 Shougo Ishio1
Shougo Ishio1 Kazutaka Kusunoki
Kazutaka Kusunoki Tadayoshi Kanao
Tadayoshi Kanao Takashi Tamura
Takashi Tamura- 1Tsukuba Research Institute, Sumitomo Forestry Co. Ltd., Tsukuba, Japan
- 2Graduate School of Environment, Life, Natural Science and Technology, Okayama University, Okayama, Japan
- 3Institute of Global Human Resource Development, Okayama University, Okayama, Japan
Acacia crassicarpa is a fast-growing leguminous tree that is widely cultivated in tropical areas such as Indonesia, Malaysia, Australia, and southern China. This tree has versatile utility in timber, furniture, and pulp production. Illumina sequencing of A. crassicarpa was conducted, and the raw data of 124,410,892 reads were filtered and assembled de novo into 93,317 unigenes, with a total of 84,411,793 bases. Blast2GO annotation, Benchmark Universal Single-Copy Ortholog evaluation, and GO-term classification produced a catalogue of unigenes for studying primary metabolism, phytohormone signaling, and transcription factors. Massive transcriptomic analysis has identified microsatellites composed of simple sequence repeat (SSR) loci representing di-, tri-, and tetranucleotide repeat units in the predicted open reading frames. Polymorphism was induced by PCR amplification of microsatellite loci located in several genes encoding auxin response factors and other transcription factors, which successfully distinguished 16 local trees of A. crassicarpa tested, representing potentially exploitable molecular markers for efficient tree breeding for plantation and biomass exploitation.
1 Introduction
Acacia is a fast-growing leguminous tree that can be harvested within a relatively short period (several years). The genetic traits for such rapid carbon fixation allow for a rapid cycle of harvesting and reforestation of tree plantations. Because of their ability to thrive in degraded soils, even under drought conditions (Pan and You, 1994), these trees are planted in large areas of Southeast Asia and Southern China (Midgley and Turnbull, 2003). Acacia crassicarpa is a preferred leguminous tree with valuable properties, such as high wood density, excellent biomass yield, low moisture content, and high combustion heat. Because of the intrinsic ability of trees to store abundant plant nutrients K+, Ca2+, and Mg2+ from the soil, their ash can become highly alkaline with a pH greater than 12 (Yusiharni and Gilkes, 2012). Large-scale burning of wood produces residual alkaline ash, which is highly sticky and causes significant mechanical damage to movable floors in biomass power plants (Fuller et al., 2019; Hallgren et al., 1999). These properties of wood species present breeding challenges that must be solved, and accurate genomic information and molecular breeding techniques are required to address these challenges.
Because traditional breeding of tree species has intrinsic limitations owing to their slow growth and naturally long lifecycles, it is highly challenging to select improved varieties that have the desired genetic traits. Molecular breeding of commercial plants requires several basic resources such as genome sequence information and annotation of genes responsible for target traits. A. crassicarpa has a relatively large genome of 1,350 Mbp (Mukherjee and Sharma, 1995), which is approximately double that of the model species Acacia mangium, at 635 Mbp (Blakesley et al., 2002). Genetic modification of A. crassicarpa is difficult without precise sequence data, such as those obtained from expressed sequence tag (EST) libraries. Efforts have been made to construct transcriptomic databases and microsatellite markers for Acacia tree species (McKinnon et al., 2018), and a few pioneering studies have identified a limited number of ESTs. For example, in A. mangium, 8,963 of 10,752 clones of a cDNA library were constructed via conventional molecular cloning and sequencing (Suzuki et al., 2011).
Massively parallel sequencing of cDNA libraries (RNA-Seq) has become a valuable tool for genome analysis, and is rapidly replacing ESTs for structural and functional genome analysis in plants (Pashley et al., 2006). The illumina-based technology permits the investigation of spliced transcripts, including alternative splicing, leading to the large-scale discovery of novel transcripts and the identification of gene boundaries at single-nucleotide resolution.
Plant somatic cells can undergo dedifferentiation to give rise to a pluripotent cell mass called a callus, which can potentially regenerate new organs or the whole plant (Sugimoto et al., 2010). Proliferative plant somatic cells also provide the opportunity to introduce foreign genes to alter their genetic traits. After genetic alteration, it is necessary to induce differentiation of callus cells into an adventitious shoot and then rooting to establish a plant body. The cellular reprogramming process involved several genetic perturbations induced by physical injury of explant and supplementation of auxin and cytokinin. Elucidation of the gene regulatory mechanism, involving several transcription factors, signaling pathway components, epigenetic alteration of chromatin, and activation of biosynthetic pathways for growth and propagation, may give us clue how to reverse the transformed callus back to the differentiated plant body.
In this study, de novo parallel sequencing was performed to characterize the transcriptome of the pluripotent state of A. crassicarpa. To our knowledge, this is the first illumination-based transcriptomic analysis of A. crassicarpa. The cultured cells provided sufficient RNA without severe mechanical shearing or chemical damage. The annotation of gene ontology (GO) terms was established with reference to the model plant Arabidopsis thaliana and the related tree Prosopis alba. Illumina sequencing data also enabled the identification of microsatellites containing simple sequence repeats (SSR). Unigenes expressing a high degree of polymorphism may reveal hidden links between genotypic diversity and physiological function, which may lead to segregation of subspecies to obtain desirable wood properties.
2 Materials and methods
2.1 Plant material and RNA isolation
Callus tissues were obtained from A. crassicarpa seedlings. The seeds were purchased from Australian Tree Seed Center (ATSC) of the Commonwealth Scientific and Industrial Research Organization. Acacia seeds were soaked in concentrated sulfuric acid overnight to burn off the surface wax, and the seed husk was then removed by washing with tap water. The seeds were sterilized with bleach (0.05% chlorine), transferred onto solid 1/2MS medium containing 0.5 ppm TDZ and 1.0 ppm IAA, and cultured for 30 days at 32°C until callus development.
For transcriptome sequencing, 0.1 g of tissue pieces were manually homogenized with a pestle and vortexed for 2 min using super beads. The total RNA was fractionated using an automated RNA extraction system (Maxwell RSC48, Maxwell Plant RNA Kit; Promega, Tokyo, Japan). RNA integrity was determined using a NanoDrop spectrophotometer and Agilent Bioanalyzer (Agilent Technologies Japan, Ltd.). For SSR analysis, 16 individual tree of A. crassicarpa were harvested from the Seed Production Area, Conn, Australia, were purchased from the ATSC.
2.2 Complementary DNA library construction and sequencing
A cDNA library was constructed from the mRNA using the Illumina TruSeq RNA Sample Preparation Kit (Illumina Inc. CA, USA). The cDNA isolated using AMPure XP beads was subjected to an end-repair process that converted the overhanging nucleotide ends into blunt ends. The blunt-end cDNAs were then incubated with adenylate to attach to the 3′ end; thus, adaptor DNA bearing a 5′-T overhang could capture the cDNA library. DNA was amplified by PCR and the PCR primer cocktail was annealed onto adaptor sequences. The Illumina next-generation sequencing protocol uses a sequencing-by-synthesis approach with four proprietary nucleotides that possess a reversible fluorophore and terminating properties. A series of images, each representing a single-base extension at a specific cluster, were recorded as the sequencing cycle was repeated at specific clusters on the flow cell surface. Raw sequence reads were deposited in the DNA Data Bank of Japan and Sequence Read Archive database (Experiment DRX208625; Analysis Run DRR218312).
2.3 Data analysis and estimation of abundance
Raw sequence data were checked for quality using the Fast QC algorithm (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The trimmomatic algorithm was then employed to trim the sequence data and to detect and remove adapter sequences (Bolger et al., 2014). This process performs in silico normalization of the total reads to reduce the number of reads that are subject to de novo assembly, thus improving the runtime of the assembly required for the following Trinity algorithm. A large volume of raw data to be analyzed by paired-end sequencing was reconstructed de novo. The relative abundance of transcripts was estimated and quantified using the RSEM 1.2.15 machine (https://www.encodeproject.org/software/rsem/) in the Trinity program (Grabherr et al., 2011).
2.4 Sequencing analysis and annotation
For sequence analysis, all unigenes were initially aligned using BLASTx (e-value < 10-5) to protein databases such as NCBI non-redundant proteins (nr) (Altschul et al., 1990), Swiss-Prot (Boeckmann et al., 2003), COG (Tatusov et al., 2000) and KEGG (Kanehisa and Goto, 2000) databases. Unigenes were then aligned using BLASTN (e-value < 10−5) to the NCBI non-redundant nucleotide (nt) nucleic acid database, and proteins with the highest sequence similarity to the given unigenes were retrieved along with their protein functional annotations. Homologous genes in A. crassicarpa were identified using a BLASTp search of putative protein sequences, with an e-value threshold of <1e–10 and > 90% identity. Annotation of the unigenes with GO terms was performed using the Blast2GO software based on BLASTX hits against the NCBI nr database (e-value < 10−5). The quality of the unigenes was evaluated via BUSCO analysis using the gVolante website (Nishimura et al., 2017), which categorizes unigenes into complete, fragmented, duplicated, and missing genes. WEGO software (Ye et al., 2018) was used to perform GO functional classification of the height of the unigenes and plot the macro-level distribution of the Acacia database. The regulation of gene expression by transcription factors (TF) is common and important for cellular response. To characterize the operational TF families underneath the dedifferentiating A. crassicarpa, 41,716 amino acid sequences deduced from the unigene sequences were subjected to transcription factor enrichment analysis in Plant TFDB search (Jin et al., 2017).
2.5 EST-SSR analysis for polymorphism
The MIcroSAtellite (MISA) identification tool ver. 2.0 in OmicX (Beier et al., 2017) was used to search for repeated nucleotide motifs. The tool predicted the presence of multiple sets of SSR in the assembled unigenes and the allele sizes of the amplified PCR products of the candidate genes were measured. The sequence search for SSRs was performed by setting the search parameters to identify at least five repeat units of SSR containing a maximum of 10 base pairs. The software requires the input file to be the sequence in which SSRs are to be found in FASTA format, and the output file contains the name of the sequence in which the SSR is detected, the repeat motif of the SSR, the number of repetitions, the start and end of the repeat, and the length of the sequence. The number, frequency, and distribution of repeats in the SSR motifs were recorded.
To obtain experimental evidence, pairs of primers were designed for the region where the SSR sequences were predicted with an intended product size of approximately 50-bp. Fluorescently labelled PCR primers were prepared using U-19 universal reverse primers (5’-gttttcccagtcacgacgt-3’) labelled with four fluorescent molecules, 6-FAM, VIC, NED, and PET. Each labelled U-19 primer was tagged with a 2-bp barcode at the 3’ end: TG for 6-FAM, AC for VIC, CA for NED, and GT for PET. Non-labelled forward primers had a pigtail (5’-gtttctt-3’) at the 5’ end. The PCR mixture was prepared at a volume of 20 µL using the Type-it Microsatellite PCR Kit (Qiagen, Germany) containing 50–100 ng of template DNA, 0.2 µM of each labelled U-19 primer, 0.2 µM of each pigtailed forward primer and 0.04 µM of each U-19-fused reverse primer. The PCR conditions were as follows: 95°C for 5 min, followed by 10 cycles of 95°C for 30 s, 58°C for 90 s with a decrease of 0.5°C in each cycle, and 72°C for 30 s, followed by 25 cycles of 95°C for 30 s, 53°C for 90 s, and 72°C for 30 s. The final extension step was performed at 60°C for 30 min. Fragment analysis was conducted on an ABI 3130xl Genetic Analyzer (Applied Biosystems) using a LIZ-600 size standard (Applied Biosystems). Allele sizes were scored using GeneMapper 4.1 (Applied Biosystems, Foster City, CA).
3 Results
3.1 Paired-end illumina sequencing and de novo assembly
Total RNA was extracted from cultured cells and mRNA was fractionated using poly T-tailed affinity beads with an automated Maxwell RSC48 System (Promega). Fractionated mRNA (59.5 μg/mL, 0.2 mL) was obtained, and cDNA was prepared and subjected to Illumina HiSeq 2000 sequencing. The raw data comprised 124,410,892 reads containing 12,565,500,092 bp, which were filtered to remove cloudy reads, low-quality reads with ambiguous N bases, reads in which >10% of the bases had Q < 20, and flanking adapter sequences (Table 1). Clean reads were assembled to recover full-length transcripts across a wide range of expression levels (Grabherr et al., 2011). After stringent quality assessment and data filtering, 123,829,386 clean reads were obtained with a total of 12,473,930,687 bases. The Q20 was 97.64%, and the guanine–cytosine (GC) content was 45.9%. Using Trinity, all clean reads were assembled de novo into 93,317 unigenes, with a total of 84,411,793 bases and an N50 length of 1,563 bp. The average unigene length 905 bp was comparable to that reported by recent studies on avocado, Persea americana (average unigene length, 988 bp; N50 = 1,050) (Vergara-Pulgar et al., 2019); raspberry, Rubus idaeus ‘Heritage’ (average, 1,168 bp; N50 = 2,046) (Travisany et al., 2019); and Salvia guaranitica L. (average, 1,039 bp; N50 = 1,603) (Ali et al., 2018).
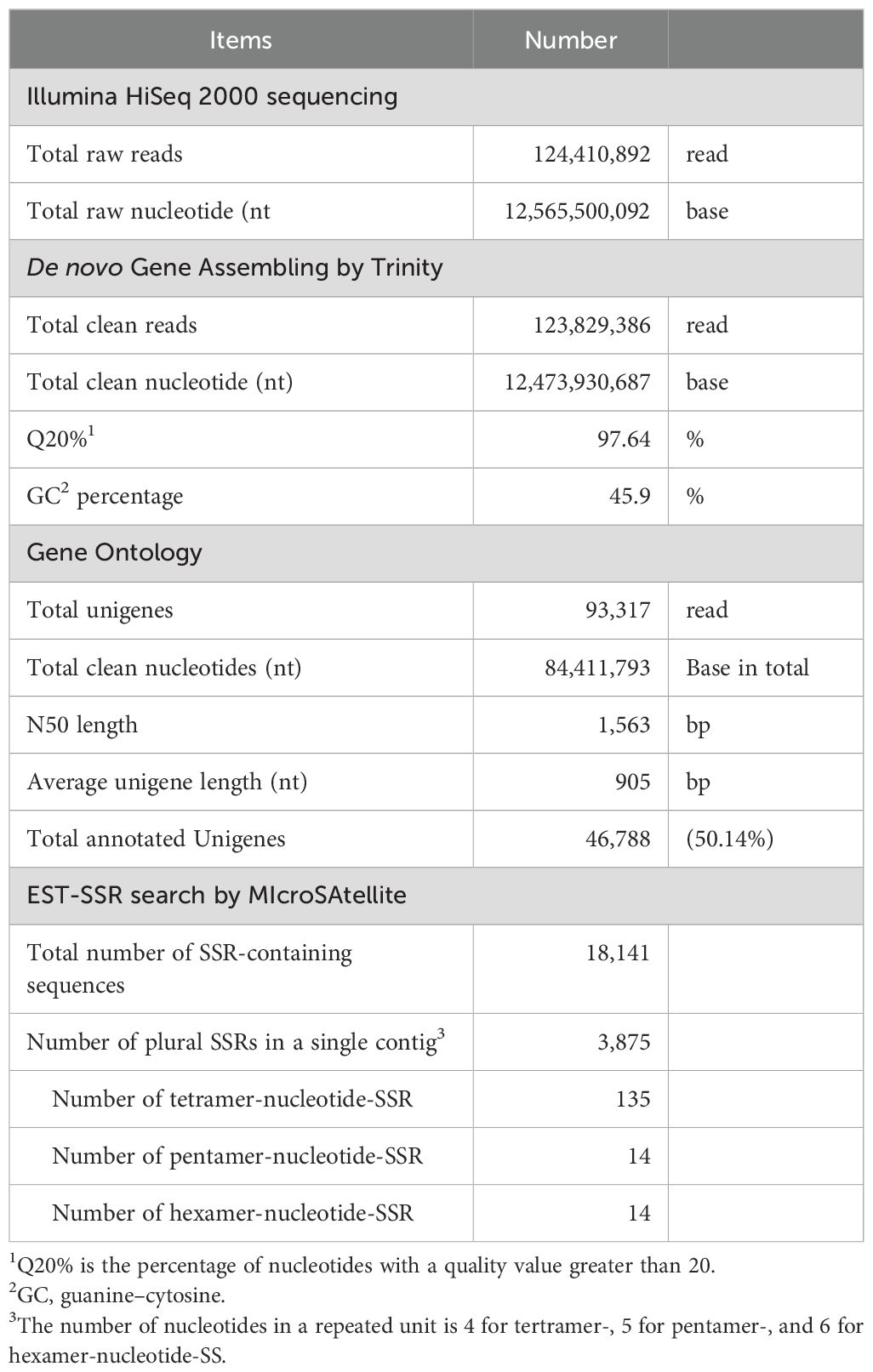
Table 1. Summary of RNA-seq Analysis of A. crassicarpa.
3.2 Quality assessment of assembled unigenes by benchmark universal single-copy ortholog analysis
The assembled unigenes were subjected to BUSCO analysis using the gVolante website (Nishimura et al., 2017) for a completeness assessment. BUSCO performs a like-for-like comparison, which allows categorization of assembled unigenes into complete, fragmented, duplicated, or missing genes. About 59% (853) of the unigenes were categorized as ‘complete,’ 23% (334) were ‘duplicate,’ 5% (68) were ‘fragmented,’ and 13% (185) were missing from the A. thaliana reference.
The term ‘complete’ refers to a unigene within two standard deviations (2σ) of the BUSCO group’s mean aligned sequence length. Unigenes outside of this limit were classified as ‘fragmented’ transcripts. The ‘duplicated’ category refers to unigenes that match multiple BUSCO reference genes, fulfilling both the ‘expected-score’ and the ‘expected-length’ cut-offs. ‘Missing’ unigenes are the ones that do not meet the expected score cut-off. The low percentage of fragmented (5%) and missing (13%) unigenes substantiated the quality of the assembled unigenes.
To further investigate the relatedness of the assembled unigenes to closely related tree species, differentially expressed genes were mapped to Acacia ESTs using all-versus-all pairwise comparison. The 93,317 unigenes identified in the present study were aligned against 81,212 genes expressed in the inner bark of Acacia koa (Lawson and Ebrahimi, 2018), 8,963 genes expressed in the secondary xylem and shoot of Acacia mangium (Suzuki et al., 2011), and 2,459 ESTs in the bark of Acacia auriculiformis × A. mangium (Yong et al., 2011). The similarities are shown in the Venn diagram (Figure 1). There were 29,707 overlapping unigenes between A. crassicarpa and the inner bark of A. koa, accounting for 44% and 37% of the assembled unigenes, respectively. The proportion of overlapping unigenes between A. crassicarpa and the secondary xylem and shoot of A. mangium was only 9%, representing a smaller library comprising 8,963 unigenes. The ESTs identified in the inner bark tissues of Acacia auriculiformis × mangium comprised a small number of 2,459 genes, and the A. crassicarpa unigenes identified in our transcriptomic library shared only 7% gene identity overlap. This indicates that the number of genes that next-generation sequencing can decode is an order of magnitude higher than the number of genes that EST sequencing can decode.
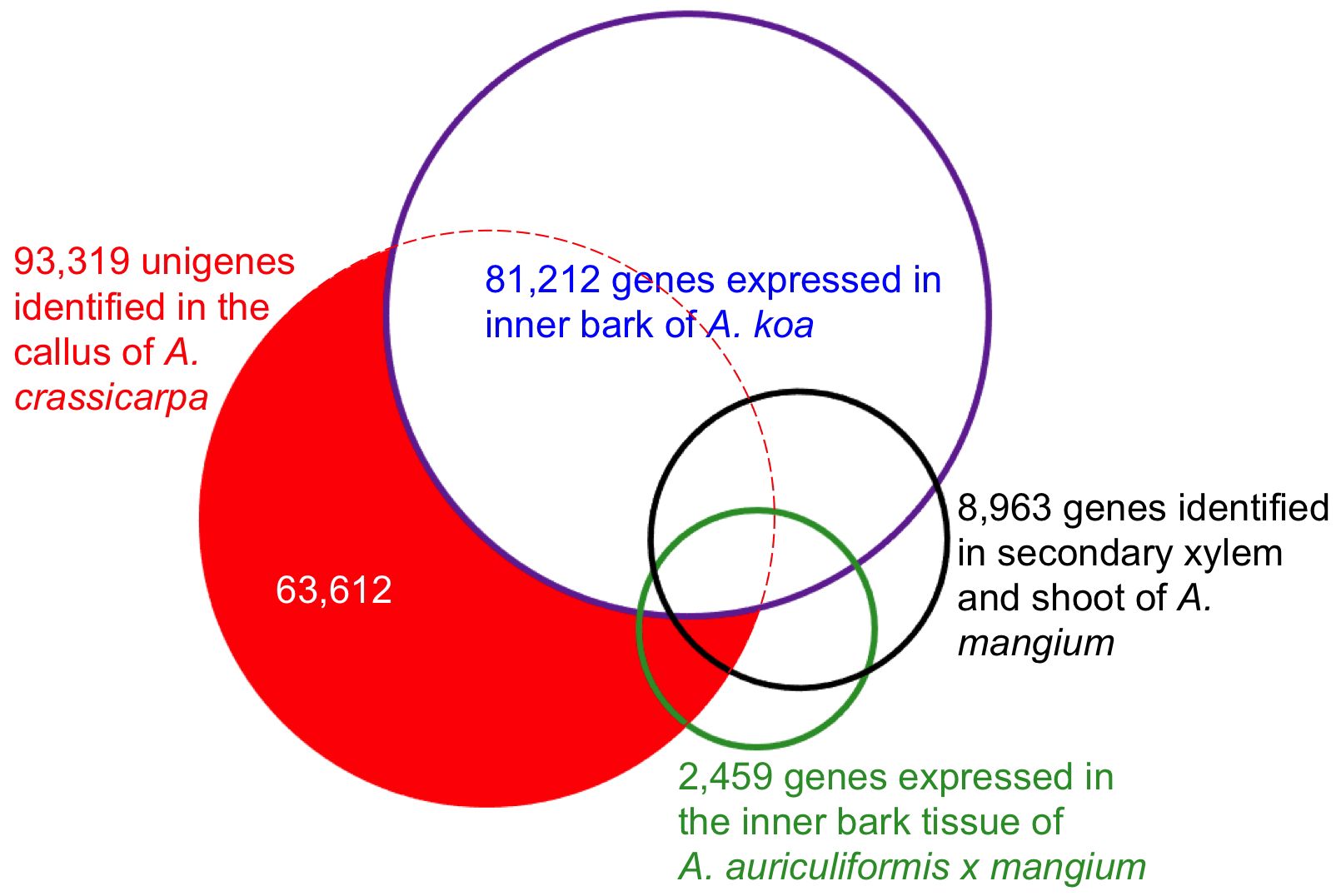
Figure 1. Overlap expressed genes in the callus of A. crassicarpa compared to the previously published EST database. Putative orthologous genes were defined by blastn search with the e-value cutoff 1xe^-10.
3.3 Functional annotation and GO-term assignment
The Blast2GO program (Conesa et al., 2005) was used to annotate the assembled 93,317 unigenes, and Gene ontology (GO) terms were assigned to 46,788 (50.14%) unigenes in reference to the NCBI nr, NCBI nt, Swiss-Prot, Clusters of Orthologous Groups (COG), and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases for annotation and validation. Base alignment with an e-value of < 10-5 was selected. To perform the GO functional classifications for all the ‘complete’ unigenes, the WEGO software (Ye et al., 2018) was used to examine the macro-level distribution of gene functions for this species. Of the complete unigenes assembled in our study, GO terms assigned by BLAST2GO were used in WEGO to categorize GO functional classifications. The Unigenes were categorized into three major categories: cellular components (31%), molecular functions (36%), and biological processes (33%). The Unigenes were assigned to cellular components (22.9%), cell parts (22.9%), membranes (17.4%), membrane parts (13.8%), and organelles (16.5%) (Figure 2). Molecular functions of the unigenes included catalytic activity (25%) and binding (21.8%). The biological processes of the unigenes included cellular (26.0%) and metabolic (26.1%) processes.
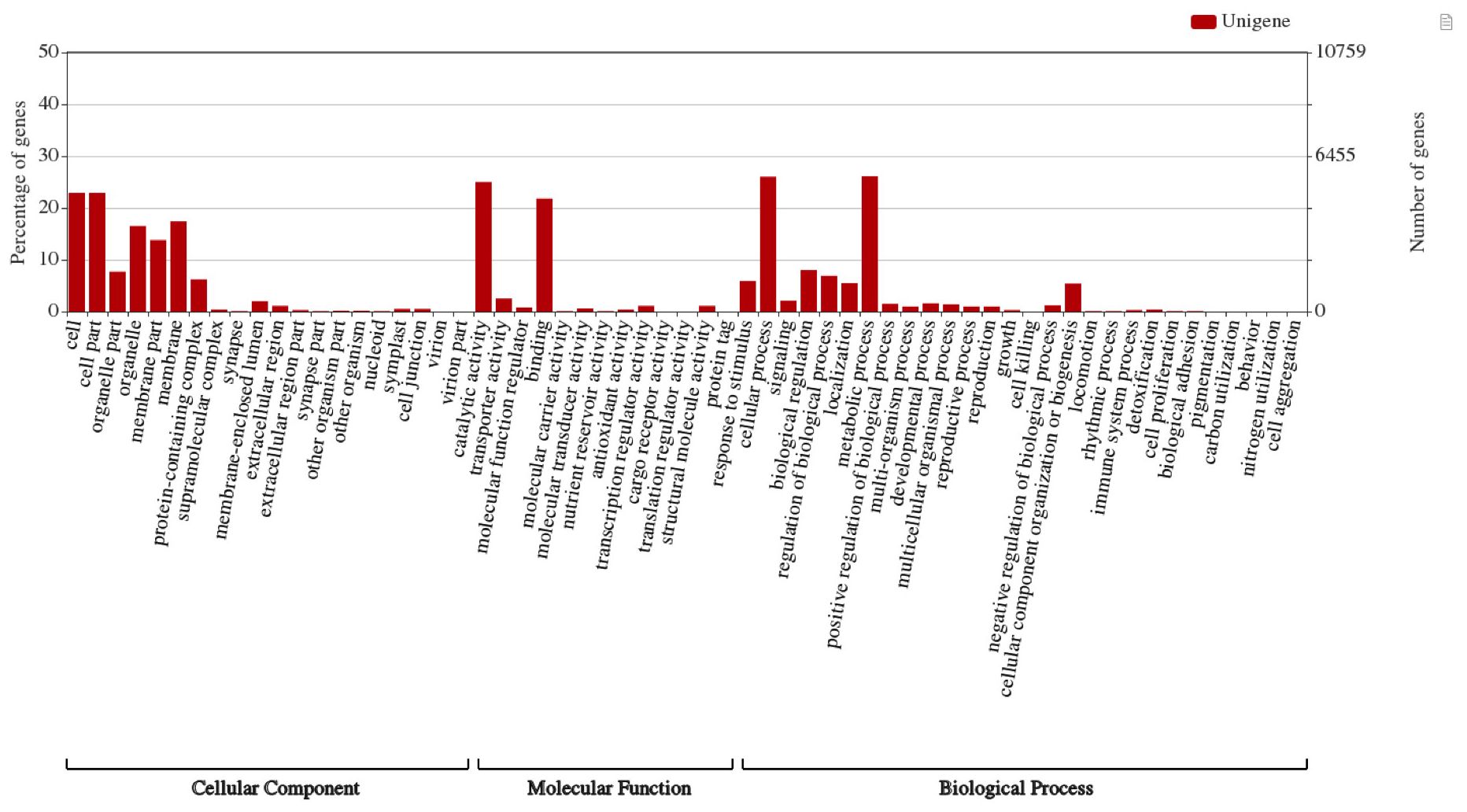
Figure 2. Gene ontology categories of assembled unigenes in A. crassicarpa callus shown by WEGO web application. GO terms were assigned in three processes regarding cellular component, molecular function and biological process.
3.4 Metabolic pathway reconstruction by KEGG mapping
The KEGG database was used to systematically assign annotated unigenes to the metabolic pathways. KEGG mapping successfully categorized 16,662 unigenes, which accounted for 35.6% of the total unigenes, in accordance with the hierarchal annotation system KEGG BRITE, into the three major protein families involved in metabolism (2,406 unigenes), genetic information processing (2,666 unigenes), and signaling and cellular process (595 unigenes). The metabolic pathways were reconstructed by mapping the functionally annotated unigenes onto pathways for carbohydrate and energy metabolism, nucleotide and amino acid metabolism, lipids, terpenes, lignin metabolism, and cofactor/vitamin biosynthesis (Figure 3). The number of genes involved in metabolic pathways was the largest for carbohydrate and energy metabolism, accounting for 666 unigenes in 35 pathways, followed by biosynthetic pathways for amino acids and nucleotides, which involved 440 unigenes in 23 pathways. The metabolic pathways for lipid, terpenoid, and lignin metabolism accounted for 277 genes in the 27 pathways. Biosynthesis of cofactors and vitamins accounted for 179 unigenes in the 12 pathways.
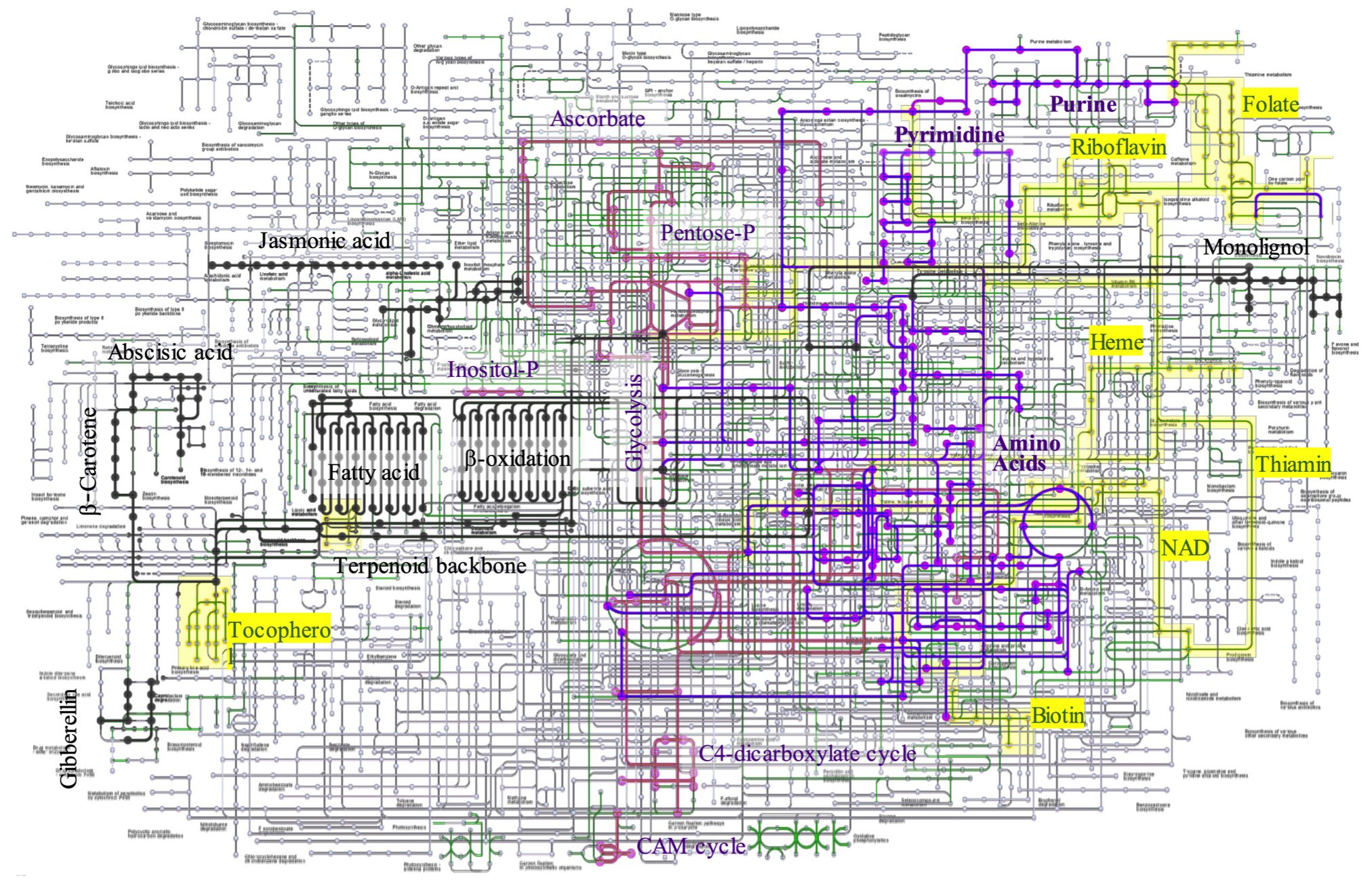
Figure 3. Metabolic pathway construction by KEGG mapping. The unigenes annotated to metabolic enzymes were reconstructed to KEGG metabolic map. Carbohydrate and Energy metabolism (pink), Nucleoside and amino acid metabolites (purple), lipid, terpen, and lignin (black), cofactors (yellow), and others (green).
Several important genes were involved in the monolignol biogenesis pathway, including phenylalanine/tyrosine ammonia-lyase [EC:4.3.1.25], trans-cinnamate 4-monooxygenase [EC:1.14.14.91], 4-coumarate-CoA ligase [EC:6.2.1.12], shikimate O-hydroxycinnamoyltransferase [EC:2.3.1.133], 5-O-(4-coumaroyl)-D-quinate 3’-monooxygenase [EC:1.14.14.96], caffeoyl-CoA O-methyltransferase [EC:2.1.1.104], cinnamoyl-CoA reductase [EC:1.2.1.44], ferulate-5-hydroxylase [EC:1.14.-.-], and caffeic acid 3-O-methyltransferase/acetylserotonin O-methyltransferase [EC:2.1.1.68 2.1.1.4]. The monolignol biosynthetic pathway is well characterized, but the coordination and regulation of cell wall formation are not well understood. Several transcription factors may be involved in the formation of lignin and wood formation (Zhang et al., 2021).
3.5 Identification of putative transcription factors
Plant Transcription Factor Database (PlantTFDB 4.0) predicted 756 transcription factors that matched the homologues of A. thaliana (E-value < 10-5) (Table 2) and classified them into 40 families. The most abundant family was the bHLH13-like protein (78 unigenes), followed by the ethylene-responsive element (ERF) (62 unigenes), MYB family (55 unigenes), WRKY DNA-binding proteins (54 unigenes), bZIP family (53 unigenes), and lysine-specific histone demethylase (53 unigenes), as well as others that were lower in number. It has been reported that major transcription factors, ERF (Ikeuchi et al., 2022), MYB (Sakamoto et al., 2022), bHLH and WRKY (Xu et al., 2023a), play pivotal roles in reprogramming in response to physical injury and hormone signaling.

Table 2. Annotation of putative transcription factors.
3.6 Unigenes involved in phytohormone signaling
Acacia crassicarpa expressed genes involved in all known plant hormone signaling pathways (Figure 3). Unigenes encoding components of auxin (AUX), gibberellin (GA), and jasmonic acid (JA) signaling were annotated. Most of the unigenes involved in cytokinin (CK) and abscisic acid (ABA) signaling were also annotated, except for the B-ARR transcription factor in CK signaling, and EIN2 and BSU1 were involved in phosphor-relaying components. Signaling components for ethylene (ET), brassinosteroids (BR), and salicylic acid (SA) were all expressed in the undifferentiated cells of A. crassicarpa.
3.7 SSR analysis for polymorphism
The assembled unigene sequences of A. crassicarpa were subjected to MISA to identify SSR representing di-, tri-, and tetra-nucleotide repeats. Dinucleotide SSRs (AG, AT, and AC) were more frequent than trinucleotide SSRs (Figure 4). Among the trinucleotide repeat motifs, the dominant repeat motif was (AAG)n, followed by (AGG)n, (AAT)n, (ATC)n, and the other trinucleotide repeats. Dinucleotide repeats occur more frequently than tri- or tetranucleotide repeat units. Among the dinucleotide repeat motifs, the most frequent repeat detected in this study was AG/CT, followed by AT/TA, and AC/GT. The least frequent dinucleotide was GC/CG. This finding is consistent with those of previous studies conducted on Stevia rebaudiana (Kaur et al., 2015), the traditional Chinese medicinal plant Epimedium sagittatum (Zeng et al., 2010) and coffee (Aggarwal et al., 2007).
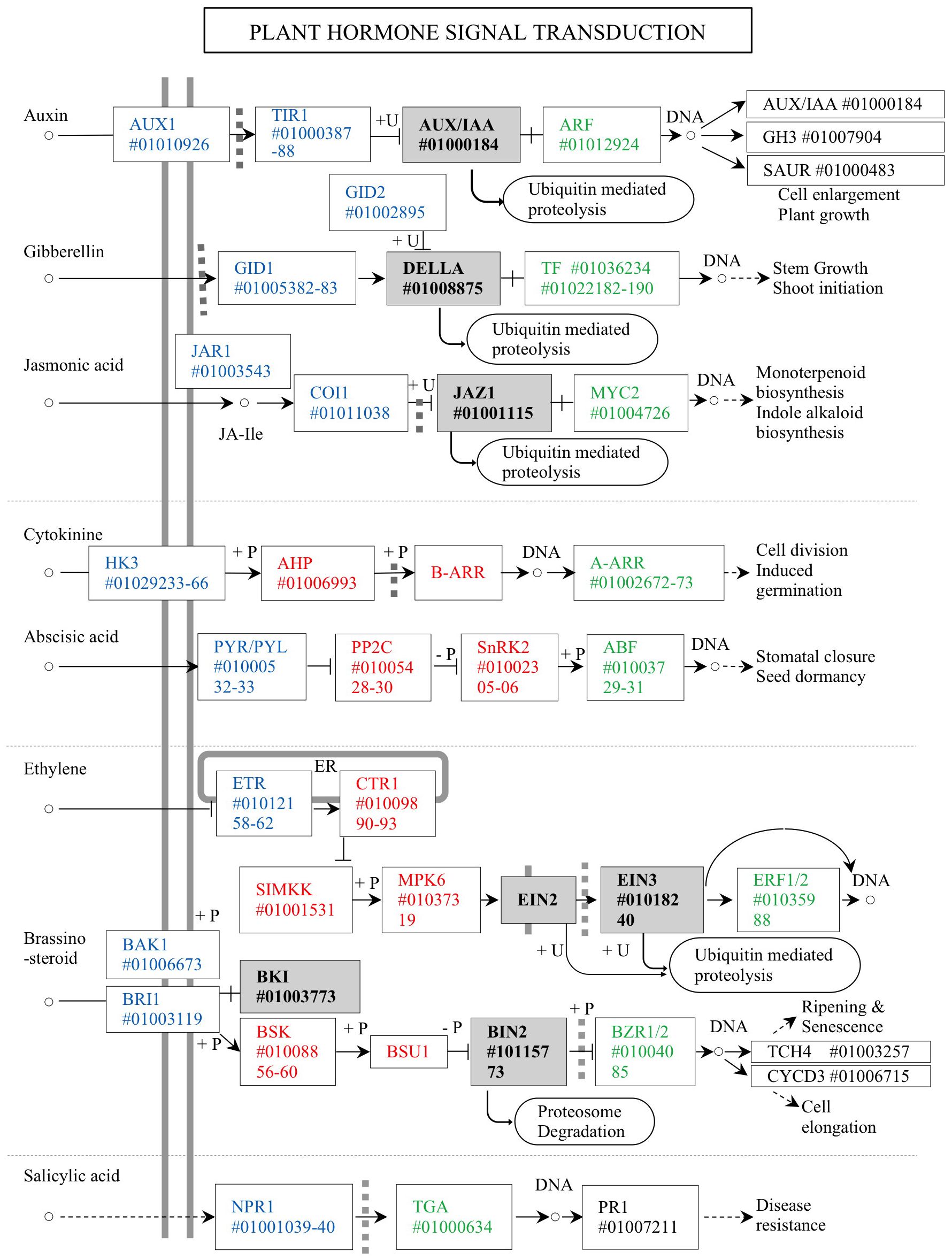
Figure 4. Unigenes annotated for the plant hormone signaling in the A. crassicarpa transcriptome. Signal transduction components are classified as ligand receptors (blue), Phospho-relay components (red), transcription factor regulators (gray), and transcription factors (green). The unigene identities are specified as ICSL numbers starting with a # symbol.
Primers were designed to detect polymorphisms by targeting unigenes that contained triplet repeats in their ORFs (Table 3). Among the 16 DNA samples from local A. crassicarpa trees, three multiple signals of 258, 287, and 290-bp were observed for the SSR marker designed for the unigene ICSL01027786 (293bp), which was annotated as auxin response factor 3 (ARF3). Three signals were also produced from the unigene ICSL01000311 (266 bp), which was annotated as ARF4 (Table 4). Other unigenes annotated as transcription factors, such as ICSL01001643 (254bp), ICSL01023085 (175 bp), and ICSL01008756 (216 bp), also produced multiplicity in SSR analysis. Unigene ICSL01001643, annotated as a putative ethylene-responsive transcription factor (Charfeddine et al., 2019), produced two PCR bands with lengths of 214 and 277 bp. Unigene ICSL01023085 encodes a proline-rich PRCC (Weterman et al., 2000) and exhibits polymorphism with four PCR bands of 197, 200, 249, and 257 bp. Unigene ICSL01008756 was annotated as tubby-like F-box protein 5 (Song et al., 2019) (Xu et al., 2016);, and its polymorphisms appeared as PCR products with lengths of 235, 238, and 241 bp. Moderate multiplicity giving signals of 179 and 182-bp was observed for unigene ICSL01034178 (154 bp), which was annotated as a pollen tube guidance gene (Li et al., 2015). The polymorphic information content (PIC), which represents the ability to detect polymorphisms, was calculated using Cervus (Kalinowski, 2007), and was high (PIC > 0.4) for ARF3, ARF4, PRCC, and Tubby-like F-box protein 5, and moderate (PIC < 0.4) for ERTF and PTG.
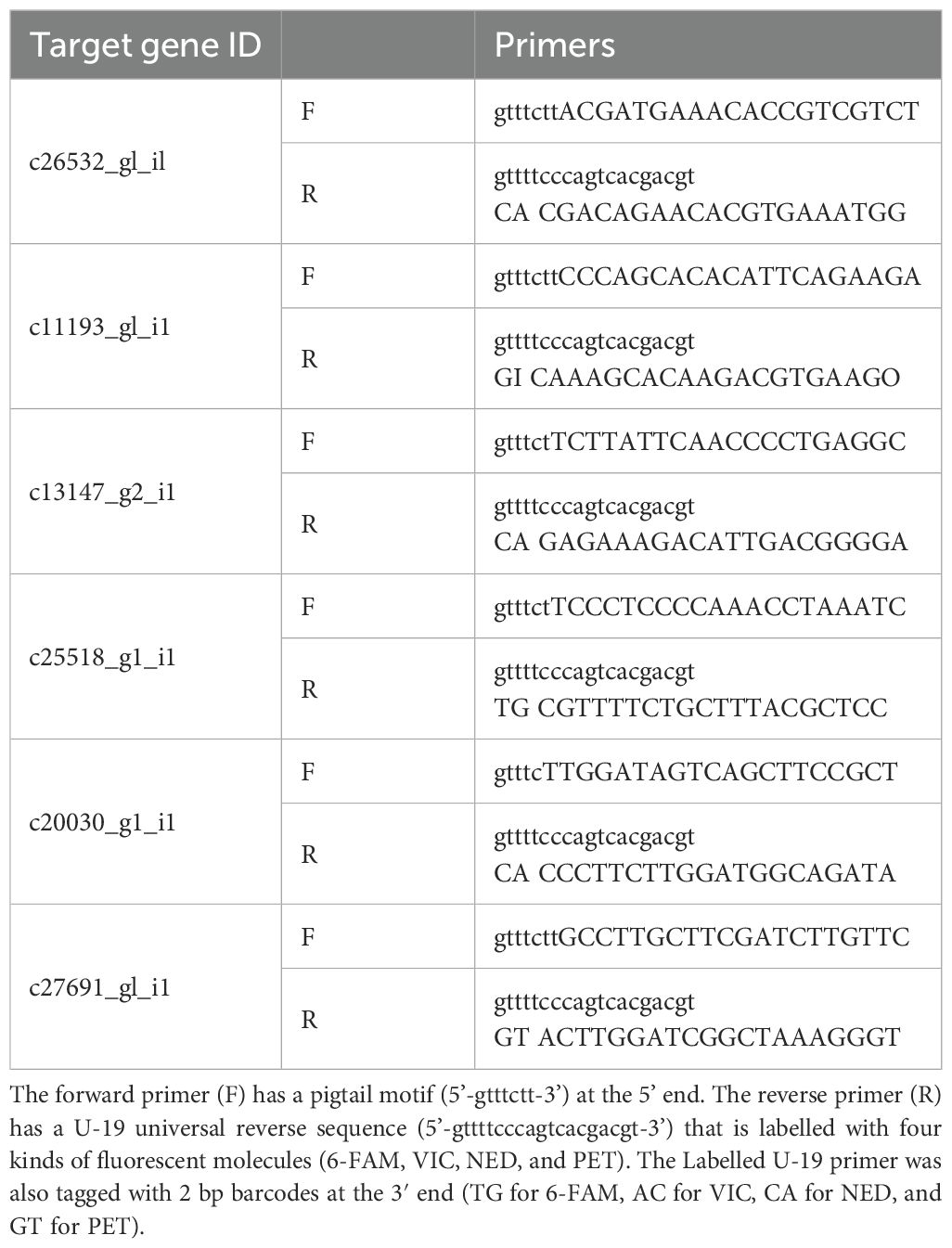
Table 3. PCR primers for SSR marker amplification.
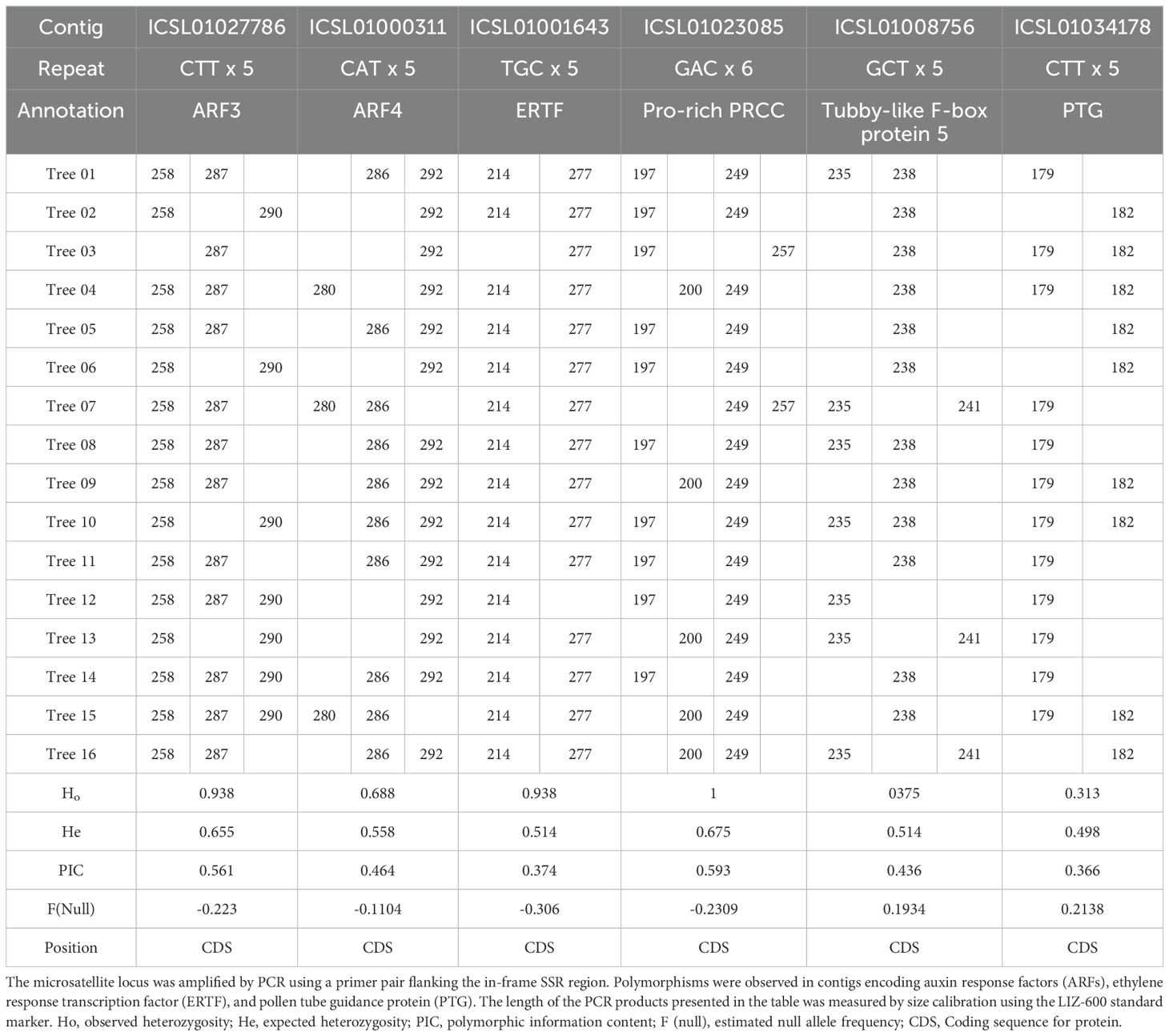
Table 4. Polymorphism represented as the length distribution of PCR products focused on SSR regions.
4 Discussion
4.1 Informative foundation for the molecular breeding of A. crassicarpa
Molecular genetics provides valuable information for developing programs for efficient tree breeding. Several commercially important traits of trees, such as fast growth, few branches, high strength, and resistance to pathogens, are likely to be controlled by multiple layers of gene regulation. Although these traits are difficult to control by environmental management such as optimized fertilization, several genes are involved in the commercially important traits of various plants. The present study reports a transcriptomic analysis utilizing next-generation sequencing, which offers an effective approach to genomic resources at a reasonable cost in a relatively short period, even for non-model tree species.
4.2 Annotation of unigenes for metabolic enzymes and transcription factors
Annotation of genes encoding metabolic enzymes and mapping on the KEGG metabolic pathway provides an atlas that describes the metabolic flow in the pluripotent cells of A. crassicarpa. Potential targets, such as those involved in monolignol biosynthesis, were among the genes involved in the primary metabolic pathways. This genetic information may offer a candidate for selective breeding and genome editing programs to increase the pulp yield and reduce pulping costs. Annotation of transcription factors also provides another set of candidates to be disrupted by genome-editing technology, thereby modifying the texture of lignin content and hormone responses, including pathogen resistance.
The cellular reprogramming process involved several genetic perturbations induced by physical injury of explant (Deng et al., 2022) (Park et al., 2023b), and supplementation of auxin and cytokinin (Asghar et al., 2023). Elucidation of the gene regulatory mechanism, involving several transcription factors (Kumar et al., 2021, Dai et al., 2020, Xu et al., 2023b), signaling pathway components (Park et al., 2023a), and epigenetic alteration of chromatin (Chen et al., 2023) may give us clue how to reverse the transformed callus back to the differentiated plant body.
4.3 Plant hormone signaling as the genetic switch
Phytohormones can provide an effective means of operating genetic switches from outside the hard bark with reproducible and quantitative responses. We considered the nature of these switches in terms of their reversibility, dose responsiveness, and mechanisms that enable crosstalk with other hormones. The three phytohormones auxin (AUX), gibberellin (GA), and jasmonic acid (JA) exploit ubiquitin-dependent proteolysis, which eliminates the specific regulator proteins that negatively control transcription factors. The elimination of negative regulators allows transcription factors to activate hormone-responsive gene expression. The switchover to the signal-responding state continues until lifted restrictions are regenerated through the biosynthesis of negative regulators. Therefore, hormone regulation via AUX, GA, and JA could provide a binary on/off switch.
Cytokinin (CK) and abscisic acid (ABA) signaling is transmitted through a phosphorylation relay between receptors and regulators. Phosphorelay-dependent regulation involves a series of phosphorylation events that lead to transcription factor activation. Phosphorylation, typically catalyzed by protein kinases, can occur in multiple steps, creating a phosphorylation gradient. The extent of phosphorylation determines the activity level of the transcription factor, and consequently, the strength of the signaling response. Accordingly, this gradient regulation allows fine-tuning of the response, enabling different levels of gene expression based on the concentration or duration of the hormone signal.
Ethylene (ET) and brassinosteroid (BR) signaling constitute the phosphorylation relay upstream; however, proteolytic removal of the transcriptional regulator is involved downstream. Such a hybrid mechanism can allow a quantitative response to intermediate signaling components, which may mediate crosstalk with other hormone signaling (Yasumura et al., 2015; Wang et al., 2020; Zhao et al., 2023), while the proteolytic switchover to gene expression provides a determinative response to hormone stimuli by the accumulated signaling of ET (Chen et al., 2022) or that of BR (Kour et al., 2021). In the systemic immune response induced by salicylic acid, the response to the SA receptor NPR1, which undergoes transformation from an oligomeric state in the cytosol to monomers upon binding to SA, activates pathogenesis-related genes in the nucleus (Palmer et al., 2019). The straightforward transmission of SA signaling allows a quick response to pathogen invasion.
4.4 SSR marker development
Individual trees may exhibit slight but discernible differences in growth rate, lignin content, stress tolerance, and pest resistance. The genetic traits underlying these characteristics may be associated with molecular genetic markers, which provides a scientific foundation for describing these tree characteristics. Short sequence repeats (SSR) are nucleotide regions that represent tandem repeat units located in coding and non-coding regions of the genome (Gupta et al., 1996).
None of the 16 subspecies of A. crassicarpa was identical in terms of the PCR products amplified by pairs of primers flanking the repeated trinucleotide sequences. The highest similarity in the EST-SSR products was observed between subspecies 05 and 11, which differed only in the PCR products from unigene ICSL01034178, which was annotated as a pollen tube guidance protein. The highest difference in polymorphism was observed between subspecies 03 and 04, which exhibited diverse multiplicities, except for ICSL01008756 and ICSL01034178. Polymorphisms in these transcription genes may exert a significant effect on the set of genes expressed under these regulators, which may be relevant to the physiological diversity of the 16 subspecies of A. crassicarpa.
Although the predominant trinucleotide repeats contained adenine-leading motifs such as AAG, AGG, AAT, and ATC (Figure 5), polymorphisms were produced when targeting repeats started with G or C, such as CTT, CAT, and GCA (Table 4). The high occurrence of repeated units does not necessarily guarantee successful targeting by SSR fingerprinting.
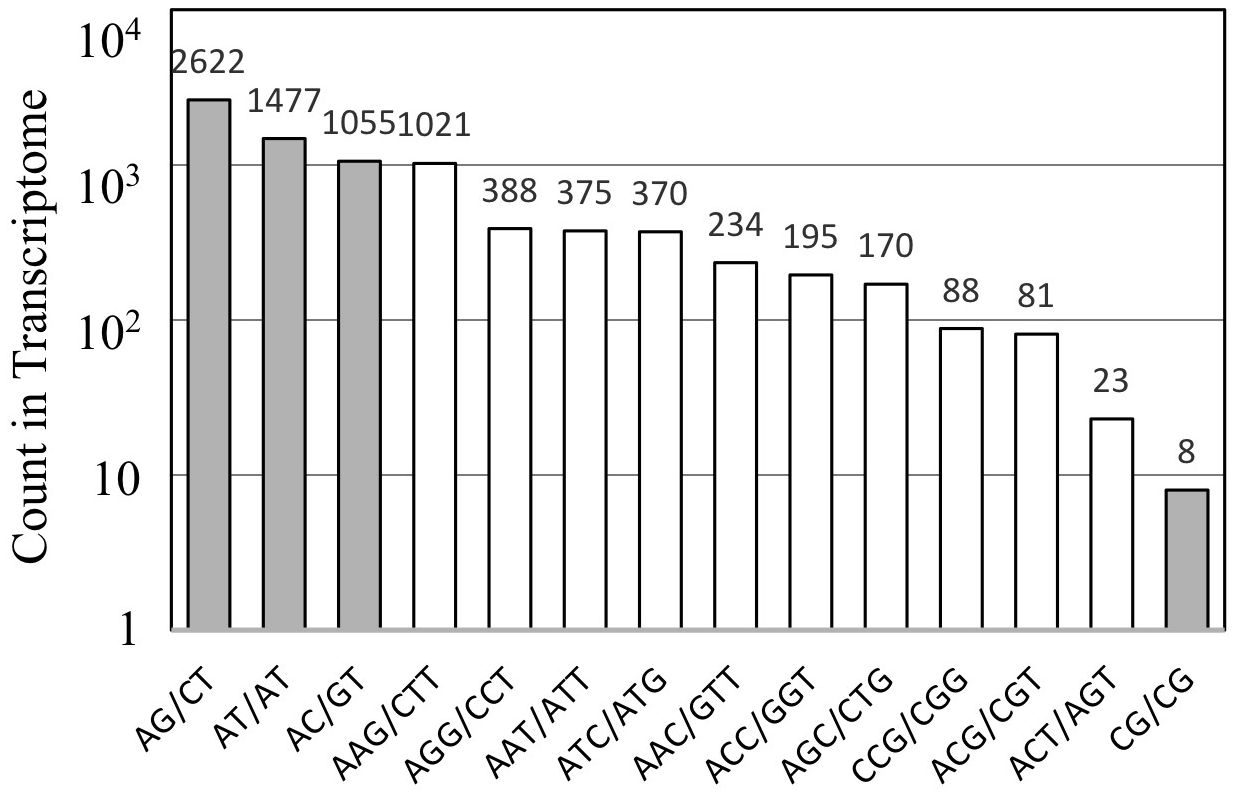
Figure 5. The distribution of SSRs identified from the A. crassicarpa transcriptome. Occurrence of repeat sequences with di- and tri-nucleotide units are designated in gray and white, respectively.
SSR fingerprinting can be produced by PCR amplification of SSR on assembled transcriptomic sequences (transcriptome-SSRs), ESTs (EST-SSRs), or genomic DNA sequences (genomic SSRs). Transcriptome SSR sequences are more accessible than genomic SSR markers because they can focus on gene-rich regions and are associated with transcription. Transcriptome-SSR markers contribute to the segregation of closely related subspecies and variants, and these markers may be used in breeding programs to improve the quality and biomass of trees as a fiber resource and to achieve the desired properties, such as the ability to lower the salt content in combustion ash when used as biomass fuel (Thiel et al., 2003; Chung et al., 2014).
In our future studies, gene co-expression networks may be constructed using time-resolved transcriptomic approaches as illustrated for maize (Du et al., 2019) soybean (Park et al., 2023a) and cotton (Fan et al., 2022), which will enable the clustering of gene expression at each phase of the cellular reprogramming process from the differentiated plant body to the pluripotent state. The molecular genetic sequence information established in the present study will assist in the molecular breeding of Acacia trees with SSR markers and functional annotation of genes and assignment to metabolic map pathways.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ddbj.nig.ac.jp/, SAMD00213802, https://www.ddbj.nig.ac.jp/, PRJDB9506, https://www.ddbj.nig.ac.jp/, DRX208625, https://www.ddbj.nig.ac.jp/, DRR218312.
Author contributions
SI: Conceptualization, Writing – review & editing. KK: Writing – review & editing, Data curation, Investigation. MN: Investigation, Writing – review & editing, Methodology. TK: Investigation, Methodology, Writing – review & editing. TT: Investigation, Conceptualization, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study received funding from Sumitomo Forestry Co. Ltd, and funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. The study was also supported by the Adaptable and Seamless Technology Transfer Program through Target-driven R&D (A-STEP) from the Japan Science and Technology Agency (JST) Grant Number JPMJTM20QX21445979. All authors declare no other competing interests.
Conflict of interest
Author SI and KK are employed by Sumitomo Forestry Co. Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aggarwal, R. K., Hendre, P. S., Varshney, R. K., Bhat, P. R., Krishnakumar, V., Singh, L. (2007). Identification, characterization and utilization of EST-derived genic microsatellite markers for genome analyses of coffee and related species. Theor. Appl. Genet. 114, 359–372. doi: 10.1007/s00122-006-0440-x
Ali, M., Hussain, R. M., Rehman, N. U., She, G. B., Li, P. H., Wan, X. C., et al. (2018). De novo transcriptome sequencing and metabolite profiling analyses reveal the complex metabolic genes involved in the terpenoid biosynthesis in Blue Anise Sage (Salvia guaranitica L.). DNA Res. 25, 597–617. doi: 10.1093/dnares/dsy028
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/s0022-2836(05)80360-2
Asghar, S., Ghori, N., Hyat, F., Li, Y., Chen, C. (2023). Use of auxin and cytokinin for somatic embryogenesis in plant: a story from competence towards completion. Plant Growth Regul. 99, 413–428. doi: 10.1007/s10725-022-00923-9
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinf. (Oxford England) 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Blakesley, D., Allen, A., Pellny, T. K., Roberts, A. V. (2002). Natural and induced polyploidy in Acacia dealbata Link. and Acacia mangium Willd. Ann. Bot. 90, 391–398. doi: 10.1093/aob/mcf202
Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M. C., Estreicher, A., Gasteiger, E., et al. (2003). The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. doi: 10.1093/nar/gkg095
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Charfeddine, M., Charfeddine, S., Ghazala, I., Bouaziz, D., Bouzid, R. G. (2019). Investigation of the response to salinity of transgenic potato plants overexpressing the transcription factor StERF94. J. Biosci. 44, 141. doi: 10.1007/s12038-019-9959-2
Chen, H., Bullock, D. A., Alonso, J. M., Stepanova, A. N. (2022). To fight or to grow: the balancing role of ethylene in plant abiotic stress responses. Plants-Basel 11, 33. doi: 10.3390/plants11010033
Chen, Y., Hung, F., Sugimoto, K. (2023). [Epigenomic reprogramming in plant regeneration: Locate before you modify]. Curr. Opin. Plant Biol. 75, 102415. doi: 10.1016/j.pbi.2023.102415
Chung, J. W., Kim, T. S., Sundan, S., Lee, G. A., Park, J. H., Cho, G. T., et al. (2014). New cDNA-SSR markers in the narrow-leaved vetch (Vicia sativa subsp nigra) using 454 pyrosequencing. Mol. Breed 33, 749–754. doi: 10.1007/s11032-013-9980-3
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Dai, X., Liu, N., Wang, L., Li, J., Zheng, X., Xiang, F., et al. (2020). MYB94 and MYB96 additively inhibit callus formation via directly repressing LBD29 expression in Arabidopsis thaliana. Plant Sci. 293, 110323. doi: 10.1016/j.plantsci.2019.110323
Deng, J., Sun, W., Zhang, B., Sun, S., Xia, L., Miao, Y., et al. (2022). GhTCE1-GhTCEE1 dimers regulate transcriptional reprogramming during wound-induced callus formation in cotton. Plant Cell. 34, 4554–4568. doi: 10.1093/plcell/koac252
Du, X., Fang, T., Liu, Y., Huang, L., Zang, M., Wang, G., et al. (2019). Transcriptome profiling predicts new genes to promote maize callus formation and transformation. Front. Plant Sci. 10, 1633. doi: 10.3389/fpls.2019.01633
Fan, Y., Tang, Z., Wei, J., Yu, X., Guo, H., Li, T., et al. (2022). Dynamic transcriptome analysis reveals complex regulatory pathway underlying induction and dose effect by different exogenous auxin IAA and 2,4-D during in vitro embryogenic redifferentiation in cotton. Front. Plant Sci. 13, 931105. doi: 10.3389/fpls.2022.931105
Fuller, A., Omidiji, Y., Viefhaus, T., Maier, J., Scheffknecht, G. (2019). The impact of an additive on fly ash formation/transformation from wood dust combustion in a lab-scale pulverized fuel reactor. Renewable Energy 136, 732–745. doi: 10.1016/j.renene.2019.01.013
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gupta, P. K., Balyan, I. S., Sharma, P. C., Ramesh, B. (1996). Microsatellites in plants: A new class of molecular markers. Curr. Sci. 70, 45–54. doi: 10.2307/24097472
Hallgren, A. L., Engvall, K., Skrifvars, B. J. (1999). Ash-induced operational difficulties in fluidised bed firing of biofuels and waste. Biomass: Growth Opportunity Green Energy Value-Added Products 1, 1365–1370. doi: 10.1016/j.fuel.2006.06.020
Ikeuchi, M., Iwase, A., Ito, T., Tanaka, H., Favero, D. S., Kawamura, A., et al. (2022). Wound-inducible WUSCHEL-RELATED HOMEOBOX 13 is required for callus growth and organ reconnection. Plant Physiol. 188, 425–441. doi: 10.1093/plphys/kiab510
Jin, J., Tian, F., Yang, D. C., Meng, Y. Q., Kong, L., Luo, J., et al. (2017). PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, D1040–D1045. doi: 10.1093/nar/gkw982
Kanehisa, M., Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kaur, R., Sharma, N., Raina, R. (2015). Identification and functional annotation of expressed sequence tags based SSR markers of Stevia rebaudiana. Turk J. Agric. For. 39, 439–450. doi: 10.3906/tar-1406-144
Kour, J., Kohli, S. K., Khanna, K., Bakshi, P., Sharma, P., Singh, A. D., et al. (2021). Brassinosteroid signaling, crosstalk and, physiological functions in plants under heavy metal stress. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.608061
Kumar, S., Ruggles, A., Logan, S., Mazarakis, A., Tyson, T., Bates, M., et al. (2021). Comparative transcriptomics of non-embryogenic and embryogenic callus in semi-recalcitrant and non-recalcitrant upland cotton lines. PLANTS-BASEL. 10, 1775. doi: 10.3390/plants10091775
Lawson, S. S., Ebrahimi, A. (2018). Development and validation of Acacia koa and A. koaia nuclear SSRs using IIlumina sequencing. Silvae Genetica 67, 20–25. doi: 10.2478/sg-2018-0003
Li, H. J., Zhu, S. S., Zhang, M. X., Wang, T., Liang, L., Xue, Y., et al. (2015). Arabidopsis CBP1 is a novel regulator of transcription initiation in central cell-mediated pollen tube guidance. Plant Cell 27, 2880–2893. doi: 10.1105/tpc.15.00370
McKinnon, G. E., Larcombe, M. J., Griffin, A. R., Vaillancourt, R. E. (2018). Development of microsatellites using next-generation sequencing for Acacia crassicarpa. J. Trop. For. Sci. 30, 252–258. doi: 10.26525/jtfs2018.30.2.252258
Midgley, S., Turnbull, J. (2003). Domestication and use of Autralian Acacias: case studies of five important species. Aust. Syst. Bot. 16, 89–102. doi: 10.1071/SB01038
Mukherjee, S., Sharma, A. K. (1995). In situ nuclear DNA variation in Australian species of Acacia. Cytobios. 75, 33–36.
Nishimura, O., Hara, Y., Kuraku, S. (2017). gVolante for standardizing completeness assessment of genome and transcriptome assemblies. Bioinformatics 33, 3635–3637. doi: 10.1093/bioinformatics/btx445
Palmer, I. A., Chen, H., Chen, J., Chang, M., Li, M., Liu, F. Q., et al. (2019). Novel salicylic acid analogs induce a potent defense response in arabidopsis. Int. J. Mol. Sci. 20, 3356. doi: 10.3390/ijms20133356
Pan, Z., You, Y. (1994). Introduction and provenance test of Acacia crassicarpa. For. Res. 7, 498–505.
Park, J., Choi, Y., Jeong, M., Jeong, Y., Han, J., Choi, H. (2023a). Uncovering transcriptional reprogramming during callus development in soybean: insights and implications. Front. Plant Sci. 14, 1239917. doi: 10.3389/fpls.2023.1239917
Park, J., Park, K., Park, S., Ko, S., Moon, K., Koo, H., et al. (2023b). WUSCHEL controls genotype-dependent shoot regeneration capacity in potato. Plant Physiol. 193, 661–676. doi: 10.1093/plphys/kiad345
Pashley, C. H., Ellis, J. R., McCauley, D. E., Burke, J. M. (2006). EST databases as a source for molecular markers: lessons from Helianthus. J. Hered 97, 381–388. doi: 10.1093/jhered/esl013
Sakamoto, Y., Kawamura, A., Suzuki, T., Segami, S., Maeshima, M., Polyn, S., et al. (2022). Transcriptional activation of auxin biosynthesis drives developmental reprogramming of differentiated cells. Plant Cell. 34, 4348–4365. doi: 10.1093/plcell/koac218
Song, Z. B., Wu, X. F., Gao, Y. L., Cui, X., Jiao, F. C., Chen, X. J., et al. (2019). Genome-wide analysis of the HAK potassium transporter gene family reveals asymmetrical evolution in tobacco (Nicotiana tabacum). Genome 62, 267–278. doi: 10.1139/gen-2018-0187
Sugimoto, K., Jiao, Y. L., Meyerowitz, E. M. (2010). Arabidopsis regeneration from multiple tissues occurs via a root development pathway. Dev. Cell 18, 463–471. doi: 10.1016/j.devcel.2010.02.004
Suzuki, S., Suda, K., Sakurai, N., Ogata, Y., Hattori, T., Suzuki, H., et al. (2011). Analysis of expressed sequence tags in developing secondary xylem and shoot of Acacia mangium. J. Wood Sci. 57, 40–46. doi: 10.1007/s10086-010-1141-2
Tatusov, R. L., Galperin, M. Y., Natale, D. A., Koonin, E. V. (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36. doi: 10.1093/nar/28.1.33
Thiel, T., Michalek, W., Varshney, R. K., Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Travisany, D., Ayala-Raso, A., Di Genova, A., Monsalve, L., Bernales, M., Martinez, J. P., et al. (2019). RNA-Seq analysis and transcriptome assembly of raspberry fruit (Rubus idaeus “Heritage”) revealed several candidate genes involved in fruit development and ripening. Sci. Hortic. 254, 26–34. doi: 10.1016/j.scienta.2019.04.018
Vergara-Pulgar, C., Rothkegel, K., González-Agüero, M., Pedreschi, R., Campos-Vargas, R., Defilippi, B. G., et al. (2019). De novo assembly of Persea americana cv. ‘Hass’ transcriptome during fruit development. BMC Genomics 20, 108. doi: 10.1186/s12864-019-5486-7
Wang, Q., Yu, F. F., Xie, Q. (2020). Balancing growth and adaptation to stress: Crosstalk between brassinosteroid and abscisic acid signaling. Plant Cell Environ. 43, 2325–2335. doi: 10.1111/pce.13846
Weterman, M. A. J., von Groningen, J. J. M., Jansen, A., van Kessel, A. G. (2000). Nuclear localization and transactivating capacities of the papillary renal cell carcinoma-associated TFE3 and PRCC (fusion) proteins. Oncogene 19, 69–74. doi: 10.1038/sj.onc.1203255
Xu, C., Chang, P., Guo, S., Yang, X., Liu, X., Sui, B., et al. (2023). Transcriptional activation by WRKY23 and derepression by removal of bHLH041 coordinately establish callus pluripotency in Arabidopsis regeneration. Plant Cell. 7, 1693. doi: 10.1093/plcell/koad255
Xu, J. N., Xing, S. S., Zhang, Z. F., Chen, X. S., Wang, X. Y. (2016). Genome-wide identification and expression analysis of the tubby-like protein family in the malus domestica genome. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01693
Yasumura, Y., Pierik, R., Kelly, S., Sakuta, M., Voesenek, L., Harberd, N. P. (2015). An ancestral role for CONSTITUTIVE TRIPLE RESPONSE1 proteins in both ethylene and abscisic acid signaling. Plant Physiol. 169, 283. doi: 10.1104/pp.15.00233
Ye, J., Zhang, Y., Cui, H., Liu, J., Wu, Y., Cheng, Y., et al. (2018). WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 46, W71–W75. doi: 10.1093/nar/gky400
Yong, S. Y. C., Choong, C. Y., Cheong, P. L., Pang, S. L., Amalina, R. N., Harikrishna, J. A., et al. (2011). Analysis of ESTs generated from inner bark tissue of an Acacia auriculiformis x Acacia mangium hybrid. Tree Genet. Genomes 7, 143–152. doi: 10.1007/s11295-010-0321-y
Yusiharni, E., Gilkes, R. (2012). Minerals in the ash of Australian native plants. Geoderma 189, 369–380. doi: 10.1016/j.geoderma.2012.06.035
Zeng, S., Xiao, G., Guo, J., Fei, Z., Xu, Y., Roe, B. A., et al. (2010). Development of a EST dataset and characterization of EST-SSRs in a traditional Chinese medicinal plant, Epimedium sagittatum (Sieb. Et Zucc.) Maxim. BMC Genomics 11, 94. doi: 10.1186/1471-2164-11-94
Zhang, B. C., Gao, Y. H., Zhang, L. J., Zhou, Y. H. (2021). The plant cell wall: Biosynthesis, construction, and functions. J. Integr. Plant Biol. 63, 251–272. doi: 10.1111/jipb.13055
Keywords: Acacia crassicarpa, illumina sequencing, polymorphism, auxin response factor, lignin
Citation: Ishio S, Kusunoki K, Nemoto M, Kanao T and Tamura T (2024) Illumina-based transcriptomic analysis of the fast-growing leguminous tree Acacia crassicarpa: functional gene annotation and identification of novel SSR-markers. Front. Plant Sci. 15:1339958. doi: 10.3389/fpls.2024.1339958
Received: 17 November 2023; Accepted: 07 August 2024;
Published: 29 August 2024.
Edited by:
Juan Du, Zhejiang University, ChinaReviewed by:
Ram Singh Purty, Guru Gobind Singh Indraprastha University, IndiaMadhab Kumar Sen, Czech University of Life Sciences Prague, Czechia
Copyright © 2024 Ishio, Kusunoki, Nemoto, Kanao and Tamura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takashi Tamura, dGt0YW11cmFAb2theWFtYS11LmFjLmpw