 Fei Li
Fei Li Kuanling Xi1†
Kuanling Xi1†
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 08 January 2024
Sec. Functional and Applied Plant Genomics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1324974
Introduction: Transmembrane 9 superfamily (TM9SF) proteins play significant roles in plant physiology. However, these proteins are poorly characterized in wheat (Triticum aestivum). The present study aimed at the genome-wide analysis of putative wheat TM9SF (TraesTM9SF) proteins and their potential involvement in response to nitrogen limitation and Bacillus amyloliquefaciens PDR1 treatments.
Methods: TraesTM9SF genes were retrieved from the wheat genome, and their physiochemical properties, alignment, phylogenetic, motif structure, cis-regulatory element, synteny, protein-protein interaction (PPI), and transcription factor (TF) prediction analyses were performed. Transcriptome sequencing and quantitative real-time polymerase reaction (qRT-PCR) were performed to detect gene expression in roots under single or combined treatments with nitrogen limitation and B. amyloliquefaciens PDR1.
Results and discussion: Forty-seven TraesTM9SF genes were identified in the wheat genome, highlighting the significance of these genes in wheat. TraesTM9SF genes were absent on some wheat chromosomes and were unevenly distributed on the other chromosomes, indicating that potential regulatory functions and evolutionary events may have shaped the TraesTM9SF gene family. Fifty-four cis-regulatory elements, including light-response, hormone response, biotic/abiotic stress, and development cis-regulatory elements, were present in the TraesTM9SF promoter regions. No duplication of TraesTM9SF genes in the wheat genome was recorded, and 177 TFs were predicted to target the 47 TraesTM9SF genes in a complex regulatory network. These findings offer valued data for predicting the putative functions of uncharacterized TM9SF genes. Moreover, transcriptome analysis and validation by qRT-PCR indicated that the TraesTM9SF genes are expressed in the root system of wheat and are potentially involved in the response of this plant to single or combined treatments with nitrogen limitation and B. amyloliquefaciens PDR1, suggesting their functional roles in plant growth, development, and stress responses.
Conclusion: These findings may be vital in further investigation of the function and biological applications of TM9SF genes in wheat.
Wheat (T. aestivum) is an important food crop consumed worldwide (Ismagul et al., 2014). Diverse factors, including drought, salinity, nutrient limitation, pest attack, and extreme temperatures, can affect the growth, development, quality, and yield of wheat and other crops (Zhang et al., 2021; Ahlawat et al., 2022; Dissanayake et al., 2022; Fang et al., 2022a; Venzhik et al., 2022; Wang et al., 2022; Yue et al., 2022; Yu et al., 2022). It is essential to examine the mechanisms involved in wheat growth under such conditions to improve productivity; it is also vital to boost wheat growth under these conditions. Recently, there has been much interest in exploring wheat transcriptome to elucidate how it responds to different types of stress. For instance, scientists have studied various abiotic stresses to find genes and transcription factors that could aid wheat in tolerating stress (Saidi et al., 2023). The transcriptome of wheat roots in response to drought has also been studied, pinpointing genes that could help drought tolerance (Xi et al., 2023). In addition, there have been some exciting developments in wheat omics (Tyrka et al., 2021; Sehgal et al., 2023; Szeliga et al., 2023), meaning there is much room for discoveries and research in wheat breeding. Studies have also demonstrated the presence of numerous gene families in the genetic material of wheat plants (Limin et al., 1997; Li et al., 2008; Ma et al., 2017; Peng and Yang, 2017; Qiao et al., 2017). However, despite some of these gene families being characterized and their functions having been identified, it is essential to investigate additional gene families to understand better how they are involved in wheat growth, development, or adaptation to unfavorable conditions.
Transmembrane 9 superfamily (TM9SF) proteins, also known as Nonaspanins or the endomembrane protein-70 (EMP70) family, belong to a family of proteins containing nine putative transmembrane domains and a sizeable non-cytoplasmic domain. The transmembrane domain is called the EMP70 domain. During evolution, a highly conserved group of TM9SFs is distributed in various eukaryotic species such as Drosophila, Dictyostelium, and Saccharomyces cerevisiae, mammals, and plants (Pruvot et al., 2010). The latest research findings suggest that when essential nutrients are deficient, TM9SFs are vital in regulating the activities of endosomes and lysosomes and are instrumental in facilitating the phagocytosis of bacteria and cellular adhesion and controlling various specific cellular functions (Pruvot et al., 2010; Perrin et al., 2015). However, despite the advances in genomic studies, TM9SFs in plants, especially wheat species, and their functional roles still need to be well studied. Identifying and elucidating the evolution and role of putative TM9SF proteins in wheat may be vital for improving agricultural properties and applications of wheat.
Studies have demonstrated that wheat cultivation is closely associated with bacterial communities that play crucial roles in its growth and immunity against diseases (Carril et al., 2021; Wang et al., 2021; Youseif et al., 2021). Among these bacterial communities, plant-growth-promoting bacteria (PGPB) are of utmost importance for various plant functions such as phosphorus (P) solubilization, phytohormone production, nitrogen (N) fixation, and siderophore synthesis. The interactions among the wheat and numerous PGPB species have been reported in previous studies, and the application of PGPB in enabling and increasing sustainable wheat biomass yields under diverse conditions such as nitrogen limitation, drought, and salinity are well documented (Smyth et al., 2011; Di Benedetto et al., 2017; Di Benedetto et al., 2019; Vannini et al., 2021). Thus, PGPB and wheat interaction may be a good tool for studying the interaction of wheat with rhizobacteria. Studies indicated that PGPB could mobilize organic and mineral-bound nutrients and are essential players in nitrogen fixation in plants; nitrogen-fixing bacteria have been reported to be involved in the improvement of root growth, reduction of N losses from agricultural ecosystems and increase of plant resilience to environmental stresses in wheat (Kant et al., 2010; Dal Cortivo et al., 2017; Di Benedetto et al., 2017; Maenhout et al., 2018; Wang et al., 2020). Our recent study reported that B. amyloliquefaciens PDR1 interacts with Arabidopsis thaliana and provides beneficial effects for this plant (Li et al., 2020). Specifically, B. amyloliquefaciens PDR1 enhanced the resistance of A. thaliana to alkaline stress by modulating the activity of plasma membrane H+-ATPase in the roots. However, to our knowledge, studies have yet to be reported on the effect of B. amyloliquefaciens PDR1 on gene regulation in wheat. In addition, no previous study has reported the involvement of the TM9SF genes in the molecular mechanisms of a PGPB effect on wheat growth, especially under nitrogen limitation conditions. Exploring this aspect may help improve practical PGPB applications in wheat cultivation.
Thus, in the present study, we identified and performed a genome-wide analysis of putative TM9SF genes via comprehensive phylogenetic, chromosomal location, gene structure, and expression change analyses under single or combined treatments with nitrogen limitation and B. amyloliquefaciens PDR1. The results of the present work offer essential data for additional functional characterization of various genes in wheat.
For the identification and characterization of the TraesTM9SF genes in the T. aestivum genome, wheat CDS, proteins, gff3, and genome data were downloaded from the wheat genome database (International Wheat Genome Sequencing Consortium (IWGSC) at https://www.wheatgenome.org/). The known TM9SF protein sequences of A. thaliana were retrieved and used to perform BLASTp search against wheat proteins at an E-value cutoff of 10−10. The retrieved putative TM9SF protein sequences were further confirmed by BLASTp against Hidden Markov Model of TM9SF (PF02990) downloaded from the protein families (Pfam) database (http://pfam.xfam.org/). We downloaded putative TM9SF protein sequences of A. thaliana (AthTM9SFs) from The Arabidopsis Information Resource (TAIR) database (https://www.arabidopsis.org/), while the putative TM9SF protein sequences of Sorghum bicolor (SbiTM9SFs), Setaria italica (SiTM9SFs), and Oryza sativa (OsTM9SFs) were retrieved from the corresponding genomes (accession number GCF_000003195.3 for S. bicolor, GCF_000263155.2 for S. italica, and GCF_001433935.1 for O. sativa) downloaded from the NCBI Genome Database (https://www.ncbi.nlm.nih.gov/genome/). The conserved domains within these putative TM9SF protein sequences were retrieved by the search against the Pfam V33.1-18271 PSSMs in the NCBI Conserved domains (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) to check for the presence of EMP70 domain (PF02990) before subsequent analyses.
The physicochemical properties of the TraesTM9SF genes, including the protein length, molecular weight (M.W), isoelectric point (pl), aliphatic index (Ai), instability index (II), theoretical net charge and Boman (Potential Protein Interaction) index were computed using the R package Peptides (Osorio et al., 2015). The subcellular localization of TraesTM9SF genes was analyzed using the CELLO online program (http://cello.life.nctu.edu.tw/, (Yu et al., 2006)). The amino acid composition of the protein sequences of TraesTM9SF genes was calculated using the Molecular MEGA7 program (Kumar et al., 2016) and visualized as a chord plot using the chordDiagram function from the circlize package in R (Gu et al., 2014). The phylogenetic analyses were achieved using the MEGA7 program to align full-length protein sequences of TraesTM9SFs, AtTM9SFs, SbTM9SFs, SiTM9SFs, and OsTM9SFs. After alignment, the sequences were used for neighbor-joining (NJ) tree construction with MEGA software with default settings, except for the bootstrap, which was adjusted to 1,000 replicates. The phylogenetic tree was visualized in MEGA7. The distribution of TraesTM9SF genes on wheat chromosomes was retrieved from the wheat genomic and annotation files using the TBtools software (Chen et al., 2020). The Sequence Identity and Similarity (SIAS) domain (http://imed.med.ucm.es/Tools/sias.html) was used for pairwise identity and similarity analysis of sequences of TraesTM9SF genes.
The chromosomal positions of TraesTM9SF genes were obtained from the wheat genomic files. The online tool Multiple Expectation Maximization for Motif Elicitation MEME (https://meme-suite.org/meme/tools/meme, (Bailey et al., 2015)) was used for the prediction of conserved motifs of TraesTM9SF protein sequences. The number of motifs to be predicted for each sequence was set to 10 motifs. The images of conserved motifs of TraesTM9SF proteins were retrieved from the MEME results and merged using Adobe Photoshop software (https://www.adobe.com).
The upstream 1500 bp fragments of each of the TraesTM9SF genes were retrieved from the genomic DNA sequences since PlantCARE (Lescot et al., 2002) recommend to use the upstream 1500 bp sequence of the translation start site as a promoter of a given gene, and as it was performed previously by other researchers (Li et al., 2018; Huang et al., 2021). These sequences were submitted to the PlantCARE (Lescot et al., 2002) database (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/) for predicting the cis-regulatory elements of the promotor sequences of TraesTM9SF genes. The visualization of cis-regulatory elements was achieved using the TBtools software (Chen et al., 2020). The counts of the cis-regulatory elements of TraesTM9SF genes were summarized and used for chord plot visualization with the chordDiagram function from the circlize package in R.
Synteny analysis and gene duplication of TraesTM9SF genes were performed in TBtools (Chen et al., 2020). The MCScanX toolkit (Wang et al., 2012) was used for identifying syntenic gene pairs of putative TM9SF genes, and the Advance Circos option in the TBtools software (Chen et al., 2020) was used for visualization.
The STRING tool (https://string-db.org/, (Szklarczyk et al., 2023)) was used for predicting the protein–protein interaction (PPI) network of TraesTM9SF proteins based on protein sequences with default parameters. The PPI network was downloaded and visualized in Cytoscape (https://cytoscape.org/, (Shannon et al., 2003)). The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation of TraesTM9SF protein sequences was achieved using the online eggNOG-mapper v2 (http://eggnog-mapper.embl.de, (Cantalapiedra et al., 2021)), and TBtools software (Chen et al., 2020) was used for their enrichment analysis.
To investigate the potential role of TraesTM9SF genes in nitrogen limitation and their potential regulation by the growth-promoting bacterial species B. amyloliquefaciens PDR1 (GenBank accession number: OR858947.1), wheat was cultured under nitrogen limitation conditions and treated with the bacterial inoculum. Briefly, healthy plump wheat seeds (T. aestivum L. cv. Chinese Spring), bred and preserved in our laboratory, were surface sterilized with 70% ethanol and then soaked in aerated water for 24 hours. The seeds were sown in pre-sterilized plastic pots (20 cm diameter, 1.0 kg commercial vermiculite, 6 seeds per pot, repeated 8 times) containing autoclaved commercial vermiculite with 400 mL half-strength Hoagland’s nutrient solution (2 mM KNO3, 1 mM KH2PO4, 1 mM MgSO4, 1 mM CaCl2, 0.1 mM Fe-EDTA, 1 mM H3BO3, 1 mM ZnSO4, 0.5 mM CuSO4, 0.3 mM Na2MoO4, and 1 mM MnCl2) every 5 days. The medium was buffered at pH 6.0 using diluted 2 M NaOH. The wheat seedlings were randomly assigned to a constant temperature in controlled greenhouse growth chambers (SRG-260D, Hangzhou Shoulian Instrument CO.,LTD) at a temperature of 26 ± 2°C, humidity of 72 ± 2%, and light levels of 800 μmol m−2 s−1 from fluorescent tubes and tungsten lighting. Nitrogen limitation and strain treatments were started after the wheat reached the three-leaf stage. For nitrogen limitation treatment, KNO3 in half-strength Hoagland’s nutrient solution was replaced by KCl for 5 days of nitrogen starvation (NS) treatment. Strain treatment was by inoculating 1 mL of B. amyloliquefaciens PDR1 bacterial suspension culture at an optical density (OD) of 0.5 OD at 600 nm near the roots of each seedling or liquid LB medium which served as a control (Li et al., 2020). The treatment groups were as follows: Control (cultivation a control soil), Control+PDR1 (control soil + bacteria treatment), N limitation (cultivation on a low nitrogen soil), and N limitation + PDR1 (low nitrogen soil + bacteria treatment). For each treatment group, the following indexes were recorded from 12 different plants: flag leaf area, plant height, number of spikelets per spike, number of culms per plant, kernel weight per spike, flag leaf dry matter, number of kernels per spike, and spike dry matter. Moreover, the total nitrogen content in roots (12 roots per treatment group) was determined using a CHNS Analyzer (CHNS-932, LECO, USA).
Finally, root samples from 12 different roots per treatment group were collected for whole transcriptome analysis of wheat grown in the above-mentioned conditions. Total RNA was extracted from the collected wheat root samples using the Invitrogen Plant RNA Purification kit (Carlsbad, CA, United States). The cDNA libraries were subsequently constructed, and after quality verification, the libraries were sequenced on the Illumina HiSeq 4000 apparatus as described in our previous studies (Gong et al., 2021). The RNA-seq reads underwent filtration to eliminate any low-quality reads and adaptor sequences, with a quality score threshold of Q > 20 being applied. The wheat genome was utilized as a reference for mapping the clean reads with the assistance of the TopHat spliced read aligner (Trapnell et al., 2012). The FPKM value was utilized via Cufflinks software to assess the expression levels of mRNAs (Trapnell et al., 2010). The expression matrix of TraesTM9SF genes was then extracted from the annotated sequencing data. Next, to identify the expression profiles of TraesTM9SF genes, Short Time-series Expression Miner (STEM, http://gene.ml.cmu.edu/stem/, (Ernst and Bar-Joseph, 2006)) was run on the transcriptome data. The pheatmap package was used for visualizing the expression profile of retrieved TraesTM9SF genes.
To validate the transcriptome data, we performed Quantitative Real-Time PCR (RT-qPCR). Total RNA isolation, reverse transcription, and PCR experiments were performed in triplicate as previously described (Gong et al., 2021), with the sequences of primers used summarized in Table 1. We employed the 2-ΔΔCt method (Livak and Schmittgen, 2001) to compute the relative gene expression of TraesTM9SF genes, using GAPDH (GenBank accession number: EF592180) as a reference housekeeping control.

Table 1 List of primers used in this study.
For in-silico data, we generated boxplots using the ggplot2 package (Wickham et al., 2019) and used the T-test to gauge differences between paired groups. For experimental data, we detected differences among treatment groups by performing a one-way ANOVA analysis followed by Tukey’s post hoc multiple comparison test. We used the GraphPad Prism 9 software from GraphPad Software in San Diego, California, United States, for these analyses. A difference was considered significant at a p-value cutoff of 0.05.
A total of 47 TraesTM9SF genes in the wheat genome were retrieved by bioinformatical approaches. These putative wheat TM9SF genes were renamed from TraesTM9SF-1 to TraesTM9SF-47 based on their position in the phylogenetic tree. With the exception of chromosomes Un (which contain clone contigs that cannot be confidently placed on a specific chromosome), 1A, 1B, 1D, 3A, 3B, and 3D, the TraesTM9SF genes were unevenly distributed on the other wheat chromosomes (Figure 1). Specifically, they were found on chromosomes 2A, 2B, 2D, 4A, 4B, 4D, 5A, 5B, 5D, 6A, 6B, 6D, 7A, 7B, and 7D (Figure 1). The largest number of TraesTM9SF genes (8 genes) was recorded on chromosome 7A, accompanied by chromosome 7B (7 genes), chromosomes 7D, 5A, and 5B (4 genes on each), chromosomes 5D, 6A, and 6B (3 genes each), chromosomes 4A and 4D (2 genes each) and chromosomes 2A, 2B, 2D, and 4B (one gene each) (Figure 1). The detailed information on the 47 TraesTM9SF genes was summarized in Supplementary Table S1, and the protein sequences were given in Supplementary Table S2. Conserved Domain Search (CD search) confirmed that all of the TraesTM9SF proteins contained two domains, which were EMP70 and EMP70 superfamily domains (Supplementary Table S3). It is worth noting that EMP70 is a specific domain, while EMP70 superfamily domains encompass a broader group of related domains.
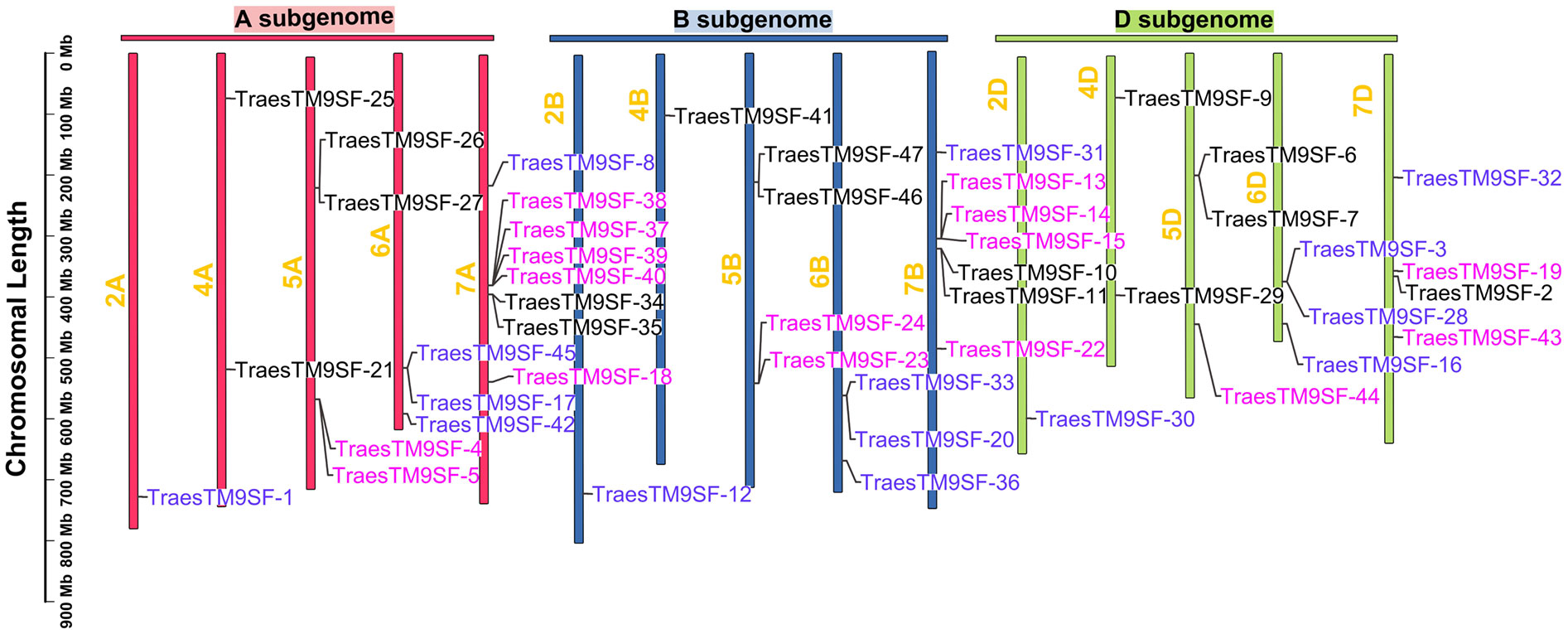
Figure 1 Chromosomal location of TraesTM9SF genes in the wheat genome.
Physiochemical analysis indicated that the TraesTM9SF genes differed in their physicochemical properties, including protein length (294 - 467 amino acids) and molecular weight ranged from 32537.32 to 48189.04 Da (Supplementary Table S1). The low value of the instability index of TraesTM9SF proteins (instability index < 40) (Supplementary Table S1) indicated that these proteins are stable (Ikai, 1980). The aliphatic index of 38.57 to 65.61 (Supplementary Table S1) indicated that TraesTM9SF proteins have moderate to high thermostability while the Boman index ranged between 1.10 and 2.25 (Additional File S1) indicated a moderate cationic charge and a moderate binding ability of TraesTM9SF proteins to other proteins (De Cena et al., 2022). The theoretical charge from 18.14 to 33.87 (Supplementary Table S1) confirmed that TraesTM9SF proteins are positively charged or cationic while the isoelectric point of 9.42 - 10.24 (Supplementary Table S1) suggested that the TraesTM9SF proteins are relatively alkaline or basic in nature. The TraesTM9SF proteins with a GRAVY index range of -0.72 to 0.09 (Supplementary Table S1) indicated that these proteins are overall relatively hydrophilic. The abundance of amino acids composing TraesTM9SF proteins was as depicted in Figure 2.
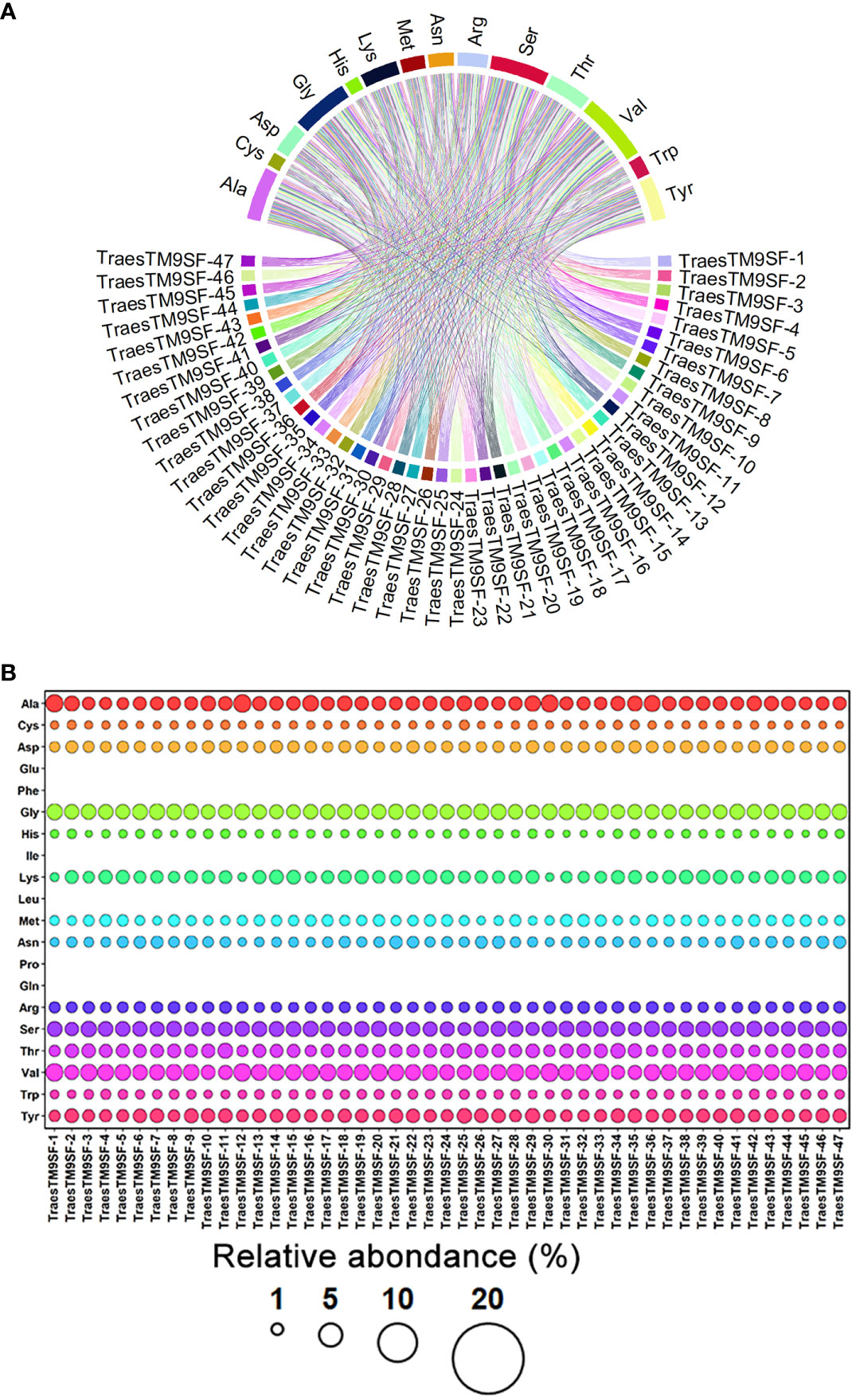
Figure 2 Amino acid composition of TraesTM9SF proteins. (A) Chord plot indicating the relationships between amino acids and different TraesTM9SF proteins. (B) Bubble plot indicating the relative abundance of amino acids in different TraesTM9SF protein sequences.
The phylogenetic analysis of putative SbiTM9SFs, SiTM9SFs, OsTM9SFs, and TraesTM9SF genes indicated that these genes were clustered into four principal groups (Figure 3). There was an uneven distribution of TraesTM9SF genes in the different groups, and some of the TraesTM9SF genes were clustered with the OsTM9SF, SiTM9SF, SbTM9SF, and AtTM9SF genes (Figure 3). These results showed a potential phylogenetic relationship between the TM9SF genes. In addition, the clustering of TraesTM9SF genes with TM9SF genes of other species (OsTM9SF, SiTM9SF, and SbTM9SF genes) indicated a possible conservation of function among these genes.
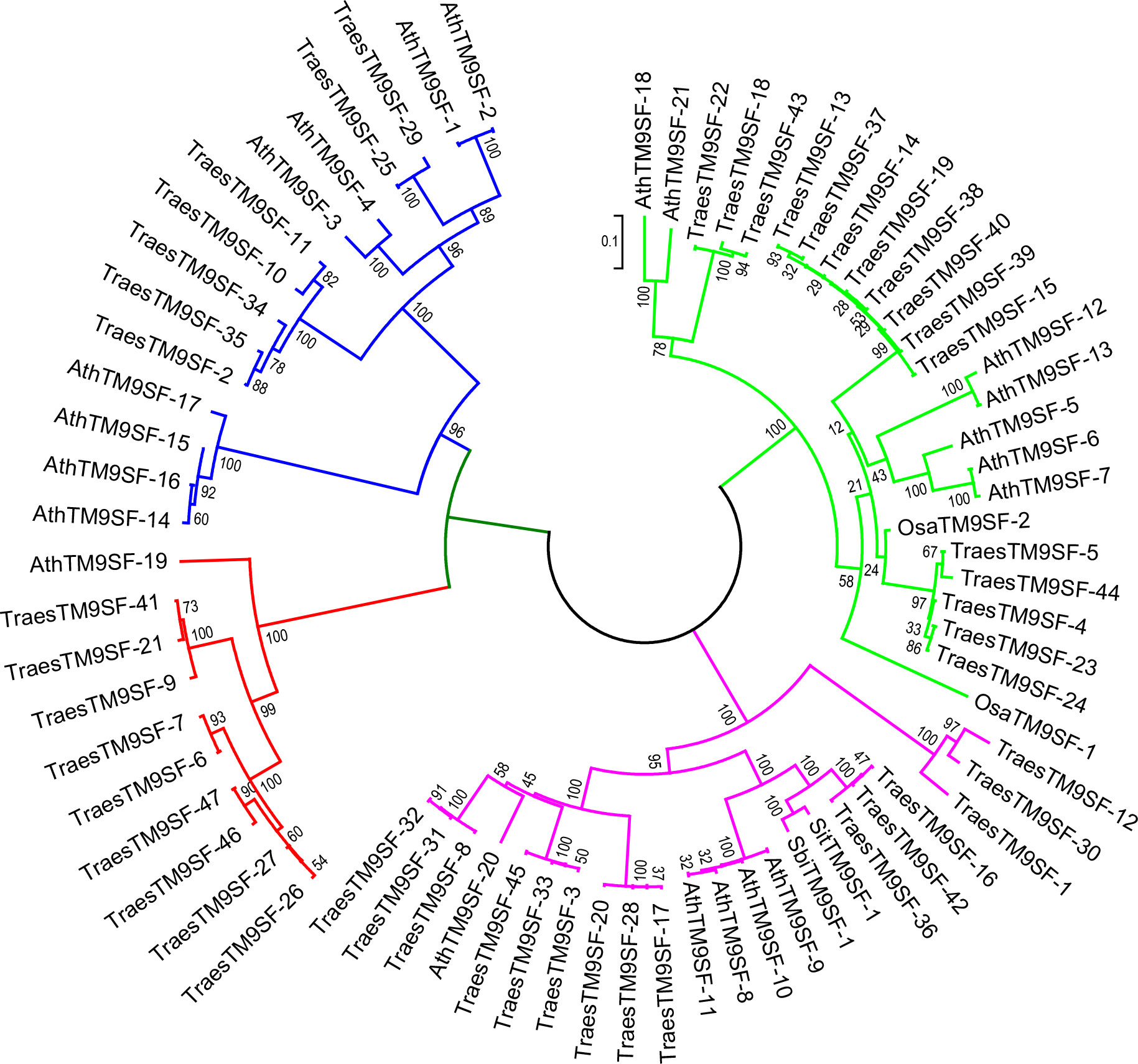
Figure 3 Neighbor joining phylogenetic tree of OsTM9SF, SiTM9SF, SbTM9SF, AtTM9SF and TraesTM9SF genes using MEGA.
To further understand the evolution of putative TM9SF genes in wheat, pairwise identity, and similarity analysis based on these genes was performed. The results (Supplementary Table S5) indicated that the similarity values ranged from 7.14% (TraesTM9SF-20/TraesTM9SF-25) to 100% (TraesTM9SF-13/TraesTM9SF-37, TraesTM9SF-38/TraesTM9SF-19 and TraesTM9SF-15/TraesTM9SF-39). The lowest similarity of 4.76% was recorded between TraesTM9SF-25 and TraesTM9SF-20 genes, while the highest identity of 99.75% was obtained between the TraesTM9SF-15 and TraesTM9SF-39 genes (Supplementary Table S5).
The relationship between the 47 TraesTM9SF genes was revealed via a phylogenetic tree, which was constructed using the NJ approach in MEGA7 (Figure 4). The fasta sequences of all TraesTM9SF proteins were summarized in Supplementary Table S4. Based on the phylogenetic relationship, TraesTM9SF proteins were divided into two main clusters in the phylogenetic tree (Figure 4). Group I was the largest group and contained 31 TraesTM9SF genes, while Group II contained 16 genes (Figure 4). Group I was divided into two subgroups, I-a and I-b; subgroup I-b was further divided into I-b-1 and I-b-2 (Figure 4). Group II was divided into subgroup II-a and subgroup II-b. In addition, the analysis of conserved motifs among the 47 TraesTM9SF genes by the Multiple Expectation Maximization for Motif Elicitation (MEME) (Bailey et al., 2009) tool indicated that each group or subgroup had a specific pattern of motifs (Figure 4). The TraesTM9SF genes in Group I were characterized by the absence of motif 7, which was present in the sequences of TraesTM9SF genes in Group II (Figure 4). The sequences in subgroup II-a were characterized by the presence of motif 5 and the absence of motif 8, while the proteins in the subgroup II-b were characterized by the absence of motif 5 and the presence of motif 8 (Figure 4). In Group I, the subgroup I-a was characterized by the absence of motif 4, while the subgroup I-b was characterized by the presence of this motif (Figure 4). In addition, the subgroup I-b-1 could be characterized by the presence of motif 9, while the subgroup I-b-2 was devoid of this motif (Figure 4). The motifs 1, 2, 3, and 6 were present on almost all of the TraesTM9SF genes (Figure 4), indicating a conserved gene structure. These observations demonstrated that TraesTM9SF genes in a given group share highly similar gene structures and the presence or absence of specific motifs can be used to classify TraesTM9SF genes into distinct subgroups. This was corroborated by their phylogenetic relationships (Figure 4).
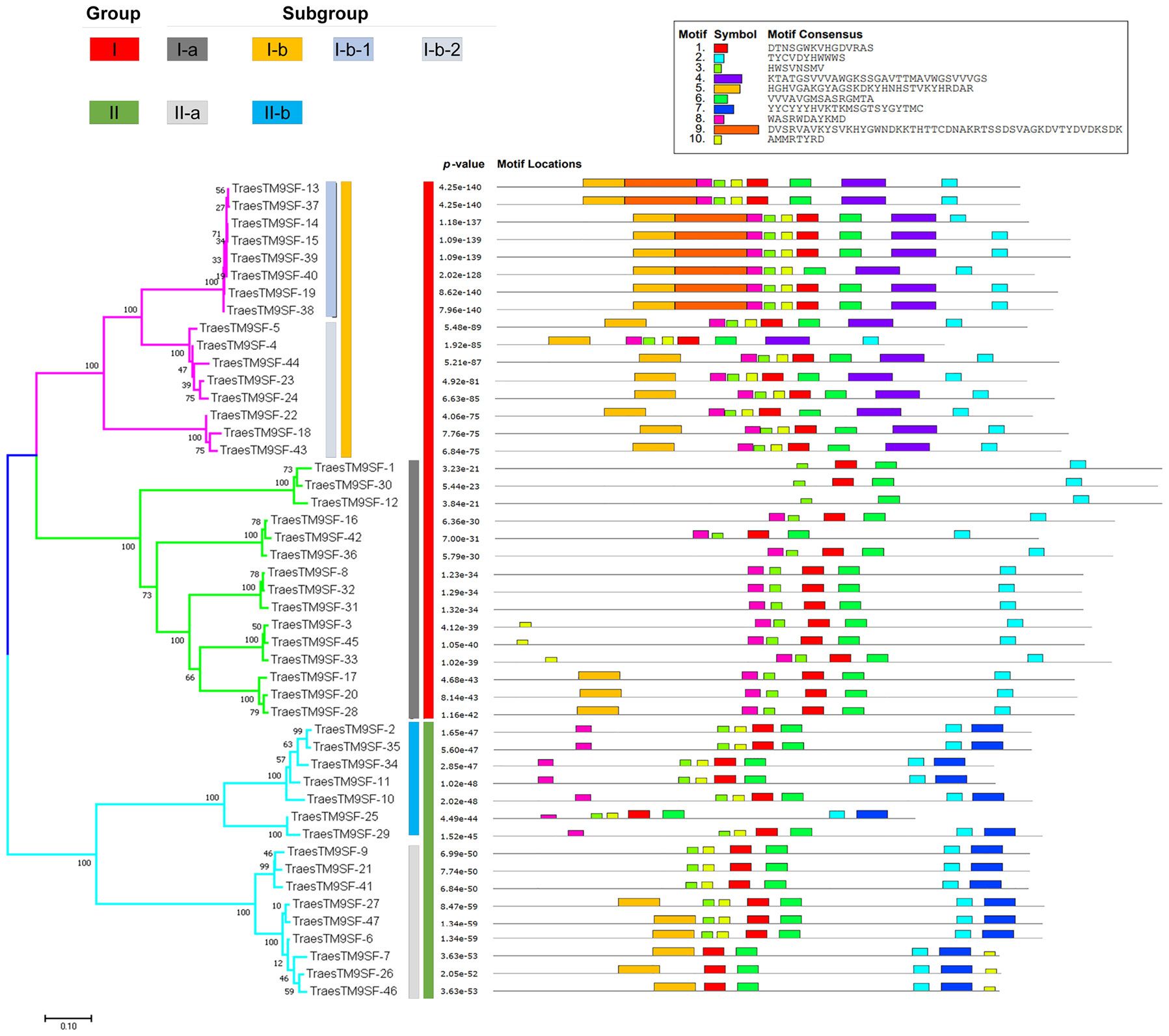
Figure 4 Motif structure and distribution on TraesTM9SF genes.
The 1500 bp upstream promoter region of each of the translated region of putative TM9SF genes in wheat was used to analyze the cis-regulatory element analysis in order to understand the role of TraesTM9SF genes. The results indicated that 54 cis-regulatory elements were present in the TraesTM9SF promoter regions (Figure 5). The light responsiveness category was the most represented and consisted of 22 cis-acting elements, including 3-AF1, 3-AF3, GT1-motif, Sp1, MBS, MBSI, MRE, CCAAT-box, ATCT-motif, Box 4, CAG-motif, chs-CMA2b, GATA-motif, GTGGC-motif, I-box, L-box, LAMP-element, TCCC-motif, TCT-motif, Box II, chs-CMA2a, and the AE-box cis-elements (Figures 5A, B). The phytohormone responsive category contained 9 cis-acting elements including TGA-element, ABRE, AuxRR-core, CGTCA-motif, TGACG-motif, P-box, GARE-motif, and TGA-box (Figures 5A, B) while the biotic/abiotic stress-related cis-elements were 29 and included WUN-motif cis-element, MBS, GC-motif, ARE, LTR, and TC-rich repeats (Figures 5A, B). The plant growth and development category comprised of 8 cis-acting elements, namely AT-rich element, Box III, CAT-box, Circadian, GCN4_motif, HD-Zip 1, O2-site, and RY-element (Figures 5A, B).

Figure 5 Cis-regulatory elements of TraesTM9SF genes. (A) Distribution of Cis-regulatory elements on TraesTM9SF Genes. (B) Heatmap showing the abundance of Cis-regulatory elements of TraesTM9SF genes.
Synteny analysis was performed to further understand the evolution and expansion mechanism of the TraesTM9SF gene family in the wheat genome. TraesTM9SF gene duplications were assessed based on a tandem or segmental duplication. The gene duplication analysis results indicated no TraesTM9SF gene pair (Figure 6), showing no duplication of TraesTM9SF genes in the wheat genome.
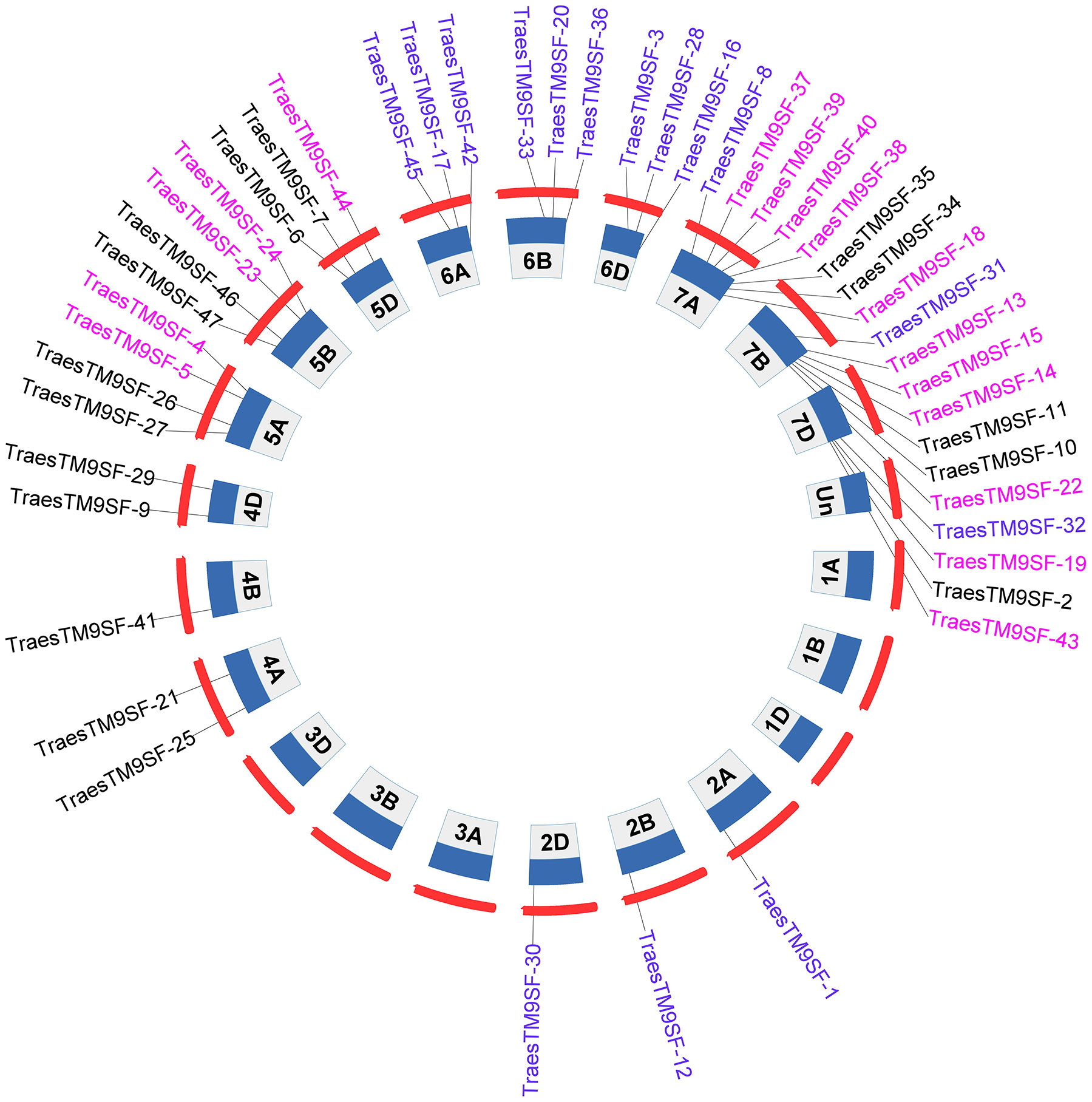
Figure 6 Synteny analysis of TraesTM9SF genes. Circos plot was used to plot gene duplications. The plot indicated no duplication of TraesTM9SF Genes.
The prediction of potential TFs targeting the 1500bp upstream region of the translated regions of putative wheat TM9SF genes indicated 776 regulations between 177 TFs and 47 genes. In addition, 98 TFs targeted TraesTM9SF genes in the input gene set under the cutoff of p-value <0.05. Figure 7 depicts the regulatory interactions between TraesTM9SF genes (in diamonds) and their TFs (in ellipses). Additionally, Figure 8 depicts the protein-protein interaction (PPI) network of TraesTM9SF genes and their TFs. The PPI network analysis indicated that the network comprised 138 nodes and 772 edges with an average number of neighbors of 11.188 (Figure 8). The functional enrichment of the TFs and TraesTM9SF proteins in the PPI network indicated that these proteins were functionally enriched in the biological processes of regulation of transcription, regulation of gene expression, regulation of molecular function, and other development-, metabolism- and cell-related processes (Supplementary Data Sheet S1). The enriched cellular components were cellular anatomical entity, nucleus, intracellular, and host cell nucleus. At the same time, the most represented molecular functions were DNA binding, DNA-binding transcription factor activity, sequence-specific DNA binding, and heterocyclic compound binding (Supplementary Data Sheet S1). The enriched pathways were plant hormone signal transduction and circadian rhythm of plants (Supplementary Data Sheet S1). The result of GO terms associated with TraesTM9SF proteins obtained from the eggNOG-mapper was depicted in Supplementary Data Sheet S2.
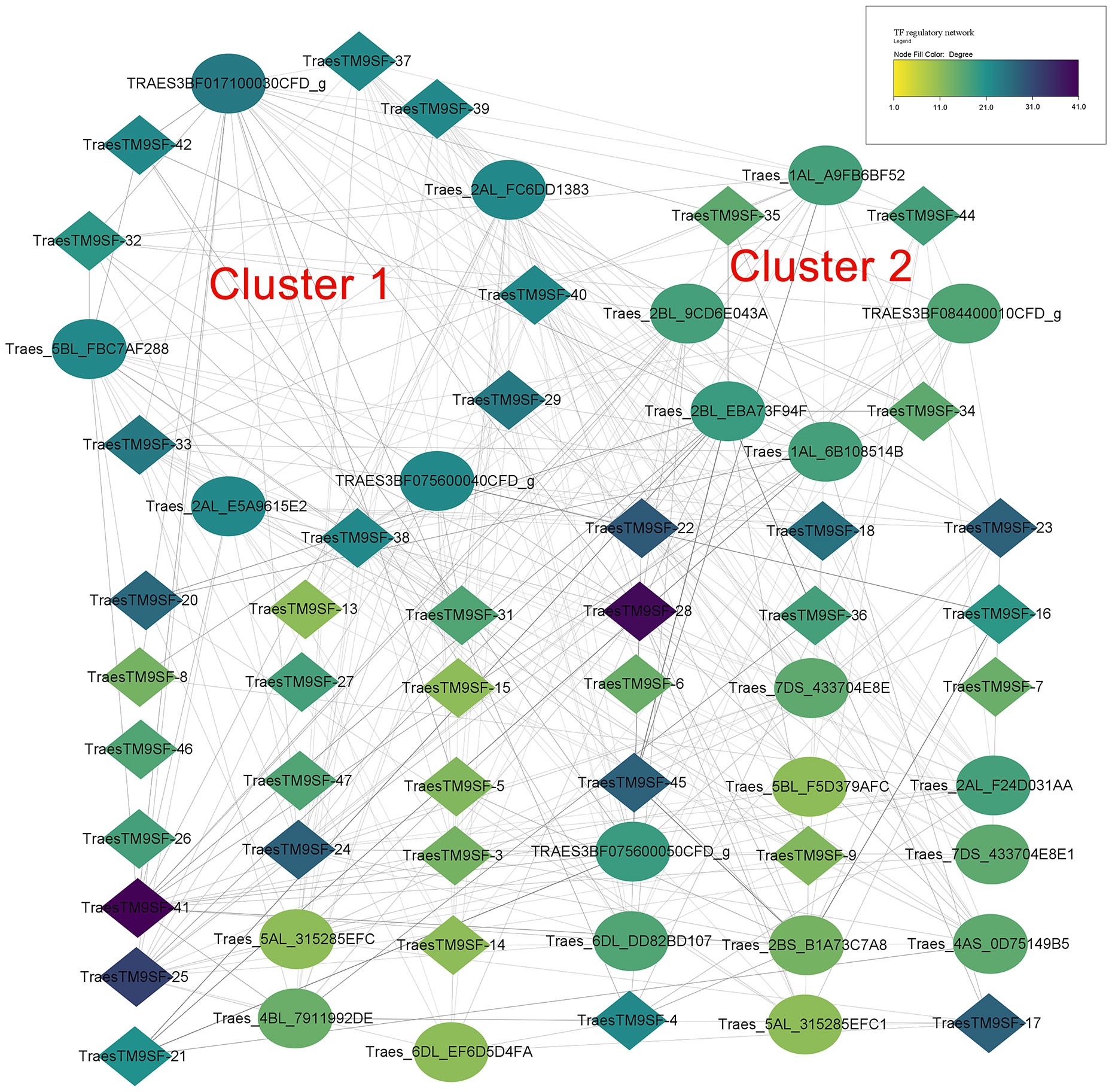
Figure 7 Regulatory network of TraesTM9SFs and their TFs. Diamonds indicate genes while the ellipses indicate TFs.
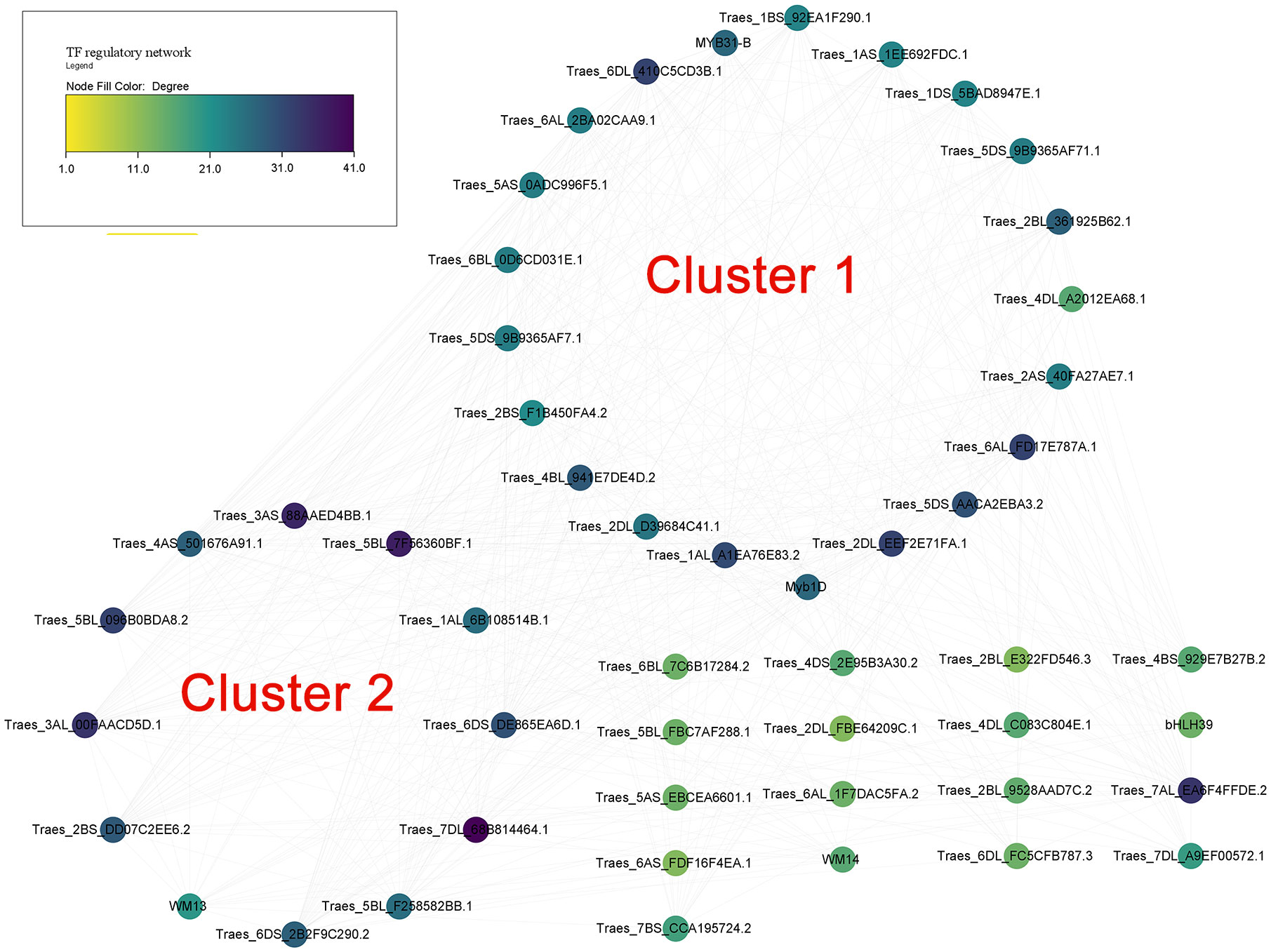
Figure 8 Protein-protein interaction (PPI) network of TraesTM9SFs and their TFs.
The functional characterization of TraesTM9SF genes has not been studied in wheat. To investigate the potential involvement of the functional role of TraesTM9SF genes in nitrogen starvation conditions and the effect of B. amyloliquefaciens PDR1, the wheat seedlings were cultivated in nitrogen limitation or combined treatment of nitrogen limitation and PDR1 strain and their relative controls. Nitrogen starvation led to deleterious changes in the plant traits (Figure 9). The growth of the wheat plant was severely affected in nitrogen limitation conditions; the measured indexes varied considerably among plants grown in normal and nitrogen limitation conditions (Figure 9). Specifically, the plants grown under nitrogen limitation conditions were shorter, and their flag leaf area and flag leaf dry matter were decreased compared to those in the normal conditions (Figure 9). In general, plants in the nitrogen limitation group had a decreased number of culms, decreased number of spikelets per spike, lower spike dry matter, lower kernel number per spike, and lower kernel weight per spike compared to the control plants (Figure 9). Moreover, the treatment with the bacterial strain PDR1 did not affect the plants grown under normal conditions but counteracted the deleterious effect of nitrogen starvation (Figure 9). Subsequently, to explore the effect of nitrogen starvation on the root transcriptome of the plant, the root samples were subjected to RNA sequencing. The heatmap showing the expression of TraesTM9SF genes in the root system of the wheat was as depicted in Figure 10A. The time series clustering was subsequently performed, and the results showed that 2 significant clustering profiles could be obtained (Figure 10B). The first cluster (profile 14) comprised 14 genes whose expression levels were increased by both the PDR1 strain and N limitation treatments with a pronounced effect for the N limitation treatment. The combined treatment with the PDR1 strain and N limitation also reversed the effect of N limitation treatment on the expression of these genes (Figures 10B, 11A, B). Interestingly, the second significant cluster (profile 7) was characterized by the upregulation of TraesTM9SF genes under nitrogen limitation conditions, but this trend was reversed by the treatment with the growth-promoting bacterial strain B. amyloliquefaciens PDR1 (Figures 10B, 11C, D; Supplementary Data Sheet S3). The genes in both profiles were considered as candidate nitrogen limitation- and PDR1-responsive TraesTM9SF genes (Supplementary Data Sheet S4). The boxplots in Figures 11B, D indicated significant differences in the expression of these genes in pairwise comparisons of treatment groups. Overall, these results showed that TraesTM9SF genes are expressed in the root system of wheat and are possibly involved in the response of this plant to biotic and abiotic stress conditions.
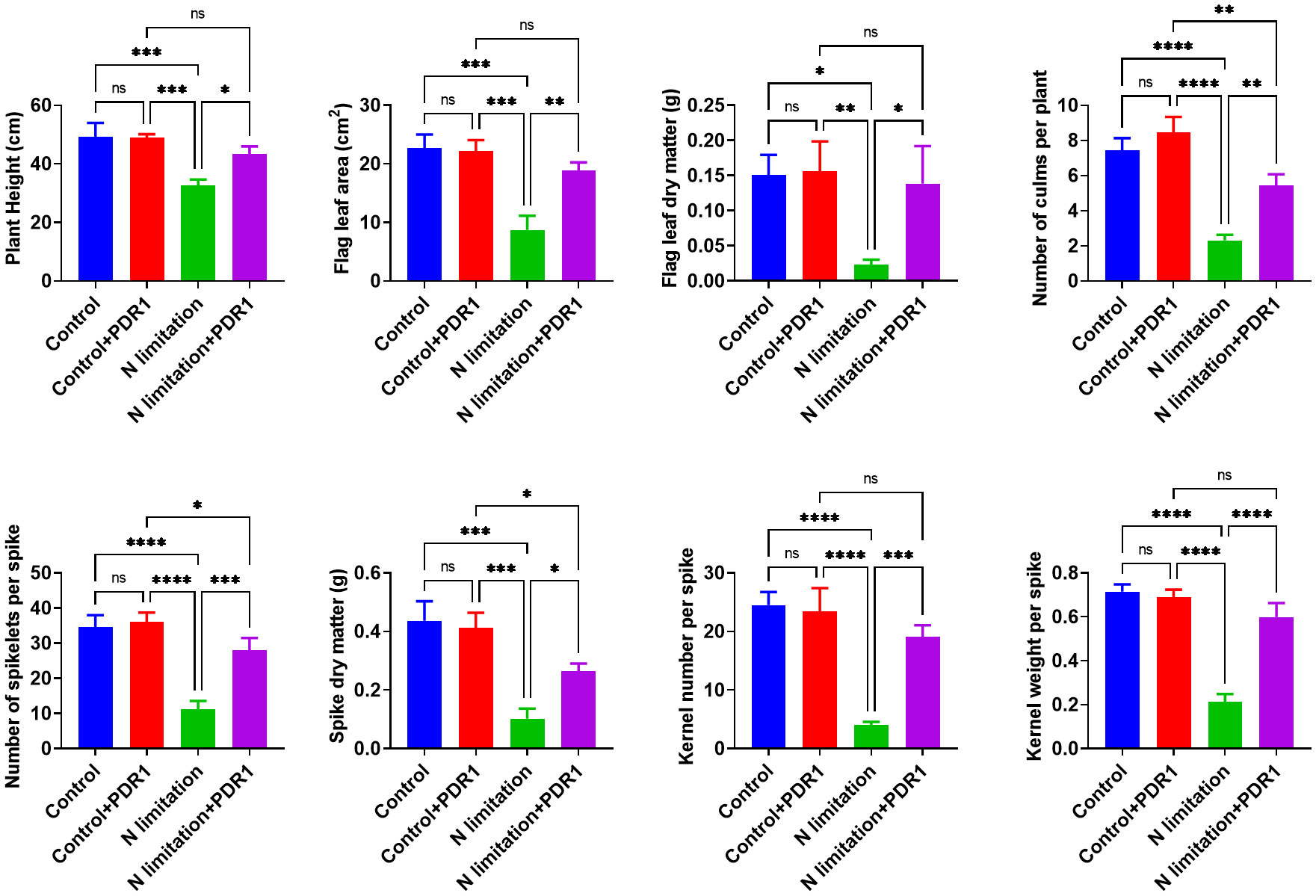
Figure 9 Determination of different indexes under stress and different treatment conditions. *p<0.05, **p<0.01, ***p<0.01 and ****p<0.01 among the compared groups; ns stands for non-significant. A total of n=12 plants per group were used and the experiments were performed in triplicate. The results are expressed as mean ± SD.
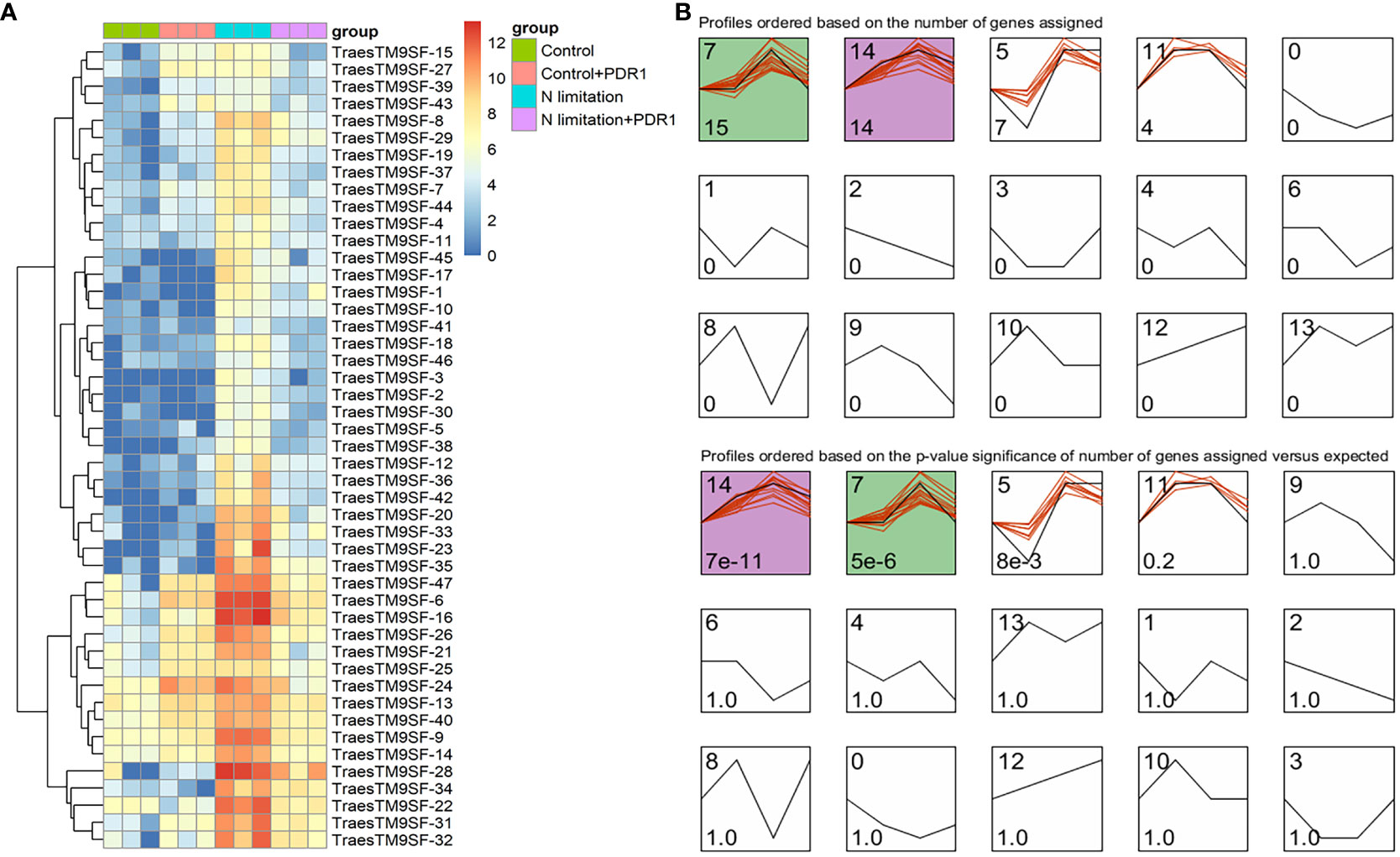
Figure 10 Transcriptome expression profile of TraesTM9SF genes by RNA sequencing under nitrogen limitation and B amyloliquefaciens PDR1 conditions. (A) Heatmap showing the expression profile of TraesTM9SF genes under different treatment conditions. (B) Clustering of TraesTM9SF genes based on their expression levels under different treatment conditions. The number in the upper right of each box corresponds to the profile number representing this cluster or group of genes while the number in the lower right indicate either the number of genes (upper panel) within each profile or the p-values (lower panel) associated with each profile.
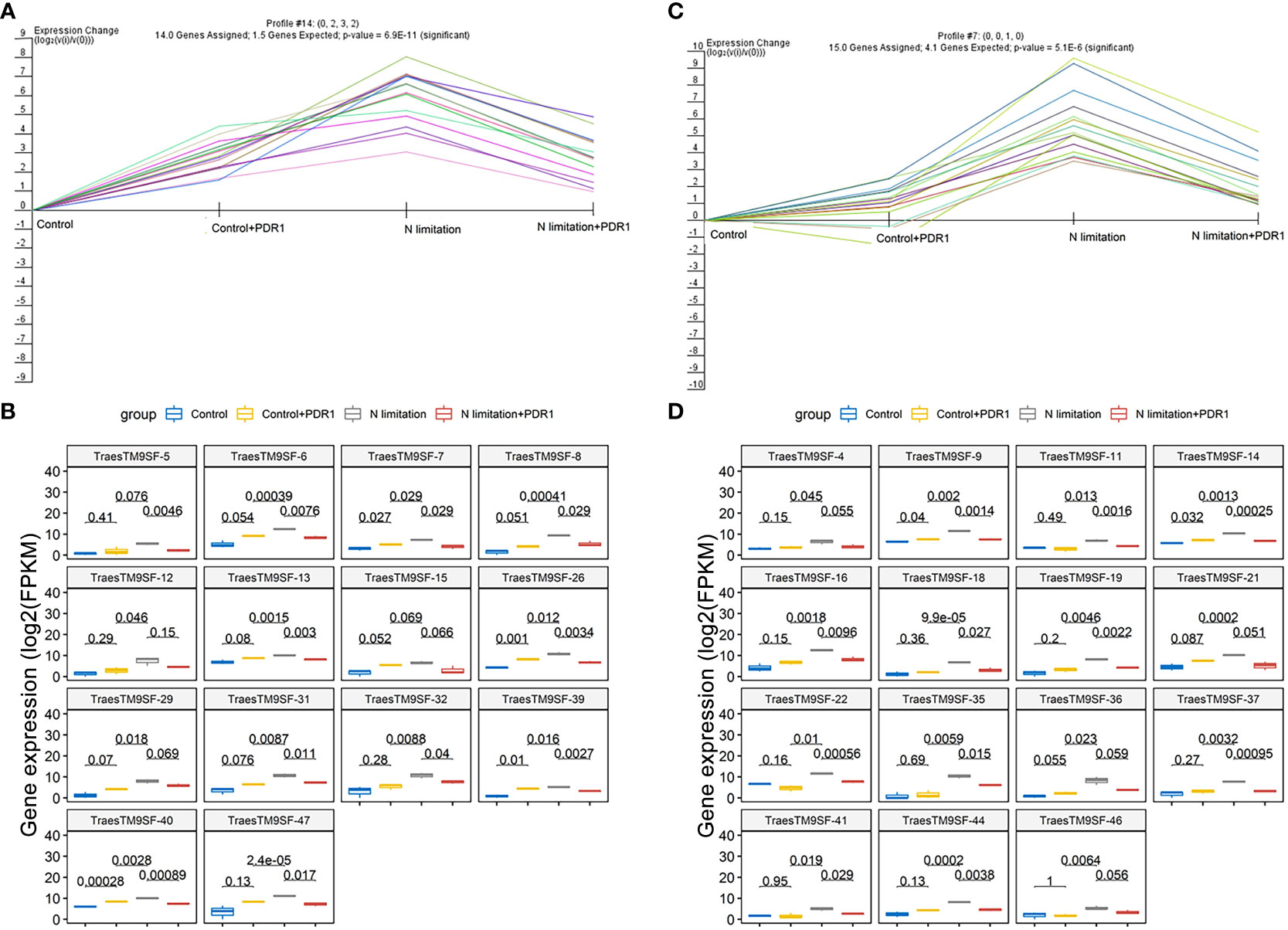
Figure 11 Transcriptome expression profile of TraesTM9SF genes in cluster 7 and cluster 14. (A) The expression profile of TraesTM9SF genes on cluster 7 under different treatment conditions. (B) Boxplots showing the expression profile changes of TraesTM9SF genes on cluster 7 under different treatment conditions. The numbers in each box represent the p-values obtained by T-test analysis among the indicated groups. (C) The expression profile of TraesTM9SF genes on cluster 14 under different treatment conditions. (D) Boxplots showing the expression profile changes of TraesTM9SF genes on cluster 14 under different treatment conditions. The numbers in each box represent the p-values obtained by T-test analysis among the indicated groups.
The TraesTM9SF-4, TraesTM9SF-5, TraesTM9SF-10, TraesTM9SF-11, TraesTM9SF-16, TraesTM9SF-17, TraesTM9SF-20, TraesTM9SF-23, TraesTM9SF-24, TraesTM9SF-28, and TraesTM9SF-44 genes were chosen for validation by qRT-PCR. After normalization with the GAPDH reference gene, the analyzed TraesTM9SF genes showed expression trends similar to that of the RNA-Seq data (Figure 12). These results showed that RNA-Seq was credible and confirmed the functional role of TraesTM9SF genes in nitrogen limitation conditions and their potential involvement in the beneficial effect of PDR1.
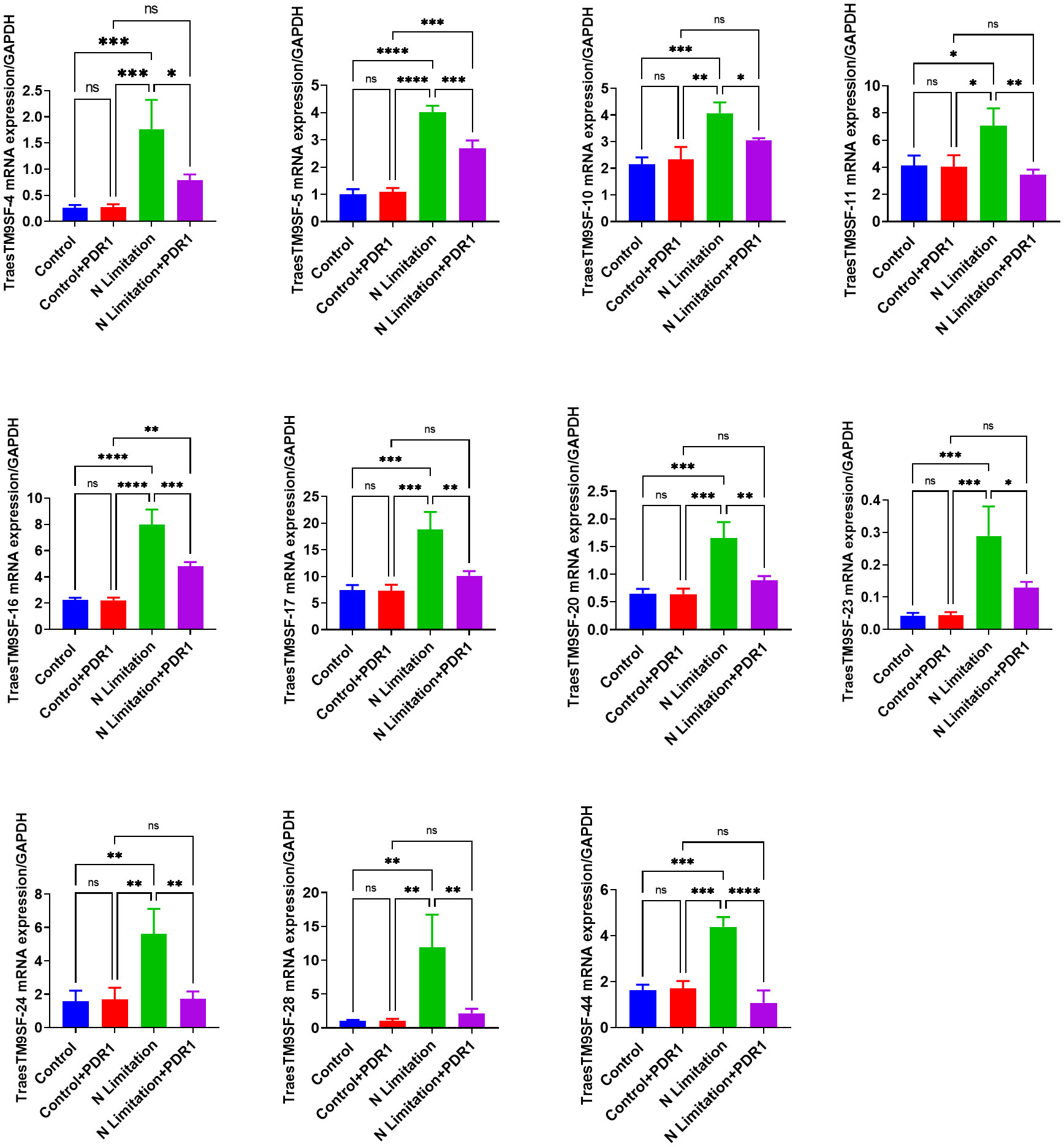
Figure 12 qRT-PCR validation of the expression of PDR1-responsive TraesTM9SF genes from transcriptome data. *p<0.05, **p<0.01, ***p<0.01 and ****p<0.01 among the compared groups; ns stands for non-significant. A total of n=12 plants per group were used and the experiments were performed in triplicate. The results are expressed as mean ± SD.
This study identified 47 TraesTM9SF genes with uneven chromosomal distribution in the wheat genome. The putative wheat TM9SF proteins contained the EMP70 and EMP70 superfamily domains and exhibited dissimilar physicochemical properties, though similarities and identities could be recorded among TraesTM9SF proteins. In addition, 37 of these TraesTM9SF proteins were most likely located in the extracellular region, whereas the other ten were located in the nucleus. The TraesTM9SF genes shared a phylogenetic relationship with the OsTM9SF, SiTM9SF, SbTM9SF, and AtTM9SF genes. Moreover, phylogenetic differences were found between TraesTM9SF genes based on the conserved motifs found on these genes. Fifty-four cis-regulatory elements were identified in the TraesTM9SF promoter regions and were mainly associated with light responsiveness, hormone response, and drought-inducibility. The gene duplication analysis results indicated no TraesTM9SF gene pair, showing no duplication of TraesTM9SF genes in the wheat genome. Moreover, 177 TFs regulating 47 TraesTM9SF genes were identified and formed a complex regulatory network with TM9SF proteins. Transcriptome analysis and validation by qRT-PCR indicated that the TraesTM9SF genes are expressed in the root system of wheat and may be potentially involved in the response of this plant to B. amyloliquefaciens PDR1 and nitrogen limitation conditions.
In this study, the total number of TraesTM9SF genes in wheat was 47, which was higher than that of A. thaliana (21 genes), rice (2 genes), S. bicolor (one gene), and S. italica (one gene). The higher number of TM9SF genes in wheat, compared to the other crops, might be due to functional importance, genetic variations, environmental adaptation or regulatory mechanisms. This finding also indicated the expansion, abundance, and potential functional diversity of the TraesTM9SF gene family in wheat. This result was in line with the findings that despite the vast number of familial genes in plant genomes, lineage-specific expansions might lead to significant discrepancies in the size of a given gene family among plant species (Panchy et al., 2016). For instance, the protein kinase superfamily has 2,532 members in Eucalyptus grandis and 426 in Chlamydomonas reinhardtii (Panchy et al., 2016). Moreover, the considerable number of TraesTM9SF genes in wheat relative to other plant species can be imputable to the complex genetic material of the wheat (Brenchley et al., 2012; Chapman et al., 2015). Polyploidy events, such as genome duplication, have been demonstrated to contribute substantially to gene family expansion and functional heterogeneity in plants (Soltis et al., 2015; Fang et al., 2022b; Mao et al., 2022). The large-scale repertoire of TraesTM9SF genes in wheat can supply the crop with various functional capacities, enabling it to adapt to various ecological niches and endure diverse abiotic and biotic stresses (Pont et al., 2013). Previous studies highlighted the pivotal role of TM9SF genes in abiotic stress responses, including drought, salinity, and heat stress (Hossain et al., 2016; Banik and Dutta, 2023). Therefore, the ability to cope with environmental challenges may be improved by the high number of TraesTM9SF genes in wheat.
Gene duplication plays a significant role in genome evolution (Magadum et al., 2013). Synteny and collinearity analyses indicated no gene duplication of TraesTM9SF genes in wheat genes, indicating that the TraesTM9SF genes might not be involved in the evolution of the wheat genome or that its implication in the genome evolution may be driven via other mechanisms. It can also be concluded that the evolutionary mechanism of the TraesTM9SF gene showed maintenance during the domestication of wheat. The finding, however, is contrary to the results of earlier studies, which have reported that gene duplications are a vital factor for genetic family expansion and plant function diversification (Fang et al., 2022b; Mao et al., 2022). The lack of gene duplication events in wheat TraesTM9SF genes poses exciting questions. Segmental or whole-genome duplications, in which wheat has undergone genetic modification over time, may have played a role in the expansion of the TraesTM9SF gene family in wheat (Panchy et al., 2016). Moreover, the TraesTM9SF gene family in wheat could have multiple copies due to retention or acquisition from related species via HGT (Dean et al., 2018; Murphy et al., 2019).
Conserved motifs play pivotal roles in the function of genes harboring them and are implicated in DNA binding, transcriptional activity, and protein interaction (Orozco and Hanley-Bowdoin, 1998). They are also involved in diverse plant functions in response to biotic and abiotic stresses (Singh and Graether, 2020). Herein, we discovered that the TraesTM9SF genes had gene structures characterized by the presence of conserved motifs, and some of these motifs were present in all of the TraesTM9SF genes. The conserved motifs are a critical functional element that can provide insight into potential roles and functional characteristics of genes (Bailey et al., 2009). Our study found conserved motifs in the TraesTM9SF gene family, indicating that certain functional domains or regions are preserved across genes. This conservation implies that these motifs can play a crucial role in the structure, function, and regulation of TraesTM9SF genes. Similar findings have been observed in other gene families where conserved features were associated with specific functional domains and protein interactions (Robson et al., 2001; Shoemaker et al., 2006).
Trans- and cis-regulatory elements are the two regulatory systems plants use to regulate gene expression (Vandepoele et al., 2009; Biłas et al., 2016; Brkljacic and Grotewold, 2017; Huang and Ecker, 2018; Ricci et al., 2019; Wang et al., 2019). The cis-regulatory elements in the promoter region are implicated in the tissue-specific, stress-responsive, and various stimulus-responsive genes (Sheshadri et al., 2016; Luján et al., 2021). In this study, we predicted 54 cis-acting elements in the promoter region of TraesTM9SF genes. These cis-regulatory components, which are mainly found to be related to the sensitivity of the light, suggested that TraesTM9SF genes could play an essential role in lighting-mediated processes like photosynthesis, photomorphogenesis, and melatonin regulation (Tan et al., 2014; Hwang et al., 2020; Zeng et al., 2022). This finding is consistent with previous studies suggesting the role of cis-acting elements in regulating light-responsive genes (Zhai et al., 2019). The cis-elements related to development, phytohormones, and metabolism were also identified, indicating the potential role of TraesTM9SF genes in hormonal regulation, development, and metabolic processes in wheat. This suggests that the TraesTM9SF genes play an important role in various developmental processes, hormone signaling pathways, and metabolic regulation in wheat. This observation aligns with the roles of various gene families involved in plant evolution, development, and metabolism (Jiang et al., 2015). Cis-acting elements involved in biotic and abiotic stresses, such as defense, stress, drought, and salinity, were also predicted, suggesting the potential of TraesTM9SF genes in regulating the response of wheat to biotic and abiotic stress, which requires further experimental validation in future studies. Thus, the TraesTM9SF genes may regulate plant response to environmental challenges and stress tolerance. The presence of stress-responsive cis-acting elements in the promoter regions of genes has been reported in several stress-responsive gene families, highlighting their importance in regulating stress-responsive gene expression (Zou et al., 2011; Sheshadri et al., 2016).
Since the role of TraesTM9SF genes has not been confirmed in wheat, we cultivated the wheat under B. amyloliquefaciens PDR1 and nitrogen limitation conditions and performed transcriptome sequencing to explore their expression patterns. We found that the TraesTM9SF genes were expressed in the root system of the wheat under normal cultivation conditions. Our results showed that in the root system, TraesTM9SF genes are expressed and seem likely to play an important role in plant growth, uptake of nutrients, or other root-related processes. After treatment with nitrogen limitation conditions, upregulation in the expression of TraesTM9SF genes was observed, indicating that these genes are responsive to nitrogen limitation. Thus, TraesTM9SF genes may play an important role in the mechanisms underlying the effect of nitrogen limitation on wheat, for example, nitrogen uptake, conversion, or recycling processes, which are needed for nutrient homeostasis and maintenance of plant growth under reduced nitrogen availability (Masclaux-Daubresse et al., 2010; Muratore et al., 2021; Zhang et al., 2023). Interestingly, the potential role of the alterations of TraesTM9SF genes due to a lack of nitrogen in increasing wheat resistance to nutrient stress, as well as optimizing nitrogen use for increased crop growth and productivity is apparent from the observations of this study. Moreover, the co-cultivation of wheat B. amyloliquefaciens PDR1 strain under nitrogen limitation conditions led to downregulation of TraesTM9SF genes, suggesting possible regulation of these genes by the beneficial bacterium under nutrient-stress conditions.
Moreover, though the PDR1 strain has been reported to be beneficial for A. thaliana (Li et al., 2020), our study is the first to report its role in reversing nitrogen limitation-induced gene dysregulation in wheat. Therefore, our work about TraesTM9SF genes in wheat has potential applications for crop breeding. The expression patterns of these genes in the root system, their response to nitrogen limitation, and their interaction with the B. amyloliquefaciens PDR1 strain suggest their potential roles in root development, nutrient stress responses, and plant-microbe interactions. Further functional characterization and molecular studies are required in wheat under various stress conditions to determine the precise mechanisms and biological function of TraesTM9SF genes.
Our study has some limitations. Although the transcript levels of some TraesTM9SF genes were responsive to nitrogen limitation and despite B. amyloliquefaciens PDR1 treatment conditions indicated that these genes might play important roles in response to nitrogen limitation or B. amyloliquefaciens, further experimental evidence is still needed to confirm their functional roles in wheat under these processes. In our future studies, we will screen one or several potential genes of the TraesTM9SF family in the RNA-seq data of wheat roots treated by nitrogen deficiency and bacterial infection and perform functional characterization of these potential genes to validate their roles in nitrogen utilization and resistance/exposure to B. amyloliquefaciens PDR1.
TM9SF proteins play important roles in the growth and development of plants; however, there has yet to be a relevant report on T. aestivum. In our study, the identification and expression profiling of putative TM9SF genes in T. aestivum was performed. Several bioinformatics analyses were carried out that include the identification of DNA/protein sequences, chromosomal localization, gene structure, calculation of genomic duplications, determination of phylogenetic groups, examination of protected motif regions, and identification of gene ontology categories. The expression patterns of TM9SF genes under nitrogen limitation and A. amyloliquefaciens PDR1 conditions were also verified experimentally. These results are essential for providing good candidate genes, which will be used for potentially breeding wheat varieties with high resistance to nitrogen limitation and response to nutrients via genetic engineering. Thus, our study provides a comprehensive basis for molecular and evolutionary characterization of TraesTM9SF genes in wheat and their role in wheat response to nitrogen limitation or B. amyloliquefaciens.
The data that support the findings of this study have been deposited into the CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number CNP0003662 (https://db.cngb.org/search/project/CNP0003662/).
FL: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. KX: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. YL: Formal Analysis, Writing – review & editing. TM: Formal Analysis, Writing – review & editing. YH: Formal Analysis, Writing – review & editing. LZ: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work was supported by the National Natural Science Foundation of China (Grant No.31960217 and 32272022), the Natural Science Foundation of Guizhou Province (Grant No.Qiankehejichu-ZK[2022] Zhongdian 033), the Joint Fund of the National Natural Science Foundation of China and the Karst Science Research Center of Guizhou (Grant No.U1812401), and the Guizhou Tobacco Company Project (Grant No. 2020XM05).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1324974/full#supplementary-material
Supplementary Table S1 | Physicochemical properties of TraesTM9SF genes.
Supplementary Table S2 | Protein sequences of TM9SF genes from different plant species.
Supplementary Table S3 | Coding domains in TraesTM9SF sequences.
Supplementary Table S4 | Subcellular localization of TraesTM9SF genes from different plant species.
Supplementary Table S5 | Similarity and identity analysis results of TraesTM9SF genes.
Supplementary Data Sheet S1 | Functional enrichment of TraesTM9SF genes and their TFs in the PPI network in String database.
Supplementary Data Sheet S2 | Functional enrichment of TraesTM9SF genes from the eggNOG-mapper analysis.
Supplementary Data Sheet S3 | List of PDR1-responsive TraesTM9SF genes under nitrogen limitation conditions in the profile 14.
Supplementary Data Sheet S4 | List of PDR1-responsive TraesTM9SF genes under nitrogen limitation conditions in the profile 7.
Ahlawat, O. P., Yadav, D., Kashyap, P. L., Khippal, A., Singh, G. (2022). Wheat endophytes and their potential role in managing abiotic stress under changing climate. J. Appl. Microbiol. 132 (4), 2501–2520. doi: 10.1111/jam.15375
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37 (Web Server issue), W202–W208. doi: 10.1093/nar/gkp335
Bailey, T. L., Johnson, J., Grant, C. E., Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43 (W1), W39–W49. doi: 10.1093/nar/gkv416
Banik, S., Dutta, D. (2023). Membrane proteins in plant salinity stress perception, sensing, and response. J. Membr Biol. 256 (2), 109–124. doi: 10.1007/s00232-023-00279-9
Biłas, R., Szafran, K., Hnatuszko-Konka, K., Kononowicz, A. K. (2016). Cis-regulatory elements used to control gene expression in plants. Plant Cell Tissue Organ Culture (PCTOC) 127 (2), 269–287. doi: 10.1007/s11240-016-1057-7
Brenchley, R., Spannagl, M., Pfeifer, M., Barker, G. L., D’Amore, R., Allen, A. M., et al. (2012). Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491 (7426), 705–710. doi: 10.1038/nature11650
Brkljacic, J., Grotewold, E. (2017). Combinatorial control of plant gene expression. Biochim. Biophys. Acta Gene Regul. Mech. 1860 (1), 31–40. doi: 10.1016/j.bbagrm.2016.07.005
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P., Huerta-Cepas, J. (2021). eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 38 (12), 5825–5829. doi: 10.1093/molbev/msab293
Carril, P., Cruz, J., di Serio, C., Pieraccini, G., Ait Bessai, S., Tenreiro, R., et al. (2021). Modulation of the wheat seed-borne bacterial community by herbaspirillum seropedicae RAM10 and its potential effects for tryptophan metabolism in the root endosphere. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.792921
Chapman, J. A., Mascher, M., Buluç, A., Barry, K., Georganas, E., Session, A., et al. (2015). A whole-genome shotgun approach for assembling and anchoring the hexaploid bread wheat genome. Genome Biol. 16 (1), 26. doi: 10.1186/s13059-015-0582-8
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi: 10.1016/j.molp.2020.06.009
Dal Cortivo, C., Barion, G., Visioli, G., Mattarozzi, M., Mosca, G., Vamerali, T. (2017). Increased root growth and nitrogen accumulation in common wheat following PGPR inoculation: Assessment of plant-microbe interactions by ESEM. Agriculture Ecosyst. Environ. 247, 396–408. doi: 10.1016/j.agee.2017.07.006
Dean, P., Sendra, K. M., Williams, T. A., Watson, A. K., Major, P., Nakjang, S., et al. (2018). Transporter gene acquisition and innovation in the evolution of Microsporidia intracellular parasites. Nat. Commun. 9 (1), 1709. doi: 10.1038/s41467-018-03923-4
De Cena, G. L., Scavassa, B. V., Conceição, K. (2022). In silico prediction of anti-infective and cell-penetrating peptides from thalassophryne nattereri natterin toxins. Pharm. (Basel) 15 (9), 1141. doi: 10.3390/ph15091141
Di Benedetto, N. A., Campaniello, D., Bevilacqua, A., Cataldi, M. P., Sinigaglia, M., Flagella, Z., et al. (2019). Isolation, screening, and characterization of plant-growth-promoting bacteria from durum wheat rhizosphere to improve N and P nutrient use efficiency. Microorganisms 7 (11), 541. doi: 10.3390/microorganisms7110541
Di Benedetto, N. A., Corbo, M. R., Campaniello, D., Cataldi, M. P., Bevilacqua, A., Sinigaglia, M., et al. (2017). The role of Plant Growth Promoting Bacteria in improving nitrogen use efficiency for sustainable crop production: a focus on wheat. AIMS Microbiol. 3 (3), 413–434. doi: 10.3934/microbiol.2017.3.413
Dissanayake, B. M., Staudinger, C., Munns, R., Taylor, N. L., Millar, A. H. (2022). Distinct salinity-induced changes in wheat metabolic machinery in different root tissue types. J. Proteomics 256, 104502. doi: 10.1016/j.jprot.2022.104502
Ernst, J., Bar-Joseph, Z. (2006). STEM: a tool for the analysis of short time series gene expression data. BMC Bioinf. 7, 191. doi: 10.1186/1471-2105-7-191
Fang, Y., Jiang, J., Hou, X., Guo, J., Li, X., Zhao, D., et al. (2022b). Plant protein-coding gene families: Their origin and evolution. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.995746
Fang, L., Yin, X., van der Putten, P. E. L., Martre, P., Struik, P. C. (2022a). Drought exerts a greater influence than growth temperature on the temperature response of leaf day respiration in wheat (Triticum aestivum). Plant Cell Environ. 45 (7), 2062–2077. doi: 10.1111/pce.14324
Gong, J., Shi, T., Li, Y., Wang, H., Li, F. (2021). Genome-Wide Identification and Characterization of Calcium Metabolism Related Gene Families in Arabidopsis thaliana and Their Regulation by Bacillus amyloliquefaciens Under High Calcium Stress. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.707496
Gu, Z., Gu, L., Eils, R., Schlesner, M., Brors, B. (2014). Circlize Implements and enhances circular visualization in R. Bioinformatics 30 (19), 2811–2812. doi: 10.1093/bioinformatics/btu393
Hossain, M. R., Bassel, G. W., Pritchard, J., Sharma, G. P., Ford-Lloyd, B. V. (2016). Trait specific expression profiling of salt stress responsive genes in diverse rice genotypes as determined by modified significance analysis of microarrays. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00567
Huang, S. C., Ecker, J. R. (2018). Piecing together cis-regulatory networks: insights from epigenomics studies in plants. Wiley Interdiscip Rev. Syst. Biol. Med. 10 (3), e1411. doi: 10.1002/wsbm.1411
Huang, B., Huang, Z., Ma, R., Chen, J., Zhang, Z., Yrjälä, K. (2021). Genome-wide identification and analysis of the heat shock transcription factor family in moso bamboo (Phyllostachys edulis). Sci. Rep. 11 (1), 16492. doi: 10.1038/s41598-021-95899-3
Hwang, O. J., Kang, K., Back, K. (2020). Effects of light quality and phytochrome form on melatonin biosynthesis in rice. Biomolecules 10 (4), 523. doi: 10.3390/biom10040523
Ikai, A. (1980). Thermostability and aliphatic index of globular proteins. J. Biochem. 88 (6), 1895–1898. doi: 10.1093/oxfordjournals.jbchem.a133168
Ismagul, A., Iskakova, G., Harris, J. C., Eliby, S. (2014). Biolistic transformation of wheat with centrophenoxine as a synthetic auxin. Methods Mol. Biol. 1145, 191–202. doi: 10.1007/978-1-4939-0446-4_15
Jiang, S. Y., Chi, Y. H., Wang, J. Z., Zhou, J. X., Cheng, Y. S., Zhang, B. L., et al. (2015). Sucrose metabolism gene families and their biological functions. Sci. Rep. 5, 17583. doi: 10.1038/srep17583
Kant, S., Bi, Y.-M., Rothstein, S. J. (2010). Understanding plant response to nitrogen limitation for the improvement of crop nitrogen use efficiency. J. Exp. Bot. 62 (4), 1499–1509. doi: 10.1093/jxb/erq297
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi: 10.1093/molbev/msw054
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30 (1), 325–327. doi: 10.1093/nar/30.1.325
Li, X. Y., Lin, E. P., Huang, H. H., Niu, M. Y., Tong, Z. K., Zhang, J. H. (2018). Molecular characterization of SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) gene family in betula luminifera. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00608
Li, F., Shi, T., Tang, X., Tang, M., Gong, J., Yi, Y. (2020). Bacillus amyloliquefaciens PDR1 from root of karst adaptive plant enhances Arabidopsis thaliana resistance to alkaline stress through modulation of plasma membrane H(+)-ATPase activity. Plant Physiol. Biochem. 155, 472–482. doi: 10.1016/j.plaphy.2020.08.019
Li, A. L., Zhu, Y. F., Tan, X. M., Wang, X., Wei, B., Guo, H. Z., et al. (2008). Evolutionary and functional study of the CDPK gene family in wheat (Triticum aestivum L.). Plant Mol. Biol. 66 (4), 429–443. doi: 10.1007/s11103-007-9281-5
Limin, A. E., Danyluk, J., Chauvin, L. P., Fowler, D. B., Sarhan, F. (1997). Chromosome mapping of low-temperature induced Wcs120 family genes and regulation of cold-tolerance expression in wheat. Mol. Gen. Genet. 253 (6), 720–727. doi: 10.1007/s004380050376
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 (4), 402–408. doi: 10.1006/meth.2001.1262
Luján, M. A., Soria-García, Á., Claver, A., Lorente, P., Rubio, M. C., Picorel, R., et al. (2021). Different cis-regulatory elements control the tissue-specific contribution of plastid ω-3 desaturases to wounding and hormone responses. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.727292
Ma, J., Yang, Y., Luo, W., Yang, C., Ding, P., Liu, Y., et al. (2017). Genome-wide identification and analysis of the MADS-box gene family in bread wheat (Triticum aestivum L.). PloS One 12 (7), e0181443. doi: 10.1371/journal.pone.0181443
Maenhout, P., Van den Bulcke, J., Van Hoorebeke, L., Cnudde, V., De Neve, S., Sleutel, S. (2018). Nitrogen limitations on microbial degradation of plant substrates are controlled by soil structure and moisture content. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.01433
Magadum, S., Banerjee, U., Murugan, P., Gangapur, D., Ravikesavan, R. (2013). Gene duplication as a major force in evolution. J. Genet. 92 (1), 155–161. doi: 10.1007/s12041-013-0212-8
Mao, K., Zhang, M., Kong, Y., Dai, S., Wang, Y., Meng, Q., et al. (2022). Origin, expansion, and divergence of ETHYLENE-INSENSITIVE 3 (EIN3)/EIN3-LIKE transcription factors during streptophytes evolution. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.858477
Masclaux-Daubresse, C., Daniel-Vedele, F., Dechorgnat, J., Chardon, F., Gaufichon, L., Suzuki, A. (2010). Nitrogen uptake, assimilation and remobilization in plants: challenges for sustainable and productive agriculture. Ann. Bot. 105 (7), 1141–1157. doi: 10.1093/aob/mcq028
Muratore, C., Espen, L., Prinsi, B. (2021). Nitrogen uptake in plants: the plasma membrane root transport systems from a physiological and proteomic perspective. Plants (Basel) 10 (4), 681. doi: 10.3390/plants10040681
Murphy, C. L., Youssef, N. H., Hanafy, R. A., Couger, M. B., Stajich, J. E., Wang, Y., et al. (2019). Horizontal gene transfer as an indispensable driver for evolution of neocallimastigomycota into a distinct gut-dwelling fungal lineage. Appl. Environ. Microbiol. 85 (15), e00988-19. doi: 10.1128/aem.00988-19
Orozco, B. M., Hanley-Bowdoin, L. (1998). Conserved sequence and structural motifs contribute to the DNA binding and cleavage activities of a geminivirus replication protein. J. Biol. Chem. 273 (38), 24448–24456. doi: 10.1074/jbc.273.38.24448
Osorio, D., Rondón-Villarreal, P., Torres, R. (2015). Peptides: a package for data mining of antimicrobial peptides. Small 12, 44–444. doi: 10.32614/RJ-2015-001
Panchy, N., Lehti-Shiu, M., Shiu, S. H. (2016). Evolution of gene duplication in plants. Plant Physiol. 171 (4), 2294–2316. doi: 10.1104/pp.16.00523
Peng, F. Y., Yang, R. C. (2017). Prediction and analysis of three gene families related to leaf rust (Puccinia triticina) resistance in wheat (Triticum aestivum L.). BMC Plant Biol. 17 (1), 108. doi: 10.1186/s12870-017-1056-9
Perrin, J., Le Coadic, M., Vernay, A., Dias, M., Gopaldass, N., Ouertatani-Sakouhi, H., et al. (2015). TM9 family proteins control surface targeting of glycine-rich transmembrane domains. J. Cell Sci. 128 (13), 2269–2277. doi: 10.1242/jcs.164848
Pont, C., Murat, F., Guizard, S., Flores, R., Foucrier, S., Bidet, Y., et al. (2013). Wheat syntenome unveils new evidences of contrasted evolutionary plasticity between paleo- and neoduplicated subgenomes. Plant J. 76 (6), 1030–1044. doi: 10.1111/tpj.12366
Pruvot, B., Laurens, V., Salvadori, F., Solary, E., Pichon, L., Chluba, J. (2010). Comparative analysis of nonaspanin protein sequences and expression studies in zebrafish. Immunogenetics 62 (10), 681–699. doi: 10.1007/s00251-010-0472-x
Qiao, L., Zhang, L., Zhang, X., Zhang, L., Li, X., Chang, J., et al. (2017). Evolution of the aux/IAA gene family in hexaploid wheat. J. Mol. Evol. 85 (3-4), 107–119. doi: 10.1007/s00239-017-9810-z
Ricci, W. A., Lu, Z., Ji, L., Marand, A. P., Ethridge, C. L., Murphy, N. G., et al. (2019). Widespread long-range cis-regulatory elements in the maize genome. Nat. Plants 5 (12), 1237–1249. doi: 10.1038/s41477-019-0547-0
Robson, F., Costa, M. M., Hepworth, S. R., Vizir, I., Piñeiro, M., Reeves, P. H., et al. (2001). Functional importance of conserved domains in the flowering-time gene CONSTANS demonstrated by analysis of mutant alleles and transgenic plants. Plant J. 28 (6), 619–631. doi: 10.1046/j.1365-313x.2001.01163.x
Saidi, M. N., Mahjoubi, H., Yacoubi, I. (2023). Transcriptome meta-analysis of abiotic stresses-responsive genes and identification of candidate transcription factors for broad stress tolerance in wheat. Protoplasma 260 (3), 707–721. doi: 10.1007/s00709-022-01807-5
Sehgal, D., Dhakate, P., Ambreen, H., Shaik, K. H. B., Rathan, N. D., Anusha, N. M., et al. (2023). Wheat omics: advancements and opportunities. Plants (Basel) 12 (3), 426. doi: 10.3390/plants12030426
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13 (11), 2498–2504. doi: 10.1101/gr.1239303
Sheshadri, S. A., Nishanth, M. J., Simon, B. (2016). Stress-Mediated cis-Element Transcription Factor Interactions Interconnecting Primary and Specialized Metabolism in planta. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01725
Shoemaker, B. A., Panchenko, A. R., Bryant, S. H. (2006). Finding biologically relevant protein domain interactions: conserved binding mode analysis. Protein Sci. 15 (2), 352–361. doi: 10.1110/ps.051760806
Singh, K. K., Graether, S. P. (2020). Conserved sequence motifs in the abiotic stress response protein late embryogenesis abundant 3. PloS One 15 (8), e0237177. doi: 10.1371/journal.pone.0237177
Smyth, E. M., McCarthy, J., Nevin, R., Khan, M. R., Dow, J. M., O’Gara, F., et al. (2011). In vitro analyses are not reliable predictors of the plant growth promotion capability of bacteria; a Pseudomonas fluorescens strain that promotes the growth and yield of wheat. J. Appl. Microbiol. 111 (3), 683–692. doi: 10.1111/j.1365-2672.2011.05079.x
Soltis, P. S., Marchant, D. B., Van de Peer, Y., Soltis, D. E. (2015). Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 35, 119–125. doi: 10.1016/j.gde.2015.11.003
Szeliga, M., Bakera, B., Święcicka, M., Tyrka, M., Rakoczy-Trojanowska, M. (2023). Identification of candidate genes responsible for chasmogamy in wheat. BMC Genomics 24 (1), 170. doi: 10.1186/s12864-023-09252-1
Szklarczyk, D., Kirsch, R., Koutrouli, M., Nastou, K., Mehryary, F., Hachilif, R., et al. (2023). The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51 (D1), D638–d646. doi: 10.1093/nar/gkac1000
Tan, D. X., Zheng, X., Kong, J., Manchester, L. C., Hardeland, R., Kim, S. J., et al. (2014). Fundamental issues related to the origin of melatonin and melatonin isomers during evolution: relation to their biological functions. Int. J. Mol. Sci. 15 (9), 15858–15890. doi: 10.3390/ijms150915858
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7 (3), 562. doi: 10.1038/nprot.2012.016
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., Van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28 (5), 511. doi: 10.1038/nbt.1621
Tyrka, M., Bakera, B., Szeliga, M., Święcicka, M., Krajewski, P., Mokrzycka, M., et al. (2021). Identification of rf genes in hexaploid wheat (Triticumaestivum L.) by RNA-seq and paralog analyses. Int. J. Mol. Sci. 22 (17), 9146. doi: 10.3390/ijms22179146
Vandepoele, K., Quimbaya, M., Casneuf, T., De Veylder, L., Van de Peer, Y. (2009). Unraveling transcriptional control in Arabidopsis using cis-regulatory elements and coexpression networks. Plant Physiol. 150 (2), 535–546. doi: 10.1104/pp.109.136028
Vannini, C., Domingo, G., Fiorilli, V., Seco, D. G., Novero, M., Marsoni, M., et al. (2021). Proteomic analysis reveals how pairing of a Mycorrhizal fungus with plant growth-promoting bacteria modulates growth and defense in wheat. Plant Cell Environ. 44 (6), 1946–1960. doi: 10.1111/pce.14039
Venzhik, Y. V., Deryabin, A. N., Popov, V. N., Dykman, L. A., Titov, A. F., Moshkov, I. E. (2022). Influence of gold nanoparticles on the tolerance of wheat to low temperature. Dokl Biochem. Biophys. 502 (1), 5–9. doi: 10.1134/s1607672922010100
Wang, X., Ji, C., Song, X., Liu, Z., Liu, Y., Li, H., et al. (2021). Biocontrol of two bacterial inoculant strains and their effects on the rhizosphere microbial community of field-grown wheat. BioMed. Res. Int. 2021, 8835275. doi: 10.1155/2021/8835275
Wang, Q., Jia, Y., Wang, Y., Jiang, Z., Zhou, X., Zhang, Z., et al. (2019). Evolution of cis- and trans-regulatory divergence in the chicken genome between two contrasting breeds analyzed using three tissue types at one-day-old. BMC Genomics 20 (1), 933. doi: 10.1186/s12864-019-6342-5
Wang, J., Li, R., Zhang, H., Wei, G., Li, Z. (2020). Beneficial bacteria activate nutrients and promote wheat growth under conditions of reduced fertilizer application. BMC Microbiol. 20 (1), 38. doi: 10.1186/s12866-020-1708-z
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40 (7), e49. doi: 10.1093/nar/gkr1293
Wang, D., Wu, X., Gao, S., Zhang, S., Wang, W., Fang, Z., et al. (2022). Systematic analysis and identification of drought-responsive genes of the CAMTA gene family in wheat (Triticum aestivum L.). Int. J. Mol. Sci. 23 (9), 4542. doi: 10.3390/ijms23094542
Wickham, H., Chang, W., Henry, L., Pedersen, T. L., Takahashi, K., Wilke, C., et al. (2019). ggplot2: create elegant data visualisations using the grammar of graphics. 2018 Vol. 2 (R package version), 173. Available at: https://CRAN.R-project.org/package=ggplot2.
Xi, W., Hao, C., Li, T., Wang, H., Zhang, X. (2023). Transcriptome analysis of roots from wheat (Triticum aestivum L.) varieties in response to drought stress. Int. J. Mol. Sci. 24 (8), 7245. doi: 10.3390/ijms24087245
Youseif, S. H., Abd El-Megeed, F. H., Humm, E. A., Maymon, M., Mohamed, A. H., Saleh, S. A., et al. (2021). Comparative analysis of the cultured and total bacterial community in the wheat rhizosphere microbiome using culture-dependent and culture-independent approaches. Microbiol. Spectr. 9 (2), e0067821. doi: 10.1128/Spectrum.00678-21
Yu, C. S., Chen, Y. C., Lu, C. H., Hwang, J. K. (2006). Prediction of protein subcellular localization. Proteins 64 (3), 643–651. doi: 10.1002/prot.21018
Yu, M., Yu, Y., Song, T., Zhang, Y., Wei, F., Cheng, J., et al. (2022). Characterization of the voltage-dependent anion channel (VDAC) gene family in wheat (Triticum aestivum L.) and its potential mechanism in response to drought and salinity stresses. Gene 809, 146031. doi: 10.1016/j.gene.2021.146031
Yue, H., Zhang, H., Su, N., Sun, X., Zhao, Q., Weining, S., et al. (2022). Integrate small RNA and degradome sequencing to reveal drought memory response in wheat (Triticum aestivum L.). Int. J. Mol. Sci. 23 (11), 5917. doi: 10.3390/ijms23115917
Zeng, W., Mostafa, S., Lu, Z., Jin, B. (2022). Melatonin-mediated abiotic stress tolerance in plants. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.847175
Zhai, Y., Peng, H., Neff, M. M., Pappu, H. R. (2019). Putative Auxin and Light Responsive Promoter Elements From the Tomato spotted wilt tospovirus Genome, When Expressed as cDNA, Are Functional in Arabidopsis. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00804
Zhang, H., Liu, L., Wang, Z., Feng, G., Gao, Q., Li, X. (2021). Induction of low temperature tolerance in wheat by pre-soaking and parental treatment with melatonin. Molecules 26 (4), 1192. doi: 10.3390/molecules26041192
Zhang, W., Zhang, T., Zhang, J., Lei, W., Zhao, L., Wang, S., et al. (2023). Low nitrogen stress promotes root nitrogen uptake and assimilation in strawberry: contribution of hormone networks. Horticulturae 9 (2), 249. doi: 10.3390/horticulturae9020249
Keywords: transmembrane 9 superfamily, gene family, wheat, nitrogen limitation, Bacillus amyloliquefaciens PDR1
Citation: Li F, Xi K, Li Y, Ming T, Huang Y and Zhang L (2024) Genome-wide analysis of transmembrane 9 superfamily genes in wheat (Triticum aestivum) and their expression in the roots under nitrogen limitation and Bacillus amyloliquefaciens PDR1 treatment conditions. Front. Plant Sci. 14:1324974. doi: 10.3389/fpls.2023.1324974
Received: 20 October 2023; Accepted: 19 December 2023;
Published: 08 January 2024.
Edited by:
Yi-Hong Wang, University of Louisiana at Lafayette, United StatesReviewed by:
Cheng Qin, Zunyi Vocational and Technical College, ChinaCopyright © 2024 Li, Xi, Li, Ming, Huang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei Li, bGlmZWkyQGd6bnUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.