 Yingfeng Niu1
Yingfeng Niu1 Chengwen Gao
Chengwen Gao- 1Yunnan Institute of Tropical Crops, National Key Laboratory for Biological Breeding of Tropical Crops, Yunnan Key Laboratory of Sustainable Utilization Research on Rubber Tree, Xishuangbanna, China
- 2Medical Research Center, The Affiliated Hospital of Qingdao University, Qingdao, China
Among the Hevea species, rubber tree (Hevea brasiliensis) is the most important source of natural rubber. In previous studies, we sequenced the complete nuclear and chloroplast genomes of Hevea species, providing an invaluable resource for studying their phylogeny, disease resistance, and breeding. However, given that plant mitochondrial genomes are more complex and more difficult to assemble than that of the other organelles, little is known about their mitochondrial genome, which limits the comprehensive understanding of Hevea genomic evolution. In this study, we sequenced and assembled the mitochondrial genomes of four Hevea species. The four mitochondrial genomes had consistent GC contents, codon usages and AT skews. However, there were significant differences in the genome lengths and sequence repeats. Specifically, the circular mitochondrial genomes of the four Hevea species ranged from 935,732 to 1,402,206 bp, with 34–35 unique protein-coding genes, 35–38 tRNA genes, and 6–13 rRNA genes. In addition, there were 17,294–46,552 bp intergenomic transfer fragments between the chloroplast and mitochondrial genomes, consisting of eight intact genes (psaA, rrn16S, tRNA-Val, rrn5S, rrn4.5S, tRNA-Arg, tRNA-Asp, and tRNA-Asn), intergenic spacer regions and partial gene sequences. The evolutionary position of Hevea species, crucial for understanding its adaptive strategies and relation to other species, was verified by phylogenetic analysis based on the protein-coding genes in the mitochondrial genomes of 21 Malpighiales species. The findings from this study not only provide valuable insights into the structure and evolution of the Hevea mitochondrial genome but also lay the foundation for further molecular, evolutionary studies, and genomic breeding studies on rubber tree and other Hevea species, thereby potentially informing conservation and utilization strategies.
Introduction
Among the species in the genus Hevea, rubber tree (Hevea brasiliensis) is the most important source of natural rubber, accounting for over 98% of the global total natural rubber production (Cheng et al., 2023). The genus Hevea comprises ten species, including H. spruceana, H. benthamiana, H. pauciflora, H. camargoana, H. brasiliensis, H. guianensis, H. microphylla, H. rigidifolia, H. nitida, and H. camporum, exclusively distributed in the Amazon basin (Priyadarshan and Goncalves, 2003). There is no biological barrier on reproduction among these species, making it possible for inter-species crossing by artificial pollination (Priyadarshan and Clement-Demange, 2004). As a result, Hevea species are considered a species complex, forming a gene pool for breeding, especially in identifying and introducing genes encoding resistance against leaf diseases (Priyadarshan, 2003).
Mitochondria are essential for several metabolic processes, including adenosine triphosphate (ATP) synthesis and cellular respiration (Meyer et al., 2019). With the popularity and rapid development of sequencing technology, increased number of plant mitochondrial genomes has been assembled. However, out of the 12,029 terrestrial plant organelle genomes assembled to date (April 2023) based on the GenBank Organelle Genome Resources, only 477 are mitochondrial genomes and the rest are chloroplast genomes. This suggests that the mitochondrial genome is more complex and more difficult to assemble than that of the other organelles (Ma et al., 2022).
Mitochondrial genomes vary widely among plant species in terms of their length, gene content, and gene order (Richardson et al., 2013; Bi et al., 2020; Lai et al., 2022). In addition, the evolution of mitochondrial genomes has been characterized by many intergenomic sequence transfers and structural rearrangements (Wu et al.; Allen, 2015; Gao et al., 2023). At the same time, mitochondrial protein-coding genes are conserved, hence a potential for addressing unresolved phylogenetic questions (Tian et al., 2006; Hao et al., 2022). With the continuous development of sequencing technology and assembly methods (He et al., 2022), more and more plant mitochondrial genomes have been sequenced. In previous studies, we sequenced the complete nuclear genome of rubber tree (Liu et al., 2020). The chloroplast genomes of four species of the genus Hevea were also sequenced (Tangphatsornruang et al., 2011; Feng et al., 2020; Niu et al., 2020a, b; Hu et al., 2022). But only one mitochondrial genome of Hevea species has been released, which needs further exploration (Shearman et al., 2014). The study on the mitochondrial genome of Hevea species will lay a foundation for further evolutionary and genomic breeding studies on rubber tree. In this study, we newly determined four Hevea mitochondrial genome sequences and performed comparative genomic analysis. This study aimed to: (1) perform a comparative analysis of the mitochondrial genome features of Hevea species; (2) identify the intergenomic sequence transfers between mitochondrial and chloroplast genomes in Hevea species; (3) reconstruct the Hevea species phylogeny using the mitochondrial gene sequences. This study will provide a better understanding of the interspecific differences in the genus Hevea and will be valuable for further research on genomic breeding studies on rubber tree.
Materials and methods
Samples collection and DNA sequencing
We collected fresh leaf samples of four Hevea species (H. benthamiana, H. camargoana, H. pauciflora, and H. spruceana) from the rubber tree germplasm resource nursery of the Chinese Academy of Tropical Agriculture Science (N 19°34′31.53″and E 109°31′17.97″). The total genomic DNA from the leaves was extracted using the CTAB method (Li et al., 2013). The genomic DNA was used for library construction. For next-generation sequencing, libraries with insert sizes of 350 bp were constructed and sequenced using the Illumina Novaseq6000 platform (Illumina, USA). For third-generation sequencing, 15 Kb fragment libraries were constructed by the PacBio RS II platform (Pacific Biosciences, Menlo Park, CA, USA). A total of 10.2–16.6 GB of raw sequencing data, of which the third-generation sequencing raw data was 5.5–10.1 GB (Supplementary Figure S1), and a mitochondrial genome depth of coverage of 138.5–489.8× was obtained from the four Hevea species.
Mitochondrial genome assembly and annotation
The short reads were filtered using Trimmomatic v0.38 (Bolger et al., 2014), and their de novo assembly was performed using SPAdes v3.5.0 with different K-mer parameters (Bankevich et al., 2012). GSAT software (He et al., 2022) was used to calculate the mitochondrial genome graph-based framework, the genomic structure maps of H. camargoana and H. spruceana mitochondria shows complex relationships between genomic sequences (Supplementary Figure S2). The obtained PacBio reads were de novo assembled using canu v2.2 and PAMT (Koren et al., 2017; Bi et al., 2024) in hifi mode, with a genome size set to 2M, and default parameters for the rest, and the draft assemblies were polished by Pilon (Walker et al., 2014). During the running of Pilon, only paired-mapped reads were used to correct, which can eliminate nuclear mitochondrial DNA segments (NUMT) as much as possible. A total of 14–110 contigs were obtained by canu assembler, the longest was 309,883 bp in length, and the average length was 53,687 bp. The circularization process consists of the following steps: A. The establishment of coherent linkages among segments, B. Extensive and uniform coverage at the sequence assembly junctions, C. The wholeness of gene segments along with consistent and meaningful coding sequences, and D. An appropriate correlation between the repetition rates in repetitive segments and the depth of sequencing, ultimately leading to the effective and comprehensive assembly of the entire genome. The assembly process was summarized in Supplementary Figure S3. At the same time, we calculated base depth and corrected the mitochondrial sequence using genotypes that matched the sequencing depth of mitochondrial sequence. The IGV software to analyze the reasonableness of sequencing read coverage and to determine if there were complete third-generation sequencing reads spanning the low-depth regions. For regions with low sequencing coverage or complex structures, validation was performed with Sanger sequencing to ensure the circular genomic sequence was accurate and complete. Specifically, the Primer Premier software was used to design the primers for each low sequencing coverage or complex structures region. The genomic DNA was used as the template to amplify the target DNA fragment. After purification and identification, PCR amplification and sequencing was performed. Subsequently, the mitochondrial genome was annotated by GeSeq (Tillich et al., 2017) and ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/) using the Hevea brasiliensis (AP014526) mitochondrial genome as the reference genome. After GeSeq annotation, the BLASTN and BLASTP alignment tools were used to identify and compare the mitochondrial sequences (Zhang et al., 2000), and the annotation results were manually modified. In addition, the tRNAs were annotated using tRNAscan-SE 2.0 (Lowe and Chan, 2016) and ARWEN (Laslett and Canback, 2008). Finally, the circular mitochondrial genome of Hevea species was mapped using Proksee (https://proksee.ca/).
Codon usage and repetitive sequences analysis
Relative synonymous codon usage values were analyzed using MEGA v7.0.26 (Kumar et al., 2016). Codon usage heatmap was plotted using the R package pheatmap v1.0.12. Next, the AT and GC skews were calculated using the formulas: AT skew = (A − T)/(A + T), and GC skew = (G − C)/(G + C), respectively. In addition, the long repeats in the mitochondrial genome were analyzed using REPuter (Kurtz et al., 2001). Finally, the simple sequence repeats (SSRs) were identified using the MISA v2.1 software (Beier et al., 2017), with 10, 6, 5, 5, 5, and 5 as the minimum numbers of repeats for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively. Tandem repeats were identified using the program Tandem Repeats Finder (TRF) (Benson, 1999).
We selected the H. benthamiana mitochondrial genome, which has the highest similarity to the rubber tree mitochondrial genome, for paired repetitive sequence detection using BLASTN, and to identify potential recombinations from these repetitive sequences. We selected two of them for PCR validation. The primers at the ends of repeats were designed by Primer Premier3 v4.1 (https://bioinfo.ut.ee/primer3-0.4.0/) (Supplementary Table S12). The PCR amplification system was 40 µL in total, consisting of 1 µL of template gDNA, 2 µL of forward and reserve primer, 20 µL of 2× Rapid Taq Master Mix, and 15 µL of ddH2O. The PCR amplification program was 94 °C denaturation for 3 min, 35 cycles of 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 60 s, and a 72 °C final extension for 5 min.
Intergenomic sequence transfer analysis
The mitochondrial genomes of the four Hevea species were searched by BLASTN against the chloroplast genomes (Zhang et al., 2000), using parameters “-evalue 1e-6 -outfmt 6”, and length > 100 bp to identify transfer fragments between the two genomes. The Intergenomic sequence transfer fragments between chloroplast and mitochondrial genomes were then mapped using Circos v0.69 software (Krzywinski et al., 2009).
Identification of RNA editing sites
The Deepred-mt (Edera et al., 2021) was used for the prediction of RNA editing sites of the mitochondrial genomes. It predicts C-to-U editing events based on the 40 nucleotides flanking a given cytidine.
Phylogenetic analysis
Phylogenetic analysis was performed for 21 Malpighiales species using Passiflora edulis as the outgroup. The mitochondrial genomes of the 21 Malpighiales species, including the four Hevea species were downloaded from the Genbank. Next, the mitochondrial protein-coding genes (atp1, atp4, atp6, atp8, atp9, ccmB, ccmC, ccmFc, ccmFn, cox1, cox2, cox3, cob, matR, nad7, nad9, nd1, nd2, nd3, nd4, nd4L, nd5, nd6, prl10, prl16, rps1, rps12, rps3, and rps4) common to all the species were extracted to construct a phylogenetic tree. The mitochondrial gene sequences of Malpighiales species were aligned using Muscle v.3.8.31 (Edgar, 2004). A phylogenetic tree was built with a maximum likelihood (ML) approach using RAxML v8.1.5 (Stamatakis, 2006) based on the best-fit substitution model (GTR+I+G) determined by jModelTest v2.1.7 (Posada, 2008). Branch support was assessed using bootstrap resampling with 1000 replicates.
Results
General features of the Hevea mitochondrial genomes
Raw mitochondrial genome sequence data were obtained from H. benthamiana (OR663908), H. camargoana (OR663909), H. pauciflora (OR663910), and H. spruceana (OR663911) and were successfully assembled as a single circular genome. The sizes of the four newly sequenced Hevea mitochondrial genomes ranged from 935,732 to 1,402,206 bp (Figure 1; Table 1), with GC contents of 44.17–44.24%. Based on the comparative analysis, the four Hevea mitochondrial genomes had 34–35 unique protein-coding genes, 35–38 tRNA genes, and 6–13 rRNA genes (Supplementary Tables S1–S4). Notably, the number of H. spruceana mitochondrial genes was less than that of the other three species, mainly because the copy number of some genes of H. spruceana (ccmB, cob, and matR et al.) was only a single copy (Supplementary Figure S4; Supplementary Table S5). The four newly sequenced Hevea mitochondrial genomes have been submitted to the GenBank with accession numbers OR663908–OR663911.
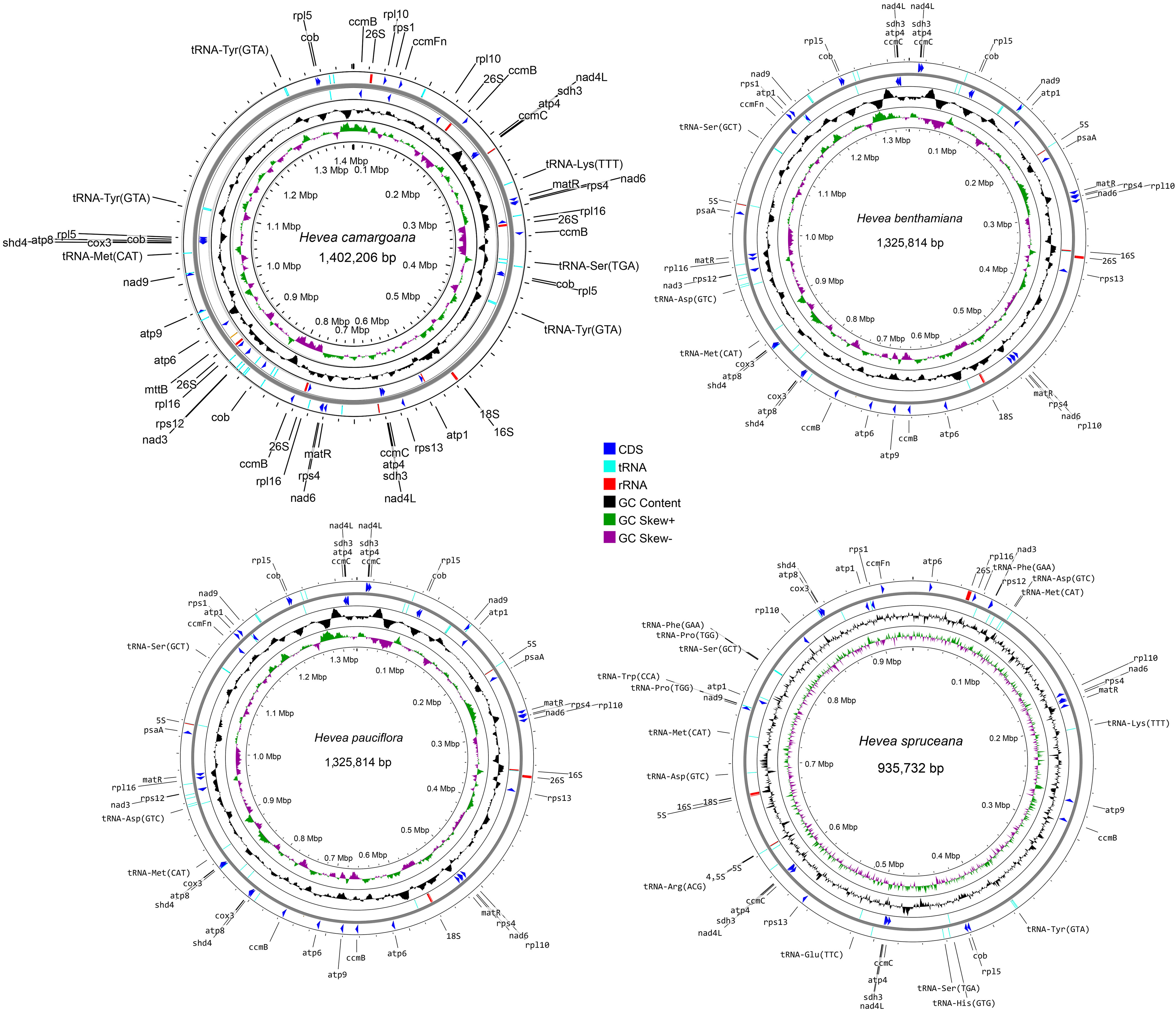
Figure 1 The mitochondrial genomes of the four Hevea species. The gene map, GC content, and GC skew are presented from the outside to the inside of the mitochondrial genome map, respectively.
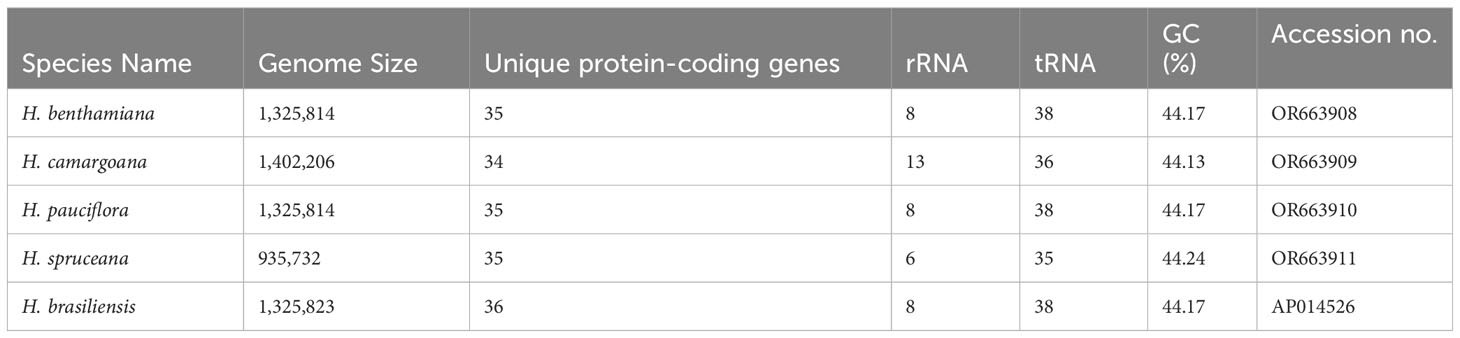
Table 1 Features of Hevea mitochondrial genomes.
Intergenomic sequence transfers between mitochondrial and chloroplast genomes
The length of the Hevea mitochondrial genomes was approximately 5.8–8.7 times that of chloroplast genomes (MT333859, MN781109, NC_059798, and NC_059799). There were 17,294–46,552 bp long common sequence fragments between the chloroplast and mitochondrial genomes in the four Hevea species, including intergenic and gene regions (Figure 2; Supplementary Table S6), which accounted for 1.9–3.5% of the mitochondrial genome and 10.7–28.9% of the chloroplast genome. The transfer sequences from the chloroplast to the mitochondrial genomes in the four Hevea species contained eight intact genes (psaA, rrn16S, tRNA-Val, rrn5S, rrn4.5S, tRNA-Arg, tRNA-Asp, and tRNA-Asn), intergenic spacers regions and partial gene sequences (psaB, ycf3, tRNA-Ile, ycf1, rrn23S, ycf1, tRNA-Ala, ycf2, atpE, ndhF, and rps16). Most of these transfer sequences are located in the IR region (50/79) of the chloroplast genomes (Supplementary Table S6).
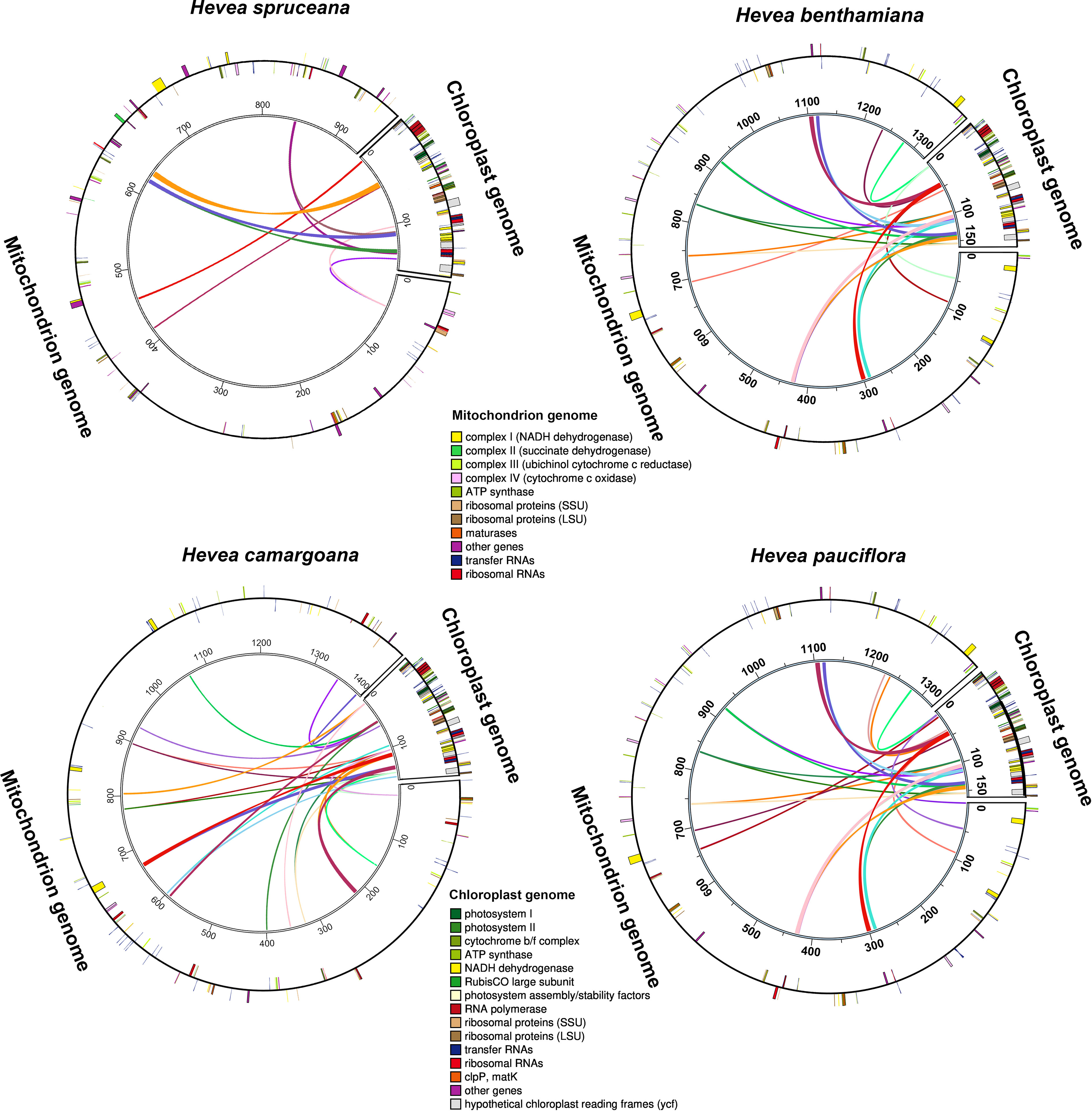
Figure 2 Schematic diagram of sequence transfer between mitochondrial and chloroplast genomes in four Hevea species. Colored lines within the circles represents intergenomic transfer sequences; genes shown outside and inside the circle were transcribed in a counterclockwise and clockwise manner, respectively.
Protein-coding genes and codon usages
The length of most Hevea mitochondrial protein-coding genes (PCGs) is conserved (Figure 3A; Supplementary Tables S1–S4). In addition, the majority strand PCGs had a negative GC and positive AT skews, while the minority strand PCGs had positive GC and negative AT skews. Notably, the PCGs GC contents and AT skews were consistent across the Hevea mitochondrial genomes (Figure 3B; Supplementary Table S7).
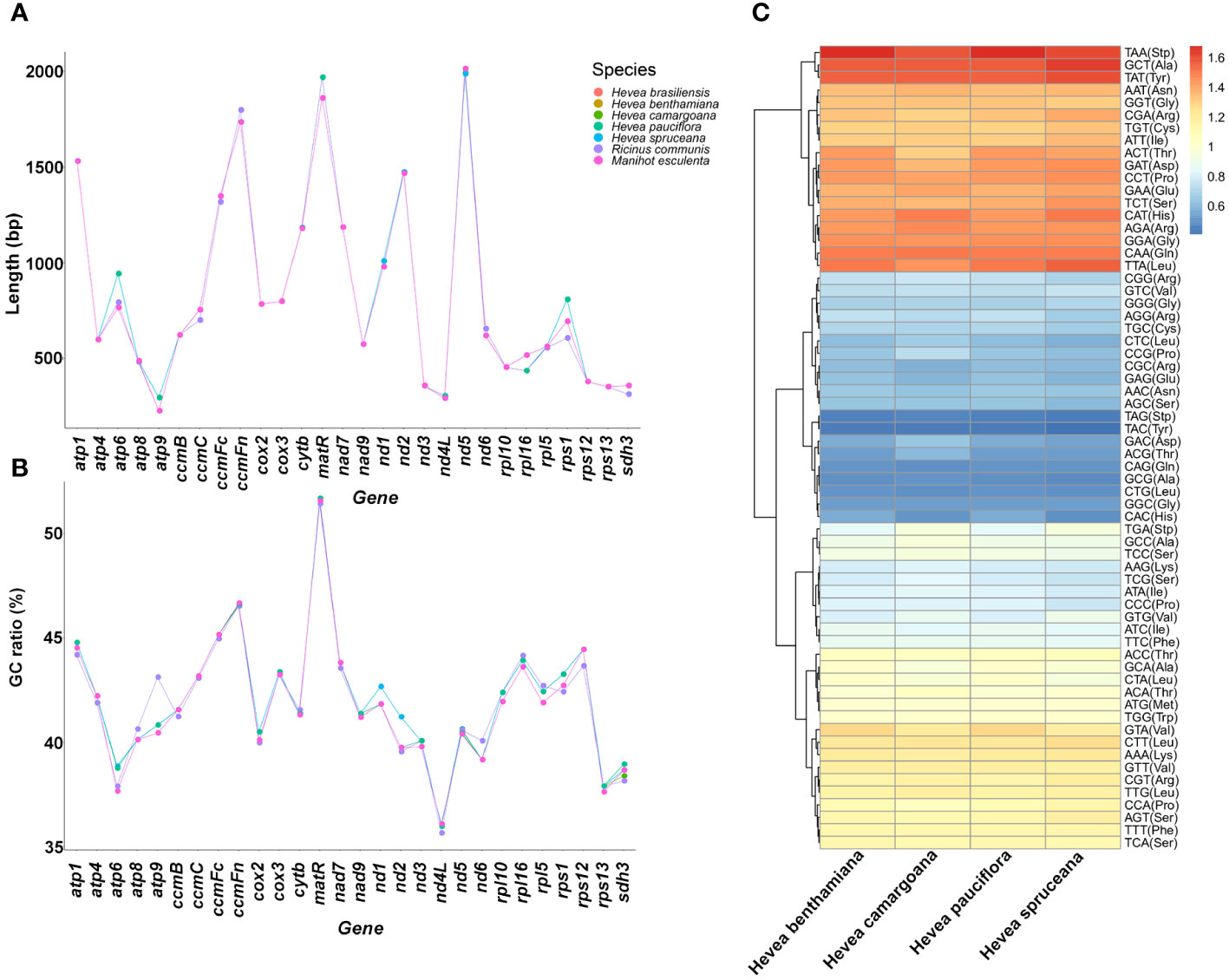
Figure 3 Protein-coding genes and the relative synonymous codon occurrence in Hevea mitochondrial genomes. (A) The length of the protein-coding genes. (B) The GC ratio of the protein-coding genes. (C) The relative synonymous codon occurrence in the protein-coding genes.
The relative synonymous codon usage analysis of the mitochondrial genome revealed that among the four Hevea species, there were 64 codons encoding 20 amino acids (Figure 3C; Supplementary Figure S5; Supplementary Table S8). The total number of codons ranged from 12,292 to 18,395, with TTT as the most frequent (456–661 instances) codon, and TGC as the least (55–87 instances) among the PCGs in the Hevea mitochondrial genomes. The codon usage pattern was largely consistent in the four Hevea mitochondrial genomes.
Analysis of repeats in the Hevea mitochondrial genomes
Analysis of the Hevea mitochondrial genome repeats revealed 453–677 long repeats with a total length of 322,471–555,902 bp, accounting for 23.0–59.4% of the mitochondrial genomes (Supplementary Table S9). Most of the long repeats were 30 to 50 bp in length, and the longest was 116,525 bp in H. benthamiana.
Tandem repeats, consisting of core repeat units are repeated many times in a tandem pattern. We detected a total of 61–93 tandem repeats in the mitochondrial genome of Hevea mitochondrial genomes with lengths between 5 and 76 bp (Supplementary Table S10).
In addition, 98–133 SSRs were identified in Hevea mitochondrial genomes, with a total size of 1,306 to 2,030 bp (Supplementary Table S11). Mononucleotide repeats were the most common, followed by di-, tri- and hexa- repeats, respectively. Most SSRs were located in the intergenic or intronic regions, except for 1–2 SSRs that were in the nad1 protein-coding region.
There were two repeat sequences that could potentially mediate the homologous recombination in H. benthamiana mitochondrial genome. We performed PCR amplification and Sanger sequencing to verify the presence of the minor conformation. The scheme of the primer design was shown in Supplementary Figure S6A and Supplementary Table S12. Both sets of specific primers were successful in amplifying fragments of the repeat sequence fragments. Upon interchanging the reverse primers, we observed that they retained the ability to amplify these repeat sequence fragments, as shown in Supplementary Figure S6B. Based on these validation outcomes, we can infer the potential types of homologous recombination in the H. benthamiana mitochondrial genome, illustrated in Supplementary Figure S6A. Both conformations A and B are loop structures, each spanning a total length of 1,325,823 base pairs, and are differentiated by the orientation of their intermediate sequences, which are either forward or reverse. Due to the presence of direct repeats, conformation C consists of loops that form a two-chromosomal structure.
Identification of RNA editing sites
We predicted a total of 436–443 potential C-to-U RNA editing sites in Hevea mitochondrial PCGs (Supplementary Table S13; Supplementary Figure S7). The predicted RNA editing sites of the mitochondrial genes of the four Hevea species are shown in Supplementary Figure S6. Among these mitochondrial PCGs, we identified 37 RNA editing sites in nad4 and 35 for ccmB gene, which were the most frequent among these PCGs. In contrast, the gene ycf3 had only 0–1 C-to-U editing sites in Hevea mitochondrial genes.
Phylogenetic analysis based on mitochondrial genome sequences
Phylogenetic analysis was performed based on 29 common mitochondrial genes, from 21 Malpighiales species. Phylogenetic analysis revealed that most branches of the phylogenetic tree had high ML bootstraps values. Four well-supported clades (C1, C2, C3, and C4 Clade) were recovered within Malpighiale. C1 Clade included family Euphorbiaceae. C2 Clade included families Rhizophoraceae and Erythroxylaceae. The Calophyllaceae and Clusiaceae families formed C3 Clade. C4 Clade included family Salicaceae. Salicaceae species formed a monophyletic clade and diverged earlier. The five Hevea species (Euphorbiaceae) were clustered together (Figure 4).
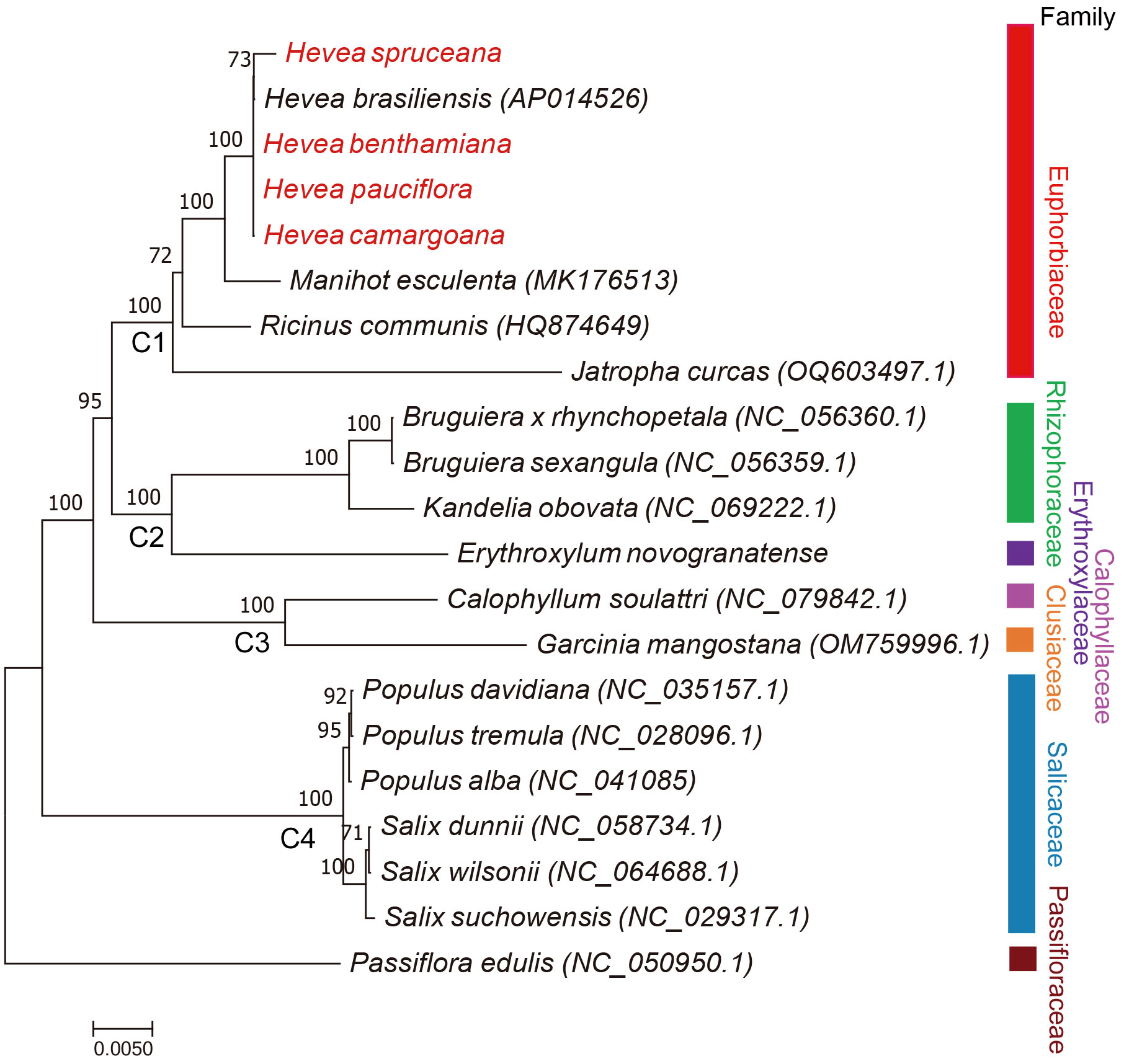
Figure 4 ML phylogenetic tree of Malpighiales species based on the common mitochondrial protein-coding genes. The numbers indicate the maximum likelihood bootstrap values (BS = 100% were not showed).
Discussion
The mitochondrial genomes of terrestrial plants have undergone dramatic structural changes that have complicated their genomic composition, making plant mitochondrial genome research relatively challenging (Morley and Nielsen, 2017; Kozik et al., 2019; Wu et al., 2022). Notably, the mitochondrial genome size varies considerably among different plant species (Wynn and Christensen, 2019; Jackman et al., 2020), with the mitochondrial genomes of Hevea species being nearly twice the Euphorbiaceae species (Rivarola et al., 2011; Tao et al., 2019). In this study, we sequenced and assembled the mitochondrial genomes of four Hevea species. Their mitochondrial genomes ranged from 935,732 bp (H. spruceana) to 1,402,206 bp (H. camargoana). However, their GC content was the same. Similar to the mitochondrial genomes of most plants, most sequences in the Hevea mitochondrial genomes are non-genes, with gene sequences ranging from 4.87% (H. spruceana) to 5.23% (H. camargoana) of the total mitochondrial genome, possibly due to the gradual increase in sequence repeats during evolution (Ma et al., 2022). In addition, most PCGs in the Hevea mitochondrial genomes use the canonical ATG as the start codon, while nad1 and rps4 genes use ACG as the start codon, a phenomenon also reported in other studies and attributed to RNA editing modifications (Sloan et al., 2018; Xu et al., 2021).
Repetitive sequences, widely found in mitochondrial genomes, are important source of information for developing markers for evolutionary and population analysis (Nishikawa et al., 2005; Liu et al., 2015; Xiong et al., 2022). Herein, there were relatively large differences in the repetitive sequences in the Hevea mitochondrial genomes. Therefore, the four newly sequenced Hevea mitochondrial genomes provide new resources on the structure and function of the Hevea mitochondrial genomes. The Hevea mitochondrial genomes contains abundant repetitive sequences, which may indicate frequent intermolecular recombination in the Hevea mitochondrial genomes. The repetitive sequences identified in this study could mediate the formation of two conformations in the H. benthamiana mitochondrial genome (Supplementary Figure S5). The specific DNA repair of mitochondrial genome may be related to these phenomena (Liu et al., 2023; Yang et al., 2023). The results of this study support the existence of multiple conformations in plant mitochondrial genomes. Intergenomic sequence transfers from chloroplast to mitochondrial genome is common during terrestrial plant evolution (Gui et al., 2016; Nguyen et al., 2020). Consistent with the findings in other genera (Niu et al., 2021, 2022), the Hevea chloroplast sequences were inserted into the mitochondrial genome at different locations. The extensive rearrangement observed within the mitochondrial genome may be attributable to intergenomic sequence transfers. This hypothesis is supported by the observation that segments of the chloroplast genome which have migrated into the mitochondria exhibit a high degree of alignment with the original chloroplast genome sequences. Furthermore, the insertion locations of these segments appear to be randomly distributed. In the Hevea mitochondrial genome, the cumulative length of these transferred fragments spans from 17,294 to 46,552 bp, a range significantly exceeding that of transfer fragments identified in other genera (Gao et al., 2023). The transferred fragments and repetitive sequences are likely responsible for the size difference among the Hevea mitochondrial genomes. The fragments transferred from the Hevea chloroplast to the mitochondrial genome included four tRNA, three rRNA, and psaA, tRNA genes, which is also a common phenomenon in angiosperms (Bi et al., 2016; Cheng et al., 2021). In addition, these transferred fragments contain partial chloroplast gene sequences that have no function in the mitochondrial genome, which may be due to the loss of function of some genes during sequence transfer. This phenomenon suggests that the sequence transfer was from the chloroplast into the mitochondrial genome (Hao and Palmer, 2009; Smith, 2011).
The genetic relationship among Hevea species has been revealed through the analysis of nuclear genes, internal ribosome transcription spacer and chloroplast genes (Cheng et al., 2009; Tangphatsornruang et al., 2011; Zhai et al., 2014). However, research on the reconstruction plant phylogeny of mitochondrial genome sequences is scarce. In this study, a phylogenetic tree was constructed using PCGs in the mitochondrial genome to explore the evolutionary relationships among Malpighiales species. The phylogenetic topology of the mitochondrial phylogenetic tree was generally consistent with previous studies (Xi et al., 2012; Endress et al., 2013). The findings from this study complement the genetic knowledge on genus Hevea, and provide a new perspective to study the evolution of Hevea species.
Conclusions
The Hevea mitochondrial genomes have consistent GC contents, codon occurrence and AT skews. However, there are significant differences in their lengths and sequence repeats. In addition, there are 17,294–46,552 bp intergenomic transfer fragments between the Hevea mitochondrial and chloroplast genomes. The evolutionary position of Hevea species was verified by phylogenetic analysis based on PCGs of the mitochondrial genome of 21 Malpighiales species. The findings from this study provide valuable genetic resources for further studies on Hevea species.
Collection of plant material
We have permission to collect Hevea species. The collection of plant material comply with relevant institutional, national, and international guidelines and legislation.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GenBank on the NCBI website at https://www.ncbi.nlm.nih.gov, using the accession numbers OQ658721–OQ658724, and accession numbers OR663908–OR663911, the raw sequencing data have been deposited at the NCBI Sequence Read Archive (SRA) under accession PRJNA946487.
Author contributions
YN and JL conceived the study, wrote and revised the manuscript. CG performed the data analyses and drafted the manuscript. All authors read and approved the final manuscript.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China (No.32060347), Yunnan Key Laboratory of Sustainable Utilization Research on Rubber Tree (202405AG340001), Key Project of Yunnan Basic Research Program (202401AS070083), Technology Innovation Talents Project of Yunnan Province (No. 202305AD160023), Natural Science Foundation of Shandong Province (No. ZR2020QH321), and Sci-tech Innovation System Construction for Tropical Crops Grant of Yunnan Province (No. RF2024).
Acknowledgments
We are grateful to Dr. Yanshi for his assistance in sample collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1234643/full#supplementary-material
References
Allen, J. F. (2015). Why chloroplasts and mitochondria retain their own genomes and genetic systems: Colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. U.S.A. 112, 10231–10238. doi: 10.1073/pnas.1500012112
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Beier, S., Thiel, T., Munch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bi, C. W., Lu, N., Xu, Y. Q., He, C. P., Lu, Z. H. (2020). Characterization and analysis of the mitochondrial genome of common bean (Phaseolus vulgaris) by comparative genomic approaches. Int. J. Mol. Sci. 21. doi: 10.3390/ijms21113778
Bi, C. W., Paterson, A. H., Wang, X. L., Xu, Y. Q., Wu, D. Y., Qu, Y. S., et al. (2016). Analysis of the complete mitochondrial genome sequence of the diploid cotton gossypium raimondii by comparative genomics approaches. BioMed. Res. Int. doi: 10.1155/2016/5040598
Bi, C., Shen, F., Han, F., Qu, Y., Hou, J., Xu, K., et al. (2024). PMAT: an efficient plant mitogenome assembly toolkit using low-coverage HiFi sequencing data. Horticult. Res. 11. doi: 10.1093/hr/uhae023
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Cheng, Y., He, X. X., Priyadarshani, S., Wang, Y., Ye, L., Shi, C., et al. (2021). Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genomics 22. doi: 10.1186/s12864-021-07490-9
Cheng, H., Song, X. M., Hu, Y. S., Wu, T. K., Yang, Q. H., An, Z. W., et al. (2023). Chromosome-level wild Hevea brasiliensis genome provides new tools for genomic-assisted breeding and valuable loci to elevate rubber yield. Plant Biotechnol. J. doi: 10.1111/pbi.14018
Cheng, H., Zhang, Y., An, Z., Huang, H. (2009). Sequence Analysis of rDNA ITS from Several Hevea plants. Chin. J. Trop. Crops 30, 1320–1324.
Edera, A. A., Small, I., Milone, D. H., Sanchez-Puerta, M. V. (2021). Deepred-Mt: Deep representation learning for predicting C-to-U RNA editing in plant mitochondria. Comput. Biol. Med. 136. doi: 10.1016/j.compbiomed.2021.104682
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Endress, P. K., Davis, C. C., Matthews, M. L. (2013). Advances in the floral structural characterization of the major subclades of Malpighiales, one of the largest orders of flowering plants. Ann. Bot. 111, 969–985. doi: 10.1093/aob/mct056
Feng, L. Y., Liu, J., Gao, C. W., Wu, H. B., Li, G. H., Gao, L. Z. (2020). Higher genomic variation in wild than cultivated rubber trees,Hevea brasiliensis, revealed by comparative analyses of chloroplast genomes. Front. Ecol. Evol. 8. doi: 10.3389/fevo.2020.00237
Gao, C., Li, T., Zhao, X., Wu, C., Zhang, Q., Zhao, X., et al. (2023). Comparative analysis of the chloroplast genomes of Rosa species and RNA editing analysis. BMC Plant Biol. 23, 318. doi: 10.1186/s12870-023-04338-0
Gui, S. T., Wu, Z. H., Zhang, H. Y., Zheng, Y. Z., Zhu, Z. X., Liang, D. Q., et al. (2016). The mitochondrial genome map of Nelumbo nucifera reveals ancient evolutionary features. Sci. Rep. 6, 11. doi: 10.1038/srep30158
Hao, J., Liang, Y. Y., Su, Y. J., Wang, T. (2022). The complete mitochondrial genome of ophioglossum vulgatum L. Is with highly repetitive sequences: intergenomic fragment transfer and phylogenetic analysis. Genes 13. doi: 10.3390/genes13071287
Hao, W. L., Palmer, J. D. (2009). Fine-scale mergers of chloroplast and mitochondrial genes create functional, transcompartmentally chimeric mitochondrial genes. Proc. Natl. Acad. Sci. U.S.A. 106, 16728–16733. doi: 10.1073/pnas.0908766106
He, W. C., Xiang, K. L., Chen, C. J., Wang, J., Wu, Z. Q. (2022). Master graph: an essential integrated assembly model for the plant mitogenome based on a graph-based framework. Briefings Bioinf. doi: 10.1093/bib/bbac522
Hu, Y. S., Huang, H. S., Liu, J. (2022). Characterization of the complete chloroplast genome of Hevea pauciflora (Euphorbiaceae), an important wild relative of the rubber tree. Mitochondr. DNA Part B-Resour. 7, 1589–1593. doi: 10.1080/23802359.2022.2115321
Jackman, S. D., Coombe, L., Warren, R. L., Kirk, H., Trinh, E., Macleod, T., et al. (2020). Complete mitochondrial genome of a gymnosperm, sitka spruce (Picea sitchensis), indicates a complex physical structure. Genome Biol. Evol. 12, 1174–1179. doi: 10.1093/gbe/evaa108
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kozik, A., Rowan, B. A., Lavelle, D., Berke, L., Schranz, M. E., Michelmore, R. W., et al. (2019). The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PloS Genet. 15. doi: 10.1371/journal.pgen.1008373
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lai, C. J., Wang, J., Kan, S. L., Zhang, S., Li, P., Reeve, W. G., et al. (2022). Comparative analysis of mitochondrial genomes of Broussonetia spp. (Moraceae) reveals heterogeneity in structure, synteny, intercellular gene transfer, and RNA editing. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1052151
Laslett, D., Canback, B. (2008). ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175. doi: 10.1093/bioinformatics/btm573
Li, J., Wang, S., Jing, Y., Ling, W., Zhou, S. (2013). A modified CTAB protocol for plant DNA extraction. Chin. Bull. Bot.
Liu, J., Shi, C., Shi, C. C., Li, W., Zhang, Q. J., Zhang, Y., et al. (2020). The chromosome-based rubber tree genome provides new insights into spurge genome evolution and rubber biosynthesis. Mol. Plant 13, 336–350. doi: 10.1016/j.molp.2019.10.017
Liu, Q., Yuan, H. Y., Xu, J. X., Cui, D. L., Xiong, G., Schwarzacher, T., et al. (2023). The mitochondrial genome of the diploid oat Avena longiglumis. BMC Plant Biol. 23. doi: 10.1186/s12870-023-04217-8
Liu, G., Zhao, Z. K., Xiao, M. L., Mason, A. S., Yan, H., Zhou, Q. H., et al. (2015). Repetitive sequence characterization and development of SSR and CMS-gene-specific markers in the Brassica mitochondrial genomes. Mol. Breed. 35. doi: 10.1007/s11032-015-0413-3
Lowe, T. M., Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Ma, Q. Y., Wang, Y. X., Li, S. S., Wen, J., Zhu, L., Yan, K. Y., et al. (2022). Assembly and comparative analysis of the first complete mitochondrial genome of Acer truncatum Bunge: a woody oil-tree species producing nervonic acid. BMC Plant Biol. 22. doi: 10.1186/s12870-021-03416-5
Meyer, E. H., Welchen, E., Carrie, C. (2019). Assembly of the complexes of the oxidative phosphorylation system in land plant mitochondria. Annu. Rev. Plant Biol. 70, 23–50. doi: 10.1146/annurev-arplant-050718-100412
Morley, S. A., Nielsen, B. L. (2017). Plant mitochondrial DNA. Front. Bioscience-Landmark 22, 1023–1032. doi: 10.2741/4531
Nguyen, V. B., Giang, V. N. L., Waminal, N. E., Park, H. S., Kim, N. H., Jang, W., et al. (2020). Comprehensive comparative analysis of chloroplast genomes from seven Panax species and development of an authentication system based on species-unique single nucleotide polymorphism markers. J. Ginseng Res. 44, 135–144. doi: 10.1016/j.jgr.2018.06.003
Nishikawa, T., Vaughan, D. A., Kadowaki, K. (2005). Phylogenetic analysis of Oryza species, based on simple sequence repeats and their flanking nucleotide sequences from the mitochondrial and chloroplast genomes. Theor. Appl. Genet. 110, 696–705. doi: 10.1007/s00122-004-1895-2
Niu, Y., Gao, C., Liu, J. (2021). Comparative analysis of the complete plastid genomes of Mangifera species and gene transfer between plastid and mitochondrial genomes. PeerJ 9, e10774. doi: 10.7717/peerj.10774
Niu, Y. F., Gao, C. W., Liu, J. (2022). Complete mitochondrial genomes of three Mangifera species, their genomic structure and gene transfer from chloroplast genomes. BMC Genomics 23. doi: 10.1186/s12864-022-08383-1
Niu, Y. F., Hu, Y. S., Liu, Z. Y., Zheng, C., Liu, J. (2020a). Complete chloroplast genome of Hevea benthamiana, a SALB-resistant relative wild species of rubber tree. Mitochondr. DNA Part B-Resour. 5, 2062–2064. doi: 10.1080/23802359.2020.1763868
Niu, Y. F., Hu, Y. S., Zheng, C., Liu, Z. Y., Liu, J. (2020b). The complete chloroplast genome of Hevea camargoana. Mitochondr. DNA Part B-Resour. 5, 607–608. doi: 10.1080/23802359.2019.1710605
Posada, D. (2008). jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Priyadarshan, P. M. (2003). Breeding Hevea brasiliensis for environmental constraints. Adv. Agron. 79, 351–400. doi: 10.1016/S0065-2113(02)79007-X
Priyadarshan, P. M., Clement-Demange, A. (2004). Breeding Hevea rubber: Formal and molecular genetics. Adv. Genet., 51–115. doi: 10.1016/S0065-2660(04)52003-5
Priyadarshan, P. M., Goncalves, P. D. S. (2003). Hevea gene pool for breeding. Genet. Resour. Crop Evol. 50, 101–114. doi: 10.1023/A:1022972320696
Richardson, A. O., Rice, D. W., Young, G. J., Alverson, A. J., Palmer, J. D. (2013). The “fossilized” mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 11. doi: 10.1186/1741-7007-11-29
Rivarola, M., Foster, J. T., Chan, A. P., Williams, A. L., Rice, D. W., Liu, X. Y., et al. (2011). Castor bean organelle genome sequencing and worldwide genetic diversity analysis. PloS One 6. doi: 10.1371/journal.pone.0021743
Shearman, J. R., Sangsrakru, D., Ruang-Areerate, P., Sonthirod, C., Uthaipaisanwong, P., Yoocha, T., et al. (2014). Assembly and analysis of a male sterile rubber tree mitochondrial genome reveals DNA rearrangement events and a novel transcript. BMC Plant Biol. 14. doi: 10.1186/1471-2229-14-45
Sloan, D. B., Wu, Z. Q., Sharbrough, J. (2018). Correction of persistent errors in arabidopsis reference mitochondrial genomes. Plant Cell 30, 525–527. doi: 10.1105/tpc.18.00024
Smith, D. R. (2011). Extending the limited transfer window hypothesis to inter-organelle DNA migration. Genome Biol. Evol. 3, 743–748. doi: 10.1093/gbe/evr068
Stamatakis, A. (2006). RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Tangphatsornruang, S., Uthaipaisanwong, P., Sangsrakru, D., Chanprasert, J., Yoocha, T., Jomchai, N., et al. (2011). Characterization of the complete chloroplast genome of Hevea brasiliensis reveals genome rearrangement, RNA editing sites and phylogenetic relationships. Gene 475, 104–112. doi: 10.1016/j.gene.2011.01.002
Tao, Q. C., Cao, J., Zhu, L., Lin, H. F. (2019). The complete mitochondrial genome of an important root crop cassava (Manihot esculenta). Mitochondr. DNA Part B-Resour. 4, 1081–1082. doi: 10.1080/23802359.2019.1586471
Tian, X. J., Zheng, J., Hu, S. N., Yu, J. (2006). The rice mitochondrial genomes and their variations. Plant Physiol. 140, 401–410. doi: 10.1104/pp.105.070060
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9. doi: 10.1371/journal.pone.0112963
Wu, Z. Q., Liao, X. Z., Zhang, X. N., Tembrock, L. R., Broz, A. (2022). Genomic architectural variation of plant mitochondria-A review of multichromosomal structuring. J. Syst. Evol. 60, 160–168. doi: 10.1111/jse.12655
Wynn, E. L., Christensen, A. C. (2019). Repeats of unusual size in plant mitochondrial genomes: identification, incidence and evolution. G3-Genes Genomes Genet. 9, 549–559. doi: 10.1534/g3.118.200948
Xi, Z. X., Ruhfel, B. R., Schaefer, H., Amorim, A. M., Sugumaran, M., Wurdack, K. J., et al. (2012). Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proc. Natl. Acad. Sci. U.S.A. 109, 17519–17524. doi: 10.1073/pnas.1205818109
Xiong, Y. L., Yu, Q. Q., Xiong, Y., Zhao, J. M., Lei, X., Liu, L., et al. (2022). The complete mitogenome of elymus sibiricus and insights into its evolutionary pattern based on simple repeat sequences of seed plant mitogenomes. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.802321
Xu, K. K., Chen, Q. P., Ayivi, S. P. G., Guan, J. Y., Storey, K. B., Yu, D. N., et al. (2021). Three Complete Mitochondrial Genomes of Orestes guangxiensis, Peruphasma schultei, and Phryganistria guangxiensis (Insecta: Phasmatodea) and Their Phylogeny. Insects 12. doi: 10.3390/insects12090779
Yang, H. Y., Chen, H. M., Ni, Y., Li, J. L., Cai, Y. S., Wang, J. H., et al. (2023). Mitochondrial Genome Sequence of Salvia officinalis (Lamiales: Lamiaceae) Suggests Diverse Genome Structures in Cogeneric Species and Finds the Stop Gain of Genes through RNA Editing Events. Int. J. Mol. Sci. 24. doi: 10.3390/ijms24065372
Zhai, W., Zhao, Y., Zhang, L. X., Li, X. J. (2014). Structural and phylogenetic analysis of Pto-type disease resistance gene candidates in Hevea brasiliensis. Genet. Mol. Res.: GMR 13, 4348–4360. doi: 10.4238/2014.June.10.2
Keywords: Hevea, mitochondrial genome, intergenomic sequence transfers, phylogenetic analysis, genome variation
Citation: Niu Y, Gao C and Liu J (2024) Mitochondrial genome variation and intergenomic sequence transfers in Hevea species. Front. Plant Sci. 15:1234643. doi: 10.3389/fpls.2024.1234643
Received: 06 June 2023; Accepted: 25 March 2024;
Published: 10 April 2024.
Edited by:
Yi-Hong Wang, University of Louisiana at Lafayette, United StatesReviewed by:
Zhiqiang Wu, Kunpeng Institute of Modern Agriculture at Foshan, ChinaYang Ni, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Hongping Deng, Southwest University, China
Copyright © 2024 Niu, Gao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Liu, bGl1amluMDZAMTI2LmNvbQ==; Chengwen Gao, Z2FvY2hlbmd3ZW5AcWR1LmVkdS5jbg==