 Xue-Min Xu
Xue-Min Xu He Xu1
He Xu1 Zhen Wei
Zhen Wei Shi-Xin Zhu
Shi-Xin Zhu- 1School of Life Sciences, Zhengzhou University, Zhengzhou, China
- 2Resource Research Institute, Henan Provincial Third Institute of Resources and Environment Investigation, Zhengzhou, China
- 3Xinjiang Institute of Ecology and Geography, Chinese Academy of Sciences, Urumchi, China
- 4College of Life Sciences, Beijing Normal University, Beijing, China
The HAP clade, mainly including Helichrysum Mill, Anaphalis DC., and Pseudognaphalium Kirp., is a major component of tribe Gnaphalieae (Asteraceae). In this clade, Anaphalis represents the largest genus of Asian Gnaphalieae. The intergeneric relationships among Anaphalis and its related genera and the infrageneric taxonomy of this genus are complex and remain controversial. However, there are few studies that have focused on these issues. Herein, based on the current most comprehensive sampling of the HAP clade, especially Anaphalis, we conducted phylogenetic analyses using chloroplast (cp) genome and nuclear ribosomal DNA (nrDNA) to evaluate the relationships within HAP clade, test the monophyly of Anaphalis, and examine the infrageneric taxonomy of this genus. Meanwhile, the morphological characters were verified to determine the circumscription and infrageneric taxonomy system of Anaphalis. Additionally, the biogeographical history, diversification processes, and evolution of crucial morphological characters were estimated and inferred. Our phylogenetic analyses suggested that Anaphalis is polyphyletic because it nested with Helichrysum and Pseudognaphalium. Two and four main clades of Anaphalis were identified in cp genome and nrDNA trees, respectively. Compared with nrDNA trees, the cp genome trees were more effective for phylogenetic resolution. After comprehensively analyzing morphological and phylogenetic evidence, it was concluded that the achene surface ornamentation and leaf base showed less homoplasy and supported the two Anaphalis lineages that were inferred from cp genome. Our biogeographical analyses based on cp genome indicated that HAP clade underwent rapid diversification from late Miocene to Pliocene. The two Anaphalis lineages appeared to have originated in Africa, then spread to Western and Southern Asia, and subsequently moved into Southwestern China forming a diversity center. The dispersal patterns of the two Anaphalis lineages were different. One dispersed around the world, except in Africa and South America. The other one dispersed to Eastern and Southeastern Asia from the ancestral origin region.
1 Introduction
The Gnaphalieae Cass. ex Lecoq & Juill. (Compositae/Asteraceae: Asteroideae) is a large tribe, with ca. 178 genera and ca. 2100 species (Smissen et al., 2020). This tribe has colonized all continents except for Antarctica and concentrates largely in southern Africa and Australia, with smaller diversity centers in the Mediterranean region and South America, while there are fewer taxa in the Northern Hemisphere (Anderberg, 1991; Bayer et al., 2007; Bergh and Linder, 2009; Nie et al., 2016). Based on the latest phylogenetic analyses, two subtribes were included in Gnaphalieae; these were a largely African-endemic Relhaniinae (124 species in 11 genera) and a much enlarged Gnaphaliinae, the latter accounting for more than 90% of the species diversity (ca. 2000 species in 167 genera) and containing six clades, Ifloga, Metalasia, Stoebe, HAP (mainly including Anaphalis DC., Pseudognaphalium Kirp., and Helichrysum Mill), FLAG (mainly including Filago Loefl., Leontopodium R.Br. ex Cass., Antennaria Gaertn., and Gamochaeta Wedd.), and Australasian clades (Smissen et al., 2020).
The genus Anaphalis represents the largest genus in the Asian Gnaphalieae, with ca. 110 species, and is distributed mainly in tropical and subtropical Asia (Chen et al., 2011; Nie et al., 2013). More than 50% of Anaphalis species occur in the Eastern Himalayas and Hengduan Mountains of Southwestern China, and most are endemic to this region (Chen et al., 2011). Anaphalis is mainly characterized by subdioecy, hermaphrodite floret sterility, and dimorphic pappus, which have been used as diagnostic characters to circumscribe this genus (Bentham and Hooker, 1873; Mueller, 1889; Beauverd, 1913; Chen et al., 1966; Drury, 1970; Ling, 1979; Anderberg, 1991; Chen et al., 2011). Some Anaphalis taxa, such as A. hancockii Maxim., A. lactea Maxim., and A. margaritacea (L.) Benth. & Hook.f., are commonly used in China as aromatic plants or herbal remedies (Ling, 1979; Sun et al., 2001; Yuan et al., 2004; Bao et al., 2009).
The circumscription of Anaphalis and its closely related genera is not clear based on morphological characters. Bentham and Hooker (1873) initially defined Anaphalis using subdioecy. Subdioecy can be defined as plants differing in the ratio of female to bisexual florets in their capitula, i.e., in some plants their capitula have few hermaphrodite florets while in others hermaphrodite florets predominate. Chen et al. (1966) studied Chinese Anaphalis and found that the ratio of female to bisexual florets in capitula was different, suggesting that this genus was evolving from monoecious to heteroecious. Drury (1970) considered subdioecy and capitulum arrangement as important characters distinguishing Anaphalis from Gnaphalium L. Ling (1979) later utilized subdioecy and pappus characters to differentiate Anaphalis from Antennaria and Leontopodium. Anderberg (1991) also emphasized that subdioecy readily distinguished Anaphalis species from closely related genera, such as Pseudognaphalium, Helichrysum, and Gnaphalium. However, Grierson (1972) suggested that the broad concept of subdioecy in Anaphalis was questionable, citing evidence from Anaphalis species in Sri Lanka with a consistent ratio of hermaphrodite florets in the capitulum, showing no evidence of subdioecy. Another significant character of Anaphalis is hermaphrodite floret sterility. Mueller (1889) employed this character to differentiate Anaphalis from its closely related genera Pseudognaphalium and Helichrysum. Beauverd (1913) supported Anaphalis as a distinct genus, noting the sterility of hermaphrodite florets in both female-predominant and hermaphrodite-predominant capitula. Ling (1979), however, argued that hermaphrodite floret sterility was not exclusive to Anaphalis, citing its presence in Antennaria and Leontopodium as well. Ling used this character to distinguish these three genera from Gnaphalium, Helichrysum, and Phagnalon Cass. Additionally, Chen et al. (2011) identified the dimorphic pappus as a crucial character distinguishing Anaphalis from its related genera Pseudognaphalium, Helichrysum, and Xerochrysum Tzvelev. Consequently, it is evident that there is no definitive diagnostic character for Anaphalis.
Recent molecular phylogenetic studies have revealed a close relationship between Anaphalis, Helichrysum, and Pseudognaphalium. However, the monophyly of Anaphalis remains uncertain. Molecular phylogenetic analyses by Glenny and Wagstaff (1997) and Breitwieser et al. (1999), focusing on nuclear ribosomal DNA (nrDNA) internal transcribed spacer (ITS), indicated a close association between Anaphalis and Pseudognaphalium. Additionally, morphological characters, such as style arm cells, anther apices, and corolla and involucral bract hairs, supported the separation of Anaphalioides (Benth.) Kirp. from Anaphalis (Glenny, 1997; Glenny and Wagstaff, 1997). Later, the phylogenetic analyses by Smissen et al. (2020) suggested Anaphalioides should be included in the Australasian clade. A clade containing two Pseudognaphalium species and the Southern African species Helichrysum patulum D.Don was supported by the analysis of chloroplast (cp) sequences (trnL intron, trnL-trnF, and matK), and this clade was associated with A. margaritacea (Ward et al., 2009). Similarly, Galbany-Casals et al. (2010) also suggested a close relationship among the three genera Helichrysum, Anaphalis, and Pseudognaphalium. Therefore, a large portion of the genus Helichrysum, along with at least parts of the genera Anaphalis and Pseudognaphalium (referred to as the HAP clade), has consistently been identified as a clade within the subtribe Gnaphaliinae (Smissen et al., 2020). Based on the nrDNA ITS and external transcribed spacer (ETS) sequences, with a focus on the Eastern Himalayan taxa of Anaphalis, Nie et al. (2013) indicated that all Anaphalis and Pseudognaphalium species nested within Helichrysum, forming the well-supported HAP clade, Anaphalis appeared close to the Mediterranean-Asian Helichrysum clade, but the monophyly of Anaphalis was weakly supported. Galbany-Casals et al. (2014) considered the phylogenetic relationships within the HAP clade with the use of two nrDNA markers (ITS and ETS) and two plastid DNA markers (ndhF and rpl32-trnL) and revealed that Achyrocline (Less.) DC., Anaphalis and Pseudognaphalium nested within the currently broadly defined Helichrysum. According to their results, Anaphalis was monophyletic based on the nrDNA markers, but in the cpDNA tree, Anaphalis was paraphyletic (Galbany-Casals et al., 2014). Consequently, additional studies are required to fully establish the composition of the HAP clade, and additional molecular data and taxa, especially regarding the Anaphalis species, should be used to consider the phylogenetic relationships within the HAP clade and examine the monophyly of Anaphalis.
The infrageneric taxonomy of Anaphalis is complex. Previous morphological and molecular studies have provided substantial insights in the taxonomic relationships within Anaphalis. The characters of involucre, capitulum, achene, and leaf have been widely used for the classification of Anaphalis. Initially, Hooker (1882) divided Anaphalis into two series based on capitulum size. Kitamura (1937) proposed two sections, Sect. Nagasawae Kitam. and Sect. Margaripes (DC.) Kitam., characterized by monoecy and dioecy, respectively. Borissova (1999) focused on capitulum number and size suggesting Sect. Anaphalis Boriss. and Sect. Polycephales Boiss. The latter section was further divided into six series (Ser. Margaripes (DC.) Boriss., Ser. Tenuicaules Boriss., Ser. Velutinae Boriss., Ser. Racemiferae Boriss., Ser. Virgatae Boriss., and Ser. Scopulosae Boiss.) (Borissova, 1999). Chen et al. (1966) indicated that A. bulleyana (Jeffrey) C.C. Chang was distinct from other Anaphalis species because of its obovate involucre, beige and membranous phyllaries, and adhesive hairs. Therefore, they divided Anaphalis into two subgenera: Subgen. Gnaphaliops Ling (including only A. bulleyana) and Subgen. Anaphalis. The latter contained two sections: Sect. Margaripes (which included seven series: Ser. Busuae Ling, Ser. Margaripes, Ser. Oxyphyllae Ling, Ser. Sinicae Ling, Ser. Suffruticosae Ling, Ser. Virgatae, and Ser. Xylorhizae Ling) and Sect. Anaphalis (which included three series: Ser. Flavescentes Ling, Ser. Nepalenses Ling, and Ser. Pannosae Ling). Based on the characters of achene surface, 17 taxa of Anaphalis from Pakistan were divided into two groups, i.e., sparse or dense papillate-clavate hair (Abid and Qaiser, 2007). Xu et al. (2021) then observed and studied achene micromorphology in 39 Anaphalis taxa from China, revealing two types of achene surface ornamentation: reticulate-claviform and ligulate protuberant, which were important for the Anaphalis infrageneric taxonomy system. In general, molecular data can provide new insights into taxonomy. However, currently only one study has focused on the molecular phylogeny of Anaphalis species. Nie et al. (2013) explored the relationships of 41 Anaphalis taxa using two nrDNA markers (ITS and ETS) and indicated that although the monophyly of Anaphalis was weakly supported, two clades within the genus were recognized. Furthermore, the phylogenetic relationships within Anaphalis corresponded to the shape of leaf base rather than the morphology of capitula and phyllaries, which were usually used for species delimitation and classification in this genus (Nie et al., 2013). Other phylogenetic studies involved only a small number of Anaphalis species (Glenny and Wagstaff, 1997; Breitwieser et al., 1999; Ward et al., 2009; Galbany-Casals et al., 2010; Galbany-Casals et al., 2014), which were insufficient for the analysis of the infrageneric taxonomy of this genus. Investigating the infrageneric relationships based on several DNA markers or a relatively small sample size may lead to erroneous and incomplete conclusions. Therefore, additional molecular data and taxa are imperative for a thorough investigation on Anaphalis relationships.
The phylogenies are inferred using cp and nuclear genes, which provide a more comprehensive phylogenetic results and can further explore the complex genetic relationships. The chloroplast, with independent genetic material, is responsible for photosynthesis and plays an important role in other aspects of plant physiology and development (Leister, 2003; Xiong et al., 2009; Wicke et al., 2011; Daniell et al., 2016; Tian et al., 2021). Compared with nuclear and mitochondrial genomes, cp genomes evolve at relatively moderate rates and has significantly contributed to phylogenetic and taxonomic studies (Dong et al., 2013; Twyford and Ness, 2017; Jiang et al., 2020; Pascual-Díaz et al., 2021; Wang et al., 2021; Zhang et al., 2021b; Chu et al., 2022; Han et al., 2022; Xu et al., 2023a). However, there are currently only 30 complete cp genomes from the HAP clade (Anaphalis, 2; Helichrysum, 4; Pseudognaphalium, 24) that have been published in the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/). Therefore, additional cp genome data must be generated and analyzed to reveal the intergeneric relationships of the HAP clade and the infrageneric relationships of Anaphalis. Compared with genomic data, DNA barcodes, such as trnL-F, rbcL, matK, trnK-matK, psbA-trnH, ITS, and ETS, are more readily available and can still be used to solve many taxonomic problems. Among these, ITS and ETS are important nrDNA markers that are widely used in phylogenetic studies (Kuzmanović et al., 2017; Vicent et al., 2017; Zhou and Zhang, 2017; Carrizo García et al., 2018; Asanuma et al., 2019; Pirani et al., 2020; Tan et al., 2020; Hashim et al., 2021; Liao et al., 2023). ITS and ETS are effective in discriminating closely related species because of their rapid evolutionary rates and higher interspecies variation (Li et al., 2011). Additionally, the incongruent phylogenetic relationships between the cp and nuclear genes suggest the existence of hybridization, incomplete lineage sorting, and/or interspecific introgression (Yang et al., 2013; Gatesy et al., 2019). Therefore, complex genetic relationships can be revealed through phylogenetic analyses based on the cp and nuclear genes. However, few studies have considered the intergeneric relationships of the HAP clade and the infrageneric relationships of Anaphalis based on both cp and nuclear genes.
In this study, we used the most comprehensive samples of the HAP clade, with a particular emphasis on Anaphalis taxa. We constructed phylogenetic trees using cp genomes and nrDNA. The objectives of this study were to (1) reconstruct a phylogeny of the HAP clade to analyze the intergeneric relationships of this clade and test the monophyly of Anaphalis; (2) resolve the relationships within Anaphalis and test previous morphological classifications of this genus; (3) estimate the biogeographical history and diversification processes of Anaphalis; and (4) infer the evolution of the crucial morphological characters.
2 Materials and methods
2.1 Taxon sampling
The sequences used in this study include both new and previously published sequences. Leaf material from 77 individuals representing 63 taxa (52 Anaphalis, 2 Euchiton Cass., 1 Gnaphalium, 2 Helichrysum, 3 Pseudognaphalium, 1 Xerochrysum, and 2 Scorzonera L.) was obtained. Most of the plant material used in this study was collected by our research group from natural populations in China. The voucher specimens were deposited in the herbarium of Zhengzhou University (ZZU; Zhengzhou, China). The plant material from a few taxa was obtained from the herbarium specimens of E (Royal Botanic Garden Edinburgh Herbarium, Edinburgh, UK), PE (Institute of Botany, Chinese Academy of Sciences, Beijing, China), and XJBI (Xinjiang Institute of Ecology and Geography, Chinese Academy of Sciences, Urumchi, China). Detailed information regarding the voucher specimens is provided in Table 1. Furthermore, the cp genome, ITS and ETS sequences (51 cp genomes, 744 ITS, and 836 ETS) of Gnaphalieae and related taxa were acquired from the NCBI database and are listed in Supplementary Tables S1, S2, respectively. Based on the phylogenetic results of Fu et al. (2016); Huang et al. (2016), Panero and Crozier (2016), Mandel et al. (2019), and Zhang et al. (2021a), Scorzonera humilis, S. radiata, Cichorium intybus L., Lactuca praevia C.D.Adams, and L. sativa L. were used as outgroups.
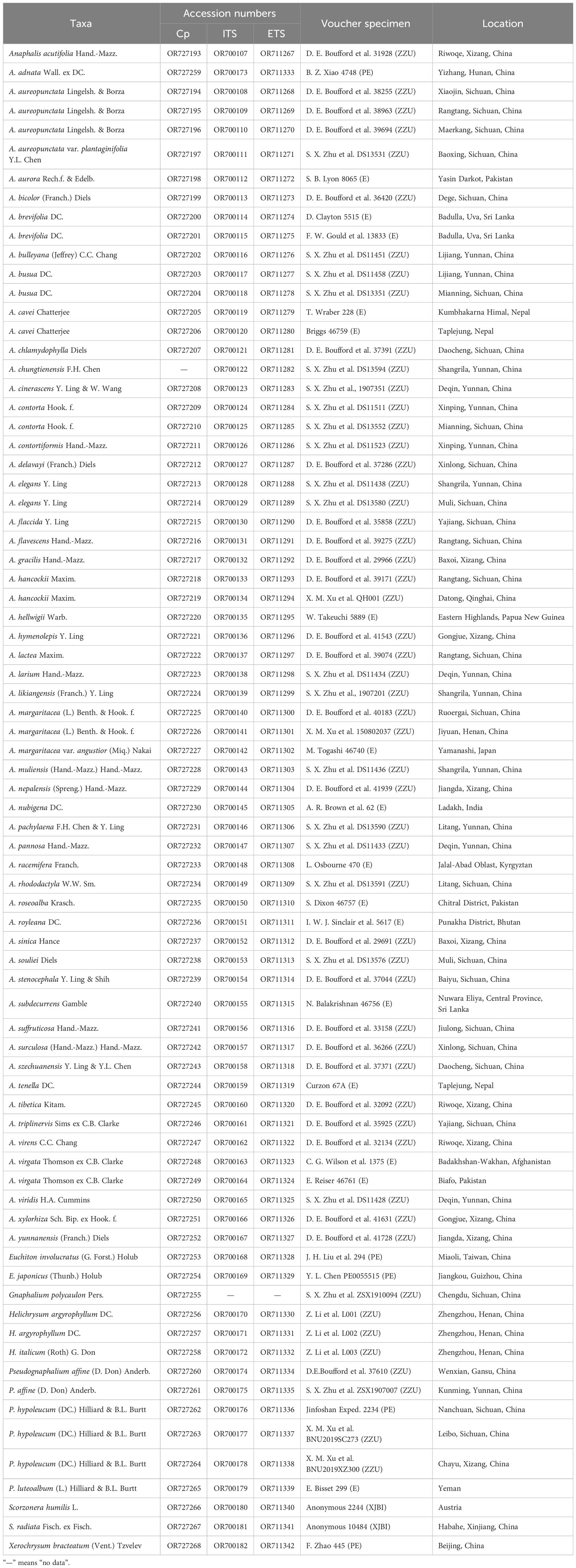
Table 1 Voucher specimens and location information, and accession numbers of cp genomes, ITS, and ETS sequences from NCBI database.
2.2 DNA extraction and sequencing, cp genome and nrDNA assembly
We used a modified cetyltrimethyl ammonium bromide (CTAB) method to extract high-quality DNA (Doyle and Doyle, 1987), which was then purified using the Wizard® DNA cleanup system (Promega, Madison, WI, USA). DNA quality was assessed using a NanoDrop spectrophotometer (Thermo Scientific, Carlsbad, CA, USA), and integrity was evaluated through electrophoresis on a 1% (w/v) agarose gel. A DNA library was prepared using the NEB Next Ultra DNA Library Prep Kit for Illumina (NEB, USA). Libraries for paired-end 150-bp sequencing were analyzed on an Illumina NovaSeq 6000 platform (Novogene Co., Ltd., Tianjin, China), to generate approximately 10 GB of data for each sample. Raw reads were filtered using SOAPnuke to remove sequencing adaptors and low-quality bases (Chen et al., 2018). The filtered reads were assembled using GetOrganelle (Jin et al., 2020) with a range of 21, 45, 65, 85, and 105 k-mers for plastomes, and 35, 85, and 115 k-mers for nrDNA. Subsequently, the ITS and ETS sequences were uploaded onto the NCBI database (the accession numbers are listed in Table 1).
2.3 Cp genome annotation
The plastome sequences were first annotated using Geneious Prime 2020.1.2 (https://www.geneious.com) by referring to the cp genome sequences of A. sinica Hance (KX148081), A. margaritacea var. yedoensis Ohwi (LC656264), and P. affine (D.Don) Anderb. (MN541094). The annotations of protein-coding sequences were then manually checked based on the open reading frame. Transfer RNA genes were verified using the online tRNAscan-SE tool with default settings (Lowe and Chan, 2016). All annotated cp genome sequences were deposited in the NCBI database (accession numbers are listed in Table 1).
2.4 Phylogenetic analysis
Phylogenetic trees were constructed based on four matrices: complete cp sequences, ITS, ETS, and the concatenated sequences of ITS and ETS. A total of 127 cp genomes (including 63 Anaphalis samples representing 52 taxa), 820 ITS sequences (including 248 Anaphalis samples representing 67 taxa), and 912 ETS sequences (including 213 Anaphalis samples representing 67 taxa) were involved in our phylogenetic analyses. Only the nrDNA sequences from the same individual were concatenated, and detailed information is shown in Supplementary Table S2. The online version of MAFFT was used to align the datasets (Katoh et al., 2019). The matrix characteristics were analyzed using MegaX (Kumar et al., 2018). The alignments of cp genome, ITS, and ETS sequences are provided in the Supplementary Material. The phylogenetic analyses were performed using the maximum likelihood (ML) and Bayesian inference (BI) methods via IQ-TREE v1.6.12 and MrBayes 3.2.2, respectively (Ronquist et al., 2012; Nguyen et al., 2015). The best-fitting model of nucleotide substitutions was determined using ModelFinder in PhyloSuite v1.2.2 (Zhang et al., 2020). The ML analyses were performed using IQ-TREE with 1,000 bootstrap replicates. The BI analysis was run for 50,000,000 generations and sampled every 5,000 generations, and the first 25% of the trees were discarded as burn-in. Trees were selected based on a 50% majority-rule consensus to estimate the posterior probabilities. The effective sample size (ESS, >200) was determined using Tracer v1.7 (Rambaut et al., 2018). Reconstructed trees were visualized using Figtree v1.4.2 (Rambaut, 2014) and TreeGraph 2 (Stöver and Müller, 2010).
2.5 Divergence time estimates
Divergence time analyses based on the cp genome matrix were performed using the BEAST v2.6.7 program (Bouckaert et al., 2019), with the Gamma site + GTR nucleotide substitution model, the yule model for tree priors, and relaxed clock log-normal model. Two nodes were calibrated: (1) The crown age of tribe Cichorieae Lam. & DC. (including Cichorium L., Scorzonera, and Lactuca L.) was calibrated based on the oldest reported Cichorium intybus type pollen fossil (early Miocene, 22.0–28.4 Mya), which was widely distributed in all Cichorieae subtribes except in Scorzonerinae (Hochuli, 1978; Tremetsberger et al., 2013; Kilian et al., 2017); (2) The earliest Ambrosia-type pollen fossil (22–30 Mya) from the Beaverhead Basin flora of Montana was used to calibrate the crown age of tribe Tageteae (including Ambrosia L., Helianthus L., and Tagetes L.) (Becker, 1969; Leopold and MacGinitie, 1972; Graham, 1996; Bergh and Linder, 2009). A Markov chain Monte Carlo (MCMC) analysis was run for 100 million generations, with sampling every 10,000 generations. Other parameters were left at the default values. The BEAST output files were examined through Tracer v1.7 (Rambaut et al., 2018) to evaluate the convergence and ensure that ESS values for all parameters were adequate and > 200. We combined the treefiles (burn-in 20%) in Logcombiner v2.6.7, and used Treeannotator v2.6.7 to produce the maximum clade credibility (MCC) tree showing mean divergence time estimates with 95% highest posterior density (HPD) intervals. The final result was visualized using Figtree v1.4.2 (Rambaut, 2014).
2.6 Diversification through time
A Bayesian analysis of macroevolutionary mixtures (BAMM v2.5.0) was performed to analyze the speciation and extinction dynamics of HAP clade (Rabosky et al., 2014a). The appropriate priors on the consensus tree were estimated using the “setBAMMpriors” function in the BAMMtools package in R Studio (Rabosky et al., 2014b). The parameters of “PRIORS” were as follows: expectedNumberOfShifts = 1; lambdaInitPrior = 0.848; lambdaShiftPrior = 0.069; muInitPrior = 0.848; and lambdaIsTimeVariablePrior = 1. The analysis was run for 10 million generations, and the MCMC chain was sampled every 1000 generations. Finally, using BAMMtools, we traced the specific clade diversification and plotted the diversification rate (speciation, extinction, and net diversification rates) through time, after discarding 10% of the generations (MCMC) as burn-in. Further, we constructed the semi-logarithmic lineage through time (LTT) plots of HAP clade using the “phytools” package in R studio (Revell, 2012) to visualize temporal variations in diversification rates (Mahler et al., 2010). Three thousand trees were randomly sampled from the converged BEAST trees of the HAP clade and were used to calculate the 95% credibility interval.
2.7 Ancestral area reconstruction
The natural distribution data of taxa in this study was assessed based on the Global Biodiversity Information Facility database (https://www.gbif.org/) and various floras (e.g., Hooker, 1882; Leopold and MacGinitie, 1972; Ling, 1979; Borissova, 1999; Chen et al., 2011). The biogeographical area codes of HAP clade were modified according to those of Gnaphalieae from Nie et al. (2016). Asia was re-coded, considering the highest species richness and endemism of Anaphalis. Seven biogeographical areas were coded as follows: (A) North America; (B) Europe to Western Asia; (C) Africa; (D) SW China; (E) South Asia; (F) Eastern and Southeastern Asia; (G) New Guinea, Australia, New Zealand, and Pacific islands. The ancestral area reconstruction was performed using the tree files generated by BEAST under the Statistical Dispersal-Vicariance-Analysis (S-DIVA) model in RASP v4.2 using the default settings (Yu et al., 2020). Random 1000 BEAST output trees were used and the maximum number of individual unit areas was set to four. To reduce geographical irrelevance from distant phylogenetic relationships, we only retained samples of the HAP clade in posterior trees and MCC tree. The analysis was applied allowing up to four areas per node.
2.8 Characteristic analyses of morphology
According to the results of Nie et al. (2013) and Xu et al. (2021), we selected two morphological characters to study their evolution in Anaphalis. These two characters and their states were achene surface ornamentation (reticulate-claviform, and ligulate protuberant) and leaf base (decurrent, non-decurrent, and cordate). The characteristics information of achene surface ornamentation and leaf base are provided in Supplementary Table S3. Ancestral state reconstruction and stochastic character mapping were performed in R Studio v4.1, using the make.simmap commands under the ER model (equal-rates model) in “phytools” package (Huelsenbeck et al., 2003; Paradis et al., 2004; Bollback, 2006; Revell, 2012), The tree file (MCC tree) generated by BEAST was used to analyze and only focused on Anaphalis. Stochastic mapping was simulated 200 times, and posterior density tree was plotted in “Phytools” package (Revell, 2012; O’Reilly and Donoghue, 2018).
3 Results
3.1 Phylogenetic relationships
A total of 228 sequences were newly generated in this study (including 76 cp genomes, 76 ITS, and 76 ETS). The sequence characteristics and nucleotide substitution models for ML and BI phylogenetic analyses of different datasets (complete cp genomes, ITS, ETS, and concatenated ITS and ETS sequences) are presented in Supplementary Table S4.
In the phylogenetic trees inferred using complete cp genomes (Figure 1A and Supplementary Figure 1), the topologies of BI and MP trees were consistent (Supplementary Figure 1). In HAP clade, Anaphalis was polyphyletic and nested with Pseudognaphalium and Helichrysum. Four clades, Clade 1 + 3 [18 taxa, ML bootstrap value (BS) = 100, Bayesian posterior probability (PP) = 1], Clade 4 (32 taxa, BS = 100, PP = 1), and two small clades (each with only one species, A. subdecurrens Gamble and A. brevifolia DC., respectively), were recognized in Anaphalis. Clade 1 + 3 and A. subdecurrens were nested with Pseudognaphalium, and Clade 4 and A. brevifolia were nested with Helichrysum.
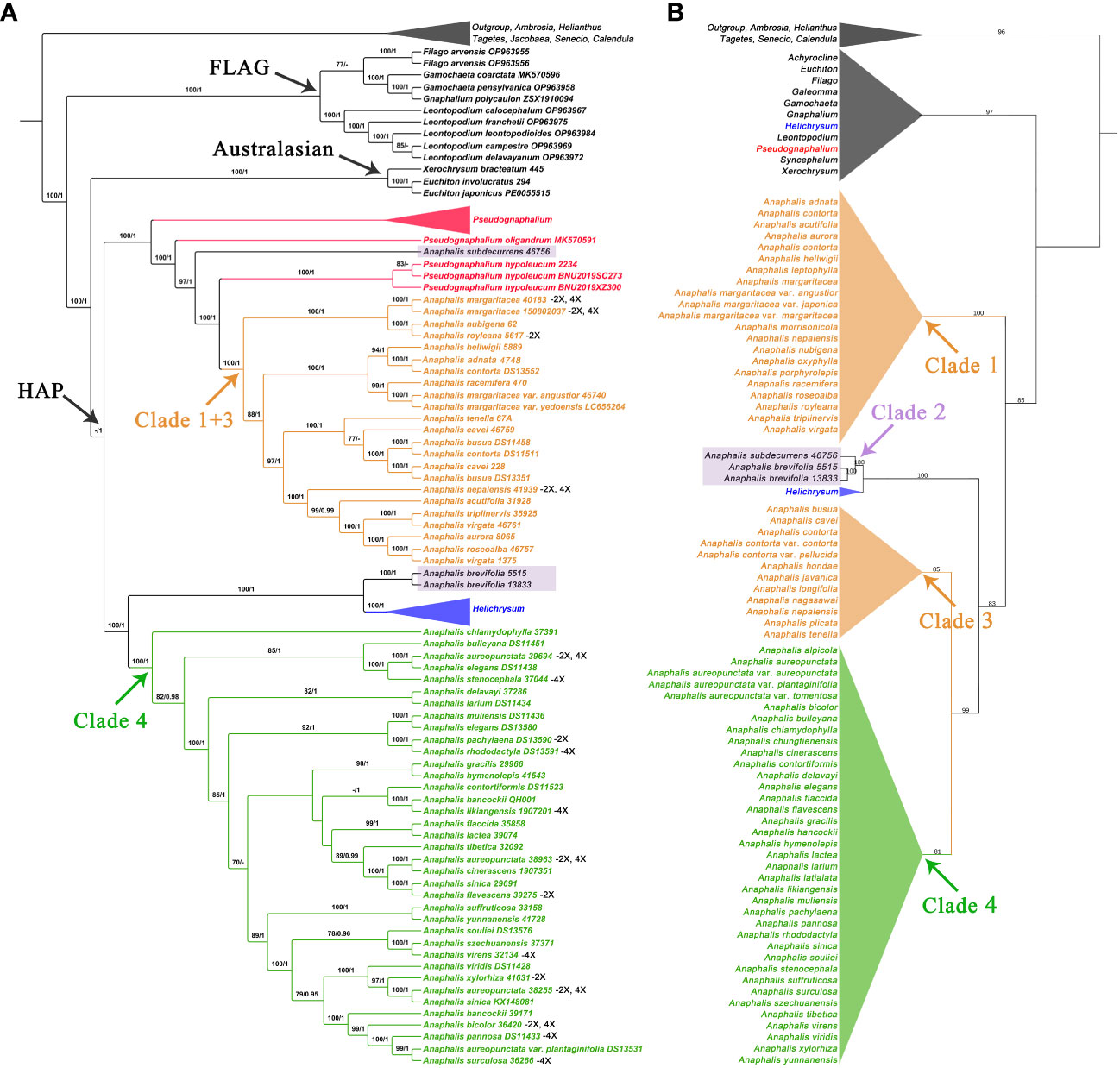
Figure 1 Comparison of phylogenetic trees inferred from the complete chloroplast genomes and the concatenated sequences of ITS and ETS. The skeletal phylogenetic trees of the HAP clade and its closely related genera are showed emphasizing the mainly clades: (A) ML tree, inferred from the complete chloroplast genomes, with bootstrap values of ML and posterior probabilities of BI shown at each node. Bootstrap values higher than 70 and posterior probabilities higher than 0.90 are indicated on branches. “-” means that the bootstrap; (B) ML tree, inferred from the concatenated sequences of ITS and ETS, with bootstrap values of ML shown at each node.
In the nrDNA trees (Figures 1B, 2 and Supplementary Figures S2-S6), the topologies of BI and MP trees were inconsistent. Additionally, the phylogenetic trees inferred from different nrDNA matrices (ITS, ETS, and concatenated ITS and ETS sequences) were also inconsistent. The genera sampled in this study, such as Pseudognaphalium, Anaphalis, Achyrocline, Gnaphalium, and Xerochrysum, all nested in Helichrysum. In the ML tree inferred from ITS (Supplementary Figure S2), Anaphalis was monophyletic, but in the other trees constructed by nrDNA, Anaphalis was polyphyletic (Figure 2 and Supplementary Figures S3-S6). Four clades were included in Anaphalis with strong support. The topological relationships among the four clades were different in different phylogenetic trees. For example, in the ML tree based on ETS (Supplementary Figure S4), Clade 2 was sister to some Helichrysum taxa and the remaining Anaphalis taxa, and Clade 1 of Anaphalis was sister to Clade 3 and Clade 4. However, in the BI tree based on ETS (Supplementary Figure S5), Clade 1 was sister to some Helichrysum taxa and the remaining Anaphalis taxa; Clade 2 was sister to Helichrysum; and Clade 3 and Clade 4 were clusted together. Among the four clades, Clade 2 included only two species: A. subdecurrens and A. brevifolia. There were 20, 11, and 36 taxa included in Clade 1, Clade 2, and Clade 4, respectively. Additionally, the samples of two species, A. contorta Hook.f. and A. nepalensis (Spreng.) Hand.-Mazz., in Anaphalis were not clustered into the same main clade (Clade 1 and Clade 3, respectively).
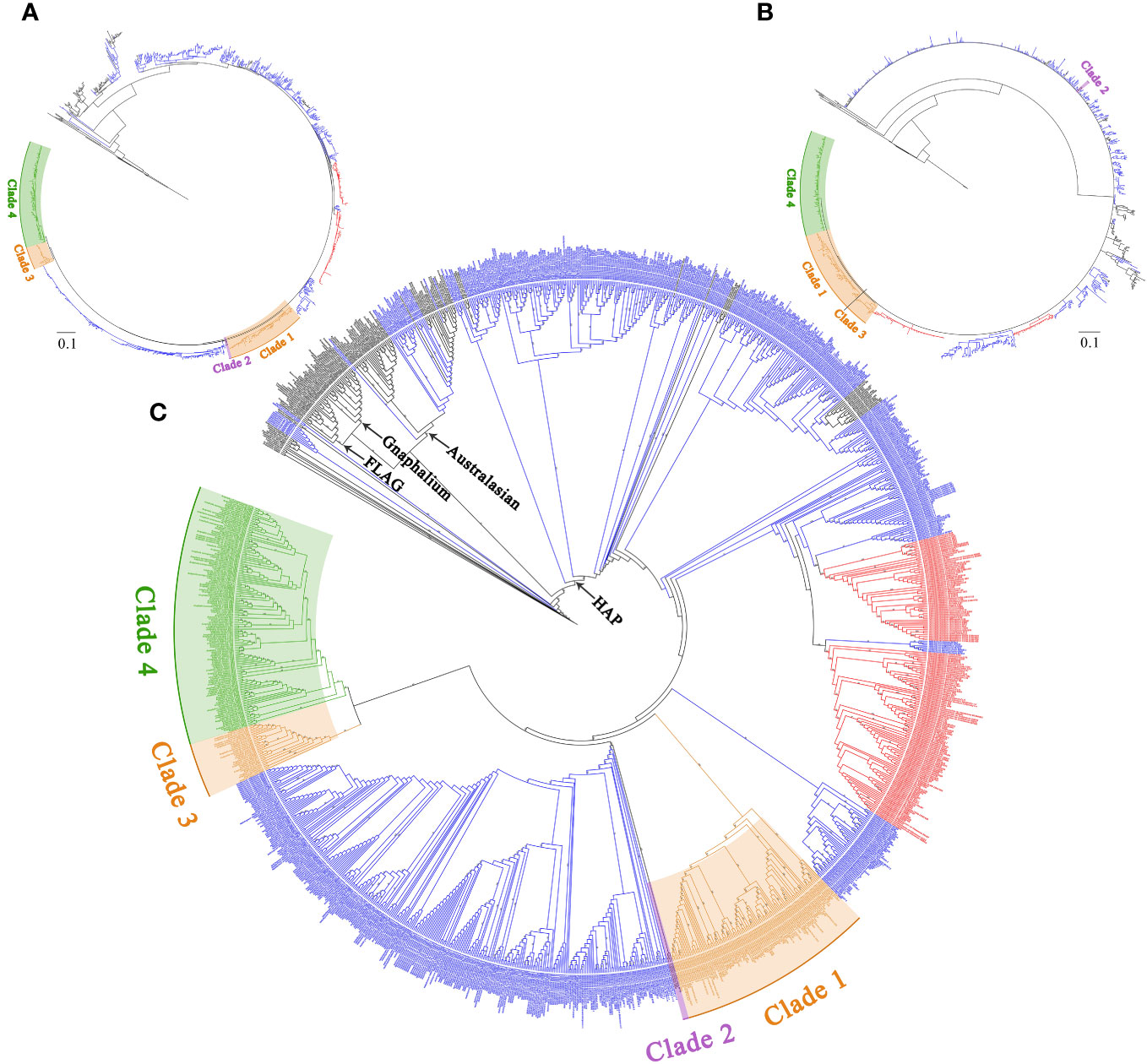
Figure 2 The complete phylogenetic trees of the HAP clade and its closely related genera are inferred from the concatenated sequences of ITS and ETS. (A) Topology of the ML tree. (B) Topology of the BI tree. (C) ML tree, with bootstrap values of ML shown at each node. Bootstrap values higher than 70 are indicated on branches.
In Anaphalis, the species in Clade 1 + 3 (cp genome tree, Figure 1A) were same as those in Clade 1 and Clade 3 (nrDNA tree, Figure 1B), and the species in Clade 4 of cp genome tree (Figure 1A) were same as those in Clade 4 of nrDNA tree (Figure 1B). In the cp genome tree, A. subdecurrens was nested in Pseudognaphalium, and A. brevifolia was sister to Helichrysum (Figure 1A). However, in the nrDNA tree, these two species were clustered together (Clade 2) and sister to Helichrysum (Figures 1B, 2).
3.2 Divergence time estimation and diversification through time
In the cpDNA date analysis (Figure 3A and Supplementary Figure S7), the ancestor of HAP clade diverged approximately 16.6 Mya (95% HPD: 10.49–22.57 Mya). In HAP clade, the MRCA of Helichrysum, A. brevifolia, and Clade 4 of Anaphalis diverged at 13.3 Mya (95% HPD: 8.2–18.66 Mya). Divergence between A. brevifolia and its sister genera, Helichrysum, was estimated to have occurred during the Miocene, at 8.69 Mya (95% HPD: 4.44–13.4 Mya). The MRCA of Clade 4 in Anaphalis diverged at 8.02 Mya (95% HPD: 5.09–11.36 Mya). Divergence among Pseudognaphalium, A. subdecurrens, and Clade 1 + 3 of Anaphalis was estimated to also have occurred during the Miocene, at 13.71 Mya (95% HPD: 8.72–18.88 Mya). The MRCA of most Pseudognaphalium species diverged at 8.65 Mya (95% HPD: 5.11–12.92 Mya). The ancestor of Clade 1 + 3 in Anaphalis diverged approximately 4.99 Mya (95% HPD: 2.53–7.64 Mya) during the Pliocene.
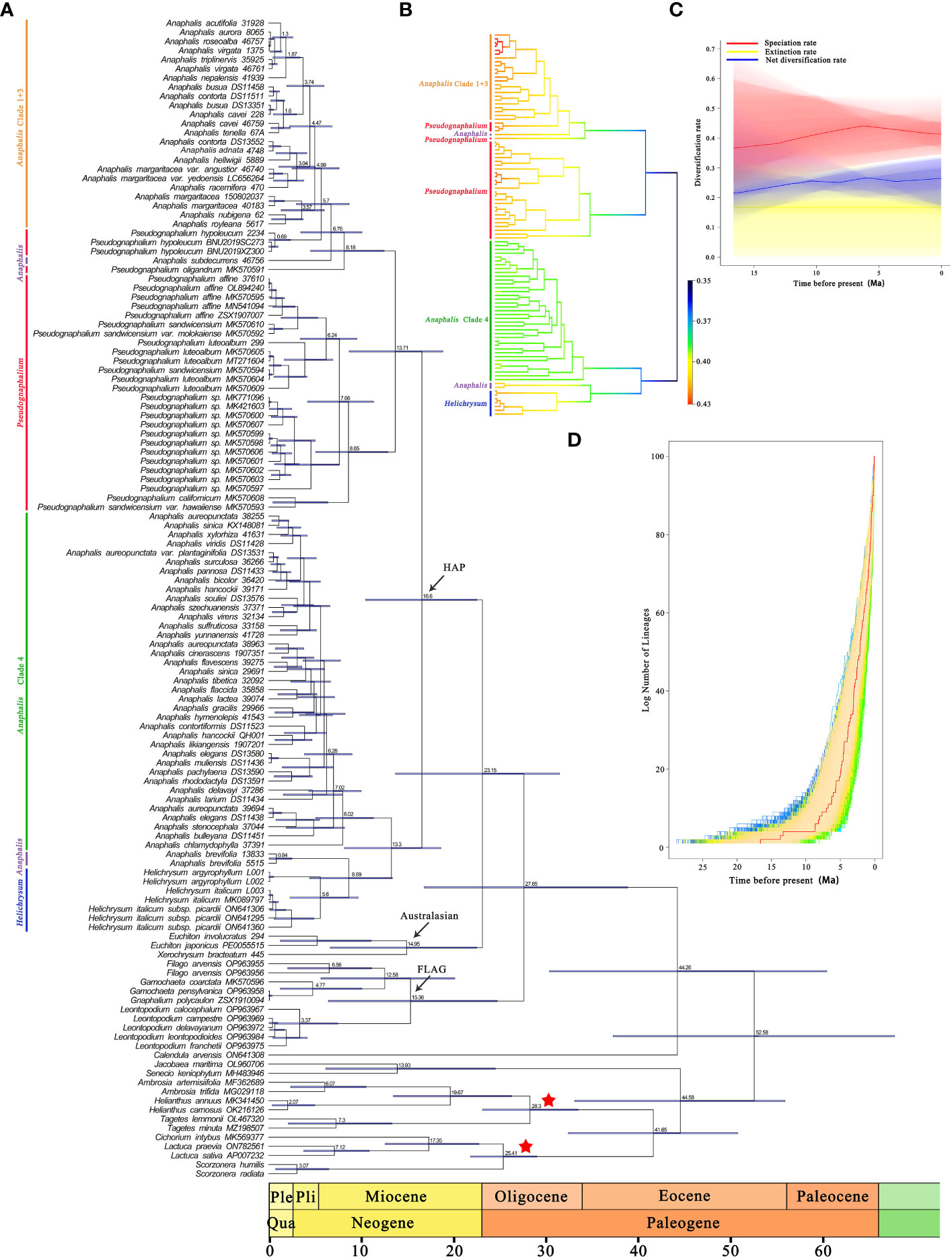
Figure 3 Divergence times estimation and BAMM analysis of HAP clade based on complete plastome sequences. (A) Time-calibrated phylogenetic tree of the HAP clade and its closely related genera, inferred with BEAST from the 127 represent samples plastid dataset. Bars on the nodes indicate the 95% HPD intervals, numbers on the bars indicate the mean age (Mya). The calibrated nodes are indicated by red asterisk. (B) Speciation rate dynamics of HAP clade estimated by BAMM. Branch color reflects the mean of the marginal posterior density of speciation rates for each segment of the branches, with rates increasing from blue to red. (C) The diversification rates from the origin of the HAP clade to the present obtained using BAMM. (D) LTT plots show the cumulative number of lineages over time of HAP clade. The red line shows the maximum clade credibility tree.
The BAMM analysis indicated that no apparent rate shift occurred in the phylogeny of the HAP clade (Figure 3B). The radiation of the HAP species began in the Miocene (ca. 16.6 Mya) at a rate of 0.35 lineages per million years. This analysis indicated that the HAP species diversification was characterized by an early relatively lower initial speciation rate, followed by a burst of speciation. Additionally, Pseudognaphalium, Helichrysum, and Clade 1 + 3 of Anaphalis had higher speciation rates than that of Clade 4 of Anaphalis. The rate-through-time plot shows that the speciation rate in the HAP clade gradually increased before ca. 6 million years, and then slightly declined (Figure 3C). The extinction rate rose in fluctuation. However, the net diversification rate in the HAP clade was constant at a low level. The semi-logarithmic LTT plot analysis corroborated the results of BAMM analysis and indicated an accelerated lineage accumulation after ca. 8 million years (Figure 3D).
3.3 Ancestral geographical range
Based on cp genomes, the ancestral area reconstruction using S-DIVA model inferred that the ancestor of HAP clade probably occurred in Africa, Southern Asia, and/or Southwestern China, (coding: CDE) (Figure 4). The MRCA of Helichrysum, A. brevifolia, and Clade 4 of Anaphalis remained in CD (Africa, Southwestern China), and/or CDE (probability = 0.5000, respectively) areas. The ancestor of Helichrysum and A. brevifolia probably occurred in Africa and/or Southern Asia (coding: CE). The ancestor of A. brevifolia occurred in Southern Asia (coding: E). The MRCA of Clade 4 of Anaphalis remained in Southwestern China (coding: D). The ancestor of Pseudognaphalium, A. subdecurrens, and Clade 1 + 3 of Anaphalis remained in the D and/or E (probability = 0.5000, respectively) areas. The MRCA of P. oligandrum, P. hypoleucum, A. subdecurrens, and Clade 1 + 3 of Anaphalis remained in the CD, CE, and/or CDE (probability = 0.3317, 0.3267, and 0.3317, respectively) areas. Then, except for P. oligandrum, the ancestor of taxa mentioned above probably occurred in the E and/or DE (probability = 0.5000, respectively) areas. The MRCA of P. hypoleucum and its sister clade, Clade 1 + 3 of Anaphalis, remained in the D and/or DE (probability = 0.5000, respectively) areas. The ancestor of Clade 1 + 3 of Anaphalis occurred in the D area.
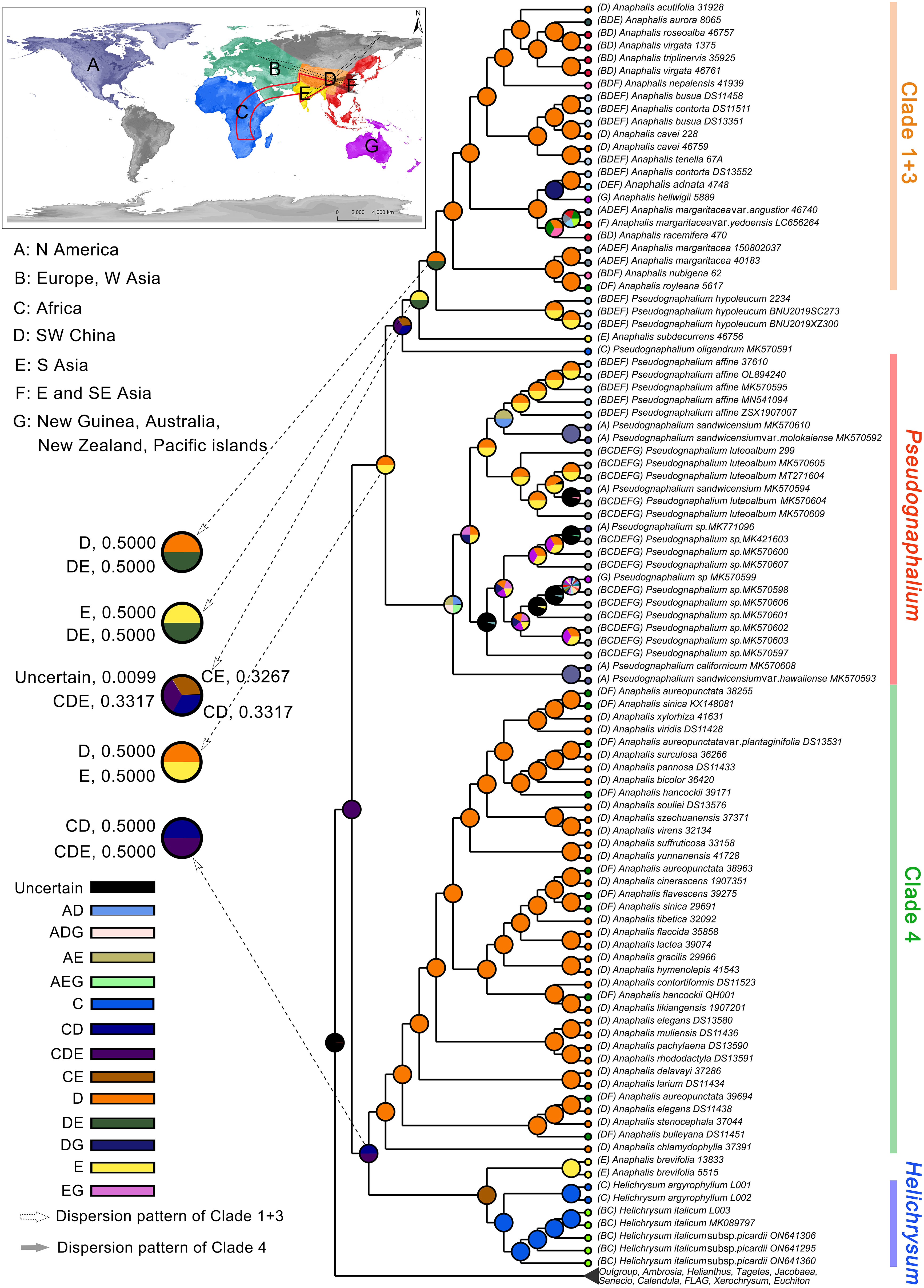
Figure 4 Ancestral area reconstruction of the HAP clade under the S-DIVA model in RASP v4.2 and using the phylogenetic tree from the BEAST analysis. Pie charts denote the ancestral areas with probability values. The map shows the coding areas in different colors.
3.4 The ancestral state reconstruction of morphological characters
The characters of achene surface ornamentation and leaf base and the reconstruction of these characters are presented in Figure 5. In Anaphalis, the taxa in Clade 4 had ligulate protuberant surface ornamentation of achene; in Clade 1 + 3, the taxa had reticulate-claviform surface ornamentation of achene, except A. royleana DC. (Figure 5). The ancestral state of achene surface ornamentation in Anaphalis was probably ligulate protuberant. And, the reticulate-claviform surface ornamentation of achene might evolve from the ligulate protuberant surface ornamentation. The leaf bases of A. brevifolia and the taxa in Clade 4 were decurrent. Most taxa in Clade 1 + 3 and A. subdecurrens had non-decurrent leaf bases. In Clade 1 + 3, A. contorta had a cordate leaf base, and A. busua DC. had a decurrent leaf base. The ancestral state of leaf base in Anaphalis was probably decurrent, and the cordate and non-decurrent leaf base might evolve from the decurrent leaf base.
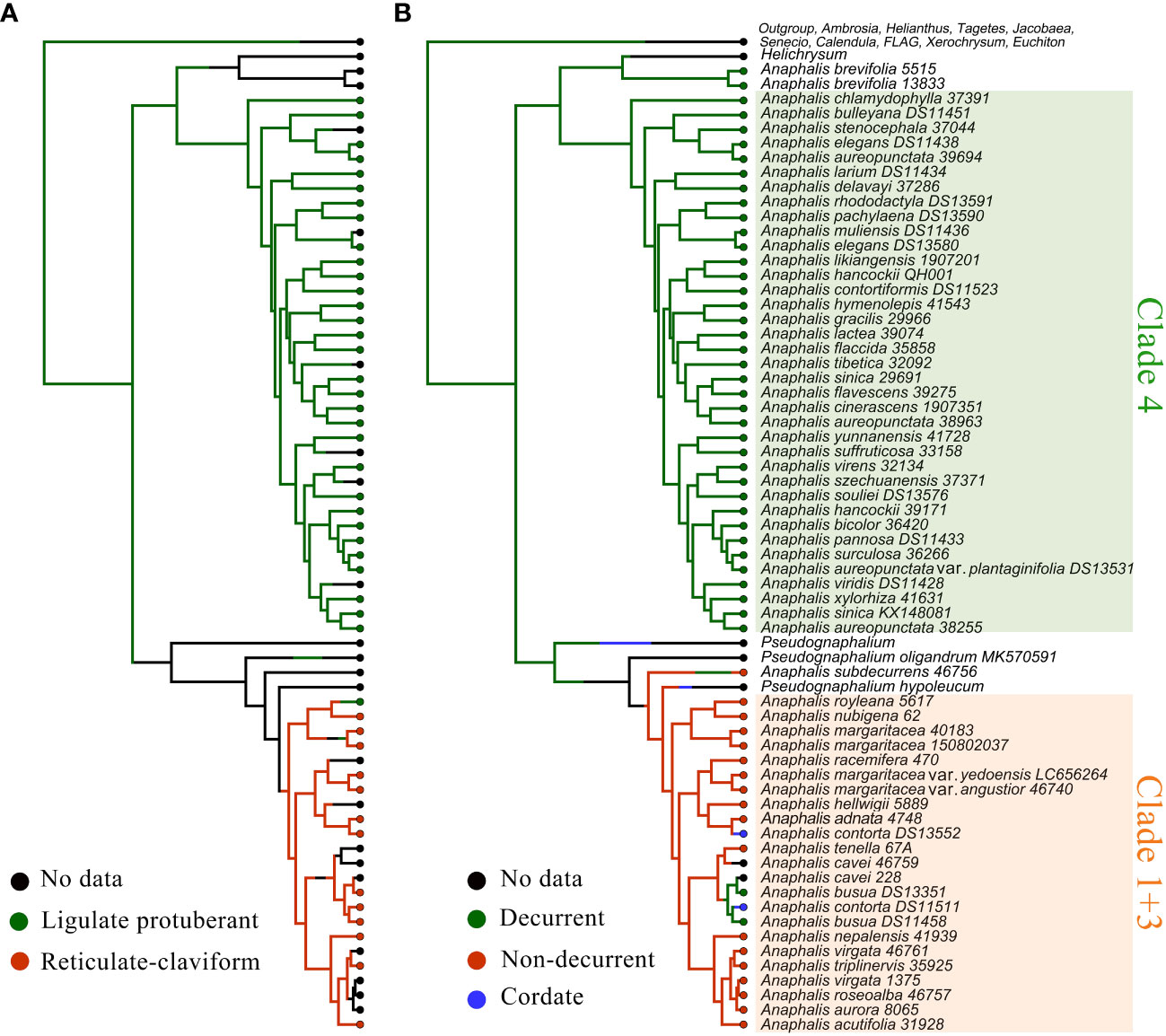
Figure 5 The ancestral state reconstruction and stochastic character mapping of morphological characters performed in R v4.1, using the make.simmap commands under ER model in phytools package. (A) Achene surface ornamentation (B) Leaf base.
4 Discussion
4.1 Topological discordance between the nrDNA and cpDNA trees
In this study, the topological trees constructed based on complete cp genomes (Figure 1A and Supplementary Figure S1) and nrDNA (Figures 1B, 2 and Supplementary Figures S2–S6) were incongruent. Various hypotheses have been proposed in previous studies to explain this phenomenon, such as ancient rapid radiation, incomplete lineage sorting, hybridization, introgression, and undersampling (Joly et al., 2009; Lo and Donoghue, 2012; Liu et al., 2017; Xu et al., 2017; Liu et al., 2020; Duan et al., 2021). These complex phylogenetic relationships are commonly found in Asteraceae (Bayer et al., 2002; Hidalgo et al., 2006; Kim et al., 2007; Barker et al., 2009; Galbany-Casals et al., 2010; Zhao et al., 2010; Jara-Arancio et al., 2017). The taxa in Clade 1 and Clade 3 of Anaphalis (nrDNA trees) were same as those in Clade 1 + 3 (cp genome trees), which suggested that the cp genetic information of these taxa was similar. However, in the nrDNA trees, Clade 3 was closely related to Clade 4 and distant from Clade 1, and the samples of A. contorta and A. nepalensis were clustered into Clade 1 and Clade 3, respectively. In previous studies, the past or present hybrid events were used to interpret the discordant phylogenetic relationships of several genera in tribe Gnaphalieae, rather than the sorting or retention of ancestral polymorphisms (Smissen et al., 2004; Smissen and Breitwieser, 2008; Galbany-Casals et al., 2010; Smissen et al., 2011). Furthermore, Smissen et al. (2011) hypothesized that Anaphalis and the Mediterranean-Asian Helichrysum had a common allopolyploid origin with possible parental species shared based on additional evidence of low-copy markers and chromosome data. However, only one Anaphalis species was represented in their study, A. margaritacea. Meng et al., 2010; Meng et al., 2014) reported the chromosome numbers of 18 Anaphalis taxa and indicated that tetraploidy is common in Anaphalis (the detailed information are shown on Figure 1), which further supported the polyploidy origin of the genus.
In the present study, we inferred that allopolyploid origin (or evolving from a polymorphic polyploid ancestral complex) and hybridization probably all occurred in Anaphalis. First, we hypothesized that there were two lineages of Anaphalis, Clade 1 + 3 and Clade 4 (excluding A. subdecurrens and A. brevifolia), based on our cpDNA analysis. The species in Clade 1 + 3 (identical to Clade 1 and Clade 3) and Clade 4 possibly evolved from Pseudognaphalium and Helichrysum, respectively, based on our cpDNA phylogenetic trees. Alternatively, the main clades of Anaphalis evolved from a polymorphic polyploid ancestral complex of Helichrysum. Then, the species in Clade 3 might have hybridized with those in Clade 4, leading to these two clades being closely related. However, this hypothesis lacked sufficient support because Clade 3 was sister to Clade 4 with strong support values instead of clustering together. The other hypothesis was that there were three lineages of Anaphalis (excluding A. subdecurrens and A. brevifolia) based on our nrDNA analysis, which possibly evolved from Helichrysum respectively. Then, the taxa in Clade 3 might have hybridized with those in Clade 1 or captured the cp from Clade 1, resulting in those taxa clustered into one clade (Clade 1 + 3) in the cpDNA phylogenetic trees. Chloroplast capture, a stochastic process that occurs in groups evolving through rapid radiation (the Asteraceae family is highly evolved and is in the stage of rapid differentiation), can influence cp genome (Rieseberg and Soltis, 1991; Bremer, 1994; Fior et al., 2013). Nevertheless, in the nrDNA tree, the samples of A. contorta and A. nepalensis were not clustered into the same main clade but rather clustered into Clade 1 and Clade 3, respectively, potentially reducing support for our hypothesis described above. Other possible reasons also could be used to explain the discordant phylogenetic relationships between nrDNA and cpDNA, such as nrDNA polymorphism, low sequence resolution, or incomplete lineage sorting, which were commonly discussed in Asteraceae (Álvarez and Wendel, 2003; Galbany-Casals et al., 2004; Ford et al., 2007; Smissen and Breitwieser, 2008; Galbany-Casals et al., 2011; Güzel et al., 2021; Thode et al., 2021). Although the exact reasons that led to these complex phylogenetic relationships are not certain with the present data, the systematic position of Clade 4 was clear. Additionally, it seems probable that more samples of Helichrysum and Pseudognaphalium are required to discover the complex phylogenetic relationships and histories.
4.2 Phylogenetic relationships within Anaphalis
Based on the current most comprehensive sampling of Anaphalis, both our phylogenetic analyses using nrDNA and cp genomes did not support the monophyly of this genus. The phylogenetic analyses based on nrDNA (ITS and ETS) by Nie et al. (2013) focusing on Anaphalis indicated that the monophyly of Anaphalis was weakly supported. Galbany-Casals et al. (2014) explored the phylogenetic relationships in Helichrysum and related genera, and seven Anaphalis taxa were included in their analyses. Similarly, the phylogenetic tree based on nrDNA also weakly supported the monophyly of Anaphalis. However, in their phylogenetic tree inferred using cpDNA markers (rpl32-trnL and ndhF), Anaphalis was not monophyletic and included two clades, both nested within Helichrysum (Galbany-Casals et al., 2014).
Additionally, our phylogenetic analysis results did not support the traditional infrageneric taxonomy system of Anaphalis. Based on the size of capitulum, Hooker (1882) divided Anaphalis into two series: series I the diameter of capitulum ca. 1/2–2/3 inch; series II, the diameter of capitulum ca. 1/6–1/3 inch. Nevertheless, our phylogenetic results did not support the monophyly of these series. For example, A. nubigena, A. royleana, A. triplinervis, and A. xylorhiza belonged to series I (Hooker, 1882), but in our phylogenetic trees, A. xylorhiza did not cluster together with the other three species (Figure 1). Borissova (1999) proposed two sections (Sect. Anaphalis and Sect. Polycephales) based on the number and the size of capitula. Sect. Anaphalis with fewer and larger capitula included two species, A. serawschanica (Winkl.) B. Fedtsch. growing on the mountain slopes of Central Asia and A. nubigena in the alpine zone of the Himalayas. Sect. Polycephales were distributed across Asia, particularly in the Himalayas, and included A. margaritacea, A. racemifera, A. roseoalba, A. virgata, etc. Obviously, A. nubigena was closely related to species of Sect. Polycephales in our phylogenetic analyses (Figure 1). Chen et al. (1966) focused on Chinese Anaphalis species and presented an infrageneric taxonomy system that included two subgenera (Subgen. Gnaphaliops and Subgen. Anaphalis), two sections, and ten series. However, the results of our phylogenetic analyses suggested that none of these were monophyletic. The Subgen. Gnaphaliops included only one species, A. bulleyana (Chen et al., 1966), but this species was not obviously separated from other Anaphalis species in our phylogenetic analyses. In the phylogenetic tree inferred by complete cp genomes (Figure 1A), A. bulleyana was clustered with A. aureopunctata, A. elegans, and A. stenocephala. In the nrDNA tree, A. bulleyana was closely related to A. aureopunctata, A. sinica, and A. delavayi (Figure 2). Two sections, Sect. Margaripes and Sect. Anaphalis, were included in Subgen. Anaphalis, mainly based on the number, size, and shape of capitula (Chen et al., 1966). Whereas, the results of our phylogenetic analysis supported that these two sections were nested with each other. For instance, A. contorta, A. margaritacea, and A. oxyphylla belonged to Sect. Margaripes; and A. acutifolia, A. nepalensis, and A. triplinervis belonged to Sect. Anaphalis (Chen et al., 1966). Nevertheless, these species were clustered into one clade in our phylogenetic analyses: Clade 1 + 3 of the cpDNA tree (Figure 1A) and Clade 1 of the nrDNA tree (Figure 1B). The same problem existed in the series proposed by Chen et al. (1966).
Recent phylogenetic analyses based on two nrDNA markers (ITS and ETS) supported two major lineages of Anaphalis that corresponded to the shape of the leaf base rather than the characters of the capitula (Nie et al., 2013). These two lineages were the “non-decurrent” clade (species with attenuate or narrowed leaf bases but never forming wings on the stem) and the “decurrent” clade (most species with decurrent leaf base and a small group with cordate leaf base). The main clades inferred by Nie et al. (2013) were mostly consistent with those of our nrDNA tree (Figure 1B). Species of the “non-decurrent” clade (Clade I and Clade II; Nie et al., 2013) were all included in Clade 1 of our nrDNA tree (Figure 1B). Three species, A. cntorta, A. plicata, and A. hondae, with cordate leaf bases were clustered with A. busua with decurrent leaf base, forming a single main clade (Clade III), which was sister to other species (Clade IV) with decurrent leaf bases (Nie et al., 2013). However, as more samples were included in our nrDNA tree, we found that the species with cordate leaf bases (A. contorta, A. hondae, and A. plicata), decurrent leaf bases (A. busua and A. nagasawai), and non-decurrent leaf bases (A. cavei, A. nepalensis, A. javanica, and A. longifolia) were all nested together and constituted one main clade (Clade 3), which was sister to other species (Clade 4) with decurrent leaf bases (Figure 1B). Additionally, our cpDNA phylogenetic tree also suggested that these species with different leaf base were nested (Figure 5B, Clade 1 + 3). As a result, the character of leaf base did not perfectly support these phylogenetic relationships. Nevertheless, compared with the characters of capitula, the genetic data suggested that the shape of leaf base appeared to show less homoplasy and was largely congruent with the phylogenetic relationships. Therefore, the significance of leaf base characteristics for infrageneric taxonomy system of Anaphalis should not be ignored. According to the characteristics of achene micro-morphology, Xu et al. (2021) divided 39 Chinese Anaphalis taxa into two groups: Group I with reticulate-claviform surface ornamentation and Group II with ligulate protuberant surface ornamentation. We compared the taxa in the two groups with those in the main clades of our phylogenetic trees. Then, we found that the taxa in Group I were all included in Clade 1 + 3 of cpDNA tree and in Clade 1 and Clade 3 of nrDNA tree. However, the taxa in Group II were divided into different main clades of our phylogenetic trees (cpDNA tree, Clade 1 + 3 and Clade 4; nrDNA tree, Clade 1 and Clade 4), because A. royleana had a different achene micro-morphology from other taxa in the same clade. Thus, similar to leaf base, although the characteristics of achene micro-morphology also did not completely support the phylogenetic relationships of Anaphalis, they are still important for the infrageneric taxonomy system of this genus because of their less homoplasy.
Therefore, compared with nrDNA trees, the cp trees were more effective for phylogenetic resolution and were supported by the morphological (achene surface ornamentation and leaf base) characters. The morphological characters evolution analysis indicated that ligulate protuberant surface ornamentation on achene was a morphological synapomorphy of Clade 4. In Clade 1 + 3, except A. royleana, the other taxa had reticulate-claviform surface ornamentation on achene. Chen and Jing (2007) inferred reticulate-claviform ornamentation was the more primitive type. However, based on our analyses, the ligulate protuberant surface ornamentation was the ancestral state of Anaphalis and evolved once in Clade 1 + 3 (Figure 5A). The presence of decurrent leaf base was a morphological synapomorphy of Clade 4. Most species of Clade 1 + 3 had non-decurrent leaf bases, and a few species had decurrent or cordate leaf bases. Decurrent leaf base was the ancestral state of Anaphalis. The characters of decurrent and cordate leaf base evolved twice in Clade 1 + 3, respectively (Figure 5B). The phylogenetically conserved pattern of leaf base in Clade 4 might be due to greater genetic constraints and/or stabilizing selection pressure favoring stasis of this character in alpine habitats, because most species of this clade were restricted to the Himalayan region. Recent phylogenetic analyses of Anaphalis (emphasis on the eastern Himalayan taxa) and Leontopodium (diversified in Himalayas and Hengduan Mountains) also suggested a higher degree of homoplasy in leaf base (Nie et al., 2013; Xu et al., 2023b).
Compared with the infrageneric taxonomy system, the interspecies relationships of Anaphalis were more complex. It was common for samples from the same species not to cluster together, such as A. aureopunctata, A. busua, A. contorta, and A. margaritacea (Figures 1A, 2). Furthermore, the inconsistent phylogenetic results inferred from different DNA data also reflected the complexity of interspecific relationships in Anaphalis. For instance, in the cpDNA phylogenetic tree, A. bulleyana was closely related to A. aureopunctata, A. elegans, and A. stenocephala (Figure 1A), whereas in the nrDNA tree, this species was closely related to A. aureopunctata and A. surculosa (Figure 2). Frequent hybridization, introgression, horizontal gene transfer, polyploidy accompanied by apomixis, or rapid radiation may all contribute to the evolutionary complexity (Zou and Ge, 2008; Liu et al., 2020). Particularly in Asteraceae, which is highly evolved and in the rapid differentiation stage, a substantial number of complex and polymorphic transitional taxa lead to several challenges in classification and phylogeny research (Bremer, 1994; Bayer et al., 2002; Hidalgo et al., 2006; Kim et al., 2007; Barker et al, 2009; Galbany-Casals et al., 2010; Zhao et al., 2010; Jara-Arancio et al., 2017). Recently, Xu et al. (2023b) indicated that the relationships within Leontopodium (Asteraceae, Gnaphalieae) were very complex and suggested that hybridization may be responsible for this phenomenon.
In Anaphalis, particular attention should be paid to three species: A. adnata Wall. ex DC., A. subdecurrens, and A. brevifolia. De Candolle (1837) first described A. adnata. Then, this species was transferred to Gnaphalium, as G. adnatum (DC.) Wall. ex Thwaites (Thwaites, 1864), which was also recognized by Ling (1979). In the Flora of China, this species was included in Pseudognaphalium, as P. adnatum (DC.) Y. S. Chen (Chen et al., 2011). Clearly, our phylogenetic results indicated that this species should be included in Anaphalis (Figures 1, 2). Furthermore, the characters of achene surface ornamentation and leaf base of this species were same as the taxa in Clade 1 + 3 of Anaphalis (Figure 5). Therefore, we suggest that P. adnatum should be considered as a synonym of A. adnata. Our phylogenetic trees showed that A. subdecurrens and A. brevifolia, which were distributed in South India and Sri Lanka, had a close relationship with Pseudognaphalium and Helichrysum (Figure 1). Subdioecy was widely used to distinguish Anaphalis from its related genera, such as Pseudognaphalium, Helichrysum, and Gnaphalium (Bentham and Hooker, 1873; Chen et al., 1966; Drury, 1970; Ling, 1979; Anderberg, 1991). However, Grierson (1972) clarified that the Anaphalis species in Sri Lanka were not subdioecious. Thus, our phylogenetic results support the use of subdioecy to distinguish Anaphalis from its related genera and the transfer of A. subdecurrens and A. brevifolia to Pseudognaphalium or Helichrysum. Nevertheless, because we did not conduct a sufficiently detailed study and investigation of the characters of these two species, it may be too early to draw such revises.
4.3 Historical biogeography
Based on cp genome data, the crown ages of the HAP clade were estimated to be 16.6 (10.49–22.57) Mya, suggesting that its early diversification occurred in the Miocene, and the two main clades of HAP diverged during the middle Miocene (Figure 3A). Based on nrDNA data, Nie et al. (2016) focusing on Gnaphalieae estimated the crown ages of HAP clade at to be approximately 15.39 (11.42–19.64) Mya, which was similar to our estimate. Our results also revealed that the three genera, Anaphalis, Pseudognaphalium, and Helichrysum, diverged in the late Miocene (Figure 3A). The Miocene was a pivotal period in the “making of the modern world” owing to specific climatic conditions (such as the overall cooling trend of the climate, the origin of modern ocean currents, and the aridification of continental interiors) and geological events (such as the uplift of the Himalayan mountains, the closure of the Tethys Ocean, and the closing of the Inter-American Seaway). This resulted in it being a key period for angiosperm phylogeny, marked by pronounced rates of dispersal (Zachos et al., 2008; Antonelli, 2009; Clark et al., 2009; Potter and Szatmari, 2009; Couvreur et al., 2011; Bacon et al., 2012; Yu et al., 2014; Nie et al., 2016; Huang et al., 2018; Dong et al., 2022). The crown age of the clade including Helichrysum and A. brevifolia was estimated to be 8.69 (4.44–13.4) Mya (Figure 3A). The crown age of the clade including P. oligandrum, P. hypoleucum, A. subdecurrens, and Clade 1 + 3 of Anaphalis was estimated to be 8.18 (4.53–12.5) Mya (Figure 3A). The crown age of the remaining Pseudognaphalium species was estimated to be 8.65 (5.11–12.92) Mya (Figure 3A). The crown ages of Clade 1 + 3 and Clade 4 of Anaphalis were estimated to be 4.99 (2.53–7.64) Mya and 8.02 (5.09–11.36) Mya, respectively (Figure 3A). These clades suggested that HAP clade underwent rapid diversification in the late Miocene to Pliocene. Our LTT plot analysis indicated an accelerated lineage accumulation after ca. 8 Mya (Figure 3D). This acceleration may be attributed to the dry habitat during late Miocene to Pliocene, providing favorable open grassland for dispersion (Palazzesi and Barreda, 2012; Nie et al., 2016). Similar cases have been found in many other flowering plants (Coleman et al., 2003; Li et al., 2009; Thiv et al., 2010; Nie et al., 2016; Pouchon et al., 2023), indicating the importance of the late Miocene to Pliocene for the assembly of modern flora. Additionally, our BAMM analysis results revealed that the HAP species experienced an early relatively lower speciation rate (middle Miocene) followed by a burst of diversification (from late Miocene to Pliocene), with a relatively constant extinction rate (Figures 3B, C). We estimated diversification rates beginning from ca. 16.6 Mya, which had a rate of 0.35 species per million years. This reported rate was similar to those found in other taxa, such as Saxifraga Tourn. ex L., Gentiana (Tourn.) L., and Delphinium L. (Jabbour and Renner, 2012; Favre et al., 2016; Ebersbach et al., 2017). Although the diversification rate of the HAP clade was not low compared with other angiosperm clades (Xing and Ree, 2017; Qian et al., 2018), it did not match the exceptionally high rate recorded in another taxa of Asteraceae, the Espeletia Mutis ex Bonpl. complex, which most likely caused by the Pleistocene climatic oscillations that triggered exceptionally rapid radiation (Pouchon et al., 2018). Regardless, our data added another empirical example of the rapid differentiation of Asteraceae plants, further demonstrating that high diversification rates occurred from the late Miocene to Pliocene.
Based on cp genomes, we were unable to determine the specific origin area of the HAP clade, likely due to the limited inclusion of Helichrysum samples. Using nrDNA and cpDNA markers and focusing on Helichrysum, Galbany-Casals et al. (2014) suggested that the HAP clade originated in the Cape region of Southern Africa and subsequently dispersed to and diversified in the Eastern and Southern Africa. The biogeographic analyses of Nie et al. (2016) and Andrés-Sánchez et al. (2019) also indicated a Southern African origin for the HAP clade. Our biogeographic analyses confirmed that the ancestor of Helichrysum occurred in Africa (Figure 4), which was consistent with the inference of Galbany-Casals et al., 2004; Galbany-Casals et al., 2014). Pseudognaphalium is a large genus with a world-wide distribution, likely resulting from recent global expansions during the late Miocene to Pliocene (Nie et al., 2016). Our results supported two separate lineages of Pseudognaphalium, one of which was nested with Anaphalis and showed an African origin, but the origin area of another independent lineage remained undefined (Figure 4). Two separate lineages of Pseudognaphalium were also inferred by Nie et al. (2016), but both nested within the Southern African HAP clade, indicating a Southern African origin. For more reliable results, future investigations should include additional taxa and molecular data of Pseudognaphalium to reveal the biogeographic pattern of this genus in the future. Obviously, the ancestral region of Anaphalis was definitively in Southwestern China, regardless of A. subdecurrens and A. brevifolia, but the dispersal patterns of the two Anaphalis lineages were markedly different (Figure 4). The two Anaphalis lineages appeared to have originated in Africa, then spread to Western and Southern Asia, and subsequently moved into Southwestern China and the pan-Himalayan region forming a diversity center (Figure 4). Then, Clade 1 + 3 of Anaphalis dispersed around the world, except in Africa and South America. The ancestral origin region and dispersal pattern of Clade 1 + 3 inferred in this study were mostly consistent with previous results. It was generally accepted that the extant Gnaphalieae originated in Southern Africa and then dispersed to other regions, often over long distances (Bergh and Linder, 2009; Ward et al., 2009; Nie et al., 2016). According to the current distribution area of Anaphalis, Galbany-Casals et al. (2014) inferred that this genus might have originated in East tropical Africa, radiated in the Himalayan range, further spread to Eastern Asia, and later reached North America, where only A. margaritacea is currently present, across the Bering Strait. Additionally, the chromosome numbers provided support for a common origin of Anaphalis and the Eurasian Helichrysum from an African ancestor (Nie et al., 2013). However, our results distinctly showed that the origin time and dispersion pattern of Clade 4 were different and unrelated to those of Clade 1 + 3. Clade 4 diverged from Helichrysum at 13.3 (8.2–18.66) Mya, whereas Clade 1 + 3 of Anaphalis diverged with Pseudognaphalium at 5.7 (3–8.73) Mya (Figures 3, 4 and Supplementary Figure S7). Moreover, the Clade 4 dispersed to Eastern and Southeastern Asia from the ancestral origin region. (Figure 4).
5 Conclusions
Our major results include: (1) Achyrocline, Anaphalis, Galeomma, Pseudognaphalium, and Syncephalum were nested with Helichrysum in the HAP clade. Anaphalis was polyphyletic and nested with Helichrysum and Pseudognaphalium. (2) Two and four mainly clades of Anaphalis were recognized in the cp genome and nrDNA phylogenetic trees, respectively. Compared with nrDNA trees, the cp trees were more effective for phylogenetic resolution, and were supported by the morphological and cp genome characters. (3) Subdioecy could be used to distinguish Anaphalis from its related genera. (4) The achene surface ornamentation and leaf base showed less homoplasy and supported the two lineages of Anaphalis inferred by cp genome. (5) The HAP clade underwent rapid diversification in the late Miocene to Pliocene. The crown ages of the two Anaphalis lineages were estimated at 4.99 (2.53–7.64) Mya and 8.02 (5.09–11.36) Mya, respectively. (6) The two Anaphalis lineages appeared to have originated in Africa, then spread to Western and Southern Asia, and subsequently moved into Southwestern China and the pan-Himalayan region forming a diversity center. The dispersal patterns of the two Anaphalis lineages were different. One dispersed around the world, except in Africa and South America. The other one dispersed to Eastern and Southeastern Asia from the ancestral origin region.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/ , OR727193-OR727268 https://www.ncbi.nlm.nih.gov/genbank/ , OR700107-OR700182 https://www.ncbi.nlm.nih.gov/genbank/ , OR700107-OR711342.
Author contributions
XX: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. HX: Investigation, Writing – original draft. ZY: Data curation, Formal analysis, Writing – original draft. ZW: Writing – review & editing. JG: Data curation, Investigation, Writing – original draft. DL: Data curation, Writing – review & editing. QL: Supervision, Writing – review & editing. SZ: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (31670188 and 31000097).
Acknowledgments
We are grateful to E (Royal Botanic Garden Edinburgh Herbarium, Edinburgh, UK), PE (Institute of Botany, Chinese Academy of Sciences, Beijing, China), and XJBI (Xinjiang Institute of Ecology and Geography, Chinese Academy of Sciences, Urumchi, China) herbariums for providing plant material for sequencing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1336229/full#supplementary-material
Supplementary Material 1 | The alignment of chloroplast (cp) genome sequences.
Supplementary Material 2 | The alignment of internal transcribed spacer (ITS) sequences.
Supplementary Material 3 | The alignment of external transcribed spacer (ETS) sequences.
Supplementary Figure 1 | Phylogenetic trees of the HAP clade and its closely related genera are inferred from the complete chloroplast genomes. (A) Topology of the ML tree. (B) Topology of the BI tree. (C) ML tree, with bootstrap values of ML and posterior probabilities of BI shown at each node. Bootstrap values higher than 70 and posterior probabilities higher than 0.90 are indicated on branches. “-” means that the bootstrap value/posterior probability is less than 70/0.90.
Supplementary Figure 2 | Phylogenetic tree of the HAP clade and its closely related genera is inferred from ITS sequences using ML method. (A) Topologies. (B) The phylogenetic tree shows bootstrap values of ML at each node. Bootstrap values higher than 70 are indicated on branches.
Supplementary Figure 3 | Phylogenetic tree of the HAP clade and its closely related genera is inferred from ITS sequences using BI method. (A) Topologies. (B) The phylogenetic tree shows posterior probabilities of BI at each node. Posterior probabilities higher than 0.90 are indicated on branches.
Supplementary Figure 4 | Phylogenetic tree of the HAP clade and its closely related genera is inferred from ETS sequences using ML method. (A) Topologies. (B) The phylogenetic tree shows bootstrap values of ML at each node. Bootstrap values higher than 70 are indicated on branches.
Supplementary Figure 5 | Phylogenetic tree of the HAP clade and its closely related genera is inferred from ETS sequences using BI method. (A) Topologies. (B) The phylogenetic tree shows posterior probabilities of BI at each node. Posterior probabilities higher than 0.90 are indicated on branches.
Supplementary Figure 6 | Phylogenetic tree of the HAP clade and its closely related genera is inferred from the concatenated sequences of ITS and ETS using BI method. The posterior probabilities of BI are showed at each node. Posterior probabilities higher than 0.90 are indicated on branches.
Supplementary Figure 7 | Time-calibrated phylogenetic tree of the HAP clade and its closely related genera, inferred with BEAST from the 127 represent samples plastid dataset. The numbers of 95% HPD intervals are showed at each node indicating the inferred age range (Mya).
References
Abid, R., Qaiser, M. (2007). Cypsela morphology of the genus Anaphalis DC. (Gnaphalieae-Asteraceae) from Pakistan. Pak. J. Bot. 39, 1897–1906.
Álvarez, I., Wendel, J. F. (2003). Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet. Evol. 29, 417–434. doi: 10.1016/s1055-7903(03)00208-2
Anderberg, A. A. (1991). Taxonomy and phylogeny of the tribe Gnaphalieae (Asteraceae). Opera Bot. 104, 1–195.
Andrés-Sánchez, S., Verboom, G. A., Galbany-Casals, M., Bergh, N. G. (2019). Evolutionary history of the arid climate-adapted Helichrysum (Asteraceae: Gnaphalieae): Cape origin and association between annual life-history and low chromosome numbers. J. Syst. Evol. 57, 468–487. doi: 10.1111/jse.12472
Antonelli, A. (2009). Have giant lobelias evolved several times independently? Life form shifts and historical biogeography of the cosmopolitan and highly diverse subfamily Lobelioideae (Campanulaceae). BMC Biol. 7, 82. doi: 10.1186/1741-7007-7-82
Asanuma, M., Zhu, S., Okura, N., Cai, S. Q., Yoshimatsu, K., Komatsu, K. (2019). Genetic polymorphism of Japanese cultivated Rheum species in the internal transcribed spacer region of nuclear ribosomal DNA. J. Nat. Med. 73, 541–554. doi: 10.1007/s11418-019-01298-4
Bacon, C. D., Baker, W. J., Simmons, M. P. (2012). Miocene dispersal drives island radiations in the palm tribe Trachycarpeae (Arecaceae). Syst. Biol. 61, 426–442. doi: 10.1093/sysbio/syr123
Bao, J. Y., Lu, Y. C., Bai, H. S. (2009). Quantitative and qualitative analysis on flavonoids in Anaphalis lactea Maxim. J. Mol. Sci. 25, 72–74.
Barker, N. P., Howis, S., Nordenstam, B., Källersjö, M., Eldenäs, P., Griffioen, C., et al. (2009). Nuclear and chloroplast DNA-based phylogenies of Chrysanthemoides Tourn. ex Medik. (Calenduleae; Asteraceae) reveal extensive incongruence and generic paraphyly, but support the recognition of infraspecific taxa in C. monilifera. S. Afr. J. Bot. 75, 560–572. doi: 10.1016/j.sajb.2009.05.006
Bayer, R. J., Breitwieser, I., Ward, J., Puttock, C. (2007). “Tribe gnaphalieae,” in The families and genera of vascular plants, vol. vol. 8 . Eds. Kadereit, J. W., Jeffrey, C. (Heidelberg: Springer), 246–283.
Bayer, R., Greber, D., Bagnall, N. (2002). Phylogeny of Australian Gnaphalieae (Asteraceae) based on chloroplast and nuclear sequences, the trnL intron, trnL/trnF intergenic spacer, matK, and ETS. Syst. Bot. 27, 801–814. doi: 10.1043/0363-6445-27.4.801
Becker, H. F. (1969). Fossil plants of the Tertiary Beaverhead Basins in southwestern Montana. Palaeontographica Abt. B. 127, 1–142.
Bergh, N. G., Linder, H. P. (2009). Cape diversification and repeated out-of-southern-Africa dispersal in paper daisies (Asteraceae–Gnaphalieae). Mol. Phylogenet. Evol. 51, 5–18. doi: 10.1016/j.ympev.2008.09.001
Bollback, J. P. (2006). SIMMAP: Stochastic character mapping of discrete traits on phylogenies. BMC Bioinf. 7, 88. doi: 10.1186/1471-2105-7-88
Borissova, A. G. (1999). “ Anaphalis ,” in Flora of the USSR, vol. vol. 25 . Ed. Shishkin, B. K. (Washington: Amerind Publishing Co. Pvt. Ltd), 337–352.
Bouckaert, R., Vaughan, T. G., Barido-Sottani, J., Duchêne, S., Fourment, M., Gavryushkina, A., et al. (2019). BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PloS Comput. Biol. 15, e1006650. doi: 10.1371/journal.pcbi.1006650
Breitwieser, I., Glenny, D. S., Thorne, A., Wagstaff, S. J. (1999). Phylogenetic relationships in Australasian Gnaphalieae (Compositae) inferred from ITS sequences. New Zeal. J. Bot. 37, 399–412. doi: 10.1080/0028825X.1999.9512644
Carrizo García, C., Basso, A. V., Leiva González, S., Gonzáles, P., Barboza, G. E. (2018). Unraveling the phylogenetic relationships of Nectouxia (Solanaceae): its position relative to Salpichroa. Plant Syst. Evol. 304, 177–183. doi: 10.1007/s00606-017-1460-5
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 7, 1–6. doi: 10.1093/gigascience/gix120
Chen, X., Jing, G. (2007). A study on fruit coats micro-morphological characters of thirty-six species of Compositae from alpine meadow in eastern Qinghai-Tibet Plateau. Pratac. Sci. 24, 9–15. doi: 10.3969/j.issn.1001-0629.2007.05.002
Chen, F. H., Ling, Y., Chen, Y. L., Shih, C., Wang, W. (1966). De genera Anaphalis DC. familiae Compositarum e florae sinicae. Acta Phytotaxon. Sin. 11, 91–112.
Chen, Y. S., Zhu, S. X., Bayer, R. J. (2011). “Gnaphalieae,” in Flora of China vol. 20–21. Eds. Wu, C. Y., Raven, P. H., Hong, D. Y. (Beijing: Science Press; St. Louis: Missouri Botanical Garden Press), 774–818.
Chu, R., Xu, X., Lu, Z., Ma, Y., Cheng, H., Zhu, S., et al. (2022). Plastome-based phylogeny and biogeography of Lactuca L. (Asteraceae) support revised lettuce gene pool categories. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.978417
Clark, J. R., Wagner, W. L., Roalson, E. H. (2009). Patterns of diversification and ancestral range reconstruction in the southeast Asian-Pacific angiosperm lineage Cyrtandra (Gesneriaceae). Mol. Phylogenet. Evol. 53, 982–994. doi: 10.1016/j.ympev.2009.09.002
Coleman, M., Liston, A., Kadereit, J. W., Abbott, R. J. (2003). Repeat intercontinental dispersal and Pleistocene speciation in disjunct Mediterranean and desert Senecio (Asteraceae). Am. J. Bot. 90, 1446–1454. doi: 10.3732/ajb.90.10.1446
Couvreur, T. L. P., Pirie, M. D., Chatrou, L. W., Saunders, R. M. K., Su, Y. C. F., Richardson, J. E., et al. (2011). Early evolutionary history of the flowering plant family Annonaceae: steady diversification and boreotropical geodispersal. J. Biogeogr. 38, 664–680. doi: 10.1111/j.1365-2699.2010.02434.x
Daniell, H., Lin, C. S., Yu, M., Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134. doi: 10.1186/s13059-016-1004-2
De Candolle, A. P. (1837). Prodromus systematis naturalis regni vegetabilis vol. 6 (Paris: Sumptibus Victoris Masson).
Dong, W., Liu, Y., Li, E., Xu, C., Sun, J., Li, W., et al. (2022). Phylogenomics and biogeography of Catalpa (Bignoniaceae) reveal incomplete lineage sorting and three dispersal events. Mol. Phylogenet. Evol. 166, 107330. doi: 10.1016/j.ympev.2021.107330
Dong, W., Xu, C., Cheng, T., Lin, K., Zhou, S. (2013). Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 5, 989–997. doi: 10.1093/gbe/evt063
Doyle, J. J., Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Drury, D. G. (1970). A fresh approach to the classification of the genus Gnaphalium with particular reference to the species present in New Zealand (Inuleae–Compositae). New Zeal. J. Bot. 8, 222–248. doi: 10.1080/0028825X.1970.10429122
Duan, L., Li, S. J., Su, C., Sirichamorn, Y., Han, L. N., Ye, W., et al. (2021). Phylogenomic framework of the IRLC legumes (Leguminosae subfamily Papilionoideae) and intercontinental biogeography of tribe Wisterieae. Mol. Phylogenet. Evol. 163, 107235. doi: 10.1016/j.ympev.2021.107235
Ebersbach, J., Schnitzler, J., Favre, A., Muellner-Riehl, A. N. (2017). Evolutionary radiations in the species-rich mountain genus Saxifraga L. BMC Evol. Biol. 17, 119. doi: 10.1186/s12862-017-0967-2
Favre, A., Michalak, I., Chen, C., Wang, J., Pringle, J. S., Matuszak, S., et al. (2016). Out-of-Tibet: the spatio-temporal evolution of Gentiana (Gentianaceae). J. Biogeogr. 43, 1967–1978. doi: 10.1111/jbi.12840
Fior, S., Li, M., Oxelman, B., Viola, R., Hodges, S. A., Ometto, L., et al. (2013). Spatiotemporal reconstruction of the Aquilegia rapid radiation through next-generation sequencing of rapidly evolving cpDNA regions. New Phytol. 198, 579–592. doi: 10.1111/nph.12163
Ford, K. A., Ward, J. M., Smissen, R. D., Wagstaff, S. J., Breitwieser, I. (2007). Phylogeny and biogeography of Craspedia (Asteraceae: Gnaphalieae) based on ITS, ETS and psbA-trnH sequence data. Taxon 56, 783–794. doi: 10.2307/25065860
Fu, Z., Jiao, B., Nie, B., Zhang, G., Gao, T., China Phylogeny Consortium (2016). A comprehensive generic-level phylogeny of the sunflower family: Implications for the systematics of Chinese Asteraceae. J. Syst. Evol. 54, 416–437. doi: 10.1111/jse.12216
Galbany-Casals, M., Andrés-Sánchez, S., Garcia-Jacas, N., Susanna, A., Rico, E., Martínez-Ortega, M. M. (2010). How many of Cassini anagrams should there be? Molecular systematics and phylogenetic relationships in the Filago group (Asteraceae, Gnaphalieae), with special focus on the genus Filago. Taxon 59, 1671–1689. doi: 10.1002/tax.596003
Galbany-Casals, M., Blanco-Moreno, J. M., Garcia-Jacas, N., Breitwieser, I., Smissen, R. D. (2011). Genetic variation in Mediterranean Helichrysum italicum (Asteraceae; Gnaphalieae): do disjunct populations of subsp. microphyllum have a common origin? Plant Biol. 13 (4), 678–687. doi: 10.1111/j.1438-8677.2010.00411.x
Galbany-Casals, M., Garcia-Jacas, N., Susanna, A., Sáez, L., Benedí, C. (2004). Phylogenetic relationships in the Mediterranean Helichrysum (Asteraceae, Gnaphalieae) based on nuclear rDNA ITS sequence data. Aust. Syst. Bot. 17, 341–350. doi: 10.1071/SB03031
Galbany-Casals, M., Unwin, M., Garcia-Jacas, N., Smissen, R. D., Susanna, A., Bayer, R. J. (2014). Phylogenetic relationships in Helichrysum (Compositae: Gnaphalieae) and related genera: Incongruence between nuclear and plastid phylogenies, biogeographic and morphological patterns, and implications for generic delimitation. Taxon 63, 608–624. doi: 10.12705/633.8
Gatesy, J., Sloan, D. B., Warren, J. M., Baker, R. H., Simmons, M. P., Springer, M. S. (2019). Partitioned coalescence support reveals biases in species-tree methods and detects gene trees that determine phylogenomic conflicts. Mol. Phylogenet. Evol. 139, 106539. doi: 10.1016/j.ympev.2019.106539
Glenny, D. (1997). A revision of the genus Anaphalioides (Asteraceae: Gnaphalieae). New Zeal. J. Bot. 35, 451–477. doi: 10.1080/0028825X.1987.10410170
Glenny, D., Wagstaff, S. (1997). Evolution and biogeography of New Zealand Anaphalis (Asteraceae, Gnaphalieae) inferred from rDNA sequences. New Zeal. J. Bot. 35, 441–449. doi: 10.1080/0028825X.1987.10410169
Graham, A. (1996). “A contribution to the geologic history of the Compositae,” in Proceedings of the international compositae conference, ke. Eds. Hind, D. J. N., Beentje, H. J. (London: Royal Botanic Gardens, Kew), 123–140.
Grierson, A. J. C. (1972). A new species of Anaphalis (Compositae) from Mexico. Notes R. Bot. Gard. Edinb 31, 389–392.
Güzel, M. E., Coşkunçelebi, K., Kilian, N., Makbul, S., Gültepe, M. (2021). Phylogeny and systematics of the Lactucinae (Asteraceae) focusing on their SW Asian centre of diversity. Plant Syst. Evol. 307, 7. doi: 10.1007/s00606-020-01719-y
Han, C., Ding, R., Zong, X., Zhang, L., Chen, X., Qu, B. (2022). Structural characterization of Platanthera ussuriensis chloroplast genome and comparative analyses with other species of Orchidaceae. BMC Genomics 23, 84. doi: 10.1186/s12864-022-08319-9
Hashim, A. M., Alatawi, A., Altaf, F. M., Qari, S. H., Elhady, M. E., Osman, G. H., et al. (2021). Phylogenetic relationships and DNA barcoding of nine endangered medicinal plant species endemic to Saint Katherine protectorate. Saudi J. Biol. Sci. 28, 1919–1930. doi: 10.1016/j.sjbs.2020.12.043
Hidalgo, O., Garcia-Jacas, N., Garnatje, T., Susanna, A. (2006). Phylogeny of Rhaponticum (Asteraceae, Cardueae-Centaureinae) and related genera inferred from nuclear and chloroplast DNA sequence data: taxonomic and biogeographic implications. Ann. Bot. 97, 705–714. doi: 10.1093/aob/mcl029
Hochuli, P. A. (1978). Palynologische untersuchungen im oligozän und untermiozän der zentralen und westlichen paratethys. Beitr. Paläontol. Österr. 4, 1–132.
Huang, J., Yang, L. Q., Yu, Y., Liu, Y. M., Xie, D. F., Li, J., et al. (2018). Molecular phylogenetics and historical biogeography of the tribe Lilieae (Liliaceae): bi-directional dispersal between biodiversity hotspots in Eurasia. Ann. Bot. 122, 1245–1262. doi: 10.1093/aob/mcy138
Huang, C. H., Zhang, C., Liu, M., Hu, Y., Gao, T., Qi, J., et al. (2016). Multiple polyploidization events across Asteraceae with two nested events in the early history revealed by nuclear phylogenomics. Mol. Biol. Evol. 33, 2820–2835. doi: 10.1093/molbev/msw157
Huelsenbeck, J. P., Nielsen, R., Bollback, J. P. (2003). Stochastic mapping of morphological characters. Syst. Biol. 52, 131–158. doi: 10.1080/10635150390192780
Jabbour, F., Renner, S. S. (2012). A phylogeny of Delphinieae (Ranunculaceae) shows that Aconitum is nested within Delphinium and that Late Miocene transitions to long life cycles in the Himalayas and Southwest China coincide with bursts in diversification. Mol. Phylogenet. Evol. 62, 928–942. doi: 10.1016/j.ympev.2011.12.005
Jara-Arancio, P., Vidal, P. M., Panero, J. L., Marticorena, A., Arancio, G., Arroyo, M. T. K. (2017). Phylogenetic reconstruction of the South American genus Leucheria Lag. (Asteraceae, Nassauvieae) based on nuclear and chloroplast DNA sequences. Plant Syst. Evol. 303, 221–232. doi: 10.1007/s00606-016-1366-7
Jiang, W., Tan, W., Gao, H., Yu, X., Zhang, H., Bian, Y., et al. (2020). Transcriptome and complete chloroplast genome of Glycyrrhiza inflata and comparative analyses with the other two licorice species. Genomics 112, 4179–4188. doi: 10.1016/j.ygeno.2020.07.007
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., de Pamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Joly, S., McLenachan, P. A., Lockhart, P. J. (2009). A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am. Nat. 174, E54–E70. doi: 10.1086/600082
Katoh, K., Rozewicki, J., Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Briefings Bioinf. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kilian, N., Sennikov, A., Wang, Z., Gemeinholzer, B., Zhang, J. (2017). Sub-Paratethyan origin and Middle to Late Miocene principal diversification of the Lactucinae (Compositae: Cichorieae) inferred from molecular phylogenetics, divergence-dating and biogeographic analysis. Taxon 66, 675–703. doi: 10.12705/663.9
Kim, S. C., Chunghee, L., Mejías, J. A. (2007). Phylogenetic analysis of chloroplast DNA matK gene and ITS of nrDNA sequences reveals polyphyly of the genus Sonchus and new relationships among the subtribe SonChinae (Asteraceae: Cichorieae). Mol. Phylogenet. Evol. 44, 578–597. doi: 10.1016/j.ympev.2007.03.014
Kitamura, S. (1937). Compositae japonicae (Pars prima). Mem. Coll. Sci. Kyoto Imp. Univ. Ser. B 13, 1–421.
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kuzmanović, N., Lakušić, D., Frajman, B., Alegro, A., Schönswetter, P. (2017). Phylogenetic relationships in Seslerieae (Poaceae) including resurrection of Psilathera and Sesleriella, two monotypic genera endemic to the Alps. Taxon 66, 1349–1370. doi: 10.12705/666.5
Leister, D. (2003). Chloroplast research in the genomic age. Trends Genet. 19, 47–56. doi: 10.1016/s0168-9525(02)00003-3
Leopold, E. B., MacGinitie, H. D. (1972). “Development and affinities of Tertiary floras in the Rocky Mountains,” in Floristics and palaeofloristics of asia and eastern north america. Ed. Graham, A. (Amsterdam: Elsevier), 147–200.
Li, Y., Dressler, S., Zhang, D., Renner, S. (2009). More Miocene dispersal between Africa and Asia—the case of Bridelia (Phyllanthaceae). Syst. Bot. 34, 521–529. doi: 10.1600/036364409789271263
Li, D., Gao, L., Li, H., Wang, H., Ge, X., Liu, J., et al. (2011). Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. U. S. A. 108, 19641–19646. doi: 10.1073/pnas.1104551108
Liao, M., Shepherd, L. D., Zhang, J., Feng, Y., Mattapha, S., Zhang, L., et al. (2023). Phylogeny, biogeography, and character evolution of the genus Sophora s.l. (Fabaceae, Papilionoideae). Mol. Phylogenet. Evol. 181, 107713. doi: 10.1016/j.ympev.2023.107713
Ling, Y. (1979). “Anaphalis DC,” in Flora reipublicae popularis sinicae, vol. vol. 75. (Beijing: The editorial committee of Flora of ChinaChinese academy of sciencesScience Press), 141–218.
Liu, B. B., Campbell, C. S., Hong, D. Y., Wen, J. (2020). Phylogenetic relationships and chloroplast capture in the Amelanchier-Malacomeles-Peraphyllum clade (Maleae, Rosaceae): Evidence from chloroplast genome and nuclear ribosomal DNA data using genome skimming. Mol. Phylogenet. Evol. 147, 106784. doi: 10.1016/j.ympev.2020.106784
Liu, X., Wang, Z., Shao, W., Ye, Z., Zhang, J. (2017). Phylogenetic and taxonomic status analyses of the Abaso section from multiple nuclear genes and plastid fragments reveal new insights into the North America origin of Populus (Salicaceae). Front. Plant Sci. 7. doi: 10.3389/fpls.2016.02022
Lo, E. Y., Donoghue, M. J. (2012). Expanded phylogenetic and dating analyses of the apples and their relatives (Pyreae, Rosaceae). Mol. Phylogenet. Evol. 63, 230–243. doi: 10.1016/j.ympev.2011.10.005
Lowe, T. M., Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Mahler, D. L., Revell, L. J., Glor, R. E., Losos, J. B. (2010). Ecological opportunity and the rate of morphological evolution in the diversification of Greater Antillean Anoles. Evolution 64, 2731–2745. doi: 10.1111/j.1558-5646.2010.01026.x
Mandel, J. R., Dikow, R. B., Siniscalchi, C. M., Thapa, R., Watson, L. E., Funk, V. A. (2019). A fully resolved backbone phylogeny reveals numerous dispersals and explosive diversifications throughout the history of Asteraceae. Proc. Natl. Acad. Sci. U. S. A. 116, 14083–14088. doi: 10.1073/pnas.1903871116
Meng, Y., Sun, H., Yang, Y. P., Nie, Z. L. (2010). Polyploidy and new chromosome counts in Anaphalis (Asteraceae: Gnaphalieae) from the Qinghai-Tibet Plateau of China. J. Syst. Evol. 48, 58–64. doi: 10.1111/j.1759-6831.2009.00061.x
Meng, Y., Yang, Y. P., Sun, H., Deng, T., Nie, Z. L. (2014). Chromosome numbers, karyotypes, and polyploidy evolution of Anaphalis species (Asteraceae: Gnaphalieae) from the Hengduan Mountains, SW China. Caryologia 67, 238–249. doi: 10.1080/0144235X.2014.974352
Mueller, F. (1889). Records of observations on Sir William MacGregor’s highland plants from New Guinea. Trans. R. Soc Vic. 1, 1–45.
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nie, Z. L., Funk, V. A., Meng, Y., Deng, T., Sun, H., Wen, J. (2016). Recent assembly of the global herbaceous flora: evidence from the paper daisies (Asteraceae: Gnaphalieae). New Phytol. 209, 1795–1806. doi: 10.1111/nph.13740
Nie, Z. L., Funk, V., Sun, H., Deng, T., Meng, Y., Wen, J. (2013). Molecular phylogeny of Anaphalis (Asteraceae, Gnaphalieae) with biogeographic implications in the Northern Hemisphere. J. Plant Res. 126, 17–32. doi: 10.1007/s10265-012-0506-6
O’Reilly, J. E., Donoghue, P. (2018). The efficacy of consensus tree methods for summarizing phylogenetic relationships from a posterior sample of trees estimated from morphological data. Syst. Biol. 67, 354–362. doi: 10.1093/sysbio/syx086
Palazzesi, L., Barreda, V. (2012). Fossil pollen records reveal a late rise of open-habitat ecosystems in Patagonia. Nat. Commun. 3, 1294. doi: 10.1038/ncomms2299
Panero, J. L., Crozier, B. S. (2016). Macroevolutionary dynamics in the early diversification of Asteraceae. Mol. Phylogenet. Evol. 99, 116–132. doi: 10.1016/j.ympev.2016.03.007
Paradis, E., Claude, J., Strimmer, K. (2004). APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Pascual-Díaz, J. P., Garcia, S., Vitales, D. (2021). Plastome diversity and phylogenomic relationships in Asteraceae. Plants 10, 2699. doi: 10.3390/plants10122699
Pirani, A., Moazzeni, H., Zarre, S., Rabeler, R. K., Oxelman, B., Pavlenko, A. V., et al. (2020). Phylogeny of Acanthophyllum s.l. revisited: An update on generic concept and sectional classification. Taxon 69, 500–514. doi: 10.1002/tax.12241
Potter, P. E., Szatmari, P. (2009). Global Miocene tectonics and the modern world. Earth-Sci. Rev. 96, 279–295. doi: 10.1016/j.earscirev.2009.07.003
Pouchon, C., Fernández, A., Nassar, J. M., Boyer, F., Aubert, S., Lavergne, S., et al. (2018). Phylogenomic analysis of the explosive adaptive radiation of the Espeletia complex (Asteraceae) in the tropical Andes. Syst. Biol. 67, 1041–1060. doi: 10.1093/sysbio/syy022
Pouchon, C., Gauthier, J., Pitteloud, C., Claudel, C., Alvarez, N. (2023). Phylogenomic study of Amorphophallus (Alismatales; Araceae): When plastid DNA gene sequences help to resolve the backbone subgeneric delineation. J. Syst. Evol. 61, 64–79. doi: 10.1111/jse.12910
Qian, C., Shi, Y., Liu, Y., Yan, X., Ma, X. (2018). Phylogenetics and dispersal patterns of Brassicaceae around the Qinghai–Tibet Plateau. J. Syst. Evol. 56, 202–217. doi: 10.1111/jse.12312
Rabosky, D. L., Donnellan, S. C., Grundler, M., Lovette, I. J. (2014a). Analysis and visualization of complex macroevolutionary dynamics: an example from Australian scincid lizards. Syst. Biol. 63, 610–627. doi: 10.1093/sysbio/syu025
Rabosky, D. L., Grundler, M., Anderson, C., Title, P., Shi, J. J., Brown, J. W., et al. (2014b). BAMMtools: an R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods Ecol. Evol. 5 (7), 701–707. doi: 10.1111/2041-210X.12199
Rambaut, A. (2014). Molecular evolution, phylogenetics and epidemiology. Available at: http://tree.bio.ed.ac.uk/software/figtree/ (Accessed May 31, 2019).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., Suchard, M. A. (2018). Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Revell, L. J. (2012). Phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210X.2011.00169.x
Rieseberg, L. H., Soltis, D. E. (1991). Phylogenetic consequences of cytoplasmic gene flow in plants. Am. J. Bot. 5, 65–84.
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Smissen, R. D., Bayer, R. J., Bergh, N. G., Breitwieser, I., Freire, S. E., Galbany-Casals, M., et al. (2020). A revised subtribal classification of Gnaphalieae (Asteraceae). Taxon 69, 778–806. doi: 10.1002/tax.12294
Smissen, R. D., Breitwieser, I. (2008). Species relationships and genetic variation in the New Zealand endemic Leucogenes (Asteraceae: Gnaphalieae). New Zeal. J. Bot. 46, 65–76. doi: 10.1080/00288250809509754
Smissen, R. D., Breitwieser, I., Ward, J. M. (2004). Phylogenetic implications of trans-specific chloroplast DNA sequence polymorphism in New Zealand Gnaphalieae (Asteraceae). Plant Syst. Evol. 249, 37–53. doi: 10.1007/s00606-004-0209-0
Smissen, R. D., Galbany-Casals, M., Breitwieser, I. (2011). Ancient allopolyploidy in the everlasting daisies (Asteraceae: Gnaphalieae): Complex relationships among extant clades. Taxon 60, 649–662. doi: 10.1002/tax.603003
Stöver, B. C., Müller, K. F. (2010). TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses. BMC Bioinf. 11, 7. doi: 10.1186/1471-2105-11-7
Sun, B., Wang, H., Lu, M., Tian, X. (2001). Volatile elements of Anaphalis margaritacea, a Chinese medicinal herb. J. Lanzhou Univ. (Nat. Sci.) 37, 66–71.
Tan, W. H., Chai, L. C., Chin, C. F. (2020). Efficacy of DNA barcode internal transcribed spacer 2 (ITS2) in phylogenetic study of Alpinia species from Peninsular Malaysia. Physiol. Mol. Biol. Pla. 26, 1889–1896. doi: 10.1007/s12298-020-00868-1
Thiv, M., Thulin, M., Hjertson, M., Kropf, M., Linder, H. P. (2010). Evidence for a vicariant origin of Macaronesian-Eritreo/Arabian disjunctions in Campylanthus Roth (Plantaginaceae). Mol. Phylogenet. Evol. 54, 607–616. doi: 10.1016/j.ympev.2009.10.009
Thode, V. A., Oliveira, C. T., Loeuille, B., Siniscalchi, C. M., Pirani, J. R. (2021). Comparative analyses of Mikania (Asteraceae: Eupatorieae) plastomes and impact of data partitioning and inference methods on phylogenetic relationships. Sci. Rep. 11, 13267. doi: 10.1038/s41598-021-92727-6
Tian, S., Lu, P., Zhang, Z., Wu, J. Q., Zhang, H., Shen, H. (2021). Chloroplast genome sequence of Chongming lima bean (Phaseolus lunatus L.) and comparative analyses with other legume chloroplast genomes. BMC Genomics 22, 194. doi: 10.1186/s12864-021-07467-8
Tremetsberger, K., Gemeinholzer, B., Zetzsche, H., Blackmore, S., Kilian, N., Talavera, S. (2013). Divergence time estimation in Cichorieae (Asteraceae) using a fossil-calibrated relaxed molecular clock. Org. Divers. Evol. 13, 1–13. doi: 10.1007/s13127-012-0094-2
Twyford, A. D., Ness, R. W. (2017). Strategies for complete plastid genome sequencing. Mol. Ecol. Resour. 17, 858–868. doi: 10.1111/1755-0998.12626
Vicent, M., Gabriel y Galán, J. M., Sessa, E. B. (2017). Phylogenetics and historical biogeography of Lomaridium (Blechnaceae: Polypodiopsida). Taxon 66, 1304–1316. doi: 10.12705/666.3
Wang, Y., Wang, S., Liu, Y., Yuan, Q., Sun, J., Guo, L. (2021). Chloroplast genome variation and phylogenetic relationships of Atractylodes species. BMC Genomics 22, 103. doi: 10.1186/s12864-021-07394-8
Ward, J., Bayer, R., Breitwieser, I., Smissen, R., Galbany, M., Unwin, M. (2009). “Gnaphalieae,” in Systematics, evolution and biogeography of the Compositae. Eds. Funk, V., Susanna, A., Stuessy, T., Bayer, R. (Vienna: IAPT), 539–588.
Wicke, S., Schneeweiss, G. M., de Pamphilis, C. W., Müller, K. F., Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Xing, Y., Ree, R. H. (2017). Uplift-driven diversification in the Hengduan Mountains, a temperate biodiversity hotspot. Proc. Natl. Acad. Sci. U. S. A. 114, E3444–E3451. doi: 10.1073/pnas.1616063114
Xiong, A. S., Peng, R. H., Zhuang, J., Gao, F., Zhu, B., Fu, X. Y., et al. (2009). Gene duplication, transfer, and evolution in the chloroplast genome. Biotechnol. Adv. 27, 340–347. doi: 10.1016/j.bioteChadv.2009.01.012
Xu, X., Liu, D., Zhu, S., Wang, Z., Wei, Z., Liu, Q. (2023a). Phylogeny of Trigonotis in China—with a special reference to its nutlet morphology and plastid genome. Plant Divers. 45, 409–421. doi: 10.1016/j.pld.2023.03.004
Xu, X., Wei, Z., Sun, J., Zhao, Q., Lu, Y., Wang, Z., et al. (2023b). Phylogeny of Leontopodium (Asteraceae) in China—with a reference to plastid genome and nuclear ribosomal DNA. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1163065
Xu, X., Wei, Z., Zhang, T., Zhu, S. (2021). Achene micro-morphology of anaphalis DC. (Gnaphalieae-asteraceae) from China. Pak. J. Bot. 53, 1883–1892. doi: 10.30848/PJB2021-5(10
Xu, B., Zeng, X. M., Gao, X. F., Jin, D. P., Zhang, L. B. (2017). ITS non-concerted evolution and rampant hybridization in the legume genus Lespedeza (Fabaceae). Sci. Rep. 7, 40057. doi: 10.1038/srep40057
Yang, H. M., Zhang, Y. X., Yang, J. B., Li, D. Z. (2013). The monophyly of Chimonocalamus and conflicting gene trees in Arundinarieae (Poaceae: Bambusoideae) inferred from four plastid and two nuclear markers. Mol. Phylogenet. Evol. 68, 340–356. doi: 10.1016/j.ympev.2013.04.002
Yu, Y., Blair, C., He, X. (2020). RASP 4: Ancestral state reconstruction tool for multiple genes and characters. Mol. Biol. Evol. 37, 604–406. doi: 10.1093/molbev/msz257
Yu, X. Q., Maki, M., Drew, B. T., Paton, A. J., Li, H. W., Zhao, J. L., et al. (2014). Phylogeny and historical biogeography of Isodon (Lamiaceae): rapid radiation in south-west China and Miocene overland dispersal into Africa. Mol. Phylogenet. Evol. 77, 183–194. doi: 10.1016/j.ympev.2014.04.017
Yuan, Y. P., Liu, Z., Jin, D. Q. (2004). Study of curing tracheitis by Anaphalis lactea Maxim. on animals. Chin. J. Comp. Med. 14, 172–175.
Zachos, J. C., Dickens, G. R., Zeebe, R. E. (2008). An early Cenozoic perspective on greenhouse warming and carbon-cycle dynamics. Nature 451, 279–283. doi: 10.1038/nature06588
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, C., Huang, C. H., Liu, M., Hu, Y., Panero, J. L., Luebert, F., et al. (2021a). Phylotranscriptomic insights into Asteraceae diversity, polyploidy, and morphological innovation. J. Integr. Plant Biol. 63, 1273–1293. doi: 10.1111/jipb.13078
Zhang, X. F., Landis, J. B., Wang, H. X., Zhu, Z. X., Wang, H. F. (2021b). Comparative analysis of chloroplast genome structure and molecular dating in Myrtales. BMC Plant Biol. 21, 219. doi: 10.1186/s12870-021-02985-9
Zhao, H., Chen, F., Chen, S., Wu, G., Guo, W. (2010). ). Molecular phylogeny of Chrysanthemum, Ajania and its allies (Anthemideae, Asteraceae) as inferred from nuclear ribosomal ITS and chloroplast trnL-F IGS sequences. Plant Syst. Evol. 284, 153–169. doi: 10.1007/s00606-009-0242-0
Zhou, X., Zhang, L. (2017). Nuclear and plastid phylogenies suggest ancient intersubgeneric hybridization in the fern genus Pyrrosia (Polypodiaceae), with a classification of Pyrrosia based on molecular and non-molecular evidence. Taxon 66, 1065–1084. doi: 10.12705/665.5
Keywords: Anaphalis, Asteraceae, biogeography, Gnaphalieae, molecular phylogenetic
Citation: Xu X-M, Xu H, Yang Z, Wei Z, Gu J-Y, Liu D-H, Liu Q-R and Zhu S-X (2024) Phylogeny, biogeography, and character evolution of Anaphalis (Gnaphalieae, Asteraceae). Front. Plant Sci. 15:1336229. doi: 10.3389/fpls.2024.1336229
Received: 10 November 2023; Accepted: 24 January 2024;
Published: 07 February 2024.
Edited by:
Gang Yao, South China Agricultural University, ChinaReviewed by:
Zelong Nie, Jishou University, ChinaRob Smissen, Manaaki Whenua Landcare Research, New Zealand
Copyright © 2024 Xu, Xu, Yang, Wei, Gu, Liu, Liu and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shi-Xin Zhu, c3h6aHVAenp1LmVkdS5jbg==