 Kaushal Pratap Singh
Kaushal Pratap Singh Preetesh Kumari
Preetesh Kumari Pramod Kumar Rai
Pramod Kumar Rai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 24 June 2024
Sec. Plant Breeding
Volume 15 - 2024 | https://doi.org/10.3389/fpls.2024.1381387
This article is part of the Research TopicManipulation of Plant Architecture for Crop ProductionView all 10 articles
Plant architecture is a crucial determinant of crop yield. The number of primary (PB) and secondary branches (SB) is particularly significant in shaping the architecture of Indian mustard. In this study, we analyzed a panel of 86 backcross introgression lines (BCILs) derived from the first stable allohexaploid Brassicas with 170 Sinapis alba genome-specific SSR markers to identify associated markers with higher PB and SB through association mapping. The structure analysis revealed three subpopulations, i.e., P1, P2, and P3, in the association panel containing a total of 11, 33, and 42 BCILs, respectively. We identified five novel SSR markers linked to higher PB and SB. Subsequently, we explored the 20 kb up- and downstream regions of these SSR markers to predict candidate genes for improved branching and annotated them through BLASTN. As a result, we predicted 47 complete genes within the 40 kb regions of all trait-linked markers, among which 35 were identified as candidate genes for higher PB and SB numbers in BCILs. These candidate genes were orthologous to ANT, RAMOSUS, RAX, MAX, MP, SEU, REV, etc., branching genes. The remaining 12 genes were annotated for additional roles using BLASTP with protein databases. This study identified five novel S. alba genome-specific SSR markers associated with increased PB and SB, as well as 35 candidate genes contributing to plant architecture through improved branching numbers. To the best of our knowledge, this is the first report of introgressive genes for higher branching numbers in B. juncea from S. alba.
Crop wild relatives (CWRs) have been established as valuable sources for incorporating genes for adaptation in crop plants under changing climatic conditions by allowing modifications in crop genetic constitution and developing high-yielding crop varieties (Hajjar and Hodgkin, 2007; Bohra et al., 2022). Among CWRs, Brassica wild relatives (BWRs) have maintained valuable genetic diversity for improving yield-related and climate-resilient traits, such as plant vigor, architecture, and the ability to withstand biotic and abiotic stresses. By enriching the gene pool of crop species, BWRs have contributed significantly by augmenting the genetic diversity of cultivated plants (Quezada-Martinez et al., 2021). The family Brassicaceae includes several wild genera, such as Brassica fruticulosa, Camelina sativa, Diplotaxis catholica, D. erucoides, Erucastrum abyssinicum, Erucastrum cardaminoides, Eruca sativa, Moricandia arvensis, and Sinapis alba. Among these, S. alba (white mustard), commonly grown as a condiment, has the highest number of PB and SB (Kumari et al., 2011; Singh et al., 2023). Interestingly, S. alba is diploid (2n= 24, SS) and has close genetic proximity to Brassica nigra L. (BB, 2n=16), a diploid ancestor of the Indian mustard (Warwick and Black, 1991).
Indian mustard (B. juncea; AABB; 2n=36) is an important oilseed crop cultivated globally for its edible oil, vegetables, and condiments (Banga and Banga, 2016). The genetic diversity of Brassica oilseed crops has become limited due to strong selection pressure for yield and quality attributes, which has made it increasingly challenging to further increase yields. Consequently, the production levels of these crops have plateaued (Singh et al., 2021a). Therefore, the enhancement of crop yield depends on the introduction of yield-related alien genes to augment the genetic basis. However, the introgression of alien genes is a tedious and time-consuming process (Kumari et al., 2020b, 2020c). Thus, the closest genera of the crop are a more reliable source for gene introduction due to the inhibition of the introduction of alien genes by intergeneric incompatibility, hybrid sterility, and reduced or absent chromosome pairing between alien and crop species and the resulting linkage drag (Bohra et al., 2022). Subsequent generations of hybrids and early backcross progenies exhibited high levels of male and female sterility due to abnormal meiosis (Lelivelt et al., 1993). In vitro fusion or protoplast fusion was initially used to introduce genes to overcome the pre- and postfertilization barriers within intergeneric hybridizations that contributed to biotic and abiotic stress tolerance in Indian mustard (Kirti et al., 1992, 1995). Moreover, genetic engineering-induced and spontaneous mutations have also been used to introduce genetic diversity. However, due to the amphidiploid nature of B. juncea, the application of reverse genetics to identify mutations that cause quantifiable phenotypic impacts is challenging. Since each phenotype is controlled by at least two mustard homoeologs with presumably redundant activities, combining mutant homoeologs from both subgenomes is necessary to change a monogenic trait (Wells et al., 2014; Emrani et al., 2015). For the trait regulated by an oligogenic or polygenic system in polyploid species, genome editing using CRISPR-Cas9 is not a realistic approach (Endo et al., 2015).
To overcome the consequences of intergeneric hybridization, we used PEG-mediated protoplast fusion to introduce new alleles of S. alba into B. juncea to avoid potential negative effects such as intergeneric incompatibility and hybrid sterility. Fortunately, we succeeded in achieving the first stable and fertile somatic hybrids of S. alba and B. juncea with proper meiosis (Kumari et al., 2018; Kumari and Bhat, 2019, 2021). These hybrids were then backcrossed with B. juncea as the recurrent parent, resulting in the first backcross progeny with a haploid set of S. alba chromosomes with a high degree of gamete viability and subsequent fertility. Among the BC1F2 generation, we identified a backcross line with the highest number of PBs (26–27) and SB (166) (Kumari et al., 2020b). The shoot branching pattern is controlled at various levels, such as node pattern, meristem determination, and axillary meristem elongation (McSteen and Leyser, 2005). Several genes regulate node patterns in A. thaliana, such as LATERAL SUPPRESSOR (LAS) (Greb et al., 2003), SHOOT MERISTEMLESS (STM) (Long et al., 1996), REVOLUTA (REV) (Talbert et al., 1995), and the REGULATORS OF AXILLARY MERISTEMS (RAX) genes (Keller et al., 2006; Muller et al., 2006). The inflorescence meristem identity is determined by the floral identity genes TERMINAL FLOWER1 (TFL1) (Bradley et al., 1997) and LEAFY (LFY) (Weigel et al., 1992). Moreover, branch elongation is regulated by several phytohormones, such as auxin, cytokinin, and abscisic acid (Ward and Leyser, 2004). Critical analyses of genes that regulate auxin signaling and transport, such as AUXIN RESISTANT1 (AXR1) (Lincoln et al., 1990; Leyser et al., 1993; Stirnberg et al., 1999) and MORE AXILLARY GROWTH (MAX) (Stirnberg et al., 2002; Sorefan et al., 2003; Booker et al., 2005; Bennett et al., 2006), have shown that these hormones play a central role in branch development.
However, a major challenge was the identification of the genes responsible for the segmental introgressions of B. juncea chromosomes, which were introduced in advance BCILs. To address this issue, we developed a set of SSR markers specific to the S. alba genome from the draft assembly and used them to genotype core sets of BCILs (Kumari et al., 2020a; Singh et al., 2021b, 2022b). Using a set of 170 monoallelic SSR markers specific to S. alba, we identified associated markers with high PB and SB by using genotypic and phenotypic data from two successive years. We utilized association mapping, which is a reliable method for addressing quantitative variations, to better understand introgressed variants (Risch and Merikangas, 1996; Nordborg and Tavaré, 2002; Gupta et al., 2014). The BCILs used in this study have euploid chromosomal counts high fertility, and are stabilized as translocation homozygotes.
Although the size of the introgressed segment likely plays a crucial role in defining BCILs, physical linkage significantly impacts the linkage disequilibrium (LD) between molecular markers (Remington et al., 2001). This provided a genetic basis for the association mapping of genes responsible for a substantial proportion of PB and SB. The extent of LD between linked markers in the complete set of BCILs was notably greater than that between unlinked markers. This study reports the results of association mapping experiments using markers specific to the donor genome and phenotypic data that were subsequently recorded from BC2F6-7 generations from two successive crop seasons. We identified closely linked markers to pinpoint the genetic regions responsible for PB and SB. The discovery of these markers will facilitate the rapid introduction of the higher branching phenotype into new germplasms, marker-assisted breeding, and selection.
We employed a subset of 86 backcross introgression lines (BCILs; BC2F6-7) of S. alba-B. juncea somatic hybrids with their parents (S. alba and B. juncea) for association studies on the number of branches in fully matured plants. These BCILs were derived by backcrossing two stable allohexaploids of B. juncea and S. alba, namely, H1 and H2, as previously reported by Kumari et al. (2018) and Kumari and Bhat (2019, 2021), with B. juncea cv. RLM-198 and NPJ-212 (Kumari et al., 2020b).
Independent field trials were conducted at the agricultural farm of the ICAR-Directorate of Rapeseed Mustard Research, Bharatpur, India, during the 2021–22 (CS-I) and 2022–23 (CS-II) crop seasons. The experimental field is situated at 77.300°E and 27.150°N. To grow the BCILs, a randomized complete block design was employed with three replications, and standard farming techniques were implemented for mustard without the use of fertilizers or fungicides. The BCILs were cultivated using 45 cm row-to-row and 30 cm plant-to-plant spacing within each row. Five plants at random from each replication of the BCILs were selected to record the phenotypic data, which included the number of PB and SB at the end of flowering.
In this study, we utilized the variability package (https://CRAN.R-project.org/package=variability; Singh and Chaudhary, 1977) in R v4.2.1 to estimate several attributes, including the standard error of the mean (SEm), the critical difference at 5% (CD), broad-sense heritability (H2), analysis of variance (ANOVA), environmental variation (EV), genotypic variation (GV), phenotypic variation (PV), coefficient of variation (CV), and frequency distribution among the 86 BCILs.
Genomic DNA was extracted from fresh young leaves of both the parent plants and the backcross progeny (BC2F6) using a modified CTAB method (Kirti et al., 1995). The extracted DNA was then quantified using a Nanodrop 8000 spectrophotometer (Thermo Fisher Scientific, USA).
Genotyping was performed using S. alba genome-specific SSR markers (Singh et al., 2022b) that were developed using a de novo whole-genome assembly (Kumari et al., 2020a). A set of 170 S. alba-specific SSR markers randomly chosen from the whole genome was used to genotype 86 BCILs of the BC2F6 generation, revealing morphological variations in PB and SB.
The genotyping study used a 10 μl PCR mixture containing 1.6 μl of template DNA (30 ng/μl), 1.1 μl of primer pairs (forward and reverse) (10 mM), 0.30 μl of dNTP mix (10 mM), 1.1 μl of MgCl2 (2.5 mM), 1.1 μl of Taq DNA polymerase buffer (10X), 0.30 μl of Taq DNA polymerase (GCC Biotech, India) (2.5 U), and 4.5 μl of nuclease-free water. The PCR conditions involved initial denaturation for 5 min at 94°C, followed by 40 cycles of denaturation at 94°C/30 s, primer annealing at 57–59°C/40 s, and primer extension at 72°C/45 s, and a final extension at 72°C for 7 min. The PCR products were separated on a 3% agarose gel and visualized using ethidium bromide staining in a gel documentation unit. The amplified bands were scored as present (1) or absent (0) for genotyping analysis (Kumari et al., 2024).
The population structure of the BCILs was analyzed using the model-based Bayesian clustering method in STRUCTURE v2.3.4. A burn-in period of 10,000 and 100,000 Markov chain Monte Carlo (MCMC) iterations was used, with k ranging from 2 to 10, to investigate the population structure (Pritchard et al., 2000). The ideal number of subpopulations (K) was determined using Evanno’s method through the use of STRUCTURE HARVESTER (Earl and VonHoldt, 2012). In TASSEL v5.0 (http://www.maizegenetics.net/), the genotypic data were utilized to calculate the genetic distance across BCILs, and a phylogenetic tree was constructed using the unweighted pair group method with arithmetic mean (UPGMA) method (Bradbury et al., 2007).
Once the K value was established, the population structure matrices (Q) were identified using STRUCTURE. TASSEL was used to estimate the kinship coefficients (K-matrix) among all genotypes (BCILs) based on similarities in the SSR markers. The association analysis between individual SSR and phenotypic values was conducted at the p-value 1e-4 using mixed linear model (MLM) and general linear model (GLM) approaches, combining K and Q matrices (Bradbury et al., 2007). Every marker used for genotyping was employed in the association analysis. A quantile–quantile (QQ) plot was used to compare the relative distributions of the observed and expected -log10(P)-values for each SSR marker–trait association. Principal component analysis (PCA) was used to stratify the population structure. Eigenvalue analysis (Patterson et al., 2006; Price et al., 2006) and a kinship matrix (Malosetti et al., 2007; Pasam et al., 2012) were used to eliminate the effects of structure on the mapping panel and relatedness between the genotypes. To assess linkage disequilibrium, r2 was calculated for each allele of the SSR marker using TASSEL software. The calculation was performed using the LD full matrix, with heterozygous calls set to missing. The r2 threshold was set at 0.1, so any SSR marker with a r2 value below that threshold was considered to have weak LD (Nyine et al., 2019). A graph in the form of an LD heatmap was generated using TASSEL to visualize the distribution pattern of the genome-wide LD decay.
The SSR markers that were found to be associated with branching traits were aligned with the S. alba genome. The 40k bp genome sequence located up- and downstream of the SSR was used for complete gene prediction through the AUGUSTUS web server (https://bioinf.uni-greifswald.de/augustus/submission.php), with A. thaliana serving as the reference organism. The genes predicted through AUGUSTUS were then annotated using BLASTP with the UniProtKB/Swiss-Prot (Swissprot_v5), nonredundant protein sequence (Nr_v5), and RefSeq protein databases. These predicted genes were also subjected to a local BLASTP for domain searches using CLC Genomics Workbench version 20.0.4 (Qiagen, USA). Furthermore, the PANNZER web server (http://ekhidna2.biocenter.helsinki.fi/sanspanz/) was utilized to predict protein functions (Toronen and Holm, 2022). To further annotate these 47 predicted genes, 1127 branching-related genes in FASTA format were retrieved from the European Nucleotide Archive (ENA). Finally, the predicted genes were subjected to BLASTN with a transcriptome assembly of H1 allohexaploid to confirm their presence in BCILs (Singh et al., 2022a).
During the CS-I and CS-II crop seasons at the ICAR-Directorate of Rapeseed Mustard Research, phenotypic data were collected from backcross introgression lines (BCILs) of S. alba and B. juncea. The PB and SB numbers were recorded and showed considerable variability among the BCILs. For example, BCIL 70 had an average minimum of 2.0 PB, while the donor parent S. alba in CS-II had an average maximum of 21.0 PB. The grand mean values for the number of PB were 6.32 ± 0.77 and 6.48 ± 0.45 for CS-I and CS-II, respectively. The donor parent S. alba had the highest average number of SB at 43.6, followed by BCILs 10 (26.2) and 83 (24), while BCILs 49 (5), 32 (6.6), 28 (7), 66, and 70 (7.2) had the lowest average number of SB. The grand mean values for the number of SB were 14.314 ± 3.06 and 14.36 ± 1.42 for CS-I and CS-II, respectively (Figures 1A, B). The cumulative effect of genotypic and environmental variance for both crop seasons resulted in the phenotypic variance of PB and SB, respectively. The phenotypic coefficient of variation (PCV) was greater than the genotypic coefficient of variation (GCV) for both traits, indicating a significant environmental influence. However, both PB and SB exhibited high broad-sense heritability (H2) values, suggesting that these traits are not influenced by environmental factors. The genetic variability for these traits was low, with genetic advancement percentages of the mean (GAM) determined to be 36.67 and 51.30 for PB and 36.55 and 69.31 for SB, respectively (Table 1). The correlation between PB and SB in both seasons was very high, as shown in the correlation heatmap (Figure 2).
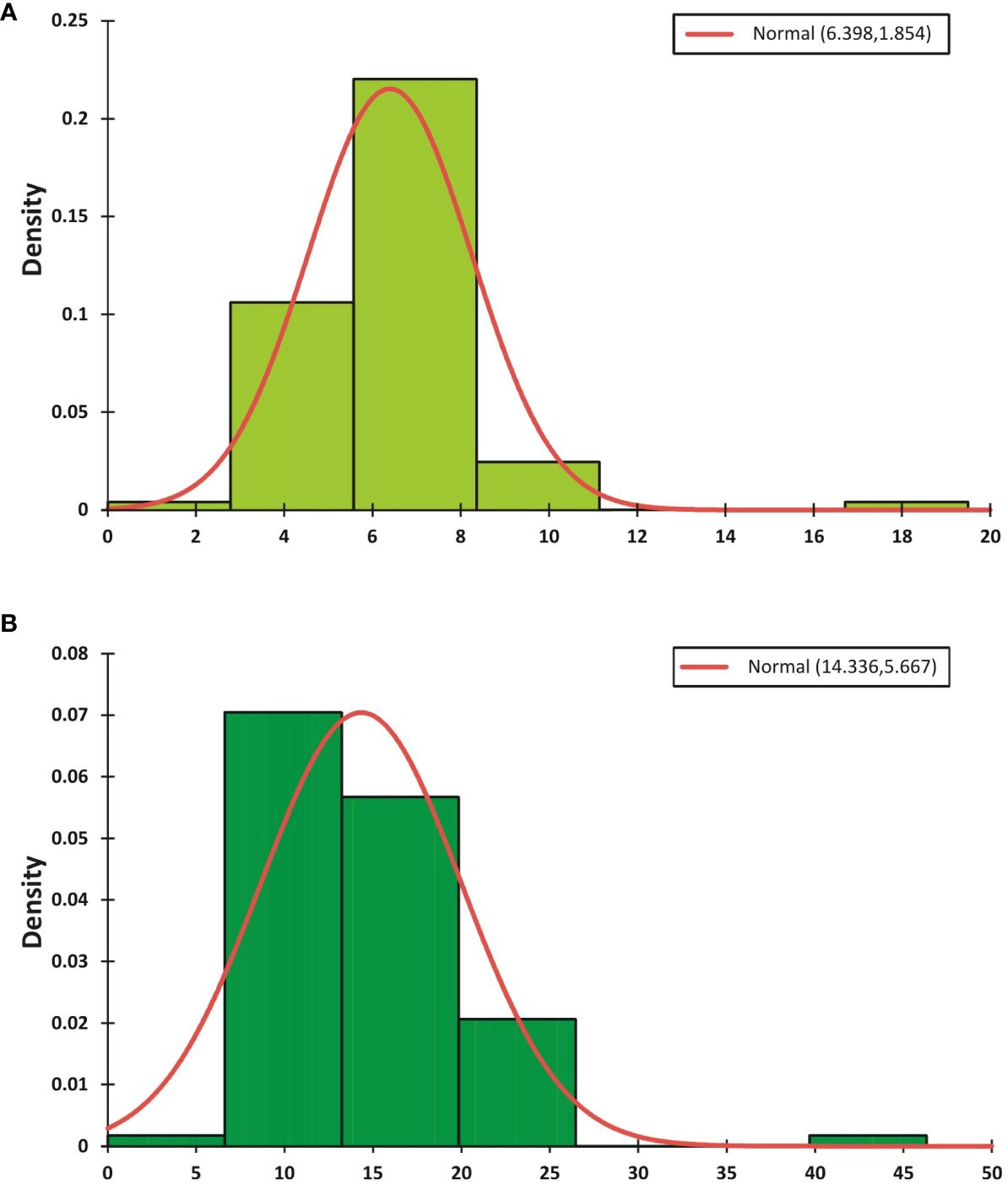
Figure 1 The mean phenotypic distributions of the PB (A) and SB (B) traits for the 2021-22 and 2022-23 crop seasons. The grand mean value and standard deviation (StDev) are presented in the top right corner of the graph.
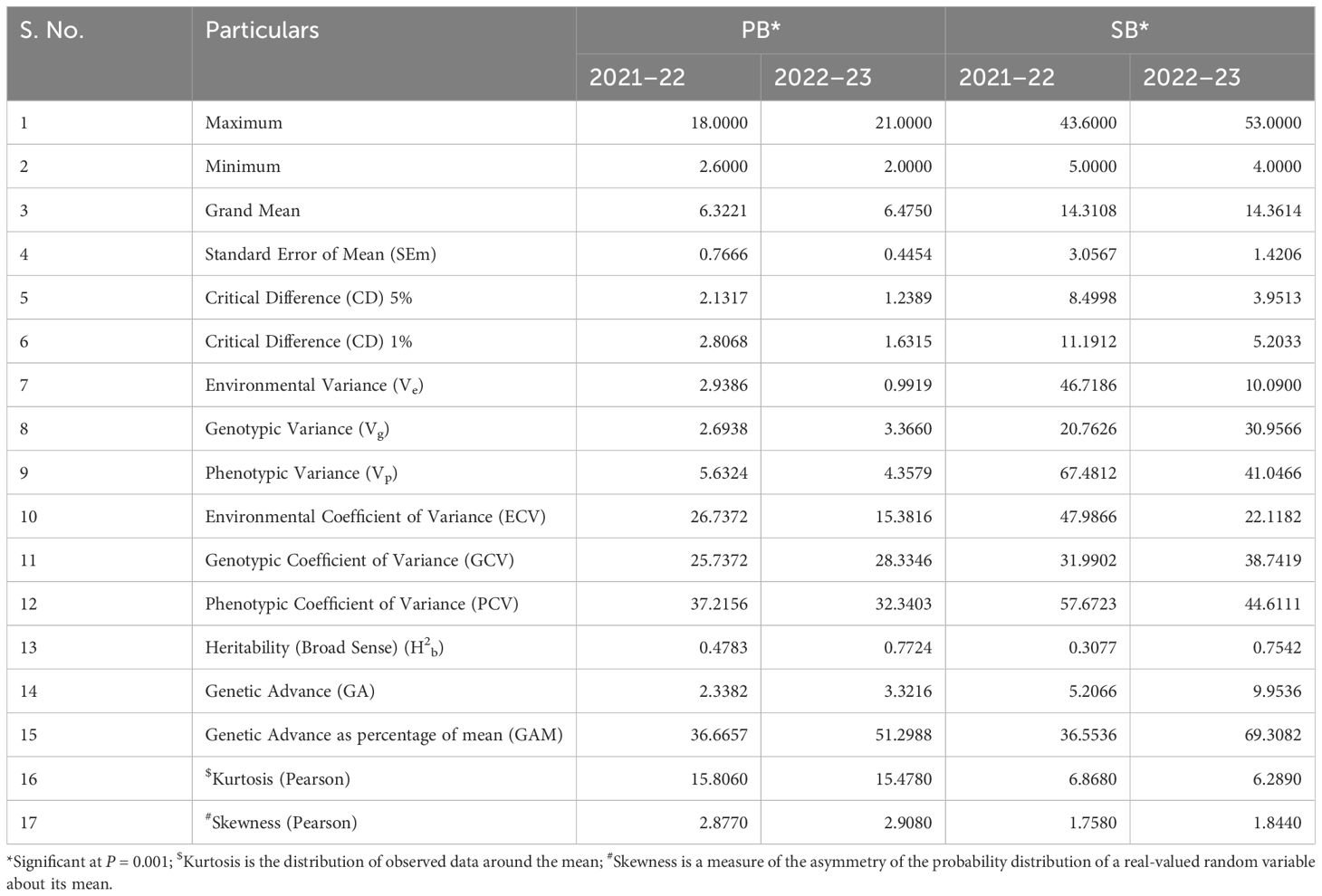
Table 1 Descriptive statistics of the primary (PB) and secondary branching (SB) traits evaluated in a set of 86 backcross lines (BCILs) of S. alba + B. juncea allohexaploids.
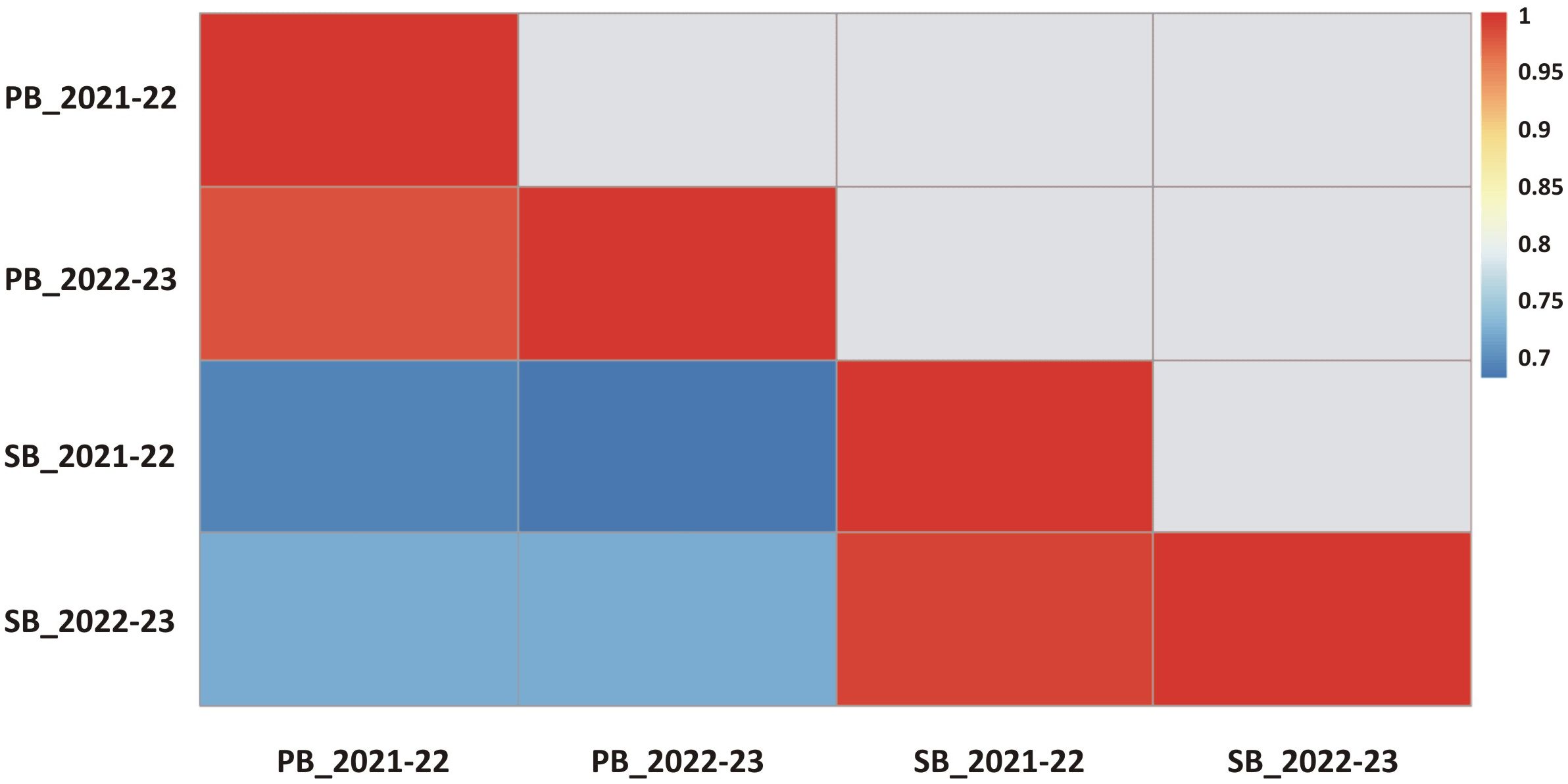
Figure 2 The correlation coefficients between branching traits for different crop seasons are presented (all coefficients were found to be statistically significant at a p value <0.01).
Population structure analysis was conducted on the association panel using 170 donor genome-specific microsatellite markers. Clustering inference was performed with possible clusters (k) ranging from 2 to 10, with five replicates for each k value. The likelihood distribution (LnP k) showed a significant change when k increased from 2 to 3, with the maximum Δk value observed at k=3 (Figures 3A, B). The Δk scheme revealed that the 86 BCILs could be grouped into three subpopulations, namely, P1 (donor group; S. alba), P2 (mixed population), and P3 (recipient parent group; B. juncea). The association panel had 11, 33, and 42 BCILs assigned to the P1, P2, and P3 populations, respectively (Figure 4A). These groups were also confirmed through kinship analysis (Figure 4B) and UPGMA-based phylogenetic analysis (Figure 4C). The phylogenetic tree constructed using the UPGMA method showed three major clades corresponding to the three groups identified by STRUCTURE. BCILs belonging to the mixed population group were distributed between the P1 and P2 subpopulations in the phylogenetic tree. Linkage disequilibrium (LD) analysis and association mapping were conducted using 170 SSR markers in a subset of 86 BCILs. The TASSEL v. 5.0 program was used to calculate LD as the squared allele frequency correlation (r2) (Bradbury et al., 2007).
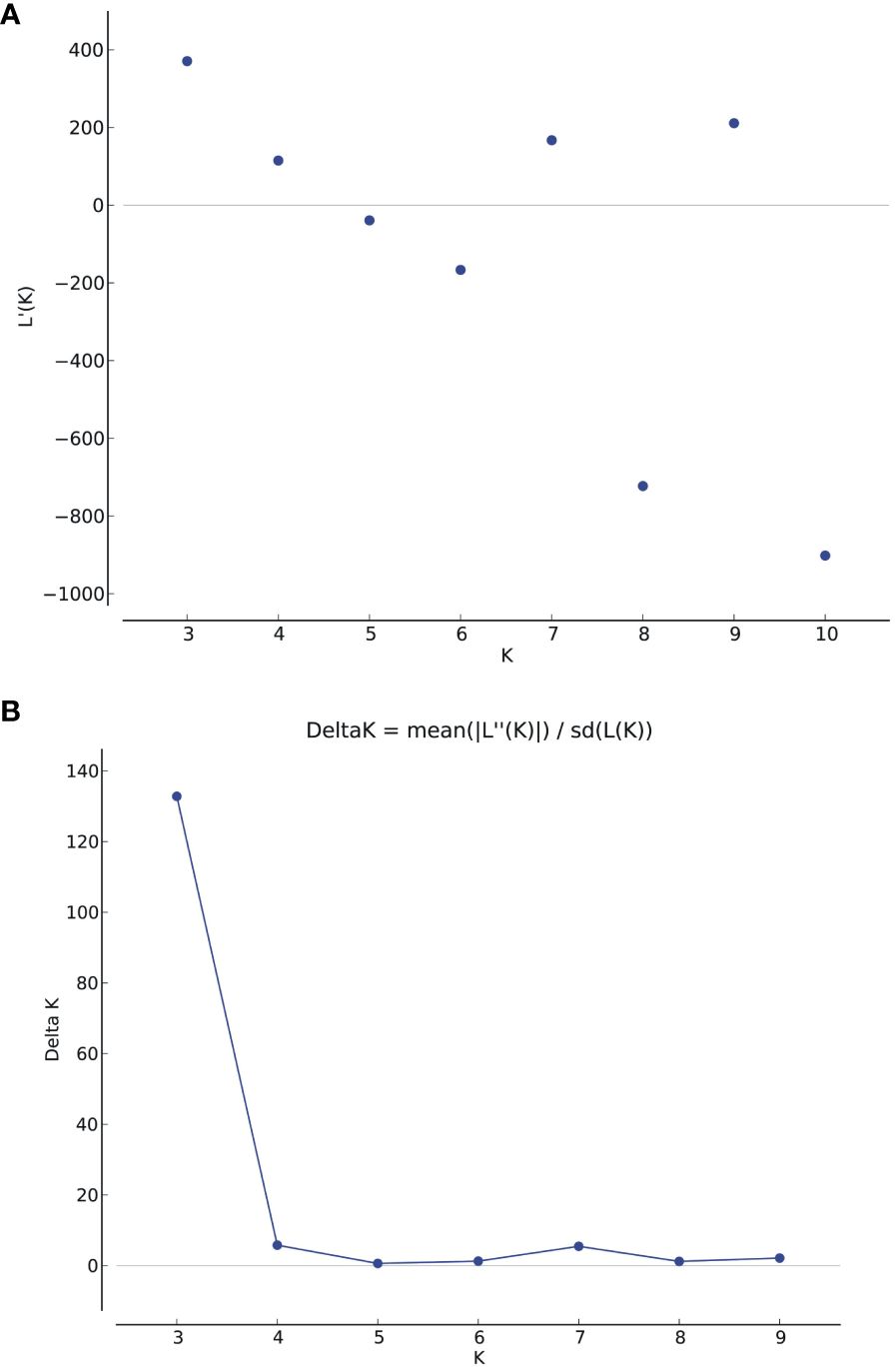
Figure 3 (A) The graph displays the estimated logarithm of the probability of data [LnP(D)] for possible clusters (k) ranging from 1 to 10; (B) Delta K is based on the rate of change in LnP (K) between successive K values.
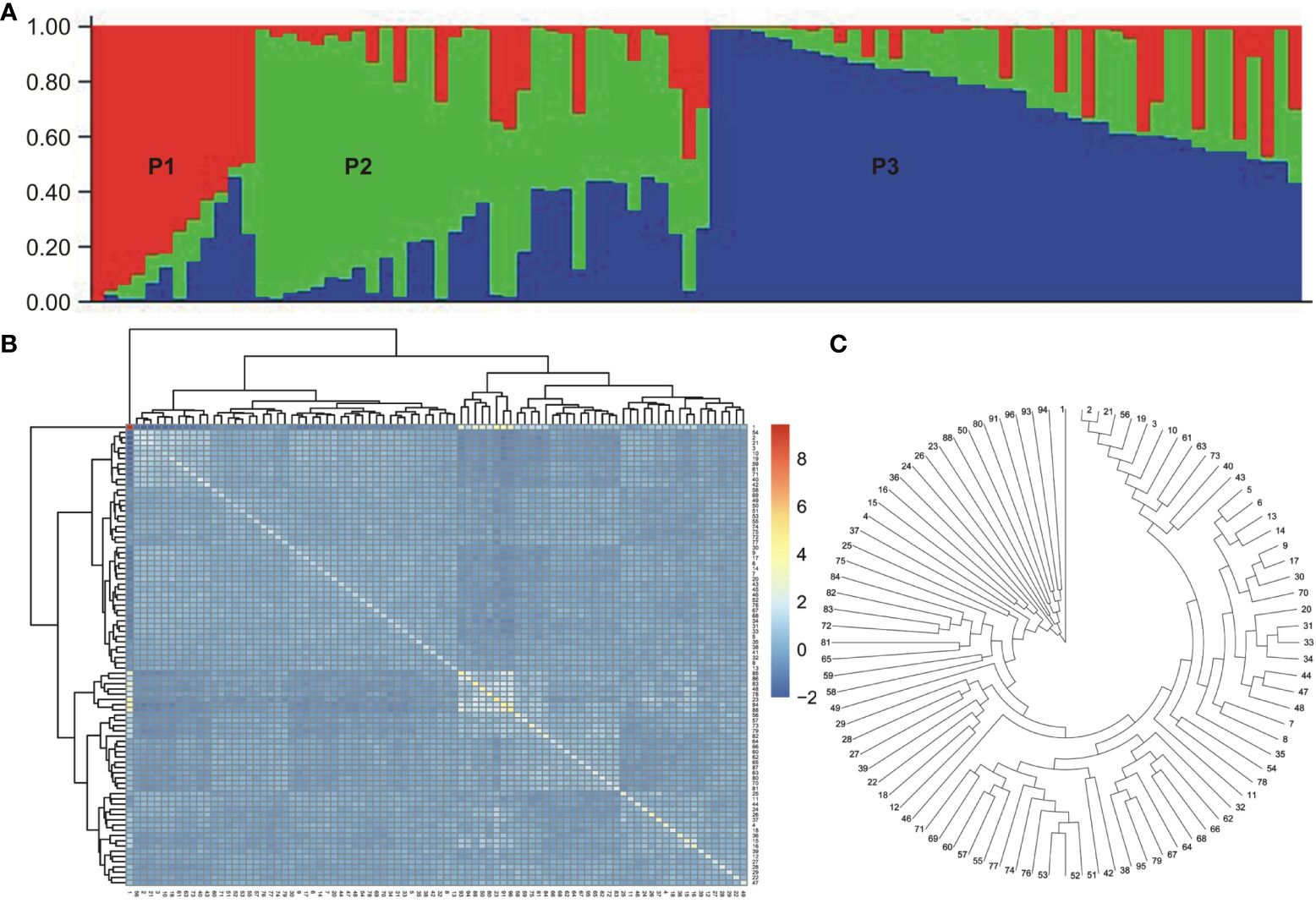
Figure 4 Population structure, Kinship analysis, and UPGMA tree for the mapping panel used in the current study. (A) Population structure. Here, different subpopulations (i.e., P1, P2, P3) are indicated using different colors. (B) Kinship dendrogram. (C) UPGMA tree.
Pairwise linkage disequilibrium (LD) was examined in the current genotyping panel using 170 SSR markers to assess the amount of LD at a sliding window size of 50, resulting in the detection of LD in 7225 locus pairs. Among these, 2535 marker pairs (35%) showed substantial LD at the r2 threshold of 0.05, while significant LD was observed for 1309 (18%) and 433 (6%) marker pairings at much higher r2 values of 0.1 and 0.2, respectively. The details of both the R2 and P values are presented in Figure 5. Additionally, principal component analysis (PCA) was conducted to assess the genetic diversity among the association panels (BCILs), with the first two PCs explaining 100% of the genetic variation. The branching trait donor parent was present in the first quadrant, while the recipient plant (B. juncea) was present in the fourth quadrant (Figure 6A). The correlation circle showed a very strong positive correlation between the PB and SB traits (Figure 6B). A total of 87 axes were identified through eigenvalue analysis of the relationships between individual BCILs in the association panel, with the first ten (1–10) principal components accounting for approximately 52% of the variation (Figure 6C). The distance matrix was calculated between the BCILs based on their genotyping data, revealing their closeness between 0 and 1, where 0 represented the highest closeness and 1 the greatest distance between BCILs. BCILs 91, 23, 96, and 93 showed distances of 0.435294, 0.458824, 0.464706, and 0.488235, respectively, from the donor parent S. alba (Line 01), while BCILs 10 (0.911765), 3 (0.929412), 19 (0.935294), 56 (0.947059), and 21 (0.964706) showed the greatest distances from the donor parent. Additionally, the distance matrix of the recipient parent (B. juncea) from the donor parent, S. alba, was 1.
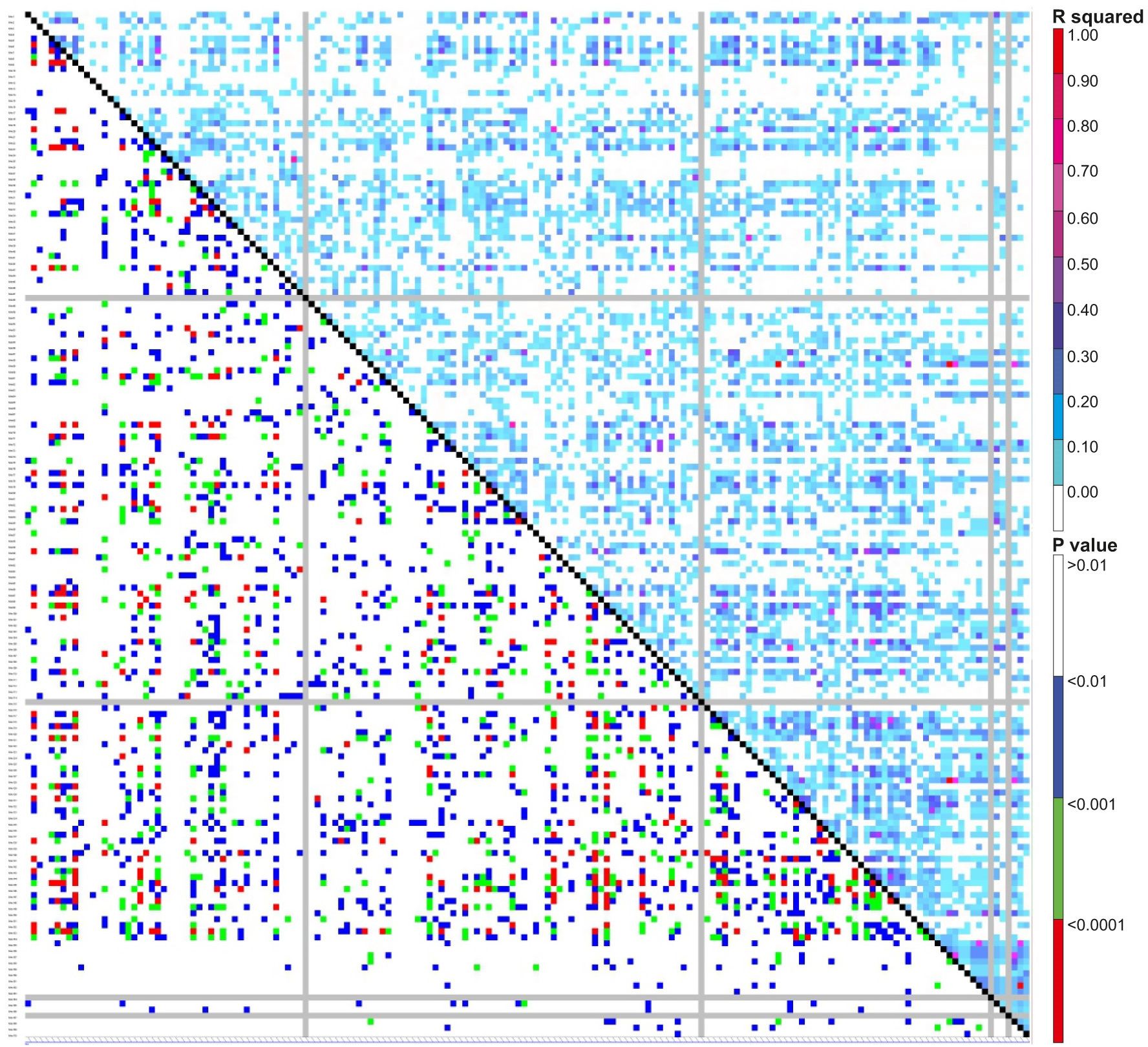
Figure 5 Graphical representation of the LD plot (linkage disequilibrium plot) for the current association mapping panel. Here, the R square and P value are shown as measures of LD (upper and lower diagonal, respectively).
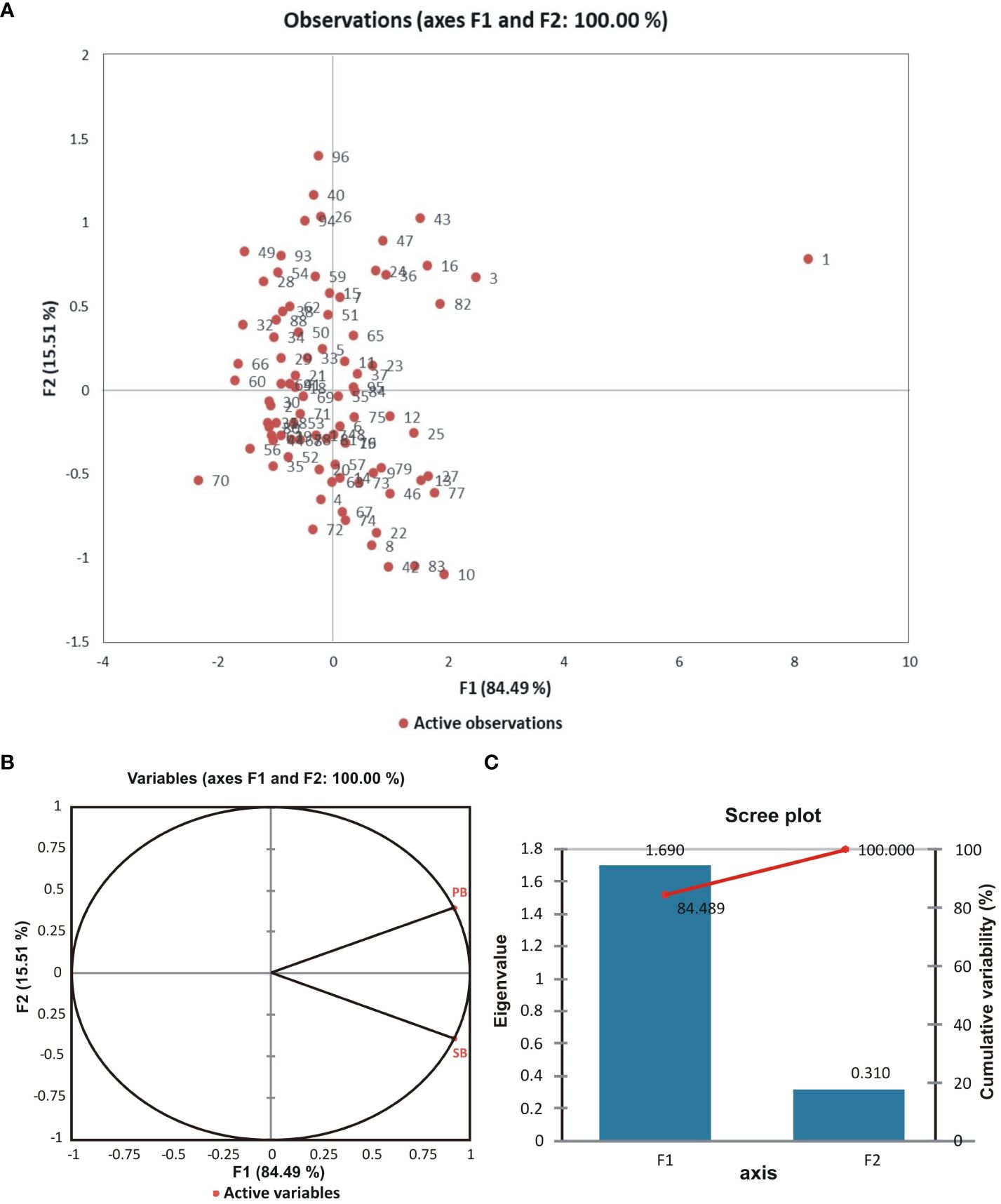
Figure 6 (A) Principal component analysis (PCA) between observations (BCILs); (B) Circle of correlation between variables (phenotypic traits) after principal component analysis (numbers on the axes represent the correlation coefficient); (C) A scree plot showing the eigenvalues of the F1 and F2 factors with their percent cumulative effects on the phenotypes.
Marker–trait associations were identified using two different statistical models, MLM and GLM. The MLM used the population structure and kinship matrix (Q + K) model, while the GLM used numerical genotypic and phenotypic data. Five associations (P<0.0001) were identified using MLM for the PB and SB traits, with five common microsatellite markers (Sa28738, Sa42751, Sa164052, Sa225736, and Sa325747) associated with both traits. These markers were distributed randomly in the association panel. These associations were recorded above the threshold value log10(P)= 4.00, with the markers showing an association with the traits at the threshold value 4.934. The total phenotypic variations explained by these markers for PB and SB were 4.55 and 28.95%, respectively. Using GLM, five common microsatellite markers were identified as being associated with the PB and SB traits, which were the same as those identified using MLM. The associations were recorded above the threshold value log10(P)= 6.4, with the markers showing an association with the traits between the threshold values of 6.482 and 10.21. The figures provided more details on the marker–trait associations identified using both models (Figures 7A, B, 8A, B).
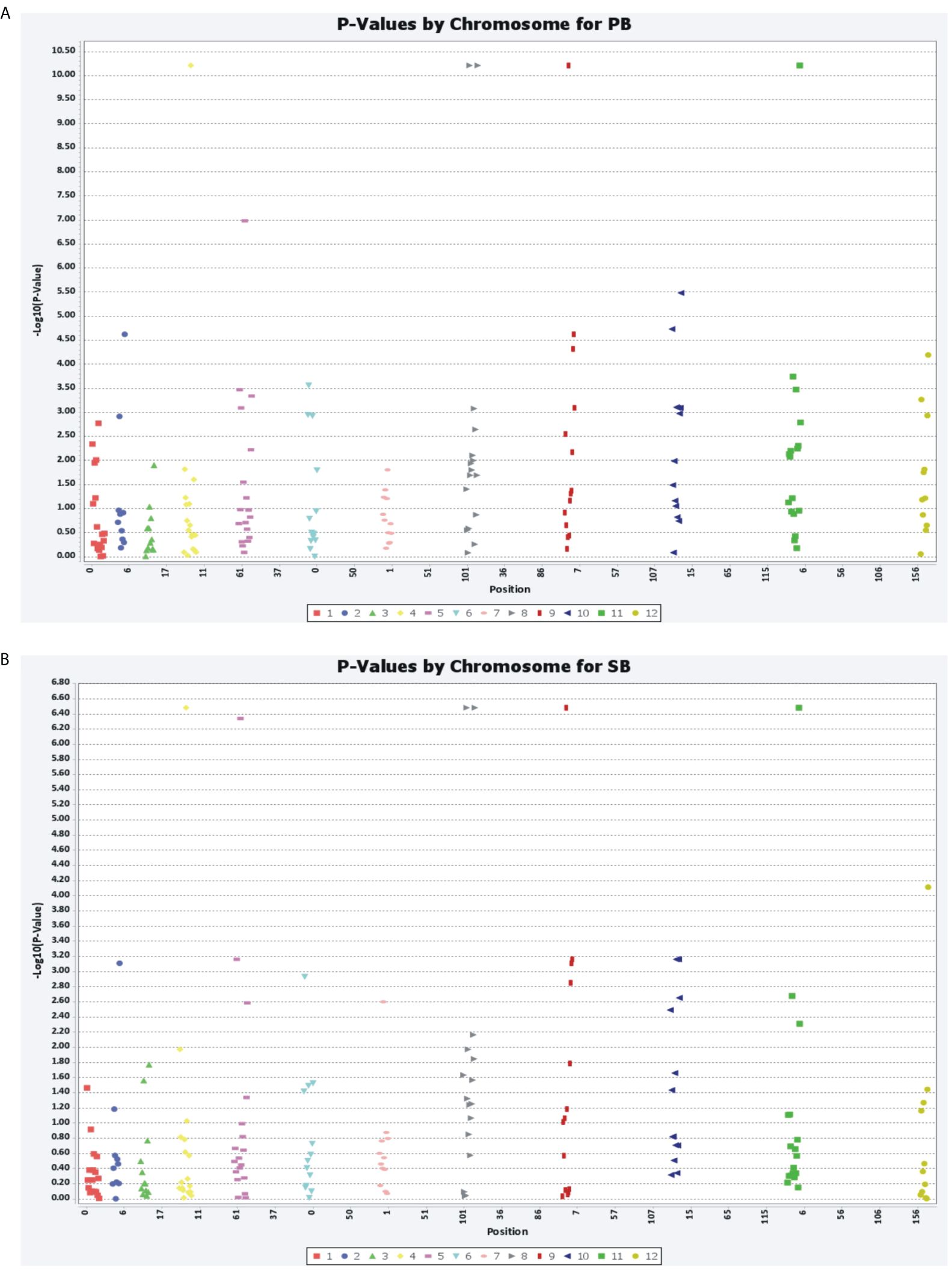
Figure 7 General linear model (GLM) showing the association of SSR markers with PB (A) and SB (B) traits.
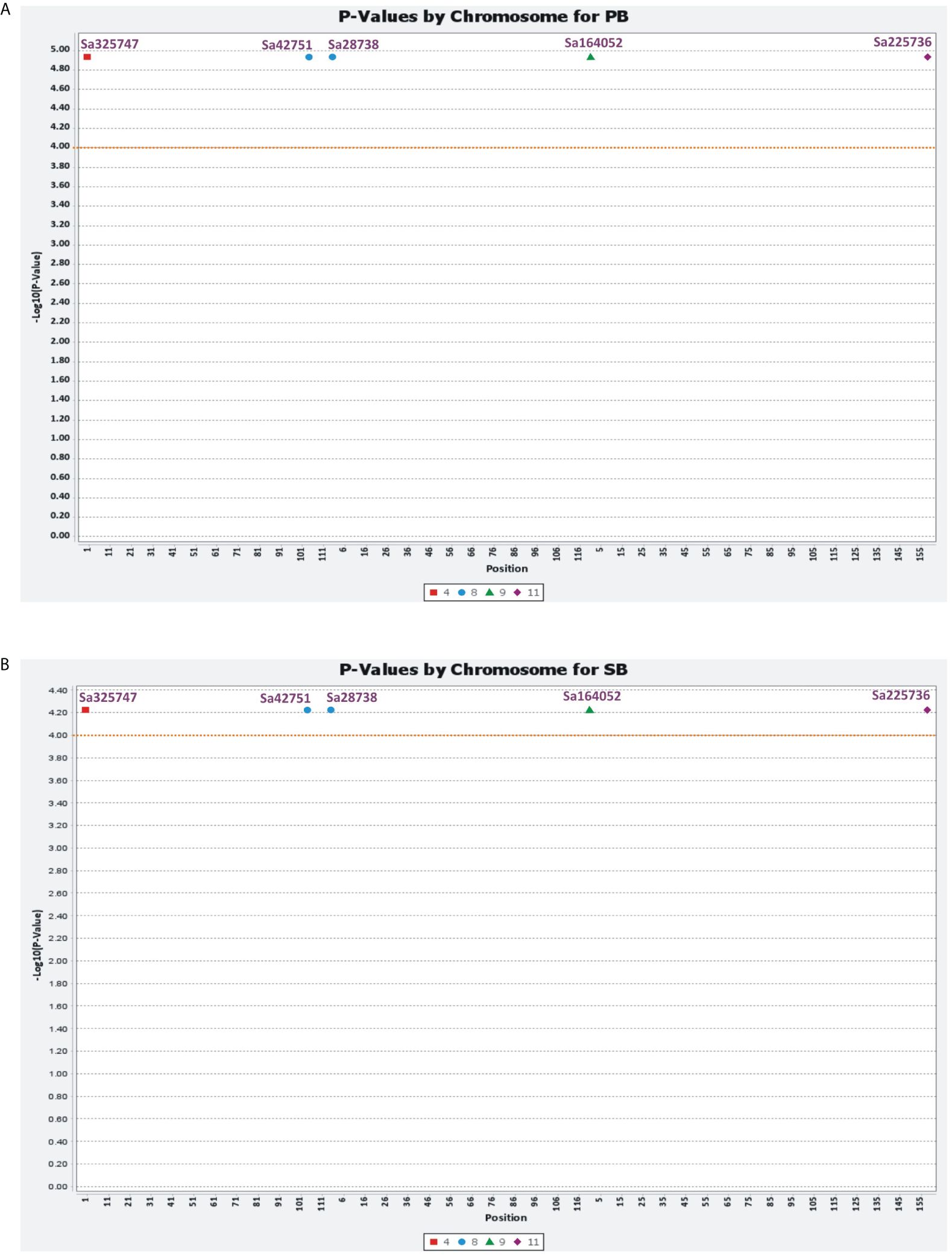
Figure 8 Mixed linear model (MLM) showing the associations of six novel SSR markers with PB (A) and SB (B) traits.
To identify candidate genes associated with the branching trait, we utilized the genome sequences of S. alba up- and downstream of 20 kb from each microsatellite marker. Our analysis revealed that the 40 kb region surrounding five significant markers contained 47 complete genes. Using the AUGUSTUS web server, we predicted gene sequences for the Sa325747, Sa42751, Sa164052, Sa225736, and Sa28738 markers, resulting in 10, 5, 12, 10, and 10 gene sequences, respectively. We then used BLASTP to identify domains and annotate the putative functions of the predicted genes with protein databases such as the Pfam domain, SwissProt, Nr, and RefSeq (Supplementary Tables S1–S4, respectively). Among the predicted genes, the majority showed significant similarity to Arabidopsis thaliana proteins in all three protein databases, with similarities ranging from 30 to 100%. Interestingly, 35 of the predicted genes showed absolute similarity to branching-related genes (1127 genes) obtained from the ENA database, with 33 genes belonging to A. thaliana and 2 genes belonging to Pisum sativum (Supplementary Table S5).
Our analysis revealed several genes with significant similarity to known regulators of the axillary meristem in A. thaliana. For instance, Gene_164052_4 and Gene_225736_1 (At5g23000) were identified as orthologous to REGULATOR OF AXILLARY MERISTEMS1 (RAX1), while Gene_164052_2 (At2g36890) was found to be orthologous to REGULATOR OF AXILLARY MERISTEMS2 (RAX2). Similarly, Gene_28738_4, Gene_164052_6, and Gene_164052_7 (At3G49690) were identified as orthologous to REGULATOR OF AXILLARY MERISTEMS3 (RAX3). We also identified a predicted gene, Gene_42751_2, as orthologous to the MORE AXILLARY GROWTH4 (MAX4) gene (At4g32810), which regulates auxin transport. Other predicted genes, such as Gene_28738_2 and Gene_28738_3 (orthologous to At1G19850), were found to play a role in the establishment of vascular and body patterns during embryonic and postembryonic development. Similarly, Gene_28738_9 and _10 were found to be orthologous to At5g03840, which controls inflorescence meristem identity. Additionally, the predicted genes, such as Gene_28738_5 and _6, Gene_225736_7, Gene_325747_1, and _3, exhibited significant similarity to genes in A. thaliana that play an active role in the transcriptional coregulation of AGAMOUS. Interestingly, Genes_28738_7 and _8 were found to be orthologous to the Pisum sativum gene AAS66906, which regulates shoot branching via physiologically defined mobile signals. Finally, Gene_42751_3 was found to be orthologous to auxin resistance protein 6 (At4g02570), which plays an active role in auxin signaling during embryonic and postembryonic development in Arabidopsis. We also identified Gene_42751_5 as having a role in carbon and nitrogen metabolism and being orthologous to the At1G53310 gene in A. thaliana.
This study identified several predicted genes that may be involved in the regulation of plant branching. Gene_164052_12 and Gene_225736_6 were found to be orthologous to ERECTA1 (ER1) (At2g26330), which plays a role in specifying organs that originate from the shoot apical meristem. Gene_164052_8 was predicted to be orthologous to APETALA1 (AP1) (At1g69120), which specifies flower meristem identity and is also required for the normal development of sepals and petals. Gene_164052_9 (At4g08150) was found to be actively expressed in the peripheral and rib zones of the shoot apical meristem but not in the leaf primordia. It is also expressed in the fourth floral whorl, particularly in the cell surrounding the transmitting tissue. Gene_225736_8, orthologous to At3g54720, encodes a glutamate carboxy peptidase and was found to be involved in ethylene-enhanced hypocotyl elongation in light. The alleles of this gene also showed an increased cotyledon number and rate of leaf initiation, along with the transformation of leaves to cotyledons, altered flowering time, and photomorphogenesis.
Gene_325747_5, orthologous to REVOLUTA (REV) (At5g60690) of A. thaliana, has been shown to regulate meristem initiation at lateral positions and is a member of a small homeodomain-leucine zipper family. Gene_325747_6 was orthologous to AINTEGUMENTA (ANT) (At4g37750), which is required for the control of cell proliferation and is actively expressed in lateral shoot organ primordia. It also regulates growth and cell numbers during organogenesis and modulates auxin biosynthesis in ovules via the regulation of YUC4. Gene_164052_11 (At1g16410) was found to be orthologous to the SUPERSHOOT1 (SPS1) gene of A. thaliana and was significantly associated with branching variation. Gene_SSR325747_2, orthologous to the At3g62980 gene of A. thaliana, regulates root and hypocotyl growth, lateral root formation, cell elongation, and gravitropism. Six predicted genes (Gene_225736_3, _4 (At2g45000), _9 (At2g42640), Gene_325747_4 (At2g45010), _7 (At2g42610), and _8 (At2g42650)) were orthologous to A. thaliana genes that have putative roles in shoot branching via unknown mechanisms. The remaining 12 genes did not show any similarity with these branching-related genes. Among these 12 genes, Gene_28738_P1 is annotated as an orthologous gene of At5G64730 of A. thaliana and is involved in cell wall synthesis, while Gene_42751_P4 (At1G11580) is involved in root growth. Gene_164052_P3 (At5G26742) is involved in chloroplast development, and Gene_164052_P10 (At4G02250) is involved in the regulation of sugar metabolism. These functions were found to be associated with branch initiation and development. Furthermore, all 47 predicted genes were subjected to BLASTN analysis of the coding genes of A. thaliana, and the results showed an absolute similarity of 45 predicted genes, while the remaining two genes showed approximately 96% similarity (Supplementary Table S6). All the genes predicted in this study were BLASTN with the transcriptome assembly of the H1 allohexaploid, one of the parents of BCILs, and the results showed an absolute similarity of these predicted genes with various coding sequences of the allohexaploid. The successful BLAST results confirmed the presence of introgressed genes controlling the number of PB and SB in the BCILs (Supplementary Table S7).
Plant architecture plays a crucial role in determining yield potential, and thus, a positive correlation between high yields and a greater number of PB and SB has been established in Indian mustard (Ehrenreich et al., 2007). Traits that have a large genetic component and a direct correlation with yield, such as the number of branches and pods per plant, number of seeds per pod, seed weight, and flowering time, can be used as suitable selection criteria. Molecular mapping has been used to analyze these morphological characteristics to identify the genes that control them. The yield potential of B. juncea has reached a plateau due to changing climatic conditions and genetic uniformity, which are likely to worsen due to various biotic and abiotic stresses, such as insects, diseases, drought, salinity, and high temperature. The expansion of agricultural land is not a feasible solution; hence, increasing crop production through higher yields is essential. The Indian mustard has a narrow genetic base, which makes it difficult to improve its yield potential through interspecific hybridization. Therefore, it is essential to investigate genetic or allelic variability to significantly improve the yield of B. juncea. Therefore, alien gene introgression is required to strengthen the genetic base of the crop Brassicas. However, it is challenging to introgress alien genes into crop species due to various barriers, including pre- and postfertilization incompatibility, poor or lack of pairing between chromosomes, hybrid sterility, and consecutive linkage drag (Kirti et al., 1992; Bohra et al., 2022). S. alba has been used as a resistance source for biotic and abiotic stresses and seed color for almost four decades (Primard et al., 1988; Hansen and Earle, 1997; Wang et al., 2005; Kumari and Singh, 2019; Kumari et al., 2020c, 2023; Singh et al., 2021a, 2022b). To date, the S. alba genome has not been utilized for the introgression of genes related to plant architecture. The identification of alien genes that govern branching phenotypes is also difficult because the introgression of alien genes has still not been reported. However, these genes were identified in the model plant A. thaliana, and it has been confirmed that these traits are controlled by multiple genes (Ehrenreich et al., 2007).
Notably, the introgression of alien genes into a crop plant is an intricate and slow process that requires a significant amount of time and effort. To address the issue of poor or lack of pairing between chromosomes of the two species, we used S. alba as a donor for genes that govern yield-contributing traits due to its potential for greater branching (Kumari et al., 2011, 2020b). S. alba and B. nigra (a diploid progenitor of B. juncea) share the common ‘Nigra’ lineage of the subtribe Brassicinae (Warwick and Black, 1991; Nelson and Lydiate, 2006), which makes them more suitable for use as protoplast fusion partners of B. juncea. In an experiment involving the S. alba and B. juncea genomes, we recorded approximately 78% genome similarity between the two genera (Singh et al., 2022b). These homologies between the S. alba and B. juncea genomes might have contributed to the successful production of the first stable and fertile somatic hybrids between B. juncea and S. alba, with adequate pairing at meiosis, which maintained the fertility of the hybrids and their backcross progenies (Kumari et al., 2018, 2020c; Kumari and Bhat, 2019, 2021). This has also allowed for the inheritance of the bushy plant architecture and higher numbers of PBs and SBs in the backcross generation (Kumari et al., 2018, 2020b; Singh et al., 2023). This, in turn, has contributed to higher yields.
The identification of S. alba introgressions in advanced backcross generations has been challenging due to the small size of the chromosomes and the presence of more heterochromatic regions, which is similar to the findings in Arabidopsis thaliana (Schweizer et al., 1987). This is because each chromosome in these species has an approximately equal amount of DNA, making it difficult to distinguish segmental introgressions in the recipient genome. Nonetheless, early generations of allohexaploids revealed S. alba introgression via in situ hybridization, which is a powerful technique for visualizing the localization of DNA sequences on chromosomes (Kumari et al., 2020b, 2020c). With the advanced generation of second backcross progeny of somatic hybrids, these BCILs carry enormous allelic diversity due to the presence of half of the haploid set of the S. alba genome, either in the form of segmental or additional chromosomal introgressions. Thus, association analysis by SNP genotyping could not be possible because of sufficient allelic variations within the core set of BCILs. As a consequence, we used a reverse genetics approach in the present study to identify the genes responsible for the greater numbers of PB and SB in a core set of 86 S. alba-B. juncea BCILs by genotyping with a set of 170 S. alba genome-specific SSR markers. However, the chromosome-wide sequence of the S. alba genome is not available to date (Kumari et al., 2020a; Singh et al., 2022b). We used these markers as an alternative approach for establishing a marker–trait association to identify the genes that contribute to the greater number of PB and SB in BCILs, which can contribute to greater yields.
The mixed linear model (MLM) and general linear model (GLM) are statistical methods commonly used in association analysis between genetic markers and phenotypic traits. The MLM approach is preferred due to its ability to control population structure and relatedness among individuals, which can lead to false positives in association analysis. The GLM approach, on the other hand, assumes independence among individuals and can produce spurious associations. In this study, both MLM and GLM were employed, and the Q and K matrices obtained from the population structure analysis and kinship analysis, respectively, were incorporated as covariates to account for the confounding effects of population structure and relatedness (Bradbury et al., 2007). It is interesting to note that despite the small size of the association panel, there was wide variation in branch numbers, indicating the presence of natural genetic diversity. The heritability of the trait was also high, suggesting that genetic factors play a significant role in determining the number of PB and SB. The identification of significant marker–trait associations using the MLM (Q + K) model provides further evidence that genetic factors influence the trait. These associations may help in identifying genomic regions or specific genes that are responsible for regulating branch numbers in BCILs.
Using multiple methods to analyze population structure and determine the most appropriate model for association mapping is a common practice in genetic studies. Population structure is responsible for the identification of numerous false-positive QTLs (Zhao et al., 2007). In this study, the Q+K model and the PCA model were considered, but the MLM (Q + K) was found to be the most appropriate for the population of BCILs being analyzed (Yu et al., 2006; Stich and Melchinger, 2009). Additionally, the use of STRUCTURE analysis, a kinship matrix, and a UPGMA tree helped to categorize the BCILs into three populations, which provided valuable information for association mapping analysis. Overall, it is important to carefully consider population structure and appropriate models when conducting association mapping studies to avoid false-positive results and to accurately identify QTLs associated with traits of interest. Indeed, the modification of plant architecture has been a major focus of crop improvement for several years and has contributed significantly to the increase in crop yield during the Green Revolution (Pingali and Raney, 2005).
Based on the MLM (Q + K) model, a total of five significant marker–trait associations were identified for PB and SB in both crop seasons. The genomic regions associated with these markers contained a total of 47 complete genes, 35 of which were identified as candidate genes for regulating the branching trait in BCILs. These genes were identified as orthologous to AINTEGUMENTA - ANT (Elliott et al., 1996; Mizukami and Fischer, 2000; Krizek et al., 2020), APETALA 1 - AP1 (Irish and Sussex, 1990; Byzova et al., 1999; Monniaux et al., 2018), RAMOSUS– RMS (Arumingtyas et al., 1992; Grbić and Bleecker, 2000; Foo et al., 2005, 2007), REGULATOR OF AXILLARY MERISTEMS - RAX (Keller et al., 2006; Muller et al., 2006), MORE AXILLARY GROWTH - MAX (Stirnberg et al., 2002; Sorefan et al., 2003; Booker et al., 2005; Bennett et al., 2006), MONOPTEROS - MP (Thomas and Gerd, 1993; Przemeck et al., 1996; Christian and Thomas, 1998), SEUSS - SEU (Bao et al., 2010; Gong et al., 2016; Huai et al., 2018), REVOLUTA - REV (Talbert et al., 1995), TERMINAL FLOWER 1 - TFL1 (Bradley et al., 1997), ALTERED MERISTEM PROGRAM 1 - AMP1 (López-García et al., 2016, 2020; Yang et al., 2018), ERECTA - ER (Godiard et al., 2003; Qu et al., 2017), SUPERSHOOT1– SPS1 (Tantikanjana et al., 2001; Ehrenreich et al., 2007), etc. genes that controlling the branching trait. The genes associated with branching traits identified in our study could be utilized in crop improvement programs through targeted genetic modifications, marker-assisted breeding (MAB), and selection (MAS) programs. The introduction of genes responsible for higher branching traits into high-yielding cultivars could lead to increased branching and consequently higher yield potential. This study demonstrated the introgression of 47 genes into B. juncea from S. alba and identified 35 of these genes as candidates involved in increased PB and SB traits (Ehrenreich et al., 2007). The identified genes were subjected to BLASTN analysis, which matched their sequences with those of the transcriptome assembly of the first stable allohexaploid Brassica (H1) strain developed from RNA-seq at the time of flowering (Singh et al., 2022a). This study highlights the potential for gene introgression to improve crop traits, such as branching, through the transfer of genetic material from related wild species.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
KS: Data curation, Investigation, Methodology, Software, Supervision, Validation, Writing – original draft. PK: Conceptualization, Data curation, Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing. PR: Resources, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1381387/full#supplementary-material
Supplementary Tables S1–18 | BLAST search results for the pfam domain (S1), UniProtKB/Swiss-Prot (S2), Nr (S3), RefSeq (S4), branching genes (BG) downloaded from the European Nucleotide Archive (ENA) (S5), coding gene sequences of Arabidopsis thaliana (S6), and coding sequences of the H1 allohexaploid transcriptome (S7) performed to annotate the predicted genes using the CLC Genomics Workbench. Details of SSR markers included in the present study for association mapping (S8).
Arumingtyas, E. L., Floyd, R. S., Gregory, M. J., Murfet, I. C. (1992). Branching in Pisum: inheritance and allelism tests with 17 ramosus mutants. Pisum. Genet. 24, 17–31.
Banga, S. S., Banga, S. (2016). Genetic diversity and germplasm patterns in Brassica juncea. Gene Pool. Divers. Crop Improv. 10, 163–186. doi: 10.1007/978-3-319-27096-8_5
Bao, F., Azhakanandam, S., Franks, R. G. (2010). SEUSS and SEUSS-LIKE transcriptional adaptors regulate floral and embryonic development in Arabidopsis. Plant Physiol. 152, 821–836. doi: 10.1104/pp.109.146183
Bennett, T., Sieberer, T., Willett, B., Booker, J., Luschnig, C., Leyser, O.. (2006). The Arabidopsis MAX pathway controls shoot branching by regulating auxin transport. Curr. Biol. 16, 553–563. doi: 10.1016/j.cub.2006.01.058
Bohra, A., Kilian, B., Sivasankar, S., Caccamo, M., Mba, C., McCouch, S. R., et al. (2022). Reap the crop wild relatives for breeding future crops. Trends Biotechnol. 40, 412–431. doi: 10.1016/j.tibtech.2021.08.009
Booker, J., Sieberer, T., Wright, W., Williamson, L., Willett, B., Stirnberg, P., et al. (2005). MAX1 encodes a cytochrome P450 family member that acts downstream of MAX3/4 to produce a carotenoid derived branch-inhibiting hormone. Dev. Cell 8, 443–449. doi: 10.1016/j.devcel.2005.01.009
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., Buckler, E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Bradley, D., Ratcliffe, O., Vincent, C., Carpenter, R., Coen, E. (1997). Inflorescence commitment and architecture in Arabidopsis. Science 275, 80–83. doi: 10.1126/science.275.5296.80
Byzova, M. V., Franken, J., Aarts, M. G., de Almeida-Engler, J., Engler, G., Mariani, C., et al. (1999). Arabidopsis STERILE APETALA, a multifunctional gene regulating inflorescence, flower, and ovule development. Genes Dev. 13, 1002–1014. doi: 10.1101/gad.13.8.1002
Christian, S. H., Thomas, B. (1998). The Arabidopsis gene MONOPTEROS encodes a transcription factor mediating embryo axis formation and vascular development. EMBO J. 17, 1405–1411. doi: 10.1093/emboj/17.5.1405
Earl, D. A., VonHoldt, B. M. (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Ehrenreich, I. M., Stafford, P. A., Purugganan, M. D. (2007). The genetic architecture of shoot branching in Arabidopsis thaliana: A comparative assessment of candidate gene associations vs. quantitative trait locus mapping. Genetics 176, 1223–1236. doi: 10.1534/genetics.107.071928
Elliott, R. C., Betzner, A. S., Huttner, E., Oakes, M. P., Tucker, W. Q., Gerentes, D., et al. (1996). AINTEGUMENTA, an APETALA2-like gene of Arabidopsis with pleiotropic roles in ovule development and floral organ growth. Plant Cell. 8, 155–168. doi: 10.1105/tpc.8.2.155
Emrani, N., Harloff, H. J., Gudi, O., Kopisch-Obuch, F., Jung, C. (2015). Reduction in sinapine content in rapeseed (Brassica napus L.) by induced mutations in sinapine biosynthesis genes. Mol. Breed. 35, 37. doi: 10.1007/s11032-015-0236-2
Endo, M., Mikami, M., Toki, S. (2015). Multigene knockout utilizing off-target mutations of the CRISPR/Cas9 system in rice. Plant Cell Physiol. 56, 41–47. doi: 10.1093/pcp/pcu154
Foo, E., Bullier, E., Goussot, M., Foucher, F., Rameau, C., Beveridge, C. A. (2005). The branching gene RAMOSUS1 mediates interactions among two novel signals and auxin in pea. Plant Cell. 17, 464–474. doi: 10.1105/tpc.104.026716
Foo, E., Morris, S. E., Parmenter, K., Young, N., Wang, H., Jones, A., et al. (2007). Feedback regulation of xylem cytokinin content is conserved in pea and Arabidopsis. Plant Physiol. 143, 1418–1428. doi: 10.1104/pp.106.093708
Godiard, L., Sauviac, L., Torii, K. U., Grenon, O., Mangin, B., Grimsley, N. H., et al. (2003). ERECTA, an LRR receptor-like kinase protein controlling development pleiotropically affects resistance to bacterial wilt. Plant J. 36, 353–365. doi: 10.1046/j.1365-313x.2003.01877.x
Gong, X., Flores-Vergara, M. A., Hong, J. H., Chu, H., Lim, J., Franks, R. G., et al. (2016). SEUSS integrates gibberellin signaling with transcriptional inputs from the SHR-SCR-SCL3 module to regulate middle cortex formation in the Arabidopsis root. Plant Physiology 170(3), 1675–1683. doi: 10.1104/pp.15.01501
Grbić, V., Bleecker, A. B. (2000). Axillary meristem development in Arabidopsis thaliana. Plant J. 21, 215–223. doi: 10.1046/j.1365-313x.2000.00670.x
Greb, T., Clarenz, O., Schafer, E., Muller, D., Herrero, R., Schmitz, G., et al. (2003). Molecular analysis of the LATERAL SUPPRESSOR gene in Arabidopsis reveals a conserved control mechanism for axillary meristem formation. Genes Dev. 17, 1175–1187. doi: 10.1101/gad.260703
Gupta, P. K., Kulwal, P. L., Jaiswal, V. (2014). Association mapping in crop plants: opportunities and challenges. Adv. Genet. 85, 109–147. doi: 10.1016/B978-0-12-800271-1.00002-0
Hajjar, R., Hodgkin, T. (2007). The use of wild relatives in crop improvement: a survey of developments over the last 20 years. Euphytica 156, 1–13. doi: 10.1007/s10681-007-9363-0
Hansen, L. N., Earle, E. D. (1997). Somatic hybrids between Brassica oleracea and Sinapis alba L. with resistance to Alternaria brassicae (Berk.) Sacc. Theor. Appl. Genet. 94, 1078–1085. doi: 10.1007/s001220050518
Huai, J., Zhang, X., Li, J., Ma, T., Zha, P., Jing, Y., et al. (2018). SEUSS and PIF4 coordinately regulate light and temperature signaling pathways to control plant growth. Mol. Plant 11, 928–942. doi: 10.1016/j.molp.2018.04.005
Irish, V. F., Sussex, I. M. (1990). Function of the apetala-1 gene during Arabidopsis floral development. Plant Cell. 2, 741–753. doi: 10.1105/tpc.2.8.741
Keller, T., Abbott, J., Moritz, T., Doerner, P. (2006). Arabidopsis REGULATOR OF AXILLARY MERISTEMS1 controls a leaf axil stem cell niche and modulates vegetative development. Plant Cell 18, 598–611. doi: 10.1105/tpc.105.038588
Kirti, P. B., Mohapatra, T., Khanna, H., Prakash, S., Chopra, V. L. (1995). Diplotaxis catholica + Brassica juncea somatic hybrids: molecular and cytogenetic characterization. Plant Cell Rep. 14, 593–597. doi: 10.1007/BF00231945
Kirti, P. B., Narasimhulu, S. B., Prakash, S., Chopra, V. L. (1992). Somatic hybridization between Brassica juncea and Moricandia arvensis by protoplast fusion. Plant Cell Rep. 11, 318–321. doi: 10.1007/BF00235090
Krizek, B. A., Blakley, I. C., Ho, Y. Y., Freese, N., Loraine, A. E. (2020). The Arabidopsis transcription factor AINTEGUMENTA orchestrates patterning genes and auxin signaling in the establishment of floral growth and form. Plant J. 103, 752–768. doi: 10.1111/tpj.14769
Kumari, P., Bhat, S. R. (2019). Allohexaploid (H2) (IC0626000 INGR18031), an Indian mustard (Brassica juncea) germplasm with heat tolerance and Resistant to Alternaria brassicae. Indian J. Plant Genet. Resour. 32, 439.
Kumari, P., Bhat, S. R. (2021). Allohexaploid (H1) (IC0628060; INGR19102), an allohexaploid (Brassica juncea + Sinapis alba) Germplasm resistant to Alternaria brassicae, Sclerotinia sclerotiorum, and tolerant to temperature. Indian J. Plant Genet. Resour. 34, 160–161.
Kumari, P., Bist, D. S., Bhat, S. R. (2018). Stable fertile somatic hybrids between Sinapis alba and Brassica juncea showing resistance to Alternaria brassicae and heat stress. Plant Cell Tissue Organ Culture 133, 77–86. doi: 10.1007/s11240-017-1362-9
Kumari, P., Singh, K. P. (2019). Characterization of stable somatic hybrids of Sinapis alba and Brassica juncea for Alternaria blight, Sclerotinia sclerotiorum resistance and heat tolerance. Indian Res. J. Ext. Edu. 19, 99–103.
Kumari, P., Singh, K. P., Bisht, D., Kumar, S. (2020b). Somatic hybrids of Sinapis alba + Brassica juncea: study of backcross progenies for morphological variations, chromosome constitution and reaction to Alternaria brassicae. Euphytica 216, 93. doi: 10.1007/s10681-020-02629-3
Kumari, P., Singh, K. P., Kumar, S., Yadava, D. K. (2020c). Development of a yellow-seeded stable allohexaploid brassica through inter-generic somatic hybridization with a high degree of fertility and resistance to Sclerotinia sclerotiorum. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.575591
Kumari, P., Singh, K. P., Rai, P. K. (2020a). Draft genome of multiple resistance donor plant Sinapis alba: an insight into SSRs, annotations and phylogenetics. PloS One 15, e0231002. doi: 10.1371/journal.pone.0231002
Kumari, P., Singh, K. P., Rai, P. K. (2023). Identification of new resistance source for Sclerotinia stem rot in backcross population of B. juncea + S. alba allohexaploids: Key to manage disease through host resistance. Euphytica 219, 76. doi: 10.1007/s10681-023-03208-y
Kumari, P., Singh, K. P., Rai, P. K. (2024). Deciphering the genetic architecture of plant height and main shoot length in backcross introgression lines of Sinapis alba + Brassica juncea allohexaploids. Euphytica 220, 3. doi: 10.1007/s10681-023-03271-5
Kumari, P., Singh, K. P., Rathore, R. K. S. (2011). Morphometric study for varietal identification of oleiferous Brassicas. Proc. Nat. Acad. Sci. India Sec-B 81, 411–415.
Lelivelt, C. L. C., Leunissen, E. H. M., Frederiks, H. J., Helsper, J. P. F. G., Krens, F. A. (1993). Transfer of resistance to the beet cyst nematode (Heterodera schachtii Schm.) from Sinapis alba L. (white mustard) to the Brassica napus L. gene pool by means of sexual and somatic hybridization. Theor. Appl. Genet. 85, 688–696. doi: 10.1007/BF00225006
Leyser, H. M., Lincoln, C. A., Timpte, C., Lammer, D., Turner, J., Estelle, M. (1993). Arabidopsis auxin-resistance gene AXR1 encodes a protein related to ubiquitin-activating enzyme E1. Nature 364, 161–164. doi: 10.1038/364161a0
Lincoln, C., Britton, J. H., Estelle, M. (1990). Growth and development of the axr1 mutants of Arabidopsis. Plant Cell 2, 1071–1080.
Long, J. A., Moan, E. I., Medford, J. I., Barton, M. K. (1996). A member of the KNOTTED class of homeodomain proteins encoded by the STM gene of Arabidopsis. Nature 379, 66–69. doi: 10.1038/379066a0
López-García, C. M., Raya-González, J., López-Bucio, J. S., Guevara-García, Á. A., López-Bucio, J. (2016). ALTERED MERISTEM PROGRAM 1 plays a role in seed coat development, root growth, and post-embryonic epidermal cell elongation in Arabidopsis. J. Plant Growth Regul. 35, 1141–1158. doi: 10.1007/s00344-016-9612-3
López-García, C. M., Ruíz-Herrera, L. F., López-Bucio, J. S., Huerta-Venegas, P. I., Peña-Uribe, C. A., de la Cruz, H. R., et al. (2020). ALTERED MERISTEM PROGRAM 1 promotes growth and biomass accumulation influencing guard cell aperture and photosynthetic efficiency in Arabidopsis. Protoplasma 257, 573–582. doi: 10.1007/s00709-019-01458-z
Malosetti, M., Ribaut, J. M., Vargas, M., Crossa, J., Boer, M., Van Eeuwijk, F. (2007). “Multi-trait multi-environment QTL modelling for drought-stress adaptation in maize,” in Scale and Complexity in Plant Systems Research: Gene-Plant-Crop Relations. Eds. Spiertz, J. H. J., Struik, P. C., Van Laar, H. H. (Springer), 23–34.
McSteen, P., Leyser, O. (2005). Shoot branching. Annu. Rev. Plant Biol. 56, 353–374. doi: 10.1146/annurev.arplant.56.032604.144122
Mizukami, Y., Fischer, R. L. (2000). Plant organ size control: AINTEGUMENTA regulates growth and cell numbers during organogenesis. Proc. Natl. Acad. Sci. U.S.A. 97, 942–947. doi: 10.1073/pnas.97.2.942
Monniaux, M., Pieper, B., McKim, S. M., Routier-Kierzkowska, A.-L., Kierzkowski, D., Smith, R. S., et al. (2018). The role of APETALA1 in petal number robustness. eLife 7, e39399. doi: 10.7554/eLife.39399
Muller, D., Schmitz, G., Theres, K. (2006). Blind homologous R2R3 Myb genes control the pattern of lateral meristem initiation in Arabidopsis. Plant Cell 18, 586–597. doi: 10.1105/tpc.105.038745
Nelson, M. N., Lydiate, D. J. (2006). New evidence from Sinapis alba L. for ancestral triplication in a crucifer genome. Genome 49, 230–238. doi: 10.1139/g05-099
Nordborg, M., Tavaré, S. (2002). Linkage disequilibrium: what history has to tell us. Trends Genet. 8, 83–90. doi: 10.1016/s0168-9525(02)02557-x
Nyine, M., Uwimana, B., Akech, V., Brown, A., Ortiz, R., Doležel, J., et al. (2019). Association genetics of bunch weight and its component traits in East African highland banana (Musa spp. AAA group). Theor. Appl. Genet. 132, 3295–3308. doi: 10.1007/s00122-019-03425-x
Pasam, R. K., Sharma, R., Malosetti, M., Eeuwijk, F. A. V., Haseneyer, G., Kilian, B., et al. (2012). Genome-wide association studies for agronomical traits in a worldwide spring barley collection. BMC Plant Biol. 12, 16. doi: 10.1186/1471-2229-12-16
Patterson, N., Price, A. L., Reich, D. (2006). Population structure and Eigen analysis. PloS Genet. 2, e190. doi: 10.1371/journal.pgen.0020190
Pingali, P., Raney, T. (2005). From the Green Revolution to the Gene Revolution: How will the Poor Fare? (Agricultural and Development Economics Division of the Food and Agriculture Organization of the United Nations (FAO - ESA). Working Papers No. 05-09.
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. doi: 10.1038/ng1847
Primard, C., Vedel, F., Mathieu, C., Pelletier, G., Chevre, A. M. (1988). Interspecific somatic hybridization between Brassica napus and Brassica hirta (Sinapis alba L.). Theor. Appl. Genet. 75, 546–552. doi: 10.1007/bf00289119
Pritchard, J. K., Stephens, M., Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1093/genetics/155.2.945
Przemeck, G. K., Mattsson, J., Hardtke, C. S., Sung, Z. R., Berleth, T. (1996). Studies on the role of the Arabidopsis gene MONOPTEROS in vascular development and plant cell axialization. Planta 200, 229–237. doi: 10.1007/BF00208313
Qu, X., Zhao, Z., Tian, Z. (2017). ERECTA regulates cell elongation by activating auxin biosynthesis in Arabidopsis thaliana. Front. Plant Sci. 8, 1688. doi: 10.3389/fpls.2017.01688
Quezada-Martinez, D., AddoNyarko, C. P., Schiessl, S. V., Mason, A. S. (2021). Using wild relatives and related species to build climate resilience in Brassica crops. Theor. Appl. Genet. 134, 1711–1728. doi: 10.1007/s00122-021-03793-3
Remington, D. L., Thornsberry, J. M., Matsuoka, Y., Wilson, L. M., Whitt, S. R., Doebley, J., et al. (2001). Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. U.S.A. 98, 11479–11484. doi: 10.1073/pnas.201394398
Risch, N., Merikangas, K. (1996). The future of genetic studies of complex human diseases. Science 273, 1516–1517. doi: 10.1126/science.273.5281.1516
Schweizer, D., Ambros, P., Gründler, P., Varga, F. (1987). Attempts to relate cytological and molecular chromosome data of Arabidopsis thaliana to its genetic linkage map. Arabidopsis Info. Service 25, 27–35.
Singh, K. P., Kumari, P., Rai, P. K. (2021a). Current status of the disease-resistant gene(s)/QTLs, and strategies for improvement in Brassica juncea. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.617405
Singh, K. P., Kumari, P., Rai, P. K. (2023). Phenotypic characterization and resistance response to Sclerotinia sclerotiorum of backcross lines developed from stable allohexaploids of Sinapis alba + Brassica juncea. Euphytica 219, 34. doi: 10.1007/s10681-023-03160-x
Singh, K. P., Kumari, P., Raipuria, R. K., Rai, P. K. (2022b). Development of genome-specific SSR markers for the identification of introgressed segments of Sinapis alba carrying genes for blight resistance in the Brassica juncea background. 3Biotech 12, 332. doi: 10.1007/s13205-022-03402-0
Singh, K. P., Kumari, P., Yadava, D. K. (2021b). Introgression and QTL mapping conferring resistance for Alternaria brassicae in the backcross progeny of Sinapis alba + Brassica juncea somatic hybrids. Plant Cell Rep. 40, 2409–2419. doi: 10.1007/s00299-021-02785-3
Singh, K. P., Kumari, P., Yadava, D. K. (2022a). Development of De-novo transcriptome assembly and SSRs in Allohexaploid Brassica with functional annotations and identification of heat-shock proteins for thermotolerance. Front. Genet. 13. doi: 10.3389/fgene.2022.958217
Singh, R. K., Chaudhary, B. D. (1977). Biometrical methods in quantitative genetic analysis (New Delhi: Kalyani publishers), 304pp.
Sorefan, K., Booker, J., Haurogne, K., Goussot, M., Bainbridge, K., Foo, E., et al. (2003). MAX4 and RMS1 are orthologous dioxygenase-like genes that regulate shoot branching in Arabidopsis and pea. Genes Dev. 17, 1469–1474. doi: 10.1101/gad.256603
Stich, B., Melchinger, A. E. (2009). Comparison of mixed-model approaches for association mapping in rapeseed, potato, sugar beet, maize, and Arabidopsis. BMC Genom. 2710, 94. doi: 10.1186/1471-2164-10-94
Stirnberg, P., Chatfield, S. P., Leyser, H. M. (1999). AXR1 acts after lateral bud formation to inhibit lateral bud growth in Arabidopsis. Plant Physiol. 121, 839–847. doi: 10.1104/pp.121.3.839
Stirnberg, P., van De Sande, K., Leyser, H. M. (2002). MAX1 and MAX2 control shoot lateral branching in Arabidopsis. Development 129, 1131–1141. doi: 10.1242/dev.129.5.1131
Talbert, P. B., Adler, H. T., Parks, D. W., Comai, L. (1995). The REVOLUTA gene is necessary for apical meristem development and for limiting cell divisions in the leaves and stems of Arabidopsis thaliana. Development 121, 2723–2735. doi: 10.1242/dev.121.9.2723
Tantikanjana, T., Yong, J. W. H., Letham, D. S., Griffith, M., Hussain, M., Ljung, K., et al. (2001). Control of axillary bud initiation and shoot architecture in Arabidopsis through the SUPERSHOOT gene. Genes Dev. 15, 1577–1588. doi: 10.1101/gad.887301
Thomas, B., Gerd, J. (1993). The role of the monopteros gene in organizing the basal body region of the Arabidopsis embryo. Development 118(2), 575–587. doi: 10.1242/dev.118.2.575
Toronen, P., Holm, L. (2022). PANNZER - a practical tool for protein function prediction. Protein Sci. 31, 118–128. doi: 10.1002/pro.4193
Wang, Y. P., Sonntag, K., Rudloff, E., Chen, J. M. (2005). Intergeneric somatic hybridization between Brassica napus L. and Sinapis alba L. J. Integr. Plant Biol. 47, 84–91. doi: 10.1111/j.1744-7909.2005.00009.x
Ward, S. P., Leyser, O. (2004). Shoot branching. Curr. Opin. Plant Biol. 7, 73–78. doi: 10.1016/j.pbi.2003.10.002
Warwick, S. I., Black, L. D. (1991). Molecular systematics of Brassica and allied genera (subtribe Brassicinae, Brassiceae)—Chloroplast genome and cytodeme congruence. Theor. Appl. Genet. 82, 839–850. doi: 10.1007/BF00231281
Weigel, D., Alvarez, J., Smyth, D. R., Yanofsky, M. F., Meyerowitz, E. M. (1992). LEAFY controls floral meristem identity in Arabidopsis. Cell 69, 843–859. doi: 10.1016/0092-8674(92)90295-N
Wells, R., Trick, M., Soumpourou, E., Clissold, L., Morgan, C., Werner, P., et al. (2014). The control of seed oil polyunsaturate content in the polyploid crop species Brassica napus. Mol. Breed. 33, 349–362. doi: 10.1007/s11032-013-9954-5
Yang, S., Poretska, O., Sieberer, T. (2018). ALTERED MERISTEM PROGRAM1 restricts shoot meristem proliferation and regeneration by limiting HD-ZIP III-mediated expression of RAP2 177(4), 1580–1594. doi: 10.1104/pp.18.00252
Yu, J., Pressoir, G., Briggs, W. H., Vroh, B. I., Yamasaki, M., Doebley, J. F., et al. (2006). Aunifed mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. doi: 10.1038/ng1702
Keywords: Brassica juncea, backcross introgression lines (BCILs), plant architecture, genome-specific SSRs, association mapping, trait-linked SSRs, candidate genes
Citation: Singh KP, Kumari P and Rai PK (2024) GWAS for the identification of introgressed candidate genes of Sinapis alba with increased branching numbers in backcross lines of the allohexaploid Brassica. Front. Plant Sci. 15:1381387. doi: 10.3389/fpls.2024.1381387
Received: 03 February 2024; Accepted: 11 June 2024;
Published: 24 June 2024.
Edited by:
Zoe Hilioti, Centre for Research and Technology Hellas (CERTH), GreeceReviewed by:
Suresh Kaushik, Independent Scientific Researcher, IndiaCopyright © 2024 Singh, Kumari and Rai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Preetesh Kumari, cHJlZXRlc2g3OUBnbWFpbC5jb20=; Pramod Kumar Rai, cHJhbW9ka3JhaTY4QGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.