 Chao Ding1
Chao Ding1 Zhao Zhang2*
Zhao Zhang2*- 1Shanxi Center for Testing of Functional Agro-Products, Shanxi Agricultural University, Taiyuan, China
- 2Beijing Key Laboratory of Development and Quality Control of Ornamental Crops, Department of Ornamental Horticulture, China Agricultural University, Beijing, China
Introduction
After decades of development, today’s Omics technologies have advanced tremendously. It is important to note, however, that the vast majority of methodologies in bioinformatics were originally developed for the study of human genetics. Unlike diploid humans, many crop genomes are polyploid, such as tetraploid potato (Sun et al., 2022), hexaploid wheat (Ling et al., 2018; Walkowiak et al., 2020), tetraploid sugarcane (Zhang et al., 2018), octoploid strawberry (Edger et al., 2019), and so on. Existing Omics methods and tools, including genome assembly strategies, algorithm, statistical methods, and efficient data processing and analysis, are far from adequate for the complex genomes of polyploid crops.
Challenges of genome assembly and annotation
Genome assembly and annotation of polyploid plants is a challenging task due to the complexity of their genomes. Although some new technological tools, such as the highly accurate long-read sequencing technologies, e.g. PacBio HiFi (Hon et al., 2020), have made it possible to accurately obtain genomic fragments that distinguish haplotypes. However, the presence of highly similar sequences tends to make assembly of repetitive regions difficult. Therefore, obtaining haplotype-resolved polyploid genomes of high quality is still a difficult task. It is worth noting that compared with allopolyploid plants, this issue is more prominent for autopolyploid plants. People have attempted to assemble the chromosome-level genome of polyploid plants using High-throughput chromosome conformation capture (Hi-C) technology (Dekker, 2006) and ALLHiC software (Zhang et al., 2019), but this approach fails in autopolyploid plants in many cases (Bao et al., 2022; Sun et al., 2022). Due to the lack of reliable bioinformatic methods, obtaining high-quality autopolyploid plant genomes can only be achieved through experimental methods. For example, the genome of autotetraploid potato was obtained by sequencing a total of 1034 individuals from a selfing population (F2 population; Bao et al., 2022), which obviously involves significant time and cost.
Furthermore, this challenge becomes even more pronounced for crops with highly heterozygous genomes. The presence of multiple alleles and variations within the genome adds an extra layer of complexity to the assembly and annotation processes. Resolving the allelic diversity and accurately assigning variations to specific loci can be particularly challenging in highly heterozygous crops. Advanced computational algorithms and innovative sequencing technologies are continually being developed to address these complexities and improve the accuracy of genome assembly and annotation in highly heterozygous crops.
Complex genome structure
Polyploid plants typically possess large genomes, e.g. approximately 16 Gb for wheat (Walkowiak et al., 2020) and 7.68 Gb for tetraploid rhubarb (Rheum officinale, a perennial medicinal herb) (Zhang et al., 2023a). For the crops that rely mainly on asexual propagation (many of which are horticultural), e.g. the tetraploid modern rose (Rosa hybrida), their genomes maintain a high level of heterozygosity due to a lack of sexual reproduction (Zhang et al., 2023b). The presence of multiple homologous copies (alleles) in the genome increases complexity to the analysis and interpretation of genomic information. Distinguishing different alleles and resolving dosage effect of gene can be challenging. Different studies have found that the expression between alleles in polyploid plants can vary significantly or may differ only slightly (Zhang et al., 2023b).
Complexity of epigenetic regulation and post-transcriptional regulation
In addition to genomic and transcriptomic complexity, polyploid plants have complex epigenetic mechanisms. Epigenetic regulation involves dynamic modifications of nucleic acids and histones, encompassing both genomic and transcriptional levels (Shen et al., 2017). Additionally, non-coding RNAs, such as miRNA, siRNA, piRNA and long non-coding RNAs, are involved in the epigenetic regulation of gene expression by targeting mRNA stability and translation (Shi et al., 2022). Epigenetic modifications may have important effects on gene expression in plants, but the complexity of the epigenetic regulatory network makes it difficult to accurately understand and interpret (Ma et al., 2015). The complexity of epigenetic regulation lies in its multilevel nature, involving interactions among DNA methylation (and methylome), histone modifications, chromatin remodeling, noncoding RNAs, and various regulatory proteins. Moreover, the three-dimensional organization of chromatin within the nucleus, refers as 3D genome, have significant implications for gene regulation and other cellular processes. Understanding the 3D genome and its dynamics provides insights into how genetic information is regulated spatially and temporally, impacting processes like plant growth, development, stress responses and so on. The interactions between different subgenomes in polyploid plants undoubtedly add another layer of complexity of their epigenetic regulation.
Reliable phenomics is necessary for effective interpretation of complex traits
In most cases, stress tolerance in plants is a complex trait. This applies not only to abiotic stress tolerance but also to resistance against biotic stress. Plant complex traits are typically determined by multiple genes and their interactions. These genes may be located on different chromosomes, making direct linkage analysis challenging, and polyploidy further exacerbates this complexity. Moreover, the effects of these genes are often subtle, resulting in minimal phenotypic differences from changes in a single gene. Therefore, reliable and high-throughput phenomics is required. Accurate assessment of phenotypes is essential for effective genetic analysis.
Data analysis require appropriate statistical models and efficient computation
The extensive data generated from polyploid plant genomics research requires complex analysis and interpretation. Challenges include standardized data processing, establishing effective analysis pipelines, and developing suitable statistical models. Additionally, integrating genomic data with phenotype information poses a further challenge in gaining deeper insights into the characteristics of polyploid plants. These tasks often rely on powerful parallel computational capabilities, and thus, developing efficient computations based on Graphics Processing Units (GPUs) can significantly enhance performance.
Prospects
In the past decade, many software tools applicable to polyploids have been developed, such as fitTetra (Voorrips et al., 2011; Zych et al., 2019) and polymapR (Bourke et al., 2018). However, genomic research on polyploid plants still faces challenges regarding complex genome structure, genome assembly and annotation, the complexity of epigenetic regulation, as well as the complexity of data analysis and interpretation (Figure 1). Overcoming these challenges requires continuous technological development and innovation, as well as enhanced collaboration and communication across different disciplines, to further advance genomic research on polyploid plants.
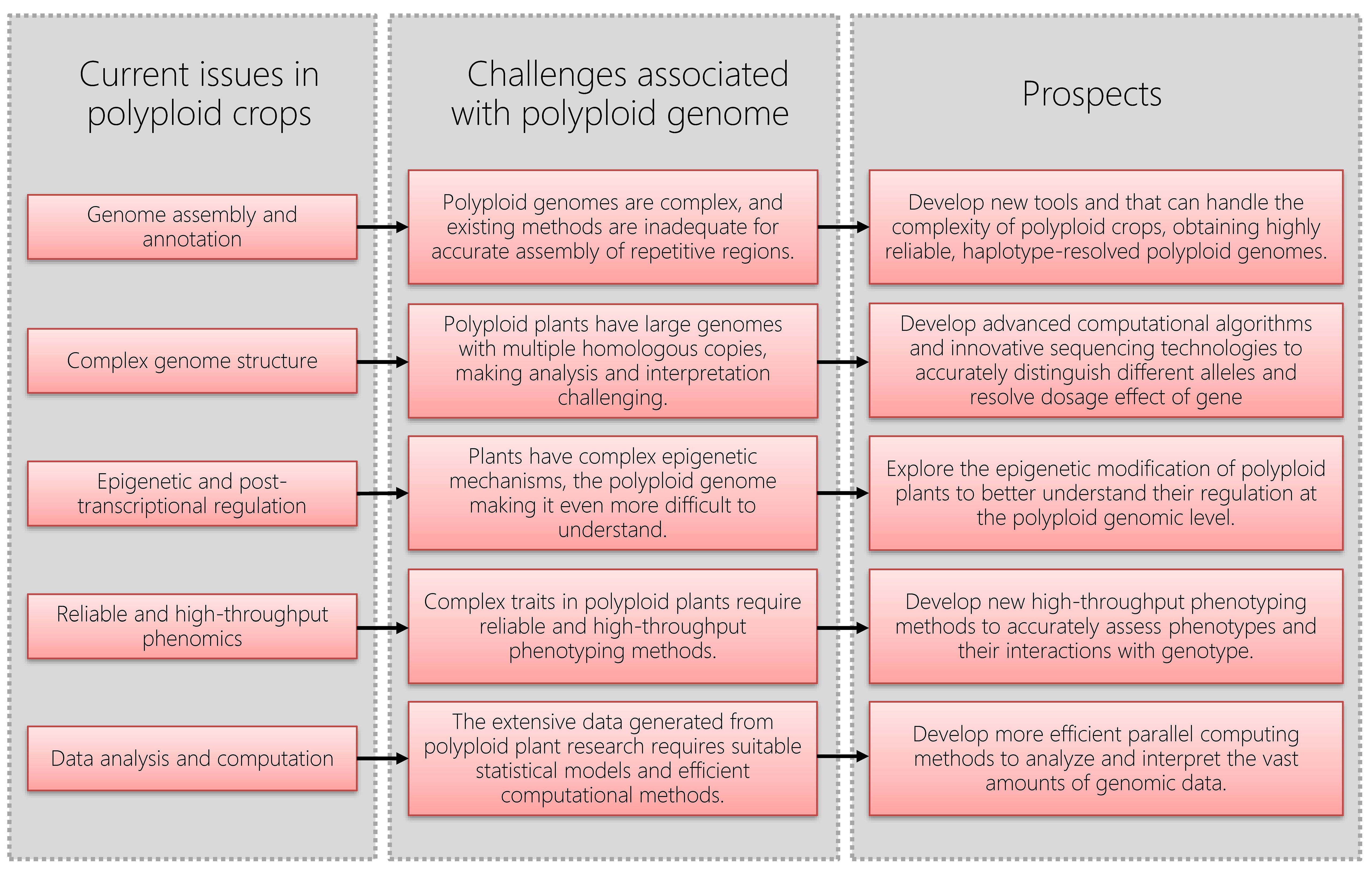
Figure 1 Challenges and prospects in polyploid crop genome research.
Author contributions
CD: Writing – original draft, Writing – review & editing. ZZ: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Special Project for Science and Technology Cooperation and Exchange of Shanxi province (Grant No.202204041101017) to Chao Ding and Zhao Zhang.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bao, Z., Li, C., Li, G., Wang, P., Peng, Z., Cheng, L., et al. (2022). Genome architecture and tetrasomic inheritance of autotetraploid potato. Mol. Plant 15 (7), 1211–1226. doi: 10.1016/j.molp.2022.06.009
Bourke, P. M., van Geest, G., Voorrips, R. E., Jansen, J., Kranenburg, T., Shahin, A., et al. (2018). polymapR-linkage analysis and genetic map construction from F1 populations of outcrossing polyploids. Bioinformatics 34 (20), 3496–3502. doi: 10.1093/bioinformatics/bty371
Dekker, J. (2006). The three ‘C’ s of chromosome conformation capture: controls, controls, controls. Nat. Methods 3 (1), 17–21. doi: 10.1038/nmeth823
Edger, P. P., Poorten, T. J., VanBuren, R., Hardigan, M. A., Colle, M., McKain, M. R., et al. (2019). Origin and evolution of the octoploid strawberry genome. Nat. Genet. 51 (3), 541–547. doi: 10.1038/s41588-019-0356-4
Hon, T., Mars, K., Young, G., Tsai, Y.-C., Karalius, J. W., Landolin, J. M., et al. (2020). Highly accurate long-read HiFi sequencing data for five complex genomes. Sci. Data 7 (1), 399. doi: 10.1038/s41597-020-00743-4
Ling, H. Q., Ma, B., Shi, X., Liu, H., Dong, L., Sun, H., et al. (2018). Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 557 (7705), 424–428. doi: 10.1038/s41586-018-0108-0
Ma, N., Chen, W., Fan, T., Tian, Y., Zhang, S., Zeng, D., et al. (2015). Low temperature-induced DNA hypermethylation attenuates expression of RhAG, an AGAMOUS homolog, and increases petal number in rose (Rosa hybrida). BMC Plant Biol. 15, 237. doi: 10.1186/s12870-015-0623-1
Shen, Y., Sun, S., Hua, S., Shen, E., Ye, C. Y., Cai, D., et al. (2017). Analysis of transcriptional and epigenetic changes in hybrid vigor of allopolyploid Brassica napus uncovers key roles for small RNAs. Plant J. 91 (5), 874–893. doi: 10.1111/tpj.13605
Shi, S., Zhang, S., Wu, J., Liu, X., Zhang, Z. (2022). Identification of long non-coding RNAs involved in floral scent of Rosa hybrida. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.996474
Sun, H., Jiao, W. B., Krause, K., Campoy, J. A., Goel, M., Folz-Donahue, K., et al. (2022). Chromosome-scale and haplotype-resolved genome assembly of a tetraploid potato cultivar. Nat. Genet. 54 (3), 342–348. doi: 10.1038/s41588-022-01015-0
Voorrips, R. E., Gort, G., Vosman, B. (2011). Genotype calling in tetraploid species from bi-allelic marker data using mixture models. BMC Bioinf. 12, 172. doi: 10.1186/1471-2105-12-172
Walkowiak, S., Gao, L., Monat, C., Haberer, G., Kassa, M. T., Brinton, J., et al. (2020). Multiple wheat genomes reveal global variation in modern breeding. Nature 588 (7837), 277–283. doi: 10.1038/s41586-020-2961-x
Zhang, H., He, Q., Xing, L., Wang, R., Wang, Y., Liu, Y., et al. (2023a). The haplotype-resolved genome assembly of autotetraploid rhubarb Rheum officinale provides insights into the genome evolution and massive accumulation of anthraquinones. Plant Commun. 100677. doi: 10.1016/j.xplc.2023.100677
Zhang, Z., Liu, Y., Yang, T., Wu, S., Sun, H., Wu, J., et al. (2023b). Haplotype-resolve genome assembly and resequencing provide insights into the origin and domestication of modern rose. bioRxiv 2006. doi: 10.1101/2023.06.02.543351
Zhang, J., Zhang, X., Tang, H., Zhang, Q., Hua, X., Ma, X., et al. (2018). Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 50 (11), 1565–1573. doi: 10.1038/s41588-018-0237-2
Zhang, X., Zhang, S., Zhao, Q., Ming, R., Tang, H. (2019). Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5 (8), 833–845. doi: 10.1038/s41477-019-0487-8
Keywords: abiotic stress, disease resistance, epigenetic regulation, post-transcriptional regulation, phenomics
Citation: Ding C and Zhang Z (2023) Effective omics tools are still lacking for improvement of stress tolerance in polyploid crops. Front. Plant Sci. 14:1295528. doi: 10.3389/fpls.2023.1295528
Received: 16 September 2023; Accepted: 18 October 2023;
Published: 27 October 2023.
Edited by:
Yi Han, Anhui Agricultural University, ChinaCopyright © 2023 Ding and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhao Zhang, emhhbmd6aGFvQGNhdS5lZHUuY24=