 Wanran Zhang
Wanran Zhang Ranran Gao
Ranran Gao Lixia Tian
Lixia Tian Zhichao Xu
Zhichao Xu- 1School of Pharmaceutical Sciences, Guizhou University, Guiyang, China
- 2College of Life Science, Northeast Forestry University, Harbin, China
- 3Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing, China
Phellodendron amurense is the essential source of bisbenzylisoquinoline alkaloids (BIAs), making it a highly valued raw material in traditional Chinese medicine. The plant’s root secondary metabolism is intricately linked to the microbial communities that surround it. However, the root-associated microbiomes of P. amurense, as well as the potential correlation between its bioactive compounds and these microbiomes, remain poorly understood. Here, the metabolic profiles of root, rhizosphere, and bulk soils of P. amurense revealed the dramatic differences in the relative content of plant-specialized metabolites. A total of 31, 21, and 0 specialized metabolites in P. amurense were identified in the root, rhizosphere soil, and bulk soil, respectively, with higher content of the seven major BIAs observed in the rhizosphere compared with that in the bulk soils. The composition of the bulk and rhizosphere microbiomes was noticeably distinct from that of the endospheric microbiome. The phylum Cyanobacteria accounted for over 60% of the root endosphere communities, and the α-diversity in root was the lowest. Targeted seven BIAs, namely, berberine, palmatine, magnocurarine, phellodendrine, jatrorrhizine, tetrahydropalmatine, and magnoflorine, were significantly positively correlated with Nectriaceae and Sphingobacteriaceae. This study has illuminated the intricate interaction networks between P. amurense root-associated microorganisms and their key chemical compounds, providing the theoretical foundation for discovering biological fertilizers and laying the groundwork for cultivating high-quality medicinal plants.
1 Introduction
Phellodendron amurense, also known as the Amur cork tree, is a tertiary relic plant and belongs to the Rutaceae family (Yu et al., 2013), widely found in Northeastern China, Inner Mongolia, the Russian Far East, southern Sakhalin, Korea, and Japan (Zhang B. Y. et al., 2023). In China, P. amurense is mainly distributed in the southeast of the Lesser Khingan, Changbai, and Wanda Mountains and in the northern Yanshan Mountains, the extreme north to Great Khingan Mountains (Yang et al., 2016). The bark of P. amurense, a traditional Chinese medicine (TCM), possesses a remarkable medicinal value and has been widely utilized in China, Japan, and Korea (Wang et al., 2015). The deciduous tree is enriched in various chemical compounds such as limonoids, alkaloids, phenolic, polysaccharides, flavonoids, phytosterols, essential oils, and fatty acids (Zhang et al., 2015). The major active ingredients are benzylisoquinoline alkaloids, including berberine, jatrorrhizine, palmatine, magnoflorine, magnocurarine, phellodendrine, and tetrahydropalmatine (Velmurugan et al., 2017; Balážová et al., 2022; Erhan et al., 2023; Feng et al., 2023), which are commonly used as anti-inflammatory, antipyretic, and antibacterial medicines, promoting blood circulation, dysentery, diuretic, hemorrhage, and blood glucose regulating (Kim et al., 2011; Steinmann et al., 2011; Velmurugan et al., 2017; Sun et al., 2019). The yellow, flexible, and tough wood of this medicinal plant boasts an excellent texture and can be used to create furniture and industrial art (Sun et al., 2015). Because of the excessive exploitation for TCM and timber, the population and habitat of wild P. amurense have significantly decreased (Ding, 2011). As a result, the Chinese government listed it as a nationally endangered wild plant species (class II) (Fu and Chin, 1992).
The P. amurense has been extensively cultivated for use in TCM and extraction of bisbenzylisoquinoline alkaloids (BIAs), but the quality of medicinal materials exhibits noticeable variations across different regions (Zhang et al., 2014; Ma et al., 2015). The quality of Chinese medicinal materials depends on the content and quantity of chemical compositions, whose accumulation is crucial for the pharmacological effects of medicinal plants (Luo et al., 2021; Xiao et al., 2022). A large number of natural products are actually produced by microbe-host interactions, which are directly or indirectly involved in the production of active ingredients (Koberl et al., 2013; Mishra et al., 2022). It also has been confirmed that the quality of traditional herbal medicines is impacted by changes in rhizomicrobiomes and endophytes (Huang et al., 2018; Ling F. et al., 2022). Plant microbiomes could significantly affect the accumulation of key medicinal components, such as alkaloids, terpenoids, and steroids (Pang et al., 2021). Stenotrophomonas in the root endosphere of Polygonum cuspidatum enhances the emodin content (Zhang Y. H. et al., 2020). Recent research has shown that plant–microbe interactions can improve biomass and tanshinone production in Salvia miltiorrhiza (Chen H. M. et al., 2018; Huang et al., 2018). To date, numerous studies have been conducted to investigate the relationship between microbiome communities and bioactive compounds found in medicinal plants (Cui et al., 2019; Zhang et al., 2020; Zhang Y. H. et al., 2020; Li et al., 2021; Cui et al., 2023; Li et al., 2023; Zhang H. et al., 2023). However, the composition characteristics and diversity of rhizosphere and root microbiome communities, as well as their relationship with the major active compounds in P. amurense, remain largely unknown.
To address the relationship between the quality of P. amurense and its root-associated microbiomes, we studied the metabolite differences among bulk soil, rhizosphere soil, and the root of P. amurense and further explore the microbial community composition characteristics, diversity, and biomarker microorganisms of the bulk soil, rhizosphere soil, and endophytic of P. amurense. Furthermore, the correlation relationship between the targeted BIAs and the root-associated microbiomes was investigated by weighted gene co-expression network analysis (WGCNA) package in R. Our research aims to offer a practical strategy for enhancing the quality of P. amurense by employing an ecological approach to manipulating the root-associated microbiomes.
2 Materials and methods
2.1 Sample collection
The root and soil of healthy P. amurense were collected from Heilongjiang Province, China (45.7662°N, 126.6247°E). Five roots (at a depth of 10–30 cm) from one P. amurense were cut off and collected as one sample. In addition, bulk soil (at a depth of 10–20 cm) was collected at a distance of 100–200 cm away from the roots. After removing loose soil from the roots, only 2 mm of rhizosphere soil remained and was collected (Edwards et al., 2015; Hilton et al., 2021). These samples were labeled as R (root of wild P. amurense), BS (bulk soil), and RS (rhizosphere soil) with five replicates, respectively. All samples were stored at −80°C. Bulk and rhizosphere soil samples were then sieved by using a 2-mm sieve. The roots of P. amurense were carefully washed and sterilized by undergoing the following operations: 70% (v/v) ethanol for 3 min, 2.5% (v/v) sodium hypochlorite (NaClO) for 5 min, and sterile water four times (Wei et al., 2021). Half of each sample was used for the analysis of microbial characteristics, whereas the other half was employed for metabolite detection. Voucher specimens labeled as PAR20221-PAR20225, PABS20221-PABS20225, and PARS20221-PARS20225 were then deposited at the College of Life Sciences, Northeast Forestry University, China.
2.2 Determination of targeted BIA metabolites
The collected root samples were dried at 60°C for 1 week, and, then, the dried materials were powered and sieved through a No. 60 mesh. An accurately weighed 0.2 g of sample of the powered materials was extracted ultrasonically with 70% aqueous methanol (2 mL) under 50°C for 1 h, with three replications. The extract was further centrifuged at 12,000 rpm for 10 min, and 1 mL of supernatant was filtered using a 0.22-µm nylon filter. As for the soil sample, 0.2 g of bulk and rhizosphere soil was extracted ultrasonically with 70% aqueous methanol (2 mL) under 50°C for 1 h, with three replications. The extract was centrifuged at 12,000 rpm for 10 min. Supernatant (1 mL) was evaporated (40°C) to dryness in vacuo, and the residue was redissolved in 0.2 mL of 70% methanol. Then, this solution was filtered using a 0.22 nylon filter.
After optimizing the experimental conditions, a method was developed using ultrahigh-performance liquid chromatography (UPLC) coupled with quadrupole time-of-flight mass spectrometry (MS) for the nontarget metabolomic profiling of P. amurense and its associated soil. Seven reference standards of phellodendrine, magnoflorine, magnocurarine, jatrorrhizine, tetrahydropalmatine, palmatine, and berberine were dissolved and then diluted with methyl alcohol to prepare a series of standard solutions of different concentrations. To ensure the robustness of the analytical method, the quality control samples were inserted into the sequence every five samples. The samples were injected into a Kinetex C18 column (2.6-μm particle size, 4.6 mm × 150 mm; Phenomenex, USA) maintained at 40°C. The gradient eluent consisted of mobile phase A (0.1% formic acid in water) and mobile phase B (acetonitrile). The gradient elution program was as follows: 0–1 min, 10% B; 1–10 min, 10% to 95% B; 10–12.3 min, 95% B; 12.3–13 min, 95% to 10% B; and 13–15 min 10% B. The flow rate used for separation was 0.5 mL/min, with an injection volume of 1 μL. The MS was equipped with an electrosprayionization (ESI) ion source and operated in the positive ion mode, in the full scan range from mass-to-charge ratio (m/z) of 100 to 2,000 with fragmentation of the five most intensive signals. The following MS conditions were used: ion spray voltage, 5,500 V; curtain gas, 25 psi; ion source gas 1, 40 psi; ion source gas 2, 60 psi; and temperature, 550°C.
The mass spectrum data were analyzed by Analyst TF1.8 Software (https://sciex.com/products/software/analyst-software/). Using the metabolome software LibraryView (AB Sciex, Shanghai, China), the samples’ metabolites were analyzed qualitatively and quantitatively. The chemical structure of the compounds was characterized on the basis of the calculated accurate masses of the molecular ions, protonated molecules, fragment ions, and retention behavior. The observed constituent ion peaks [M]+/[M+H]+/[M+NH4]+ provide reliable information that can confirm the molecular weights and structures of the compounds. The relative contents (after taking log values) of compounds in different samples were displayed by R package pheatmap (v1.0.12).
2.3 DNA extraction and sequencing
Metagenomic DNA from root, rhizosphere soil, and bulk soil samples was extracted using the HiPure Soil DNA Kits (Magen, Guangzhou, China) according to the manufacturer’s recommendations. The DNA concentration and purity were accurately measured using the NanoDrop 2000 spectrophotometer prior to performing agarose (1% w/v) gel electrophoresis. The purified DNA was used to amplify either the fungal Internal Transcribed Spacer 2 (ITS2) region ITS3_KYO2 (5′-GATGAAGAACGYAGYRAA-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) (Guo et al., 2017) or the V3-V4 region of the bacterial 16S ribosomal RNA (rRNA) gene 341F (5′-CCT ACG GGNGGC WGC AG-3′) and 806R (5′-GGAC TAC HVGGG TAT CTA AT-3′) (Toju et al., 2012). The PCR-negative controls consisted of ddH2O instead of DNA template. All samples were subjected to the following PCR conditions: initial denaturation at 95°C for 2 min, followed by 27 cycles of denaturation at 98°C for 10 s, annealing at 62°C for 30 s, extension at 68°C for 30 s, and a final extension at 68°C for 10 min. The paired-end approach was utilized for sequencing by using the Illumina Novaseq 6000 sequencing platform.
2.4 Characteristic analysis of the microbial communities
To obtain high-quality clean reads, raw reads were further filtered using FASTP (v0.18.0) (Chen S. F. et al., 2018). Paired-end clean reads were merged into raw tags using FLASH (v1.2.11) (Magoc and Salzberg, 2011) with a minimum overlap of 10 bp and mismatch error rates of 2%. The clean tags were clustered into operational taxonomic units (OTUs) of ≥ 97% similarity using UPARSE (v9.2.64) (Edgar, 2013), a high-accuracy and high-throughput OTU clustering pipeline. In each cluster, the tag sequence with the highest abundance was selected as the representative sequence.
The stacked bar plot of the community composition was visualized in the ggplot2 package (v2.2.1) (Wicham, 2011), and circular layout representations of species abundance at the genus level were created using circos (v0.69-3) (Krzywinski et al., 2009). To evaluate the diversity of species and community of the bacterial and fungal microorganisms in roots, rhizosphere soil, and bulk soil of P. amurense, alpha-diversity based on the diversity index and beta-diversity based on the weighted UniFrac distance matrix were conducted, respectively. Chao1, Abundance-based Coverage Estimator (ACE), Shannon, and Simpson index were calculated using QIIME (v1.9.1) (Caporaso et al., 2010), and PCoA (principal coordinate analysis) (Dixon, 2003) was generated using the Vegan package (v2.5.3) and ggplot2 package (v2.2.1) of R project. The biomarker features in bacterial and fungal microorganism communities of P. amurense were evaluated through Linear discriminant analysis Effect Size (LEfSe) (Segata et al., 2011).
2.5 Correlation analyses of the main active compounds and microbial communities
WGCNA was performed to evaluate the correlation between the secondary metabolites in roots and their associated microbial communities. After obtaining the absolute contents of seven key BIAs in the bulk soil, rhizosphere soil, and roots of P. amurense, combined with the abundance of bacteria and fungi OTU in the bulk soil, rhizosphere soil, and roots of P. amurense, co-expression network modules were generated by WGCNA package in R. Eigengene value was calculated for each module to search the family-level microorganisms associated with BIA metabolism in P. amurense. The co-expression modules were obtained using automatic network construction function (blockwiseModules) with default parameters; apart from the soft threshold power of 18, networkType was unsigned, TOMtype was unsigned, mergeCutHeight was 0.1, and minModuleSize was 15. In addition, the Pearson correlation coefficient was also calculated. Microorganisms and metabolites with a correlation greater than 0.9 and P < 0.01 were selected, and the correlation network was drawn using Cytoscape (v3.9.1, https://cytoscape.org/).
3 Results
3.1 Distribution of targeted metabolites in the roots and rhizosphere soils of P. amurense
To uncover the distribution characteristics of main active compounds of bulk, rhizosphere soil, and root of P. amurense, the targeted metabolomics was employed, which focuses on the analysis of specific categories of metabolites with more selectivity and sensitivity (Roberts et al., 2012). Obvious differences in the quantity and relative content of specialized metabolites were observed at the different compartments of P. amurense. A total of 31 metabolites were detected and identified (Table 1, Figure 1A). These metabolites include 28 alkaloids, two limonoids, and one quinic acid. Among the 28 alkaloids, 23 are BIAs, two are indole-type, two are quinoline-type, and one is another type of alkaloid. The 23 BIAs can be classified into three groups: three aporphine alkaloids, seven benzylisoquinoline alkaloids, and 13 protoberberine alkaloids (Supplementary Table S1, Supplementary Information). The number of BIAs is the greatest among the 28 alkaloids, accounting for 82% of all the BIA metabolites. In addition, 31 specialized compounds were characterized in the root of P. amurense, whereas 21 were shared in the rhizosphere soil. No specialized metabolites of P. amurense were detected in the bulk soil (Figure 1B). There was a significantly higher content of the 21 commonly specialized compounds in the roots compared with that in the rhizosphere soil.
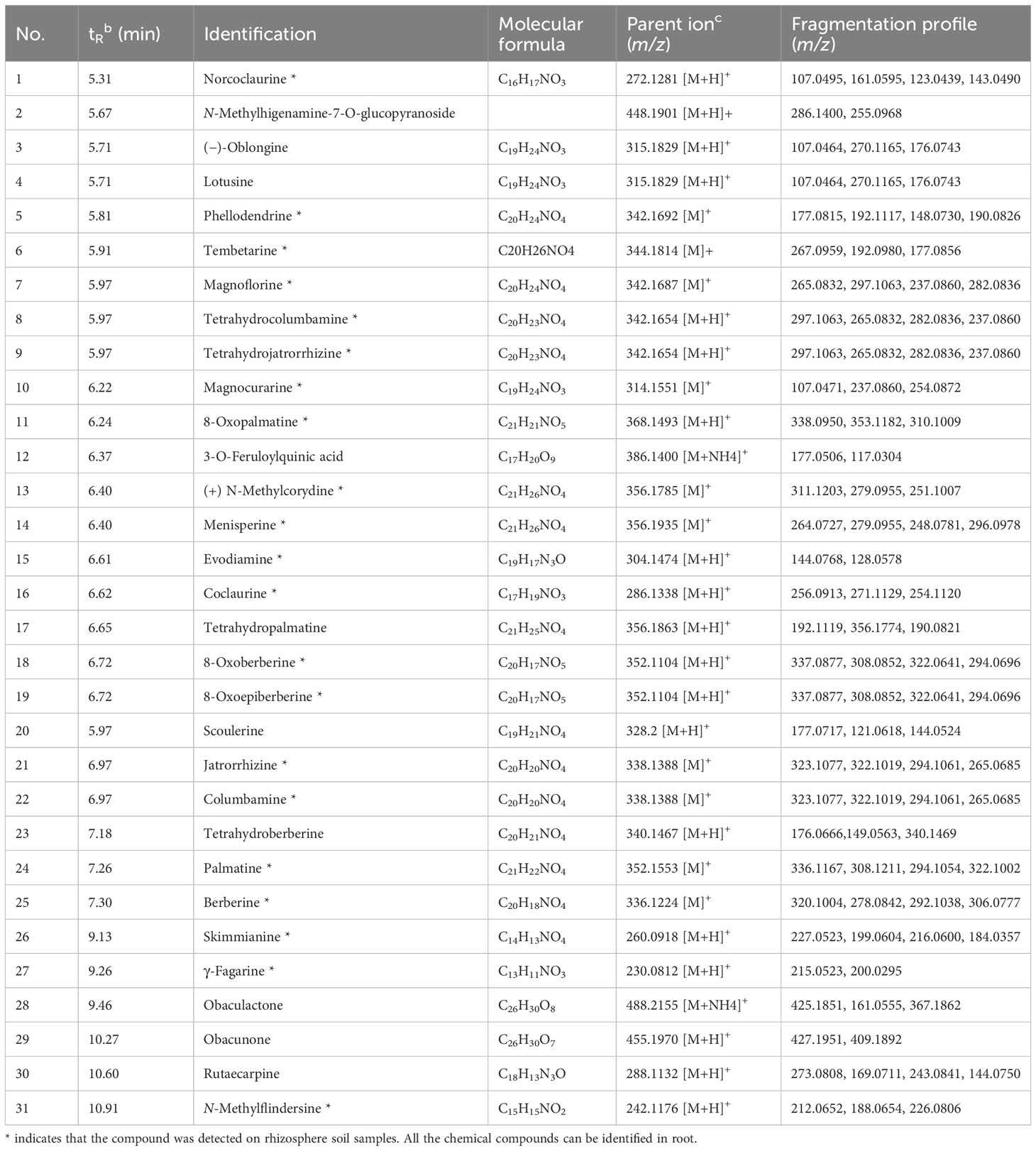
Table 1 UPLC-ESI-MS/MS identification of the compounds contained in roots and rhizosphere soil of P. amurense in a positive mode.
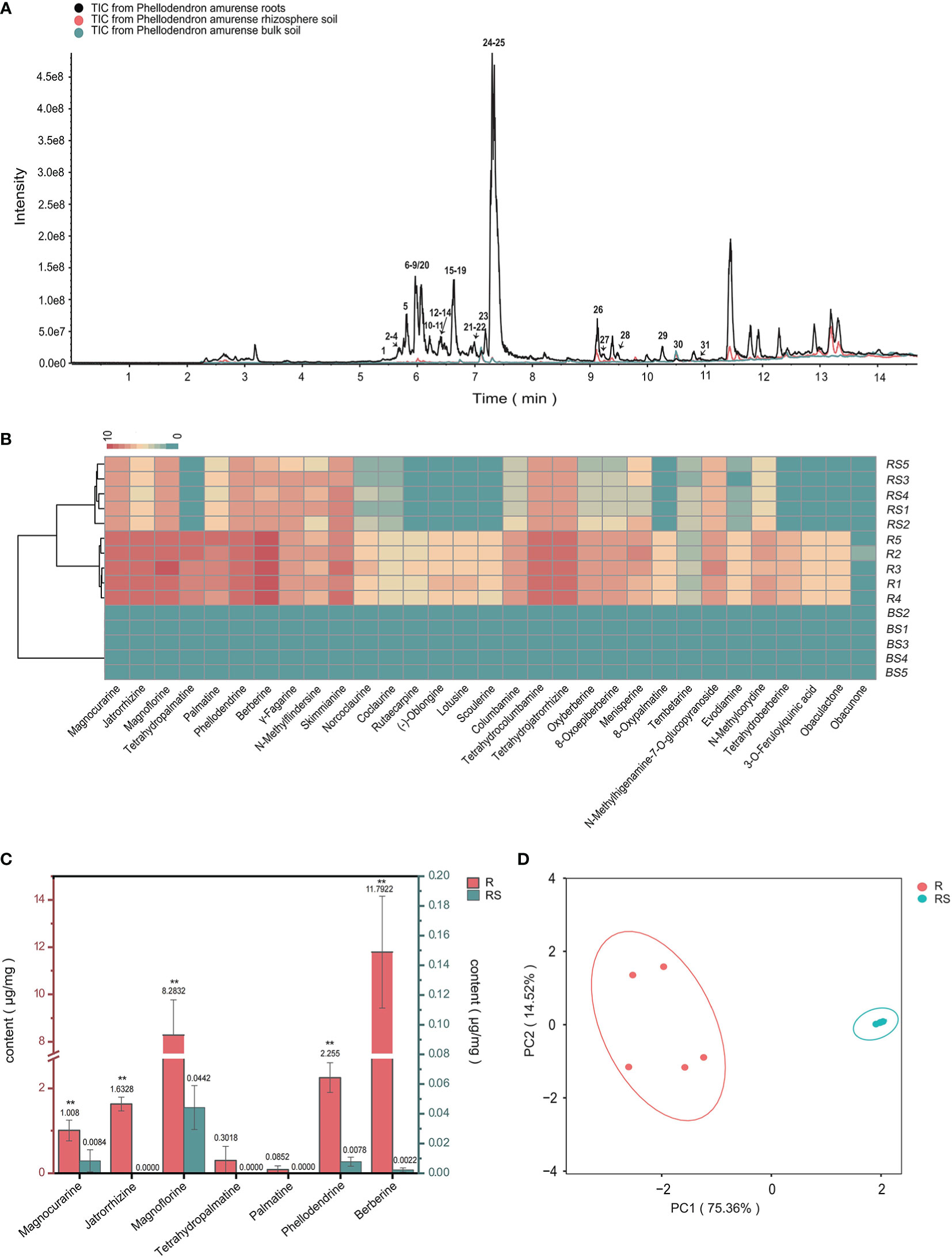
Figure 1 Metabolic analyses of Phellodendron amurense roots (R), rhizosphere soil (RS), and bulk soil (BS). (A) ESI-MS total ion chromatogram profiles of the P. amurense extracts. (B) UPLC-ESI-MS/MS identification of the compounds contained in 15 samples of P. amurense is shown in heatmap. (C) The histogram of seven main alkaloids in P. amurense roots and rhizosphere soil. The red scale (left) is used to measure the absolute content of active ingredients in roots, and, similarly, the blue scale (right) is used to measure the absolute content of bioactive compounds in rhizosphere soil. *p < 0.05 and **p < 0.01. (D) Principal component analysis (PCA) of roots and rhizosphere soil.
To determine the major chemical compounds present in the rhizosphere soil and roots of P. amurense, we used UPLC-MS/MS to analyze the content of seven BIAs (phellodendrine, magnoflorine, magnocurarine, jatrorrhizine, tetrahydropalmatine, palmatine, and berberine), which have been shown to have well pharmacological activity. The highest contents of magnoflorine and berberine were found in the roots, whereas magnoflorine, magnocurarine, and phellodendrine were the main compounds in the rhizosphere soil (Figure 1C). The content of the seven BIAs in roots was significantly higher than that in rhizosphere soil, and these BIAs in roots and rhizosphere soil were, respectively, clustered in two groups, suggesting well sample repetition (Figure 1D).
3.2 Composition of P. amurense root-associated microbial communities
To understand the differences in microbial composition among the bulk, rhizosphere soil, and root, statistical analyses were performed on different taxonomic levels. By using a cutoff of >97% sequence identity, high-quality reads were clustered into bacterial 15,950 OTUs, 16,471 OTUs, and 4,452 OTUs of bulk, rhizosphere soil, and root, respectively. Regarding fungi, high-quality reads were clustered into 3,359 OTUs, 3,526 OTUs, and 1,503 OTUs of bulk, rhizosphere soil, and root, respectively. Rarefaction curve analysis revealed that all samples were nearly parallel to the X-axis, suggesting that the obtained sequences adequately depict the overall bacterial and fungal diversity (Supplementary Figure S1, Supplementary Information). A taxonomic histogram showed that the composition of P. amurense root-associated microbial communities varied along with the compartments. At the bacterial phylum level, the microbial composition analysis revealed that Firmicutes, Acidobacteriota, and Proteobacteria exhibited the highest abundance in bulk soil, whereas Actinobacteriota, Proteobacteria, and Acidobacteriota were enriched in rhizosphere soil (Figure 2A). Notably, the root microbiota was characterized by a prevalence of Cyanobacteria, Proteobacteria, and Actinobacteriota. A marked increase in the proportion of the dominant Cyanobacteria species was observed in roots. At the genus level, the predominant bacterial genera in bulk soil were Bacillus (21.68%), Paenibacillus (11.52%), and RB41 (7.90%). In rhizosphere soil, Pseudomonas (6.23%), RB41 (3.63%), and Catenulispora (2.10%) exhibited the highest proportions. Similarly, Pseudomonas (4.76%), Steroidobacter (1.46%), and Bacillus (0.34%) were identified as the top three bacterial genera in the root (Figure 2B).
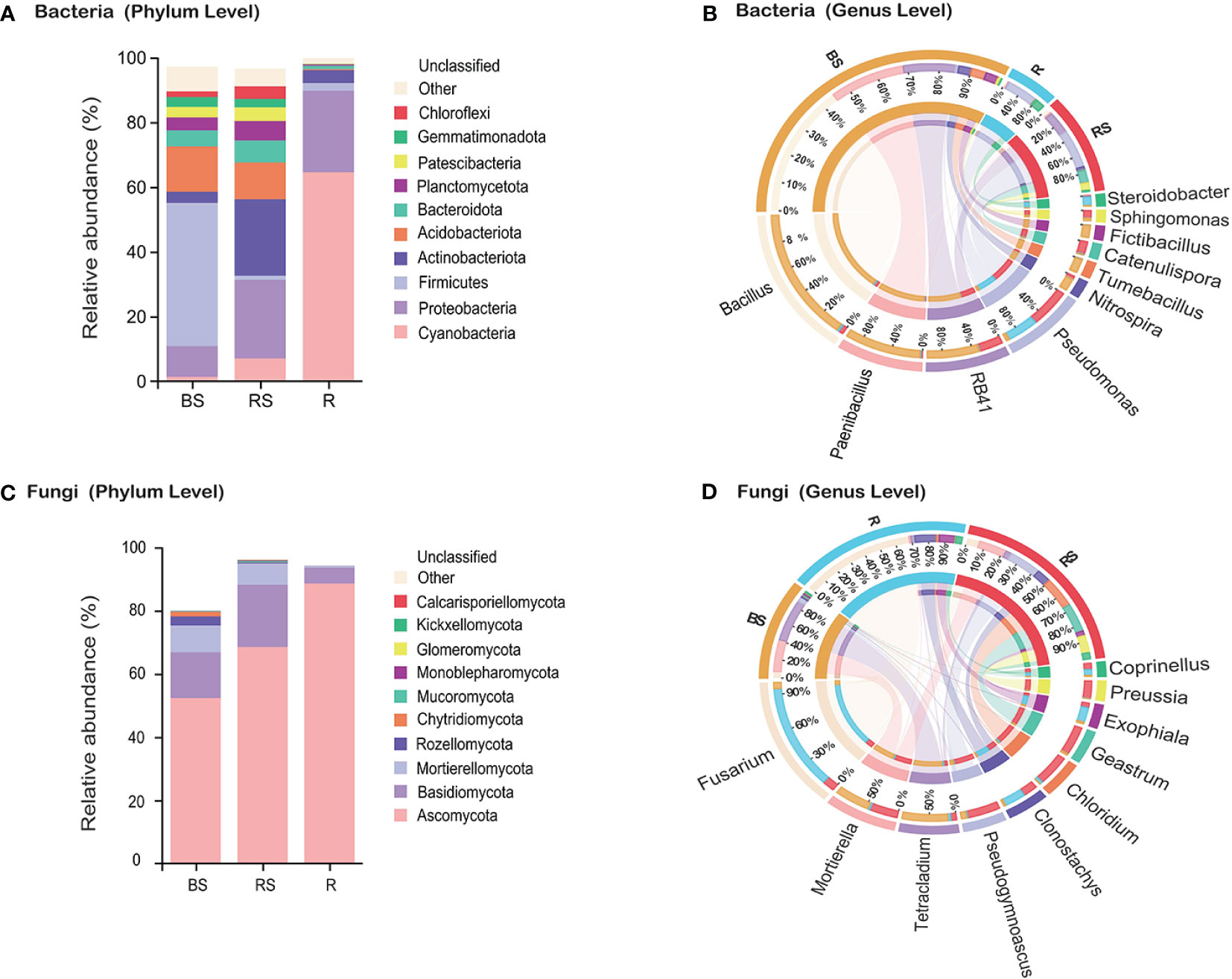
Figure 2 Microbiota community composition of bacteria and fungi in Phellodendron amurense roots (R), rhizosphere soil (RS), and bulk soil (BS). (A, C) The relative abundance of bacteria and fungi at the phylum levels among different samples. (B, D) Circos plot showing the distribution proportion of core microbiota at the genus levels among different samples.
Regarding fungi, the phyla Ascomycota, Basidiomycota, and Mortierellomycota were identified as the most abundant, aligning with a globally observed soil pattern (Figure 2C). Analysis of the relative abundance of fungal genera indicated higher levels of Fusarium (BS, 1.71%; RS, 3.04%; and R, 27.71%), Clonostachys (BS, 0.50%; RS, 3.51%; and R, 5.15%), and Exophiala (BS, 0.51%; RS, 1.26%; and R, 3.89%) in the roots compared with that in the bulk soil and rhizosphere soil (Figure 2D). Conversely, the relative abundance of fungal genera Mortierella (BS, 8.48%; RS, 6.79%; and R, 0.52%), Tetracladium (BS, 11.04%; RS, 1.37%; and R, 0.68%), and Pseudogymnoascus (BS, 1.33%; RS, 7.90%; and R, 0.25%) demonstrated higher levels in the rhizosphere soil and bulk soil compared with that in the root.
3.3 Diversity of P. amurense root-associated microbial communities
The α-diversity index serves as a quantitative metric for assessing species richness within microbial communities, with higher diversity being considered advantageous for overall soil health. Analysis of α-diversity revealed significant variations among sampling compartments (bulk, rhizosphere soil, and root), demonstrating higher diversity in the bulk soil and rhizosphere soil compared with that in root. The Simpson, Chao1, ACE, and Shannon indices for both bacteria and fungi exhibited a sequential decrease in the order of bulk, rhizosphere soil, and root, with the α-diversity in root being the lowest (Figures 3A, B). Kruskal–Wallis analysis further confirmed α-diversity significant differences (p < 0.01) among bulk, rhizosphere soil, and root for both bacteria and fungi (Supplementary Figure S2, Supplementary Information). For β-diversity, PCoA using Bray–Curtis distance assessed the compositional relatedness of bacterial and fungal communities. Results indicated distinct separation of microbiome communities across different sampling compartments. In the case of bacteria, PCoA1 and PCoA2 accounted for 52.45% and 21.38% of the total variance, respectively, with the cumulative variance of PCoA contributing to 73.83% (Figure 3C). Similarly, for fungi, PCoA1 and PCoA2 explained 32.91% and 16.81% of the total variance, respectively (Figure 3D). The PCoA analysis underscored pronounced differences in the community composition of bacteria and fungi among bulk, rhizosphere soil, and root. Collectively, these findings emphasize the distinct microbial community compositions across the sampled compartments, underscoring the ecological heterogeneity within the studied soil system.
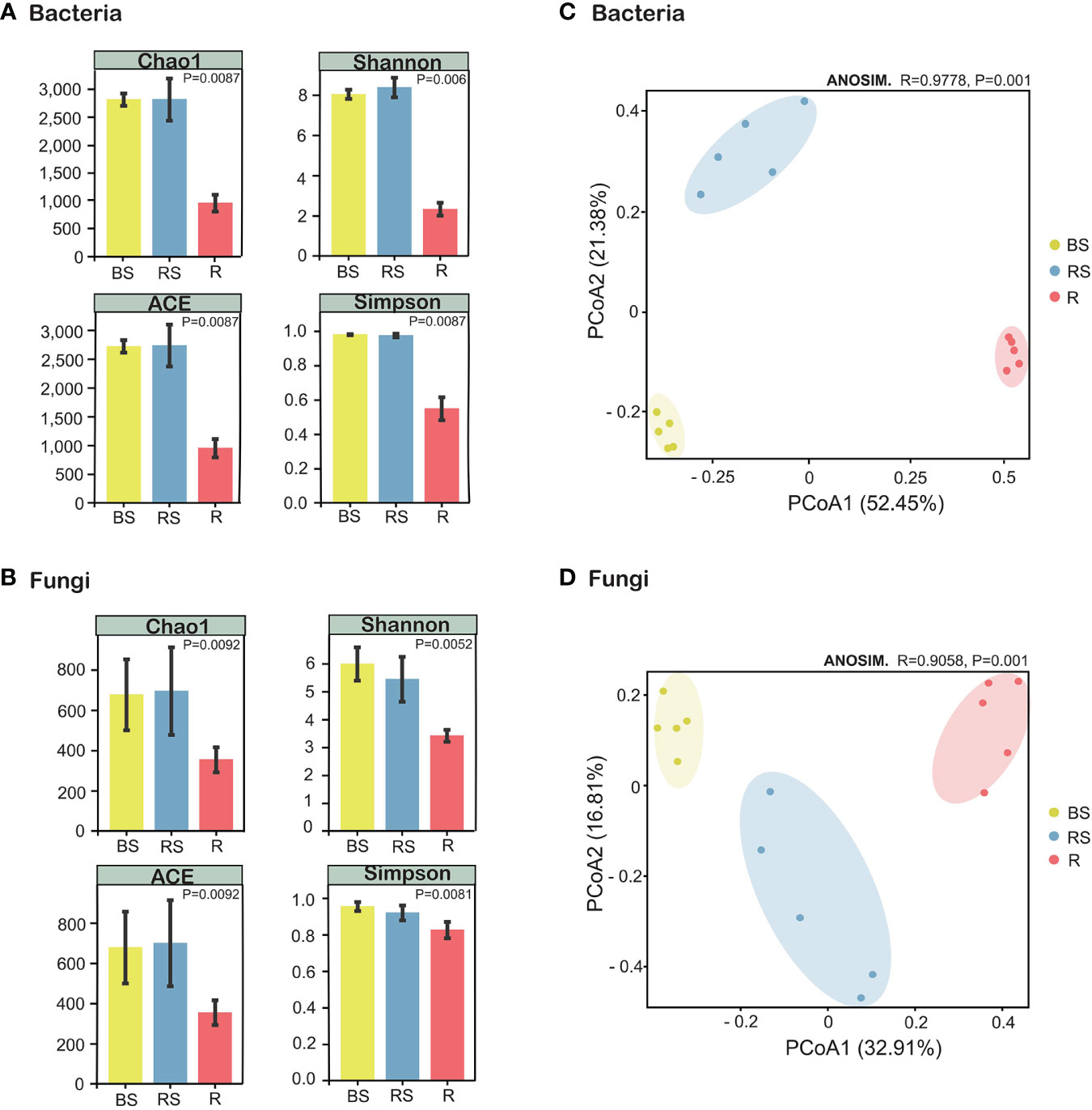
Figure 3 Alpha- and beta-diversity indices of the microorganisms in Phellodendron amurense roots (R), rhizosphere soil (RS), and bulk soil (BS). (A) Chao1 index, ACE index, Shannon index, and Simpson index of the bacterial communities. (B) Chao1 index, ACE index, Shannon index, and Simpson index of the fungal communities. (C) Principal coordinate analysis (PCoA) of the bacterial communities. (D) PCoA plot of the fungal communities.
3.4 Potential biomarkers of P. amurense root-associated microbial communities
The LEfSe methods were employed to discern features exhibiting significant differential abundance among bulk, rhizosphere soil, and root, subsequently determining biomarker microbiota at the family level with a stringent criterion of linear discriminant analysis (LDA) score > 4 (Supplementary Figure S3). For bacteria (Figure 4A), the LDA scores for Bacillaceae and Paenibacillaceae were most elevated in bulk soil, whereas those for Micrococcaceae and Micropepsaceae peaked in rhizosphere soil. Notably, no bacterial biomarkers were annotated at the family level in roots with an LDA score exceeding 4. In the fungi (Figure 4B), the family-level LDA scores revealed distinct biomarkers. Helotiaceae and Mortierellaceae exhibited markedly higher scores in bulk soil, whereas Geastraceae and Pseudeurotiaceae dominated in rhizosphere soil. On the other hand, roots featured a higher abundance of Nectriaceae and Bionectriaceae. Consequently, we identified 17 biomarker microbial families, namely, Bacillaceae, Paenibacillaceae, Pyrinomonadaceae, Gemmatimonadaceae, Alicyclobacillaceae, Alicyclobacillaceae, Micrococcaceae, Micropepsaceae, Catenulisporaceae, Helotiaceae, Mortierellaceae, Leptosphaeriaceae, Geastraceae, Pseudeurotiaceae, Nectriaceae, Bionectriaceae, and Herpotrichiellaceae. These biomarkers contribute to the distinctive microbial signatures characterizing bulk, rhizosphere soil, and root in the studied soil ecosystem.
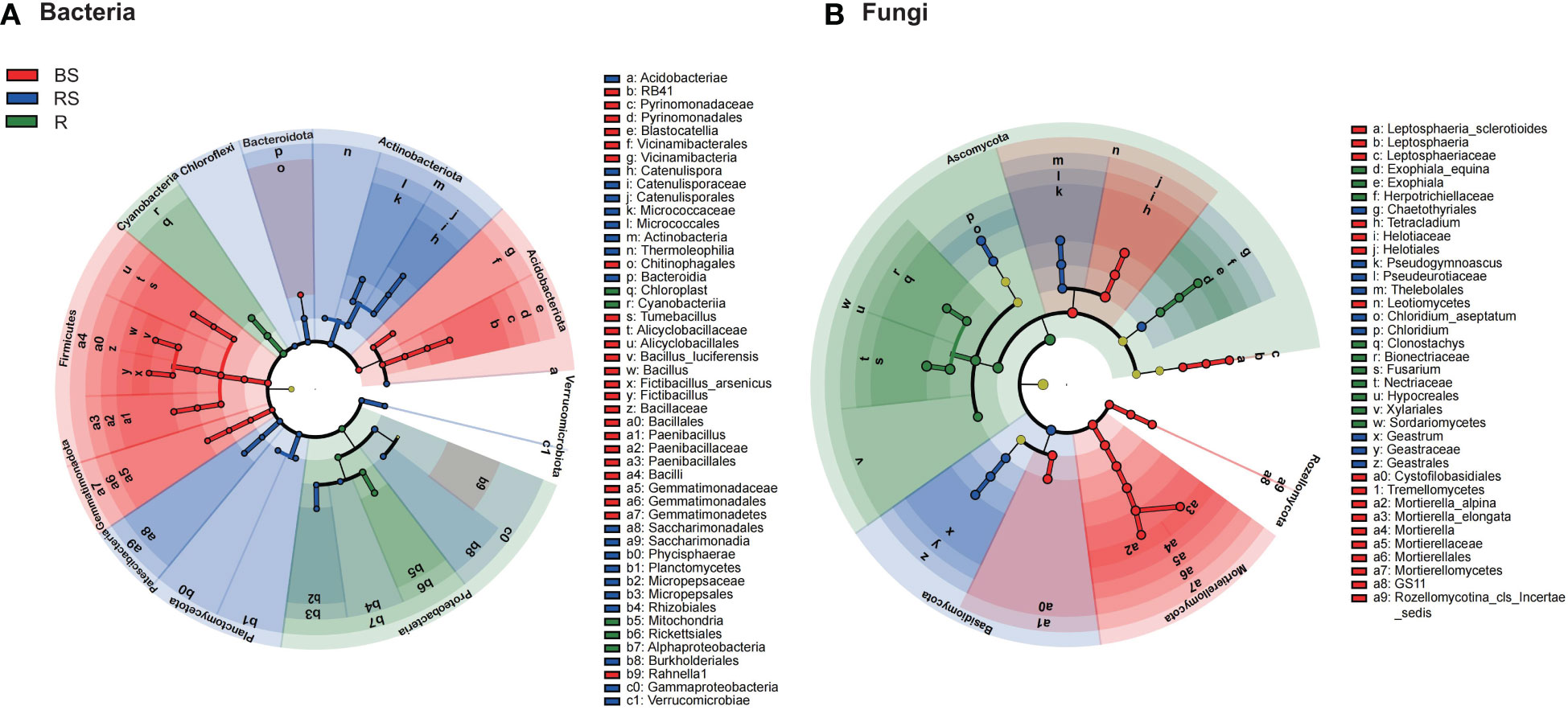
Figure 4 The cladograms of taxa with significant differences in Phellodendron amurense roots (R), rhizosphere soil (RS), and bulk soil (BS). (A, B) Circular tree plot generated from LEfSe analysis of bacteria and fungi with a linear discriminant analysis (LDA) score higher than 4.0 and p-values less than 0.05.
3.5 Correlation analyses of the main active compounds and microbial communities
To unravel the correlation between microbiome communities and the principal bioactive chemical compounds in P. amurense, a total of 4,717 bacterial OTUs and 1,344 fungal OTUs from roots, rhizosphere soil, and bulk soil, alongside data on the seven main alkaloids, were utilized to construct a WGCNA network. This analysis identified 30 and 16 co-expression modules for bacteria and fungi, respectively, based on their similar expression patterns (Figures 5A, B). In bacteria, MEpurple is significantly related to Tetrahydropalmatine. Additonally, the module MEdarkgrey is significantly correlated with Magnoflorine; And the module MEturquoise is significantly related to Palmatine (Figure 5C). In fungi, MEmagenta is significantly related to Tetrahydropalmatine and the module MEred is significantly related to the Palmatine (Figure 5D). Subsequently, correlation analysis between key microorganisms within these modules and the seven BIAs of P. amurense was conducted, and the networks were visualized using Cytoscape. Regarding bacteria, all seven benzylisoquinoline alkaloids displayed significantly positive correlations (p < 0.01) with Streptosporangiaceae and Sphingobacteriaceae (Figure 5E). However, Chitinophagaceae showed a negative correlation with tetrahydropalmatine, whereas Paenibacillaceae exhibited negative correlations with tetrahydropalmatine, berberine, and jatrorrhizine. In fungi, berberine, palmatine, jatrorrhizine, phellodendrine, tetrahydropalmatine, magnocurarine, and magnoflorine exhibited significantly positive correlations (p < 0.01) with Nectriaceae and Cunninghamellaceae (Figure 5F). These findings provide insights into the intricate interplay between microbial communities and the biosynthesis of bioactive compounds in P. amurense.
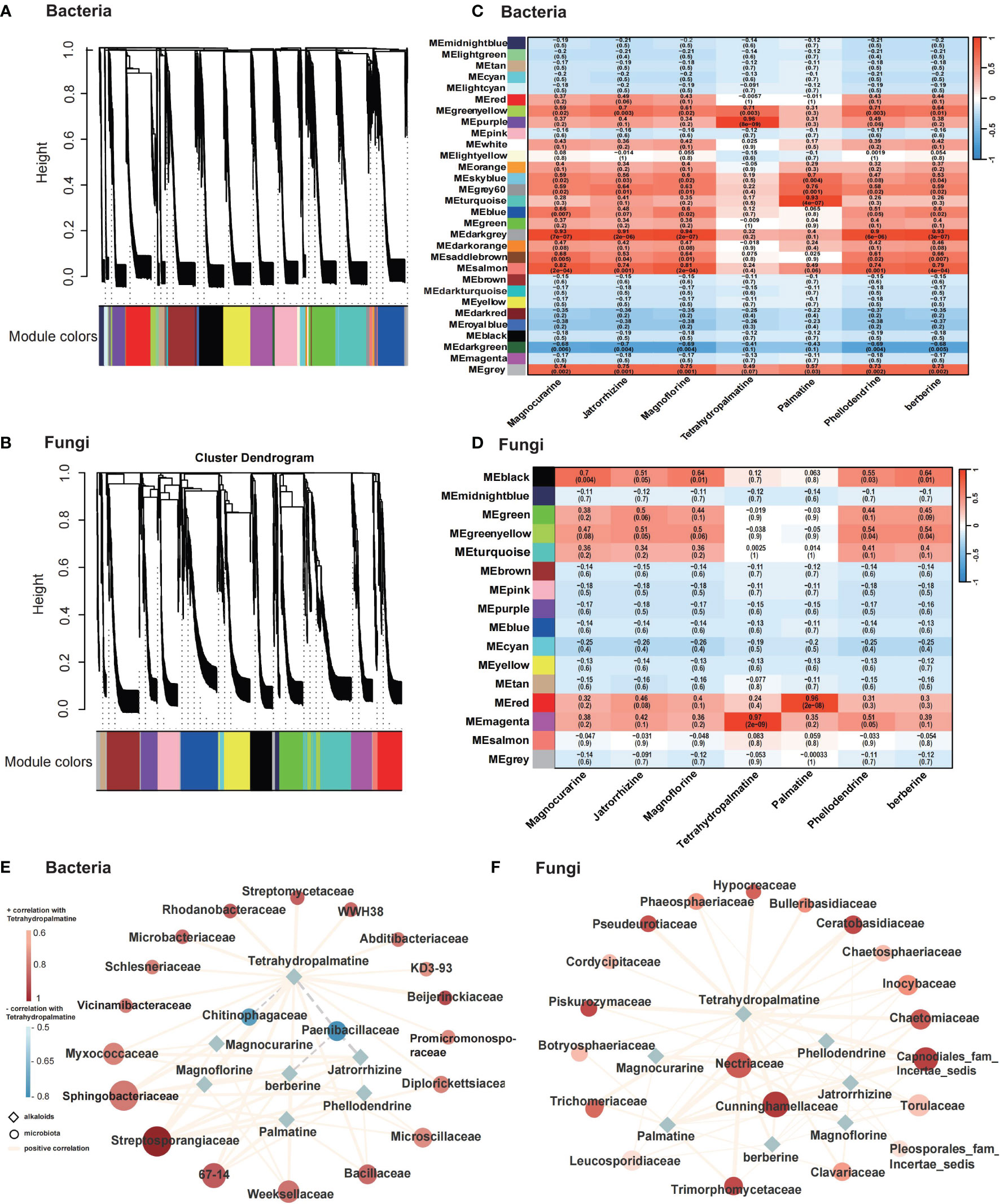
Figure 5 Microbial and metabolic correlation analysis across roots (R), rhizosphere soil (RS), and bulk soil (BS) samples of Phellodendron amurense. (A, B) Dendrogram showing co-expression modules (clusters) identified by weighted correlation network analysis (WGCNA) across plant root compartments and bulk soil. (C, D) Heatmap showing module–alkaloid correlations of bacteria and fungi in P. amurense. Each row corresponds to a module indicated by different colors, represents a cluster of OTUs with similarity over 90%. Each column corresponds to an alkaloid. (E, F) Metabolism of main alkaloids and top 20 putative bacteria and fungi microorganisms’ regulatory network (P < 0.01). The solid line (orange) and dotted line (gray) indicate positive and negative correlations, respectively. The shape of the node denotes the components used in this study (ellipse, microbiome; diamond, alkaloids), and the color indicates the degree of correlation with tetrahydropalmatine.
4 Discussion
P. amurense may exudate specialized BIA metabolites into rhizosphere soil. The rhizosphere refers to the narrow zone of soil that is influenced by root secretions. The present study revealed that the relative content of root alkaloids was higher than that in rhizosphere soil, with bulk soil being not detected. In a study of pepper greenhouse cultivation, it was observed that metabolites were differ between rhizosphere soil and bulk soil, with the majority of metabolites being more abundance in rhizosphere soil compared with that in bulk soil (Song et al., 2020). We observed the similar results in P. amurense, and these differences were primarily observed in plant alkaloid metabolites, which were attributed to the biological properties of P. amurense. P. amurense mainly accumulates BIAs, especially in protoberberine-type alkaloids, including berberine, palmatine, jatrorrhizine, phellodendrine, and tetrahydropalmatine (Wang et al., 2015). This finding supported that plant root-released metabolites play an essential role in the metabolites profiles in rhizosphere soil (Zhang H. L. et al., 2020).
Various studies have demonstrated that medicinal plants can impact their microbial composition by releasing different types of metabolites (Hu et al., 2018; Nakayasu et al., 2021; Sugiyama, 2021). The root-associated bacterial and fungal communities were found to be distinctly different in the three soil-root compartments. This could be due to the fact that the root exudates of P. amurense have a greater impact on rhizosphere bacteria than on bulk soil bacteria. The higher input of rhizodeposits into the rhizosphere may lead to significant shift in the bacterial community. The fungi communities associated with the roots in P. amurense were similar to previous studies, mainly including Ascomycota, Basidiomycota, and Mortierellomycota, regardless of plant species (Muller et al., 2016; Trivedi et al., 2020; Liu et al., 2023). In addition, the α-diversity of soil bacteria and fungi was higher in the bulk soil than that in the rhizosphere, with the lowest diversity found in the roots. This result is consistent with other studies that explain how plants select their ecological niche (Trivedi et al., 2020; Xiong et al., 2021; Liu et al., 2022). This may be due to the fact that bulk soil can be compared to a “microbial seed bank,” serving as a reservoir from which rhizosphere microorganisms are recruited. As a result, the diversity of rhizosphere microorganisms is reduced (Ling N. et al., 2022).
In the present study, the phylum Cyanobacteria constituted more than 60% of the root endosphere communities. It is noteworthy that some Cyanobacteria have the ability to fix nitrogen through photosynthesis, and inner diazotrophic bacteria have been well documented in rice, maize, sugarcane, and some gramineous energy plants (Roesch et al., 2007; Thaweenut et al., 2010; Zhang et al., 2019; Purwani et al., 2021). Nitrogen-fixing microorganism is a kind of microorganism that can convert nitrogen into ammonia. Nitrogen-fixing microorganism plays an important role in nature and can convert nitrogen in the atmosphere into ammonia that can be absorbed and utilized by plants, which plays an important role in maintaining ecological balance and promoting plant growth. Alkaloids are a class of nitrogen-containing organic compounds existing in organisms, which usually have complex structures and diverse biological activities. Alkaloid components are formed by the amino acid pathway, and amino acids decarboxylated into amines, which are converted into alkaloids through a series of chemical reactions (methylation, oxidation, reduction, rearrangement, etc.). Therefore, it is speculated by our study that nitrogen-fixing microorganisms may provide nitrogen source for alkaloid biosynthesis by fixing more nitrogen, resulting in a higher alkaloid content in phellodendron amurense. However, this hypothesis requires further experimental verification.
In this study, significant relationships were observed between specific microbes and the contents of bioactive compounds. Nectriaceae was significantly positively correlated with seven BIAs, including berberine, palmatine, jatrorrhizine, phellodendrine, tetrahydropalmatine, magnocurarine, and magnoflorine. Nectriaceae was also the biomarker of root compared with that of bulk and rhizosphere soil in LEfSe analysis. It has been reported that members of Nectriaceae family are commonly found in various environments, where they play significant socio-economic roles in agriculture, industry, and medicine. The majority of these species are soil-borne saprobes or weak to virulent, facultative, or obligate plant pathogens, whereas some are facultatively fungicolous or insecticolous (Lombard et al., 2015).
Furthermore, seven BIAs showed a significant positive correlation with Sphingobacteriaceae. These bacteria are commonly found in nature, particularly in soils, oceans, and freshwater, due to their ability to utilize a variety of organic compounds and to thrive and survive under low-nutrient conditions. Sphingobacteriaceae, a member of Sphingobacteriales, plays a significant role as a plant growth-promoting rhizobacteria that can enhance the growth of rice and tomato (Asaf et al., 2018). In addition, it serves as an eco-friendly biological resource for decontaminating polluted areas while promoting the growth of plants facing environmental disturbances (Khan et al., 2014; Kim et al., 2019). In the next step, inoculation experiments will be conducted to clarify the mechanism by which the specific microbes stimulate the production of plant specialized metabolites in P. amurense.
5 Conclusions
Here, we investigated the rhizosphere soil and root metabolites of the medicinal plant P. amurense, as well as their microbiome characteristics, and decoded the relationship between the microbial communities and the seven root-derived specialized metabolites. These metabolites varied significantly among the P. amurense compartments. The composition of the rhizosphere microbiome significantly differed from that of the endospheric microbiome. Seven benzylisoquinoline alkaloids (berberine, palmatine, jatrorrhizine, phellodendrine, tetrahydropalmatine, magnocurarine, and magnoflorine) were significantly positive correlated with Nectriaceae and Cunninghamellaceae. In summary, this study has revealed a complex interplay between root-associated microorganisms and plant secondary metabolites, offering a potential strategy for enhancing the industrial and pharmacological value of P. amurense.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: BioProject, PRJNA1066709 and PRJNA1066243.
Author contributions
LT: Conceptualization, Funding acquisition, Supervision, Writing – review & editing. ZX: Conceptualization, Funding acquisition, Resources, Supervision, Writing – review & editing. RG: Data curation, Funding acquisition, Methodology, Writing – review & editing. WZ: Formal Analysis, Investigation, Validation, Visualization, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research has benefited from National Natural Science Foundation of China (Grant No.82360754, 82274037, U23A20511), Fundamental Research Funds for the Central public welfare research institutes (ZZ16-ND-10-02), Guizhou University introduced talent research project (Gui Da Ren Ji He Zi (2022) 57), National Key R&D Program of China (2023YFC3504800), Key project at central government level: The ability establishment of sustainable use for valuable Chinese medicine resources (2060302).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1363063/full#supplementary-material
References
Asaf, S., Khan, A. L., Khan, M. A., Al-Harrasi, A., Lee, I. J. (2018). Complete genome sequencing and analysis of endophytic Sphingomonas sp. LK11 and its potential in plant growth. 3 Biotech. 8, 389. doi: 10.1007/s13205-018-1403-z
Balážová, L., Kurhajec, S., Kello, M., Bedlovičová, Z., Zigová, M., Petrovová, E., et al. (2022). Antiproliferative effect of Phellodendron amurense Rupr. Based on angiogenesis. Life 12, 767. doi: 10.3390/life12050767
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chen, H. M., Wu, H. X., Yan, B., Zhao, H. G., Liu, F. H., Zhang, H. H., et al. (2018). Core microbiome of medicinal plant Salvia miltiorrhiza seed: a rich reservoir of beneficial microbes for secondary metabolism? Int. J. Mol. Sci. 19, 672. doi: 10.3390/ijms19030672
Chen, S. F., Zhou, Y. Q., Chen, Y. R., Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cui, J., Gong, Y., Vijayakumar, V., Zhang, G., Wang, M., Wang, J., et al. (2019). Correlation in chemical metabolome and endophytic mycobiome in Cynomorium songaricum from different desert locations in China. J. Agric. Food Chem. 67, 3554–3564. doi: 10.1021/acs.jafc.9b00467
Cui, X. S., Meng, T. T., Zhu, N., Li, X., Leng, F. F., Wang, Y. G. (2023). Composition, function and driving factors of microbial communities in rhizosphere soil and root endophyte of Codonopsis pilosula in different years. Rhizosphere 27, 100712. doi: 10.1016/j.rhisph.2023.100712
Dixon, P. (2003). VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x
Edgar, C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/Nmeth.2604
Edwards, J., Johnson, C., Santos-Medellín, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U.S.A. 112, E911–E920. doi: 10.1073/pnas.1414592112
Erhan, S. E., Pârvu, A. E., Ciorîţă, A., Putri, A. A., Molina, A. J., Pârvu, M., et al. (2023). Chemical composition and anti-inflammatory effect of Phellodendron amurense Rupr. stem bark extract. Not. Bot. Horti Agrobot. Cluj Napoca. 51, 13306. doi: 10.15835/nbha51313306
Feng, Y., Zheng, Y. G., Xin, X. M., Guo, Q. H., Hao, Y., Li, C. H., et al. (2023). Study on chemical constituents and fingerprints of Phellodendron amurense Rupr. J. Sep. Sci. 46, 2300151. doi: 10.1002/jssc.202300151
Fu, L., Chin, C. (1992). China plant red data book: rare and endangered plants (Beijing: Science Press).
Guo, M. J., Wu, F. H., Hao, G. G., Qi, Q., Li, R., Li, N., et al. (2017). Bacillus subtilis improves immunity and disease resistance in rabbits. Front. Immunol. 8. doi: 10.3389/fimmu.2017.00354
Hilton, S., Picot, E., Schreiter, S., Bass, D., Norman, K., Oliver, A. E., et al. (2021). Identification of microbial signatures linked to oilseed rape yield decline at the landscape scale. Microbiome 9, 19. doi: 10.1186/s40168-020-00972-0
Hu, L. F., Robert, C. A. M., Cadot, S., Zhang, X., Ye, M., Li, B. B., et al. (2018). Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 9, 2738. doi: 10.1038/s41467-018-05122-7
Huang, W. J., Long, C. L., Lam, E. (2018). Roles of plant-associated microbiota in traditional herbal medicine. Trends Plant Sci. 23, 559–562. doi: 10.1016/j.tplants.2018.05.003
Khan, A. L., Waqas, M., Kang, S. M., Al-Harrasi, A., Hussain, J., Al-Rawahi, A., et al. (2014). Bacterial endophyte sphingomonas sp. LK11 produces gibberellins and IAA and promotes tomato plant growth. J. Microbiol. 52, 689–695. doi: 10.1007/s12275-014-4002-7
Kim, J. H., Huh, J. E., Baek, Y. H., Lee, J. D., Choi, D. Y., Park, D. S. (2011). Effect of Phellodendron amurense in protecting human osteoarthritic cartilage and chondrocytes. J. Ethnopharmacol. 134, 234–242. doi: 10.1016/j.jep.2010.12.005
Kim, Y. J., Lim, J., Sukweenadhi, J., Seok, J. W., Lee, S. W., Park, J. C., et al. (2019). Genomic characterization of a newly isolated rhizobacteria Sphingomonas panacis reveals plant growth promoting effect to rice. Biotechnol. Bioprocess Eng. 24, 119–125. doi: 10.1007/s12257-018-0386-2
Koberl, M., Schmidt, R., Ramadan, E. M., Bauer, R., Berg, G. (2013). The microbiome of medicinal plants: diversity and importance for plant growth, quality and health. Front. Microbiol. 4. doi: 10.3389/fmicb.2013.00400
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Li, Y. M., Jin, J., Li, P. R., Wang, Q., Xu, L. L., Wei, G. H., et al. (2023). Regional variations and plant compartments shape the community structures of the endophytic microbiome and secondary metabolites of Astragalus mongholicus. Ind. Crops Prod. 192, 116037. doi: 10.1016/j.indcrop.2022.116037
Li, Y. M., Liu, Y., Zhang, H., Yang, Y., Wei, G. H., Li, Z. F. (2021). The composition of root-associated bacteria and fungi of Astragalus mongholicus and their relationship with the bioactive ingredients. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.642730
Ling, F., Egan, S., Zhuang, Y. R., Chang, L. R., Xiao, L. Y., Yang, Q., et al. (2022). Epimicrobiome shifts with bleaching disease progression in the brown seaweed Saccharina japonica. Front. Mar. Sci. 9. doi: 10.3389/fmars.2022.865224
Ling, N., Wang, T. T., Kuzyakov, Y. (2022). Rhizosphere bacteriome structure and functions. Nat. Commun. 13, 836. doi: 10.1038/s41467-022-28448-9
Liu, Y., Li, D., Gao, H., Li, Y. H., Chen, W. M., Jiao, S., et al. (2022). Regulation of soil micro-foodwebs to root secondary metabolites in cultivated and wild licorice plants. Sci. Total Environ. 828, 154302. doi: 10.1016/j.scitotenv.2022.154302
Liu, J. P., Tang, Y. J., Bao, J. S., Wang, H. K., Chen, M. Y., Peng, F., et al. (2023). Integrated microbiome and metabolomics analysis reveal a closer relationship between soil metabolites and bacterial community than fungal community in pecan plantations. Land Degrad. Dev. 34, 2812–2824. doi: 10.1002/ldr.4649
Lombard, L., Merwe, N. A. V., Groenewald, J. Z., Crous, P. W. (2015). Generic concepts in nectriaceae. Stud. Mycol. 80, 189–245. doi: 10.1016/j.simyco.2014.12.002
Luo, H., Zhao, Y., Hua, H., Zhang, Y., Zhang, X. B., Fang, Q. M., et al. (2021). Research progress on quality assurance of genuine Chinese medicinal in Sichuan. Chin. Med. 16, 19. doi: 10.1186/s13020-021-00428-z
Ma, L., Li, J. Q., Hu, Y. D. (2015). Determination of berberine in Phellodendron amurense from different sites of Changbai Mountain. J. For. Res. 26, 201–207. doi: 10.1007/s11676-014-0013-9
Magoc, T. J., Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mishra, A. K., Sudalaimuthuasari, N., Hazzouri, K. M., Saeed, E. E., Shah, I., Amiri, K. M. A. (2022). Tapping into plant-microbiome interactions through the lens of multi-omics Techniques. Cells 11 (20), 3254. doi: 10.3390/cells11203254
Muller, D. B., Vogel, C., Bai, Y., Vorholt, J. A. (2016). The plant microbiota: systems-level insights and perspectives. Annu. Rev. Genet. 50, 211–234. doi: 10.1146/annurev-genet-120215-034952
Nakayasu, M., Ohno, K., Takamatsu, K., Aoki, Y., Yamazaki, S., Takase, H., et al. (2021). Tomato roots secrete tomatine to modulate the bacterial assemblage of the rhizosphere. Plant Physiol. 186, 270–284. doi: 10.1093/plphys/kiab069
Pang, Z. Q., Chen, J., Wang, T. H., Gao, C. S., Li, Z. M., Guo, L. T., et al. (2021). Linking plant secondary metabolites and plant microbiomes: a review. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.621276
Purwani, J., Pratiwi, E., Sipahutar, I. A. (2021). The effect of different species of cyanobacteria on the rice yield and nitrogen use efficiency under different levels of nitrogen fertilizer on alluvial West Java. IOP Conf. Ser.: Earth Environ. Sci. 648, 12196. doi: 10.1088/1755-1315/648/1/012196
Roberts, L. D., Souza, A. L., Gerszten, R. E., Clish, C. B. (2012). Targeted metabolomics. Curr. Protoc. Mol. Biol. 98, 30.2.1–30.2.24. doi: 10.1002/0471142727.mb3002s98. Chapter 30: Unit 30.2.1-30.2.24.
Roesch, L. F. W., Camargo, F. A. O., Bento, F. M., Triplett, E. W. (2007). Biodiversity of diazotrophic bacteria within the soil, root and stem of field-grown maize. Plant Soil. 302, 91–104. doi: 10.1007/s11104-007-9458-3
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. doi: 10.1186/gb-2011-12-6-r60
Song, Y., Li, X. N., Yao, S., Yang, X. L., Jiang, X. (2020). Correlations between soil metabolomics and bacterial community structures in the pepper rhizosphere under plastic greenhouse cultivation. Sci. Total Environ. 728, 138439. doi: 10.1016/j.scitotenv.2020.138439
Steinmann, D., Baumgartner, R. R., Heiss, E. H., Bartenstein, S., Atanasov, A. G., Dirsch, V. M., et al. (2011). Bioguided isolation of (9Z)-octadec-9-enoic acid from Phellodendron amurense Rupr. and identification of fatty acids as PTP1B Inhibitors. Planta Med. 78, 219–224. doi: 10.1055/s-0031-1280377
Sugiyama, A. (2021). Flavonoids and saponins in plant rhizospheres: roles, dynamics, and the potential for agriculture. Biosci. Biotechnol. Biochem. 85, 1919–1931. doi: 10.1093/bbb/zbab106
Sun, Y., Lenon, G. B., Yang, A. W. H. (2019). Phellodendri cortex: a phytochemical, pharmacological, and pharmacokinetic review. Evid. Based Complement. Altern. Med. 2019, 7621929. doi: 10.1155/2019/7621929
Sun, M. L., Xu, L. J., Peng, Y. L., Liu, T., Zhang, Y. H., Zhou, Z. Q. (2015). Multiscale analysis of the contents of palmatine in the Nature populations of Phellodendron amurense in northeast China. J. For. Res. 27, 265–272. doi: 10.1007/s11676-015-0200-3
Thaweenut, N., Hachisuka, Y., Ando, S., Yanagisawa, S., Yoneyama, T. (2010). Two seasons’ study on nifH gene expression and nitrogen fixation by diazotrophic endophytes in sugarcane (Saccharum spp. hybrids): expression of nifH genes similar to those of rhizobia. Plant Soil. 338, 435–449. doi: 10.1007/s11104-010-0557-1
Toju, H., Tanabe, A. S., Yamamoto, S., Sato, H. (2012). High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PloS One 7, e40863. doi: 10.1371/journal.pone.0040863
Trivedi, P., Leach, J. E., Tringe, S. G., Sa, T. M., Singh, B. K. (2020). Plant-microbiome interactions: from community assembly to plant health. Nat. Rev. Microbiol. 18, 607–621. doi: 10.1038/s41579-020-0412-1
Velmurugan, N., Kalpana, D., Cho, J. Y., Lee, Y. S. (2017). Chemical composition and antioxidant capacity of the aqueous extract of Phellodendron amurense. J. For. Res. 29, 1041–1048. doi: 10.1007/s11676-017-0532-2
Wang, W. C., Li, Q. Y., Liu, Y. H., Chen, B. B. (2015). Ionic liquid-aqueous solution ultrasonic-assisted extraction of three kinds of alkaloids from Phellodendron amurense Rupr and optimize conditions use response surface. Ultrason. Sonochem. 24, 13–18. doi: 10.1016/j.ultsonch.2014.10.009
Wei, G. F., Ning, K., Zhang, G. Z., Yu, H. B., Yang, S. M., Dai, F., et al. (2021). Compartment niche shapes the assembly and network of Cannabis sativa-associated microbiome. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.714993
Wicham, H. (2011). ggplot2. Wiley interdisciplinary reviews: computational statistics. Focus article. 3, 180–185. doi: 10.1002/wics.147
Xiao, Q., Mu, X. L., Liu, J. S., Li, B., Liu, H. T., Zhang, B. G., et al. (2022). Plant metabolomics: a new strategy and tool for quality evaluation of Chinese medicinal materials. Chin. Med. 17, 45. doi: 10.1186/s13020-022-00601-y
Xiong, C., Singh, B. K., He, J. Z., Han, Y. L., Li, P. P., Wan, L. H., et al. (2021). Plant developmental stage drives the differentiation in ecological role of the maize microbiome. Microbiome 9, 171. doi: 10.1186/s40168-021-01118-6
Yang, H. S., Li, X. P., Liu, D. J., Chen, X. B., Li, F. H., Qi, X. L., et al. (2016). Genetic diversity and population structure of the endangered medicinal plant Phellodendron amurense in China revealed by SSR markers. Biochem. Syst. Ecol. 66, 286–292. doi: 10.1016/j.bse.2016.04.018
Yu, J. H., Chen, C. M., Han, S. J., Guo, X. R., Yuan, S. S., Wang, C. J., et al. (2013). Development and characterization of polymorphic microsatellite loci in Phellodendron amurense (Rutaceae). Appl. Plant Sci. 1, 1200321. doi: 10.3732/apps.1200321
Zhang, H. L., Huang, M., Zhang, W. H., Gardea-Torresdey, J. L., White, J. C., Ji, R., et al. (2020). Silver nanoparticles alter soil microbial community compositions and metabolite profiles in unplanted and cucumber-planted soils. Environ. Sci. Technol. 54, 3334–3342. doi: 10.1021/acs.est.9b07562
Zhang, J. Y., Liu, Y. X., Zhang, N., Hu, B., Jin, T., Xu, H. R., et al. (2019). NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 37, 676–684. doi: 10.1038/s41587-019-0104-4
Zhang, H., Xia, Y., Cui, J. L., Ji, X., Miao, S. M., Zhang, G., et al. (2023). The composition characteristics of endophytic communities and their relationship with metabolites profile in Ephedra sinica under wild and cultivated conditions. Environ. Sci. pollut. Res. 30, 95648–95659. doi: 10.1007/s11356-023-29145-w
Zhang, Y. H., Xu, L. J., Qiu, J. J., Sun, M. L., Xia, C. M., Zhou, Z. Q., et al. (2014). Provenance variations in berberine content of Phellodendron amurense, a rare and endangered medicinal plant grown in Northeast China. Scand. J. For. Res. 29, 725–733. doi: 10.1080/02827581.2014.978885
Zhang, Y., Zhang, Z. P., Liu, H. T., Zhang, B. G., Liao, Y. H., Zhang, Z. (2015). Quantitative and chemical fingerprint analysis for quality evaluation of the dried bark of wild Phellodendron amurense Rupr. based on HPLC-DAD-MS combined with chemometrics methods. Anal. Methods 7, 2041–2049. doi: 10.1039/c4ay02827a
Zhang, Y. H., Zheng, L. L., Zheng, Y., Xue, S., Zhang, J. X., Huang, P., et al. (2020). Insight into the assembly of root-associated microbiome in the medicinal plant Polygonum cuspidatum. Ind. Crops Prod. 145, 112163. doi: 10.1016/j.indcrop.2020.112163
Zhang, B. Y., Zou, H., Chen, B. R., Zhang, X. Y., Kang, X., Wang, C., et al. (2023). Optimizing the distribution pattern of species under climate change: the protection and management of Phellodendron amurense in China. Front. Ecol. Evol. 11. doi: 10.3389/fevo.2023.1186627
Keywords: Phellodendron amurense, bisbenzylisoquinoline alkaloids, microbial communities, 16S rRNA, ITS
Citation: Zhang W, Gao R, Tian L and Xu Z (2024) Integrated microbiome and metabolomics analysis reveal the relationship between plant-specialized metabolites and microbial community in Phellodendron amurense. Front. Plant Sci. 15:1363063. doi: 10.3389/fpls.2024.1363063
Received: 29 December 2023; Accepted: 29 January 2024;
Published: 21 February 2024.
Edited by:
Chun Sui, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Mingkun Huang, Lushan Botanical Garden (CAS), ChinaMengmeng Liu, Hebei University, China
Copyright © 2024 Zhang, Gao, Tian and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lixia Tian, dGx4bWlhb0AxNjMuY29t; Zhichao Xu, emN4dUBuZWZ1LmVkdS5jbg==