
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 31 January 2024
Sec. Functional and Applied Plant Genomics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1320638
This article is part of the Research Topic Genetics, Genomics and Breeding of Plant Architecture, Biomass, Grain Quality and Grain Yield Traits in Rice and Wheat View all 15 articles
 Foster Kangben1†
Foster Kangben1† Sonika Kumar1†
Sonika Kumar1† Zhigang Li1
Zhigang Li1 Avinash Sreedasyam2
Avinash Sreedasyam2 Chris Dardick3
Chris Dardick3 Don Jones4
Don Jones4 Christopher A. Saski1*
Christopher A. Saski1*Introduction: Plants can adapt their growth to optimize light capture in competitive environments, with branch angle being a crucial factor influencing plant phenotype and physiology. Decreased branch angles in cereal crops have been shown to enhance productivity in high-density plantings. The Tiller Angle Control (TAC1) gene, known for regulating tiller inclination in rice and corn, has been found to control branch angle in eudicots. Manipulating TAC1 in field crops like cotton offers the potential for improving crop productivity.
Methods: Using a homolog-based methodology, we examined the distribution of TAC1-related genes in cotton compared to other angiosperms. Furthermore, tissue-specific qPCR analysis unveiled distinct expression patterns of TAC1 genes in various cotton tissues. To silence highly expressed specific TAC1 homeologs in the stem, we applied CRISPR-Cas9 gene editing and Agrobacterium-mediated transformation, followed by genotyping and subsequent phenotypic validation of the mutants.
Results: Gene duplication events of TAC1 specific to the Gossypium lineage were identified, with 3 copies in diploid progenitors and 6 copies in allotetraploid cottons. Sequence analysis of the TAC1 homeologs in Gossypium hirsutum revealed divergence from other angiosperms with 1-2 copies, suggesting possible neo- or sub-functionalization for the duplicated copies. These TAC1 homeologs exhibited distinct gene expression patterns in various tissues over developmental time, with elevated expression of A11G109300 and D11G112200, specifically in flowers and stems, respectively. CRISPR-mediated loss of these TAC1 homeologous genes resulted in a reduction in branch angle and altered petiole angles, and a 5 to 10-fold reduction in TAC1 expression in the mutants, confirming their role in controlling branch and petiole angles. This research provides a promising strategy for genetically engineering branch and petiole angles in commercial cotton varieties, potentially leading to increased productivity.
The Tiller Angle Control (TAC1) gene is duplicated in the Gossipum lineage and regulates branch angle with significant A-subgenome expression bias. Manipulating this gene in Upland cotton can potentially improve productivity through high-density planting.
Improving crop performance involves various factors such as optimized light capture, carbon assimilation, and photosynthetic efficiency to achieve higher yield (Kant et al., 2012; Murchie and Burgess, 2022). The precise manipulation of plant architecture, particularly branching, can significantly enhance crop management and productivity. By controlling branching and limiting plant-to-plant interference, farmers can increase planting density, photosynthesis and carbon assimilation, boost yield, and improve mechanization efficiency (McGarry et al., 2016; Fladung, 2021).
Plant architecture is defined by the spatial configuration and morphological traits of its aerial and rooting structures in three dimensions. This fundamental feature has undergone modification throughout crop domestication and is crucial to the plant’s adaptability and productivity (Yang and Hwa, 2008; Cai et al., 2016). Plant architecture is the outcome of an intricate interplay between diverse regulatory mechanisms encompassing genetic programming, various hormone signaling pathways, and response to environmental cues such as light, gravity, and mechanical forces. A plant’s form, stem and leaf arrangement, and overall growth patterns are all influenced by these factors (Reinhardt and Kuhlemeier, 2002; Li et al., 2022; Sun et al., 2022). In angiosperms, variations in branch angles and other structures, such as leaves and branches, are viewed as adaptive strategies in modulating light interception efficiency. The ability to intercept light under varying conditions can also affect plant growth patterns and structures (Duursma et al., 2012). A key aspect of the success of the Green Revolution was the genetic modification of cereal crops, such as wheat, to improve their productivity and growth potential through breeding and selection for short, robust stems. This led to the development of wheat varieties that could withstand damage from wind and rain (Peng et al., 1999; Reinhardt and Kuhlemeier, 2002; Song and Zhang, 2009). Likewise, the architecture and yield of corn has been substantially improved through selection for reduced tillering, upright leaf angles, and increased apical dominance compared to its ancestor, teosinte (Whipple et al., 2011; Schmidt et al., 2016). Modern breeding has focused on the development of elite cultivars with erect, compact stem architecture, upright leaf angles, reduced branching, high harvest indices, and increased seed yield (Li et al., 2013). It is now commonly recognized that branches oriented vertically near the meristem and more horizontally at the lower canopy, are considered ideal for optimal light interception and correlated with higher yields (Kaggwa-Asiimwe et al., 2013; Sun et al., 2022). Using modern tools, plant architecture, including branch and leaf orientation, can now be manipulated with genome editing tools such as CRISPR to optimize light exposure and water uptake.
The Tiller Angle Control 1 (TAC1) gene has been identified as a crucial regulator of upright lateral organ orientation in rice, corn, and rapeseed (Yu et al., 2007; Ku et al., 2011; Li et al., 2017). TAC1 belongs to a small gene family, known as the IGT family, which controls the orientation of organ growth by inhibiting response to gravity in various plants, including grasses, dicots, and trees. Along with TAC1, the IGT family includes LAZY and DRO subfamilies that promote upward orientation of branches and downward growth of lateral roots by facilitating auxin redistribution (Duvick, 2005; Yu et al., 2007; Ku et al., 2011; Li et al., 2017). Although LAZY and DRO mechanisms have been extensively studied, little is known about TAC1, which has been suggested as a negative regulator of LAZY (Hollender et al., 2020). Model species such as Arabidopsis and rice have a single copy of the TAC1 gene (Guseman et al., 2017; Hollender et al., 2020). The loss of TAC1 function leads to a broom or pillar-like plant architecture, characterized by vertically oriented branches, leaves, inflorescence, flower buds, and tillers (Dardick et al., 2013). Under optimal light conditions the expression of TAC1 is up-regulated, potentially via the constitutive photomorphogenesis (COP1) gene, resulting in wider branch angles that enhance photoreception efficiency (Waite and Dardick, 2018).
Plants exhibiting loss of TAC1 phenotypes occupy less space which allows for a higher planting density corresponding to an increased yield. Loss of function of TAC1 results in a more inclined orientation of branch growth when plants are grown in prolonged darkness (Waite and Dardick, 2018; Hollender et al., 2020). In Arabidopsis and peach TAC1 is primarily expressed in the apical shoots and upper sections of the stem, as well as the upper laterals. TAC1 expression in rice is temporal peaking at 60 days after sowing and declining to a minimum at 100 days after sowing, especially during the heading stage. This leads to a decrease in leaf shading and increases photosynthetic efficiency (Yu et al., 2007; Dardick et al., 2013).
Cotton is a widely grown crop, covering 5% of the world’s farmland and valued for its fiber, seeds, and oil. It’s vital to the U.S. economy, which is the top cotton exporter and ranks third in production after China and India (Wang et al., 2012; Yuan et al., 2015; Shahbandeh, 2021). One approach to maximizing cotton yield is through the augmentation of planting densities (Kaggwa-Asiimwe et al., 2013; Khan et al., 2020). Current recommendations based on elite cotton varieties range from 20,000 to 45,000 cotton plants per acre (ppa). Plant populations exceeding 60,000 or falling below 20,000 ppa can result in management challenges and significant reductions in yield potential (Adams et al., 2019). Conversely, leading cotton seed suppliers often advocate for a plant density of 50,000 plants ha-1 to achieve optimal yields per hectare, resulting in a range of 700 to 1000 kg/ha. Higher plant density per unit area confers numerous advantages, including increased yield due to a greater number of plants within a given space, efficient utilization of resources such as water, nutrients, and sunlight, decreased weed growth and soil erosion, improved pest management, and enhanced microclimate conditions (Kaggwa-Asiimwe et al., 2013; Adams et al., 2019; Basu and Parida, 2021). However, under poor soil and seed conditions, higher planting population densities of up to 129,000 plants ha-1 have been reported to achieve similar yields (Norton et al., 1995; Fok, 1998). The expected cotton yield is contingent upon various factors such as the specific cotton variety cultivated, land availability, soil type, climatic conditions, planting density, available resources, nutrition, and management practices.
One possible explanation for the limited increase in yield observed in high-density cotton plantings could be linked to the prevalent spreading or horizontally oriented branching patterns seen in most cotton cultivars. In fact, small acreage farmers opt for crop varieties with narrower branch angles that can support higher plant density per area while also facilitating mechanical field management. Cotton plants exhibit two types of branches: monopodial and sympodial. Monopodial branches, also known as vegetative branches, originate from the main stem and continue to grow indefinitely throughout the plant’s development. Monopodial branches primarily grow in a nearly upright position. In contrast, sympodial branches, or fruiting branches, are determinate and bear the inflorescence, which eventually develops into the cotton bolls. Sympodial branches typically grow almost laterally from the main stem and are largely responsible for bearing the reproductive buds (squares). Fruiting branch angles ranging from 36.86° to 64.56° have been reported (Shao et al., 2022). With multiple branch types, cotton plants can allocate resources efficiently to both vegetative growth and reproduction (Gore, 1935; McGarry et al., 2016). Altered branch orientation for both branches could potentially impact cotton plant architecture. Given the desire for cotton varieties with narrow branch angles, it is critical that we improve our understanding of how monopodial and sympodial branch angles are regulated and identify strategies to develop improved germplasm with branch angles better suited for very high planting densities.
This study aimed to analyze the gene content and variation of TAC1 in the genomes of diploid and allotetraploid cottons as well as other Angiosperms. Our goals were to: (I) identify TAC1 orthologs in cotton species and other plant species; (II) determine homeologous gene expression profiles of TAC1 in various tissues (stem, leaf, flower, fiber, meristem and root) in the allotetraploid cotton genotype Coker312; (III) functionally profile via CRISPR/CAS9 knockout the homeologous TAC1 copies that are the most expressed in stem tissue; and (IV) develop gene editing strategies to modify cotton branch angles.
To uncover potential TAC1-related genes we conducted a blast search and motif analysis of the PpeTAC1 coding sequence from Prunus persica in the full genome sequences of Gossypium species (G. hirsutum, G.raimondii, G. arboreum, G. barbadense, G. darwinii, G.tomentosum) and other selected plants. For G. hirsutum we used the widely used and well-annotated Coker312 genotype as it is known for its regenerative capacity (Kumar et al., 2021). Coding and protein sequences were obtained from Phytozome, and the BLOSUM62 matrix was used in the blast search with an expected value of 1e-5. Amino acid sequences were concatenated for species with multiple copies using the concatenate feature in Geneious Prime software version 2023.2.1. Multiple alignments were performed with MUSCLE v5.1 (Edgar, 2004), and phylogenetic trees were constructed using the RAxML GAMMA GTR and the Neighbor-Joining method with 5,000 bootstrap replicates (Stamatakis, 2014; Kozlov et al., 2019). Genomic data for the analysis of most of the crops was retrieved from Phytozome Genome Database (Goodstein et al., 2012)
Samples of tissue were collected 14 days after anthesis (dpa) and immediately frozen. The tissue was then ground into a fine powder using a pestle and mortar in liquid nitrogen. Total RNA was extracted from 100mg of the tissue from leaf (basal, midsection and apical part), stem (between the 5th and 15th node and sliced into smaller pieces), meristem (apical part at 30cm downward), root (bulk roots and root tips and homogenized), flower (day before bloom), and fiber (14dpa) of the Coker 312 cotton genotype using a modified CTAB protocol method (Kumar et al., 2021). The purity and concentration of the extracted RNA were measured using a Nanodrop 8000 UV-Vis Spectrophotometer (Thermo Scientific).
Six differentially expressed TAC1 genes (GhCoker.A08G143500, GhCoker.D08G158700, GhCoker.A11G109300, GhCoker.D11G112200, GhCoker.A12G131200 and GhCoker.D12G137700) and three biological replicates each of leaf, stem, meristem, root, flower and fiber of Coker312 were used for qPCR. For mutants three biological replicates of stem tissue were used for qPCR analysis. For cDNA synthesis 1 ug of total RNAs was extracted from leaf, stem, meristem, root, flower, and fiber. The first strand of cDNA was synthesized using the M-MuLV reverse transcriptase (New England Biolabs, USA) and primed by d(T)25-VN following the manufacturer’s instructions. qPCR of gene transcripts was carried out using an iCycler iQ system (Bio-Rad, Hercules, CA, USA) in 20µL of PCR reaction solution using the Luna Universal qPCR Master Mix, New England Biolabs, USA, the SYBR®Green method was used for running the qPCR. Conditions for thermal cycling included initial denaturation at 95°C for 120 s, followed by 45 cycles of 95°C for 20 s, 60°C 30 s, and 72°C for 20s. Lastly a unique melting curve was performed from 60.0°C to 95.0°C in 0.5°C increments to amplify distinctive PCR product. One reference gene, GhPP2A1 was used for normalization of the expression data. The reference gene was chosen because of its uniform expression between cells of different tissues and under variable experimental conditions (Artico et al., 2010). The Ct values of four technical samples for each of the three biological replications were used to estimate the relative expression of genes using the 2−ΔΔCt equation (Schmittgen and Livak, 2008). Except for the reference genes, specific primer pairs were designed from conserved coding sequence and listed in Supplementary Table S1.
The modified binary vector pCSbar (a bar gene cassette was cloned into EcoRI-HindIIIsites of pAMBIA1300) integrating spCAS9 genes and two sgRNAs cassettes were prepared for cotton transformation (Supplementary Figure S1). The spCAS9 driven by a dual 35S promoter was cloned into EcoRI site of pCSbar, and then the two synthesized sgRNAs (Synbio, NJ, USA) were integrated into the AvrII site of pCSbar-Cas9, and the result was in the final construct. The two crRNAs targeted the flanking sequences of a 128-base pair region specific to the second exon of the GhCoker.D11G112200 and GhCoker.A11G109300 driven by AtU6-26t and AtU6-29t promoters, respectively. PAM sequences are highlighted. The two crRNAs share 100% similarities to conserved regions of the two homologs GhCoker.D11G112200 and GhCoker.A11G109300 but low similarities to the other four homeologs of GhCoker.A08G143500.1, GhCoker.D08G158700.1, GhCoker.D12G137700.1 and GhCoker.A12G131200.1 (Supplementary Table S2).
The Agrobacterium (EHA105) mediated transformation and plant regeneration protocol was adopted from a standard transformation publication (Jin et al., 2012). The regenerated plants were transplanted into one-gallon pots (Dillen Products, Middlefield, OH) containing commercial potting mixture soil (Fafard 3-B Mix, Fafard Inc., Anderson, SC, USA) and were developed under the greenhouse system conditions (Biosystems Research Complex, Clemson University) for morphological observation and materials harvesting. Three independent transgenic events (tac1-72, -73 and -74, verified by PCR of bar and Cas9) showing the phenotype were selected to verify the genotype. The total DNA obtained from each was subjected to PCR. The primer pair: F-AGATGGGCTTGCACGAAATGTTAAG and R-CGTTTTTGGCAGGAAGAGRAGATG were carefully selected to amplify the region overlapping the target sequences of the two crRNAs in the two homologous. The PCR products were cloned into pGEM-T-Easy vector and sequenced using standard Sanger sequencing techniques. The sequencing was conducted at the Genomics Core, Biosciences at Arizona State University. The chromatograms and sequence analysis were conducted in Geneious Prime (Supplementary Figure S2, Supplementary Table S3). To confirm the tac1-73 knockout genotype, isolated gDNA was sequenced at Hudson Alpha Institute of Biotechnology, 2x150bp paired-end reads at an average coverage of 12.58X across the genome. Reads were trimmed using Trimmomatic (Bolger et al., 2014) to remove Illumina adaptor sequences, and aligned to the G. hirsutum ‘Coker312’ genome version 1.1 (Gossypium hirsutum Coker v1.1, DOE-JGI, http://phytozome.jgi.doe.gov/info/GhirsutumCoker_v1_1) using Burrow-Wheeler Alignment (bwa-mem) (Li, 2013). The aligned BAM file for the tac1-73 knockout sample was investigated using Integrative Genomics Viewer (IGV) to verify the Crispr-Cas9 target sites (Robinson et al., 2011). BAM files were prepared for genotyping using samtools to eliminate duplicate reads (Danecek et al., 2021). Off-target single nucleotide polymorphisms were genotyped using samtools integrated with Varscan2 mpileup2snp (Koboldt et al., 2012; Danecek et al., 2021). Investigation of possible off-target indels or large structural variants was performed with Varscan2 mpileup2indel and Delly’s structural variant caller (Koboldt et al., 2012; Rausch et al., 2012; Danecek et al., 2021). Guide RNAs were aligned to the Coker312 genome with BLASTN (wordsize=7) and only found 8 hits with identity, Supplementary Table S4. Bedtools (Quinlan and Hall, 2010) intersect was used to determine that no variants were found in these regions when intersected with the variant call file (.VCF), data not shown.
Branch and petiole angles were measured manually with a protractor and digitally in Adobe Photoshop to estimate the angle between the main stem to the branch and petiole (BioRender.com, 2023), Supplementary Figure S3 (Sun et al., 2016a). Data was collected from five representative sympodial branches and two monopodial branches per plant and computed mean branch and petiole angles similar to (Li et al., 2017). We also collected data on branch length, plant height, boll count. Data was collected from the T0 mutants and wild type Coker 312 plants.
Stable integration of the CRISPR/Cas9 T-DNA was determined by screening the T1 mutants using the BAR gene selectable marker. The BAR gene has been extensively utilized and researched as a positive selectable marker for herbicide resistance, providing plants with resistance to phosphinothricin (PPT), the active ingredient in the broad-spectrum herbicide known as Basta. This functionality facilitates the elimination of non-transformed individuals and the selective advancement of transformants in plant regeneration processes (Thompson et al., 1987; Rathore et al., 1993; Hahn et al., 2017).
The analysis of variance was performed using JMP Pro version 16.1 (SAS Institute, Cary, NC, USA). Mean separation was assessed through a one-way ANOVA with Tukey’s HSD test (p<0.05).
Copy number and phylogenetic analysis was conducted to examine the distribution of cotton TAC1 orthologs in a range of representative angiosperms, encompassing both species with known single copies (e.g. Prunus persica, Zea mays, Vitus vinifera, Vigna unguiculata, Theobroma cacao, Medicago trunculata, and Eucalyptus grandis) and those with two copies (Glycine max and Populus trichocarpa), Table 1. Analysis revealed that TAC1 gene copy number in Medicago truncatula, a basal eudicot species, is most divergent from V. unguiculata in the phylogenetic tree (Figure 1). Our findings revealed a separate clade for the Rosaceae family members, Prunus persica (1 copy) and Malus domestica (2 copies), respectively. The analysis also showed a separate clade for the Fabaceae family member V. unguiculata and G. max both with 1 copy each. Notably, we detected an additional TAC1 copy in the lineage containing the diploid cotton progenitor species G. arboreum and G. raimondii, each possessing three TAC1 copies, indicating a TAC1 duplication event specific to this lineage (Figure 1). As anticipated, within the Gossypium clade, the TAC1 gene copy number remained consistent, with three copies in both the A and D subgenomes in all examined allotetraploid cotton species (G. hirsutum and G. tomentosum) examined (Figure 1; Table 2). As expected, the A-subgenome diploid progenitor, Gossypium arboreum, and the D-subgenome diploid progenitor, Gossypium raimondii, are ancestral Table 1; Figure 1. Furthermore, the phylogenetic analysis also revealed a close relationship between T.cacao (cocoa) and the clade containing the cotton lineages highlighting the relationship of both Theobroma cacao and Gossypium spp. within the Malvaceae family.
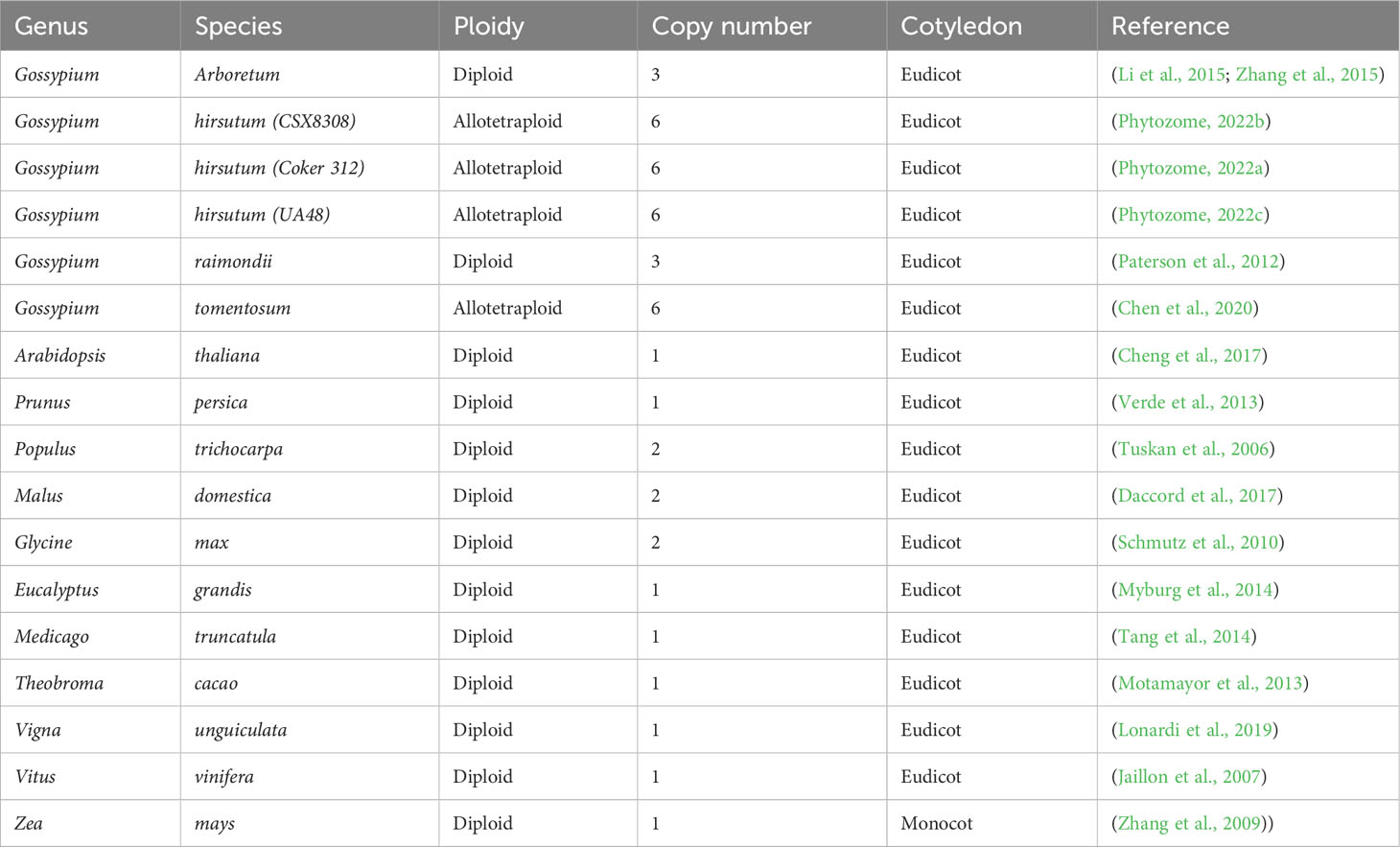
Table 1 List of putative TAC1 orthologs in cotton and other plant species.
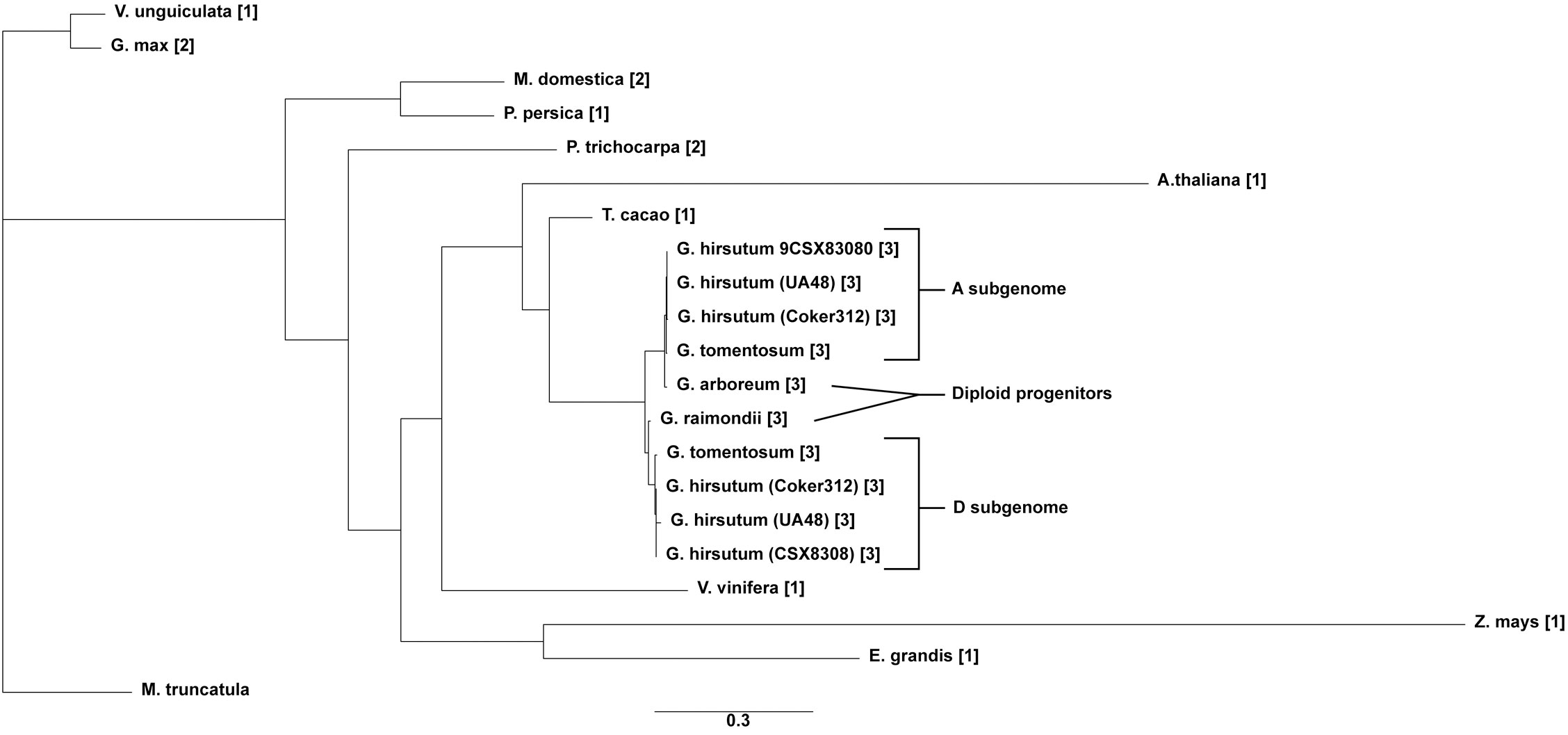
Figure 1 The phylogenetic tree of the TAC1 gene in cotton and other plant species. The tree was constructed by RAxML GAMMA GTR using the Neighbor-Joining method with 5000 bootstrap replicates (Stamatakis, 2014; Kozlov et al., 2019). The bar indicates an evolutionary distance of 2.0%.
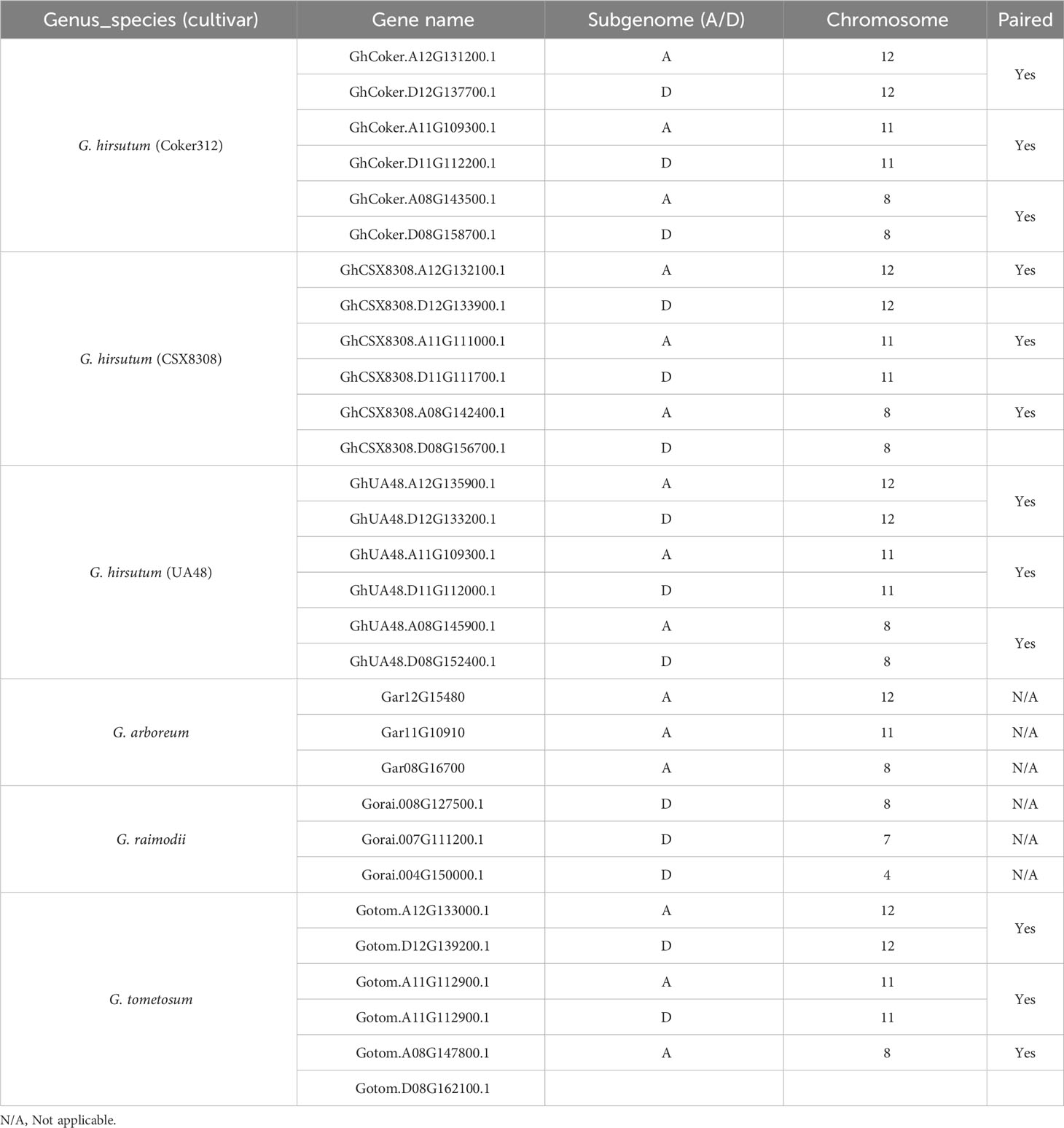
Table 2 TAC1 gene distribution in cotton.
The TAC1 gene is close to 300 amino acids in length and is predicted to contain an NAD-dependent protein deacetylase domain from the PantherDB (Mi and Thomas, 2009), Figure 2. A multiple sequence alignment of TAC1 from cotton and various plant species is only modestly similar with an average percent identity of 33% and only 30 (9%) identical sites (Figure 2). Alignments revealed the presence of conserved domains of the TAC1 gene in other plant species, such as the IGT conserved motif (Figure 2), which is known to have an impact on vertical shoot growth in a broad range of plant species (Roychoudhry and Kepinski, 2015; González-Arcos et al., 2019). Similarity, homeologs also differed significantly among the TAC1 copies. For example, the A11/D11 homeologs had high similarity at 96.8% at the amino acid level, while the A12/D12 and A08/D08 homeologs were 73% identical (Figure 3A; Supplementary Table S5). The regions of conserved sequence previously reported (Dardick et al., 2013) are highlighted as four domains and are conserved among all the homeologs, Figure 3A. Examination of the GhTAC1 phylogeny within the allotetraploid shows A08/D08 and A11/D11 homeologous gene pairs showed separate clades forming from the A12/D12 gene pairs (Figure 3B).

Figure 2 Multiple protein sequence alignment of predicted amino acid sequences of TAC1 orthologs cotton, peach, Arabidopsis, cacao, soybean, and maize. Highly conserved residues are highlighted in black with the IGT conserved domain indicated in red box at the 67-69bp position, which is typical of the IGT gene family.
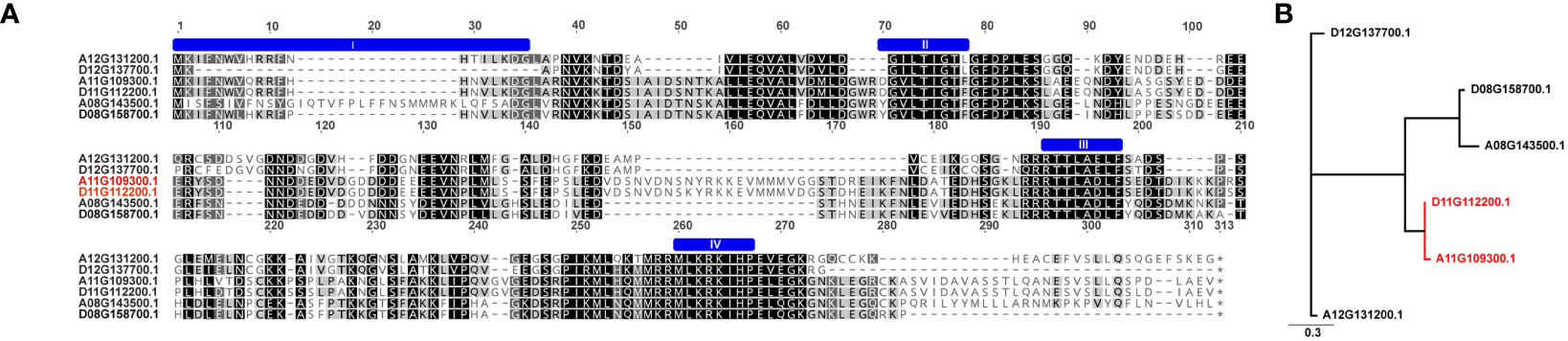
Figure 3 Multiple sequence alignment and dendrogram of TAC1 copies in (G) hirsutum (Coker312) and peach. (A) Six GhTAC1 protein sequences alignment. The previously reported 4 conserved domains (Dardick et al., 2013) are annotated by a blue “bar” above the sequence. (B) The homeologous copies on A12/D12 clade have the highest sequence identity to the single prunus copy. The six homologs within Coker312 are also quite diverged with only ~50% pairwise identity.
To determine tissue-specific expression patterns of the six GhTAC1 genes, we performed qPCR analysis in the Coker 312 cotton genotype at various stages of reproductive and vegetative growth utilizing gene and homoeolog-specific primers. Various TAC1 homeologs displayed subgenome expression bias (A-subgenome dominance in all experiments) and differential expression in various cotton tissue sources (Figure 4A; Table 3). In vegetative tissues (leaf, stem, meristem, and roots) significant expression with subgenome bias was observed for the A11 TAC1 homeolog (GhCoker.A11G109300) in stem tissue with over 10-fold expression compared to the other five homeologous copies (Figure 4B; Table 3). In leaf tissue GhCoker.A12G131200 was highly expressed compared to the other five homeologs with about a 2-fold expression compared to the second gene (GhCoker.D08G158700) that was highly expressed (Figure 4B and Table 3). Fairly low relative expression levels were observed in meristem and root tissue of all GhTAC1 homeologous copies (Figure 4C; Table 3) which was expected, particularly in roots. Interestingly, in flower and fiber tissues, high expression levels were observed for GhCoker.A11G109300 (dominant copy) and GhCoker.D11G112200, respectively. The expression level of GhCokerA11.G109300 was over 7-fold higher in flower tissue and 20-fold higher in fiber compared to the other gene copies (Figure 4D; Table 3). The presence of multiple copies of the GhTAC1 gene, along with its notably high expression in the flower tissue, may be the reason for the nearly horizontal growth pattern of the flower.
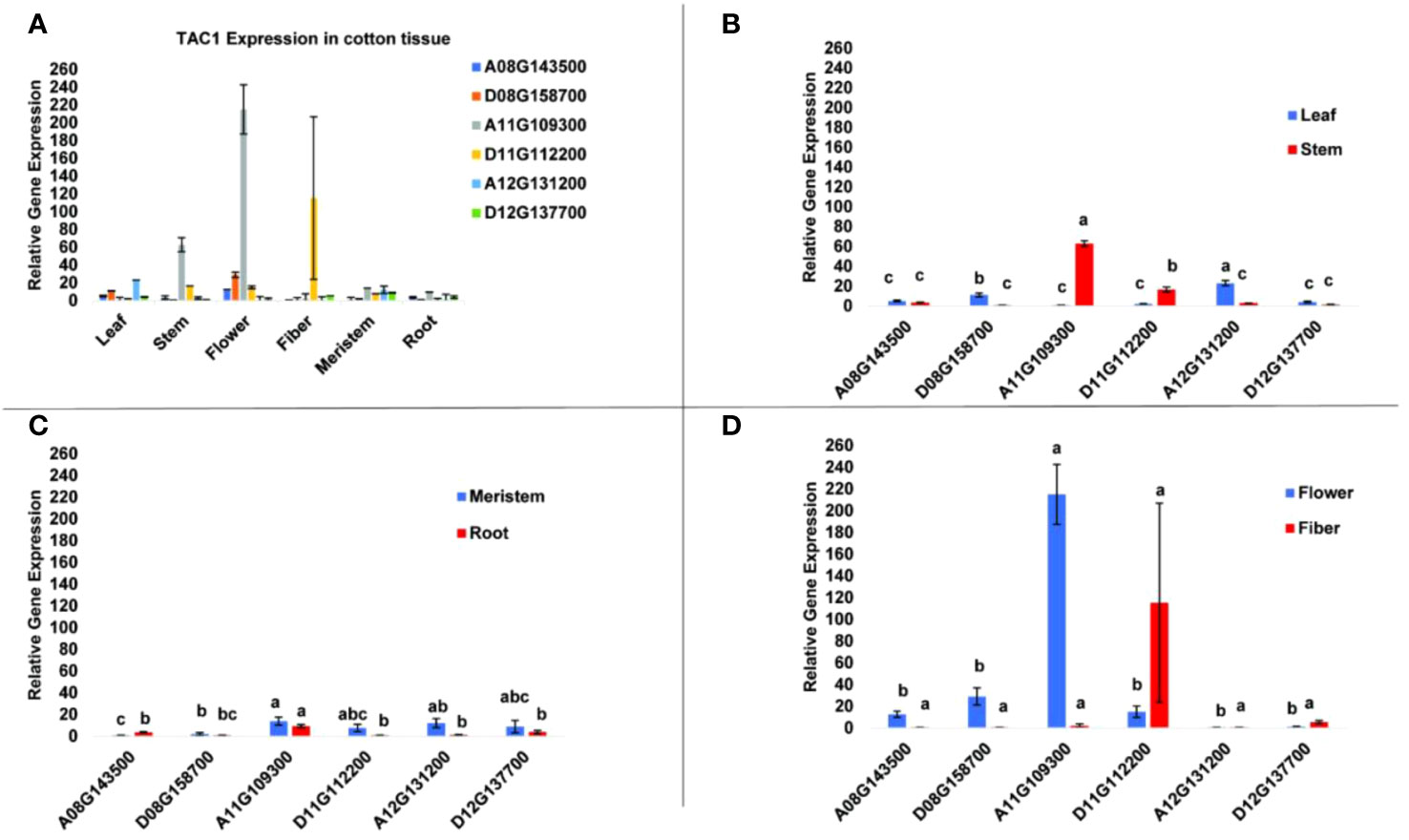
Figure 4 (A) Expression profiles of putative TAC1 homologs in various cotton tissue. (B) leaf and stem, (C) meristem and root, (D) flower and fiber tissues. RT-qPCR was performed using template cDNA primed by Poly T(25) from total RNA that was isolated from 100 mg of tissue. The statistical difference between groups was determined by one-way ANOVA with a post-hoc Tukey HSD Test. Means not sharing the same letter are statistically significantly different (P < 0.05). All the data presented as the mean ± SD (n=3).
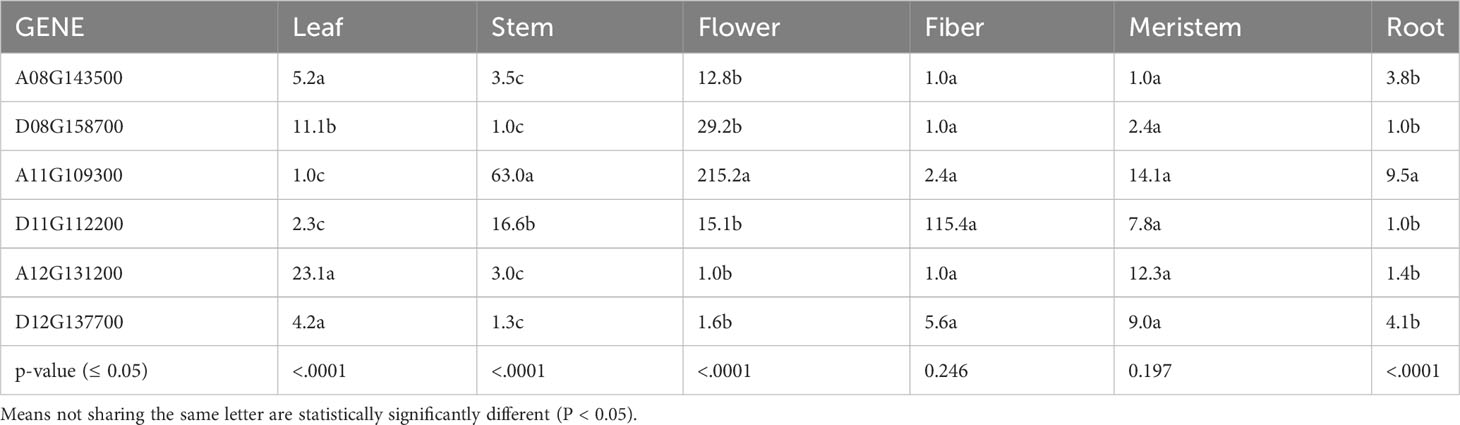
Table 3 Analysis of variance of TAC1 expression in multiple cotton tissues.
The dual guide RNAs used in this study to create a deletion were designed to target a 128-base pair (bp) region specific to the second exon of the GhCoker.D11G112200 and GhCoker.A11G109300 homeologous gene pair, Supplementary Table S6. Two types of mutations were observed at the target sites of SpCas9-edited lines that include a 94-base pair (bp) deletion (detected in most of the edited lines) and an 89bp inversion flanked by a 6bp and 10bp deletion, respectively, Figures 5A, B, Supplementary Table S7. The 94 bp deletion was found in both A and D subgenomes and included the PAM sequence, Figure 5A. In this event, the A and D subgenomes were clearly distinguished by numerous homeologous SNPs, arrow in Figure 5A. The 89 bp inversion flanked by 2 deletions that include approximately half of the gRNA1 and gRNA2 target sites was found only in the D-subgenome TAC1 copy, (Figure 5B). The two edited lines with both 94bp deletions or a mosaic of the 94bp deletion and the 89bp inversion displayed the same branch/petiole angle inclination phenotype, Supplementary Figure S3.
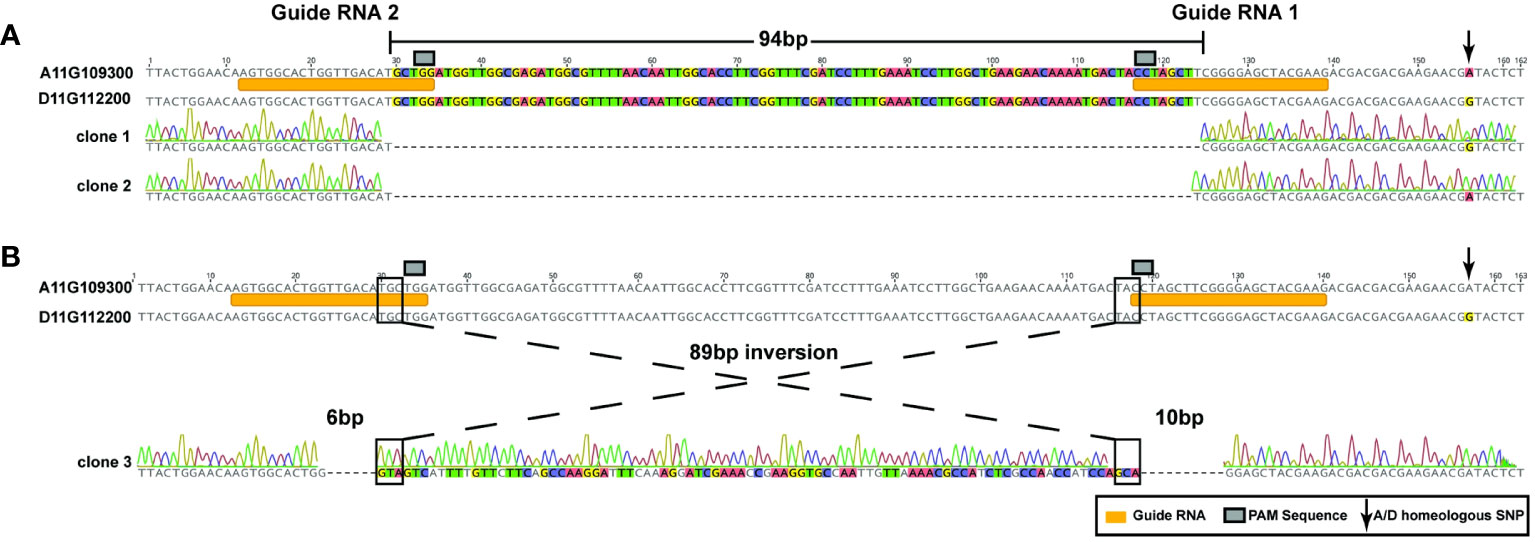
Figure 5 2 types of homeologous GhTAC1 edits by the CRISPR/Cas9 system in Coker312. (A) A 94-bp deletion that includes the PAM motif and a portion of the guide RNA. This deletion was found in the following events: tac1-72 (A and D subgenome) and in the A-subgenome of tac1-73 and tac1-74. (B) An 89bp inversion flanked by 6bp and 10bp deletions that include the PAM sequence and a portion of each guide RNA, respectively.
The T1 seeds that were generated were sown to identify mutants and screen for individuals that may segregate with the genome editing reagents and the TAC1 deletion. Among these mutants, tac1-73 and tac1-74 exhibited a favorable segregation ratio of 3:1, thus indicating a single copy (Figure 6). In our study, all 24 seeds of both tac1-73 and tac1-74 variants exhibited germination. Following screening for the presence of CAS9 in these mutants, 18 seedlings of tac1-73 and 17 of tac1-74 survived. These results verify that the knockout strategy for the mutants developed was successful and could be further validated by the phenotype observed five weeks after emergence (Figure 6A). The T1 plants of tac1-73 showed columnar branch formation as depicted in more inclined branch angles (Figure 6B) compared to wild type Coker 312 (Figure 6C).

Figure 6 (A) Representative images depicting columnar phenotype inclination in wild type (Coker 312) vs tac1-73 (T1 generation). (B) Branch angle measurements of wild type (Coker 312) and (C) Branch angle measurements and tac1-73 (T1 generation).
To assess the expression profiles of TAC1 genes and specifically investigate the knockout effects on the homoeologous gene pairs GhCoker.A11G109300 and GhCoker.D11G112200 in TAC1 mutant lines (tac1-72, tac1-73, and tac1-74) in comparison to the wild-type Coker 312, a quantitative polymerase chain reaction (qPCR) analysis was conducted using stem tissue samples. Homoeolog-specific primers were designed to target the 128bp knockout region within the second exon of GhCoker.A11G109300 and GhCoker.D11G112200 homoeologous gene pair. Remarkably, all three mutant lines exhibited significantly reduced expression levels of both GhCoker.A11G109300 and GhCoker.D11G112200 homoeologous gene pairs (Figure 7). Notably, when compared to the wild-type Coker 312, the tac1-73 and tac1-74 mutants displayed very low expression, with reductions of approximately 10-fold and 5-fold in tac1-73 and tac1-74 mutants, respectively. However, the tac1-72 mutant demonstrated a more moderate 2-fold reduction in expression. Based on the gene-specific expression patterns, GhCoker.A11G109300 (the dominant copy) exhibited lower expression levels in all three mutant plant lines when compared to GhCoker.D11G112200, except in tac1-72 where GhCoker.D11G112200 showed lower expression in comparison to GhCoker.A11G109300 (Figure 7).
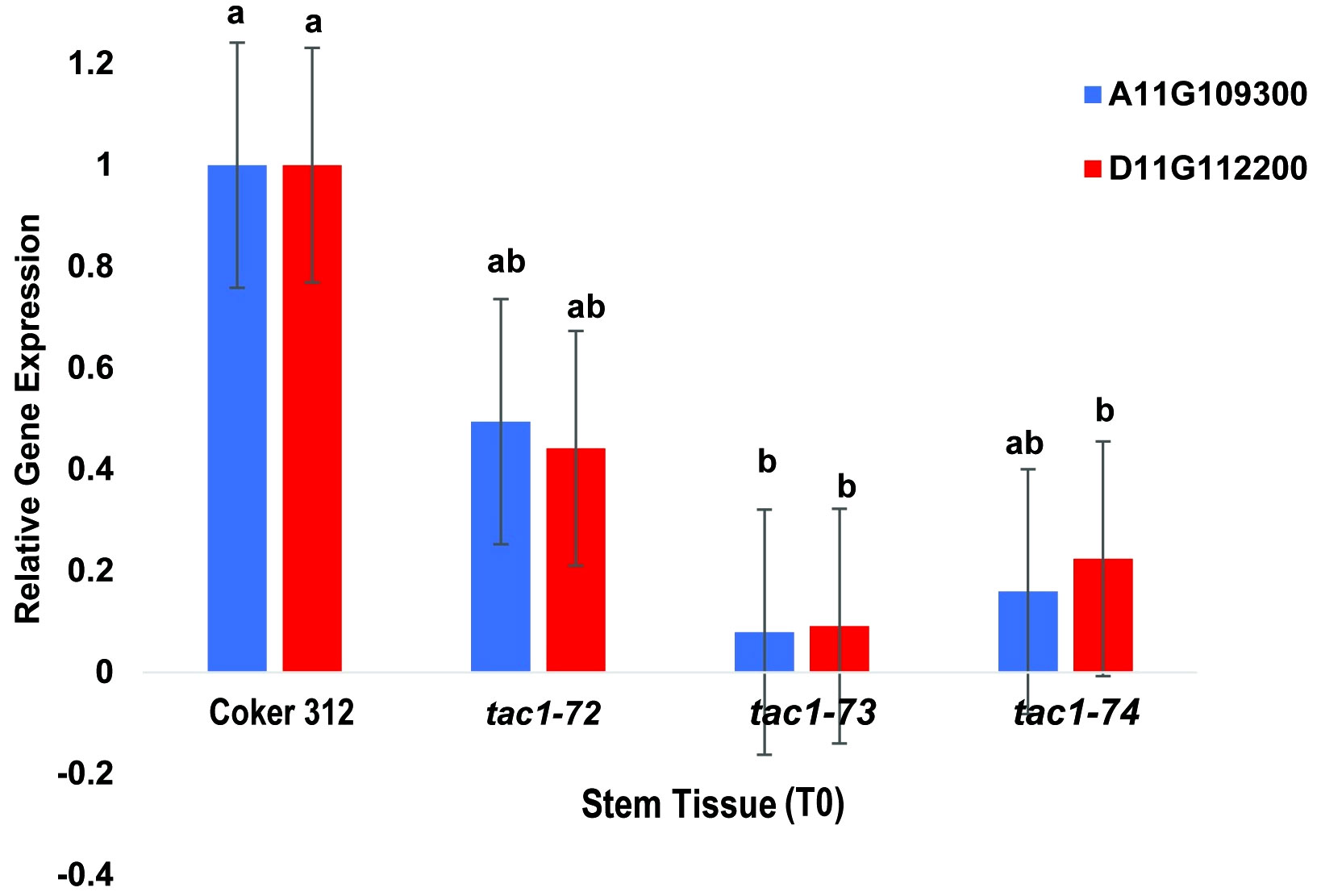
Figure 7 qPCR results of GhTAC1 expression in stem tissue from wild type Coker 312 and tac1-72, tac1-73, and tac1-74. Error bars represent standard deviations of three biological replicates. Means not sharing the same letter are statistically significantly different (P < 0.05).
To explore the role of GhTAC1 in branch angle, the A11/D11 homoeologs were targeted because of their high levels of expression in stem tissue. Transgenic plants harboring targeted knockouts of the A11/D11 homoeologous gene pair were generated. Plants with the validated GhTAC1 (A11/D11) knockouts (mutant) at the T0 stage displayed significant differences in branch inclination angles when compared to wild type Coker 312 plants (Figure 8A). Branch inclination angles were measured at flower bud formation. Mean branch inclination angles widely differed among the transgenic A11/D11 knockouts (transgenic positive plant) and wild type Coker 312 for both monopodial and sympodial branches (Figures 8A–C). The phenotyping results revealed significant differences in branch inclination angles among the different plant variants. For sympodial branches the mutant exhibited an angle of 51.2° while the wild-type Coker 312 measured 74.6°. On the other hand, no significant differences were observed in monopodial branch angles, with the mutant measuring 47.5° and the wild type Coker 312 being 57.5°. (see Figure 8B; Table 4). Regarding petiole angles, the mutant showed narrower angles in sympodial branches compared to wild type Coker 312. However, no significant difference was found in monopodial branches (see Figure 8C; Table 5). Furthermore, it was noted that the mutant plants displayed increased branching and leaf biomass in the basal region while displaying fewer branches and leaves in the apical region though not significantly different from the wild-type Coker 312 plant (Supplementary Figures S4-S6). To assess the impact of GhTAC1 on cotton morphology, measurements were taken for boll count, plant height, and branch length. The analysis revealed no significant differences in these variables between the wild-type Coker 312 and the mutants (Table 6).
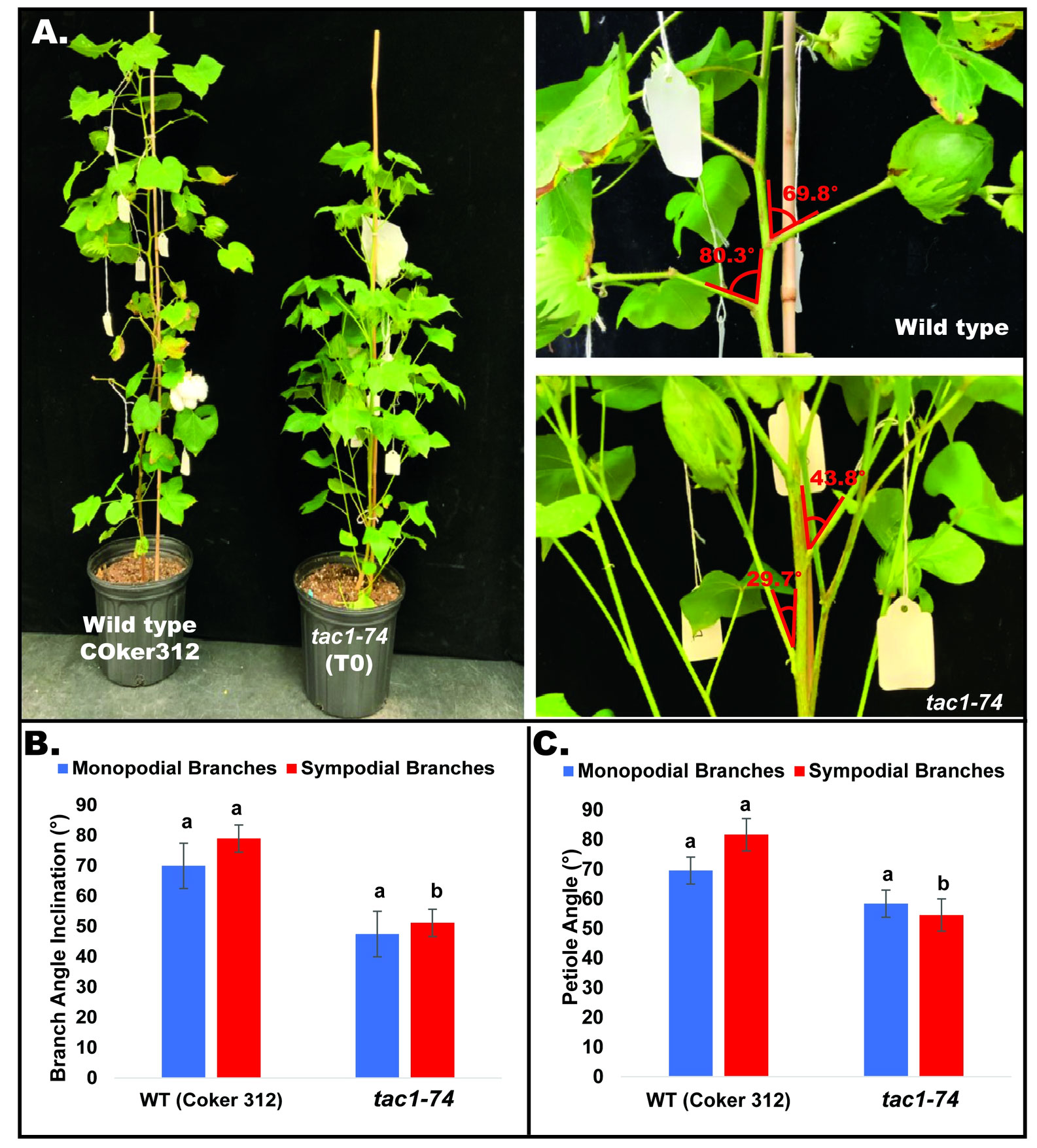
Figure 8 (A) Representative images depicting columnnar phenotype inclination in wild type (Coker 312) and mutant plant. (B) Mean branch angle measurements of wild type (Coker 312) and mutant plant (tac1-74). (C) Mean petiole angle measurements of wild type (Coker 312) and mutant plant (tac1-74). The statistical difference between groups was determined by one-way ANOVA with a post-hoc Tukey HSD Test. Means not sharing the same letter are statistically significantly different (P < 0.05). Errors bars represent SD.
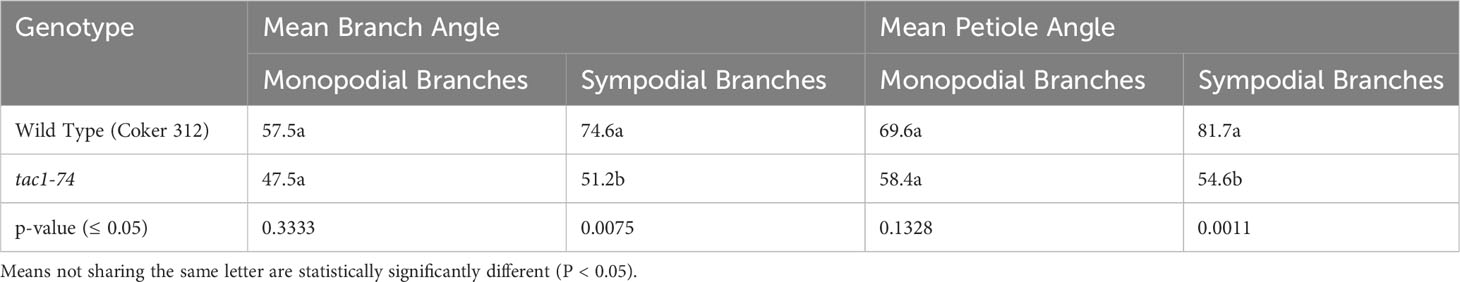
Table 4 Analysis of variance for branch angle inclination and petiole Angle.
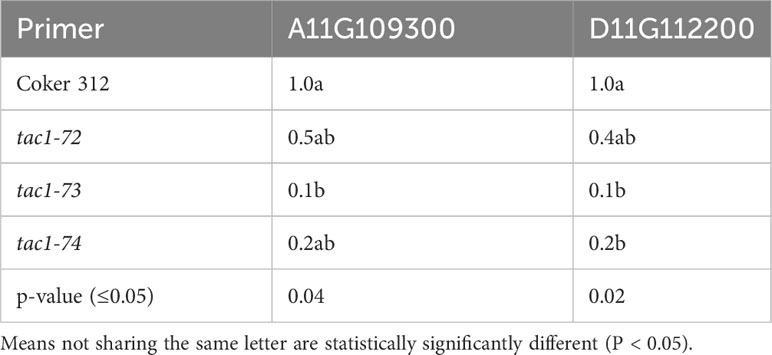
Table 5 Analysis of variance for TAC1 expression in stem tissue of wild type (Coker 312) and mutants (tac1-72 tac1-73 and tac1-74).
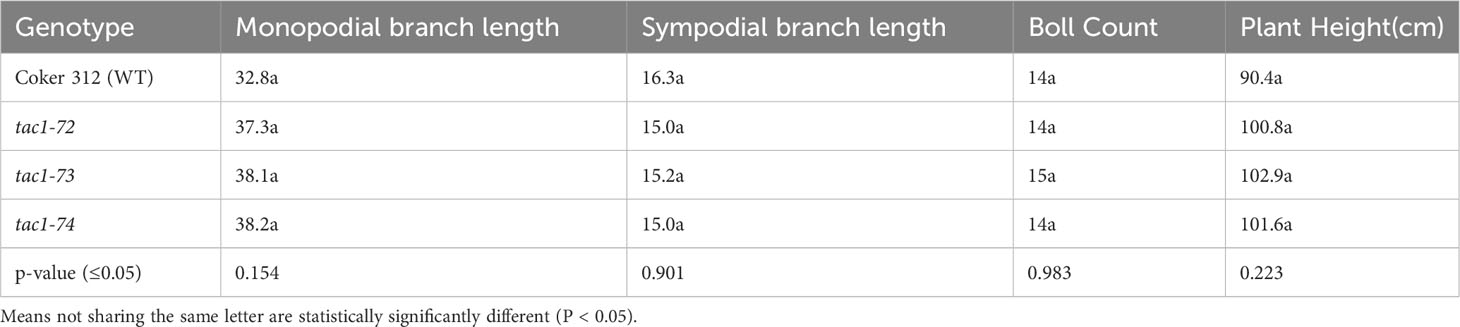
Table 6 Analysis of variance for some morphological traits of wild type (Coker 312) and mutants (tac1-72 tac1-73 and tac1-74).
Upland cotton is a commonly cultivated fiber crop for the textile industry. Meeting the increasing food and fiber demands of the rising global population and addressing unpredictable climatic shifts requires enhancing crop productivity on the same or reduced land area, thus using the same or fewer resources. Combining higher-density planting with improved photosynthetic capacity is a promising approach to achieving this objective. This would maximize resource utilization while optimizing the plant’s energy capture and conversion efficiency resulting in higher crop yields with resource efficiency, thereby benefiting growers and consumers. Understanding the impact of plant architecture on cotton growth is paramount to enable the breeding of cotton varieties with enhanced architectural traits (Kaggwa-Asiimwe et al., 2013; Hollender et al., 2018).
The genetic modification of plant architecture can offer several advantages such as increased carbon assimilation, improved light utilization in dense crop canopies, heightened mechanical process efficiency, decreased susceptibility to insects and diseases by reducing canopy humidity, and the potential for higher yields. The growth and yield of cotton plants are significantly impacted by light penetration, especially in dense cotton fields where the upper parts of the plant receive most of the light. This uneven distribution of light within the branch canopy critically impacts optimal growth and yield. Light distribution is influenced by various factors, such as genetics, environment, and management practices (Mao et al., 2014; Yao et al., 2017). Cotton varieties with columnar canopies tend to have an open structure that facilitates better radiance interception and light penetration throughout the canopy (Chapepa et al., 2020). The positioning of bolls is also crucial for yield, with the first position bolls proportionally contributing to plant yield due to their larger size and weight. To produce these bolls, older leaves must produce enough photosynthates, which require even and adequate light penetration throughout the plant (Jiang et al., 2012; Hollender et al., 2018; Huang et al., 2022). Therefore, developing cotton cultivars with modified plant architecture can significantly improve crop performance and have far-reaching benefits for the cotton industry. Here, we evaluate the genomic content, phylogenetic distribution, and functional implications of TAC1 in cotton.
Phylogenetic analysis of TAC1 revealed between 1 and 2 copies in representative diploid Angiosperms, except for the diploid progenitors of the Gossypium lineage (G. arboreum and G. raimondii, which contained 3 copies, Figure 1; Table 1, indicating that the TAC1 gene duplication is specific to this lineage. In Prunus, a species with a single TAC1 copy, altered expression of this gene resulted in pleiotropic shoot phenotypes. For example, silencing of TAC1 resulted in plum trees with severely vertical branch orientations, while overexpression resulted in trees with more horizontal branch orientations. Collectively, alteration of TAC1 in Prunus species leads to pleiotropic shoot phenotypes (Dardick et al., 2013; Hollender et al., 2020). Similarly in Arabidopsis plants with a single copy of the TAC1 gene, mutant plants display a more inclined branch angle relative to the wild type (Hollender et al., 2020). A study on the functional characterization of TAC1 in Populus trichocarpa (poplar), which contains two copies of the gene (Potri.014G102600 and Potri.002G175300), revealed that knocking out these genes resulted in narrower leaf angles and upright shoot growth (Fladung, 2021). In Malus domestica, two members of the TAC1 gene (MdTAC1a and MdTAC1b) have been identified. Subcellular localization analysis of MdTAC1a showed that it is detected in the nucleus and cell membrane, while MdTAC1b is detected only in the cell membrane. Both genes play a role in regulating branch inclination in Malus domestica, and they are highly expressed in the shoot tips and vegetative buds of weeping cultivars. However, they exhibit weak expression in columnar cultivars (Li et al., 2022).
Our results revealed three copies in the diploid progenitor cotton species (G. arboreum and G. raimondii) indicating the duplication event supersedes the polyploidization event in allotetraploid cotton and underscores the potential importance retaining this gene duplication event in success of both the wild and domesticated allotetraploid cottons (Cronn et al., 1999; Meng et al., 2020). Visual comparisons of cotton plants with various Angiosperms reveals that most cotton genotypes have near horizontal branches which could be attributed to the presence of additional TAC1 alleles. However, certain naturally occurring mutant cotton varieties exhibit more acute branch angles compared to their wild relatives, which could be a result of gene regulation ((Ji et al., 2021). Moreover, previous studies have identified two types of cotton mutants, known as cluster branching (cl1) in G. hirsutum (Upland cotton) and short branch (cl2) in G. barbadense (Pima cotton), which exhibit a distinct phenotype called “nulliplex-branch” (nb). These mutants display determinate sympodial growth (Kearney, 1930; Pathak and Singh, 1975). The cl1 mutant is associated with a recessive allele located on chromosome D07, while the cl2 mutant is associated with a recessive allele located on chromosome A07 (Stephens, 1955; Endrizzi and Ray, 1992). The nulliplex-branch mutants demonstrate unique characteristics, such as flowers developing directly from leaf axils on the main stem or occasionally from a short branch with a single node. These phenotypes typically lack fruiting branches and possess a compact plant structure. As a result, they are highly suitable for high-density planting and mechanical harvesting. Additionally, they mature early, and their growth does not require chemical regulation or manual pruning, which reduces labor inputs (Silow, 1946; McGarry et al., 2016; Sun et al., 2016b; Si et al., 2018; Chen et al., 2019; Huang et al., 2022; Sun et al., 2022).
Sequence similarity analysis of a representative TAC1 gene in cotton showed expected regions of high identity when compared to other species (cacao, peach, soybean, Arabidopsis, and maize), Figure 2. Protein family analysis of TAC1 identified an NAD-Dependent protein deacetylase HST1-like domain (Panther 38366). This functional domain (histone deacetylation) is involved in telomeric silencing and methylation maintenance. Other studies have shown that LAZY and DRO interact with a protein that has an RCC1 (regulator of chromatin condensation) domain. The strong sequence similarity between the TAC1 gene in Arabidopsis and the wild diploid cotton species Gossypium raimondii confirms the functional role of TAC1 in wild cotton. This finding supports the notion that domestication processes resulted in targeted artificial selection for agronomic traits, including yield, pest resistance, fiber length, compact branching, and reduced growth and maturity periods. The multiple alignment of TAC1 amino acid sequences (Figure 2) also showed structural similarities in the IGT domain, suggesting that these ancient IGT genes play a consistent role in determining shoot growth angle orientation (Dardick et al., 2013; Dong et al., 2016; Montesinos et al., 2021).
An analysis of the six homeologous copies of TAC1 in the Coker312 genotype revealed intriguing differences in the level of homeologous and homologous similarity among the six TAC1 copies at the sequence identity level. Notably, the A11/D11 copies of TAC1 exhibited a 96.8% identity, while their similarity with homologs on chromosome 8 or 12 was only up to 58% (Supplementary Table 1). We observed that this pair of TAC1 homologs displayed dominant tissue-specific expression when compared to the other homologs. Additionally, their expression patterns displayed subgenome bias as shown in Figures 3A, B, 4A. The A08/D08 and A12/D12 copies exhibited a sequence identity of approximately 74% between homeologs, respectively (refer to Supplementary Table 1 for details). Significantly, the expression patterns of A11 and A12 TAC1 homologs in various vegetative tissues, including leaf, stem, meristem, and root, revealed a distinct prevalence of expression in leaf and stem tissues (Figure 4). Previous studies have demonstrated that TAC1 exhibits increased expression in actively growing vegetative buds, despite its low expression levels in meristem tissue (Xu et al., 2017). The low expression of TAC1 in root tissue aligns with its regulatory role in upper branches of plants, with minimal or no expression in lower branches (Dardick et al., 2013). Our analysis showed a notable difference in the expression levels of A11 and D11 TAC1 copies, with A11 exhibiting significantly high expression in flower tissue and D11 displaying the highest expression in fiber tissue. These results highlight the subgenome expression bias and suggest that the functional domains of the A11/D11 coding sequence (CDS) may be redundant, while their regulatory regions (5’ and 3’ UTRs and promoters) could be differentially activated in various tissues and developmental stages (Figure 4). Previous studies have indicated that TAC1 exhibits high expression in reproductive structures such as flower buds, which may explain the horizontal growth of the flower and its subsequent development into fiber (Dardick et al., 2013) The A12/D12 copies displayed higher expression levels in vegetative tissues, with the A12 copy showing subgenome expression bias over D12 in leaf, stem, meristem, and root tissues, indicating the possibility of functional redundancy or sub-functionalization with a role in leaf angle.
Targeted knockout of the A11/D11 homeologous TAC1 copies resulted in a cotton plant with an induced columnar phenotype with distinct branch and petiole inclination, Figure 6; Supplementary Figure S5 and S6. Overall, the mutant plants consistently exhibited narrower measurements in terms of branch inclination angle and petiole angle for sympodial branches. However, there were no notable differences observed in the measurements of monopodial branches and petiole angle for both the mutant and wild type Coker 312 (Figures 6B, C). No variations were observed in some morphological traits, including branch length (monopodial and sympodial branches), plant height, and boll count (Table 6). Sequence analysis of the mutants from three independent events with columnar branching (tac1-72, tac1-73, and tac1-74) revealed a high efficiency of the CRISPR-induced mutations (Figure 5A, B) resulting in hemizygous/homozygote mutations with large deletions and a fragment rearrangement (Supplementary Tables S6, S7). In tac1-72 and tac1-73 mutants (Figure 5A) a 94bp deletion was observed in both A11/D11 homeologs. Interestingly, the analysis of tac1-74 mutant revealed an 89bp inversion flanked by two deletions at the target site in D11 and the 94bp deletion in A11 (Figure 6B). These mutations altered the GhTAC1 gene function, rendering it non-functional, and subsequently led to the same observed columnar phenotype in all independent events. These findings align with previous studies in other Eudicots which have shown a correlation between mutations in TAC1 and the development of a nearly erect plant architecture phenotype (Dardick et al., 2013; Hollender et al., 2018; Hollender et al., 2020; Fladung, 2021; Dutt et al., 2022; Li et al., 2022). Intriguingly, we noted an enhanced canopy structure in the mutants accompanied by a significant increase in the number of leaves, auxiliary buds, and subtending leaves, particularly in the basal region of the plants compared to the apical region (see Supplementary Figures S5, S6). This pattern is characteristic of columnar plant phenotypes which have the potential to improve light distribution within the plant canopy (Chapepa et al., 2020). Validation of the expression of both A11/D11 homeologs in stem tissue revealed a decrease in expression levels for A11/D11 pairs. This decline in expression for both gene pairs contributes to the columnar phenotype observed in the mutants. These findings are consistent with earlier research which indicates that the loss/mutation of TAC1 gene function results in decreased expression in mutant varieties (Dardick et al., 2013; Hollender et al., 2020).
The segregation analysis of T1 progeny derived from the mutants, particularly tac1-73 and tac1-74, revealed a notably favorable segregation ratio, emphasizing a single copy and a confirmation supported by the presence of resistance to the bar gene selectable marker (Supplementary Figures S5, S6). These findings provide strong evidence of transformation efficiency.
Several studies have indicated that the canopy microenvironment plays a critical role in regulating not only the distribution of light but also the temperature and relative humidity within the canopy. These factors collectively influence the radiation received by plants (Yang et al., 2014; Chapepa et al., 2020). The findings of this study are consistent with previous studies in peach and poplar, where mutant varieties with loss of function of the TAC1 gene showed pronounced nearly upright growth of branches (Dardick et al., 2013; Fladung, 2021). The potential benefits of upright branch growth in peach, poplar, and other woody Eudicots include high density planting, increased yield, automated harvesting, and reduced pruning (Dardick et al., 2013; Fladung, 2021). Harnessing knowledge of plant architecture offers the potential for ultimately enhancing cotton productivity and increasing higher yields.
Plant architecture undergoes dynamic changes throughout their development. Our study revealed that the TAC1 gene is duplicated and unique to the Gossypium lineage within the Angiosperms. Interestingly, we observed a correlation between the number of native TAC1 copies and the horizontal branching pattern, with cotton exhibiting the most pronounced lateral growth among the Angiosperms. We also confirmed CRISPR-mediated knockout of the highly expressed copies of the TAC1 gene (A11G109300 and D11G112200) led to a significant reduction in both branch and petiole angles. The results of this study demonstrate the potential of gene editing technology to introduce novel traits into high-value crops, such as cotton, that would otherwise not exist naturally because of the presence of multiple subgenomes. These findings have significant implications for improving planting density, understanding optimal light interception and the correlation with yield potential, and enhancing our understanding of the evolution and domestication of the Gossypium lineage.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
FK: Investigation, Writing – original draft, Methodology. SK: Methodology, Writing – original draft, Supervision, Writing – review & editing. ZL: Supervision, Writing – review & editing, Data curation, Formal Analysis. AS: Data curation, Formal Analysis, Writing – review & editing. CD: Conceptualization, Formal Analysis, Writing – original draft, Writing – review & editing. DJ: Resources, Writing – review & editing. CS: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by Cotton Incorporated project number 20-655.
The authors extend appreciation to Stephen Parris, Lauren Carneal, Meredith Cobb, Quinn LaFave and Taylor Martin for assistance in branch angle data collection and maintenance of experimental plants in the greenhouse facility.
Author DJ was employed by the company Cotton Incorporated.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1320638/full#supplementary-material
Supplementary Figure 1 | Dual guide RNA design
Supplementary Figure 2 | The total DNA amplified using primer pair: F-AGATGGGCTTGCACGAAATGTTAAG and R-CGTTTTTGGCAGGAAGAGRAGATG. The PCR products were cloned into pGEM-T-Easy vector and sequenced using standard Sanger sequencing techniques. The sequence alignments were conducted by Geneious Prime. For alignment, the sequences of A11G109300 and D11G112200 are from Coker312 whole genome sequence, the sequences of A1_Sp6 to A18_Sp6 are from sanger sequencing results. The 95-bp deletion showed in Clone A3-A4, the 95-bp deletion showed in Clone A1, A2, clone A6, A7, clone A10-A12, A14-A16 and A18; the one bp insertion (A) found in Clone A8 and A13; the 89-bp inversion detected in Clone A9 and A17. The highlighted line arrow indicated SNP between A11G109300 and D11G112200.
Supplementary Figure 3 | A cartoon image describing the branch and petiole angle was measured in the study. In measuring branch angles, a line was drawn between the branch point and 2cm down the branch, and the angle with respect to the line and stem was recorded. For petiole angle measurements, a line was drawn tangent to the branch and 1cm back from the last petiole on the branch, and the angle of this line with respect to the angle of the stem was recorded (Adapted from "Cotton Plant", Adapted from "Cotton Plant" by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates.
Supplementary Figure 4 | Branch and leaf count at the apical part (at 45cm height)
Supplementary Figure 5 | Images of mutant plants (tac1-72, tac1-73, and tac1-74)
Supplementary Figure 6 | Representative images illustrating the comparison between Wild Type Coker 312 and T1 generation of mutant plants (tac1-73 and tac1-74) at vegetative growth (before squaring).
USDA-ARS, United States Department of Agriculture-Agricultural Research Services; BLAST, Basic Local Alignment Search Tool; CAS9, CRISPR-associated protein 9; CDS, Coding Sequence; COP1, Constitutive Photomorphogenesis; CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats; CTAB, Cetyltrimethyl Ammonium Bromide; TAC1, Tiller Angle Control 1.
Adams, C., Thapa, S., Kimura, E. (2019). Determination of a plant population density threshold for optimizing cotton lint yield: A synthesis. Field Crops Res. 230, 11–16. doi: 10.1016/j.fcr.2018.10.005
Artico, S., Nardeli, S. M., Brilhante, O., Grossi-de-Sa, M. F., Alves-Ferreira, M. (2010). Identification and evaluation of new reference genes in Gossypium hirsutumfor accurate normalization of real-time quantitative RT-PCR data. BMC Plant Biol. 10 (1), 49. doi: 10.1186/1471-2229-10-49
Basu, U., Parida, S. K. (2021). Restructuring plant types for developing tailor-made crops. Plant Biotechnol. J. 21 (6), 1106–1122. doi: 10.1111/pbi.13666
BioRender.com. (2023). Cotton plant. Available at: https://app.biorender.com/biorender-templates (Accessed 2023).
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Cai, G., Yang, Q., Chen, H., Yang, Q., Zhang, C., Fan, C., et al. (2016). Genetic dissection of plant architecture and yield-related traits in Brassica napus. Sci. Rep. 6 (1), 21625. doi: 10.1038/srep21625
Chapepa, B., Mudada, N., Mapuranga, R. (2020). The impact of plant density and spatial arrangement on light interception on cotton crop and seed cotton yield: an overview. J. Cotton Res. 3 (1), 18. doi: 10.1186/s42397-020-00059-z
Chen, W., Yao, J., Li, Y., Zhao, L., Liu, J., Guo, Y., et al. (2019). Nulliplex-branch, a TERMINAL FLOWER 1 ortholog, controls plant growth habit in cotton. Theor. Appl. Genet. 132 (1), 97–112. doi: 10.1007/s00122-018-3197-0
Chen, Z. J., Sreedasyam, A., Ando, A., Song, Q., De Santiago, L. M., Hulse-Kemp, A. M., et al. (2020). Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement. Nat. Genet. 52 (5), 525–533. doi: 10.1038/s41588-020-0614-5
Cheng, C.-Y., Krishnakumar, V., Chan, A. P., Thibaud-Nissen, F., Schobel, S., Town, C. D. (2017). Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 89 (4), 789–804. doi: 10.1111/tpj.13415
Cronn, R. C., Small, R. L., Wendel, J. F. (1999). Duplicated genes evolve independently after polyploid formation in cotton. Proc. Natl. Acad. Sci. U.S.A. 96 (25), 14406–14411. doi: 10.1073/pnas.96.25.14406
Daccord, N., Celton, J.-M., Linsmith, G., Becker, C., Choisne, N., Schijlen, E., et al. (2017). High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 49 (7), 1099–1106. doi: 10.1038/ng.3886
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. GigaScience 10 (2), 1–4. doi: 10.1093/gigascience/giab008
Dardick, C., Callahan, A., Horn, R., Ruiz, K. B., Zhebentyayeva, T., Hollender, C., et al. (2013). PpeTAC1 promotes the horizontal growth of branches in peach trees and is a member of a functionally conserved gene family found in diverse plants species. Plant J. 75 (4), 618–630. doi: 10.1111/tpj.12234
Dong, H., Zhao, H., Xie, W., Han, Z., Li, G., Yao, W., et al. (2016). A novel tiller angle gene, TAC3, together with TAC1 and D2 largely determine the natural variation of tiller angle in rice cultivars. PloS Genet. 12 (11), e1006412. doi: 10.1371/journal.pgen.1006412
Dutt, M., Mahmoud, L. M., Nehela, Y., Grosser, J. W., Killiny, N. (2022). The Citrus sinensis TILLER ANGLE CONTROL 1 (CsTAC1) gene regulates tree architecture in sweet oranges by modulating the endogenous hormone content. Plant Sci. 323, 111401. doi: 10.1016/j.plantsci.2022.111401
Duursma, R. A., Falster, D. S., Valladares, F., Sterck, F. J., Pearcy, R. W., Lusk, C. H., et al. (2012). Light interception efficiency explained by two simple variables: a test using a diversity of small- to medium-sized woody plants. New Phytol. 193 (2), 397–408. doi: 10.1111/j.1469-8137.2011.03943.x
Duvick, D. N. (2005). “The Contribution of Breeding to Yield Advances in maize (Zea mays L.),” in Advances in agronomy (Ames, Iowa: Academic Press), 83–145.
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 5 (1), 113. doi: 10.1186/1471-2105-5-113
Endrizzi, J., Ray, D. (1992). Mapping of the cl1, R1, yg1, and Dw loci in the long arm of chromosome 16 of cotton. J. Heredity 83 (1), 1–5. doi: 10.1093/oxfordjournals.jhered.a111148
Fladung, M. (2021). Targeted CRISPR/cas9-based knock-out of the rice orthologs TILLER ANGLE CONTROL 1 (TAC1) in poplar induces erect leaf habit and shoot growth. Forests 12 (12), 1615. doi: 10.3390/f12121615
Fok, M. A. C. (1998). “Cotton yield stagnation addressing a common effect of various causes,” in World cotton research conference (U.S.A. Ccic: ICAC), 5–11.
González-Arcos, M., Fonseca, M. E., Zandonadi, D., Peres, L., Arruabarrena, A., Silva ferreira, D., et al. (2019). A loss-of-function allele of a TAC1-like gene (SlTAC1) located on tomato chromosome 10 is a candidate for the Erectoid leaf (Erl) mutation. Euphytica 215, 1–16. doi: 10.1007/s10681-019-2418-1
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40 (Database issue), D1178–D1186. doi: 10.1093/nar/gkr944
Gore, U. R. (1935). Morphogenetic studies on the inflorescence of cotton. Botanical Gazette 97 (1), 118–138. doi: 10.1086/334540
Guseman, J. M., Webb, K., Srinivasan, C., Dardick, C. (2017). DRO1 influences root system architecture in Arabidopsis and Prunus species. Plant J. 89 (6), 1093–1105. doi: 10.1111/tpj.13470
Hahn, F., Mantegazza, O., Greiner, A., Hegemann, P., Eisenhut, M., Weber, A. P. M. (2017). An efficient visual screen for CRISPR/cas9 activity in arabidopsis thaliana. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00039
Hollender, C. A., Hill, J. L., Jr., Waite, J., Dardick, C. (2020). Opposing influences of TAC1 and LAZY1 on lateral shoot orientation in arabidopsis. Sci. Rep. 10 (6051), 1–13. doi: 10.1038/s41598-020-62962-4
Hollender, C. A., Waite, J. M., Tabb, A., Raines, D., Chinnithambi, S., Dardick, C. (2018). Alteration of TAC1 expression in Prunus species leads to pleiotropic shoot phenotypes. Hortic. Res. 5, 26. doi: 10.1038/s41438-018-0034-1
Huang, X., Liu, H., Ma, B. (2022). The current progresses in the genes and networks regulating cotton plant architecture. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.882583
Jaillon, O., Aury, J. M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., et al. (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449 (7161), 463–467. doi: 10.1038/nature06148
Ji, G., Liang, C., Cai, Y., Pan, Z., Meng, Z., Li, Y., et al. (2021). A copy number variant at the HPDA-D12 locus confers compact plant architecture in cotton. New Phytol. 229 (4), 2091–2103. doi: 10.1111/nph.17059
Jiang, J., Tan, L., Zhu, Z., Fu, Y., Liu, F., Cai, H., et al. (2012). Molecular evolution of the TAC1 gene from rice (Oryza sativa L.). J. Genet. Genomics 39 (10), 551–560. doi: 10.1016/j.jgg.2012.07.011
Jin, S.-x., Liu, G.-z., Zhu, H.-g., Yang, X.-y., Zhang, X.-l. (2012). Transformation of upland cotton (Gossypium hirsutum L.) with gfp gene as a visual marker. Journal of Integrative Agriculture 11 (6), 910–919. doi: 10.1016/s2095-3119(12)60081-1
Kaggwa-Asiimwe, R., Andrade-Sanchez, P., Wang, G. (2013). Plant architecture influences growth and yield response of upland cotton to population density. Field Crops Res. 145, 52–59. doi: 10.1016/j.fcr.2013.02.005
Kant, S., Seneweera, S., Rodin, J., Materne, M., Burch, D., Rothstein, S., et al. (2012). Improving yield potential in crops under elevated CO2: Integrating the photosynthetic and nitrogen utilization efficiencies. Front. Plant Sci. 3. doi: 10.3389/fpls.2012.00162
Khan, N., Han, Y., Xing, F., Feng, L., Wang, Z., Wang, G., et al. (2020). Plant density influences reproductive growth, lint yield and boll spatial distribution of cotton. Agronomy 10 (1), 14. doi: 10.3390/agronomy10010014
Koboldt, D. C., Zhang, Q., Larson, D. E., Shen, D., McLellan, M. D., Lin, L., et al. (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22 (3), 568–576. doi: 10.1101/gr.129684.111
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35 (21), 4453–4455. doi: 10.1093/bioinformatics/btz305
Ku, L., Wei, X., Zhang, S., Zhang, J., Guo, S., Chen, Y. (2011). Cloning and characterization of a putative TAC1 ortholog associated with leaf angle in maize (Zea mays L.). PloS One 6 (6), e20621. doi: 10.1371/journal.pone.0020621
Kumar, S., Ruggles, A., Logan, S., Mazarakis, A., Tyson, T., Bates, M., et al. (2021). Comparative transcriptomics of non-embryogenic and embryogenic callus in semi-recalcitrant and non-recalcitrant upland cotton lines. Plants (Basel) 10 (9), 1–16. doi: 10.3390/plants10091775
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997, 1–13.
Li, F., Fan, G., Lu, C., Xiao, G., Zou, C., Kohel, R. J., et al. (2015). Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 33 (5), 524–530. doi: 10.1038/nbt.3208
Li, Y., Tan, X., Guo, J., Hu, E., Pan, Q., Zhao, Y., et al. (2022). Functional Characterization of MdTAC1a Gene Related to Branch Angle in Apple (Malus x domestica Borkh.). Int. J. Mol. Sci. 23 (3), 1–17. doi: 10.3390/ijms23031870
Li, H., Zhang, L., Hu, J., Zhang, F., Chen, B., Xu, K., et al. (2017). Genome-wide association mapping reveals the genetic control underlying branch angle in rapeseed (Brassica napus L.). Front. Plant Sci. 8, 1054. doi: 10.3389/fpls.2017.01054
Li, Y.-h., Zhao, S.-C., Ma, J.-X., Li, D., Yan, L., Li, J., et al. (2013). Molecular footprints of domestication and improvement in soybean revealed by whole genome re-sequencing. BMC Genomics 14 (1), 579. doi: 10.1186/1471-2164-14-579
Lonardi, S., Muñoz-Amatriaín, M., Liang, Q., Shu, S., Wanamaker, S. I., Lo, S., et al. (2019). The genome of cowpea (Vigna unguiculata [L.] Walp.). Plant J. 98 (5), 767–782. doi: 10.1111/tpj.14349
Mao, L., Zhang, L., Zhao, X., Liu, S., van der Werf, W., Zhang, S., et al. (2014). Crop growth, light utilization and yield of relay intercropped cotton as affected by plant density and a plant growth regulator. Field Crops Res. 155, 67–76. doi: 10.1016/j.fcr.2013.09.021
McGarry, R. C., Prewitt, S. F., Culpepper, S., Eshed, Y., Lifschitz, E., Ayre, B. G. (2016). Monopodial and sympodial branching architecture in cotton is differentially regulated by the Gossypium hirsutum Single Flower Truss and Self-Pruning Orthologs. New Phytol. 212 (1), 244–258. doi: 10.1111/nph.14037
Meng, F., Pan, Y., Wang, J., Yu, J., Liu, C., Zhang, Z., et al. (2020). Cotton duplicated genes produced by polyploidy show significantly elevated and unbalanced evolutionary rates, overwhelmingly perturbing gene tree topology. Front. Genet. 11. doi: 10.3389/fgene.2020.00239
Mi, H., Thomas, P. (2009). PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 563, 123–140. doi: 10.1007/978-1-60761-175-2_7
Montesinos, A., Dardick, C., Rubio-Cabetas, M. J., Grimplet, J. (2021). Polymorphisms and gene expression in the almond IGT family are not correlated to variability in growth habit in major commercial almond cultivars. PloS One 16 (10), e0252001. doi: 10.1371/journal.pone.0252001
Motamayor, J. C., Mockaitis, K., Schmutz, J., Haiminen, N., Iii, D. L., Cornejo, O., et al. (2013). The genome sequence of the most widely cultivated cacao type and its use to identify candidate genes regulating pod color. Genome Biol. 14 (6), r53. doi: 10.1186/gb-2013-14-6-r53
Murchie, E. H., Burgess, A. J. (2022). Casting light on the architecture of crop yield. Crop Environ. 1 (1), 74–85. doi: 10.1016/j.crope.2022.03.009
Myburg, A. A., Grattapaglia, D., Tuskan, G. A., Hellsten, U., Hayes, R. D., Grimwood, J., et al. (2014). The genome of Eucalyptus grandis. Nature 510 (7505), 356–362. doi: 10.1038/nature13308
Norton, E. R., Silvertooth, J. C., Stedman, S. W. (1995). "Plant population evaluation for upland cotton" (University of Arizona (Tucson, AZ: College of Agriculture).
Paterson, A. H., Wendel, J. F., Gundlach, H., Guo, H., Jenkins, J., Jin, D., et al. (2012). Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492 (7429), 423–427. doi: 10.1038/nature11798
Pathak, R. S., Singh, R. B. (1975). Genetic analysis of the duplicate loci, cluster and short branch in. Theor. Appl. Genet. 46 (6), 281–287. doi: 10.1007/BF00281150
Peng, J., Richards, D. E., Hartley, N. M., Murphy, G. P., Devos, K. M., Flintham, J. E., et al. (1999). ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature 400 (6741), 256–261. doi: 10.1038/22307
Phytozome (2022a) Gossypium hirsutum Coker v1.1 (Department of Energy's Joint Genome Institute). Available at: http://phytozome.jgi.doe.gov/info/GhirsutumCoker_v1_1 (Accessed September10, 2022).
Phytozome (2022b) Gossypium hirsutum CSX8308 v1.1 (Department of Energy's Joint Genome Institute). Available at: http://phytozome.jgi.doe.gov/info/GhirsutumCSX8308_v1_1 (Accessed September 10, 2022).
Phytozome (2022c) Gossypium hirsutum UA48 v1 (Department of Energy's Joint Genome Institute). Available at: http://phytozome.jgi.doe.gov/info/GhirsutumUA48_v1_1 (Accessed September 10, 2022).
Quinlan, A. R., Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26 (6), 841–842. doi: 10.1093/bioinformatics/btq033
Rathore, K. S., Chowdhury, V. K., Hodges, T. K. (1993). Use of bar as a selectable marker gene and for the production of herbicide-resistant rice plants from protoplasts. Plant Mol. Biol. 21 (5), 871–884. doi: 10.1007/bf00027118
Rausch, T., Zichner, T., Schlattl, A., Stütz, A. M., Benes, V., Korbel, J. O. (2012). DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28 (18), i333–i339. doi: 10.1093/bioinformatics/bts378
Reinhardt, D., Kuhlemeier, C. (2002). Plant architecture. EMBO Rep. 3 (9), 846–851. doi: 10.1093/embo-reports/kvf177
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29 (1), 24–26. doi: 10.1038/nbt.1754
Roychoudhry, S., Kepinski, S. (2015). Shoot and root branch growth angle control-the wonderfulness of lateralness. Curr. Opin. Plant Biol. 23, 124–131. doi: 10.1016/j.pbi.2014.12.004
Schmidt, J. E., Bowles, T. M., Gaudin, A. C. M. (2016). Using ancient traits to convert soil health into crop yield: impact of selection on maize root and rhizosphere function. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00373
Schmittgen, T. D., Livak, K. J. (2008). Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3 (6), 1101–1108. doi: 10.1038/nprot.2008.73
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463 (7278), 178–183. doi: 10.1038/nature08670
Shahbandeh, M. (2021) Cotton yield Per Harvested acre in the U.S. 2020 (Statista). Available at: https://www.statista.com/statistics/191494/cotton-yield-per-harvested-acre-in-the-us-since-2000/ (Accessed April, 20 2023).
Shao, P., Peng, Y., Wu, Y., Wang, J., Pan, Z., Yang, Y., et al. (2022). Genome-wide association study and transcriptome analysis reveal key genes controlling fruit branch angle in cotton. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.988647
Si, Z., Liu, H., Zhu, J., Chen, J., Wang, Q., Fang, L., et al. (2018). Mutation of SELF-PRUNING homologs in cotton promotes short-branching plant architecture. J. Exp. Bot. 69 (10), 2543–2553. doi: 10.1093/jxb/ery093
Silow, R. A. (1946). Evidence on chromosome homology and gene homology in the amphidiploid new world cottons. J. Genet. 47 (2), 213–221. doi: 10.1007/BF02986788
Song, X., Zhang, T. (2009). Quantitative trait loci controlling plant architectural traits in cotton. Plant Sci. 177 (4), 317–323. doi: 10.1016/j.plantsci.2009.05.015
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi: 10.1093/bioinformatics/btu033
Stephens, S. G. (1955). LINKAGE IN UPLAND COTTON. Genetics 40 (6), 903–917. doi: 10.1093/genetics/40.6.903
Sun, Q., Du, X., Cai, C., Long, L., Zhang, S., Qiao, P., et al. (2016b). To be a flower or fruiting branch: insights revealed by mRNA and small RNA transcriptomes from different cotton developmental stages. Sci. Rep. 6 (1), 23212. doi: 10.1038/srep23212
Sun, C., Wang, B., Wang, X., Hu, K., Li, K., Li, Z., et al. (2016a). Genome-wide association study dissecting the genetic architecture underlying the branch angle trait in rapeseed (Brassica napus L.). Sci. Rep. 6 (1), 33673. doi: 10.1038/srep33673
Sun, Q., Xie, Y., Li, H., Liu, J., Geng, R., Wang, P., et al. (2022). Cotton GhBRC1 regulates branching, flowering, and growth by integrating multiple hormone pathways. Crop J. 10 (1), 75–87. doi: 10.1016/j.cj.2021.01.007
Tang, H., Krishnakumar, V., Bidwell, S., Rosen, B., Chan, A., Zhou, S., et al. (2014). An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genomics 15 (1), 312. doi: 10.1186/1471-2164-15-312
Thompson, C. J., Movva, N. R., Tizard, R., Crameri, R., Davies, J. E., Lauwereys, M., et al. (1987). Characterization of the herbicide-resistance gene bar from Streptomyces hygroscopicus. EMBO J. 6 (9), 2519–2523. doi: 10.1002/j.1460-2075.1987.tb02538.x
Tuskan, G. A., DiFazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., et al. (2006). The genome of black cottonwood, populus trichocarpa (Torr. & Gray). Science 313 (5793), 1596–1604. doi: 10.1126/science.1128691
Verde, I., Abbott, A. G., Scalabrin, S., Jung, S., Shu, S., Marroni, F., et al. (2013). The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 45 (5), 487–494. doi: 10.1038/ng.2586
Waite, J. M., Dardick, C. (2018). TILLER ANGLE CONTROL 1 modulates plant architecture in response to photosynthetic signals. J. Exp. Bot. 69 (20), 4935–4944. doi: 10.1093/jxb/ery253
Wang, K., Wang, Z., Li, F., Ye, W., Wang, J., Song, G., et al. (2012). The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 44 (10), 1098–1103. doi: 10.1038/ng.2371
Whipple, C. J., Kebrom, T. H., Weber, A. L., Yang, F., Hall, D., Meeley, R., et al. (2011). grassy tillers1 promotes apical dominance in maize and responds to shade signals in the grasses. Proc. Natl. Acad. Sci. U.S.A. 108 (33), E506–E512. doi: 10.1073/pnas.1102819108
Xu, D., Qi, X., Li, J., Han, X., Wang, J., Jiang, Y., et al. (2017). PzTAC and PzLAZY from a narrow-crown poplar contribute to regulation of branch angles. Plant Physiol Biochem 118, 571–578. doi: 10.1016/j.plaphy.2017.07.011
Yang, X. C., Hwa, C. M. (2008). Genetic modification of plant architecture and variety improvement in rice. Heredity 101 (5), 396–404. doi: 10.1038/hdy.2008.90
Yang, G.-z., Luo, X.-j., Nie, Y.-c., Zhang, X.-L. (2014). Effects of plant density on yield and canopy micro environment in hybrid cotton. J. Integr. Agric. 13 (10), 2154–2163. doi: 10.1016/S2095-3119(13)60727-3
Yao, H., Zhang, Y., Yi, X., Zuo, W., Lei, Z., Sui, L., et al. (2017). Characters in light-response curves of canopy photosynthetic use efficiency of light and N in responses to plant density in field-grown cotton. Field Crops Res. 203, 192–200. doi: 10.1016/j.fcr.2016.12.018
Yu, B., Lin, Z., Li, H., Li, X., Li, J., Wang, Y., et al. (2007). TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J. 52 (5), 891–898. doi: 10.1111/j.1365-313X.2007.03284.x
Yuan, D., Tang, Z., Wang, M., Gao, W., Tu, L., Jin, X., et al. (2015). The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci. Rep. 5 (1), 17662. doi: 10.1038/srep17662
Zhang, L., Chia, J.-M., Kumari, S., Stein, J. C., Liu, Z., Narechania, A., et al. (2009). A genome-wide characterization of microRNA genes in maize. PloS Genet. 5 (11), e1000716. doi: 10.1371/journal.pgen.1000716
Keywords: branch angle, CRISPR, plant architecture, petiole angle, tac1, upland cotton
Citation: Kangben F, Kumar S, Li Z, Sreedasyam A, Dardick C, Jones D and Saski CA (2024) Phylogenetic and functional analysis of tiller angle control homeologs in allotetraploid cotton. Front. Plant Sci. 14:1320638. doi: 10.3389/fpls.2023.1320638
Received: 12 October 2023; Accepted: 20 December 2023;
Published: 31 January 2024.
Edited by:
Reyazul Rouf Mir, Sher-e-Kashmir University of Agricultural Sciences and Technology, IndiaReviewed by:
Upendra Kumar, M. J. P. Rohilkhand University, IndiaCopyright © 2024 Kangben, Kumar, Li, Sreedasyam, Dardick, Jones and Saski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher A. Saski, c2Fza2lAY2xlbXNvbi5lZHU=
†These authors contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.