs), and three in T. spelta (TsEs). The 24 E-class genes were classified into three subgroups using a phylogenetic approach. All genes were highly expressed in spikes, and most were only highly expressed at the floret meristem stage. The effects of TaSEP5-A on flowering and growth cycles were confirmed in homologous mutants and transgenic Arabidopsis thaliana. The E-class genes were able to regulate the growth cycle of Arabidopsis. Finally, we confirmed the interactions between TaSEP5-A and other wheat E-class genes based on yeast two-hybrid assays.
Discussion: Our findings provide information regarding the E-class genes in wheat and will potentially promote the application of these genes in wheat improvement.
1 Introduction
Wheat (Triticum aestivum L.) is a vital cereal crop cultivated worldwide. It serves as a major source of daily caloric intake for over one-fifth of the global population and accounts for approximately 28% of global cereal production (Tu and Li, 2020). The projected global population will reach 9.9 billion by 2050 (Population Reference Bureau, 2020; https://www.stats.gov.cn/). An annual growth rate of 2% in wheat production is required to meet the increasing demand. Consequently, a comprehensive understanding of the genes associated with functional traits assumes critical significance in the molecular breeding of wheat, presenting substantial implications for yield and quality enhancement.
Flower development has been the focus of extensive research, particularly in dicotyledonous plants such as Arabidopsis thaliana and Plantago asiatica (Jack, 2004). Those investigations have contributed to our overall comprehension of floral organ development in higher plants, leading to the proposition of the ABCDE model, which delineates the classification of plant floral organs based on the functions of A-, B-, C-, D-, and E-class genes (Zahn et al., 2006). According to the ABCDE model, the first floral whorl is specified by the A- and E-class genes, governing sepal development. In the subsequent whorl, petal development is regulated by the A-, B-, and E-class genes. The third whorl is marked by the actions of B-, C-, and E-class genes, determining stamen formation. In the fourth whorl, carpel differentiation is controlled by the C- and E-class genes. Finally, the D- and E-class genes are responsible for ovule development within the pistil.
Notably, the E-class genes play crucial roles in the morphological construction of all floral organs. In Arabidopsis, these genes encompass four members: SEPALLATA1 (SEP1), SEP2, SEP3, and SEP4, which display partially redundant functions in the development of petals, stamens, and carpels (Ditta et al., 2005; Honma and Goto, 2001; Pelaz et al., 2000). Through the course of evolution, the diversification of the E-class genes has contributed to substantial changes in floral morphology among terrestrial plants (Irish and Litt, 2005). Studies on monocotyledonous plants like rice (Oryza sativa L.), similar to those on common wheat, have categorized the E-class genes into SEP and OsMADS1 clades. The simultaneous knockout of all the four rice SEP genes resulted in the homologous transformation of all floral organs, except for the lemmas, giving rise to leaf-like structures. Conversely, the OsMADS1 mutant phenotype in rice exhibited leaf-like lemmas and paleas (Agrawal et al., 2005; Malcomber and Kellogg, 2004). Moreover, duplication of the E-class genes potentially contributes to the increased complexity of regulatory mechanisms underlying floral organ formation (Cui et al., 2010). Therefore, the identification and functional analysis of the E-class genes in crops is essential for improving our understanding of floral development processes in these plants, thereby enhancing crop yields.
Spike development plays a crucial role in determining wheat yield (Kellogg et al., 2001). A wheat spike is composed of multiple florets (Kong et al., 2022). In common wheat, the process of inflorescence development occurs in distinct stages. Initially, the inflorescence meristem forms a specialized structure known as the spikelet meristem, which is found in the axils of leaves. Subsequently, the spikelet meristem differentiates into floret meristems, giving rise to floral organs (Sakuma et al., 2019). Each spike typically contains six to eight florets, with some located at the top. Internally, each floret comprises protective structures called the lemma and palea (Liu et al., 2021; Park et al., 2014). The lemma is derived from the axillary meristem, and the palea develops from the primordial leaf formed by the axillary meristem. A wheat flower consists of a pistil, three stamens, and two vestigial leaf-like structures called lodicules. The pistil is composed of three fused carpels, while the stamens consist of filaments and anthers. During flowering, the lodicules swell, which causes the lemma and pedicel to separate, facilitating the release of pollen from anthers onto the receptive stigma (Dobrovolskaya et al., 2015; Li et al., 2019). Overall, spike development in common wheat is closely linked to the development of floral organs, including the lemma, palea, stamens, and pistil (Ikeda et al., 2007; Ikeda-Kawakatsu et al., 2012). The proper establishment of these floral structures is regulated by the E-class genes, which play a critical role in spike and floral development (Yoshida et al., 2013).
Common wheat is an allohexaploid plant species with a genetic makeup of 2n = 6x = 42. It consists of three homoeologous sub-genomes (A, B, and D) (Dubcovsky and Dvorak, 2007; Lev-Yadun et al., 2000). This characteristic makes it an excellent model for studying sub-functionalization and polyploidy in plants (Mcfadden and Sears, 1946). Cultivated wheat as we know it today emerged approximately 8,500–9,000 years ago through hybridization events involving the domesticated tetraploid ancestors T. turgidum (AABB) and T. dicoccoides (AABB) and the diploid donor of the D sub-genome, Aegilops tauschii (DD) (Jia et al., 2013; Luo et al., 2017; Marcussen et al., 2014; Shewry and Hey, 2015). The identity of one diploid donor, T. urartu (A sub-genome), has been confirmed in one of the sub-genomes of the tetraploid ancestor (Avni et al., 2017; Maccaferri et al., 2019; Zhao et al., 2017). However, the exact origin of the B sub-genome, which is the cytoplasmic donor of common wheat, remains uncertain. It is either extinct, yet to be discovered, or part of a polyphyletic origin (Levy and Feldman, 2022; Li et al., 2021; Tsunewaki et al., 2019). Common wheat quickly spread globally from its origin in the Fertile Crescent, becoming a staple food for humans (Dubcovsky and Dvorak, 2007; Lémin et al., 2019; Mayrose et al., 2011; Mirzaghaderi and Mason, 2017). This expansion may have been facilitated by allopolyploidy, which promotes genetic variation, novel trait acquisition, intergenomic interactions, and mutational buffering, thus satisfying the yield and quality requirements of human agriculture (Murat et al., 2017; Rice et al., 2019; Salse et al., 2008; Wang et al., 2015).
Despite significant advancements in plant genomics, there remains a crucial research gap in understanding the comprehensive evolutionary and functional dynamics of E-class genes in wheat (Kong et al., 2022). In this study, we utilized machine learning algorithms to optimize the classification and functional prediction of genes (Homology Analysis, Domain Analysis, Gene Co-expression Network Analysis, etc.). Due to the highly specific functions of these genes, it is not possible to identify them using a single-domain Hidden Markov Model. Therefore, we employed a homologous gene search approach to identify these genes. We identified 15 E-class genes (TaEs) in common wheat, the result is consistent with previous research findings (Kong et al., 2022), but our approach treats E-class genes as a cohesive gene family, aimed at uncovering novel insights, one in A. tauschii (AtE), one in T. urartu (TuE), two in T. turgidum (TtEs), two in T. dicoccoides (TdEs), and three in T. spelta (TsEs). We conducted a comprehensive investigation into the evolutionary and structural characteristics of the TaEs through phylogenetic, gene domain and conserved motif, chromosomal distribution, and collinearity analyses. Furthermore, we determined expression patterns and functions of the TaEs using RNA-seq data and cis-element analysis. We performed Arabidopsis transformation experiments to validate the functions of the target genes. Finally, we confirmed the interactions among the E-class genes using a yeast two-hybrid assay. Our study fills this critical research gap by conducting a thorough gene family analysis of the E genes, providing novel insights into their evolutionary dynamics within wheat. By elucidating the evolutionary journey of E genes, we offer a unique perspective on the evolutionary history of wheat, contributing significantly to the fields of plant genetics and evolution research.
2 Materials and methods
2.1 Identification of the E-class genes in common wheat and its relatives
The protein sequences for the E-class genes of Arabidopsis and proteomic data of common wheat were obtained from the Ensembl Plant database (https://plants.ensembl.org/index.html). They were used to perform a BLASTP (Basic Local Alignment Search Tool Protein) search against the common wheat protein database using an Expect value (e value) of 10−5 (Lei et al., 2021). The same method was employed to determine the E-class genes of ancestral/closely related species of common wheat, including A. tauschii, T. dicoccoides, T. spelta, T. turgidum, and T. urartu). Values for molecular weight (Mw), isoelectric point (pI), instability index (II), aliphatic index (AI), and grand average of hydropathicity (GRAVY) of the identified E-class proteins were calculated using the ExPASy online software (http://web.expasy.org/protparam/) (Wilkins et al., 1999), whereas their subcellular localization was predicted using the Cell Ploc 2.0 online tool (http://www.csbio.sjtu.edu.cn) (Yang, 2005). Homologous gene pairs were identified based on chromosomal location and sequence similarity. Molecular selection effects were assessed based on calculations of the non-synonymous substitution/synonymous substitution (dN/dS) values using the codeml program in the PAML package (Guo et al., 2017). The genes with dN and dS values of 0 were eliminated, as this may be attributable to sequence saturation or sequence mismatches (Barberis et al., 2021). Genes with dN/dS values >1 were designated positively selected genes (PSGs), and those with dN/dS values <1 were considered negatively selected genes (NSGs).
2.2 Phylogenetic, motif, and domain analyses
To gain further understanding of the evolutionary relationships among the E-class genes, we compared the genes in Arabidopsis, rice, common wheat and its ancestral/closely related species (Larkin et al., 2007). Full-length alignment of the E-class gene sequences was performed using MUSCLE, and neighbor-joining (NJ) trees were constructed using the MEGA X software with a bootstrap value of 1,000 (Kumar et al., 2016). Conserved motifs were predicted for each E-class protein sequence using the MEME tool (http://meme-suite.org/), with the maximum number of motifs set to 10 (Bailey et al., 2015). Duplicates were eliminated, and all other parameters were set to default values. The conserved domains of the E-class proteins in common wheat and its ancestral/closely related species were identified using the NCBI-CDD (https://www.ncbi.nlm.nih.gov/cdd/) database (Lu et al., 2020; Mistry et al., 2021), and the results of motif and domain analyses were visualized using TBtools software (Chen et al., 2021).
2.3 Promoter analysis and miRNA prediction
The 2,000 base pair (bp) genomic DNA sequences upstream of the TaE transcription start sites were obtained from the common wheat reference genome IWGSC_v1.1 in the PlantCare database (http://bioinformatice.psb.ugent.be/webtools/plantcare/) (Lescot et al., 2002). They were used to identify cis-regulatory elements. cDNA sequences of the TaEs were used as candidate targets for predicting potential miRNAs. Using the psRNATarget Server (https://www.zhaolab.org/psRNATarget/) with default parameters, we then searched for candidate targets by screening the mature sequences of common wheat miRNAs obtained from the miRbase database (http://www.mirbase.org/) (Dai et al., 2018). The linkage of predicted miRNAs with corresponding target genes was displayed by Cytoscape software (Otasek et al., 2019).
2.4 Chromosomal distribution and collinearity analysis
Using the genome annotation files, we applied the Basic Circos program in TBtools to localize and visualize the identified E-class genes on chromosomes in the common wheat genome. The homology of the E-class genes in common wheat and its related wild species was then analyzed using the MCScanX and Dual Synteny Plotter tools (https://github.com/CJ-Chen/TBtools).
2.5 TaE expression and protein–protein interaction network analyses
To gain insight into the spatiotemporal expression patterns of the identified TaEs, we downloaded the expression data of the target genes in five wheat tissues (roots, stems, leaves, spikes, and kernels) at different stages of spike development from the WheatOmics (http://202.194.139.32/) database (Li et al., 2018; Ma et al., 2021; Pfeifer et al., 2014). Differentially expressed genes were identified based on the fragments per kilobase of transcript per million mapped reads values for each TaE, and expression levels were visualized by constructing a heatmap using the pheatmap R package. Then, the interaction networks were identified based on orthologous genes in wheat and Arabidopsis using the AraNetV2 (http://www.inetbio.org/aranet/) and STRING (http://string-db.org/cgi) databases, and the predicted interaction network was displayed with Cytoscape.
2.6 Cloning of target genes and transformation of Arabidopsis
Target gene sequences for PCR amplification were obtained from the Ensembl Plant database (https://plants.ensembl.org/index.html), and gene-specific primers were designed using Oligo 7 software (https://www.oligo.net/). The PCR reactions were carried out in a reaction mixture (20 μL) containing 10 μL of 2× Phanta Max MasterMix (Vazyme, Nanjing, China), 1.5 μL of cDNA, 0.5 μL each of forward and reverse primers, and 7.5 μL of enzyme-free water. The PCR amplification protocol consisted of an initial denaturation step at 98°C for 30 s, followed by 34 cycles of denaturation at 98°C for 10 s, annealing at 60°C for 20 s, and extension at 68°C for 1 min. A final extension step was performed at 68°C for 7 min. The resulting PCR products were separated on a 0.75% agarose gel by electrophoresis at 100 V for 30 min. The desired target bands were excised from the gel. The recovered PCR products were then ligated into the pMD™18-T vector (TaKaRa Bio, Kusatsu, Japan). The ligated products were subjected to restriction digestion with XbaIand KpnI(NEB, Massachusetts, American) and ligated into the overexpression vector pCAMBIA1300 (Abcam, Cambridge, UK). The recombinant vectors carrying target genes were transformed into Agrobacterium tumefaciens GV3101 cells (COOLABER, Beijing, China). The wild-type (WT) Arabidopsis Columbia ecotype and the AT3G61120 (TaSEP5-A homolog) mutant Arabidopsis (SALK_203944C) were used as the transformation recipients. Arabidopsis seedlings were cultivated under at 24°C with a 16-h light and 8-h dark photoperiod. Upon maturity, seeds were collected from transgenic Arabidopsis plants as the T0 generation and grown on half-strength Murashige and Skoog medium containing 50 µg/mL hygromycin. Subsequently, plants were transferred to the soil. T1 generation seeds were harvested and similarly cultivated until T3 generation seeds were obtained. The genetic function of the transgenic plants was verified using the T3 generation Arabidopsis. The WT Arabidopsis transformed T3 (WT-OE), WT, mutant (MT), and transformed mutant T3 (MT-OE) seeds were cultivated in the soil. Three plantings were performed at a weekly interval. The number of days to flowering and days to maturation in these lines were determined. The morphology of fully blooming Arabidopsis flowers was observed using a stereomicroscope (SZX7; Olympus, Tokyo, Japan).
2.7 Yeast two-hybrid assay
The cloning vectors for WLHS1-A, TaSEP3-A, TaSEP6-A, TaSEP4-A, and TaSEP4-B were constructed using the same methods used to construct the cloning vectors for the target genes. Gene TaSEP5-A was ligated into the bait vector (pGADT7), whereas WLHS1-A, TaSEP3-A, TaSEP6-A, TaSEP4-A, and TaSEP4-B were ligated separately into respective prey vectors (pGBKT7). To ensure the accuracy of the experiment, we also generated several control vectors. The pGBKT-53 and pGADT7 vectors were used to co-transform yeast as positive controls. Combinations of constructed bait and blank prey vectors, as well as constructed prey and blank bait vectors, were used as negative controls. Assays involved pairing each co-transforming yeast with different combinations of each constructed bait and prey vector. The transformed yeasts were cultured on double-dropout (SD/-His-Leu) and quadruple-dropout (SD/-His-Leu-Tre-Lys) media. Protein–protein interactions (PPI) were determined based on the growth status of the yeast. The primers used for amplification are shown in Supplementary Table S1.
3 Results
3.1 Identification of the E-class genes in common wheat and its relatives
A total of 15 E-class genes were identified in common wheat by BLASTP. Three (WLHS1-A, WLHS1-B, and WLHS1-D), six (TaSEP3-A, TaSEP3-B, TaSEP3-D, TaSEP5-A, TaSEP5-B, and TaSEP5-D), and six (TaSEP6-A, TaSEP6-B, TaSEP6-D, TaSEP4-A, TaSEP4-B, and TaSEP4-D) genes were located on homoeologous group chromosomes 4, 5, and 7, respectively (Table 1). Using the same method, one (AtSEP3), one (TuSEP3), two (TdSEP3-A and TdSEP3-B), two (TtSEP3-Aand TtSEP3-B), and three (TsSEP3-A, TsSEP3-B, and TsSEP3-D) E-class genes were identified in A. tauschii (DD), T. urartu (AA), T. dicoccoides (AABB), T. turgidum (AABB), and T. spelta (AABBDD), respectively (Table 1). We considered species names and chromosomal location for gene nomenclature.
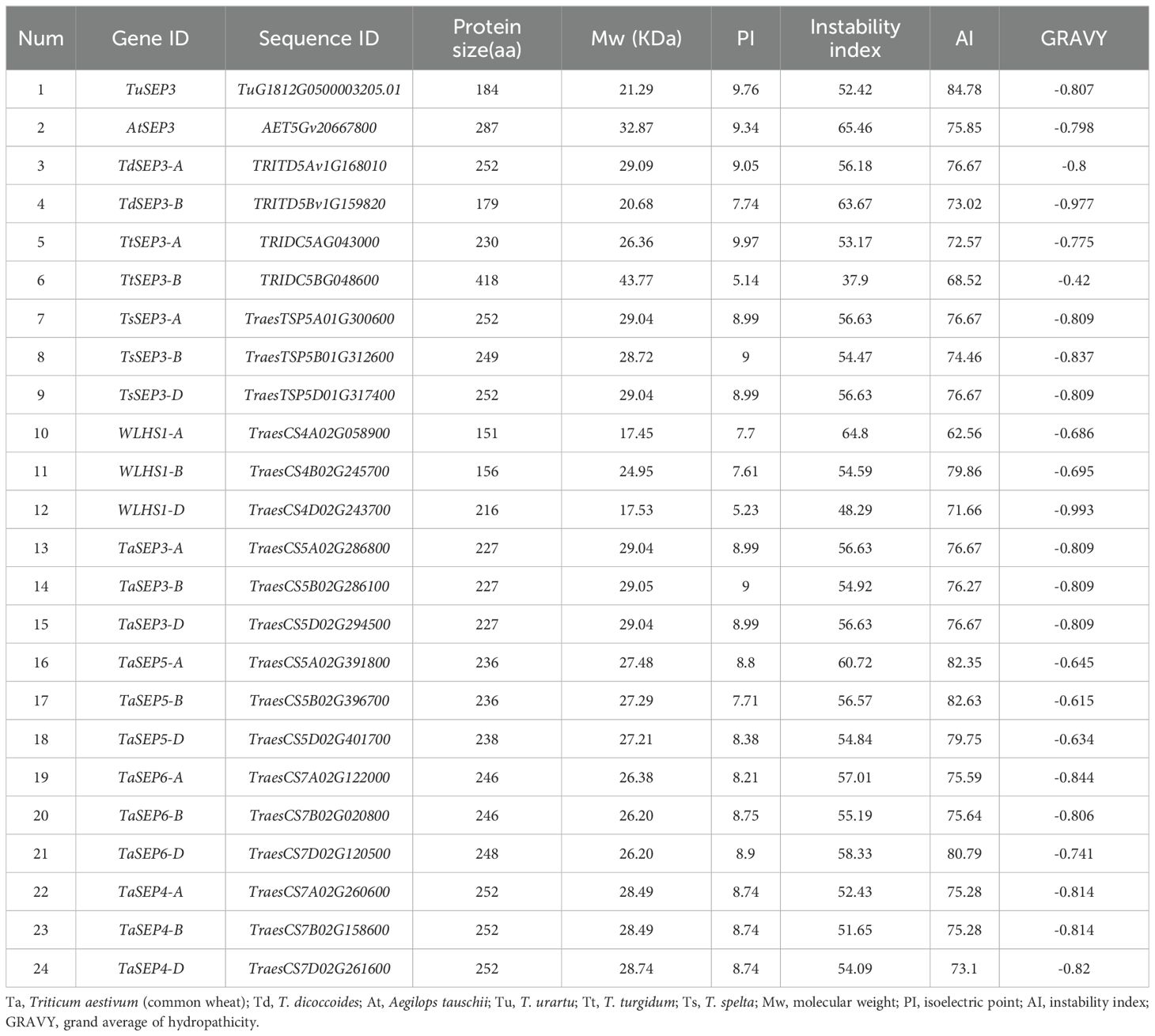
Table 1. Nomenclature and characteristics of the putative E-class proteins in wheat and its ancestral species.
The characteristics of the 24 E-class genes in six wheat species are shown in Table 1. The coding amino acid lengths of these genes in common wheat ranged from 151–252, with a mean size of 227 amino acids, and the molecular weights ranged from 17.5–29.1 kDa. Among the genes in the progenitors/wild relatives, the coding amino acid lengths ranged from 179–418, with a mean size of 247 amino acids, and the molecular weights ranged from 20.1–43.8 kDa. The predicted pI values of TaE proteins, ranging from 5.23–9.00 (mean: 8.30), differed significantly from those of progenitors/wild relatives (5.14–9.97, mean: 8.77). In contrast, no significant difference was found between the II (mean: 55.78), AI (mean: 76.27), or GRAVY (mean: -0.7689) values of common wheat and those of its progenitors/wild relatives (II mean: 55.03; AI mean: 75.75; GRAVY mean: −0.7862). The negative GRAVY score obtained for all E-class genes indicates that the encoded proteins are hydrophilic in nature. Furthermore, prediction of subcellular localization revealed that all the E-class genes are expressed in the nucleus.
3.2 Phylogenetic, motif, and domain analyses of the E-class family genes
We constructed a neighbor-joining tree to examine the phylogenetic relationships and evolutionary patterns of the E-class genes in common wheat and its ancestors. The E-class genes were categorized into three subgroups (Figure 1), among which subgroup I contained four genes (3 TaEs and 1 OsE), subgroup II, eight (6 TaEs and 2 OsEs), and subgroup III, 20. All the E-class genes in the progenitors/wild relatives of common wheat were placed in subgroup III, indicating that there is no significant genetic divergence among these genes.
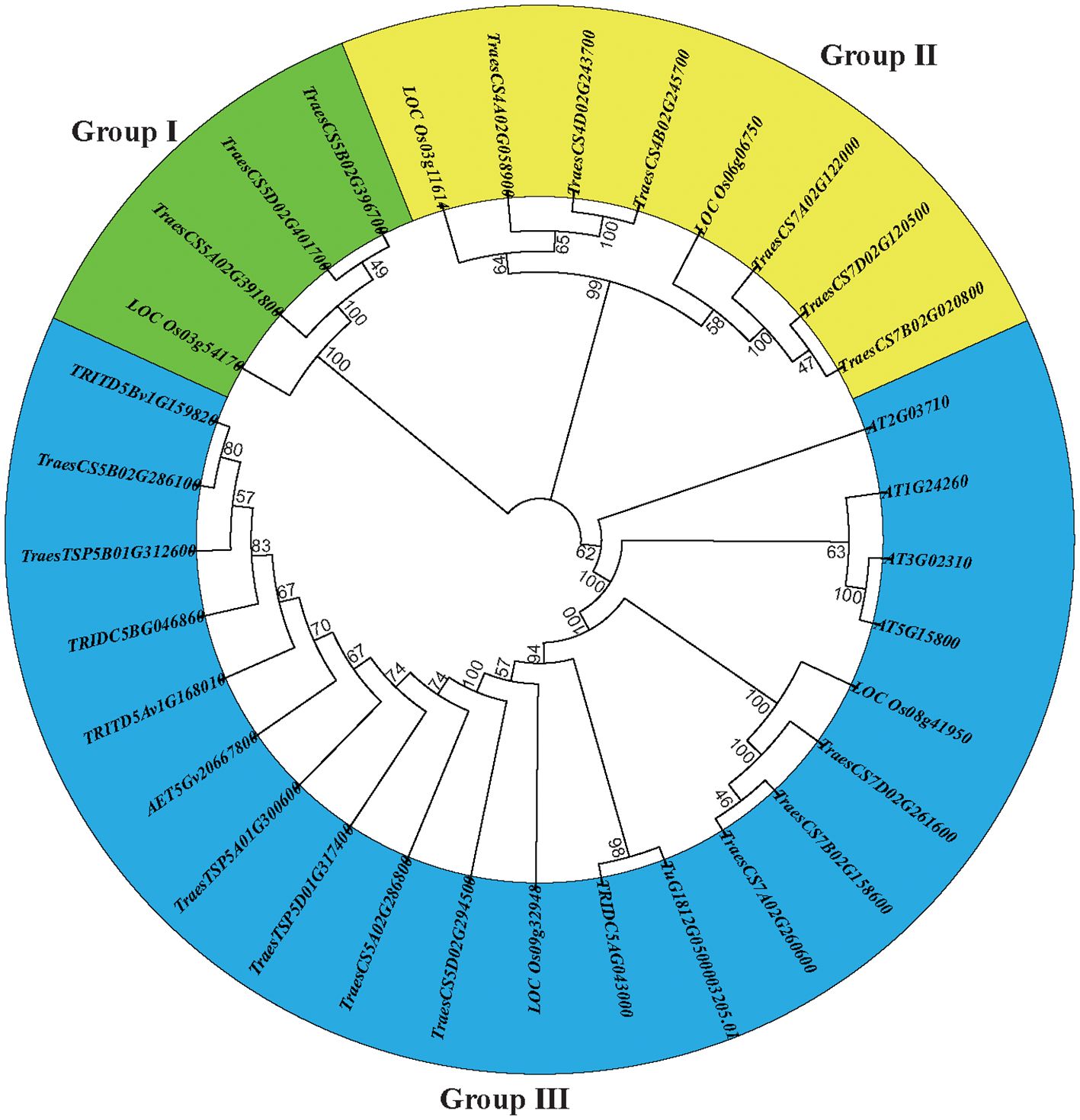
Figure 1. A dendrogram of the E-class genes constructed based on the neighbor-joining method using MEGA X with 1000 bootstrap replications.
Based on an analysis of motifs in the 24 E-class genes from the six wheat species (Figure 2A), we identified a total of 10 motif types. Motifs 3 and 4 were present in 23 (all but WLHS1-A), and motifs 1 and 2 in 22 E-class genes. Motif 8 was specific to subgroup I. All genes in subgroup I had five motifs. Motif 10 was specific to subgroup II, with the number of motifs ranging from 3 to 6. Motif 7 was specific to subgroup III. Motif 5 was detected in 12 genes. Motifs 6 and 9 were present in 9 genes each within subgroup III (Figure 2B). The E-class genes in different subgroups vary in conserved motif distributions, which might suggest a conserved function of different subfamilies. Three types of conserved domains (MADS-MEF2, K-Box, and HD-ZIP_N) were identified in the six wheat species. K-Box domains were distributed in 23 E-class genes (all but WLHS1-A), MADS-MEF2 in 22 (all but WLHS1-D and Td-5A-E1), and HD-ZIP_N in 33 genes in subgroup I (Figure 2C). Most homologous genes were characterized by similar motif and domain compositions, indicating that these genes probably do not differ significantly with respect to sequence or function.
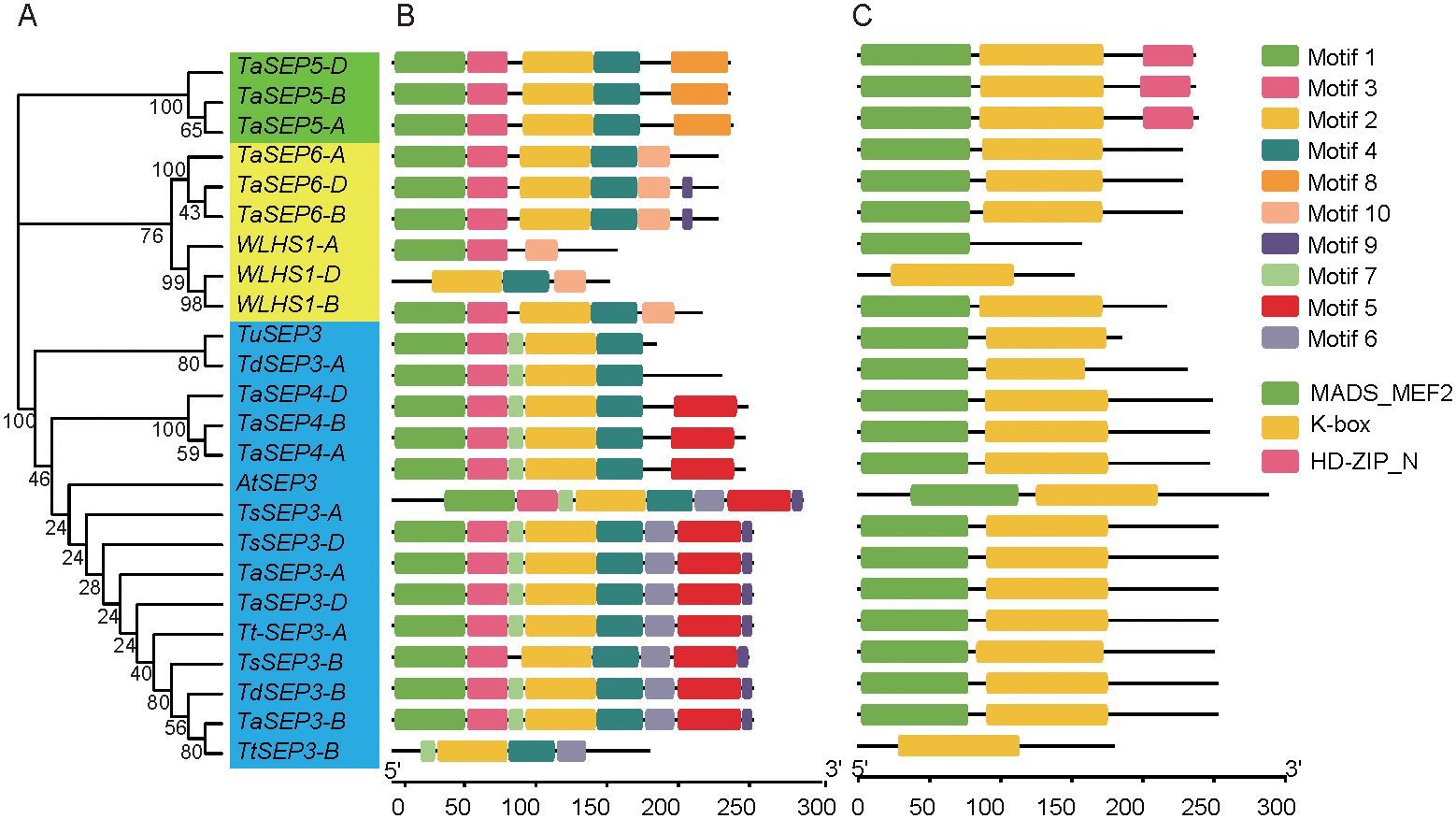
Figure 2. A dendrogram of the E-class genes, conserved motifs, and gene structure domains of the E-class genes in wheat and its ancestral species. (A) A dendrogram constructed based on the full-length amino acid sequence of the E-class genes in wheat and its ancestral species. (B) The conserved motifs of the E-class genes based on their phylogenetic relationship were identified using the MEME software. (C) The 10 types of motifs are denoted by different colors. The green, yellow, and pink rectangles represent the domains.
3.3 Chromosomal distribution of E-class genes in the six wheat species
The TaEs were located on nine chromosomes of the homoeologous group 4, 5, and 7. The TaEs number of A, B and D subgenomes were all five, with one or two on each chromosome (Figure 3). The TaEs on 5 and 7 chromosomes are homologous copies of those on the wheat subgenomes A, B, and D, whereas the TaEs on chromosome 4 are not.In this regard, the specific retention or loss of homologous copies may represent evolutionary signatures of chromosomal interactions and functional divergence. The E-class genes were only located on group 5 chromosomes in the progenitors and wild relatives of common wheat. However, synteny analysis illustrated that all the E-class genes in the six wheat species had collinear relationships (Figure 3).
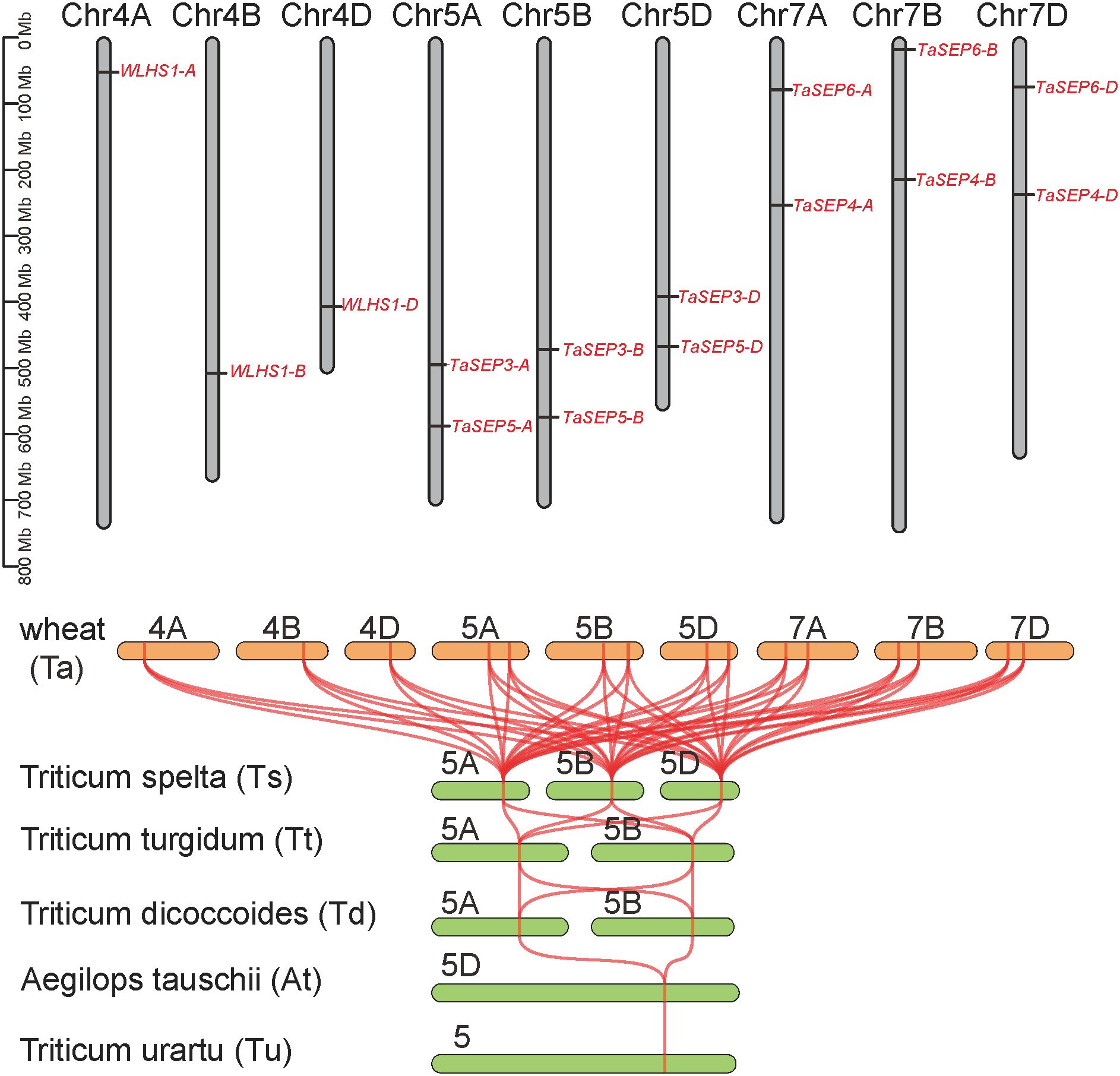
Figure 3. Evolution of the E-class genes in common wheat. Chromosome localization of E-class genes in the wheat genome. Collinearity analysis of the E-class genes in wheat and its ancestral species. Tu, Triticum urartu; At, Aegilops tauschii; Td, T. dicoccoides; Tt, T. turgidum; Ts, T. spelta; Ta, T. aestivum.
Molecular selection effects were assessed by comparing the dN/dS values of homologous E-class genes. No PSGs were detected in any of the groups (Supplementary Table S1). A total of 20, 15, and 11 NSGs were found in the wheat subgenome A (TaA) vs. its ancestral/closely related species subgenomes A (Tu-A-E, Ts-A-E, Td-A-E, and Tt-A-E), wheat subgenome B (TaB) vs. its ancestral/closely related species subgenomes B (Ts-B-E, Td-B-E, and Tt-B-E), as well as wheat subgenome D (TaD) vs. its ancestral/closely related species subgenomes D (At-D-E, Ts-D-E, Td-D-E, and Tt-D-E). The dN/dS values of Ta-E-A vs. Ta-E-B, Ta-E-A vs. Ta-E-D, and Ta-E-B vs. Ta-E-D were 0.3344, 0.3606, and 0.1915, respectively, and their NSGs are all 5. These results suggest that the E-class gene family have underwent purifying selective pressure during evolution and is rather conservative.
3.4 Cis-acting regulatory element analysis of TaEs
To better understand the transcriptional regulation and potential functions of TaEs, we predicted the cis-elements of the promoter regions for these genes and thereby identified 10 classes of functional cis-elements (Supplementary Table S3, Figure 4). The light-responsive element (G-Box), which is closely associated with the function of the class-E genes as key regulators of flower development, was detected in all the E-class genes. In total, 45 hormone-responsive elements were identified in these promoter regions, including 14 abscisic acid-responsive elements, 12 jasmonic acid-responsive elements (e.g., CGTCA and TGACG), nine auxin-responsive elements (AuxRR-core), five gibberellin-responsive elements (TATC), and five salicylic acid-responsive elements (TCA). These findings revealed that TaEs contain at least two hormone-related elements in their promoter regions, suggesting their extensive involvement in hormone-mediated metabolic processes and signal transduction networks in different growth and developmental processes of common wheat. We also detected stress-related elements, such as low-temperature-responsive elements; MYB binding sites involved in drought induction, defense, and stress response (TC-rich repeats); wound response elements (WUN-motif); elements associated with plant growth, such as those regulating plant meristem (CAT-box) and endosperm (GCN4) growth; and circadian elements. These findings indicate that the class-E genes play important roles not only in responding to biotic and abiotic stresses but also in the development and growth of common wheat.
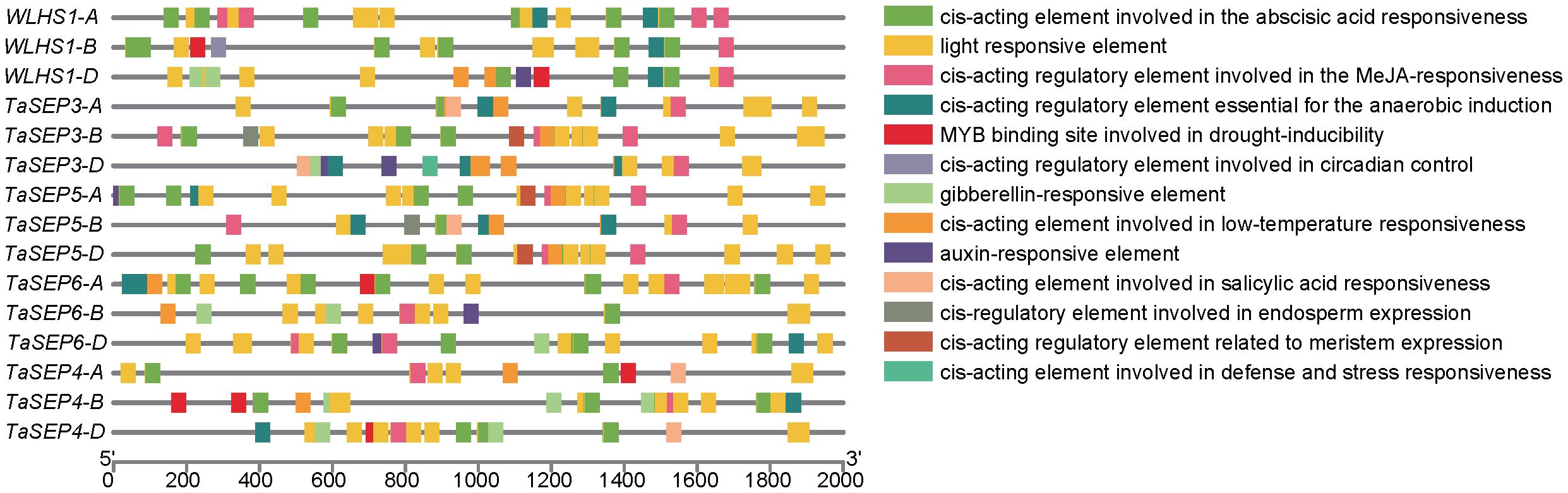
Figure 4. Chromosomal distribution of the TaE genes and cis-element analysis of the TaEs. Each E-class gene was mapped to its chromosomal location based on its physical position in the wheat genome. The chromosome number is labeled at the top of each chromosome. The scale bar is in mega bases (Mb). Different colors represent the 13 types of functional modules.
3.5 Expression and functional analysis of TaE in Arabidopsis
Expression pattern analysis showed that all the TaEs were highly expressed in common wheat spikes (Figure 5A). In addition, 13 E-class genes (excluding WLHS1-A and TaSEP6-B) were highly expressed in stems. In particular, TaSEP5-A, TaSEP5-B, and TaSEP5-D were characterized by high expression from the double-ridge stage to the floret meristem stage, and the other E-class genes showed specific expression patterns in the floret meristem stage of spike development (Figure 5B).
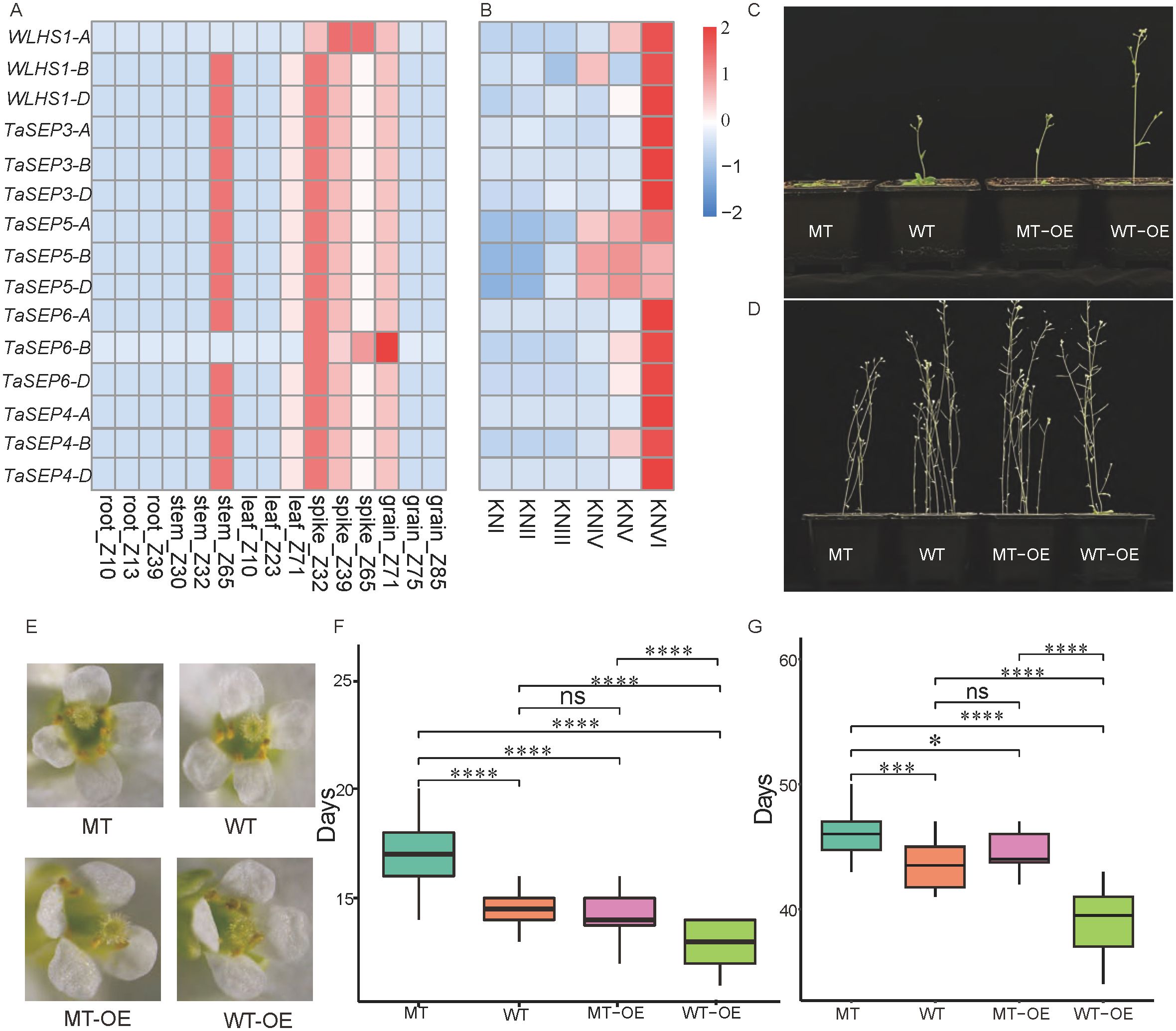
Figure 5. The expression levels of 15 selected TaEs, E-class genes in different parts of wheat, and E-class genes in Arabidopsis thaliana. (A) Expression of the E-class genes in different parts of wheat. (B) Expression of the E-class genes at different stages of wheat spike development. KNI, vegetative stage; KNII, elongation stage; KNIII, single-ridge stage; KNIV, double-ridge stage; KNV, glume primordium differentiation stage; and KNVI, floret differentiation stage. (C) The flowering status (single planting) of four Arabidopsis lines during the flowering period of the wild-type (WT) Arabidopsis. (D) The maturity status of the four Arabidopsis lines during the maturation of the WT Arabidopsis. (E) The morphology of flowers of the four Arabidopsis lines at full bloom. (F, G) Comparison of days to flowering and maturity in the four Arabidopsis lines. *, *** and **** represent significance at p < 0.05, 0.001 and 0.0001, respectively. “ns” stands for not significant (Student’s t-test).
To further characterize the functions and regulatory mechanisms of this gene family in common wheat, the effects of TaSEP5-A on spike development were investigated by transgenic and mutant (AT3G61120) Arabidopsis plants. The transgenic line was characterized by more rapid flowering. By the time that the WT Arabidopsis had produced its first flower, the transgenic line (WT-OE) had already produced several, the mutant line (MT) had yet to bolt, and the mutant overexpression line (MT-OE) had a phenotype like that of the WT (Figure 5C). The number of days to flowering for MT (16 d) was significantly longer than those for WT (13.5 d) and MT-OE plants (14 d) (P < 0.001), whereas that for WT-OE plants (10 d) was significantly shorter than those for MT-OE and WT plants (P < 0.001). No significant difference in days to flowering was detected between the MT-OE and WT plants (Figure 5F). Although different Arabidopsis lines did not vary significantly in morphology at maturity (Figure 5D), the days to flowering and maturity of the WT, MT-OE, MT, and WT-OE lines showed a similar trend (Figure 5G). Stereomicroscopic observations revealed that the different Arabidopsis lines were characterized by comparable flower morphologies at full bloom. We detected no evidence of developmental abnormalities or aberrations (Figure 5E).
3.6 miRNA prediction and protein–protein interactions
miRNA prediction analysis revealed four TaEs to be regulated by three miRNAs, among which two genes, TaSEP6-B and TaSEP4-A, were targeted by a single miRNA (tae-miR2275-3p), potentially a key interaction determining the effects of the E-class genes (Supplementary Figure S1A). According to PPI predictions, the E-class proteins are characterized by many intergroup interactions (Supplementary Figure S1B), as well as interactions with TaMADS, which are key transcription factors controlling flower development in wheat. Given that all the E-class genes identified in the progenitors and wild relatives of common wheat are located on chromosome 5, we selected E-class genes on chromosome 5 in common wheat for analysing intragroup interactions (Figure 6). Transformed yeast was cultured using double- and quadruple-dropout media. Survival status was assessed to determine whether TaSEP5-A interacts with other genes. Thus, we established that TaSEP5-A interacts with WLHS1-A, TaSEP3-A, TaSEP6-A, and TaSEP4-A, but not TaSEP4-B. These findings indicate that TaSEP5-A may have synergistic interactions with WLHS1-A, TaSEP3-A, TaSEP6-A, and TaSEP4-A in specific biological processes or signaling pathways, whereas interactions with TaSEP4-B may be absent or regulated by other factors.

Figure 6. Yeast two-hybrid assay. (A) Negative control group. (B) Positive control group. (C) Experimental group. SD/-2: SD/-His-Leu, SD/-4: SD/-His-Leu-Tre-Lys. AD: pGADT7, BD: pGBKT7.
4 Discussion
4.1 The number of homologous relationships between the E-class genes in common wheat and its progenitors and wild relatives reveal its evolutionary history
An important milestone in studying wheat evolution is the determination of the number of chromosomes in wheat (Thompson and Hake, 2009), which evidences that wheat consists of a polyploid series, including diploid (2n = 2x = 14), tetraploid (2n = 4x = 28), and hexaploid (2n = 6x = 42) wheats (Liu et al., 2021). The evolution of common wheat is assumed to have involved two rounds of autoploidization, and its genome can be arranged into seven homologous groups and three sub-genomes, with a high degree of synteny between the sub-genomes (Suárez-Baron et al., 2017).
Based on a homology comparison, 24 E-class genes were identified in the six wheat species. Phylogenetic analysis showed that the E-class genes in common wheat were distributed across all the three sub-groups, whereas those detected in the progenitors/wild relatives were found exclusively in subgroup III. Furthermore, we established that the ploidy relationship of the number of E-class genes among the progenitors/wild relatives of common wheat was consistent with the ploidy relationship of their chromosome sets. For example, the number of E-class genes in T. dicoccoides (AABB) was twice that of T. urartu (AA). These findings thus indicate that E-class genes among the progenitors/wild relatives of common wheat have a common ancestor and that the number of E-class genes accumulated with an increase in chromosome sets throughout the long-term evolution (Tsaftaris et al., 2011). During such evolution, gene duplication is the main driving force for the expansion of gene families in different species, which mainly involves segmental, tandem, and whole-genome duplication. Contrastingly, in the ancestral and closely related species of common wheat, increase in the number of E-class genes is assumed to be due to the accumulation of chromosomes over long-term evolution, rather than gene duplication (Chen et al., 2020).
The E-class genes in common wheat proved to be variously distributed on chromosomes 4 (3 genes), 5 (6 genes), and 7 (6 genes). The genes on chromosomes 5 and 7 were all homologous copies among the sub-genomes, whereas WLHS1-A, WLHS1-B, and WLHS1-D on chromosome 4 were not. This finding accordingly provides an evidence for a crossover event occurring on chromosome 4 during meiosis (Miftahudin et al., 2004). The E-class genes in the progenitors and wild relatives of common wheat were all located on chromosome 5. Thus, based on the established evolutionary process, we anticipated that the E-class genes in common wheat would similarly all be on chromosome 5 rather than distributed across multiple chromosomes (i.e., 4, 5, and 7) (Chen et al., 2020; Przewieslik-Allen et al., 2021; Zhao et al., 2021). Moreover, the number of E-class genes in cultivated wheat was markedly higher than those in its ancestral and closely related species. Taken together, these findings indicate that gene duplication occurred in response to selective pressure during the evolution of common wheat. Consistent with this scenario, the findings of previous studies have provided evidence to indicate that a large-scale structural rearrangement events occurred on chromosomes 4, 5, and 7 in common wheat (Chen et al., 2020).
In summary, by examining the evolution of E-class genes in common wheat and its ancestral/closely related species, We established the evolutionary process of common wheat and identified structural rearrangement events that occurred during this process through machine learning-based clustering tree construction and synteny analysis. These findings will provide new insights for assessing species evolution, whereby evolution can be inferred by studying the evolutionary history of a single class of key trait-related genes. Previous studies using phylogenetic analysis of key genes TtBtr1-A and TtBtr1-B clearly demonstrated that cultivated wheat has a monophyletic origin from wild wheat in the northern Levant. Further research on the newly discovered premature stop codon mutation in gene Ppd-D1 reveals it as the first example of convergent evolution during the process of environmental adaptation in crops (Zhao et al., 2023).
4.2 E-class genes regulate flower spike development in common wheat
It has previously been established that the E-class genes are expressed primarily in the flowers and fruits of plants (Chahtane et al., 2023). In rice, the expression of OsMADS7 and OsMADS8 is limited to inflorescences and developing kernels (Cui et al., 2010; Yoshida et al., 2013). During flower development, these two genes are expressed in the floral meristems that give rise to the lodicule and stamen primordia. The expression of these genes is subsequently detected in the stamens and pistils in the lodicules until maturity (Peréz-Mesa et al., 2019). The E-class genes are highly expressed in petals of Nelumbo (Liu et al., 2021), specifically expressed in anthers and ovaries of Aristolochia clematitis (Peréz-Mesa et al., 2019; Yu et al., 2017), and preferentially expressed in carpels, petals, and stamens of Carica papaya (Lee et al., 2014). Consistently, we established that all the E-class genes in common wheat were highly expressed from the double-ridge stage to the floret meristem stage, suggesting their potential activity during these stages.
With respect to the involvement of E-class genes in flower development, knockout of the E-class genes OsMADS7 and OsMADS8 in rice gave rise to plants characterized by marked phenotypic variability, including late flowering, the homologous transition of calyces, stamens, and carpels to palea/lemma-like organs, and a loss of floral determinacy (Liu et al., 2013). Similarly, in response to the knockout of all E-class genes in rice, all floral organs, with the exception of lemmas, were homologously transformed into leaf-like organs in flowers (Liu et al., 2013). Similar results were observed in Arabidopsis (Zhao et al., 2020). Our analysis of floral development in mutant, WT, and transgenic Arabidopsis revealed significant differences among these lines at both anthesis and maturity. These results provide evidence that E-class genes can contribute to the regulation of flowering and maturation in Arabidopsis. Considering that the deletion of E-class genes can considerably influence the floral morphology in rice and Arabidopsis, we also performed stereomicroscopic observations of the floral morphology of different Arabidopsis lines during the blooming period. However, no significant changes were noted among these lines, which is consistent with the findings of previous studies (Zhao et al., 2020).
The relevance of E-class genes in regulating plant flowering processes underscores their potential application in crop improvement strategies, particularly within wheat cultivation. In-depth investigations into how these genes influence wheat floral organ development enable precise control over flowering timing and organ formation, thereby enhancing yield stability across different growing seasons and environmental conditions. Integrating these genes into wheat breeding programs facilitates the development of new varieties with enhanced stress resilience and adaptability. Amidst increasing climate variability and stress pressures, leveraging E-class genes to regulate wheat flowering and yield traits aids in breeding more robust and productive crop varieties to address global food security challenges. Elucidating the mechanisms of E-class genes in wheat provides theoretical support for developing novel genetic improvement strategies. Precision editing of these genes or introduction of superior alleles accelerates wheat breeding efforts, enhancing efficiency and success rates in genetic improvement.
4.3 Interactions among the E-class genes of common wheat
Through a homology-based approach, we identified many interactions among the E-class proteins of common wheat and between E-class and MADS proteins. MADS proteins regulate spike development in common wheat and thus influence overall yield (Kyoko et al., 2004). Floral homeotic MADS-domain transcription factor SEPALLATA3 from A. thaliana acts as a PPI controlling flower development, because it contains leucine residues at intermolecular and intramolecular interfaces (Florian et al., 2017). This finding accordingly provides further evidence for the function of the E-class genes. In addition, we predicted the miRNAs targeting TaEs and accordingly identified four TaEs regulated by three miRNAs, with one gene targeted by two. This indicates that the TaEs are involved in a complex regulatory network. Moreover, these miRNA–TaE associations provide valuable information that could contribute to the miRNA-mediated genetic engineering of TaEs. Collectively, the integration of TaE PPI networks and miRNA-mediated pathways will contribute to a better understanding of the pathways associated with the mechanisms of action for the E-class genes in common wheat.
4.4 The novelty and limitations of the method
We employed a homology-based sequence comparison method, originally validated in Arabidopsis thaliana, to identify E-class genes in wheat, confirming findings consistent with prior studies (Kong et al., 2022). Additionally, we extended this method to five ancestral/closely related species, systematic analysis of E-class genes in wheat filling a critical research gap and providing new insights into the evolutionary dynamics of E-class genes in wheat using machine learning. Our comprehensive analysis also offers implications for the application of E-class genes in wheat breeding. Despite advancements in studying biological gene families, we recognize limitations in our experimental approach, specifically in functional gene exploration by transient expression in Arabidopsis rather than in wheat, which may impact accuracy and reliability. Future studies are warranted to address these challenges with more precise and in-depth analyses to validate our discoveries. Future research is necessary to validate and elucidate the functional mechanisms of E-class genes through wheat transformation experiments and multi-omics approaches. In conclusion, while acknowledging these limitations, we believe our study’s innovative perspectives and findings make significant contributions to the understanding of the wheat E-class gene family, laying a solid foundation for future research in this field.
5 Conclusions
Although E-class genes have been widely established to play key roles in regulating flower development in a broad range of plants, their identities and molecular functions in wheat are still poorly understood. We comprehensively analyzed the E-class genes in common wheat by using machine learning. We showed that TaE accumulation has occurred as a consequence of genome polyploidization and gene duplication during evolution. In addition, we confirmed the function of E-class genes based on expression analysis and Arabidopsis transformation experiments. Interacting E-class genes in common wheat were identified based on the interaction network prediction and yeast two-hybrid assays. Our findings indicate that certain TaEs have miRNA-binding sites and may accordingly be regulated by miRNAs. Although these findings provide a solid basis for understanding the potential functional roles of the E-class genes in wheat, many unresolved issues remain regarding wheat flower development. Accordingly, the potential mechanisms whereby TaEs are regulated warrant further investigation.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
XB: Data curation, Investigation, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. PQ: Investigation, Software, Validation, Visualization, Writing – review & editing. HL: Investigation, Validation, Writing – original draft. YS: Data curation, Writing – original draft. JG: Writing – review & editing, Funding acquisition, Supervision. KD: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Natural Science Foundation of China (31901541).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1419437/full#supplementary-material
Supplementary Figure 1 | Gene interactions. (A) A schematic representation of the regulatory network relationships between the putative miRNAs and their targeted TaE genes. (B) Predicted protein-protein interaction networks of TaE proteins with other wheat proteins using STRING tool.
References
Agrawal, G. K., Abe, K., Yamazaki, M., Miyao, A., Hirochika, H. (2005). Conservation of the E-function for floral organ identity in rice revealed by the analysis of tissue culture-induced loss-of-function mutants of the OsMADS1 gene. Plant Mol. Biol. 59, 125–135. doi: 10.1007/s11103-005-2161-y
Avni, R., Nave, M., Barad, O., Baruch, K., Twardziok, S. O., Gundlach, H., et al. (2017). Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 357, 93–97. doi: 10.1126/science.aan0032
Bailey, T. L., Johnson, J., Grant, C. E., Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43, W39–W49. doi: 10.1093/nar/gkv416
Barberis, E., Marengo, E., Manfredi, M. (2021). Protein subcellular localization prediction. Methods Mol. Biol. 2361, 197–212. doi: 10.1007/978-1-0716-1641-3_12
Chahtane, H., Lai, X. G., Rieu, P., Arnoux-Courseaux, M., Cancé, C., Marondedze, C., et al. (2023). Flower development in Arabidopsis. Methods Mol. Biol. 2686, 3–38. doi: 10.1007/978-1-0716-3299-4_1
Chen, C. J., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y. H., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cui, R. F., Han, J. K., Zhao, S. Z., Su, K. M., Wu, F., Du, X. Q., et al. (2010). Functional conservation and diversification of class E floral homeotic genes in rice (Oryza sativa). Plant J. 61, 767–781. doi: 10.1111/j.1365-313X.2009.04101.x
Dai, X. B., Zhuang, Z. H., Zhao, P. X. (2018). psRNATarget: a plant small RNA target analysis server, (2017 release). Nucleic Acids Res. 46, W49–WW54. doi: 10.1093/nar/gky316
Dobrovolskaya, O., Pont, C., Sibout, R., Martinek, P., Badaeva, E., Murat, F., et al. (2015). FRIZZY PANICLE drives supernumerary spikelets in bread wheat. Plant Physiol. 167, 189–199. doi: 10.1104/pp.114.250043
Dubcovsky, J., Dvorak, J. (2007). Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 316, 1862–1866. doi: 10.1126/science.1143986
Guo, Y., Liu, J., Zhang, J. F., Liu, S. Y., Du, J. C. (2017). Selective modes determine evolutionary rates, gene compactness and expression patterns in Brassica. Plant J. 91, 34–44. doi: 10.1111/tpj.13541
Honma, T., Goto, K. (2001). Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 409, 525–529. doi: 10.1038/35054083
Ikeda, K., Ito, M., Nagasawa, N., Kyozuka, J., Nagato, Y. (2007). Rice ABERRANT PANICLE ORGANIZATION 1, encoding an F-box protein, regulates meristem fate. Plant J. 51, 1030–1040. doi: 10.1111/j.1365-313X.2007.03200.x
Ikeda-Kawakatsu, K., Maekawa, M., Izawa, T., Itoh, J., Nagato, Y. (2012). ABERRANT PANICLE ORGANIZATION 2/RFL, the rice ortholog of Arabidopsis LEAFY, suppresses the transition from inflorescence meristem to floral meristem through interaction with APO1. Plant J. 69, 168–180. doi: 10.1111/j.1365-313X.2011.04781.x
Irish, V. F., Litt, A. (2005). Flower development and evolution: gene duplication, diversification and redeployment. Curr. Opin. Genet. Dev. 15, 454–460. doi: 10.1016/j.gde.2005.06.001
Jack, T. (2004). Molecular and genetic mechanisms of floral control. Plant Cell 16 Suppl, S1–17. doi: 10.1105/tpc.017038
Jia, J. Z., Zhao, S. C., Kong, X. Y., Li, Y. R., Zhao, G. Y., He, W. M., et al. (2013). Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496, 91–95. doi: 10.1038/nature12028
Kong, X. C., Wang, F., Geng, S. F., Guan, J. T., Tao, S., Jia, M. L., et al. (2022). The wheat AGL6-like MADS-box gene is a master regulator for floral organ identity and a target for spikelet meristem development manipulation. Plant Biotechnol. J. 20, 75–88. doi: 10.1111/pbi.13696
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Lee, M. J., Yang, W. J., Chui, C. T., Chen, J. J., Chen, F. C., Chang, L. S. (20142014). Isolation and characterization of the papaya MADS-box E-class genes, CpMADS1 and CpMADS3, and a TM6 lineage gene CpMADS2. Genet. Mol. Res. 13, 5299–5312. doi: 10.4238/2014.July.24.9
Lei, P. Z., Wei, X. L., Gao, R. T., Huo, F. L., Nie, X. J., Tong, W., et al. (2021). Genome-wide identification of PYL gene family in wheat: evolution, expression and 3D structure analysis. Genomics 113, 854–866. doi: 10.1016/j.ygeno.2020.12.017
Lémin, S., Scornavacca, C., Dainat, J., Burgarella, C., Viader, V., Ardisson, M., et al. (2019). Pervasive hybridizations in the history of wheat relatives. Sci. Adv. 5, eaav9188. doi: 10.1126/sciadv.aav9188
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Levy, A. A., Feldman, M. (2022). Evolution and origin of bread wheat. Plant Cell 34, 2549–2567. doi: 10.1093/plcell/koac130
Lev-Yadun, A., Gopher, A., Abbo, S. (2000). The cradle of agriculture. Science 288, 1602–1603. doi: 10.1126/science.288.5471.1602
Li, Y. P., Fu, X., Zhao, M. C., Zhang, W., Li, B., An, D. G., et al. (2018). A genome-wide view of transcriptome dynamics during early spike development in bread wheat. Sci. Rep. 18, 15338. doi: 10.1038/s41598-018-33718-y
Li, C. X., Lin, H. Q., Chen, A., Lau, M., Jernstedt, J., Dubcovsky, J. (2019). Wheat VRN1, FUL2 and FUL3 play critical and redundant roles in spikelet development and spike determinacy. Development 146, null. doi: 10.1242/dev.175398
Li, L. F., Zhang, Z. B., Wang, Z. H., Li, N., Sha, Y., Wang, X. F., et al. (2021). Genome sequences of the five Sitopsis species of Aegilops and the origin of polyploid wheat B-subgenome. Mol. Plant 15(2022), 488–503. doi: 10.1016/j.molp.2021.12.019
Liu, Y., Cui, S. J., Wu, F., Yan, S., Lin, X. L., Du, X. Q., et al. (2013). Functional conservation of MIKC*-Type MADS box genes in Arabidopsis and rice pollen maturation. Plant Cell 25, 1288–1303. doi: 10.1105/tpc.113.110049
Liu, Z. X., Zhang, D. S., Zhang, W. W., Xiong, L., Liu, Q. Q., Liu, F. L., et al. (2021). Molecular cloning and expression profile of class E genes related to sepal development in Nelumbo nucifera. Plants (Basel) 10. doi: 10.3390/plants10081629
Lu, S. N., Wang, J. Y., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48, D265–D268. doi: 10.1093/nar/gkz991
Luo, M. C., Gu, Y. Q., Puiu, D., Wang, H., Twardziok, S. O., Deal, K. R., et al. (2017). Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551, 498–502. doi: 10.1038/nature24486
Ma, S. W., Wang, M., Wu, J. H., Guo, W. L., Chen, Y. M., Li, G. W., et al. (2021). WheatOmics: A platform combining multiple omics data to accelerate functional genomics studies in wheat. Mol. Plant 14, 1965–1968. doi: 10.1016/j.molp.2021.10.006
Maccaferri, M., Harris, N. S., Twardziok, S. O., Pasam, R. K., Gundlach, H., Spannagl, M., et al. (2019). Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 51, 885–895. doi: 10.1038/s41588-019-0381-3
Malcomber, S. T., Kellogg, E. A. (2004). Heterogeneous expression patterns and separate roles of the SEPALLATA gene LEAFY HULL STERILE1 in grasses. Plant Cell 16, 1692–1706. doi: 10.1105/tpc.021576
Marcussen, T., Sandve, S. R., Heier, L., Spannagl, M., Pfeifer, M., Jakobsen, K. S., et al. (2014). Ancient hybridizations among the ancestral genomes of bread wheat. Science 345, 1250092. doi: 10.1126/science.1250092
Mayrose, I., Zhan, S. H., Rothfels, C. J., Magnuson-Ford, K., Barker, M. S., Rieseberg, L. H., et al. (2011). Recently formed polyploid plants diversify at lower rates. Science 333, 1257. doi: 10.1126/science.1207205
Mcfadden, E. S., Sears, E. R. (1946). The origin of Triticum spelta and its free-threshing hexaploid relatives. J. HERED 37, 81–107. doi: 10.1093/oxfordjournals.jhered.a105590
Miftahudin, D., Ross, K., Ma, X. F., Mahmoud, A. A., Layton, J., Milla, M. A., et al. (2004). Analysis of expressed sequence tag loci on wheat chromosome group 4. Genetics 168, 651–663. doi: 10.1534/genetics.104.034827
Mirzaghaderi, G., Mason, A. S. (2017). Revisiting pivotal-differential genome evolution in wheat. Trends Plant Sci. 22, 674–684. doi: 10.1016/j.tplants.2017.06.003
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419. doi: 10.1093/nar/gkaa913
Murat, F., Armero, A., Pont, C., Klopp, C., Salse, J. (2017). Reconstructing the genome of the most recent common ancestor of flowering plants. Nat. Genet. 49, 490–496. doi: 10.1038/ng.3813
Otasek, D., Morris, J. H., Bouças, J., Pico, A. R., Demchak, B. (2019). Cytoscape Automation: empowering workflow-based network analysis. Genome Biol. 20, 185. doi: 10.1186/s13059-019-1758-4
Park, S. J., Eshed, Y., Lippman, Z. B. (2014). Meristem maturation and inflorescence architecture–lessons from the Solanaceae. Curr. Opin. Plant Biol. 17, 70–77. doi: 10.1016/j.pbi.2013.11.006
Pelaz, S., Ditta, G. S., Baumann, E., Wisman, E., Yanofsky, M. F. (2000). B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature 405, 200–203. doi: 10.1038/35012103Pelaz
Peréz-Mesa, P., Suárez-Baron, H., Ambrose, B. A., González, F., Pabón-Mora, N. (2019). Floral MADS-box protein interactions in the early diverging angiosperm Aristolochia fimbriata Cham. (Aristolochiaceae: Piperales). Evol. Dev. 21, 96–110. doi: 10.1111/ede.12282
Pfeifer, M., Kugler, K. G., Sandve, S. R., Zhan, B. J., Rudi, H., Hvidsten, T. R., et al. (2014). International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345, 1251788. doi: 10.1126/science.1251788
Przewieslik-Allen, A. M., Wilkinson, P. A., Burridge, A. J., Winfield, M. O., Dai, X., Beaumont, M., et al. (2021). The role of gene flow and chromosomal instability in shaping the bread wheat genome. Nat. Plants 7, 172–183. doi: 10.1038/s41477-020-00845-2
Rice, A., Šmarda, P., Novosolov, M., Drori, M., Glick, L., Sabath, N., et al. (2019). The global biogeography of polyploid plants. Nat. Ecol. Evol. 3, 265–273. doi: 10.1038/s41559-018-0787-9
Sakuma, S., Golan, G., Guo, Z., Ogawa, T., Tagiri, A., Sugimoto, K., et al. (2019). Unleashing floret fertility in wheat through the mutation of a homeobox gene. Proc. Natl. Acad. Sci. U.S.A. 116, 5182–5187. doi: 10.1073/pnas.1815465116
Salse, J., Bolot, S., Throude, M., Jouffe, V., Piegu, B., Quraishi, U. M., et al. (2008). Identification and characterization of shared duplications between rice and wheat provide new insight into grass genome evolution. Plant Cell 20, 11–24. doi: 10.1105/tpc.107.056309
Shewry, P. R., Hey, S. J. (2015). The contribution of wheat to human diet and health. Food Energy Secur 4, 178–202. doi: 10.1002/fes3.64
Suárez-Baron, H., Pérez-Mesa, P., Ambrose, B. A., González, F., Pabón-Mora, N. (2017). Deep into the Aristolochia flower: expression of C, D, and E-Class genes in Aristolochia fimbriata (Aristolochiaceae). J. Exp. Zool B Mol. Dev. Evol. 328, 55–71. doi: 10.1002/jez.b.22686
Thompson, B. E., Hake, S. (2009). Translational biology: from Arabidopsis flowers to grass inflorescence architecture. Plant Physiol. 149, 38–45. doi: 10.1104/pp.108.129619
Tsaftaris, A., Pasentsis, K., Makris, A., Darzentas, N., Polidoros, A., Kalivas, A., et al. (2011). The study of the E-class SEPALLATA3-like MADS-box genes in wild-type and mutant flowers of cultivated saffron crocus (Crocus sativus L.) and its putative progenitors. J. Plant Physiol. 168, 1675–1684. doi: 10.1016/j.jplph.2011.03.015
Tsunewaki, K., Mori, N., Takumi, S. (2019). Experimental evolutionary studies on the genetic autonomy of the cytoplasmic genome "plasmon" in the Triticum (wheat)-Aegilops complex. Proc. Natl. Acad. Sci. U.S.A. 116, 3082–3090. doi: 10.1073/pnas.1817037116
Tu, M., Li, Y. (2020). Toward the genetic basis and multiple QTLs of kernel hardness in wheat. Plants (Basel) 9, null. doi: 10.3390/plants9121631
Wang, X., Wang, J., Jin, D., Guo, H., Lee, T. H., Liu, T., et al. (2015). Genome alignment spanning major Poaceae lineages reveals heterogeneous evolutionary rates and alters inferred dates for key evolutionary events. Mol. Plant 8, 885–898. doi: 10.1016/j.molp.2015.04.004
Wilkins, M. R., Gasteiger, E., Bairoch, A., Sanchez, J. C., Williams, K. L., Appel, R. D., et al. (1999). Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552. doi: 10.1385/1-59259-584-7:531
Yang, Z. H. (2005). The power of phylogenetic comparison in revealing protein function. Proc. Natl. Acad. Sci. U.S.A. 102, 3179–3180. doi: 10.1073/pnas.0500371102
Yoshida, A., Sasao, M., Yasuno, N., Takagi, K., Daimon, Y., Chen, R., et al. (2013). TAWAWA1, a regulator of rice inflorescence architecture, functions through the suppression of meristem phase transition. Proc. Natl. Acad. Sci. U.S.A. 110, 767–772. doi: 10.1073/pnas.1216151110
Yu, L. H., Wu, J., Zhang, Z. S., Miao, Z. Q., Zhao, P. X., Wang, Z., et al. (2017). Arabidopsis MADS-Box transcription factor AGL21 acts as environmental surveillance of seed germination by regulating ABI5 expression. Mol. Plant 10, 834–845. doi: 10.1016/j.molp.2017.04.004
Zahn, L., Feng, B., Ma, H. (2006). Beyond the ABC-model: regulation of floral homeotic genes. Adv. Bot. Res. 44, 163–207. doi: 10.1016/S0065-2296(06)44004-0
Zhao, X. B., Guo, Y. F., Kang, L. P., Yin, C. B., Bi, A. Y., Xu, D. X., et al. (2023). Population genomics unravels the Holocene history of bread wheat and its relatives. Nat. Plants 9, 403–419. doi: 10.1038/s41477-023-01367-3
Zhao, P. X., Miao, Z. Q., Zhang, J., Chen, S. Y., Liu, Q. Q., Xiang, C. B. (2020). Arabidopsis MADS-box factor AGL16 negatively regulates drought resistance via stomatal density and stomatal movement. J. Exp. Bot. 71, 6092–6106. doi: 10.1093/jxb/eraa303
Zhao, H. B., Xie, D., Huang, L., Zhang, S. J., Luo, J. T., Jiang, B., et al. (2021). Integrating the physical and genetic map of bread wheat facilitates the detection of chromosomal rearrangements. J. Integr. Agric. 20, 2333–2342. doi: 10.1016/S2095-3119(20)63289-0
Keywords: wheat, E-class genes, evolution, flower, interaction
Citation: Bai X, Qiao P, Liu H, Shang Y, Guo J and Dai K (2024) Genome-wide identification of the E-class gene family in wheat: evolution, expression, and interaction. Front. Plant Sci. 15:1419437. doi: 10.3389/fpls.2024.1419437
Received: 18 April 2024; Accepted: 14 August 2024;
Published: 03 September 2024.
Edited by:
Andrés J. Cortés, Colombian Corporation for Agricultural Research (AGROSAVIA), ColombiaReviewed by:
Tian Li, Chinese Academy of Agricultural Sciences, ChinaJindong Liu, Chinese Academy of Agricultural Sciences, China
Copyright © 2024 Bai, Qiao, Liu, Shang, Guo and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keli Dai, MjEwMDEzQHN4YXUuZWR1LmNu; Jie Guo, bnhnajExMTUzMjZAc3hhdS5lZHUuY24=
†These authors share first authorship