 Peilin Wang
Peilin Wang Mubashir Abbas
Mubashir Abbas Jianhan He3†
Jianhan He3† Hongmei Cheng
Hongmei Cheng Huiming Guo
Huiming Guo- 1Nanfan Research Institute, Chinese Academy of Agricultural Sciences (CAAS), Sanya, Hainan, China
- 2Biotechnology Research Institute, Chinese Academy of Agricultural Sciences, Beijing, China
- 3Institute of Cereal and Oil Crops, Hebei Academy of Agriculture and Forestry Sciences, Hebei Key Laboratory of Crop Genetics and Breeding, Shijiazhuang, Hebei, China
- 4Yazhouwan National Laboratory, Sanya, Hainan, China
Cotton production faces challenges in fluctuating environmental conditions due to limited genetic variation in cultivated cotton species. To enhance the genetic diversity crucial for this primary fiber crop, it is essential to augment current germplasm resources. High-throughput sequencing has significantly impacted cotton functional genomics, enabling the creation of diverse mutant libraries and the identification of mutant functional genes and new germplasm resources. Artificial mutation, established through physical or chemical methods, stands as a highly efficient strategy to enrich cotton germplasm resources, yielding stable and high-quality raw materials. In this paper, we discuss the good foundation laid by high-throughput sequencing of cotton genome for mutant identification and functional genome, and focus on the construction methods of mutant libraries and diverse sequencing strategies based on mutants. In addition, the important functional genes identified by the cotton mutant library have greatly enriched the germplasm resources and promoted the development of functional genomes. Finally, an innovative strategy for constructing a cotton CRISPR mutant library was proposed, and the possibility of high-throughput screening of cotton mutants based on a UAV phenotyping platform was discussed. The aim of this review was to expand cotton germplasm resources, mine functional genes, and develop adaptable materials in a variety of complex environments.
1 Introduction
The landscape of genomics has undergone a profound transformation in recent years, thanks to the rapid progress in high-throughput genome sequencing technologies. These advancements have paved the way for a thorough examination of entire genomes, enabling the identification of genes and their associated biological functions (Ng and Kirkness, 2010). Consequently, the scientific community has been actively engaged in characterizing genes crucial for development, cellular processes, and responses to diverse stresses (Ng and Kirkness, 2010). Traditionally, the understanding of gene function relied on two distinct yet complementary approaches: forward genetics and reverse genetics. Forward genetics entails identifying phenotype variations arising from spontaneous or induced mutations. Subsequently, the responsible mutant genes and their functions are identified and characterized (Jill, 2016). Initially, this method leveraged naturally occurring mutants with specific traits to uncover and study the genes governing those traits (Schneeberger and Weigel, 2011). However, with the advent of cost-effective high-throughput genome sequencing, it has become more practical to intentionally induce mutations and explore their effects on gene function (Johannes and Schmitz, 2019).
To conduct comprehensive high-throughput functional research on plant genes, it is crucial to have an extensive collection of mutant materials covering a diverse range of plant species. Leveraging whole-genome sequence data and employing reverse genetics methods enable large-scale screening of mutant libraries, facilitating the systematic study and annotation of all genes within a genome (Hagen, 2000). The completion of whole-genome sequencing for the model plant Arabidopsis thaliana in 2000 (Arabidopsis Genome Initiative, 2000), coupled with the subsequent establishment of an Arabidopsis mutant library (Greene et al., 2003), marked a significant breakthrough that propelled genomics research into a new era. Similarly, the sequencing of the rice genome in 2002 (Yu et al., 2002) and the creation of the rice mutant library in 2006 (Li et al., 2006) ushered in unprecedented advancements in genomics research.
Cotton, serving as a primary source of renewable fiber globally, plays a crucial role in the textile manufacturing industry. The cotton genus encompasses 46 diploid (2n=2x=26) species and seven tetraploid (2n=4x=52) species. The diploid cotton species are thought to share a common ancestor, with genomes categorized into eight groups: A, B, C, D, E, F, G, and K. The tetraploid cotton species originated from hybridization between the A genome species G. arboreum (A2) and the D genome species G. raimondii (D5) (Huang et al., 2021). Significant strides have been made in cotton genomics in recent years. The draft genome of the diploid cotton G. raimondii (D5) (Wang et al., 2012), G. arboreum (A2) in 2014 (Li et al., 2014). G. hirsutum (AD), a tetraploid cotton species, was fully sequenced, along with the development of a high-density genetic map (Wang et al., 2015). These groundbreaking achievements in cotton genomics, including the sequencing of the complete cotton genome, have ushered in a new era in cotton functional genomics research.
The creation of cotton mutant libraries through diverse approaches is imperative given the notable progress in functional genomics. Diversifying cotton germplasm resources not only presents significant potential for generating stable, high-quality cotton materials, but also serves as the foundation for enhancing crop productivity, improving fiber quality, and developing varieties with increased resistance to biotic and abiotic stresses.
The main goals of this review are to thoroughly analyze the establishment of cotton mutant libraries and the sequencing methodologies utilized, as well as to emphasize the discovery of new germplasm resources and gene functions. Additionally, we suggest prospective directions for cotton mutant research and explore the methods used in creating cotton mutant libraries, encompassing the induction of mutations using ethyl methanesulfonate (EMS), a widely employed mutagen in plant genetics.
Our discussion covers an assessment of the advantages and limitations of EMS mutagenesis, along with an examination of the strategies used to screen and characterize mutants exhibiting altered traits. Moreover, we underscore the efficacy of next-generation sequencing technologies in scrutinizing the mutant population, pinpointing causative mutations, and unraveling the functional consequences at the genomic level.
2 Recent advances in cotton genome sequencing and EMS mutant library development
Cotton is a versatile natural textile fiber with great importance in the global economy. The genus Gossypium consists of 45 diploid (2n = 2x = 26) and 5 tetraploid (2n = 4x = 52) species. The tetraploid cotton species are believed to have originated from the interspecific hybridization between an A-genome diploid species (e.g. G. herbaceum or G. arboreum) and a D-genome diploid species (G. raimondii), eventually leading to the emergence of at least 5 allotetraploid AD genome species (Huang et al., 2021). Allopolyploid cotton, which includes the primary cotton species G.hirsutum and G. barbadense, is believed to have emerged in the last 1–2 million years through hybridization and subsequent polyploidization events between the progenitors of the A- and D-subgenomes (Wendel et al., 2009). The global cotton production is dominated by G. hirsutum due to its high yield potential, moderate fiber quality, and broad adaptability. In contrast, G. barbadense, renowned for its high-quality, long-staple cotton fiber, plays a vital role as a key raw material for high-grade textile production, despite its lower yield potential (Hu et al., 2019). However, the constrained environmental adaptability of G. barbadense poses a significant barrier to its widespread cultivation.
In nature, the continuous interplay of natural and artificial selection leads to the emergence of a pool of spontaneous mutations. While this pool contains a considerable number of mutations, the intricate genetic background of these mutants presents challenges for researchers. The complexity, coupled with the low frequency of spontaneous mutations, poses difficulties for high-throughput and large-scale functional genomics research. This challenge is particularly pronounced in tetraploid cotton, where natural mutations are scarce, significantly impeding the progress of functional genomics development.
The efforts to enhance functional genomics efficiency have been accelerated by advancements in genome sequencing, which over the past two decades, has led to the development of genetically modified cotton varieties with increased resistance to insects and herbicides (Guo et al., 2015; Yu et al., 2016). This progress is a testament to the collaborative efforts within the scientific community, as exemplified by the application of comprehensive whole-genome sequences from model organisms like Arabidopsis and rice. Such foundational work has significantly contributed to consortium-based cotton genome research, spearheaded by initiatives like the Cotton Genome Consortium strategic plan in 2007 (Chen et al., 2007). This plan aimed to sequence the less complex diploid genomes, which would have direct implications for the more complicated tetraploid cotton species. The prioritization of the D-genome species G. raimondii for complete sequencing was a calculated step toward this goal. By 2012, the release of the draft genome sequence of G. raimondii marked a pivotal achievement, setting the stage for subsequent characterization of the larger A-diploid and AD-tetraploid cotton genomes, thereby bridging the gap between initial sequencing efforts and the broader application to cotton genomics (Paterson et al., 2012; Wang et al., 2012). This continuum of research and development underscores the interconnected nature of genomic studies, where each phase builds upon the previous, culminating in a comprehensive understanding that benefits functional genomics research.
Diploid species, such as the D-genome (G. raimondii) and A-genome (G. arboreum or G. herbaceum), are considered likely ancestors of the prominent cotton fiber-producing species, G. hirsutum and G. barbadense (Wendel, 1989). Consequently, G. raimondii took the lead as the first species to undergo whole-genome sequencing, with an estimated genome size of approximately 740 Mb (Paterson et al., 2012; Wang et al., 2012). In comparison, the genome assembly of G. arboreum is more than twice the size of G. raimondii (Li et al., 2014). The draft genome of diploid G. arboreum was completed in 2014, employing next-generation sequencing technologies (NGS, Illumina) (Huang et al., 2021).
The successful sequencing and assembly of diploid cotton genomes laid the foundation for unraveling the complexities of allotetraploid cotton species. In 2015, draft genomes of Gossypium hirsutum TM-1 were published, utilizing various sequencing platforms such as Illumina and PacBio RSII, along with assembly strategies like 10× genomics+BioNano+Hi-C and PacBio+Hi-C (Li et al., 2015; Zhang et al., 2015). Concurrently, the genome of Gossypium barbadense was also sequenced using similar methods (Liu et al., 2015; Yuan et al., 2015).
Recent studies have further refined the assembly of cultivated allotetraploid cotton genomes, including G. hirsutum and G. barbadense, resulting in high-quality assemblies with improved centromeric regions (Wang M, et al., 2019; Hu et al., 2019). These advancements have paved the way for understanding genomic differences between these two tetraploid cotton species.
Additionally, the genome of Gossypium turneri was sequenced using PacBio long reads, Hi-C, and Bionano optical mapping technologies. This effort contributed to the correction of minor assembly errors in the previous G. raimondii genome assembly (Udall et al., 2019). The new de novo genome assembly of G. raimondii and its close relative G. turneri was achieved at the chromosome level, enhancing accuracy and correctness compared to the earlier Sanger sequencing-based assembly.
These breakthroughs in cotton genome sequencing hold promise for improving the accuracy and translation of genomics in cotton breeding and genetics.
The recent successful sequencing of multiple cotton genomes has greatly expedited research in cotton functional genomics and population genetics. This achievement has enabled the identification of crucial agronomic trait genes through genome-wide association studies and map-based cloning techniques, thus advancing our understanding of cotton genetics and enhancing breeding efforts. However, mapping data from diverse materials to a single reference genome may lead to the loss of important variants, including presence/absence variation (PAV) and copy number variation (CNV) (Golicz et al., 2016). Consequently, the construction of a population pan-genome through the direct analysis of individual genome sequences presents notable advantages.
The core genome (Core-genome), encompassing genome sequences present in all individuals, is compared with the variable genome (Variable-genome), which includes sequences found in only some individuals (Liu et al., 2020). The crop pan-genome plays a pivotal role in uncovering genetic variations lost during domestication and improvement. This allows breeders to harness core genome variation and rare variation in each material, thereby providing genetic resources to enhance traits such as yield, quality, resistance, and adaptability (Abbas et al., 2022). In 2021, the inaugural cotton pan-genome of allotetraploid cotton was released, featuring the most diverse variation data-set to date, followed by the pangenome of diploid cotton species (Wang et al., 2022; He et al., 2024). It sheds light on the genomics foundations of cotton domestication and improvement across multiple scales, offering valuable insights into essential cotton traits from a pan-genome perspective (Li et al., 2021).
Moreover, Wang et al. (2018) revealed alternative splicing (AS) in polyploid cotton through Pacific Biosciences single-molecule long-read isoform sequencing (Iso-Seq), offering new insight into the complexities and regulation of AS. The swift evolution of sequencing technology, marked by a myriad of sequencing methods and increased precision, robustly bolsters cotton functional genomics research. The creation of mutant populations for broader scientific utility in research and breeding pursuits stands out promising and effective strategy.
In general, prior to large-scale sequencing of genomes, the identification of mutants was limited from a single trait to a single functional gene. However, after the vigorous development of sequencing, large-scale genome-wide mutation identification appeared, which greatly enriched the mutant library and mutant materials, and also greatly promoted the development of forward genetics.
3 Genome sequencing revolutionizes plant mutagenesis and crop improvement
Genome sequencing has transformed plant breeding by providing powerful tools like MutMap, MutMap-Gap, Mut-Ren-Seq, and whole-genome sequencing to identify causal mutations underlying desirable traits (Takagi et al., 2015; Chaudhary et al., 2019). Mapping-by-sequencing methods, combining bulked-segregant analysis with NGS data alignment to a reference genome, allow faster gene cloning and candidate gene identification, eliminating the need for extensive marker saturation and physical mapping (Mascher et al., 2014). High-quality reference genomes are crucial for these approaches, providing the genomic context to infer mutation order and position.
Genome sequencing has also enabled precise genome editing tools like CRISPR/Cas9 for targeted mutagenesis in crop plants, offering a more directed approach than random chemical or physical mutagenesis (Nerkar et al., 2022). Mutation rate and pattern analysis from sequencing data has revealed insights like lower mutation frequencies in genes under strong selective pressure, informing effective mutation breeding strategies (Chaudhary et al., 2019). The integration of genome sequencing with mutagenesis techniques has significantly improved functional genomics research in plants. Kubo et al. (2022) employed whole-genome sequencing (WGS) to analyze an MNU-mutagenized rice mutant library, demonstrating the effectiveness of MNU mutagenesis for in silico screening. Sun et al. (2024) used CRISPR/Cas9 and WGS to create and analyze insect-resistant cotton mutants, elucidating molecular pathways contributing to resistance. He et al. (2023) highlighted the potential of CRISPR-based methods for large-scale gene knockout screening and uncovering gene functions and regulatory networks. Wang et al. (2023) combined EMS mutagenesis with exome capture sequencing to create a cataloged wheat mutant library, identifying novel allelic variations in abiotic stress response genes. Zhao et al. (2020) constructed a genome-wide ihpRNA library in Brassica napus using RCA-mediated technology, enabling efficient gene silencing and phenotype identification in this allopolyploid species. Bi et al. (2023) demonstrated the construction of a transcription factor mutagenesis population in tomato using a pooled CRISPR/Cas9 plasmid library and WGS analysis. These studies collectively showcase how genome sequencing has revolutionized mutagenesis approaches, enabling the creation of extensive mutant collections and the understanding of gene functions and regulatory networks underlying important traits. This has significantly accelerated crop improvement efforts by providing insights into the genetic basis of desirable characteristics.
4 Advancing crop breeding through mutagenesis
Genetic transformation in crops, such as cotton, is labor-intensive and time-consuming, with a single transgenic event frequently lasting more than a year. Physicochemical mutagenesis has emerged as a viable alternative, eliminating the need for plant genetic transformation and tissue culture techniques.
Mutagenesis techniques, which mix physical and chemical agents, have proven beneficial in crop breeding. Chemical mutagens, such as alkylating agents (e.g., Ethyl Methanesulfonate, Sodium Azide), offer novel methods for creating genetic variations, allowing breeders to research novel traits while boosting crop adaptability, yield, and quality.
Recent advances in genomics technology have expanded the application of mutational breeding. Site-directed mutagenesis, particularly using the CRISPR/Cas9 system, has shown useful in creating targeted modifications in numerous crops, leading to improvements in agronomic metrics such as yield, quality, and stress tolerance (Chen et al., 2017).
To generate superior cultivars, crop breeders typically use induced mutagenesis, which employs physical mutagens such as radiation and chemical mutagens such as EMS. It considerably improves genetic diversity for desired qualities such as stress tolerance and biofortification. Mutagenized populations have been developed for major agricultural species like rice, maize, wheat, and barley (Jankowicz-Cieslak et al., 2017).
Advances in sequencing technologies have made it easier to identify and exploit beneficial mutations induced by mutagenesis. Cloning and inserting desirable mutant alleles into elite germplasm is becoming more feasible, providing a speedy and cost-effective method for producing genetic diversity and improving agriculturally important traits.
In cotton breeding, physical mutagens such as radiation and chemical mutagens such as EMS have been widely used to create mutations that result in heat-resistant, early-maturing, and high-yielding cotton varieties (Zhao et al., 2022). The CRISPR/Cas9 system has emerged as an effective technique for targeted mutagenesis in cotton, allowing for precise gene editing at high efficiency. Mutagenesis in wild cotton species can result in useful traits that can be introduced into cultivated cotton through genetic study and breeding (Kushanov et al., 2022).
In general, mutagenesis, both random and targeted, has proven to be an effective technique for crop breeders to expand genetic diversity and improve agriculturally necessary traits, addressing the global challenge of ensuring food security in the face of rising population and climate change.
5 EMS-induced mutant libraries in cotton
In the realm of cotton research, notable advancements have been achieved in the development of EMS-induced mutant libraries within diverse cotton species, such as G. arboreum (Kong et al., 2017), G. barbadense (Abid et al., 2020), G. hirsutum (Lian et al., 2020; Wei et al., 2022), and G. herbaceum (Kumar et al., 2022). It is noteworthy that the primary focus of these events centered on conventional cultivated G. hirsutum varieties. The key steps and considerations regarding mutagenesis in cotton are outlined below.
5.1 Selecting suitable plant materials
The initial phase entails the meticulous selection of appropriate plant materials for mutagenic treatment, placing a strong emphasis on ensuring a high seed germination rate. Significantly, EMS mutagenesis demonstrates broad applicability across various cotton germplasms.
5.2 Optimizing EMS concentration and treatment duration
The subsequent crucial step involves conducting preliminary experiments to ascertain the optimal concentration of EMS and the necessary treatment duration. It’s crucial to note that due to the distinctive chemical properties of EMS, utilizing a concentration that is too low may result in an insufficient mutation rate, whereas excessively high concentrations can induce plant mortality and impede seed germination (Abid et al., 2020).
5.3 Determining lethal dose
To strike the right balance, researchers commonly calculate the lethal dose (LD50) based on a 50% survival rate under conditions conducive to viable seed production. However, this calculation can pose challenges when dealing with genotypes that do not yield fertile seeds, a phenomenon observed in some instances (Ke et al., 2019; Abid et al., 2020).
5.4 Diverse outcomes based on EMS concentration
It’s important to recognize that EMS mutagenesis produces diverse outcomes upon the concentration of EMS applied. Reported concentrations span from as low as 0.3% to as high as 5%, with the majority of experiments falling within the range of 1% to 3%. Despite endeavors to attain an LD50 of 50%, the inherent toxicity of EMS and developmental abnormalities in certain mutant strains often lead to a final M1 generation acquisition rate below 50% (Kong et al., 2017; Lian et al., 2020).
5.5 Variability in cotton material sensitivity to EMS
Furthermore, the sensitivity of different cotton materials to EMS significantly influences the frequency of EMS mutagenesis. In terms of treatment concentration, certain studies have employed concentrations as low as 0.3%, while others have reached up to 5%. Nonetheless, most experiments utilize concentrations ranging from 1% to 3%. Irrespective of LD50 calculations, it’s noteworthy that the final M1 generation acquisition rate typically falls below 50% due to EMS toxicity and the occurrence of developmental anomalies (Kong et al., 2017; Lian et al., 2020).
5.6 A Plethora of mutant phenotypes
EMS mutagenesis induces a diverse range of mutant phenotypes, influencing various aspects of cotton plants. These variations encompass plant type, leaf and flower shapes, anthers, flower buds, cotton boll characteristics, fiber quality, length, color, as well as resistance to adverse conditions and diseases (refer to Supplementary Table S1).
5.7 Stabilizing mutant traits
Following the mutagenic treatment, sown seeds frequently yield numerous abnormal plants in the M1 generation. Consequently, multiple generations of breeding become imperative to stabilize mutant traits. Subsequently, the agronomic traits of the mutagenized population are meticulously examined at various growth stages, including cotyledon, seedling, bud, flowering, boll, and spitting stages, to pinpoint variants with distinct phenotypes (Figure 1).
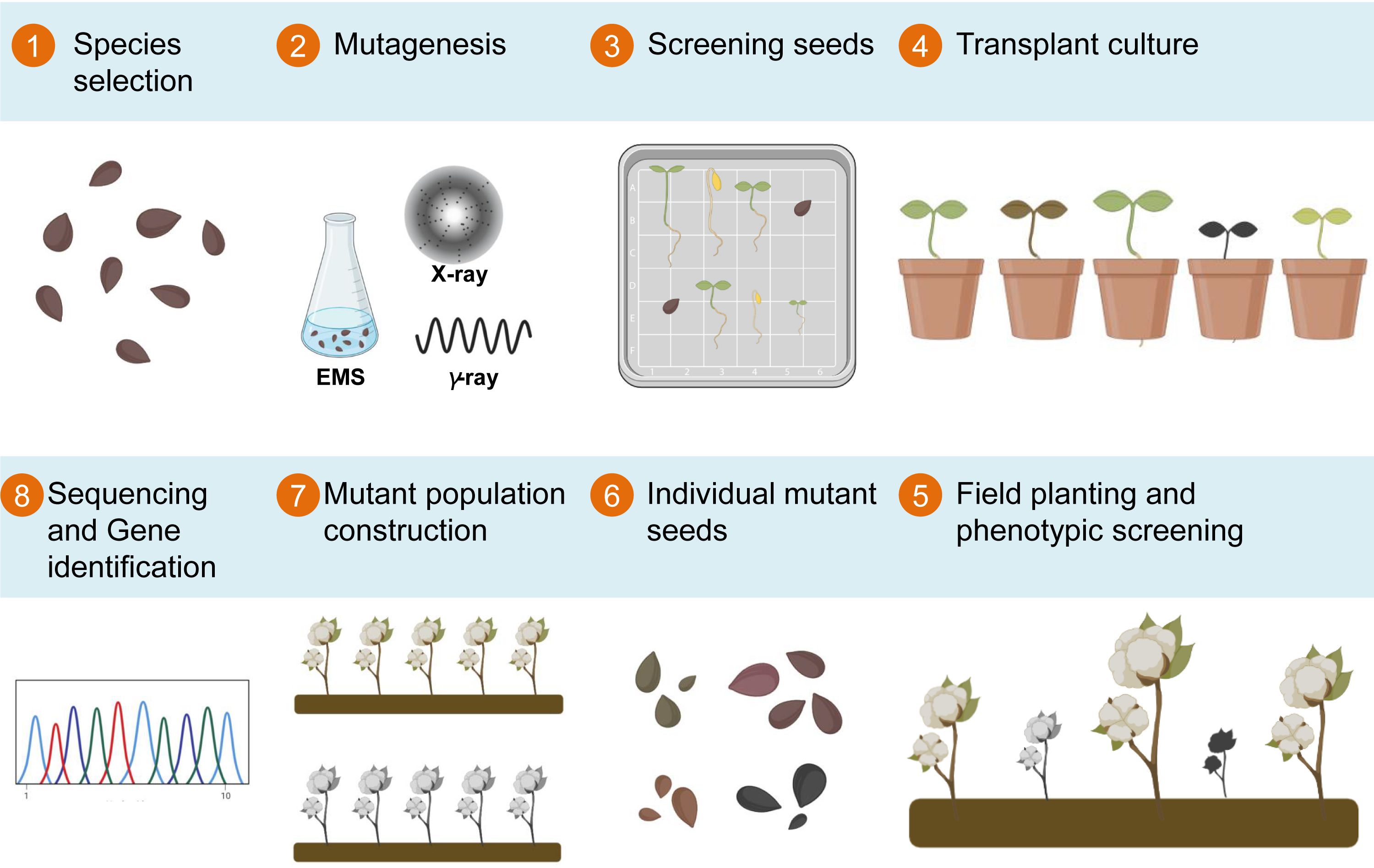
Figure 1 Flowchart illustrating the process of establishing EMS-induced mutant libraries in cotton. The process starts with seed selection (Step 1), followed by the application of a suitable mutagenic treatment, such as EMS, X-ray, or γ-ray (Step 2). Mutated seed screening eliminates damaged and inviable seeds (Step 3), and germinated seeds are transplanted into small pots (Step 4). The selected seedlings are then transplanted to the field for phenotype observation (Step 5), while seeds from plants exhibiting desired traits are harvested (Step 6), forming the mutant population. This mutant population serves as the foundation for mutant sequencing and subsequent gene mapping (Steps 7 and 8, respectively), facilitating comprehensive genetic analysis and gene discovery.
6 Radiation-induced mutagenesis in cotton breeding
Radiation mutagenesis has played a pivotal role in cotton breeding by inducing favorable traits such as heat resistance and early maturation. Gamma-ray irradiation of cotton seeds has been widely employed for this purpose (Maluszynski et al., 1995). A notable example is the development of the ‘Lumian1’ cotton variety, which is distinguished for its high and consistent yield. This variety was derived from hybrid progenies resulting from X-ray radiation mutagenesis applied to ‘Zhongmian 2’ and ‘1195’ lines (Morgan et al., 1996).
Similarly, to establish a mutant library centered on the cotton inbred line G. hirsutum L. TM-1 (Texas Marker-1), linear electron acceleration-based radiation mutagenesis has been employed. Extensive studies have confirmed the stability of TM-1 as a cotton inbred line (Zhao et al., 2022). Interestingly, a distinct investigation on upland cotton pollen grains exposed to 60Co γ-ray gamma-ray revealed noteworthy alterations in their internal structure. While the surface of the pollen remained unaffected, the interior structure underwent remarkable changes, including thinning and irregularities in the interior wall, depolymerization of the endoplasmic reticulum, increased pollen grain inclusions, and a reduced number of pollen tubes in the style. Consequently, these changes resulted in decreased germination rates in the M1 progeny (Yue and Zou, 2012).
Furthermore, the mutant line ‘Zhonghuzhi PI 935’ (referred to as “PI 935”) was derived from G. hirsutum cv. Liaomian No. 9 through the utilization of 60Co gamma-ray mutagenesis. PI 935 not only exhibited favorable traits similar to the original cultivar, such as growing period, drought tolerance, lint color, and fiber quality but also demonstrated higher lint output and yield. Remarkably, compared to control cultivars like Junmian No. 1 or Xinluzhong No. 5, PI 935 displayed a significantly elevated lint output of approximately 47.3%, making it a promising choice for cotton production (He et al., 2001).
These advancements underscore the potential of radiation mutagenesis in cotton breeding, providing opportunities to enhance cotton fiber properties, yield, and various agronomic traits, thereby contributing to sustainable and improved cotton cultivation.
Although both chemical and physical mutagenesis can bring about mutations in the genome, in contrast, chemical mutagenesis, especially EMS mutagenesis, is currently the most commonly used strategy in plants, mainly based on its relatively simple and rapid operation and good balance of LD50 and mutation frequency.
7 High-throughput sequencing for mutant gene identification
Traditionally, geneticists relied on map-based cloning for the identification of mutant genes, a method involving the labor-intensive process of associating markers with the mutant phenotype and precisely mapping the candidate genes. However, this approach requires large populations and an extensive array of markers, making it time-consuming (Kole and Gupta, 2004). The emergence of high-throughput sequencing has revolutionized mutant gene identification in plants, offering several methods summarized here (Figure 2) and their corresponding applications (Supplementary Table S2).
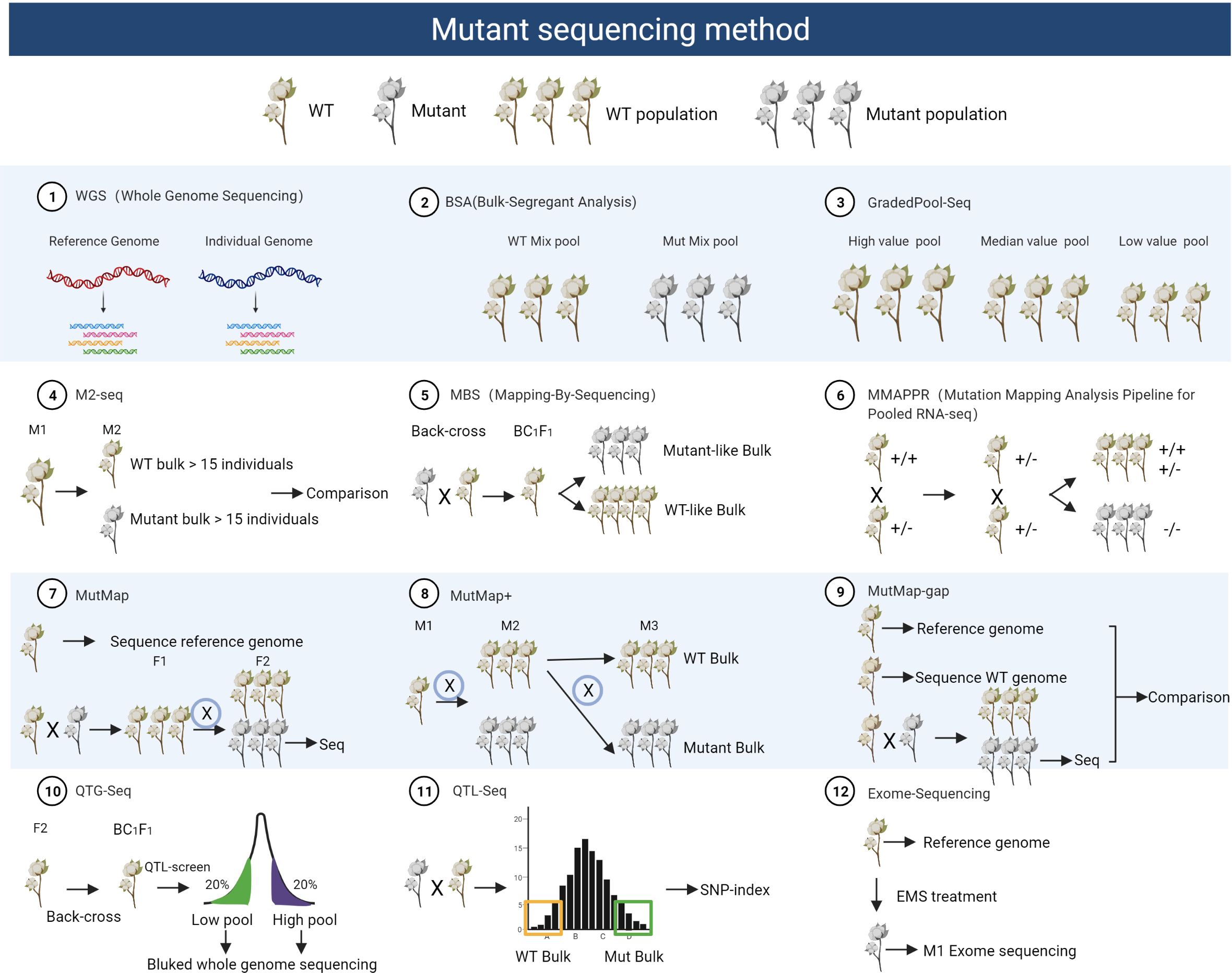
Figure 2 Schematic diagram of different mutant sequencing methods. This figure outlines the steps involved in mutant sequencing methods, from constructing mutant populations to selecting materials and employing various techniques, including Whole Genome Sequencing (WGS), Bulk Segregant Analysis (BSA), Graded Pool-Seq, M2-Seq, Mutant Bulk Segregation (MBS), MMAPPR, MutMap, MutMap+, Mutmap-gap, QTG-Map, QTL-Seq, and Exome Sequencing. Each method serves distinct research purposes, from comprehensive genomic analysis (WGS) to the identification of mutations linked to specific traits (BSA), graded phenotyping (Graded Pool-Seq), or tracking M2 mutations (M2-Seq). These methods offer flexibility in characterizing mutant populations and uncovering genetic variations.
One effective approach is Bulked-Segregant Analysis (BSA), which utilizes next-generation sequencing (NGS) to swiftly map genes in plants. BSA facilitates the identification of molecular markers closely linked to causal genes responsible for specific traits (Michelmore et al., 1991). The process involves constructing a segregating population, generating DNA pools from progenies with contrasting phenotypes, and genotyping them with polymorphic molecular markers. These markers guide the further analysis of the linkage between the obtained marker and the target gene position in a known molecular map or chromosome. Cotton research has successfully employed BSA to map important genes (Zhu et al., 2017; Wei et al., 2022).
Building upon BSA, the MutMap method replaces conventional markers with SNPs and utilizes resequencing to directly analyze SNP polymorphisms. This approach involves crossing mutant individuals with their wild-type counterparts to create an F2 population. From this population, mutant phenotypes are selected, and only the DNA from the hybrid pool undergoes high-throughput sequencing. The concept of SNP-index, a commonly used BSA positioning method, is integral to MutMap. It involves fitting SNP-index values to identify genomics intervals associated with the trait, as demonstrated in locating the rice leaf color mutant gene (Abe et al., 2012).
In contrast, MMAPPR (Mutation Mapping Analysis Pipeline for Pooled RNA-seq) deviates from MutMap by sequencing the RNA of extreme individuals within the mutant pool rather than resequencing DNA. This method proves advantageous for larger genomes. It introduces the Euclidean distance (ED) algorithm for trait association analysis within hybrid progeny populations lacking parents. Background noise reduction is achieved through ED power value fitting using LOESS (locally estimated scatterplot smoothing), as exemplified in the analysis of zebrafish cardiovascular mutants (Hill et al., 2013).
MutMap+, an extension of MutMap, ingeniously selects heterozygous individuals at mutation sites for selfing when unexplored mutant individuals cannot develop normally and die at the seedling stage. By sequencing both extreme pools, this method effectively reduces background noise. It also introduces the △SNP-index algorithm for determining trait correlation areas, as demonstrated in locating the rice light green leaf color mutation and validating the function of the OsNAP6 gene’s function (Fekih et al., 2013).
The Mutmap-Gap method combines MutMap and de novo assembly to address gene mutations at missing reference gene sites. It involves comparing the wild-type and reference genome to obtain the studied genotype reference genome. Then, using EMS mutagenesis, MutMap analysis identifies the SNP-index map’s peak region genes. If no related genes are found, it implies the mutation site is in the strain-specific gene region. Unmapped reads are de novo assembled to identify potential new genes. This method successfully isolated the blast-resistant gene Pii in rice (Takagi et al., 2013a).
QTL-seq complements BSA mapping by focusing on quantitative traits in plants. Specifically, this method selects the reference sequence from the parent with the trait of interest and calculates the SNP-index for trait association using a confidence interval algorithm. The rice blast fungus resistance was mapped using this method (Takagi et al., 2013b). Similarly, QTG-Seq combines genetic map QTL mapping, backcrossing progeny background selection, and QTL-seq to fine-tune the positioning of genes regulating quantitative or qualitative traits and determine the number of main effect sites. It utilizes the F2 genetic map for preliminary mapping, followed by the selection of specific BC1F1 progeny for further analysis and fine positioning. This approach was exemplified in corn plant height, mapping it to the candidate gene Zm00001d020874 (Zhang et al., 2019). Moreover, GradedPool-Seq is an improvement over traditional QTL-seq. Instead of using only two extreme pools, it introduces intermediate phenotype individuals to create “high-value,” “median-value,” and “low-value” groups. Using the Ridit algorithm to calculate allele frequency, this method significantly enhances positioning accuracy. GradedPool-Seq was employed to locate the heterosis QTL GW3p6 (Wang et al., 2019).
Mapping-By-Sequencing (MBS) has accelerated the identification of pathogenic mutations in tomatoes. MBS, reliant on whole-genome sequencing, is a rapid method that takes only 6–12 months from mutant isolation to pathogenic mutation identification. The process involves screening EMS mutants to isolate plants with pathogenic phenotypes, generating an F2 population, and identifying mutated genes through whole-genome sequencing. Utilizing MBS, a yellow-colored tomato fruit mutant was analyzed, revealing a point mutation in the carotenoid pathway gene PSY1 (Garcia et al., 2016).
In maize and wheat, Exome-Sequencing combines EMS mutagenesis, exon capture, and next-generation sequencing to create mutant libraries. This approach has led to the identification of numerous mutation sites causing amino acid changes, contributing significantly to our understanding of the genetics of these plants’ (Krasileva et al., 2017; Lu et al., 2018).
M2-seq provides a rapid and cost-effective tool for identifying candidate causal mutation sites. It eliminates background mutations through the comparison of M2 populations, and the ΔSNP-index method facilitates effective identification of causal mutations. This approach successfully pinpointed the candidate gene Glyma.08G193200 in soybean M2 mutant populations (Zhou et al., 2021).
Various sequencing methods for mutant analysis have been demonstrated, with extensive application and validation in model plants like Arabidopsis and rice. Researchers can select the most suitable method based on their specific research objectives and the traits under investigation. For instance, QTL-seq is well-suited for qualitative and quantitative traits influenced by major genes, MutMap is valuable for the analysis of mutagenized mutants, MutMap+ is particularly useful for early lethal mutations or strains that cannot be outcrossed, and MutMap-Gap is designed for scenarios where the target gene is not present in the reference genome (Figure 2, Supplementary Table S2).
Some of these sequencing strategies are original protocols, and some are based on the original methods to improve and expand to meet the needs of different mutant libraries, and in general, these diverse sequencing techniques have significantly accelerated mutant gene identification in plants. They not only enhance the efficiency of gene mapping but also facilitate a deeper understanding of the genetic basis of various traits, contributing to advancements in plant breeding and genetic research. The choice of method ultimately depends on the specific requirements and nature of the mutant analysis, allowing researchers to employ these tools effectively in their investigations.
8 Genes identified by cotton EMS mutant library
Cotton has lags behind model plants like Arabidopsis and rice in terms of whole-genome sequencing, primarily due to its complex polyploid genome. However, recent advancements in functional genomics and mutagenesis research have facilitated the identification of numerous genes and candidate genes associated with key traits in cotton, such as leaf color, plant type, and fiber development (Supplementary Table S3).
In relation to leaf color-related genes, researchers conducted crosses on the Sumian 22 mutant population, observing a 3:1 separation ratio of medium green and green plants in the F2 generation. A specific 0.34 Mb hypermutation interval was identified on the mutant D10 chromosome, encompassing 31 genes. Remarkably, among these genes, only ABCI1 exhibited significantly lower expression levels in mutants compared to the wild type (WT). Simultaneously, the levels of Mg-protoporphyrin IX, prochlorophyll lactone, chlorophyll a, and b in the mutant were markedly reduced in line with the downregulation of ABCI1. Furthermore, a critical A to T mutation was identified at -317 bp from the start codon of ABCI1 in the mutant genome sequence. This mutation likely inhibits ABCI1 transcription, leading to the green mutation in Sumian 22. The reduced transport of protoporphyrin IX to plastids ultimately hinders the synthesis of Mg-protoporphyrin IX, protochlorophyll lactone, and chlorophyll, explaining the observed green phenotype (Gao et al., 2021).
Another leaf color-related gene, GhCHLI, was identified through bulked segregant analysis-next-generation sequencing and virus-induced gene silencing strategies. A single nucleotide conversion at position 1366bp (G to A) resulted in the substitution of lysine (K) with arginine (R) at the 361st amino acid, causing the observed change in leaf color (Zhu et al., 2017). Additionally, mapping was used to locate the virescent gene v1, GhChlI, which exhibited a non-synonymous nucleotide mutation (G1082A) in its 1269bp coding region. This mutation replaced arginine (R) with lysine (K) in the third exon of Gh_D10G0283. Interestingly, both mutations involve a single amino acid change and impact essential amino acids crucial for optimal growth, nitrogen balance, and fundamental metabolic processes, including photosynthesis, chloroplast biogenesis, and maintenance mechanisms (Alban et al., 2014).
Similarly, for plant type-related genes, researchers successfully cloned the G. barbadense axillary flowering (GbAF) mutant gene and the upland cotton cluster branch (cl1) mutant gene. Notably, the substitution of aspartic acid (Asp) at position 73 with asparagine (Asn) in the deduced amino acid sequence of GbAF led to cotton bolls growing directly on the main plant stem. Dynamic variations in GhSFT and GhSP levels played a pivotal role in regulating meristems between monopodial and sympodial programs within a single plant (McGarry et al., 2016). These findings suggest that cotton orthologs of SFT and SP genes can be harnessed to enhance cotton plant architecture (Si et al., 2018).
In the realm of fiber-related genes, a short fiber phenotype mutant was identified within the EMS mutant library, linked to a tetrapeptide repeat-like superfamily protein encoded by Ghir_A12G008870. Gene silencing of Ghir_A12G008870 significantly reduced the fiber length in the WT cotton line MD15 (Fang et al., 2020). Furthermore, researchers discovered a recessive tufted-fuzzless seed mutant on chromosome D04, with a genome interval of approximately 411 kb. Seven genes in this region showed significant differential expression between the tufted lint-free mutant and the wild type. Ghir_D04G019490 emerged as the prime candidate gene due to its proximity to the SNP marker D04_549, which displayed the highest LOD score association with the fuzzless phenotype. Although the exact function of Ghir_D04G019490 remains unknown, this study suggests its involvement in down hair fiber development (Naoumkina et al., 2021). Additionally, researchers characterized a chemically-induced short fiber mutant cotton line, Ligon-lintless-y (liy), controlled by a single recessive locus that affected multiple traits, including plant height and fiber length and maturity. Three candidate genes (2700, 477, and 3260) were identified, showing significant up-regulation in liy at different stages. Gene set enrichment analysis unveiled substantial alterations in various metabolic pathways, such as carbohydrate, cell wall, hormone metabolism, and transport, during liy fiber development (Naoumkina et al., 2017). Additionally, fiber gene expression analysis of 20 selected miRNAs revealed differential expression profiles in short fiber mutants compared to the WT during fiber development, reflecting distinct transcript regulation in mutant lines compared to WT fiber cells. Four miRNA families exhibited significant correlations with fiber length across 11 diverse upland cotton lines (Naoumkina et al., 2016).
The discovery of Ligon lintless-1 (Li1) mutant revealed significantly differentially expressed transcription factors AS2, YABBY5, and KANDI-like in mutant tissues compared to WT tissues. Notably, several down-regulated genes in the mutant leaf transcriptome were related to fiber development, encompassing heat shock protein families, cytoskeleton arrangement, cell wall synthesis, energy metabolism, H2O2 metabolism-related genes, and WRKY transcription factors (Ding et al., 2014). Moreover, in the mutant GhACT17DM from Li1 plants, the substitution of Gly65 with valine on the nucleotide-binding domain of GhACT17D influenced F-actin polymerization. Compared to the wild-type control, actin filaments in Li1 fibers exhibited higher growth and shrinkage rates, reduced filament skewness, increased filament density, and parallel arrangements (Cao et al., 2021). Similarly, Mapping-by-sequencing unveiled a 22-bp deletion in a pentatricopeptide repeat (PPR) gene that was entirely linked to the immature fiber phenotype in a large F2 plant population and absent in all 163 cultivated varieties tested (Thyssen et al., 2016).
Although the physicochemical mutagenesis technology is relatively simple and it is relatively easy to construct mutant populations, the mutagenesis process is difficult to control, and often a mutant contains more point mutations, which may be caused by the joint action of multiple point mutations, and the mutation phenotype appears. Moreover, after physicochemical mutagenesis, the plant genome may also undergo the rearrangement or deletion of large DNA fragments, and may also promote the transposition of the reverse poson, which will make it more difficult to identify functional genes. At present, most of the functional genes identified by mutants are clearly regulated and observable mutations(leaf color, plant type, fiber, etc.), which also misses a large part of the mutant genes.
9 Future directions
9.1 Embracing the future: the promise of T-DNA and CRISPR mutant libraries
The insertion mutant library is mainly a mutant library constructed by insertion mutagenesis, and the inserted elements are mainly transposon or T-DNA, and the transposon insertion mutant library and T-DNA insertion mutant library can be created accordingly. Generally, the efficiency of insertion mutagenesis is high, and it has been widely used in the construction of mutant libraries, which has played an important role in functional genomics research. When Agrobacterium is used for transformation, through the Ti plasmid, the T-DNA carrying foreign genes infects the plant, through a complex biochemical process, crosses the nuclear membrane, enters the nucleus, and is randomly integrated into the nuclear genome. T-DNA integrated into the nuclear genome can be passed on more stably to the next generation. Different locations where T-DNA is inserted into the genome can cause different genetic mutations, resulting in mutants with different phenotypes. At present, the T-DNA transformation systems of Arabidopsis thaliana and rice are relatively mature (Gelvin, 2017; Gong et al., 2021), the library of insertion mutants constructed from T-DNA insertions has played a huge role in the corresponding functional genomics research. However, in some other species, such as maize and cotton, an efficient Agrobacterium transformation system has not yet been established, and the Agrobacterium-mediated T-DNA transformation process is time-consuming and expensive, and there are often chimeras in the mutants, which increases the difficulty of using T-DNA to construct a mutant library in other species.
The future of cotton research is shifting towards the CRISPR mutant library as a promising avenue (Figure 3). Currently, various mutants have been screened for resistance genes against different adversities. In addition to the EMS mutant library, other cotton mutant libraries, including the T-DNA insertion mutant library, transposon mutation library, physical mutagenesis via radiation mutants, and the recent CRISPR mutant library (Jia et al., 2012; Zhang et al., 2007; Wang et al., 2013; Ramadan et al., 2021), have been developed. However, the number of mutant cotton plants obtained remains limited. Among the various libraries, the CRISPR mutant library emerges as the foremost choice for prospective mutant library construction, as illustrated in Figure 4. Its unique capability to induce targeted and accurate gene mutations presents a compelling advantage, setting it apart from the EMS library, where more than 90% of mutations originate from intergenic regions. In rice, the researchers (Lu et al., 2017; Meng et al., 2017) constructed a total of 91,004 targeted loss-of-function mutants and 12,802 genes mutant libraries, respectively, and although each vector could knock out more than one target gene, it still carried out a considerable scale of transformation. Almost impossible to finish for cotton. In cotton, researchers used a high-throughput gene editing system to create a library of cotton insect resistance gene mutants, but the genes covered by the library were very limited (Sun et al., 2024). In fact, for cotton research, the bottleneck of CRISPR library creation is not the CRISPR technology itself, but the deficiency of the cotton transformation system, which is difficult to transform and has a long cycle, and the only way at present is to transform 2–3 plasmids together to improve the base of the library. Therefore, for cotton and some plants with difficult transformation, the EMS is still the best strategy to obtain large-scale mutagenic resources.
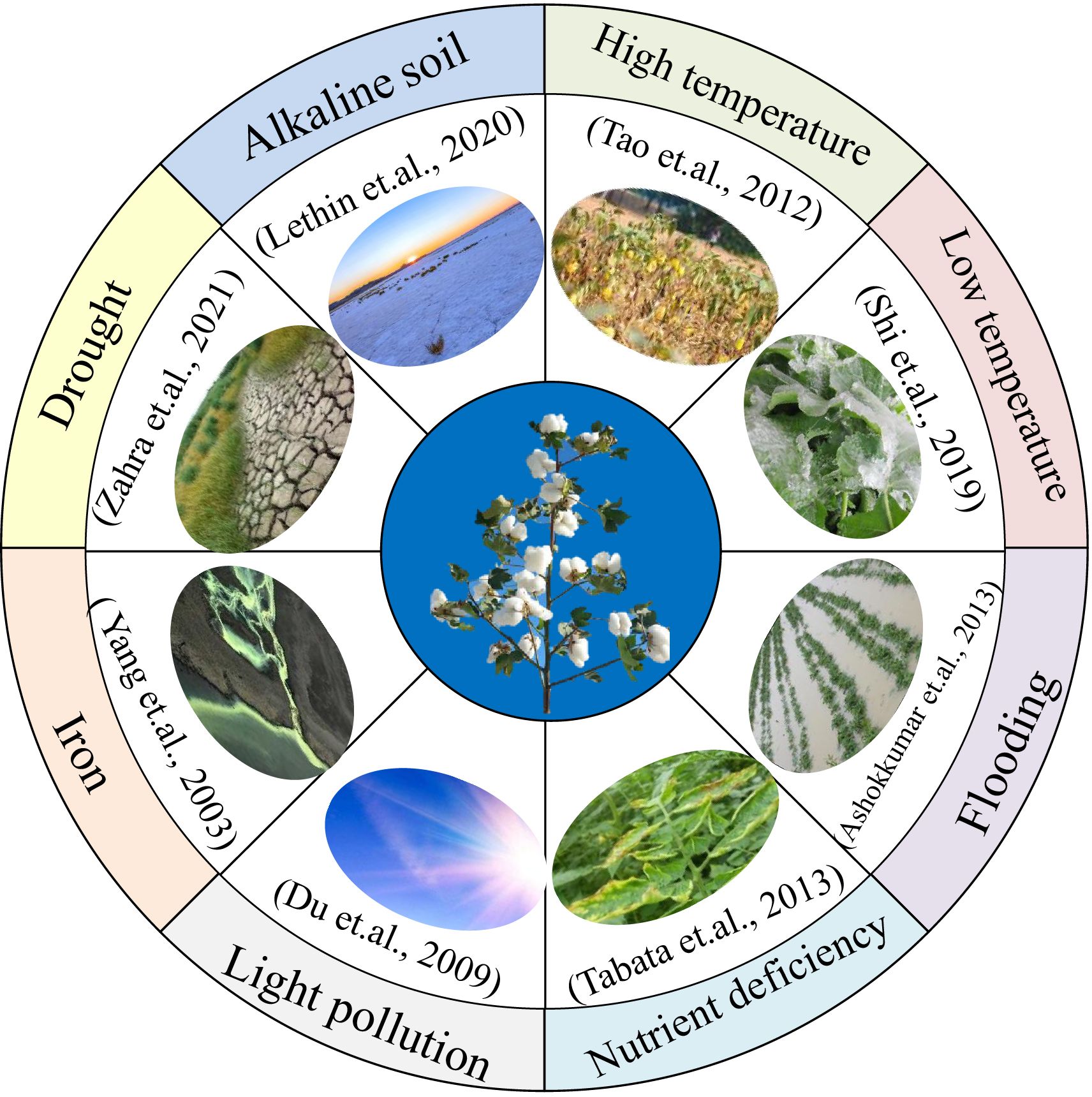
Figure 3 Adaptability of Mutant Plants in Diverse and Challenging Environments This figure highlights the adaptability of mutant plants in diverse and challenging environmental conditions, shedding light on their practical applications to complex scenarios. Mutant materials have proven invaluable in research and applications related to various environmental stressors, including drought, alkaline soil conditions, extreme temperature fluctuations (both high and low), flooding events, nutrient deficiencies, and light pollution. Additionally, these mutants have demonstrated resilience in coping with iron-related challenges. Mutant plants are crucial tools for exploring and enhancing crop adaptability to multiple environmental stresses.
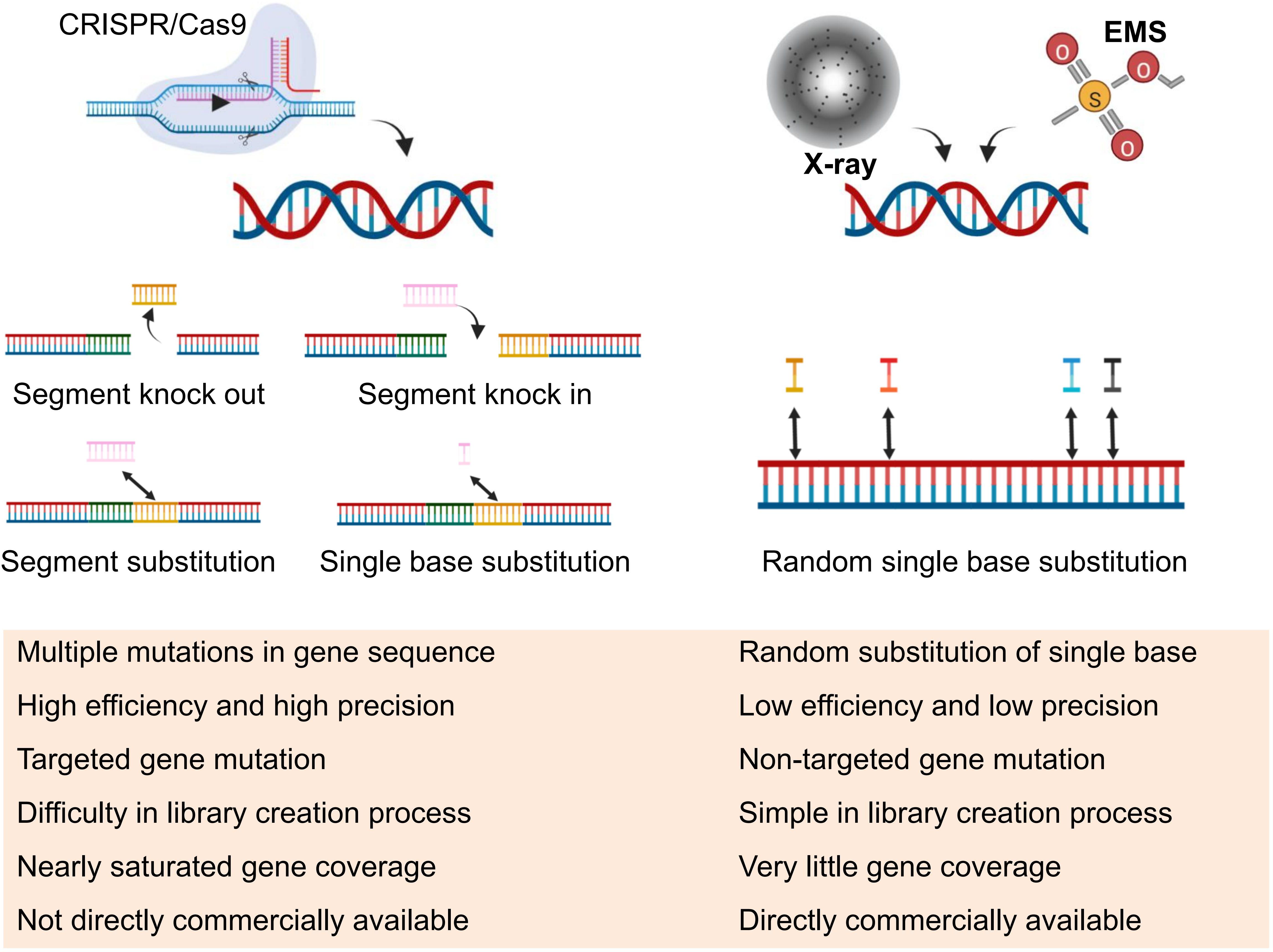
Figure 4 Comparison of CRISPR and Mutagenesis Libraries. In this figure, a concise comparison is presented between CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and other mutagenesis libraries, outlining key differences implementation and utilization, offering insights into the distinct approaches employed in genetic research and manipulation.
In the realm of cotton genomics, the shortage of resources for reverse genetics in polyploid materials presents a challenge. Conventional cultivated cotton, a heterotetraploid species with a short evolutionary history, features two highly similar sub-genomes with functional redundancy, hindering the identification and selection of recessive mutation traits (Zhang, 2013). Additionally, cotton transformation is a complex and time-consuming process. Establishing a mutant library encompassing numerous genes is crucial for advancing cotton genomics and enriching cotton germplasm resources. Currently, research on mutant cotton germplasm primarily focuses on identifying phenotypic variations, an approach that often overlooks recessive mutations. Therefore, to drive future progress, employing precise sequencing methods to uncover recessive mutations and construct a near-saturation mutant library for cotton is imperative. This endeavor promises to unlock hidden genetic diversity and expedite progress in cotton genomics research.
9.2 Harnessing genetic diversity and innovation
Mutation breeding is an innovative agricultural approach that holds significant promise for the development of stress-resilient cotton varieties. The core elements of this promising future include the integration of diverse germplasm, the utilization of advanced mutagenic sources, the application of cutting-edge phenotyping techniques via unmanned aerial vehicles (UAVs) (Ye et al., 2023), and the adoption of super pangenome strategies for comprehensive genomic analysis (Figure 5). Using a super-pangenome constructed by using different species of cotton at the genus level, representing historical cotton germplasm resources, can also increase the chances of detecting meaningful genetic variants induced by artificial selection.
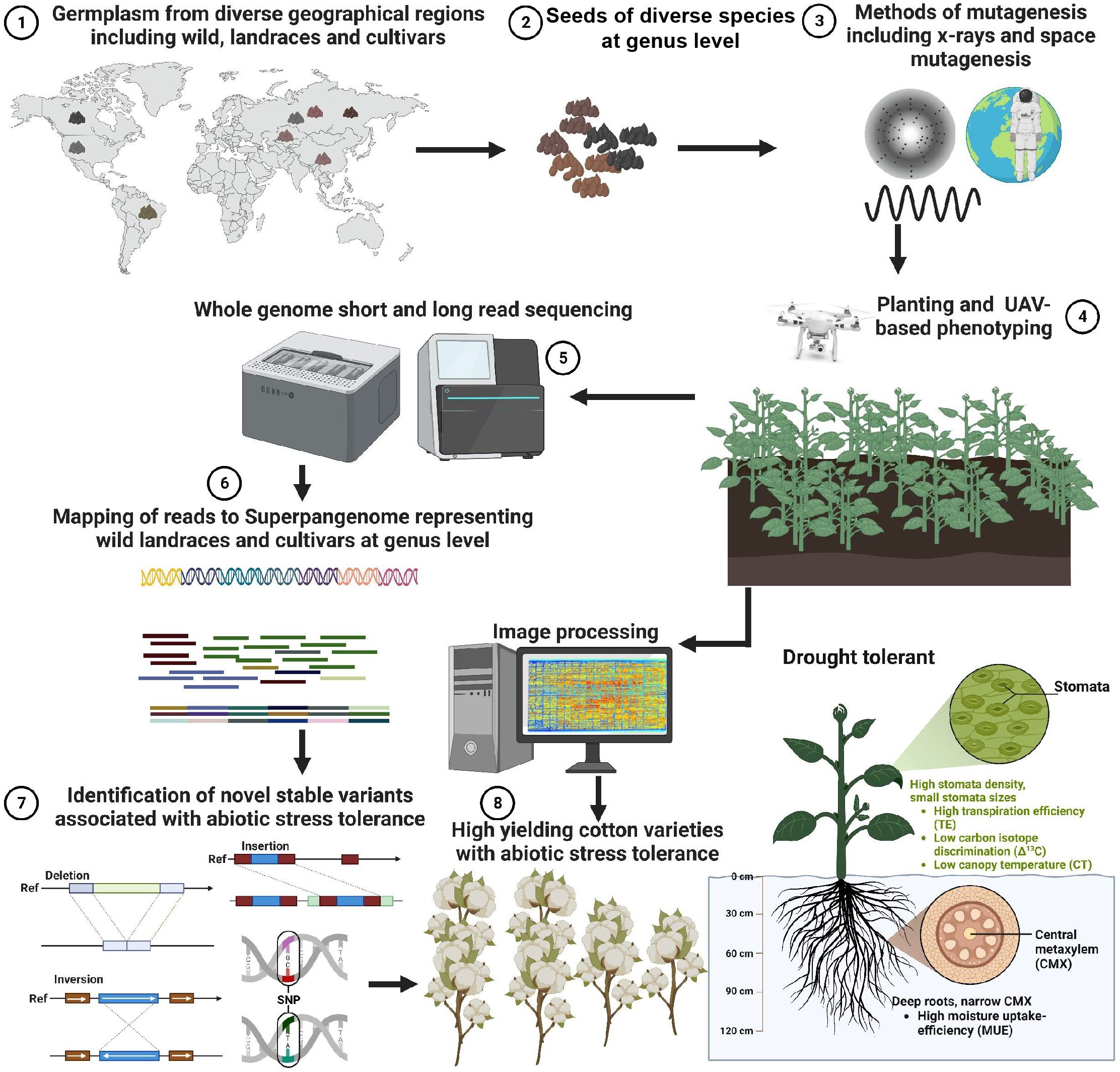
Figure 5 Harnessing Mutation Breeding for Enhanced Stress Tolerance in Cotton Varieties. Mutation breeding offers a promising strategy for the development of cotton varieties resilient to abiotic stress while maintaining high yield potential. To achieve this, a diverse collection of cotton germplasm encompassing wild type, landrace, and cultivar varieties from different global regions is selected at the genus level. Mutations are induced in this germplasm pool through either space-based or x-ray-based mutagenic sources. Subsequently, unmanned aerial vehicle-based (UAV) phenotyping techniques are employed to screen the mutants for specific desired traits. Plants exhibiting the desired phenotypes undergo comprehensive whole-genome sequencing using both long-read and short-read technologies. Notably, emerging super pangenome strategies (Shang et al., 2022) can be employed for read mapping, enabling comprehensive analysis instead of relying on a single linear reference genome. This innovative approach facilitates the identification of novel insertions, deletions, inversions, and single nucleotide polymorphisms (SNPs) associated with stress tolerance, particularly in the context of drought stress induced by climate change.
One critical aspect for advancing mutation breeding lies in the careful selection of a wide-ranging and diverse array of germplasm. This includes wild types, landraces, and cultivars sourced from various regions across the globe. By combining such diversity with the deliberate introduction of mutations, crop adaptability to fluctuating environmental conditions can be significantly enhanced.
Advanced mutagenic sources, such as space-based and x-ray-based techniques, are poised to play a pivotal role in shaping the future of mutation breeding. These sources efficiently induce mutations, allowing for the creation of novel genetic variations that can confer stress tolerance in cotton.
The future of mutation breeding also entails the use of cutting-edge technology for efficient phenotyping. Unmanned aerial vehicles equipped with state-of-the-art phenotyping techniques are set to transform the screening of mutant populations on a large scale. This innovation will enable researchers to swiftly identify and select plants exhibiting the desired phenotypes, thereby expediting the breeding process.
Similarly, comprehensive whole-genome sequencing is vital for unraveling the genetic underpinnings of mutant plants. The adoption of super pangenome strategies, as demonstrated in recent research, opens new horizons for genus-level genetic analysis. This approach not only captures genetic diversity but also reveals genomic complexity, aiding in the identification of novel genetic variants associated with stress tolerance.
Illustrative case studies in rice (Shang et al., 2022) and barley (Jayakodi et al., 2020) underscore the potential of super pangenome strategies. These studies highlight the benefits of pangenomes in capturing genetic diversity and identifying genetic variation, in particular, where frequent inversions were identified from germplasms.
In conclusion, the future of mutation breeding in cotton has the potential to significantly enhance its resilience to environmental challenges while sustaining high productivity levels. This can be achieved through the strategic utilization of diverse germplasms, advanced mutagenic sources, UAV-based phenotyping, the adoption of pangenome strategies, and the application of novel algorithms and machine learning techniques to accurately detect induced mutations, distinguishing them from spontaneous mutations, and thereby uncovering their functional impact on plant phenotypes.
Author contributions
PW: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. MA: Investigation, Writing – review & editing. JH: Investigation, Writing – review & editing. LZ: Investigation, Writing – review & editing. HC: Investigation, Writing – review & editing. HG: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors declare that this study received funding from the Nanfan special project, CAAS (Grant Nos. ZDXM2303 and YBXM14) and Win all Hi-tech Seed Co., Ltd. (GMLM2023). The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1400201/full#supplementary-material
Supplementary Table 1 | EMS mutants in cotton.
Supplementary Table 2 | Plant mutant sequencing methods.
Supplementary Table 3 | Functional genes identified by mutants in cotton.
References
Abbas, M., Abid, M. A., Meng, Z., Abbas, M., Wang, P., Lu, C., et al. (2022). Integrating advancements in root phenotyping and genome-wide association studies to open the root genetics gateway. Physiologia Plantarum 174, e13787. doi: 10.1111/ppl.13787
Abe, A., Kosugi, S., Yoshida, K., Natsume, S., Takagi, H., Kanzaki, H., et al. (2012). Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 30, 174–178. doi: 10.1038/nbt.2095
Abid, M. A., Wang, P., Zhu, T., Liang, C., Meng, Z., Malik, W., et al. (2020). Construction of Gossypium barbadense mutant library provides genetic resources for cotton germplasm improvement. Int. J. Mol. Sci. 21 (18), 6505. doi: 10.3390/ijms21186505
Alban, C., Tardif, M., Mininno, M., Brugière, S., Gilgen, A., Ma, S., et al. (2014). Uncovering the protein lysine and arginine methylation network in Arabidopsis chloroplasts. PloS One 9, e95512. doi: 10.1371/journal.pone.0095512
Arabidopsis Genome Initiative (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815. doi: 10.1038/35048692
Bi, M., Wang, Z., Cheng, K., Cui, Y., He, Y., Ma, J., et al. (2023). Construction of transcription factor mutagenesis population in tomato using a pooled CRISPR/Cas9 plasmid library. Plant Physiol. Biochem. 205, 108094. doi: 10.1016/j.plaphy.2023.108094
Cao, Y., Huang, H., Yu, Y., Dai, H., Hao, H., Zhang, H., et al. (2021). A modified actin (Gly65Val substitution) expressed in cotton disrupts polymerization of actin filaments leading to the phenotype of ligon lintless-1 (Li1) mutant. Int. J. Mol. Sci. 22, 3000. doi: 10.3390/ijms22063000
Chaudhary, J., Deshmukh, R., Sonah, H. (2019). Mutagenesis approaches and their role in crop improvement. Plants 8, 467. doi: 10.3390/plants8110467
Chen, X., Lu, X., Shu, N., Wang, S., Wang, J., Wang, D., et al. (2017). Targeted mutagenesis in cotton (Gossypium hirsutum L.) using the CRISPR/Cas9 system. Sci. Rep. 7, 44304. doi: 10.1038/srep44304
Chen, Z. J., Scheffler, B. E., Dennis, E., Triplett, B. A., Zhang, T., Guo, W., et al. (2007). Toward sequencing cotton (Gossypium ) genomes. Plant Physiol. 145, 1303–1310. doi: 10.1104/pp.107.107672
Ding, M., Jiang, Y., Cao, Y., Lin, L., He, S., Zhou, W., et al. (2014). Gene expression profile analysis of Ligon lintless-1 (Li1) mutant reveals important genes and pathways in cotton leaf and fiber development. Gene. 535, 273–285. doi: 10.1016/j.gene.2013.11.017
Fang, D. D., Naoumkina, M., Thyssen, G. N., Bechere, E., Li, P., Florane, C. B. (2020). An EMS-induced mutation in a tetratricopeptide repeat-like superfamily protein gene (Ghir_A12G008870) on chromosome A12 is responsible for the liy short fiber phenotype in cotton. Theor. Appl. Genet. 133, 271–282. doi: 10.1007/s00122–019-03456–4
Fekih, R., Takagi, H., Tamiru, M., Abe, A., Natsume, S., Yaegashi, H., et al. (2013). MutMap+: genetic mapping and mutant identification without crossing in rice. PloS One 8, e68529. doi: 10.1371/journal.pone.0068529
Gao, J., Shi, Y., Wang, W., Wang, Y. H., Yang, H., Shi, Q. H., et al. (2021). Genome sequencing identified novel mechanisms underlying virescent mutation in upland cotton Gossypium hirsutum. BMC Genomics 22, 498. doi: 10.1186/s12864–021-07810-z
Garcia, V., Bres, C., Just, D., Fernandez, L., Tai, F. W., Mauxion, J. P., et al. (2016). Rapid identification of causal mutations in tomato EMS populations via mapping-by-sequencing. Nat. Protoc. 11, 2401–2418. doi: 10.1038/nprot.2016.143
Gelvin, S. B. (2017). Integration of agrobacterium T-DNA into the plant genome. Annu. Rev. Genet. 51, 195–217. doi: 10.1146/annurev-genet-120215–035320
Golicz, A. A., Batley, J., Edwards, D. (2016). Towards plant pangenomics. Plant Biotechnol. J. 14, 1099–1105. doi: 10.1111/pbi.12499
Gong, W., Zhou, Y., Wang, R., Wei, X., Zhang, L., Dai, Y., et al. (2021). Analysis of T-DNA integration events in transgenic rice. J. Plant Physiol. 266, 153527. doi: 10.1016/j.jplph.2021.153527
Guo, S., Wang, Y., Sun, G., Jin, S., Zhou, T., Meng, Z., et al. (2015). Twenty years of research and application of transgenic cotton in china. Sci.Agi. Sin. 48, 3372–3387. doi: 10.3864/j.issn.0578-1752.2015.17.005
Greene, E. A., Codomo, C. A., Taylor, N. E., Henikoff, J. G., Till, B. J., Reynolds, S. H., et al. (2003). Spectrum of chemically induced mutations from a large-scale reverse-genetic screen in Arabidopsis. Genetics. 164, 731–740. doi: 10.1093/genetics/164.2.731
Hagen, J. B. (2000). The origins of bioinformatics. Nat. Rev. Genet. 1, 231–236. doi: 10.1038/35042090
He, X., Qi, Z., Liu, Z., Chang, X., Zhang, X., Li, J., et al. (2024). Pangenome analysis reveals transposon-driven genome evolution in cotton. BMC Biol. 22, 92. doi: 10.1186/s12915–024-01893–2
He, J., Zeng, C., Li, M. (2023). Plant functional genomics based on high-throughput CRISPR library knockout screening: A perspective. Advanced Genet. (Hoboken N.J.) 5, 2300203. doi: 10.1002/ggn2.202300203
He, Z. P., Zhou, Q. Q., Xu, S. P., Xu, J., Shi, W. J., Zhu, W. J. (2001). Higher lint percent and drought tolerant cotton line selected by radiation breeding. Shi yan sheng wu xue bao. 34, 51–54. doi: 10.1007/978–1-4020–5578-2_31
Hill, J. T., Demarest, B. L., Bisgrove, B. W., Gorsi, B., Su, Y. C., Yost, H. J. (2013). MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 23, 687–697. doi: 10.1101/gr.146936.112
Hu, Y., Chen, J., Fang, L., Zhang, Z., Ma, W., Niu, Y., et al. (2019). Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 51, 739–748. doi: 10.1038/s41588–019-0371–5
Huang, G., Huang, J. Q., Chen, X. Y., Zhu, Y. X. (2021). Recent advances and future perspectives in cotton research. Annu. Rev. Plant Biol. 72, 437–462. doi: 10.1146/annurev-arplant-080720–113241
Jankowicz-Cieslak, J., Mba, C., Till, B. J. (2017). “Mutagenesis for crop breeding and functional genomics,” in Biotechnologies for plant mutation breeding: protocols, New York NY: Springer 3–18.
Jayakodi, M., Padmarasu, S., Haberer, G., Bonthala, V. S., Gundlach, H., Monat, C., et al. (2020). The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 588, 284–289. doi: 10.1038/s41586-020-2947-8
Jia, P., Ding, L., Zhou, B., Guo, H., Gao, F. (2012). Construction of a T-DNA insertional mutant library for verticillium dahliae kleb. and analysis of a mutant phenotype. Cotton Science. 24, 62–70. doi: 10.7124/bc.000239
Jill Harrison, C. (2016). Development and genetics in the evolution of land plant body plans. Philos. Trans. R Soc. Lond B Biol. Sci. 372, 20150490. doi: 10.1098/rstb.2015.0490
Johannes, F., Schmitz, R. J. (2019). Spontaneous epimutations in plants. New Phytol. 221, 1253–1259. doi: 10.1111/nph.15434
Ke, C., Guan, W., Bu, S., Li, X., Deng, Y., Wei, Z., et al. (2019). Determination of absorption dose in chemical mutagenesis in plants. PloS One 14, e0210596. doi: 10.1371/journal.pone.0210596
Kole, C., Gupta, P. K. (2004). Genome mapping and map based cloning. Plant Breeding., 257–299. doi: 10.1007/978–94-007–1040-5_11
Kong, D., Qu, L., Zhang, X., Liu, J., Wang, P., Li, F. (2017). Optimization of EMS mutagenesis condition and screening of mutants in gossypium arboretum l. Cotton Sci. 29, 336–344. doi: 10.11963/1002-7807.KDPLFG.20170703
Krasileva, K. V., Vasquez-Gross, H. A., Howell, T., Bailey, P., Paraiso, F., Clissold, L., et al. (2017). Uncovering hidden variation in polyploid wheat. Proc. Natl. Acad. Sci. U S A. 114, E913–E921. doi: 10.1073/pnas.1619268114
Kubo, T., Yamagata, Y., Matsusaka, H., Toyoda, A., Sato, Y., Kumamaru, T. (2022). Whole-genome sequencing of rice mutant library members induced by N-methyl-N-nitrosourea mutagenesis of fertilized egg cells. Rice (New York N.Y.) 15, 38. doi: 10.1186/s12284–022-00585–1
Kumar, U., Chavan, N. S., Sawant, S. V., et al. (2022). Evaluation and characterization of EMS induced mutant population of gossypium herbaceum. Vegetos 35, 1036–1046. doi: 10.1007/s42535-022-00385-4
Kushanov, F. N., Komilov, D. J., Turaev, O. S., Ernazarova, D. K., Amanboyeva, R. S., Gapparov, B. M., et al. (2022). Genetic analysis of mutagenesis that induces the photoperiod insensitivity of wild cotton Gossypium hirsutum subsp. purpurascens. Plants 11, 3012. doi: 10.3390/plants11223012
Li, F., Fan, G., Lu, C., Xiao, G., Zou, C., Kohel, R. J., et al. (2015). Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 33, 524–530. doi: 10.1038/nbt.3208
Li, F., Fan, G., Wang, K., Sun, F., Yuan, Y., Song, G., et al. (2014). Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 46, 567–572. doi: 10.1038/ng.2987
Li, S. C., Yang, L., Deng, Q. M., Wang, S. Q., Wu, F. Q., Li, P. (2006). Phenotypic characterization of a female sterile mutant in rice. J. Integr. Plant Biol. 48, 307–314. doi: 10.1111/j.1744–7909.2006.00228.x
Li, J., Yuan, D., Wang, P., Wang, Q., Sun, M., Liu, Z., et al. (2021). Cotton pan-genome retrieves the lost sequences and genes during domestication and selection. Genome Biol. 22, 119. doi: 10.1186/s13059–021-02351-w
Lian, X., Liu, Y., Guo, H., Fan, Y., Wu, J., Guo, H., et al. (2020). Ethyl methanesulfonate mutant library construction in Gossypium hirsutum L. for allotetraploid functional genomics and germplasm innovation. Plant J. 103, 858–868. doi: 10.1111/tpj.14755
Liu, Y., Du, H., Li, P., Shen, Y., Peng, H., Liu, S., et al. (2020). Pan-genome of wild and cultivated soybeans. Cell. 182, 162–176.e13. doi: 10.1016/j.cell.2020.05.023
Liu, X., Zhao, B., Zheng, H. J., Hu, Y., Lu, G., Yang, C. Q., et al. (2015). Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites. Sci. Rep. 5, 14139. doi: 10.1038/srep14139
Lu, X., Liu, J., Ren, W., Yang, Q., Chai, Z., Chen, R., et al. (2018). Gene-indexed mutations in maize. Mol. Plant 11, 496–504. doi: 10.1016/j.molp.2017.11.013
Lu, Y., Ye, X., Guo, R., Huang, J., Wang, W., Tang, J., et al. (2017). Genome-wide targeted mutagenesis in rice using the CRISPR/cas9 system. Mol. Plant 10, 1242–1245. doi: 10.1016/j.molp.2017.06.007
Maluszynski, M., Ahloowalia, B., Sigurbjörnsson, B. (1995). Application of in vivo and in vitro mutation techniques for crop improvement. Euphytica. 85, 303–315. doi: 10.1007/bf00023960
Mascher, M., Jost, M., Kuon, J. E., Himmelbach, A., Aßfalg, A., Beier, S., et al. (2014). Mapping-by-sequencing accelerates forward genetics in barley. Genome Biol. 15, 1–15. doi: 10.1186/gb-2014-15-6-r78
McGarry, R. C., Prewitt, S. F., Culpepper, S., Eshed, Y., Lifschitz, E., Ayre, B. G. (2016). Monopodial and sympodial branching architecture in cotton is differentially regulated by the Gossypium hirsutum SINGLE FLOWER TRUSS and SELF-PRUNING orthologs. New Phytol. 212, 244–258. doi: 10.1111/nph.14037
Meng, X., Yu, H., Zhang, Y., Zhuang, F., Song, X., Gao, S., et al. (2017). Construction of a genome-wide mutant library in rice using CRISPR/cas9. Mol. Plant 10, 1238–1241. doi: 10.1016/j.molp.2017.06.006
Michelmore, R. W., Paran, I., Kesseli, R. V. (1991). Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. U.S.A. 88, 9828–9832. doi: 10.1073/pnas.88.21.9828
Morgan, W., Day, J., Kaplan, M., McGhee, E., Limoli, C. (1996). Genomic instability induced by ionizing radiation. Radiat. Res. 146, 247. doi: 10.2307/3579454
Naoumkina, M., Bechere, E., Fang, D. D., Thyssen, G. N., Florane, C. B. (2017). Genome-wide analysis of gene expression of EMS-induced short fiber mutant Ligon lintless-y ( liy ) in cotton ( Gossypium hirsutum L.). Genomics. 109, 320–329. doi: 10.1016/j.ygeno.2017.05.007
Naoumkina, M., Thyssen, G. N., Fang, D. D., Bechere, E., Li, P., Florane, C. B. (2021). Mapping-by-sequencing the locus of EMS-induced mutation responsible for tufted-fuzzless seed phenotype in cotton. Mol. Genet. Genomics 296, 1041–1049. doi: 10.1007/s00438–021-01802–0
Naoumkina, M., Thyssen, G. N., Fang, D. D., Hinchliffe, D. J., Florane, C. B., Jenkins, J. N. (2016). Small RNA sequencing and degradome analysis of developing fibers of short fiber mutants Ligon-lintles-1 (Li 1 ) and -2 (Li 2 ) revealed a role for miRNAs and their targets in cotton fiber elongation. BMC Genomics 17, 360. doi: 10.1186/s12864–016-2715–1
Nerkar, G., Devarumath, S., Purankar, M., Kumar, A., Valarmathi, R., Devarumath, R., et al. (2022). Advances in crop breeding through precision genome editing. Front. Genet. 13, 880195. doi: 10.3389/fgene.2022.880195
Ng, P. C., Kirkness, E. F. (2010). Whole genome sequencing. Methods Mol. Biol. 628, 215–226. doi: 10.1007/978–1-60327–367-1_12
Paterson, A. H., Wendel, J. F., Gundlach, H., Guo, H., Jenkins, J., Jin, D., et al. (2012). Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature. 492, 423–427. doi: 10.1038/nature11798
Ramadan, M., Alariqi, M., Ma, Y., Li, Y., Liu, Z., Zhang, R., et al. (2021). Efficient CRISPR/Cas9 mediated Pooled-sgRNAs assembly accelerates targeting multiple genes related to male sterility in cotton. Plant Methods 17, 16. doi: 10.1186/s13007–021-00712-x
Schneeberger, K., Weigel, D. (2011). Fast-forward genetics enabled by new sequencing technologies. Trends Plant Sci. 16, 282–288. doi: 10.1016/j.tplants.2011.02.006
Shang, L., Li, X., He, H., Yuan, Q., Song, Y., Wei, Z., et al. (2022). A super pan-genomic landscape of rice. Cell Res. 32, 878–896. doi: 10.1038/s41422-022-00685-z
Si, Z., Liu, H., Zhu, J., Chen, J., Wang, Q., Fang, L., et al. (2018). Mutation of SELF-PRUNING homologs in cotton promotes short-branching plant architecture. J. Exp. Bot. 69, 2543–2553. doi: 10.1093/jxb/ery093
Sun, L., Alariqi, M., Wang, Y., Wang, Q., Xu, Z., Zafar, M. N., et al. (2024). Construction of host plant insect-resistance mutant library by high-throughput CRISPR/cas9 system and identification of A broad-spectrum insect resistance gene. Advanced Sci. (Weinheim Baden-Wurttemberg Germany) 11, e2306157. doi: 10.1002/advs.202306157
Takagi, H., Abe, A., Yoshida, K., Kosugi, S., Natsume, S., Mitsuoka, C., et al. (2013b). QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 74, 174–183. doi: 10.1111/tpj.12105
Takagi, H., Tamiru, M., Abe, A., Yoshida, K., Uemura, A., Yaegashi, H., et al. (2015). MutMap accelerates breeding of a salt-tolerant rice cultivar. Nat. Biotechnol. 33, 445–449. doi: 10.1038/nbt.3188
Takagi, H., Uemura, A., Yaegashi, H., Tamiru, M., Abe, A., Mitsuoka, C., et al. (2013a). MutMap-Gap: whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol. 200, 276–283. doi: 10.1111/nph.12369
Thyssen, G. N., Fang, D. D., Zeng, L., Song, X., Delhom, C. D., Condon, T. L., et al. (2016). The immature fiber mutant phenotype of cotton (Gossypium hirsutum) is linked to a 22-bp frame-shift deletion in a mitochondria targeted pentatricopeptide repeat gene. G3 (Bethesda). 6, 1627–1633. doi: 10.1534/g3.116.027649
Udall, J. A., Long, E., Hanson, C., Yuan, D., Ramaraj, T., Conover, J. L., et al. (2019). De Novo Genome Sequence Assemblies of Gossypium raimondii and Gossypium turneri. G3 (Bethesda). 9, 3079–3085. doi: 10.1534/g3.119.400392
Wang, S., Chen, J., Zhang, W., Hu, Y., Chang, L., Fang, L., et al. (2015). Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes. Genome Biol. 216, 108. doi: 10.1186/s13059–015-0678–1
Wang, M., Li, J., Qi, Z., Long, Y., Pei, L., Huang, X., et al. (2022). Genomic innovation and regulatory rewiring during evolution of the cotton genus Gossypium. Nat. Genet. 54, 1959–1971. doi: 10.1038/s41588–022-01237–2
Wang, D., Li, Y., Wang, H., Xu, Y., Yang, Y., Zhou, Y., et al. (2023). Boosting wheat functional genomics via an indexed EMS mutant library of KN9204. Plant Commun. 4, 100593. doi: 10.1016/j.xplc.2023.100593
Wang, M., Wang, P., Liang, F., Ye, Z., Li, J., Shen, C., et al. (2018). A global survey of alternative splicing in allopolyploid cotton: landscape, complexity and regulation. New Phytol. 217 (1), 163–178. doi: 10.1111/nph.14762
Wang, C., Tang, S., Zhan, Q., Hou, Q., Zhao, Y., Zhao, Q., et al. (2019). Dissecting a heterotic gene through GradedPool-Seq mapping informs a rice-improvement strategy. Nat. Commun. 10, 2982. doi: 10.1038/s41467–019-11017-y
Wang, M., Tu, L., Yuan, D., Zhu, D., Shen, C., Li, J., et al. (2019). Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 51, 224–229. doi: 10.1038/s41588–018-0282-x
Wang, K., Wang, Z., Li, F., Ye, W., Wang, J., Song, G., et al. (2012). The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 44, 1098–1103. doi: 10.1038/ng.2371
Wang, X. H., Zhao, X. M., Ye, K., Chen, W. W., Mei, Y., Wang, H. (2013). Application and advance in the space flight mutation breeding in agriculture. J. Anhui Agri Sci. 41, 9575–9576, 9598. doi: 10.1109/icbeb.2012.365
Wei, Y., Liu, Y., ABID, M. A., Xiao, R., Liang, C., Meng, Z., et al. (2022). Rich variant phenotype of gossypium hirsutum l. saturated mutant library provides resources for cotton functional genomics and breeding. Ind. Crops Products 186. doi: 10.1016/j.indcrop.2022.115232
Wendel, J. F. (1989). New World tetraploid cottons contain Old World cytoplasm. Proc. Natl. Acad. Sci. U.S.A. 86, 4132–4136. doi: 10.1073/pnas.86.11.4132
Wendel, J. F., Brubaker, C., Alvarez, I., Cronn, R., Stewart, J. M. (2009). “Evolution and natural history of the cotton genus,” in Genetics and genomics of cotton, Springer, New York, NY 3–22. doi: 10.1007/978–0-387–70810-2_1
Ye, Y., Wang, P., Zhang, M., Abbas, M., Zhang, J., Liang, C., et al. (2023). UAV-based time-series phenotyping reveals the genetic basis of plant height in upland cotton. Plant J. doi: 10.1111/tpj.16272
Yu, J., Hu, S., Wang, J., Wong, G. K., Li, S., Liu, B., et al. (2002). A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science. 296, 79–92. doi: 10.1126/science.1068037
Yu, L. H., Wu, S. J., Peng, Y. S., Liu, R. N., Chen, X., Zhao, P., et al. (2016). Arabidopsis EDT1/HDG11 improves drought and salt tolerance in cotton and poplar and increases cotton yield in the field. Plant Biotechnol. J. 14 (1), 72–84. doi: 10.1111/pbi.12358
Yuan, D., Tang, Z., Wang, M., Gao, W., Tu, L., Jin, X., et al. (2015). The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci. Rep. 5, 17662. doi: 10.1038/srep17662
Yue, J., Zou, J. (2012). Study of radiation effects on upland cotton (Gossypium hirsutum L.) pollen grain irradiated by 60Co-gamma ray. J. Agri Sci. 4, 7. doi: 10.5539/jas.v4n7p85
Zhang, B. (2013). Agrobacterium-mediated transformation of cotton. Methods Mol. Biol. 958, 31–45. doi: 10.1007/978–1-4939–8952-2_2
Zhang, T., Hu, Y., Jiang, W., Fang, L., Guan, X., Chen, J., et al. (2015). Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537. doi: 10.1038/nbt.3207
Zhang, H., Wang, X., Pan, Q., Li, P., Liu, Y., Lu, X., et al. (2019). QTG-seq accelerates QTL fine mapping through QTL partitioning and whole-genome sequencing of bulked segregant samples. Mol. Plant 12, 426–437. doi: 10.1016/j.molp.2018.12.018
Zhao, Z., Liu, Z., Zhou, Y., Wang, J., Zhang, Y., Yu, X., et al. (2022). Creation of cotton mutant library based on linear electron accelerator radiation mutation. Biochem. Biophys. Rep. 30, 101228. doi: 10.1016/j.bbrep.2022.101228
Zhao, S., Luo, J., Zeng, X., Li, K., Yuan, R., Zhu, L., et al. (2020). Rolling Circle Amplification (RCA)-Mediated Genome-Wide ihpRNAi Mutant Library Construction in Brassica napus. Int. J. Mol. Sci. 21, 7243. doi: 10.3390/ijms21197243
Zhou, H., Tang, K., Li, G., Liu, W., Yu, H., Yuan, X., et al. (2021). A robust and rapid candidate gene mapping pipeline based on M2 populations. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.681816
Keywords: cotton, sequencing, diversity, mutant library, germplasm resources
Citation: Wang P, Abbas M, He J, Zhou L, Cheng H and Guo H (2024) Advances in genome sequencing and artificially induced mutation provides new avenues for cotton breeding. Front. Plant Sci. 15:1400201. doi: 10.3389/fpls.2024.1400201
Received: 13 March 2024; Accepted: 10 June 2024;
Published: 02 July 2024.
Edited by:
Junhua Peng, Spring Valley Agriscience Co., Ltd, Jinan, ChinaReviewed by:
Zhaoen Yang, Institute of Cotton Research, Chinese Academy of Agricultural Sciences, ChinaZhengwen Liu, Chinese Academy of Agricultural Sciences (CAAS), China
Si Huan, Tobacco Research Institute, Chinese Academy of Agricultural Sciences, China
Copyright © 2024 Wang, Abbas, He, Zhou, Cheng and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peilin Wang, d2FuZ3BlaWxpbjE5QDEyNi5jb20=; Huiming Guo, Z3VvaHVpbWluZ0BjYWFzLmNu
†These authors have contributed equally to this work