
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 08 January 2024
Sec. Functional and Applied Plant Genomics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1327009
This article is part of the Research TopicOmics Applications in Agriculture Systems: Unveiling Functionality and PracticalityView all 6 articles
 Jiin Kim1†‡
Jiin Kim1†‡ Hoyeol Jang2†Sun Mi Huh1‡Ara Cho2,3‡Bomi Yim1Seung-Hoon Jeong2Haneul Kim2Hee-Ju Yu1,4*‡
Hoyeol Jang2†Sun Mi Huh1‡Ara Cho2,3‡Bomi Yim1Seung-Hoon Jeong2Haneul Kim2Hee-Ju Yu1,4*‡ Jeong-Hwan Mun2*‡
Jeong-Hwan Mun2*‡Accumulation of anthocyanins in the taproot of radish is an agronomic trait beneficial for human health. Several genetic loci are related to a red skin or flesh color of radish, however, the functional divergence of candidate genes between non-red and red radishes has not been investigated. Here, we report that a novel genetic locus on the R2 chromosome, where RsMYB1.1 is located, is associated with the red color of the skin of radish taproot. A genome-wide association study (GWAS) of 66 non-red-skinned (nR) and 34 red-skinned (R) radish accessions identified three nonsynonymous single nucleotide polymorphisms (SNPs) in the third exon of RsMYB1.1. Although the genotypes of SNP loci differed between the nR and R radishes, no functional difference in the RsMYB1.1 proteins of nR and R radishes in their physical interaction with RsTT8 was detected by yeast-two hybrid assay or in anthocyanin accumulation in tobacco and radish leaves coexpressing RsMYB1.1 and RsTT8. By contrast, insertion- or deletion-based GWAS revealed that one large AT-rich low-complexity sequence of 1.3–2 kb was inserted in the promoter region of RsMYB1.1 in the nR radishes (RsMYB1.1nR), whereas the R radishes had no such insertion; this represents a presence/absence variation (PAV). This insertion sequence (RsIS) was radish specific and distributed among the nine chromosomes of Raphanus genomes. Despite the extremely low transcription level of RsMYB1.1nR in the nR radishes, the inactive RsMYB1.1nR promoter could be functionally restored by deletion of the RsIS. The results of a transient expression assay using radish root sections suggested that the RsIS negatively regulates the expression of RsMYB1.1nR, resulting in the downregulation of anthocyanin biosynthesis genes, including RsCHS, RsDFR, and RsANS, in the nR radishes. This work provides the first evidence of the involvement of PAV in an agronomic trait of radish.
Understanding the genetic bases of agronomically important traits and mining of related genes is a focus of crop science and agriculture. Genomic studies of crop species have focused on the construction of genome assemblies and systemic analyses of genetic resources. The availability of high-quality reference genomes and cost-effective sequencing and genotyping techniques has not only enabled investigation of genome architecture but has also accelerated population genomics and genome-wide association studies (GWAS) of crop species. The discovery of genetic variations by comparing genomes has enabled the evaluation of genomic differences between genotypes and the identification of trait-related genes. GWASs typically analyze single nucleotide polymorphisms (SNPs), however, SNPs do not capture all of the genomic variation that results in phenotypic differences (Saxena et al., 2014; Alseekh et al., 2021). GWASs based on insertion or deletion (InDel) polymorphisms have identified structural variations (SVs), which contribute to genetic diversity and are determinants of genome structure and function (reviewed in Yuan et al., 2021). SVs are defined as ≥50 bp modifications of DNA between genotypes or individuals. There are several types of SVs including copy number variations (CNVs), presence/absence variations (PAVs), InDels, inversions, and translocations. SVs can change the effects of mutations in multiple parts of cis-regulatory regions on gene expression (Alonge et al., 2020) or lead to phenotypic variation in a species as a result of gene loss, gene duplication, the generation of novel genes, and complex rearrangements (Cook et al., 2020). In several plant species, SVs regulate agronomic traits, such as fruit shape in tomato (Xiao et al., 2008) and peach (Guan et al., 2021), fruit texture in apple (Wu et al., 2023), fruit color in apple (Espley et al., 2009) and peach (Guo et al., 2020), leaf shape in rapeseed (Hu et al., 2018), and resistance to biotic stress in maize (Zuo et al., 2015), sorghum (Gobena et al., 2017), and rice (Ashikawa et al., 2008).
Raphanus sativus (radish, 2n = 18) belongs to the genus Raphanus in the Brassicaceae family. It is closely related to the diploid Brassica species (B. rapa, B. nigra, and B. oleracea) and originated from the Brassica B genome lineage (Cho et al., 2022). Divergence of R. sativus from the wild species (R. raphanistrum) likely occurred during the Quaternary glaciation beginning 2.4 million years ago (Mya), presumably as a result of climate change (Zhang et al., 2021). R. sativus is an ancient domesticated species native to both the Mediterranean region and Eastern Asia (Becker, 1962; George and Evans, 1981; Kaneko and Matsuzawa, 1993). Targeted breeding linked with domestication has led to the development of a variety of radish lines and cultivars. R. sativus is a commercially important root vegetable crop that is cultivated globally, accounting for 2% of the total global production of vegetables; the fresh-radish market was worth 1264 million USD in 2021 (Kopta and Pokluda, 2013; Verified Market Research, 2023) Genomics tools and plant collections are essential for radish research and breeding. Genome sequencing has yielded at least 16 genome assemblies of Raphanus species. A comparison of genome assemblies revealed that the genome size of radish ranged from 430–515 Mb with approximately 42,000–53,000 predicted genes, the numbers of which did not deviate from the average by >6% (Zhang et al., 2021; Cho et al., 2022), suggesting genomic variation between genotypes. A Raphanus pan-genome study using 11 genome assemblies reported an average of 5.6–10.1 SNPs and 1.2–2.3 InDels per kb between each pair of genomes and 7–11% of genes were potentially affected by SVs. The genes affected by variation were enriched in disease-resistance genes and gibberellin-related genes, suggesting that genetic variation contributes to phenotypic divergence and the adaptive evolution of radish. In parallel with genome sequencing and genotyping tools, conservation of genetic resources is ongoing. The collections reported to date include the Radish Core Collection of 125 accessions from South Korea (Lee et al., 2018; Huh et al., 2023), the National Plant Germplasm System collection of 152 accessions from the United States (Arro and Labate, 2022), and the Core Germplasm Collection comprising 217 accessions from China (Li et al., 2023).
Anthocyanins are natural antioxidant phenolic pigments found in many plant species. In the red radish taproot, anthocyanins accumulate in the vacuoles of skin or flesh cells. The radish anthocyanins are raphanusins, i.e., glucosylated pelagonidin anthocyanins acylated with 4-coumaric acid (Ishikura and Hayashi, 1963; Giusti et al., 1999). Anthocyanins are produced in a branch of the phenylpropanoid pathway, which is involved in the biosynthesis of several flavonoids. One molecule of 4-coumaroyl-CoA is condensed with three molecules of malonyl-CoA by chalcone synthase (CHS) to form naringenin chalcone, which is converted into naringenin by chalcone isomerase. Naringenin is converted into dihydroflavonols by hydroxylation via flavonoid 3’-hydroxylase and flavonoid 3,’ 5’-hydroxylase. Dihydroflavonols are reduced by dihydroflavonol 4-reductase (DFR) to form leucoanthocyanidins. Oxidation of leucoanthocyanidins by anthocyanidin synthase (ANS) generates anthocyanidins, which are highly unstable. Flavonoid glucosyltransferase rapidly catalyzes glycosylation of C3 in ring C or other positions of anthocyanidins to form anthocyanins. O-Methyltransferase and anthocyanin acyltransferase methylate and acylate, respectively, the hydroxyl groups of rings A and B (reviewed by Zha and Koffas, 2017). The expression of structural genes of the phenylpropanoid pathway is coordinated and regulated transcriptionally by a ternary transcription factor (TF) complex named the MBW complex, which is made up of R2R3-MYB TF, basic helix-loop-helix (bHLH) TF, and tryptophan-aspartic acid repeat protein WDR or WD40 (Hichri et al., 2011). The MBW complex interacts with the MYB-recognition element (ANCNNCC) and the bHLH-recognition element (CACN[A/C/T][G/T]) in the promoter regions of the structural genes of the anthocyanin biosynthesis pathway (Zhu et al., 2015). In radish, RsMYB1 promotes anthocyanin biosynthesis by interacting with RsTT8 (Kim et al., 2021). At least three RsMYB1, namely RsMYB1.1 (Liu et al., 2019a), RsMYB1.3 (Tao et al., 2022), and RsMYB1.4 (Yi et al., 2018; Kim et al., 2021), have been identified by genetic mapping, molecular genetics, and SNP GWAS of red radish. Although RsMYB1.3 and RsMYB1.4 of red radish reportedly interact with RsTT8 to modulate anthocyanin biosynthesis in radish taproot (Kim et al., 2021; Tao et al., 2022), whether their counterpart genes in non-red radish are functional is unknown.
According to the annotations of gene models in the Rs2.0 radish genome assembly of cv. WK10039 (Cho et al., 2022), the genes R2.009390 (Rs094840 of Rs1.0) and R7.017240 (Rs388430 of Rs1.0) encode RsMYB1.1 and RsMYB1.4, respectively. These are candidate genes related to a red skin or flesh color, however, their functions in WK10039, which has a taproot with white skin and flesh, are unknown. We performed SNP and InDel GWASs of 100 accessions, which identified a genetic locus associated with the red skin color of radish taproot. This locus encompasses RsMYB1.1, which has sequence variation between non-red-skinned (nR) and red-skinned (R) radishes. There was no functional difference between the RsMYB1.1 proteins of nR and R radishes. By contrast, a PAV in the promoter region of RsMYB1.1 in nR radish negatively regulated the transcription of the coding sequence (CDS), thus suppressing anthocyanin biosynthesis. This is the first report that the PAV in the promoter region of RsMYB1.1 rather than the presence of specific type of RsMYB1.1 is responsible for the red color of the skin of radish taproot.
Seeds of each radish inbred line and tobacco (Nicotiana benthamiana and N. tabacum) were germinated on horticultural potting soil (Nongwoo Bio, Suwon, South Korea) in a growth chamber set at 22°C under a 16 h light/8 h dark cycle and 60% humidity. Radish seedlings were transferred to an open field 1 week after germination and grown for 1 to 2 months. Tobacco seedlings were grown in a growth chamber under the same conditions. Genomic DNA (gDNA) was extracted from the leaves of 1-month-old plants using the DNeasy Plant Maxi Kit (Qiagen, Germantown, MD, USA). Total RNA was extracted from plant tissues using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) and treated with DNase using the TURBO DNA-Free Kit (Thermo Fisher Scientific). The quality and quantity of extracted nucleic acids were analyzed using the Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA).
The skin color of the taproot of radish accessions was examined 50 to 70 days after planting in the field. For genotype data, we used previously generated whole-genome resequencing data (Kim et al., 2016; Huh et al., 2023). Briefly, approximately 23-fold the average genome coverage (the estimated R. sativus genome size is 510.8 Mb) of the quality filtered paired-end (PE) Illumina HiSeq reads from 100 accessions were mapped to the Rs2.0 reference genome (Cho et al., 2022) using BWA-MEM 0.7.12 (Li, 2013) with the parameters of k13, c1000, r10, v1, mapping quality 40, and 99.99% probability. Duplicate alignments were removed using the Picard v2.2.4 package (http://broadinstitute.github.io/picard). For SNP calling, the GATK v3.7.0 toolkit (Mckenna et al., 2010) was used. The HaplotypeCaller and GenotypeGVCFs modules were used to call variant sites per sample, generate a per-sample gVCF, and convert variant calls in gVCF into VCF format. The VariantFiltration module with parameters of FS >1.0 and MQ <40 was used to filter variants. VCFtools v0.1.13 (Danecek et al., 2011) with parameters of DP >10 and GQ <20 was used for additional filtering. The SelectVariants module was used to generate a subset VCF file of SNPs. For InDel calling, Pindel v0.2.5b9 (Ye et al., 2009) with parameters of r false, t false, x 4, and w 3 was used. The resulting SNP and InDel variants (1 bp to 3 kb) were further filtered using the variation positions on the chromosomes and PLINK v1.90 software (Purcell et al., 2007) with parameters of mind 0.2 and maf 0.1. GWAS was performed using GEMMA v0.98.5 (Zhou and Stephens, 2012) with the gk 1 parameter for calculating the centered relatedness matrix of SNP and InDel variants with phenotype, respectively, and the lmm 4 parameter for association tests with three univariate linear mixed models (Wald, likelihood ratio, and score tests). The significant and suggestive thresholds values of P were set at 0.05 and 1 per the number of SNPs and InDels used in the GWAS (Guo et al., 2017). Manhattan and Quantile-Quantile (Q-Q) graphs based on P-values were plotted using the ggplot2 package (http://ggplot2.org).
The primers used are listed in Supplementary Table S1. The target genome regions of each accession were reconstructed by reference-guided assembly. Illumina reads from each accession mapped to the target regions were extracted and assembled into a consensus sequence using Geneious Prime 2023.1.2 (Biomatters, Auckland, New Zealand) with the default parameters. The assembled sequences were multiple-aligned with the reference sequence of WK10039 and genotyping primers were designed based on the aligned sequences using the tools in Geneious Prime. For InDel genotyping, PCR amplification was performed using the gt-pRsMYB1.1 primer pair, which targeted the conserved region of the RsMYB1.1 promoter, followed by cloning and sequencing. For SNP genotyping, multiplex-PCR primer sets including amplification (gt-RsMYB1.1-E3) and extension (e1 to e3) primers were designed to target the third exon of RsMYB1.1 using MassARRAY Typer software (Agena, San Diego, CA, USA). Protein structures were predicted using AlphaFold Protein Structure Database (AlphaFold DB, released on May, 2020) (Jumper et al., 2021) and viewed through YASARA (Land and Humble, 2018). PCR amplification, single-base extension, and genotyping of target loci were performed using the MassARRAY system according to the manufacturer’s instructions (Agena). The CDSs of RsMYB1.1 and RsTT8 were synthesized and cloned into the pTwist-ENTR vector by Twist Bioscience (South San Francisco, CA, USA). The entry plasmids were recombined with the pK7WG2D vector in the presence of LR Clonase II (Thermo Fisher Scientific) to obtain the binary constructs for overexpression of the RsMYB1.1 and RsTT8 CDSs under the control of the CaMV 35S promoter. To construct the promoter::GUS reporter fusions, the 5’-flanking regions of the RsMYB1.1 genes were amplified from the gDNAs of WK10039 (3.2 kb) and CUR034 (1.9 kb) using Phusion High Fidelity DNA polymerase (Thermo Fisher Scientific) with the c-pMYB1.1nR-full and c-pMYB1.1R-full primers, respectively. For promoter deletion analysis, the c-pMYB1.1nR-d1 to c-pMYB1.1nR-d4 primer pairs were used to amplify fragments of the RsMYB1.1 promoter of WK10039. The resulting PCR amplicons were purified by agarose gel electrophoresis, cloned into the All-in-One vector (Biofact, Daejeon, South Korea), and confirmed by sequencing. The cloned fragments were digested with EcoRI/NcoI and ligated into the pCAMBIA3301 vector. The resulting binary vector constructs were inserted into Agrobacterium tumefaciens strain GV3101.
The CDSs of RsMYB1.1 and RsTT8 were amplified from the synthetic fragments described above using the y-RsMYB1.1, y-RsTT8nR, and y-RsTT8R primer pairs and were inserted in-frame into the prey vector pGADT7 and the bait vector pGBKT7. Pairwise interactions between RsMYB1.1 and RsTT8 were screened in the yeast strains AH109 and PBN204, which were sequentially transformed by the bait vector and the prey vector. Transformed yeasts were cultured on SD Leu– Trp– Ade– or His– medium for AH109 and SD Leu– Trp– Ura– or Ade– medium for PBN204. Colonies positive for PBN204 were confirmed via filter assay using X-gal as the substrate for β-galactosidase.
Transgenes were delivered into plant cells using the syringe infiltration method. For leaf infiltration, A. tumefaciens GV3101 culture with an optical density at 600 nm (OD600) of 0.8 in infiltration medium (10 mM MES [pH 5.6], 10 mM MgCl2, and 100 mM acetosyringone) was infiltrated into the abaxial side of fully expanded radish and tobacco leaves by pressing the tip of a 1 mL syringe without a needle. For coexpression of two independent vector constructs, an equal volume of A. tumefaciens suspension with an OD600 of 0.8 in infiltration medium was mixed before infiltration. For root-section transformation, the Agrobacterium cultures with an OD600 of 1.0 were suspended in 0.5× MS medium containing 2% sucrose, 150 mM acetosyringone, and 0.01% Tween 20 (pH 5.8), and infiltrated into 1 mm thick radish taproot sections using a 1 mL syringe without a needle. Typically, multiple injections were performed on a single leaf or taproot section to expand the infiltrated parts. The Agrobacterium-infiltrated plant materials were incubated for 3 (GUS analysis) to 5 (anthocyanin quantification) days in a growth chamber and were used to evaluate gene expression. Histochemical and fluorometric analyses of GUS activity were performed as described previously (Alwen et al., 1992) with modifications. For GUS histochemical staining, plant materials were incubated in phosphate buffer (100 mM NaPO4 [pH 7.0], 0.5 mM potassium ferricyanide, 0.5 mM potassium ferrocyanide, 10 mM EDTA [pH 8.0], and 0.1% Triton X-100), containing 1 mM 5-bromo-4-chloro-3-indolylglucuronide (X-Gluc; Sigma-Aldrich, Saint Louis, MO, USA) as the substrate, at 37°C for 24 h. Photographs of histochemically stained sections were obtained without fixation using a digital camera (Nikon, Tokyo, Japan). For fluorometric assays, 50 mg plant material ground in liquid nitrogen was incubated in 0.5 mL GUS extraction buffer (50 mM NaH2PO4 [pH 7.0], 10 mM EDTA, 0.1% Triton X-100, 0.1% sarcosyl, and 10 mM β-mercaptoethanol) containing 4-methylumbelliferyl-β-D-glucuronide (MUG; Sigma-Aldrich) as the substrate at 37°C for 1 h. Fluorescence was measured with excitation at 365 nm and emission at 455 nm using an Infinite M200 PRO Plate Reader (Tecan, Männedorf, Switzerland). GUS values were expressed as nmol 4-methylumbelliferone (4-MU; Sigma-Aldrich) min–1 mg–1 soluble protein.
Radish roots were sampled from 50-day-old plants grown in the field and tobacco leaves were harvested 5 days after Agrobacterium infiltration. For quantification of total anthocyanin levels, 0.3 g ground tissue in liquid nitrogen was added to 1 mL 1% (v/v) HCl methanol and incubated for 24 h at 25°C with gentle stirring in the dark. The anthocyanin solution was centrifuged at 4°C and 13,000 × g for 15 min, and the supernatants were subjected to measurement of absorbance at wavelengths of 530, 620, and 650 nm using an SM 1200 UV-Vis Spectrophotometer (AZZOTA Scientific, Claymont, DE, USA). Anthocyanin levels were calculated from three biological replicates using cyanidin-3-glucoside as the reference, according to the following formula (Giusti and Wrolstad, 2001): [(A530 − A620) − 0.1 × (A650 − A620)] × MW × DF × 1000 × ∋; where A is supernatant absorbance, wavelengths are indicated as subscripts, MW is the molecular weight of cyanidin-3-glucoside (449.2 g/mol), DF is a dilution factor, and ∋ is the cuvette optical path length (1 cm). Expression values of genes were obtained from previously generated mRNA sequencing (mRNA-seq) data (Jeong et al., 2016). For quantitative PCR (qPCR) analysis, first-strand cDNA was synthesized using total RNA extracted from root tissues with the Revert Aid First-Strand cDNA Synthesis Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. cDNAs were diluted 10-fold and qPCR was performed using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific), primer pairs designed to amplify target genes, and the StepOne Plus™ Real-Time PCR System (Thermo Fisher Scientific). The 2-ΔΔCt method was used to quantify gene expression with RsActin7-2 as the reference. To statistically test the difference in gene expression, the independent t-test was performed using Excel.
In total, 100 genotypes, including 83 inbred lines of the core collection (Huh et al., 2023) and 17 previously analyzed genotypes (Kim et al., 2016), were subjected to GWAS of red taproot skin color (Supplementary Table S2). In all, 34 genotypes had red skins; the others were white, white-green, or black (Figure 1A). The R radishes contained 118.2 mg anthocyanins per 100 g fresh weight (FW) of root on average whereas nR radishes had no anthocyanins (Huh et al., 2023). For genome sequences, we obtained approximately 1170 Gb clean data from 100 genotypes. The average read mapping rate and depth were 98.7% and 26.4-fold, respectively. The coverage per genotype using Rs2.0 as the reference genome was 73.6–98.2%. Most unmapped regions (83.6%) in the Rs2.0 were repetitive, heterochromatic regions. The initial variation calls identified 34,380,875 SNPs by GATK and 1,825,271 InDels by Pindel. After filtering and variant calling, 794,484 SNPs and 308,433 InDels were selected to investigate the association between phenotypes and variation.
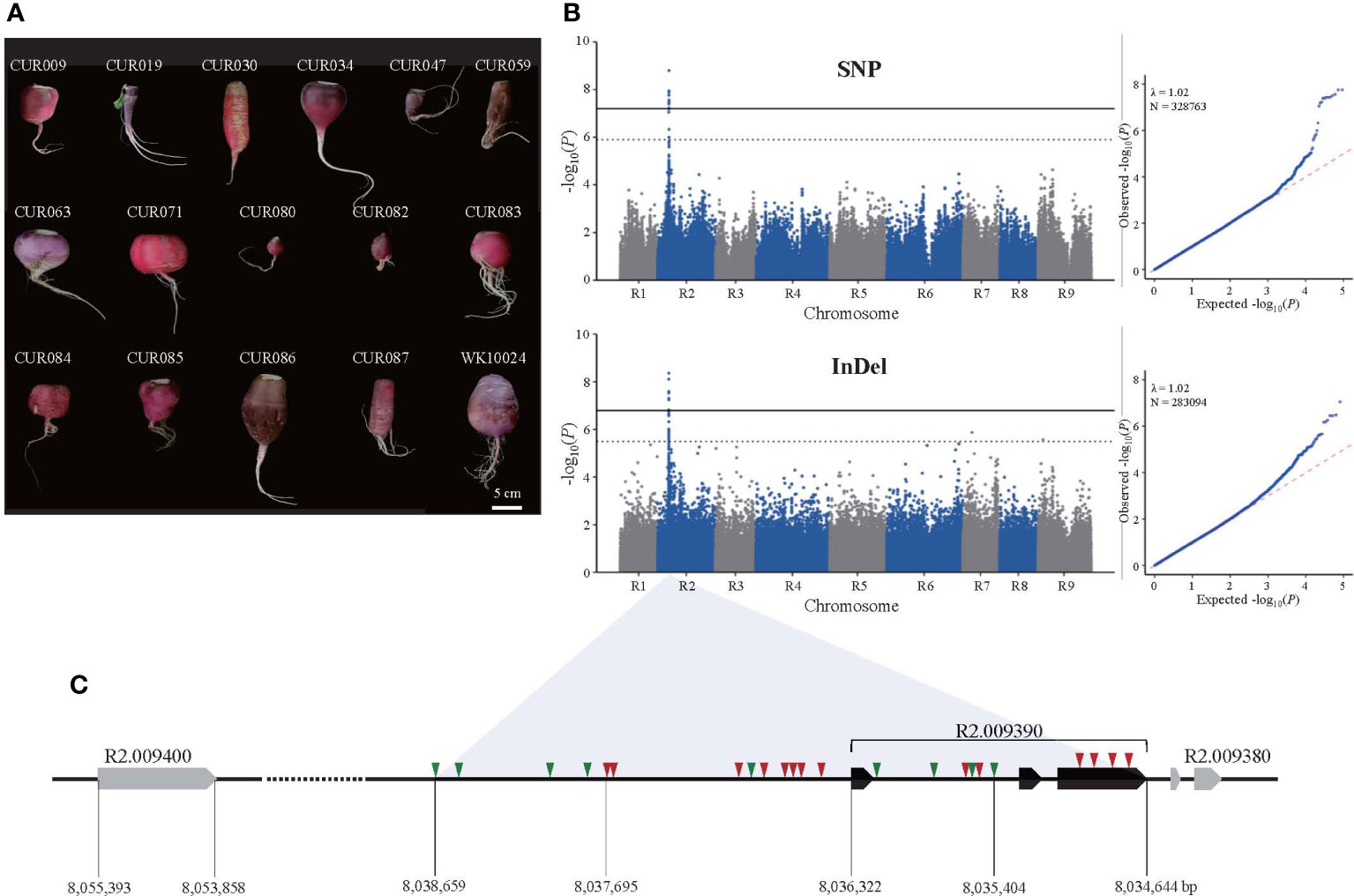
Figure 1 Identification of genomic loci associated with the taproot skin color of radish by GWAS. (A) Photographs of selected red-skinned radish accessions in the core collection. (B) Manhattan and Q-Q plots of SNP (upper panel) and InDel (lower panel) GWAS. Solid and dotted lines in Manhattan plots depict thresholds for significant and suggestive, respectively. (C) Variation in the candidate genome region on the R2 chromosome. Triangles represent SNP (red) and InDel (green) variation.
The GWAS showed that taproot skin color was associated with a region on the R2 chromosome that spans 7.95 to 8.10 Mb (Figure 1B). The SNP-based GWAS identified 127 SNPs associated with the trait, among which 14 SNPs reached the suggestive threshold (–log10(P) = 5.90). Interestingly, 12 of 14 suggestive SNPs were located in five genes in the genomic region, and the others were in the intergenic region (Supplementary Table S3). The lead SNP (–log10(P) = 8.80) and one significant SNP (–log10(P) = 7.44) were located in the promoter and the third exon of R2.009390 (MYB1.1). We also annotated suggestive SNPs in the coding regions of R2.009320 (methyltransferase), R2.009350 (bHLH), R2.009400 (DELLA protein RGL1), and R2.009420 (pentatricopeptide repeat-containing protein). The InDel-based GWAS identified 259 associated InDels, 21 of which reached the suggestive threshold (–log10(P) = 5.49). All of the suggestive InDels were intergenic except two in introns of R2.009320 (2 bp deletion) and R2.009350 (1 bp deletion). In all, 43 InDel loci were identified in the genomic region near R2.009390, including the promoter and intron (Supplementary Table S4). The size differences in these InDels ranged from 1 to 42 bp, indicating high sequence variation. Unfortunately, none reached the suggestive threshold for taproot skin color. Based on the role of MYB1.1 in anthocyanin biosynthesis, we selected R2.009390, named RsMYB1.1, and nearby sequences as a candidate region associated with taproot skin color (Figure 1C).
An in-depth sequence comparison of candidate regions between nR and R radishes was performed using the consensus sequences assembled from the short read sequences of each genotype. In multiple alignment analyses of the corresponding sequences, only RsMYB1.1 and its nearby sequences showed conserved patterns of SNP and InDel variation. In the third exon of RsMYB1.1, one synonymous SNP locus (Rs:8,034,806 A/G) and three group-specific nonsynonymous SNP loci (allele1, R2:8,034,922 C/A; allele 2, R2:8,034,895 A/T; allele 3, R2:8,034,850 A/G) having different encoded amino acids (allele 1, 158 Q/K; allele 2, 167 N/Y; allele 3, 182 T/A) between the nR and the R radishes were identified (Figure 2A). Group-specific SNP types of the loci were conserved in the core collection accessions and the commercial cultivars. Genotyping of the SNP loci using the MassARRAY markers showed that all of the nR radishes had C, A, and A, whereas R radishes had A, T, and G for alleles 1, 2, and 3, respectively (Figure 2B; Table 1). Protein structure prediction with AlphaFold DB showed that amino acid changes due to nonsynonymous SNPs may affect C-terminal structure of the RsMYB1.1 proteins (Supplementary Figure S1).
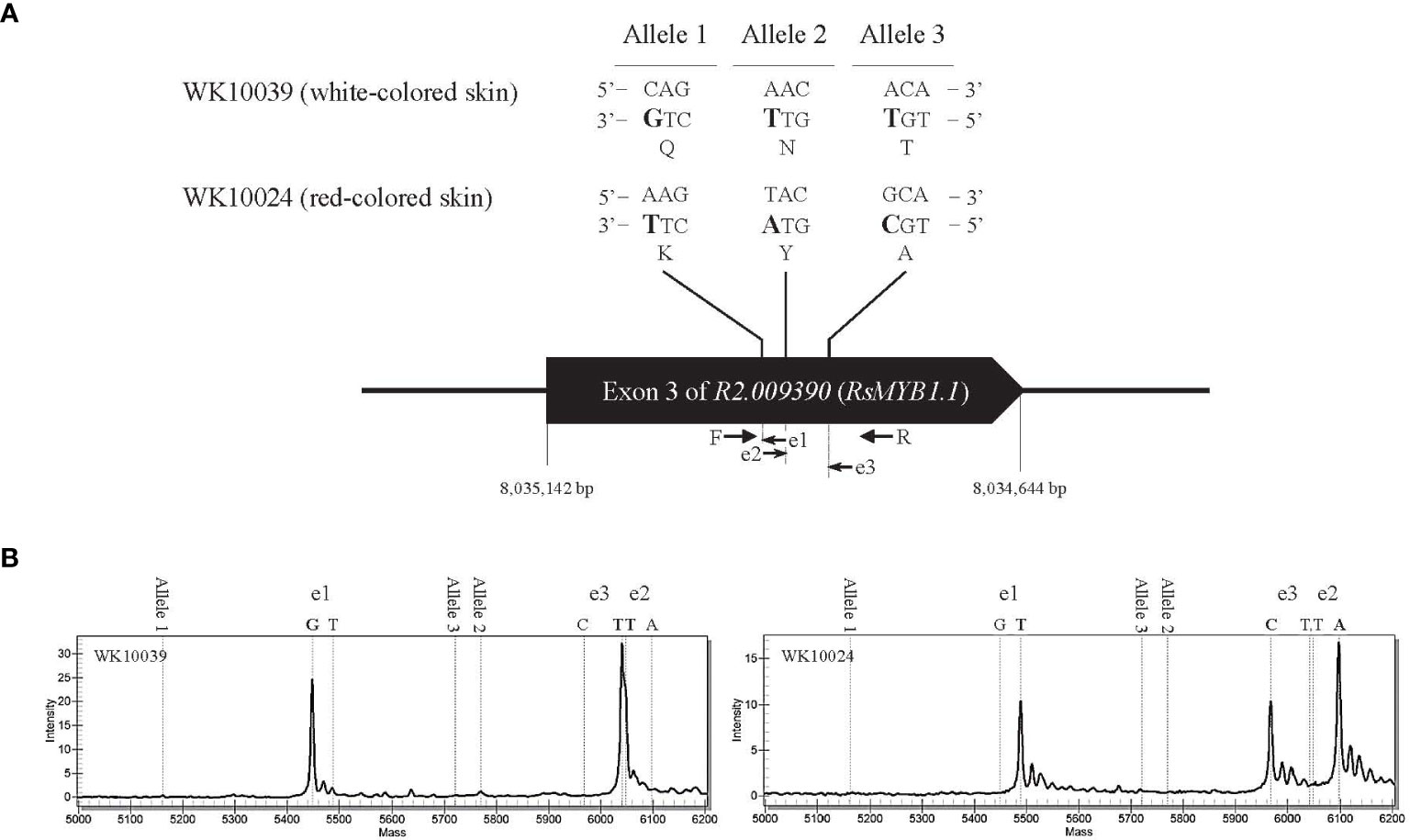
Figure 2 Genotyping of nonsynonymous SNP alleles in the third exon of R2.009390. The target regions of three SNP alleles showing nonsynonymous changes between the nR radish WK10039 and the R radish WK10024 were amplified by PCR and genotyped using the MassARRAY markers. (A) Nucleotide and amino acid sequences of SNP alleles. Arrows indicate primers for MassAARY genotyping. (B) MassARRAY spectrograms of WK10039 (left) and WK10024 (right). Peaks in the spectrograms represent nucleotides extending from the target alleles.
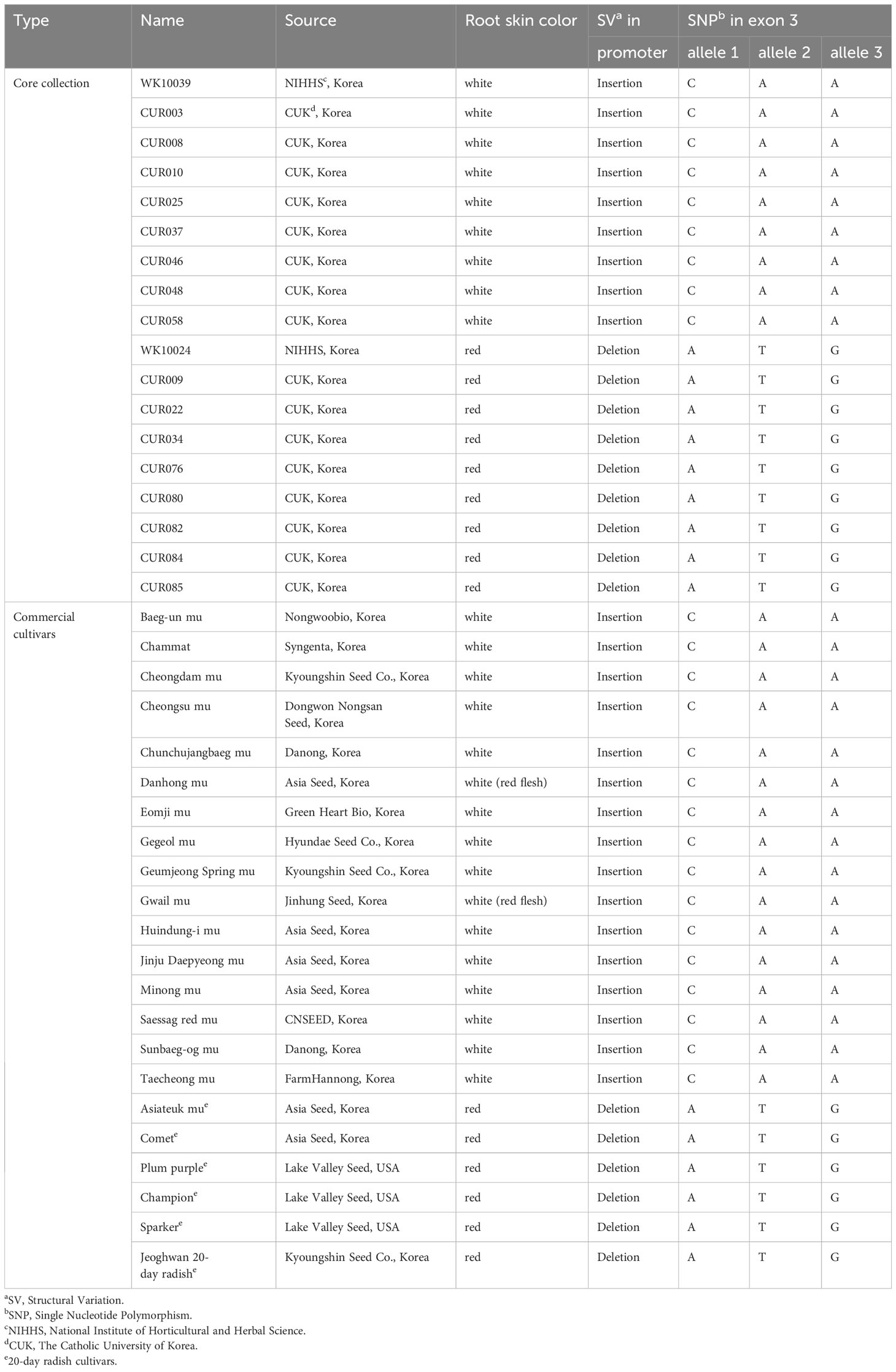
Table 1 Genotypes of SV and SNP loci in the promoter and coding sequences of RsMYB1.1 genes of core collection accessions and commercial cultivars.
The promoter region of RsMYB1.1 showed group-specific SVs. We extracted the promoter region (approximately 4 kb in length from the start codon) of each RsMYB1.1 gene. Multiple alignments of the sequences showed that the nR radishes had an insertion sequence of average size 1.3 kb in the upstream 1.4 kb region from the start codon of RsMYB1.1 (–2777 to –1437 bp of RsMYB1.1 in WK10039). By contrast, the R radishes had no insertion sequences in the corresponding promoter regions (Supplementary Figure S2). This InDel variation was also detected in genome assemblies (Zhang et al., 2021), in which seven genotypes with white- or black-skinned taproots had insertions but only Rs06 (red-skinned taproot) had a deletion in the promoter region (Supplementary Figure S3). Agarose gel electrophoresis of PCR amplicons from the promoter region of RsMYB1.1 demonstrated that the nR radishes had 1.3 to 2 kb insertions in the target region, which was absent in all of the R radishes, indicating a PAV of SV (Figure 3). We named it the R. sativus insertion sequence (RsIS).
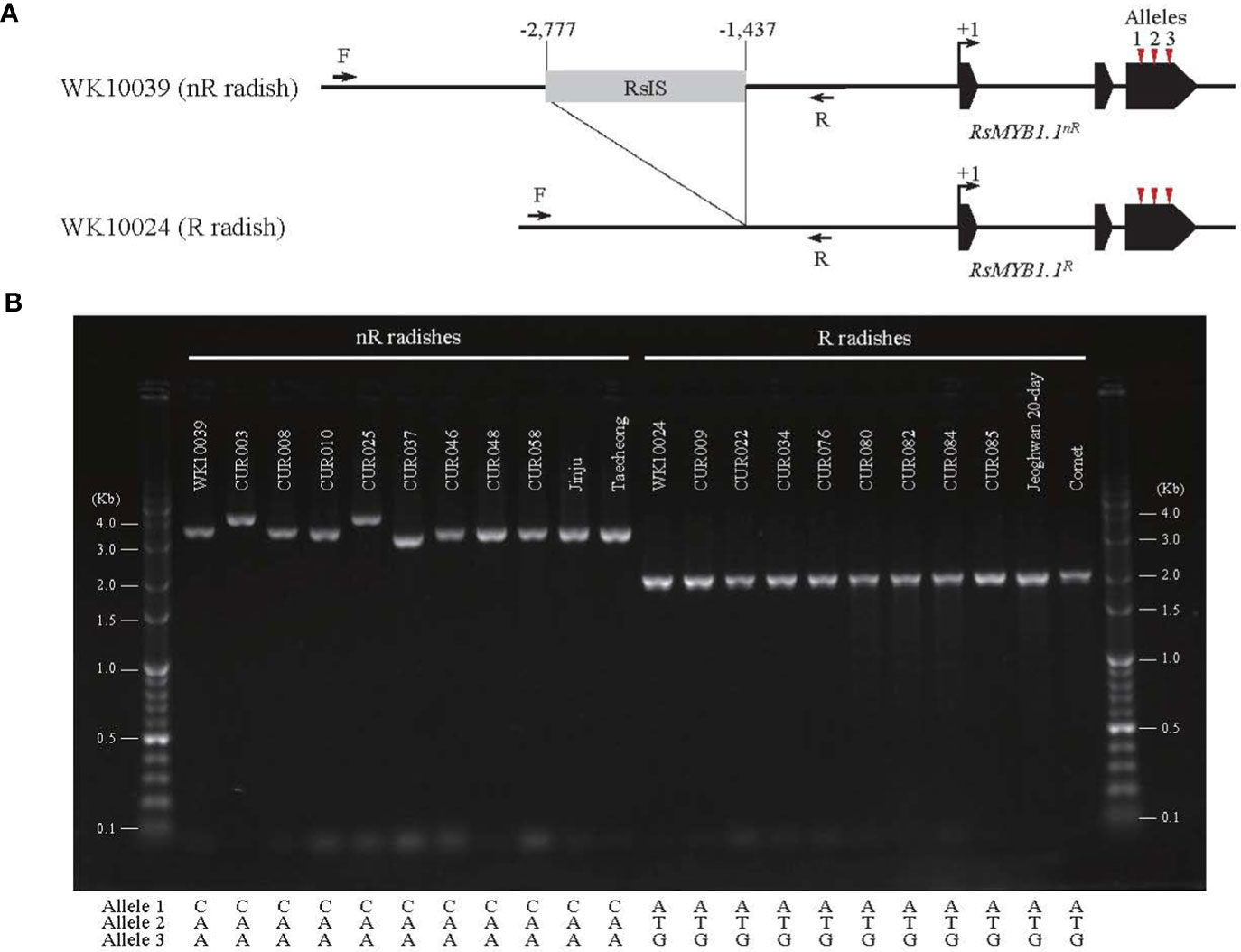
Figure 3 The PAV in the promoter region of RsMYB1.1. (A) The 1.3 kb RsIS is inserted 1.4 kb upstream from the start codon of RsMYB1.1 of WK10039; the corresponding region of WK10024 has no RsIS. Arrows indicate primers for PCR amplification. Numbers are upstream positions from the start codon (+1). (B) Agarose gel electrophoresis of PCR amplicons from the promoter region of RsMYB1.1 of radish accessions. Only the non-red-skinned (nR) radishes have RsIS insertions with length variation. The positions and sequences of nonsynonymous SNP alleles for each accession are also shown.
The RsIS of WK10039 was highly rich in AT (78.7% AT content). The RsIS is a low-complexity DNA sequence of a biased composition containing many simple sequences repeat units. It contained 167 repeat units of 11 to 21 nucleotides and a number of predicted secondary hairpin structures (Supplementary Figure S4). We analyzed the sequence using the Plant Repeat Database, however, neither specific matches to the known repeat elements including DNA transposons, retroelements, and long terminal repeats, nor repeated patterns were identified. A BLASTN search (cutoff of 1E–10 and query and subject coverages of 50%) of the RsIS of WK10039 against radish, diploid Brassica, and Arabidopsis genomes revealed that this sequence was specifically amplified in both the cultivar and wild radish genomes and had CNVs (132 to 183 copies) (Supplementary Table S5). RsIS had 144 BLAST matches distributed on nine chromosomes of WK10039. Sequence analysis revealed that RsIS consists of two regions. The front 700 bp is a conserved region between the BLAST matched sequences, showing 74.4–85.3% nucleotide identity, and the rear 600 bp is a variable region with <50% nucleotide identity. Most of the RsIS-like sequences (60%) were in intergenic regions, 43 copies were in the promoter regions (up to 3 kb upstream of genes) of 43 genes, and 15 copies were in the introns of 14 genes (Supplementary Table S6). Interestingly, mRNA-seq analysis showed that the expression levels of the genes harboring RsIS-like sequences in their promoter regions were low in a variety of tissues (Supplementary Table S7). The average TMM values of 41 of 43 genes were <50. The transcription of RsMYB1.1 was markedly downregulated in all the investigated tissues of WK10039 with an average TMM value of 1.2.
We examined the expression profiles of three key transcription factors (RsMYB1.1, R9.018870 encoding RsTT8, and R4.018220 encoding RsTTG1) of the MBW complex and three anthocyanin biosynthesis genes (R2.030580 encoding RsCHS, R9.040290 encoding RsDFR, and R2.039640 encoding RsANS) in the roots of nR and R radishes by qPCR (Figure 4). The expression levels of RsMYB1.1 were significantly lower in the eight nR radishes than in the eight R radishes (p < 0.01). Insertion of the RsIS in the promoter region of RsMYB1.1 likely suppresses the transcription of RsMYB1.1 in nR radishes. Considering the stability of root character, the availability of sufficient seeds for experiments, and available genome sequences, we selected three nR radishes (CUR010, WK10039, and Jinju) and three R radishes (CUR034, CUR082, and WK10024) for further analysis. Similar to RsMYB1.1, RsTT8, RsCHS, RsDFR, and RsANS were expressed only in the R radishes. By contrast, RsTTG1 was expressed at similar levels in the nR and R radishes. These results are consistent with the anthocyanin levels of the nR and R radishes in the core collection accessions (Huh et al., 2023); therefore, downregulation of the anthocyanin biosynthesis pathway genes in the nR radishes reduces the anthocyanin accumulation in the root, resulting in non-red-colored skin. The downregulation of not only RsMYB1.1 but also RsTT8 may be involved in the suppression of anthocyanin biosynthesis in the root of nR radishes.
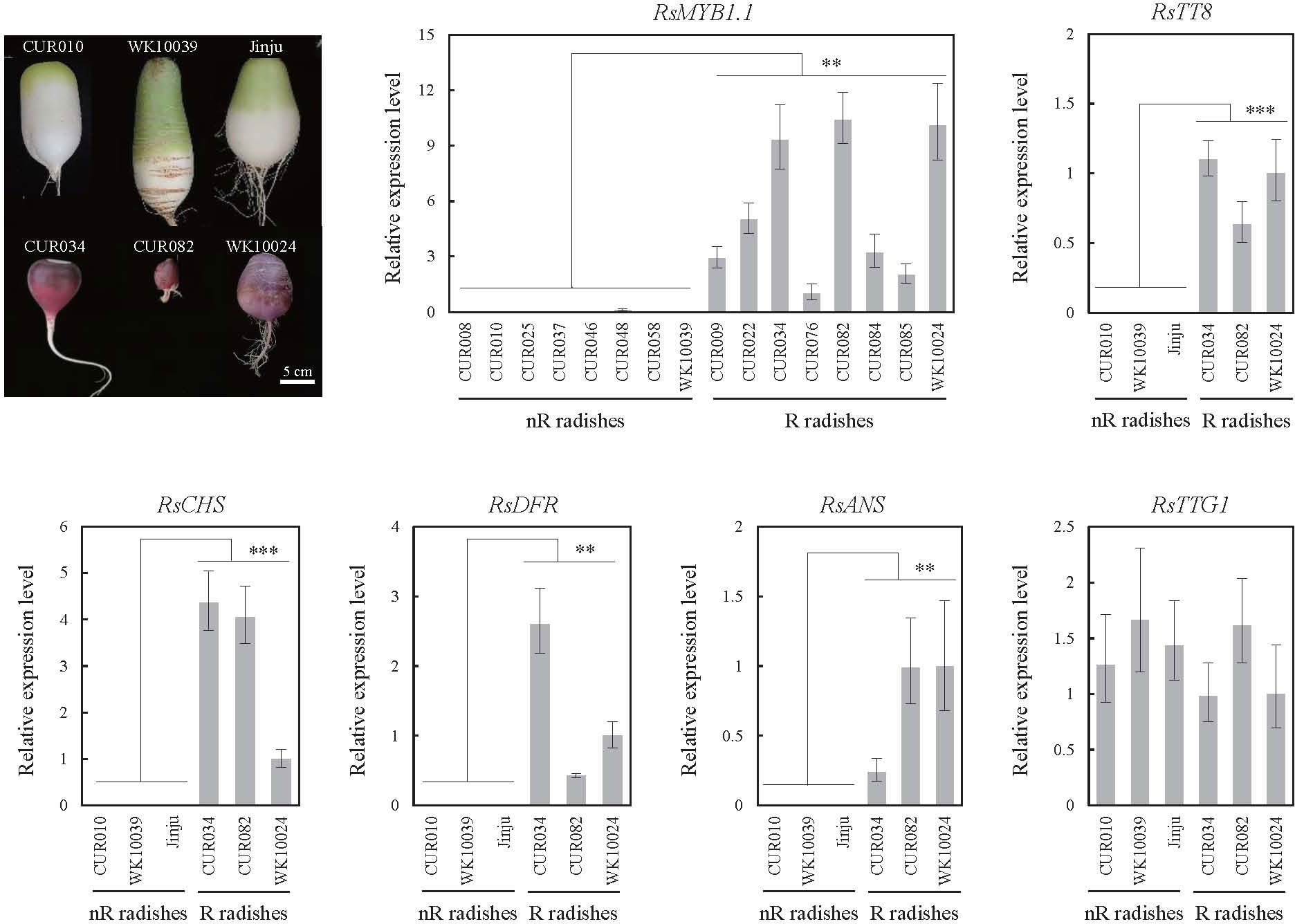
Figure 4 Expression levels of the MBW complex (RsMYB1.1, RsTT8, and RsTTG1) and selected structural genes (RsCHS, RsDFR, and RsANS) of the anthocyanin biosynthesis pathway in the radish accessions as determined by qPCR. Root tissues were harvested 42 days after germination. Comparative cycle threshold (2–ΔΔCt) values represent relative expression calculated using the CUR076 (RsMYB1.1) and WK10024 (other genes) samples as a reference. Error bars depict the standard deviations of three independent biological replicates. Asterisks represent statistical significance (**p < 0.01, ***p < 0.001) between nR and R radishes by t-test.
To examine the physical interaction between RsMYB1.1 and RsTT8, we performed a pairwise yeast two-hybrid (Y2H) assay (Figure 5). The synthesized full-length CDSs of RsMYB1.1 and RsTT8 of WK10039 (nR radish; RsMYB1.1nR and RsTT8nR) and CUR034 (R radish; RsMYB1.1R and RsTT8R) were cloned into bait pGBKT7 and prey pGADT7 vectors. The identities of the two proteins between WK10039 and CUR034 were 98.0% (RsMYB1.1) and 98.5% (RsTT8). The bait and prey vectors were cotransformed into PBN204 yeast, which was cultured on synthetic dropout medium lacking leucine, tryptophan, and uracil or adenine. Pairwise coexpression of RsMYB1.1 and RsTT8 of WK10039 and CUR034 led to the growth of yeast colonies, indicating that RsMYB1.1 of WK10039 and CUR034 interact with RsTT8 of WK10039 and CUR034 and vice versa. A filter assay confirmed the interactions among these proteins. Both RsMYB1.1 of WK10039 and CUR034 showed self-transcriptional activation but RsTT8 did not. RsMYB1.1 and RsTT8 showed a similar interaction in AH109 yeast (data not shown). Therefore, RsMYB1.1 of nR and R radishes may contribute similarly to the formation of the MBW complex. Nevertheless, the downregulation of RsMYB1.1 likely suppresses anthocyanin biosynthesis in nR radishes.
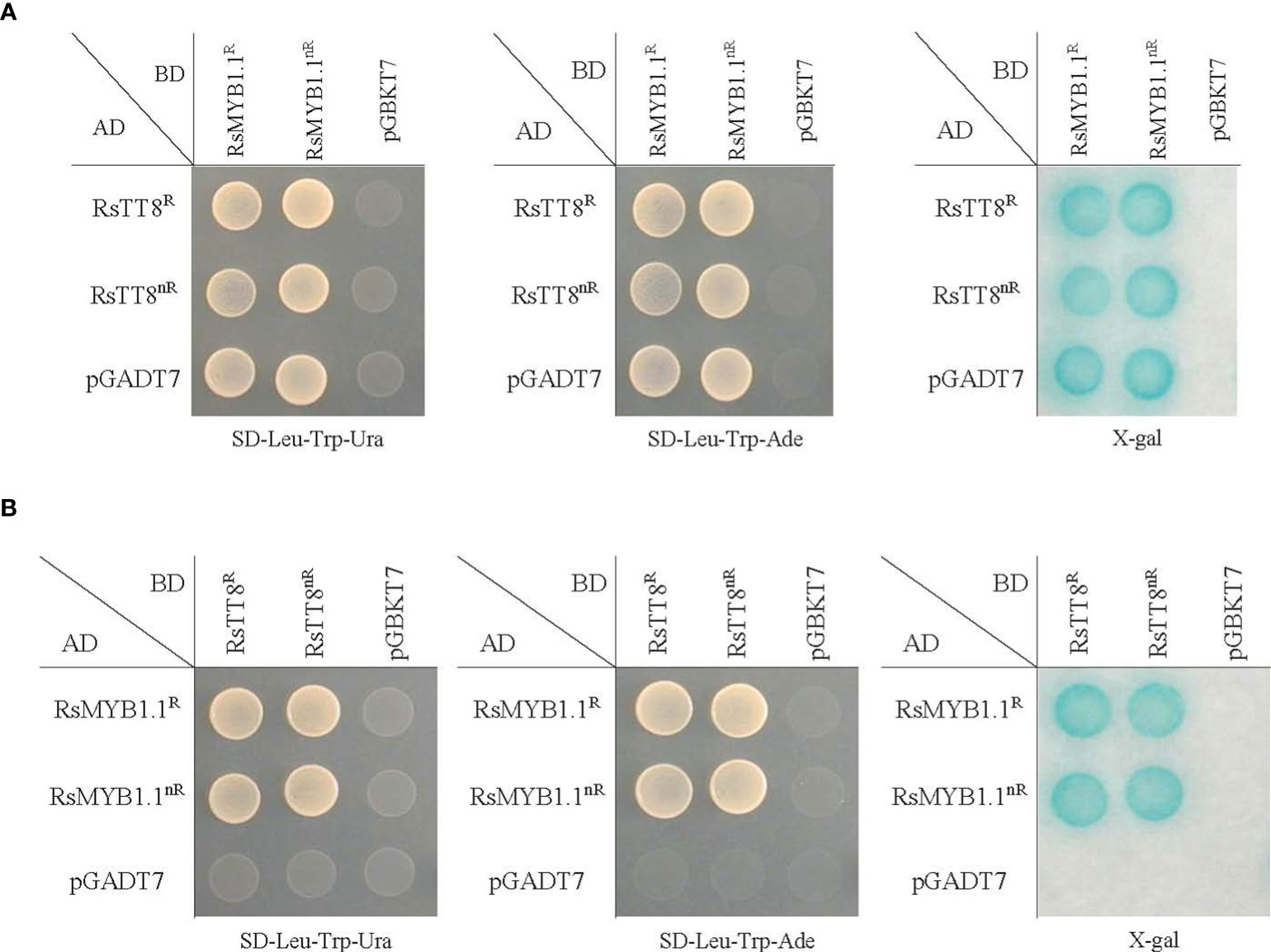
Figure 5 Pairwise interaction between RsMYB1.1 and RsTT8 in the yeast two-hybrid (Y2H) assay. The prey vector pGADT7 (AD) and the bait vector pGBKT7 (BD) are Y2H vectors without an insert. RsMYB1.1 and RsTT8 of WK10039 (nR) and CUR034 (R) were cloned into pGADT7 and pGBKT7. PBN204 yeast was cotransformed with the bait (BD) and prey (AD) vectors. Interactions between the two proteins were identified in both cases using RsMYB1.1 as the bait and RsTT8 as the prey (A) and RsMYB1.1 as the prey and RsTT8 as the bait (B).
To determine whether the RsMYB1.1 proteins of nR and R radishes are functionally active in anthocyanin biosynthesis, we expressed CDSs of RsMYB1.1nR and RsMYB1.1R individually or together with RsTT8nR or RsTT8R in N. benthamiana leaves under the control of the 35S promoter and measured the anthocyanin contents 5 days after Agrobacterium infiltration (Figure 6). Transient overexpression of individual RsMYB1.1 or RsTT8 hardly changed leaf color and led to an accumulation of a small amount of anthocyanin (2.7–8.3 mg/100 g FW) in tobacco leaves, whereas expression of the nR radish genes resulted in slightly greater accumulation of anthocyanins than the R radish genes. Coexpression of RsMYB1.1nR with RsTT8nR resulted in a pale-red leaf and the highest accumulation of anthocyanins (30.1 mg/100 g FW), which was approximately 5.3- and 1.6-fold higher than caused by the expression of RsMYB1.1nR and coexpression of RsMYB1.1R with RsTT8R, respectively. Interestingly, the combinations of RsMYB1.1nR with RsTT8R and RsMYB1.1R with RsTT8nR also increased the accumulation of anthocyanins (23.4–28.3 mg/100 g FW) compared to the expression of RsMYB1.1 or RsTT8 individually. Coexpression of RsMYB1.1nR with RsTT8nR also increased anthocyanin accumulation in WK10039 and N. tabacum leaves (Supplementary Figure S5). Therefore, RsMYB1.1nR has no functional defect compared to RsMYB1.1R despite three different amino acid residues as a result of SNPs in its CDS, and coexpression of RsMYB1.1 with RsTT8 increased anthocyanin biosynthesis. The proposed role of RsIS in the promoter region of RsMYB1.1nR needs to be confirmed by expressing genes under the control of the native promoter.
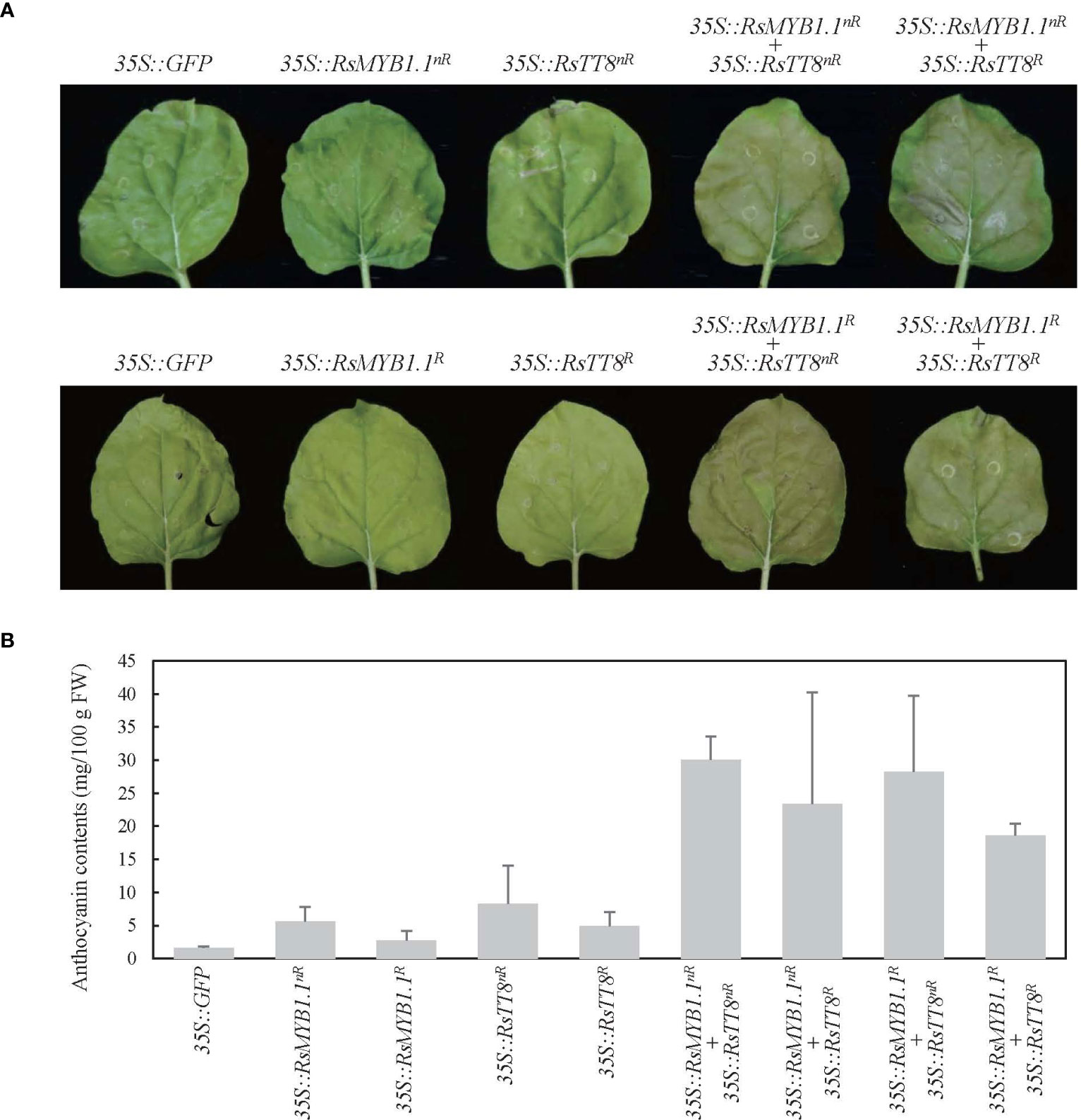
Figure 6 Transient expression of RsMYB1.1 and RsTT8 in tobacco leaves. (A) RsMYB1.1 and RsTT8 of WK10039 (nR) and CUR034 (R) under the control of the 35S promoter were expressed individually or coexpressed in N. benthamiana leaves. Photographs were obtained 5 days after Agrobacterium infiltration. (B) Total anthocyanin levels in Agrobacterium-infiltrated tobacco leaves. Error bars depict the standard deviations of three independent biological replicates.
The function of the RsIS in RsMYB1.1nR expression was investigated following the transient transformation of radish root sections by Agrobacterium infiltration. To determine whether the RsIS negatively regulates the activity of the RsMYB1.1nR promoter, we cloned the promoters of RsMYB1.1nR and RsMYB1.1R and generated seven promoter::GUS fusion constructs (Figure 7A). The P1 to P4 constructs included the 3.2 kb native promoter of RsMYB1.1nR and successive deletions of the RsIS region. The P5 and P6 constructs contained 5’-upstream 0.6 kb and 3’-downstream 1.4 kb fragments of the RsMYB1.1nR promoter, respectively. The P7 construct has the 1.9 kb native promoter of RsMYB1.1R, which corresponds to P1 except for the RsIS. Three independently transformed root sections of WK10039 were examined to analyze the effect of the RsIS on gene expression.
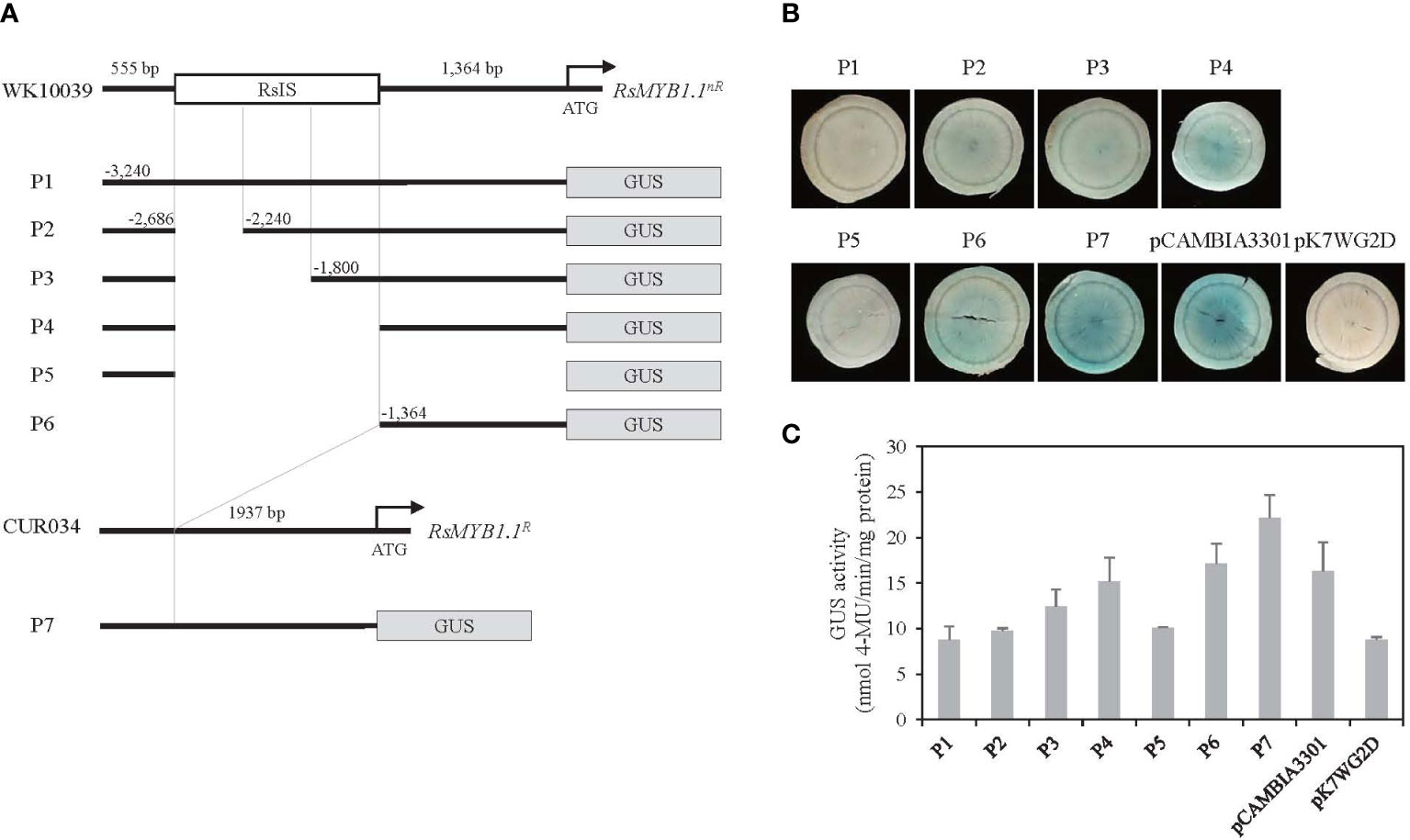
Figure 7 Histochemical analysis of transient GUS expression in radish taproot section. (A) Schematics of RsMYB1.1 promoter::GUS fusion constructs. P1 to P6 are successive deletion constructs for pRsMYB1.1nR of WK10039. P7 is a construct for pRsMYB1.1R of CUR034. The white box is the RsIS in pRsMYB1.1 of WK10039 and the gray boxes are the GUS coding regions. ATG is the start codon. Numbers are upstream positions from the start codon. (B) Photographs of GUS-stained WK10039 taproot sections obtained 3 days after Agrobacterium infiltration. GUS staining was performed for 24 h. (C) GUS activity expressed as nmol 4-methyl-umbelliferone min–1 mg–1 soluble protein. Error bars depict the standard deviations of three independent biological replicates.
No GUS expression was detected in the P1 and P5 constructs whereas strong GUS staining was observed in the P7 construct, indicating that the promoter of RsMYB1.1nR is inactive (Figure 7B). However, the expression of RsMYB1.1nR promoter::GUS was altered by deletion of the RsIS. Very faint GUS staining was detected in the P2 construct, in which one third of the RsIS was deleted. The P3 construct, in which two thirds of the RsIS was deleted, showed slightly increased GUS staining compared to the P2 construct. Moreover, distinct staining was observed in the P4 construct, in which the whole RsIS was deleted. Similar GUS staining was detected in the P6 construct, which consisted of 1.4 kb of the core promoter of RsMYB1.1nR lacking the RsIS. A fluorometric GUS assay indicated that deletion of RsIS from the promoter of RsMYB1.1nR (P4 and P6) enhanced GUS expression approximately 1.7- to 2.0-fold relative to P1, which reached 69–77% of the expression level of RsMYB1.1R (P7) (Figure 7C). Therefore, deletion of the RsIS was correlated with increased expression of the GUS reporter gene, indicating that the RsIS negatively regulates the promoter activity of RsMYB1.1nR.
Unraveling the genetic basis of agronomic traits is a challenge in agriculture and crop science. Advances in genome sequencing techniques and the availability of an increasing number of quality reference genome assemblies have enabled GWASs with the aim of discovering novel genes in diverse crop species. We identified a genetic element that negatively regulates the skin color of radish taproot by performing an InDel GWAS using the Rs2.0 genome assembly, high-depth resequencing data, and phenotype data of 100 radish accessions. We identified a highly variable region in the promoter of RsMYB1.1 on the R2 chromosome. Although we detected three nonsynonymous SNPs in the third exon of RsMYB1.1 between nR and R radishes, the RsMYB1.1 proteins of nR and R radishes had no functional differences. This suggests that nonsynonymous SNPs in the coding sequence of RsMYB1.1 are likely to have arisen as a result of selective sweep during breeding of cultivars. By contrast, insertion of 1.3 kb of the AT-rich RsIS in the promoter region of RsMYB1.1nR significantly downregulated its expression, suppressing anthocyanin biosynthesis and the growth of nR radishes. Successive deletions of RsIS from promoter sequences indicated that its removal restores the activity of the RsMYB1.1nR promoter. Therefore, this type of PAV could be the genetic basis of the phenotypic difference between nR and R radishes.
The sizes of plant genomes tended to increase during evolution as a result of, for example, occasional polyploidy and the accumulation of repetitive sequences. In the tribe Brassiceae of the family Brassicaceae, radish and its closely related diploid Brassica species (B. rapa, B. nigra, and B. oleracea) underwent triplication of their entire genomes, resulting in genomes approximately 4.2- to 4.6-fold larger than A. thaliana. Differential lineage-specific amplification of repetitive sequence elements is likely to be responsible for the variation in size and function in the radish and Brassica genomes. Radish arose from the Brassica B genome lineage and is a sibling species of B. nigra rather than B. rapa and B. oleracea (Cho et al., 2022). The most important differences between the radish and B. nigra genomes are the proportions of repetitive sequences and uniquely amplified elements. The repetitive sequence fraction of the radish genome is 38%, which is smaller than that of B. nigra (49–54%) but similar to those of B. rapa (38%) and B. oleracea (39%). Although the predominant repetitive sequences in the radish and B. nigra genomes are Ty3/Gypsy retrotransposons, Ale family Ty1/Copia retrotransposons have been amplified in the chromosomes and centromeres of B. nigra as a result of recent nested transposition events (Perumal et al., 2020). In this study, a low-complexity sequence, RsIS, was amplified only from the radish genome. Based on the number of short repeat units in the RsIS, we hypothesize that replication slippage or slipped-strand mispairing may have resulted in misalignment of DNA strands during the replication of short, repeated DNA sequences, leading to the emergence of the RsIS. Chromosome rearrangement, homologous exchange, and unequal crossing-over in the radish lineage after splitting of the radish and B. nigra genomes 11.1 Mya may have contributed to the distribution of the RsIS throughout the chromosomes of radish.
Lineage-specific amplification of repetitive sequences can be a major source of insertions, resulting in PAVs with profound effects on phenotypic and genomic variation. In the sorghum genome, the major components of PAVs are mobile elements and repeat sequences (Zhang et al., 2014). Insertion of TEs can affect the expression of nearby genes, and regulate gene function in diverse ways such as cis up- or downregulation of transcription by insertion at the promoter, exon, intron, and downstream regions, and trans production of short interfering RNAs (siRNAs) via two RNA-directed DNA methylation pathways (Gill et al., 2021). Similarly, the insertion of low-complexity AT-rich repetitive sequences may modify the expression of nearby genes by altering the promoter, coding, and downstream regions or the topology of the chromatin strand. We detected CNVs (132 to 183 copies) of RsIS among the radish genomes sequenced to date. Identification of RsIS as a PAV indicates that several RsIS diverged as low-frequency PAVs, leading to novel variants and accelerating the generation of new genetic elements that negatively regulate phenotypic traits. These results are consistent with the findings of a radish pan-genome study, suggesting that insertion is more important than deletion in the SVs in Raphanus genomes (Zhang et al., 2021). Because AT-rich DNA is concentrated in the nucleosome-free regions (NFRs) associated with the transcription start sites of most genes (Lorch et al., 2014), RsIS may disturb the transcription of RsMYB1.1nR by acting as a redundant NFR at the wrong position or changing the topology of the chromatin strand. Additionally, AT-rich sequences are known to interact with regulatory proteins and especially non-coding RNAs involved in transcriptional regulation (Kato et al., 2023). These interactions can in turn fine-tune gene expression levels in response to cellular signals. Further study is needed to determine whether RsIS influences the transcription of genes or the conformation of chromatin.
RsMYB1 is a radish ortholog of AtPAP1 (AT1G56650) which encodes a key transcription factor involved in anthocyanin accumulation in A. thaliana. Three or four RsMYB1 paralogs have been identified on the R2, R5, and R7 chromosomes of radish. RsMYB1.1 (R2.009390) is one of three RsMYB1 paralogs in the genome of WK10039; the others are RsMYB1.2 (R5.050800) and RsMYB1.4 (R7.017240); RsMYB1.3, which was identified as Rsa10033919 on the R7 chromosome of cv. XYB36–2 (Liu et al., 2019a), was absent in the WK10039 genome. In the cv. XYB36–2 genome, RsMYB1.3 and RsMYB1.4 are separated by approximately 1.97 Mb on the R7 chromosome and are more closely related than the other two paralogs (Supplementary Figure S6). To date, three RsMYB1 paralogs (RsMYB1.1, RsMYB1.3, and RsMYB1.4) have been suggested as candidate genes related to anthocyanin biosynthesis in radish, however, which is the key determinant of red taproot depends on the accessions analyzed. QTL-Seq combined with linkage analysis suggested RsMYB1.1 as a candidate gene for the purple skin of the taproot of cv. CX16Q-25–2, however, the function of this gene and its transcriptional regulation are unclear (Liu et al., 2019a). RsMYB1.3 reportedly regulates anthocyanin biosynthesis in cv. Bordeaux (Lim et al., 2016) and cv. Xinlimei (Liu et al., 2019b), both of which have taproots with red flesh. The involvement of RsMYB1.4 in anthocyanin synthesis has been reported in the red-skinned radish cv. Lian Yan No. 1 (Yi et al., 2018). In addition, QTL-seq and transcriptome sequencing of the red-fleshed radish cv. MTH01 (Wang et al., 2020) and a molecular genetic study of the red-skinned radish cv. HongPi (Kim et al., 2021) have identified RsMYB1.4. Interestingly, a short insertion in the exon of RsMYB1.3 and epigenetic regulation of the promoter region of RsMYB1.4 disrupts anthocyanin accumulation. Because RsMYB1.1 and RsMYB1.3/RsMYB1.4 are located on different chromosomes and are separated into distinct phylogenetic clades, we hypothesize that RsMYB1.1 and RsMYB1.3/RsMYB1.4 have different functions in the tissues of the radish taproot. We propose that the function of RsMYB1.1 is restricted to the skin, compared to the flesh and skin for RsMYB1.3/RsMYB1.4. This suggests that the RsMYB1 paralog that regulates anthocyanin synthesis in radish taproot is genotype dependent. It is important to categorize radish genetic resources according to the expression of RsMYB1 paralogs to enable targeted breeding of radish.
Anthocyanin biosynthesis is a multistep process transcriptionally regulated by the MBW complex, which interacts with MYB- and bHLH-recognition elements in the promoter regions of anthocyanin biosynthesis genes (Zhu et al., 2015). Our findings show that the expression of RsMYB1.1 must be coordinated with that of RsTT8 for the accumulation of anthocyanins in radish and tobacco tissues. Irrespective of what nR and R radish lines the two proteins originated from, coexpression of RsMYB1.1 and RsTT8 is necessary for anthocyanin biosynthesis; overexpression of each gene individually did not result in anthocyanin biosynthesis. Given the lack of RsTT8nR expression in the taproots of nR radishes and the absence of any structural variation in the promoter, CDS, and downstream region of RsTT8 between nR and R radishes, it is probable that the transcriptional regulation of RsTT8nR is different from that of RsMYB1.1nR. According to previous studies of transcription factor genes in anthocyanin pathways (Oikawa et al., 2015; Zhang et al., 2019; Wang et al., 2020; Yang et al., 2021), remote transcriptional regulators, post-transcriptional regulation, or epigenetic mechanisms may influence the expression of RsTT8nR. Genome-wide miRNA sequencing or estimation of the degree of methylation of RsTT8nR and its flanking sequences by McrBC-PCR and bisulfite sequencing of nR radish will provide insight into the regulatory mechanism of RsTT8nR expression. Our hybridization experiment of radish accessions with red-skin/white-flesh taproots and white-skin/red-flesh taproots demonstrated that the red-skin color is inherited as a single dominant trait whereas red flesh is unstable and maternally influenced (unpublished data). Therefore, RsTT8 may act as a maternal regulator of flesh color. In A. thaliana, fatty acid accumulation in seeds and triploid seed development are maternally controlled by TT8 (Chen et al., 2014; Zumajo-Cardona et al., 2023). Characterization of the functions of RsTT8 and RsMYB1.1 may enable the development of a method to control taproot pigmentation for use in radish breeding programs.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
JK: Formal Analysis, Resources, Writing – original draft. HJ: Data curation, Formal Analysis, Methodology, Software, Writing – review & editing. SH: Data curation, Resources, Writing – review & editing. AC: Data curation, Resources, Software, Validation, Visualization, Writing – review & editing. BY: Methodology, Resources, Writing – review & editing. S-HJ: Data curation, Resources, Writing – review & editing. HK: Data curation, Resources, Writing – review & editing. H-JY: Conceptualization, Data curation, Funding acquisition, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing. J-HM: Conceptualization, Data curation, Funding acquisition, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Research Foundation of Korea (grant number NRF-2017R1D1A1B06029741 to J-HM and NRF-2021R1F1A1049631 to H-JY) and Rural Development Administration (PJ015773 to J-HM).
We would like to thank the SEEDERS (Daejeon, South Korea) for technical support of data analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1327009/full#supplementary-material
Alonge, M., Wang, X., Benoit, M., Soyk, S., Pereira, L., Zhang, L., et al. (2020). Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell 182, 145–161.e123. doi: 10.1016/j.cell.2020.05.021
Alseekh, S., Kostova, D., Bulut, M., Fernie, A. (2021). Genom-wide association studies: assessing trait characteristics in model and crop plants. Cell Mol. Life Sci. 78, 5743–5754. doi: 10.1007/s00018-021-03868-w
Alwen, A., Moreno, R., Vicente, O., Heberle-Bors, E. (1992). Plant endogenous β-glucuronidase activity: how to avoid interference with the use of the E. coli β-glucuronidase as a reporter gene in transgenic plants. Transgenic Res. 1, 63–70. doi: 10.1007/BF02513023
Arro, J., Labate, J. (2022). Genetic variation in a radish (Raphanus sativus L.) geodiversity collection. Genet. Resour Crop Evol. 69, 163–171. doi: 10.1007/s10722-021-01212-6
Ashikawa, I., Hayashi, N., Yamane, H., Kanamori, H., Wu, J., Matsumoto, T., et al. (2008). Two adjacent nucleotide-binding site-leucine-rich repeat class genes are required to confer Pikm-specific rice blast resistance. Genetics 180, 2267–2276. doi: 10.1534/genetics.108.095034
Becker, G. (1962). “Rettich and Radies (Raphanus sativus)” in Handbuch der Pflanzenzüchtung, eds. Roemer, T. H., Rudorf, W. (Berlin und Hamburg: Paul Parey), 23–78.
Chen, M., Xuan, L., Wang, Z., Zhou, L., Li, Z., Du, X., et al. (2014). TRANSPARENT TESTA8 inhibits seed fatty acid accumulation by targeting several seed development regulators in Arabidopsis. Plant Physiol. 165, 905–916. doi: 10.1104/pp.114.235507
Cho, A., Jang, H., Baek, S., Kim, M.-J., Yim, B., Huh, S., et al. (2022). An improved Raphanus sativus cv. WK10039 genome localizes centromeres, uncovers variation of DNA methylation and resolves arrangement of the ancestral Brassica genome blocks in radish chromosomes. Theor. Appl. Genet. 135, 1731–1750. doi: 10.1007/s00122-022-04066-3
Cook, G., Benton, M., Akerley, W., Mayhew, G., Moehlenkamp, C., Raterman, D., et al. (2020). Structural variation and its potential impact on genome instability: novel discoveries in the EGFR landscape by long-read sequencing. PloS One 15, e0226340. doi: 10.1371/journal.pone.0226340
Danecek, P., Auton, A., Abecasis, G., Albers, C., Banks, E., Depristo, M., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Espley, R., Brendolise, C., Chagné, D., Kutty-Amma, S., Green, S., Volz, R., et al. (2009). Multiple repeats of a promoter segment causes transcription factor autoregulation in red apples. Plant Cell 21, 168–183. doi: 10.1105/tpc.108.059329
George, R., Evans, D. (1981). A classification of winter radish cultivars. Euphytica 30, 483–492. doi: 10.1007/BF00034013
Gill, R., Scossa, F., King, G., Golicz, A., Tong, C., Snowdon, R., et al. (2021). On the role of transposable elements in the regulation of gene expression and subgenomic interactions in crop genomes. Crit. Rev. Plant Sci. 40, 157–189. doi: 10.1080/07352689.2021.1920731
Giusti, M., Rodriguez-Saona, L., Wrolstad, R. (1999). Molar absorptivity and color characteristics of acylated and non-acylated pelargonidin-based anthocyanins. J. Agr Food Chem. 47, 4631–4637. doi: 10.1021/jf981271k
Giusti, M., Wrolstad, R. (2001). “Characterization and measurement of anthocyanins by UV-visible spectroscopy” in Current protocols in food analytical chemistry. Eds. Ronald, E., Terry, E., Haejung, A., Eric, A., Michael, H., Davis, S., Steven, J., Charles, F., Peter, S. (Hoboken, NJ: John Wiley and Sons, Inc.), F1.2.1–F1.2.13.
Gobena, D., Shimels, M., Rich, P., Ruyter-Spira, C., Bouwmeester, H., Kanuganti, S., et al. (2017). Mutation in sorghum LOW GERMINATION STIMULANT 1 alters strigolactones and causes Striga resistance. Proc. Natl. Acad. Sci. U.S.A. 114, 4471–4476. doi: 10.1073/pnas.1618965114
Guan, J., Xu, Y., Yu, Y., Fu, J., Ren, F., Guo, J., et al. (2021). Genome structure variation analyses of peach reveal population dynamics and a 1.67 Mb causal inversion for fruit shape. Genome Biol. 22, 13. doi: 10.1186/s13059-020-02239-1
Guo, J., Cao, K., Deng, C., Li, Y., Zhu, G., Fang, W., et al. (2020). An integrated peach genome structural variation map uncovers genes associated with fruit traits. Genome Biol. 21, 258. doi: 10.1186/s13059-020-02169-y
Guo, Y., Huang, Y., Hou, L., Ma, J., Chen, C., Ai, H., et al. (2017). Genome-wide detection of genetic markers associated with growth and fatness in four pig populations using four approaches. Genet. Sel Evol. 49, 21. doi: 10.1186/s12711-017-0295-4
Hichri, I., Barrieu, F., Bogs, J., Kappel, C., Delrot, S., Lauvergeat, V. (2011). Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 62, 2465–2483. doi: 10.1093/jxb/erq442
Hu, L., Zhang, H., Yang, Q., Meng, Q., Han, S., Nwafor, C., et al. (2018). Promoter variations in a homeobox gene, BnA10.LMI1, determine lobed leaves in rapeseed (Brassica napus L.). Theor. Appl. Genet. 131, 2699–2708. doi: 10.1007/s00122-018-3184-5
Huh, S., Cho, A., Yim, B., Kim, J., Jeong, J., Jang, H., et al. (2023). Characterization of agronomic traits and genomic diversity in a newly-assembled radish core collection. Crop Sci. doi: 10.1002/csc1002.21135
Ishikura, N., Hayashi, K. (1963). Chromatographic separation and characterization of the component anthocyanins in radish root. Studies on anthocyanins, XXXVIII. Bot. Mag Tokyo 76, 6–13. doi: 10.15281/jplantres1887.76.6
Jeong, Y.-M., Kim, N., Ahn, B., Oh, M., Chung, W.-H., Chung, H., et al. (2016). Elucidating the triplicated ancestral genome structure of radish based on chromosome-level comparison with the Brassica genomes. Theor. Appl. Genet. 129, 1357–1372. doi: 10.1007/s00122-016-2708-0
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi: 10.1038/s41586-021-03819-2
Kaneko, Y., Matsuzawa, Y. (1993). “"Radish (Raphanus sativus L.),",” in Genetic improvement of vegetable crops. Eds. Kalloo, G., Bergh, B. O. (Oxford: Pergamon Press Ltd), 487–510.
Kato, M., Chen, Z., Das, S., Wu, X., Wang, J., Li, A., et al. (2023). Long non-coding RNA lncMGC mediates the expression of TGF-β-induced genes in renal cells via nucleosome remodelers. Front. Mol. Biosci. 10, 1204124. doi: 10.3389/fmolb.2023.1204124
Kim, D.-H., Lee, J., Rhee, J., Lee, J.-Y., Lim, S.-H. (2021). Loss of the R2R3 MYB transcription factor RsMYB1 shapes anthocyanin biosynthesis and accumulation in Raphanus sativus. Int. J. Mol. Sci. 22, 10927. doi: 10.3390/ijms222010927
Kim, N., Jeong, Y., Jeong, S., Kim, G., Baek, S., Kwon, Y., et al. (2016). Identification of candidate domestication regions in the radish genome based on high-depth resequencing analysis of 17 genotypes. Theor. Appl. Genet. 129, 1797–1814. doi: 10.1007/s00122-016-2741-z
Kopta, T., Pokluda, R. (2013). Yields, quality and nutritional parameters of radish (Raphanus sativus) cultivars when grown organically in the Czech Republic. Hortic. Sci. 40, 16–21. doi: 10.17221/27/2012-HORTSCI
Land, H., Humble, M. (2018). YASARA: A tool to obtain structural guidance in biocatalytic investigations. Methods Mol. Biol. 1685, 43–67. doi: 10.1007/978-1-4939-7366-8_4
Lee, Y.-J., Mun, J.-H., Jeong, Y.-M., Joo, S.-H., Yu, H.-J. (2018). Assembly of a radish core collection for evaluation and preservation of genetic diversity. Hortic. Environ. Biotechnol. 59, 711–721. doi: 10.1007/s13580-018-0079-y
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 1303.3997v2. doi: 10.48550/arXiv.1303.3997
Li, X., Cui, L., Zhang, L., Huang, Y., Zhang, S., Chen, W., et al. (2023). Genetic diversity analysis and core germplasm collection construction of radish cultivars based on structure variation markers. Int. J. Mol. Sci. 24, 2554. doi: 10.3390/ijms24032554
Lim, S.-H., Song, J.-H., Kim, D.-H., Kim, J., Lee, J.-Y., Kim, Y.-M., et al. (2016). Activation of anthocyanin biosynthesis by expression of the radish R2R3-MYB transcription factor gene RsMYB1. Plant Cell Rep. 35, 641–653. doi: 10.1007/s00299-015-1909-3
Liu, T., Wang, J., Wu, C., Zhang, Y., Zhang, X., Li, X., et al. (2019a). Combined QTL-Seq and traditional linkage analysis to identify candidate genes for purple skin of radish fleshy taproots. Front. Genet. 10, 460684. doi: 10.3389/fgene.2019.00808
Liu, T., Zhang, Y., Zhang, X., Sun, Y., Wang, H., Song, J., et al. (2019b). Transcriptome analyses reveal key genes involved in skin color changes of 'Xinlimei' radish taproot. Plant Physiol. Biochem. 139, 528–539. doi: 10.1016/j.plaphy.2019.04.006
Lorch, Y., Maier-Davis, B., Konberg, R. (2014). Role of DNA sequence in chromatin remodeling and the formation of nucleosome-free regions. Genes Dev. 28, 2492–2497. doi: 10.1101/gad.250704.114
Mckenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Oikawa, T., Maeda, H., Oguchi, T., Yamaguchi, T., Tanabe, N., Ebana, K., et al. (2015). The birth of a black rice gene and its local spread by introgression. Plant Cell 27, 2401–2414. doi: 10.1105/tpc.15.00310
Perumal, S., Koh, C., Jin, L., Buchwaldt, M., Higgins, E., Zheng, C., et al. (2020). A high-contiguity Brassica nigra genome localizes active centromeres and defines the ancestral Brassica genome. Nat. Plants 6, 929–941. doi: 10.1038/s41477-020-0735-y
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Saxena, R., Edwards, D., Varshney, R. (2014). Structural variations in plant genomes. Brief. Funct. Genom 13, 296–307. doi: 10.1093/bfgp/elu016
Tao, J., Li, S., Wang, Q., Yuan, Y., Ma, J., Xu, M., et al. (2022). Construction of a high-density genetic map based on specific-locus amplified fragment sequencing and identification of loci controlling anthocyanin pigmentation in Yunnan red radish. Hortic. Res. 9, uhab031. doi: 10.1093/hr/uhab031
Verified Market Research (2023) Global fresh radish market size by type (round root radish, elongated root radish), by application (supermarkets/hypermarkets, convenience stores), by geography scope and forecast. Available at: https://www.verifiedmarketresearch.com/product/fresh-radish-market/ (Accessed September 1, 2023).
Wang, Q., Wang, Y., Sun, H., Sun, L., Zhang, L. (2020). Transposon-induced methylation of the RsMYB1 promoter disturbs anthocyanin accumulation in red-fleshed radish. J. Exp. Bot. 71, 2537–2550. doi: 10.1093/jxb/eraa010
Wu, M., Luo, Z., Cao, S. (2023). Promoter variation of the key apple fruit texture related gene MdPG1 and the upstream regulation analysis. Plants (Basel) 12, 1452. doi: 10.3390/plants12071452
Xiao, H., Jiang, N., Schaffner, E., Stockinger, E., van der Knaap, E. (2008). A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 319, 1527–1530. doi: 10.1126/science.1153040
Yang, K., Han, H., Li, Y., Ye, J., Xu, F. (2021). Significance of miRNA in enhancement of flavonoid biosynthesis. Plant Biol. 24, 217–226. doi: 10.1111/plb.13361
Ye, K., Schulz, M., Long, Q., Apweiler, R., Ning, Z. (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. doi: 10.1093/bioinformatics/btp394
Yi, G., Kim, J.-S., Park., J., , Shin, H., Yu, S., Park., S., et al. (2018). MYB1 transcription factor is a candidate responsible for red root skin in radish (Raphanus sativus L.). PloS One 13, e0204241. doi: 10.1371/journal.pone.0204241
Yuan, Y., Pe, B., Batley, J., Edwards, D. (2021). Current status of structural variation studies in plants. Plant Biotechnol. J. 19, 2153–2163. doi: 10.1111/pbi.13646
Zha, J., Koffas, M. (2017). Production of anthocyanins in metabolically engineered microorganisms: Current status and perspectives. Synth Syst. Biotechnol. 2, 259–266. doi: 10.1016/j.synbio.2017.10.005
Zhang, L., Hu, J., Han, X., Li, J., Gao, Y., Richards, C., et al. (2019). A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 10, 1–13. doi: 10.1038/s41467-019-09518-x
Zhang, X., Liu, T., Wang, J., Wang, P., Qiu, Y., Zhao, W., et al. (2021). Pan-genome of Raphanus highlights genetic variation and introgression among domesticated, wild, and weedy radishes. Mol. Plant 14, 2032–2055. doi: 10.1016/j.molp.2021.08.005
Zhang, L., Luo, H., Liu, Z., Zhao, Y., Luo, J., Hao, D., et al. (2014). Genome-wide patterns of large-size presence/absence variants in sorghum. J. Integr. Plant Biol. 56, 24–37. doi: 10.1111/jipb.12121
Zhou, X., Stephens, M. (2012). Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821–824. doi: 10.1038/ng.2310
Zhu, Z., Wang, H., Wang, Y., Guan, S., Wang, F., Tang, J., et al. (2015). Characterization of the cis elements in the proximal promoter regions of the anthocyanin pathway genes reveals a common regulatory logic that governs pathway regulation. J. Exp. Bot. 66, 3775–3789. doi: 10.1093/jxb/erv173
Zumajo-Cardona, C., Aguirre, M., Castillo-Bravo, R., Mizzotti, C., Marzo, M., Banfi, C., et al. (2023). Maternal control of triploid seed development by the TRANSPARENT TESTA 8 (TT8) transcription factor in Arabidopsis thaliana. Sci. Rep. 13, 1316. doi: 10.1038/s41598-023-28252-5
Keywords: radish, red skin color, anthocyanin, GWAS, presence/absence variation, RsMYB1.1, promoter
Citation: Kim J, Jang H, Huh SM, Cho A, Yim B, Jeong S-H, Kim H, Yu H-J and Mun J-H (2024) Effect of structural variation in the promoter region of RsMYB1.1 on the skin color of radish taproot. Front. Plant Sci. 14:1327009. doi: 10.3389/fpls.2023.1327009
Received: 24 October 2023; Accepted: 18 December 2023;
Published: 08 January 2024.
Edited by:
Dan Zhu, Qingdao Agricultural University, ChinaReviewed by:
Madhab Kumar Sen, Czech University of Life Sciences Prague, CzechiaCopyright © 2024 Kim, Jang, Huh, Cho, Yim, Jeong, Kim, Yu and Mun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hee-Ju Yu, eXVoZWVqdUBjYXRob2xpYy5hYy5rcg==; Jeong-Hwan Mun, bXVuamhAbWp1LmFjLmty
†These authors have contributed equally to this work
‡ORCID: Jilin Kim, https://orcid.org/0009-0003-6222-7761
Sun Mi Huh, https://orcid.org/0009-0004-2454-1028
Ara Cho, https://orcid.org/0000-0002-5791-6795
Hee-Ju Yu, https://orcid.org/0000-0002-0123-8665
Jeong-Hwan Mun, https://orcid.org/0000-0003-4330-3637
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.