
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 12 January 2024
Sec. Functional and Applied Plant Genomics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1295186
This article is part of the Research Topic Economic Plant Genome and Database Construction and Research View all 10 articles
 Lingyun Wan1,2,3†
Lingyun Wan1,2,3† Qiulan Huang4†
Qiulan Huang4† Cui Li2,3Haixia Yu1,2,3Guiyu Tan1,2,3
Cui Li2,3Haixia Yu1,2,3Guiyu Tan1,2,3 Shugen Wei1,2,3
Shugen Wei1,2,3 Ahmed H. El-Sappah4,5
Ahmed H. El-Sappah4,5 Suren Sooranna3,6Kun Zhang1,2,3Limei Pan1,2,3Zhanjiang Zhang1,2,3
Suren Sooranna3,6Kun Zhang1,2,3Limei Pan1,2,3Zhanjiang Zhang1,2,3 Ming Lei2,3*
Ming Lei2,3*Centella asiatica (L.) Urban is a well-known medicinal plant which has multiple pharmacological properties. Notably, the leaves of C. asiatica contain large amounts of triterpenoid saponins. However, there have only been a few studies systematically elucidating the metabolic dynamics and transcriptional differences regarding triterpenoid saponin biosynthesis during the leaf development stages of C. asiatica. Here, we performed a comprehensive analysis of the metabolome and transcriptome to reveal the dynamic patterns of triterpenoid saponin accumulation and identified the key candidate genes associated with their biosynthesis in C. asiatica leaves. In this study, we found that the key precursors in the synthesis of terpenoids, including DMAPP, IPP and β-amyrin, as well as 22 triterpenes and eight triterpenoid saponins were considered as differentially accumulated metabolites. The concentrations of DMAPP, IPP and β-amyrin showed significant increases during the entire stage of leaf development. The levels of 12 triterpenes decreased only during the later stages of leaf development, but five triterpenoid saponins rapidly accumulated at the early stages, and later decreased to a constant level. Furthermore, 48 genes involved in the MVA, MEP and 2, 3-oxidosqualene biosynthetic pathways were selected following gene annotation. Then, 17 CYP450s and 26 UGTs, which are respectively responsible for backbone modifications, were used for phylogenetic-tree construction and time-specific expression analysis. From these data, by integrating metabolomics and transcriptomics analyses, we identified CaHDR1 and CaIDI2 as the candidate genes associated with DMAPP and IPP synthesis, respectively, and CaβAS1 as the one regulating β-amyrin synthesis. Two genes from the CYP716 family were confirmed as CaCYP716A83 and CaCYP716C11. We also selected two UGT73 families as candidate genes, associated with glycosylation of the terpenoid backbone at C-3 in C. asiatica. These findings will pave the way for further research on the molecular mechanisms associated with triterpenoid saponin biosynthesis in C. asiatica.
Centella asiatica (L.) Urban, a stoloniferous perennial herb of the Umbelliferae family found in tropical and sub-tropical areas (Nguyen et al., 2019), is a well-known medicinal plant used in China, India and Southeast Asia (Bhavna and Jyoti, 2011; Weng et al., 2012). It has been registered in different Pharmacopoeia of the world such as China, India and Europe (Brinkhaus et al., 2000; Kalita et al., 2015). Experimental and clinical investigations show that C. asiatica is useful in the treatment of varicose veins, eczema, wound healing disturbances, intellectual disability, ulcer, lupus and striae gravidarum (Liu et al., 2008; Prakash et al., 2017). This is attributed to an enormous amount of triterpenoid saponins, mainly asiaticoside, madecassoside, centelloside and sceffoleoside, which are collectively known as centelloids in C. asiatica (Matsuda et al., 2001; James and Dubery, 2009). Therefore, the triterpenoid saponins of C. asiatica are vital resources for their important and unique medicinal values. Identification of the relevant biosynthetic pathways will thus be helpful for our further understanding and utilization of these triterpenoid saponins. However, most of the previous studies on these compounds have only focused on their properties, bioactivities and pharmacology, and only a few genes involved in the triterpenoid saponin biosynthesis in C. asiatica have been identified, such as HMG-CoA reductase (Kalita et al., 2018), farnesyl diphosphate synthase (Kim et al., 2005a), squalene synthase (Kim et al., 2005b), oxidosqualene cyclase (Kim et al., 2005c), CYP450s (Fukushima et al., 2011; Kim et al., 2018) and UGTs (Kim et al., 2017; Han et al., 2022) and so on. Therefore, the biosynthetic pathways involved have yet to be fully characterized and need to be further investigated at the molecular level.
It is reported that there are more than 300 types of triterpenoid carbon skeletons (Xu et al., 2004; Thimmappa et al., 2014), whose chemical structures are formed from triterpene aglycones (sapogenins) and one or more sugar groups connected by glycosidic bonds (Phillips et al., 2006). Further evidences have supported the notion that there are similar routes of the triterpenoid saponin biosynthesis in different plants (Augustin et al., 2011; Cárdenas et al., 2019), and the core of triterpenoid saponin biosynthetic pathway is well-understood (Xu et al., 2022).
The whole triterpenoid saponin pathway involves three major stages: an initial stage, followed by terpenoid backbone construction and the modification stage (Sawai and Saito, 2011; Zhao et al., 2022). In the first stage, isopentenyl diphosphate (IPP) and its isomer dimethylallyl diphosphate (DMAPP), are formed from two independent pathways: IPP biosynthesis by initially condensing three units of acetyl-CoA in the cytosolic mevalonate (MVA) pathway (Huang et al., 2014), and DMAPP biosynthesis starting from the condensation of pyruvate and phosphoglyceraldehyde in the plastidial methylerythritol phosphate (MEP) pathway (Luo et al., 2016). IPP can be partially isomerized into DMAPP by IPP isomerase (IDI) (Wen et al., 2017). When the terpenoid backbone is constructed, farnesyl diphosphate synthase (FPS) condenses and catalyzes IPP and DMAPP into farnesyl pyrophosphate (FPP). Subsequently, two FPP molecules form squalene by the action of squalene synthase (SS). Under the action of squalene epoxidase (SE), epoxidation of squalene at the second and third carbon positions yields 2,3-oxidosqualene, which is a direct precursor to the construction of the triterpenoid backbones (Haralampidis et al., 2002). Next, oxidosqualene cyclases (OSCs) catalyze the cyclitization of 2,3-oxidosqualene into various triterpenoid backbones, which is thus a critical step of the triterpenoid biosynthesis pathway (Aharoni et al., 2006). During the modification stage, the triterpenoid backbones are chemically modified by cytochrome P450 monooxygenases (CYP450s) through a series of hydroxylation/oxidation reactions and then these are changed by UDP-glycosyltransferases (UGTs) through glycosylation reactions, all of which finally result in the formation of various triterpenoid saponins (Seki et al., 2015; Yang et al., 2018).
Transcriptomics is an effective technical means of mining the key regulatory genes associated with the active ingredients of biosynthetic pathways (Kuwahara et al., 2019). In an earlier study, an analysis of transcriptome sequences of leaves of C. asiatica by Illumina (Solexa sequencing technology) was conducted, and this contributed to the mining of the targeted genes related to triterpenoid saponin biosynthesis (Sangwan et al., 2013). The development of long-read sequencing technologies and assembly pipelines has allowed the completion of the whole genome assembly of C. asiatica at the chromosome-scale (Pootakham et al., 2021). These complete, accurate and contiguous representative genome sequences can provide valuable references to further understand the triterpenoid saponin biosynthesis in C. asiatica. Additionally, the metabolomics database can then be used to provide information regarding the accumulation of various secondary metabolites during the biosynthetic pathways of bioactive ingredient formation (Saito and Matsuda, 2010), and it can also provide reference information for gene mining.
The comprehensive analysis of transcriptomics and metabolomics has been widely applied to delineate the relationships between the dynamic changes of secondary metabolites and the differential expression of corresponding genes during the bioactive ingredients biosynthetic pathways in numerous medicinal plants, such as Panax notoginseng (Wei et al., 2018), Sapindus mukorossi (Xu et al., 2022), Platycodon grandiflorus (Chang et al., 2022), Dendrobium huoshanense (Yuan et al., 2022) and Carthamus tinctorius (Wang et al., 2021). The medicinal properties of C. asiatica are largely attributed to the centelloids that present predominantly in the leaves of C. asiatica (Sangwan et al., 2013). However, there have been few studies systematically elucidating the metabolic dynamics and transcriptional differences regarding the triterpenoid saponin biosynthesis during the leaf developmental stages of C. asiatica based on triterpenoid saponin metabolism and transcriptomics data.
In this study, we identified the key candidate genes and metabolites by analyzing the expression patterns of differentially expressed genes (DEGs) by transcriptomics and the differentially accumulated metabolites (DAMs) via metabolomics in C. asiatica leaves at different growth stages. This study will pave the way for future research on the molecular mechanisms associated with triterpenoid saponin biosynthesis in C. asiatica.
The plants of C. asiatica were artificially cultivated in the greenhouse at the Guangxi Botanical Garden of Medicinal Plants. The plants were cultivated using an illumination intensity of 300 µmol m-2 s-1, a relative humidity of 60 ± 2% at 26 ± 2°C with a light/dark cycle of 16/8 h. The leaves of C. asiatica were collected from four growth stages and representative examples are shown in Supplementary Figure S1. The samples were at the 10, 20, 30 and 40 day leaf-ages with three replicates at each stage and were designated as JXC1-1, JXC1-2, JXC1-3, JXC2-1, JXC2-2, JXC2-3, JXC3-1, JXC3-2, JXC1-3, JXC4-1, JXC4-2 and JXC4-3, respectively. The leaf age was determined by attaching a label to each leaf indicating the date when the leaf first emerged. The number of leaves collected at each stage was subjected to their size, and all the samples were immediately frozen by using liquid nitrogen and then stored at −80°C for subsequent extraction of RNA and metabolites.
A wide targeted metabolomics analysis was conducted on the samples from each leaf growth stage to explore the dynamic patterns of metabolites during the developmental stages of C. asiatica leaves. This analysis was carried out by MetWare Biotechnology Co., Ltd., Wuhan, China.
Initially, the leaf samples were freeze-dried by using a vacuum freeze-dryer (Scientz-100F, Ningbo, China), and then zirconia beads were added and the mixture were grinded in a mixer mill (MM 400, Retsch, Haan, Deutschland) for 1.5 min, followed by 70% methanol extraction. The suspension was centrifuged (at 2,000 rpm for 3 min) (Anpel, Shanghai, China), and then filtered through a 0.22 μm econofilter. The filtrates were analyzed using a UPLC-ESI-MS/MS system (UPLC, SHIMADZU Nexera X2, MS, Applied Biosystems 4500 Q TRAP). An Agilent SB-C18 column (1.8 µm, 2.1 mm×100 mm) (California, USA) was used for separation at 40°C. Mobile phases of solvents A and B were used and these were composed of pure water with 0.1% formic acid and acetonitrile with 0.1% formic acid, respectively. The gradient conditions were as follows: Phase B started with 5%, followed by 95% at 0 ~ 10 min, and maintained at 95% for another 1.0 min, and finally 5% at 11.1 ~ 14 min,. For each sample, the injection volume was 4 μL at a flow rate of 0.35 mL/min. The effluent was collected and assessed after connection to electrospray ionization (ESI)-triple quadrupole-linear ion trap (QTRAP)-MS.
The operating parameters for ESI were as follows: the source temperature was 550°C and the ion spray voltage (IS) was set to 5,500 V (positive ion mode)/-4,500 V (negative ion mode). The ion source gases I and II as well as the curtain gas were set at 50, 60 and 25 psi, respectively, and the collision-activated dissociation was set to high. Ten and 100 μmol/L polypropylene glycol solutions were used to tune the instrument and to perform mass calibration in the triple quadrupole (QQQ) as well as the linear ion trap modes, respectively. Scans of the QQQ were acquired in the form of multiple reaction monitoring (MRM) experiments with nitrogen as the collision gas and this was set to medium. The declustering potential (DP) and collision energy (CE) for each MRM transition were performed after further optimizations. A specific set of MRM transitions was used to monitor each period based on the metabolites found to be eluted within that period.
To assess the metabolite diversity between and within groups, all the metabolic data were subjected to multivariate statistical analysis, including unsupervised principal component (PCA) and orthogonal partial least squares discriminant analyses (OPLS-DA). After evaluation of these results, the metabolites with |Log2 (FC)| ≥ 1, p-value < 0.05, and variable importance in the project (VIP) ≥ 1 were identified as DAMs.
RNA-seq was carried out by MetWare Biotechnology Co., Ltd. (Wuhan, China). Total RNA was extracted from the four stages of C. asiatica leaves and its quality was assessed by running on 1% agarose gels and using the NanoPhotometer® spectrophotometer (IMPLEN, CA, USA). RNA concentration was measured using the Qubit® RNA Assay Kit in Qubit®2.0 Flurometer (Life Technologies, CA, USA). RNA integrity was determined using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). One μg of RNA from each sample was used for sample preparation. cDNA libraries were generated using the NEBNext®UltraTM RNA Library Prep Kit purchased from Illumina® (NEB, USA) by following the manufacturer’s instructions. Raw sequence reads with nucleotides with uncertain base information as well as those with low-quality sequence reads were filtered using FASTP v 0.19.3 to produce clean reads. These clean reads were then mapped to the whole genome of C. asiatica (unpublished data) to acquire the genes using HISAT v2.1.0.
The expression levels of the genes were calculated after normalization to the number of fragments per kilobase of transcript per million fragments (FPKM). Genes were further analyzed using functional annotations and pathway analyses to seven public databases: KEGG, KOG, GO, Pfam, Tremble, NR and Swiss-Prot. The levels of DEGs were analyzed using the original count data from each sample which was obtained by their expression quantification with DESeq2 software. Those genes with Benjamini-Hochberg-adjusted p value < 0.05 and |log2 (FC)| > 1 were identified as DEGs.
Two methods were used for gene identification of the CYP450 and UGT families in the C. asiatica genome. Firstly, the protein sequences from the chromosome-scale genome of C. asiatica (unpublished data) were used to construct a local protein database. The known protein sequences of Arabidopsis thaliana for 242 CYP450s and 117 UGTs were collected from the Arabidopsis Information Resource (TAIR, https://www.arabidopsis.org/). These were used to search the local protein database of C. asiatica by using BLASTP, which is a basic local environment alignment search tool for proteins. The cut-off E-value used was 10-10. Subsequently, the Hidden-Markov Model (HMM) files for P450.hmm (PF00067) and UGT.hmm (PF00201) which were downloaded from the Pfam database (https://pfam.xfam.org/), were used to search the local protein database of C. asiatica to find all predicted CYP450 and UGT family members via the HMMER 3.0 (https://www.hmmer.org/) program with a cut-off E-value of 10-10. After integrating the results of the above two methods, the remaining sequences were assumed as the candidate CYP450 and UGT family members of C. asiatica, after removing the redundant sequences. The candidate family members were further verified via PfamScan (e-value = 0.001, https://www.ebi.ac.uk/Tools/pfa/pfamscan/).
Phylogenetic analysis was performed to align the full-length protein sequences of CYP450 and UGT from Arabidopsis and CYP450 and UGT from C. asiatica by using the MEGA software (version 7.0) with default parameters, respectively. A phylogenetic tree was constructed by using the MEGA software with the 1,000-replicate neighbor-joining (NJ) method and the results were illustrated by using the interactive tree of life online software (https://itol.embl.de/help.cgi).
To better outline relationships of the data between the genes and related metabolites involved in triterpenoid saponin biosynthesis, weighted gene coexpression network analysis (WGCNA) and Pearson correlation analysis were conducted with the default parameters in the free online data analysis, Metware Cloud Platform (https://cloud.metware.cn). The soft threshold, the network heatmap plot of genes, the module heatmap and the trait-related modules were constructed by using WGCNA. The correlation results obtained from Pearson correlation analysis were mapped in a correlation cluster heatmap.
Eighteen DEGs associated with the triterpenoid saponin biosynthesis were selected for real-time quantitative PCR (RT-qPCR) analysis relative to the reference gene, CaGAPDH. cDNA was synthesized using the SuperScript® III Reverse Transcriptase kit by following the manufacturer’s protocols. RT-qPCR was performed on a Bio-Rad CFX96 RT-qPCR platform (Bio-Rad Laboratories, Hercules, CA, USA) using SYBR® Premix Ex Taq™ kit (Takara, Dalian, China) according to the manufacturer’s protocols. The relative expression levels of DEGs were calculated using the 2-ΔΔCT method and then normalized against CaGAPDH. The primers used are listed in Supplementary Table S1.
In order to evaluate the metabolite variations in the leaves at different growth stages, a wide targeted metabolomics analysis was performed using samples collected at four time-points during leaf development. A clustering heatmap analysis was performed from the whole metabolite database, which showed that the three replicates used in this study had a high similarity (Supplementary Figure S2A). PCA analysis also showed a distinct separation in leaf samples from the four different stages, indicating that these metabolites presented a dynamic change pattern with the leaf growth (Supplementary Figure S2B). These results indicated that the metabolite detection methods used in this study were reliable.
A total of 802 metabolites were obtained, mainly containing terpenoids (68), flavonoids (84), alkaloids (44), amino acids and their derivatives (96), phenolic acids (135), nucleotides and their derivatives (49), lignans (18), coumarins (26), tannins (1), organic acids (68) and lipids (108) (Supplementary Table S2). To further understand the complexity of the different metabolites at each leaf growth stage, these metabolites were divided into four clusters by using a k-mean clustering method. Terpenoids were mainly found in clusters 3 and 2, with the highest accumulation of metabolites belonging to these clusters being from JXC1 and JXC2, respectively (Figure 1).
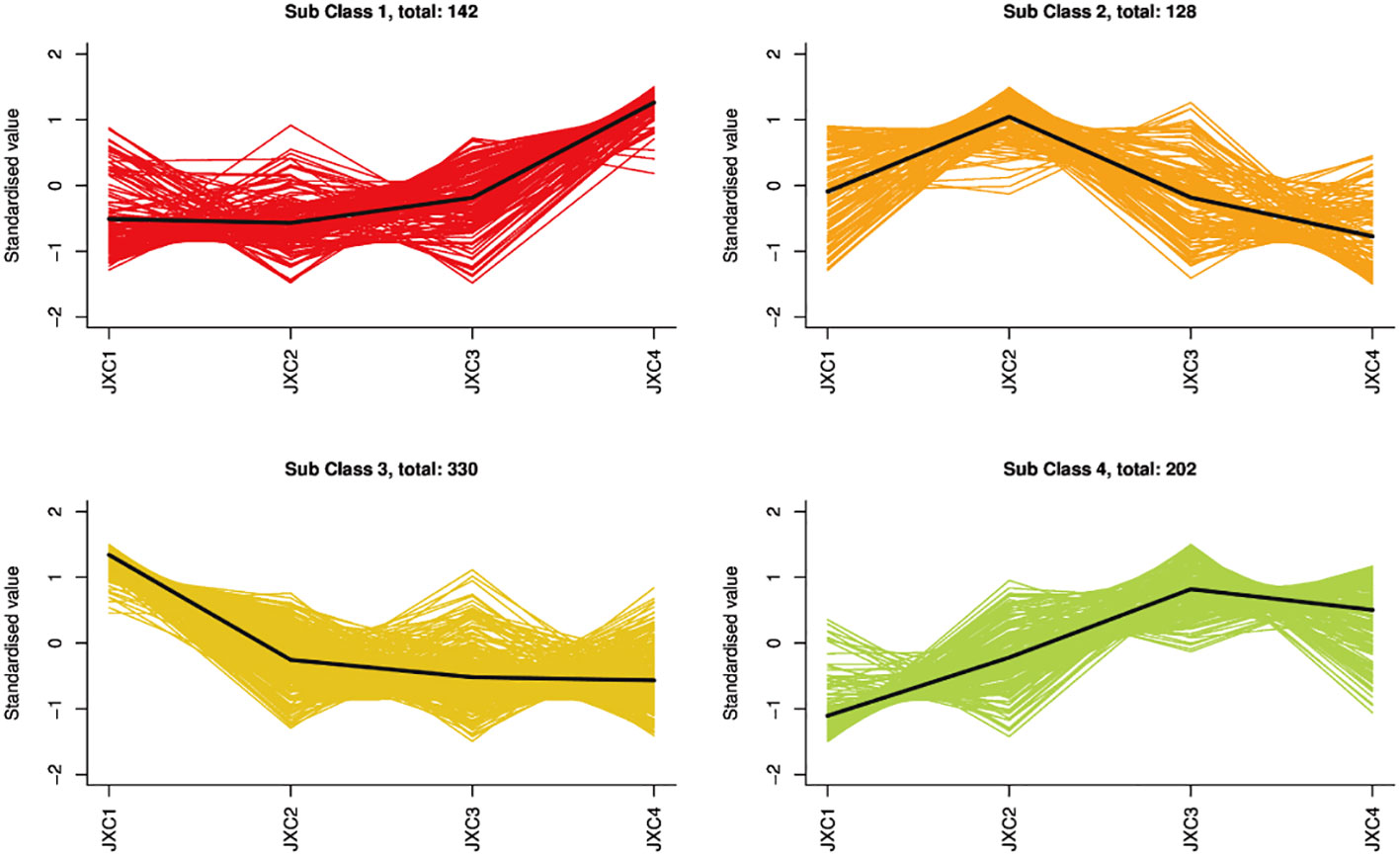
Figure 1 The k-means analysis of metabolites.
The 263 metabolites were mapped to the reference pathways according to KEGG annotations, and these were annotated to 96 pathways into three categories. A total of 91 pathways were associated with metabolism, and three and two pathways were involved with environmental information and genetic information processing, respectively. For the “metabolism” term, the top priority was “global and overview maps” (228 metabolites), followed by “amino acid metabolism” (89 metabolites), “carbohydrate metabolism” (69 metabolites) and “biosynthesis of other secondary metabolites” (67 metabolites) (Supplementary Figure S3A).
The key precursors in the biosynthesis of terpenoids, including DMAPP (Wmjp001948), IPP (Wmjn000963) and β-amyrin (Wmjn007463), were detected and they showed differential accumulation in the C. asiatica leaves at the four different growth stages (Supplementary Table S3). Gradual increased trends were observed in their contents from JXC1 to JXC4 with a peak at JXC4 (Supplementary Table S3).
In the downstream metabolites of triterpenoid saponin biosynthesis, a total of 32 triterpenes and 14 triterpenoid saponins were tentatively detected (Supplementary Table S3). Hierarchical cluster analysis (HCA) based on the relative levels of triterpenes (Supplementary Figure S4A) and triterpenoid saponins (Supplementary Figure S4B) in the C. asiatica leaves at the four growth stages showed that the levels of most of these compounds were markedly changed during leaf development, and this further confirmed the PCA results. Additionally, a total of 22 triterpenes were found to be differentially accumulated (Supplementary Table S4), which were divided into 7 groups based on their accumulation levels. The 12 triterpenes in cluster A had their levels significantly decreased from stage JXC3, and three of these compounds in cluster B showed high accumulations at stage JXC3 (Supplementary Figure S5A). A total of eight triterpenoid saponins showed differential accumulation (Supplementary Table S4), which were divided into four groups. In cluster A, the five triterpenoid saponins showed high accumulation at stage JXC1 and then tapered off from stages JXC2 to JXC4. For cluster B, the accumulation of one triterpenoid saponin showed a downtrend trend, with the lowest level seen at stage JXC3, but then it increased by stage JXC4. Cluster C was similar to cluster B, where lower levels were seen at stages JXC2 and JXC3. The content of triterpenoid saponins in cluster D appeared to increase continuously during leaf growth (Supplementary Figure S5B).
In this study, in order to explore gene expression patterns during the four different stages of leaf development, and to identify genes participating in the biosynthetic pathways of triterpenoid saponins at the transcriptional level, 12 cDNA libraries, in batches of three, were prepared and analyzed. The 12 leaf samples generated 56.59 million high-quality clean reads which constituted 84.89 GB of cDNA sequence data. The Q20 and Q30 values ranged from 98.09 to 98.46% and 94.03 to 95.00%, respectively, and the GC content ranged from 43.50 to 44.92%. The reads in each library were aligned to the C. asiatica reference genome with an average match ratio of 82.71% (Table 1).
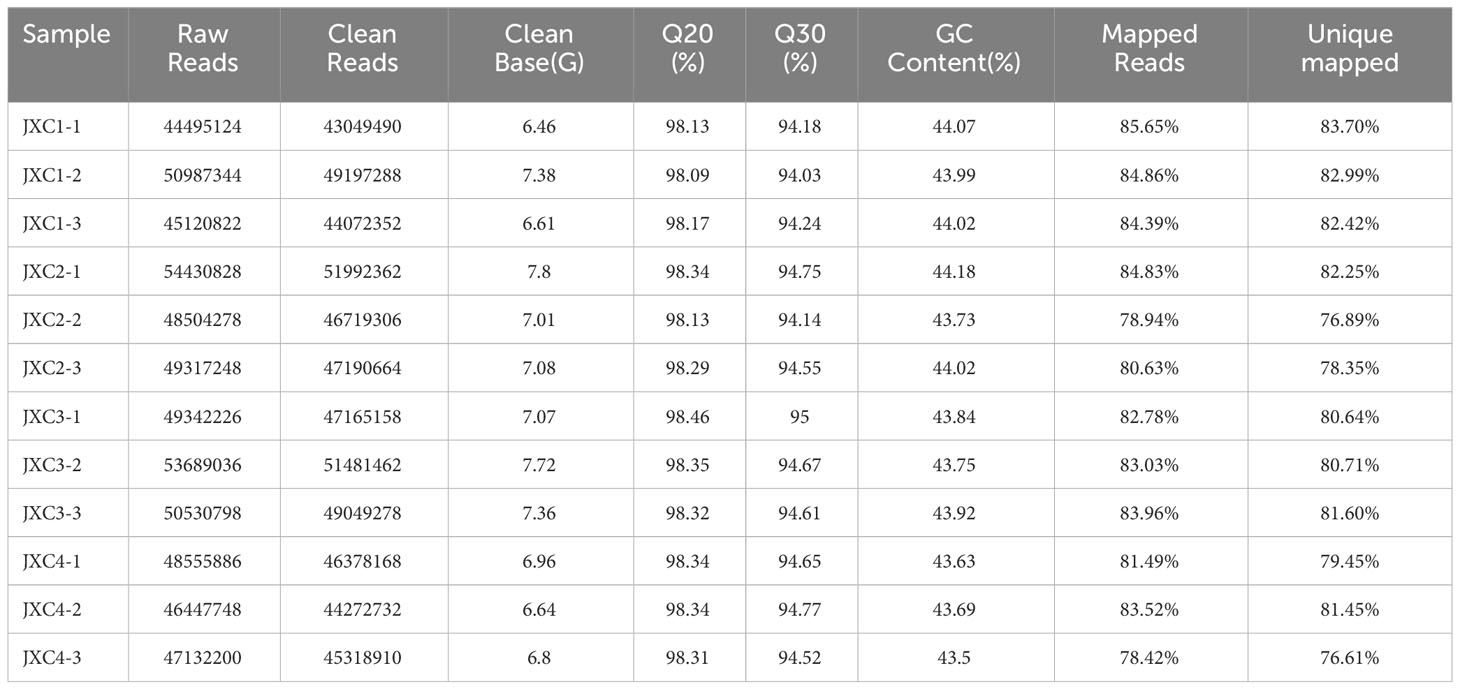
Table 1 Summary statistics of the RNA-seq results.
A total of 22,594 genes expressed were obtained in at least one sample with FPKM > 0 (Supplementary Table S5). The PCA results showed most of the samples at the same stages were highly correlated, indicating that the gene expression patterns were also highly reproducible (Supplementary Figure S6). Six comparative groups were constructed to demonstrate the differentiation at the transcriptional level at different growth stages of C. asiatica leaves: JXC1_vs_JXC2, JXC1_vs_JXC3, JXC1_vs_JXC4, JXC2_vs_JXC3, JXC2_vs_JXC4 and JXC3_vs_JXC4, and a total of 3889 DEGs were identified (Supplementary Table S6). Among these DEGs, the numbers of up- and down-regulated DEGs for each comparison group are shown in Table 2. There were more down-regulated DEGs than up-regulated ones in all the compared groups except for JXC3_vs_JXC4. Compared with JXC4, JXC3 contained a smaller number of DEGs, with 253 and 238 up- and down-regulated DEGs, respectively. The highest number of DEGs were identified in the JXC1_vs_JXC4 comparison, which showed 945 and 1,960 up- and down-regulated DEGs, respectively. In summary, there were significant differences in the number of DEGs presented in the six comparative groups.
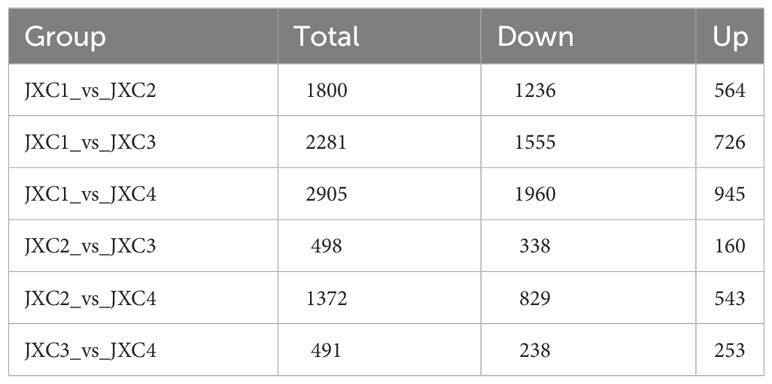
Table 2 The counts of differentially expressed genes.
The functions of DEGs were annotated by GO enrichment analysis, which were enriched within three main ontologies: biological process (BP), cellular component (CC) and molecular function (MF). Finally, the 3,389 DEGs of all samples were annotated to 3,623 GO terms (Supplementary Figure S7). Among them, the BP category contained 2,151 GO terms (59.37%), of which the major terms were the cellular (GO: 0009987), metabolic (GO: 0008152), single-organism (GO: 0044699) and biological processes (GO: 0050789)). In addition, some of these DEGs also responded to stimuli (GO: 0050896), biological regulation (GO: 0065007) and localization (GO: 0051179). The CC category contained 387 GO terms, including the membrane (GO: 0016020 and 0044425), cell (GO: 0005623 and 0044464) and organelle (GO: 0043226). The MF category contained 1,085 GO terms (29.95%), including mainly catalytic (GO: 0003824), binding (GO: 0005488), transporter (GO: 0005215) and nucleic acid binding transcription factor activities (GO: 0001071).
Moreover, the DEGs were also mapped to the KEGG pathways by using the KEGG database. The 3,389 DEGs in all samples were annotated to 126 KEGG pathways into five categories. There were three, four, 18, two and 99 pathways for cellular processes, environmental information processing, genetic information processing, organismal systems and metabolism, respectively (Supplementary Figure S3B). The main enriched pathways associated with secondary metabolism in each comparison group in our study were: biosynthesis of secondary metabolites (ko01110), metabolic pathways (ko01100), sesquiterpenoid and triterpenoid biosynthesis (ko00940), terpenoid backbone biosynthesis (ko00900), phenylpropanoid biosynthesis (ko00940) and flavonoid biosynthesis (ko00941). These enriched pathways provided valuable information for mining candidate genes associated with triterpenoid saponin biosynthesis.
To further explore the molecular mechanism of triterpene and triterpenoid saponin biosynthesis in the C. asiatica leaves, the expression levels of genes associated with their biosynthesis were analyzed. From the transcriptomics analysis, we found 48 genes were associated with the terpenoid backbone biosynthetic pathways, all of which could be classified into the MVA, MEP and 2, 3-oxidosqualene pathways (Figure 2).

Figure 2 The proposed pathways for triterpenoid saponin biosynthesis in C. asiatica. The expression levels of the gene encoding enzymes that catalyze each step of the triterpenoid saponin biosynthesis pathway are shown.
The KEGG pathway analysis showed that a total of 12 genes encoding six enzymes were associated with the MVA pathway: two acetyl-CoA C-acetyltransferase (AACTs), two 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) synthetases (HMGSs), four HMG-CoA reductases (HMGRs), one mevalonate kinase (MVK), two phosphomevalonate kinases (PMKs) and one metholvalic-5-diphosphate decarboxylase (MVD). The MEP pathway, starting with pyruvate and glyceraldehyde3-phosphate, consisted of 17 genes encoding eight enzymes: four 1-deoxy-D-xylulose-5-phosphate (DXP) synthetases (DXSs), two DXP reductoisomerases (DXRs), one 4-cytidine diphosphate-2-C-methyl-D-erythritol (CDP-ME) synthetase (MCT), 1 CDP-ME kinase (CMK), one 2-C-methyl-D-erythritol-2,4-cyclic phosphate synthetase (MDS), two (E)-4-hydroxy-3-methyl-2-butenyl-pyrophosphate (HMBPP) synthetases (HDSs), two HMBPP reductases (HDRs) and four isopentenyl pyrophosphate (IPP) isomerases (IDIs). The expression levels were highest in JXC1 for most of these genes, except for CaAACT2, CaMVK and CaPMK1 which were involved in the MVP pathway and for CaDXS4, CaDXR2, CaMDS, CaHDS2, CaHDR1 and 3 CaIDIs involved in the MEP pathway.
Additionally, 19 genes encoding six enzymes were involved in carbocyclic biosynthesis: seven geranyl pyrophosphate (GPP) synthetases (GPSs), three farnesyl pyrophosphate(FPP) synthetase (FPSs), one squalene synthase (SS), four squalene epoxidases (SEs), three β-amyrin synthases (βASs) and one dammarenediol-II synthase (DDS). However, these genes had variable expression levels at the different developmental stages.
Through our genome-wide analysis, we identified 231 CYP450s in the C. asiatica. By using the CYP450 reference database, they were classified into 9 clades, 41 families, with 48.05 and 51.95% being the A- and non-A-types of CYP450s, respectively (Supplementary Table S7). For further analysis of their functional roles and evolutionary relationships in C. asiatica, 231 CYP450s along with 242 CYP450s from A. thaliana were employed in generating phylogenetic trees for the A- (Supplementary Figure 8A) and non-A-types (Supplementary Figure 8B) CYP450s, respectively. All the A-types of CYP450s were represented by the CYP71 clans. These consisted of 111 genes belonging to 18 families, which were CYP71, 73, 75-79, 81-P84, 89, 93, 98, 701, 703, 706 and 712. The members of non-A-type CYP450s were divided into eight clans according to the phylogenetic tree, including five single-family clans (CYP51, 74, 710, 97 and 711) and three multifamily clans (CYP72, 85 and 86). CYP72, 85 and 86 were the largest three clans, containing 48, 37 and 27 CYP450s, respectively.
By using genome-wide analysis, we identified 140 UGTs in the C. asiatica. We aligned the C. asiatica UGT amino acid sequences with the 117 UGTs obtained from Arabidopsis and was able to construct a phylogenetic tree (Supplementary Table S8). We ultimately classified these UGTs into 15 groups and 20 families, among which three with amino acid sequences less than 40% identities to any representative sequences could not be classified. The UGT85 family of Group G was the largest family with 19 genes, and the next largest group was the UGT73 family of Group D with 18 genes (Supplementary Figure 9).
It has been reported that the members of the CYP85, CYP72 and CYP71 families are primary CYP450s which are associated with triterpenoid saponin biosynthesis in dicotyledonous plants. We identified five CYP716s of CYP85 family genes (one CYP716C gene, two CYP716E genes and two CYP716A genes), six CYP72 family genes (two CYP72A genes and four CYP714E genes) and six CYP71D genes by using the transcriptomic data obtained from C. asiatica. According to the results of the phylogenetic analysis (Figure 3A), Casi0095280.1, Casi0032820.1 and Casi0108660.1 had 100.00, 100.00 and 87.62% identities to CaCYP716A83, CaCYP716C11 and CaCYP716A86, respectively. Casi0014680.1 and Casi0066970.1 exhibited 100.00 and 69.58% sequence identities with CaCYP716E41, respectively. Casi0218490.1, Casi0218500.1, Casi0163190.1 and Casi0217210.1 exhibited 94.79, 79.23, 42.27 and 40.32% sequence identities with CaCYP714E19, respectively. Casi0080830.1 and Casi0080840.1 exhibited 64.63 and 63.85% sequence identities with KsCYP72A397, respectively.
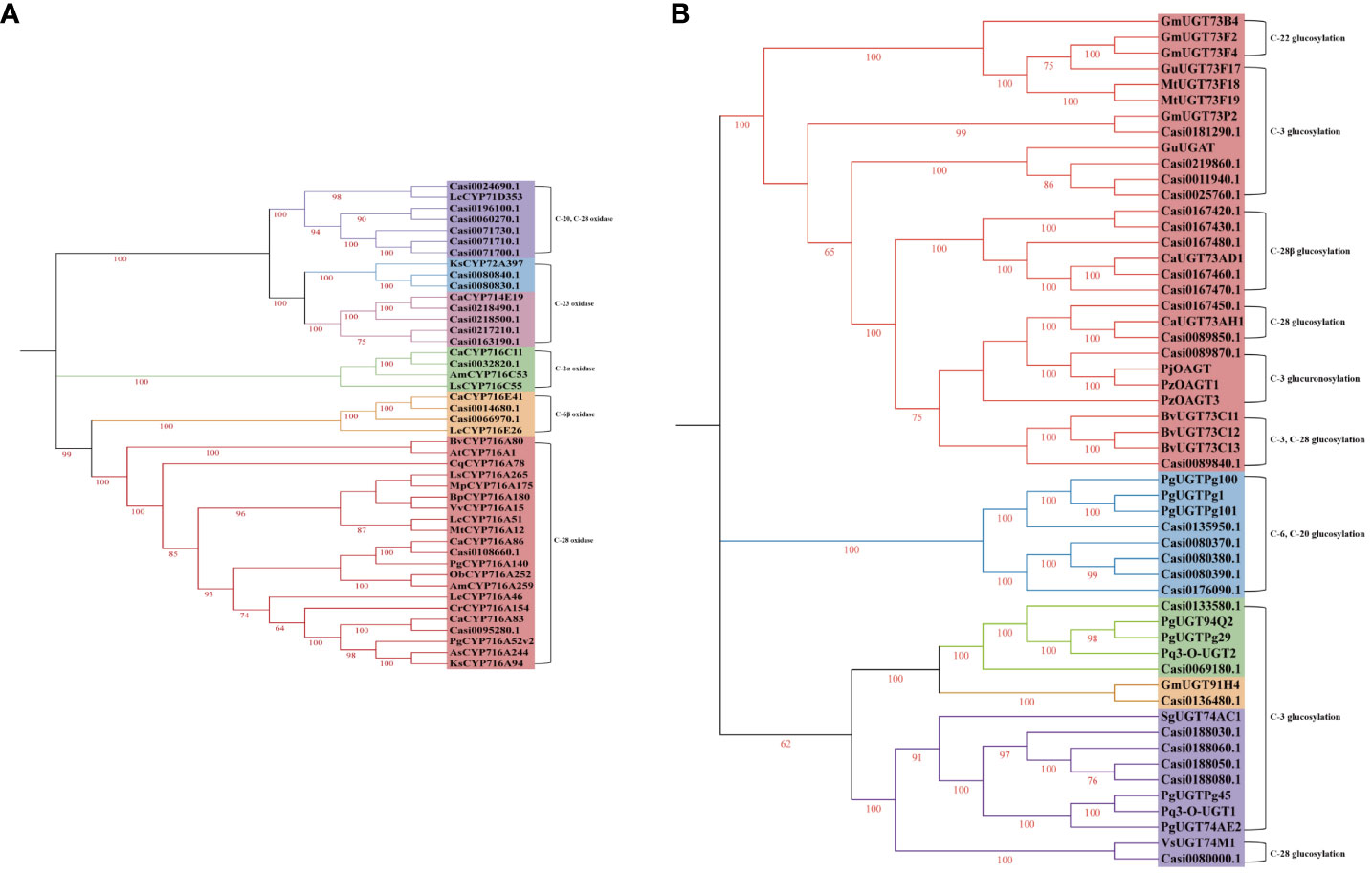
Figure 3 The phylogenetic tree of previously characterized triterpenoid biosynthesis CYP450s (A), UGTs (B) and those CYP450s (A) and UGTs (B) isolated from C. asiatica in this study.
In recent studies, several UGT family members (UGT71, UGT73, UGT74, UGT91 and UGT94) were shown to be associated with triterpenoid saponin biosynthesis. Here, we screened five UGT71, 13 UGT73, five UGT74, one UGT91 and two UGT94 family gene members via the transcriptomic data obtained from C. asiatica. According to the results of the phylogenetic analysis (Figure 3B), Casi0167460.1, Casi0167470.1, Casi0167480.1, Casi0167430.1 and Casi0167420.1 exhibited 99.60, 85.11, 72.95, 52.92 and 47.29% sequence identities with CaUGT73AD1, respectively. Casi0089850.1 and Casi0167450.1 exhibited 100.00 and 64.88% sequence identities with CaUGT73AH1, respectively.
To identify genes related to triterpenoid saponin biosynthesis, low-expressed genes (FPKM value ≤ 0) were filtered out, and a total of 15,808 genes obtained were used for the construction of gene co-expression networks by WGCNA. Our results showed that a soft threshold power of 16 was the lowest power with a scale-free topology model fit index of 0.90 and relatively high mean connectivity (Figure 4A). Gene clustering dendrogram was constructed and detected 13 gene co-expression modules labeled with different colors. Genes in the same module indicated that their expression patterns were similar (Figures 4B, C).
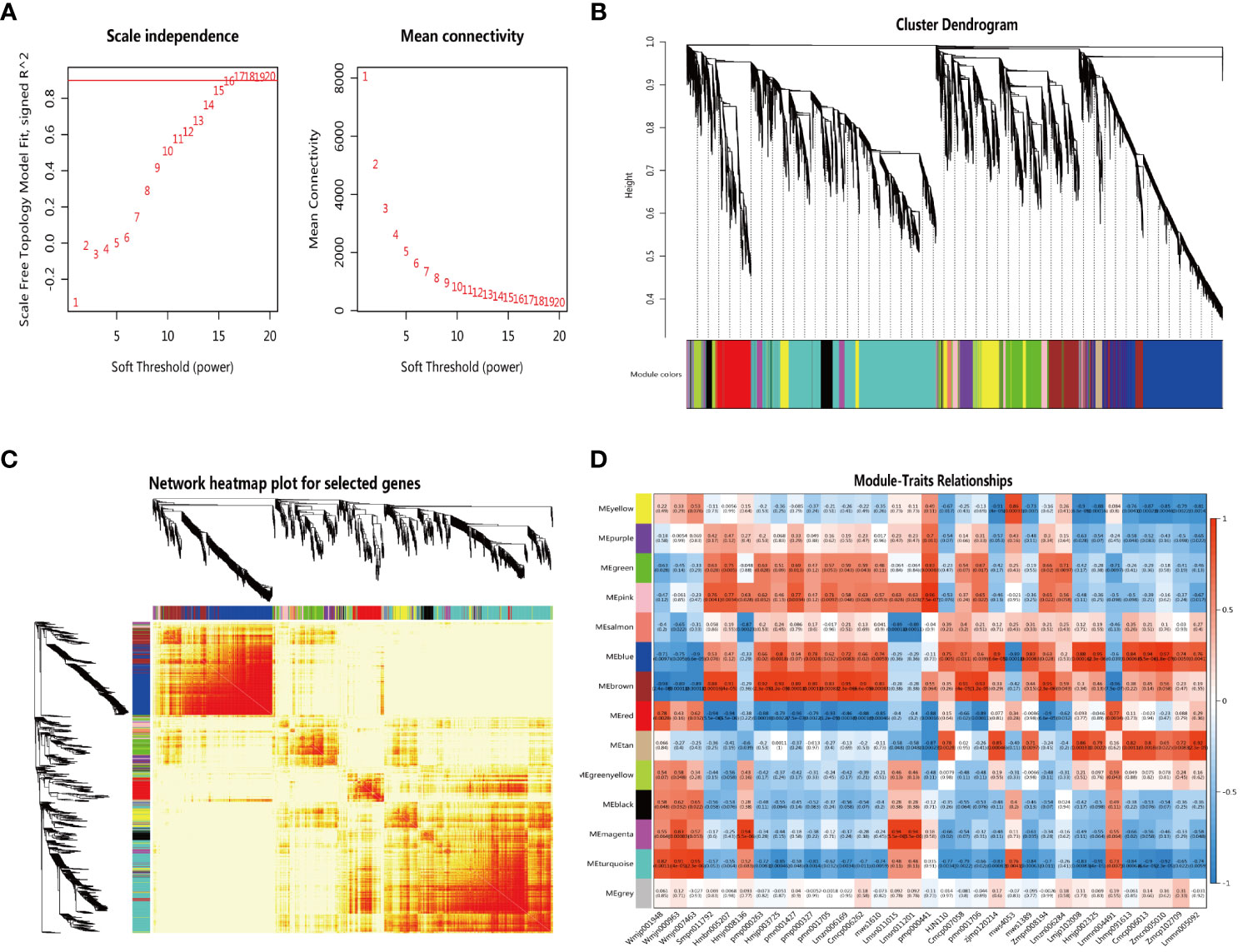
Figure 4 The network topology analysis for different soft threshold powers (A) The gene clustering dendrogram with adjacency-based dissimilarity, together with assigned module colors (B) The construction of module heatmap (C) The construction of the relationships between modules and traits (D).
To determine the correlation relationship between gene co-expression modules and 33 metabolites involved in the triterpenoid saponin biosynthesis, the heatmap of module-traits relationships was constructed (Figure 4D). We observed that the ‘turquoise’ module had the highest correlation with three key precursors in the biosynthesis of terpenoids, DMAPP (Wmjp001948) (r = 0.82, p = 0.0011), IPP (Wmjn000963) (r = 0.91, p < 0.001) and β-amyrin (Wmjn007463) (r = 0.95, p < 0.001). The ‘brown’ module was associated with 18 triterpenes and had the highest correlation with 13 triterpenes (r > 0.83, p < 0.001). The ‘blue’ module was associated with 7 triterpenoid saponins, of which 5 had the highest correlation (r > 0.84, p < 0.001).
The expression levels of the 68 candidate genes and the metabolite concentrations involved in triterpenoid saponin biosynthesis were analyzed by Pearson correlation (Figure 5). The results indicated that DMAPP (Wmjp001948), IPP (Wmjn000963) and β-amyrin (Wmjn007463), the key precursors in the biosynthesis of terpenoids, were significantly and positively correlated with HDR1 (Casi0219000.1) (r = 0.78, p = 0.0026), IDI2 (Casi0022220.1) (r = 0.79, p = 0.0023) and CaβAS1 (Casi0015080.1) (r = 0.91, p < 0.001). Our results showed that Casi0095280.1 and Casi0108660.1 had 100.00 and 87.62% identities to CaCYP716A83 and CaCYP716A86, respectively, and these CYPs catalyzed α-amyrin/β-amyrin to ursolic acid/oleanolic acid. Ursolic acid (mws4053) was positively correlated with Casi0095280.1. In addition, corosolic (mws1610) and maslinic acids (Zmpn008194) showed positive correlations with Casi0032820.1, which had 100.00% identity to CaCYP716C11. Finally, we found that saikosaponin L (Zmcp102709) and medicagenic acid-3-O-glucosyl-(1,6)-glucosyl-(1,3)-glucoside (Lmmn004491) were significantly and positively correlated with Casi0219860.1 (r = 0.85, p = 0.0004) and Casi0011940.1 (r = 0.93, p < 0.001), respectively, both of which were members of UGT73 family and had high degrees of identity with GuUGA.
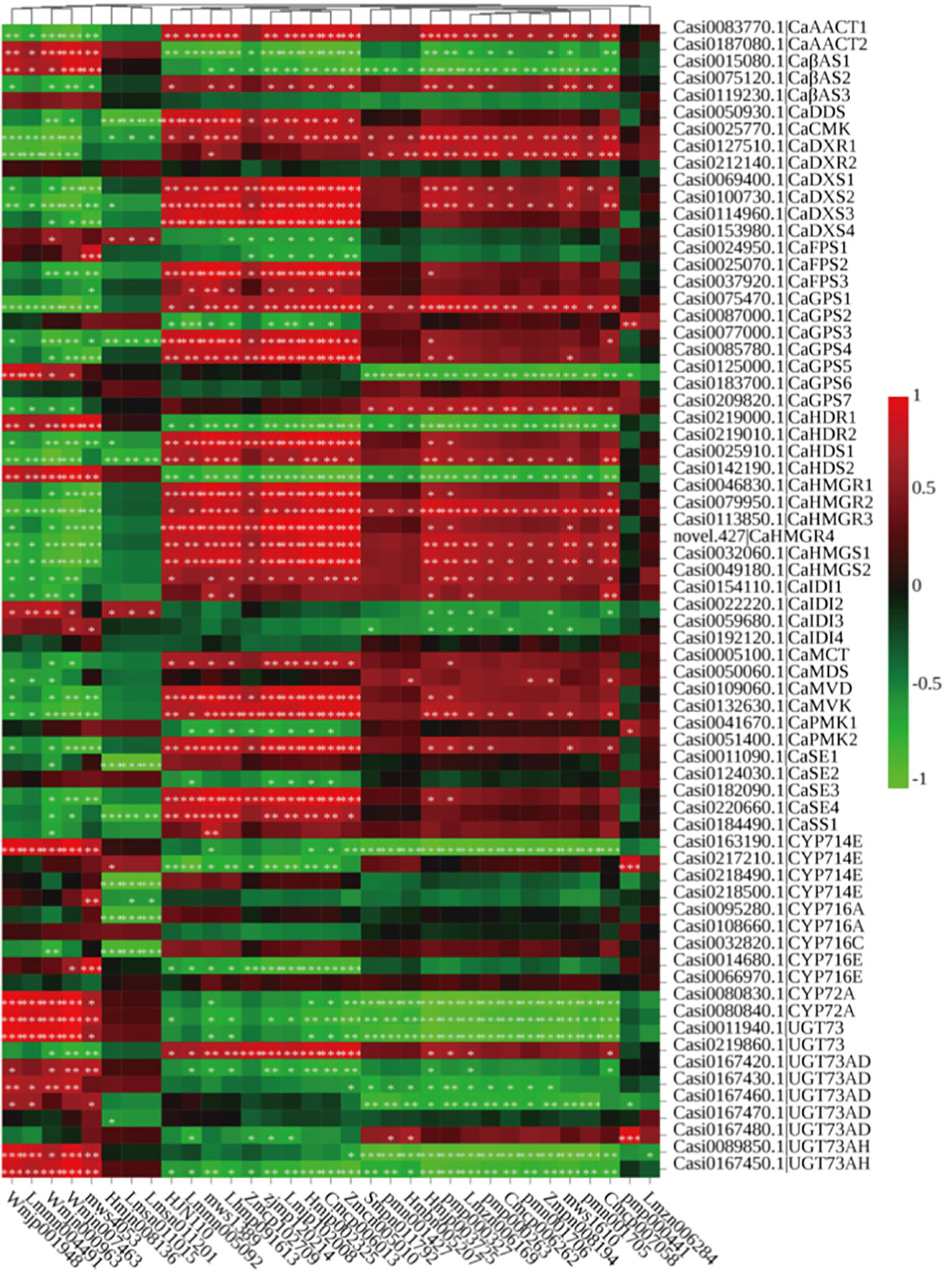
Figure 5 The Pearson correlation analysis between genes and metabolites in the triterpenoid saponin pathway. Significant ones p < 0.05 marked with *, p < 0.01 marked with **, p < 0.001 marked with ***.
To further verify the transcriptomics data obtained, the relative expression levels of 18 genes associated with triterpene and triterpenoid saponin biosynthesis in C. asiatica were selected for analysis by RT-qPCR. These included six genes (CaAACT1, CaHMGS1, CaHMGR2, CaMVK, CaPMK2 and CaMVD) involved in the MVP pathway, four genes (CaDXS1, CaDXR1, CaHDS1 and CaHDR2) involved in the MEP pathway, four genes (CaGPS1, CaFPS2, CaSE3 and CaβAS1) involved in the terpenoid backbone construction stage and four genes (CaCYP714E, CaCYP716A, CaCYP716C and CaUGT73) involved in terpenoid backbone modification. The results showed that the majority of these genes exhibited consistent expression patterns with the transcriptomics data (Figure 6), which supported the analysis we performed.

Figure 6 The qRT-PCR analysis of the potential genes involved in triterpenoid saponin biosynthesis.
The triterpenes and triterpenoid saponins of C. asiatica have been regarded as its essential bioactive compounds due to their important medicinal values in the treatment of various chronic disorders (Prakash et al., 2017; Puttarak et al., 2017). However, previous studies regarding these compounds from C. asiatica have only focused on their chemical structures, bioactivity assessments and cultivation (Prasad et al., 2019). Relatively, few studies regarding the accumulation of metabolites as well as the molecular mechanisms of their biosynthesis in C. asiatica have been conducted. Therefore, it is important to assess the biosynthetic pathways of triterpenes and triterpenoid saponins in C. asiatica in order to achieve the optimal production of these bioactive compounds from the plants. The majority of the triterpenes and triterpenoid saponins are known to be predominantly accumulated in the leaves of C. asiatica (Sangwan et al., 2013). Here, we identified the metabolites and genes of C. asiatica leaves at different growth stages based on transcriptomics and metabolomics data, in order to gain novel insights into the accumulation and biosynthesis of triterpenes and triterpenoid saponins.
Structurally, the triterpenes and triterpenoid saponins in the C. asiatica belong to the terpenoid group, and they have a similar precursor synthesis pathway to those of the other terpenoids. These are thought to be derived through a cross-talk between the MVA and MEP pathways (Patel et al., 2016; Li et al., 2022). We identified 12 genes in the MVA pathway and these involved six key enzymes and 17 genes in the MEP pathway as well as the involvement of eight other key enzymes from our transcriptomics analysis. These genes were further analyzed in combination with the metabolomics analysis, and the expression levels of CaHDR1 (Casi0219000.1) and CaIDI2 (Casi0022220.1) were found to be consistent with the concentrations of DMAPP (Wmjn000963) and IPP (Wmjp001948), respectively.
HDR, the limiting enzyme in the MEP pathway, catalyzes HMBPP into a mixture of 5:1 IPP and DMAPP (Adam et al., 2002). Overexpression of the HDR genes in Arabidopsis has been shown to increase the production of IPP and DMAPP (Botella-Pavía et al., 2010). Both IPP and DMAPP are the essential precursors at regulatory branching points in the biosynthesis of terpenoids (Sun et al., 2010), but the relatively unreactive IPP can be interconverted to its highly electrophilic isomer, DMAPP, by the catalysis of IDI. This compound provides both the electrophilic primer substrate and the condensation substrate for terpenoid biosynthesis (Chen et al., 2012). Therefore, the function of IDI is considered to be the critical first step that controls the overall biosynthesis of all terpenoids (Ramos-Valdivia et al., 1997; Zhu et al., 2014). Previous studies reported that overexpression of the IDI gene generated an accumulation of several related terpenoids by acting downstream in the MEP pathway (Kajiwara et al., 1997; Sun et al., 1998). All these reports suggested that IDI may be a target enzyme for regulating terpenoid biosynthesis (Chen et al., 2012). Our results also showed that the up-regulation of CaHDR1 and CaIDI2 promoted the accumulation of IPP and DMAPP, suggesting that these genes play pivotal roles in the regulation of the biosynthesis of these compounds in C. asiatica.
The OSCs are considered to play key roles in the formation of the different triterpenoid skeletons (Basyuni et al., 2007), because they are the key initial steps for catalyzing 2,3-oxidosqualene cyclization into diverse forms of the triterpenoid skeletons (Mao et al., 2023). This is supported by experiments whereby the enzyme activities were altered and differential effects on triterpene synthesis were observed (Morita et al., 2000). The down-regulation of the expression of the β-AS gene in Panax ginseng hairy roots has also been shown to reduce β-amyrin and oleanane-type ginsenoside (Zhao et al., 2015). In addition, α-AS from Pisum sativum, a multifunctional triterpene synthase, generated α- and β-amyrins by heterologous expression in yeast (Iturbe-Ormaetxe et al., 2003). Similarly, in our study, the expression level of CaβAS1 (Casi0015080.1) was consistent with the concentration of β-amyrin (Wmjn007463). In addition, the sequence of CaβAS1 had a high identity (79.79%) with the known C. asiatica βAS, which indicated that CaβAS1 may catalyze 2,3-oxidosqualene to β-amyrin in C. asiatica.
The triterpenoid skeletons result from carboxyl modification by the CYP450s and then the saccharide side chains are normally introduced by the UGTs leading to the biosynthesis of different triterpene saponins (Xu et al., 2022). The members of CYP450s associated with triterpenoid saponin biosynthesis are mainly the CYP85, CYP72 and CYP71 families. For instance, CYP716s of the CYP85 family catalyze the successive three-step oxidation of α-amyrin, lupel and β-amyrin at C-28 (Shang and Huang, 2019). CYP72s (CYP72A and CYP714E) catalyze the hydroxylation of oleanolic acid/ursolic acid at C-23 (Han et al., 2018) and the members of CYP71D subfamily are involved in the successive three-step oxidation of 20-hydroxylupeol in Lotus japonicus to produce 20-hydroxybetulinic acid (Krokida et al., 2013). It has been shown that CYP716As catalyze the successive three-step oxidation of the α-amyrin/β-amyrin/lupeol backbone to form hydroxyl, aldehyde and carboxyl moieties at C-28 (Xu et al., 2022). In this study, two CYP716As, Casi0095280.1 and Casi0108660.1, showed 100.00 and 87.62% identities to CaCYP716A83 and CaCYP716A86, respectively, and these can catalyze α-amyrin/β-amyrin to ursolic acid/oleanolic acid (Miettinen et al., 2017). Our results also showed that ursolic acid (mws4053) was more abundant at the 20 and 40 day stages of leaf development, which was similar to the expression pattern of Casi0095280.1. In addition, Casi0032820.1 had 100.00% identity to CaCYP716C11, which is known to catalyze the hydroxylation of ursolic acid/oleanolic acid/6β-hydroxy-oleanolic acid at C-2α to corosolic acid/maslinic acid/6β-hydroxy-maslinic acid (Miettinen et al., 2017). In our study, corosolic (mws1610) and maslinic acids (Zmpn008194) from our metabolomics data also showed they accumulated at the 10 and 20-day stages of leaf development, which was consistent with the up-regulation of Casi0032820.1 at JXC1 and JXC2.
It was reported that CaCYP716E41 can catalyze the hydroxylation of ursolic acid/oleanolic acid/maslinic acid at C-6β to 6β-hydroxy-ursolic acid/6β-hydroxy-oleanolic acid/6β-hydroxy-maslinic acid in C. asiatica (Miettinen et al., 2017). Although the products of CaCYP716E41 were not found in our metabolomics data, Casi0014680.1 and Casi0066970.1 exhibited 100.00% and 69.58% sequence identities with CaCYP716E41, respectively, according to our results of the phylogenetic analysis. Therefore, Casi0066970.1 may also be a candidate gene involved in triterpene biosynthesis in C. asiatica.
We also found that four candidate CYP72As (Casi0218490.1, Casi0218500.1, Casi0163190.1 and Casi0217210.1) exhibited 94.79, 79.23, 42.27 and 40.32% sequence identities with CaCYP714E19, respectively. In addtion, Casi0080830.1 and Casi0080840.1 exhibited 64.63 and 63.85% sequence identities with KsCYP72A397, respectively, and it catalyzes the hydroxylation of ursolic acid/oleanolic acid at C-23. This suggested that these CYP72As may catalyze ursolic acid/oleanolic acid to hederagenin/23-hydroxy ursolic acid in C. asiatica (Han et al., 2018; Liu et al., 2019). We also found an accumulation of hederagenin (Hmbn005207). However, the change in expression pattern of this compound was significantly and negatively correlated with Casi0163190.1, Casi0080830.1 and Casi0080830.1. A previous study had shown similar results, where it was found that CqCYP716A15 levels were negatively correlated with the triterpene content of Chenopodium quinoa (Zhao et al., 2022). These data inferred that CYP72As had multiple regulatory roles during the biosynthesis of triterpenes in C. asiatica.
The UGT73 clan is the best candidate family for triterpenoid saponin biosynthesis. We identified five genes (Casi0167460.1, Casi0167470.1, Casi0167480.1, Casi0167430.1 and Casi0167420.1) belonging to the UGT73 clan which showed high degrees of identity with CaUGT73AD1. This enzyme transfers glucuronic acid at C-28β of asiatic acid/madecassic acid to form 28β-Glc-asiatic acid/28β-Glc-madecassic acid (De Costa et al., 2017), suggesting that they catalyze the glucuronosylation of the C28β-hydroxyl group for the biosynthesis of triterpenoid saponins in C. asiatica. In addition, the other two members of the UGT73 clan (Casi0089850.1 and Casi0167450.1) exhibited 100.00 and 64.88% sequence identities with CaUGT73AH1, respectively. This enzyme is known to transfer glucuronic acid at C-28 of asiatic acid/UDP-Glc to form 28-Glc-asiatic acid (Kim et al., 2017). This indicates that these genes may catalyze the glucuronosylation of the C28-hydroxyl group for the biosynthesis of triterpenoid saponins in C. asiatica. However, the expression levels of the above seven UGT73 candidates were not significantly different during the different stages of leaf development. We found 10 triterpenoid saponins glycosylated at the C-28 position from metabolomics data, most of which also showed no significant difference during the different developmental stages. Therefore, these candidate UGT73 genes and their key intermediates involved in the triterpenoid saponin biosynthetic pathway of C. asiatica need to be studied further.
Our study also found two candidate UGT73 DEGs (Casi0219860.1 and Casi0011940.1) which catalyzed glucuronic acid at the C-3 position and showed high degrees of identity to GuUGA (Xu et al., 2016). The expression pattern of Casi0219860.1, which showed a high expression level and accumulation at the 10 day-stage of leaf development, was consistent with the level found in saikosaponin L (Zmcp102709). In addition, the expression pattern of Casi0011940.1, which showed high expression levels and accumulation at the 30 and 40 day-stages of leaf development, was consistent with the level of medicagenic acid-3-O-glucosyl-(1,6)-glucosyl-(1,3)-glucoside (Lmmn004491). This result infers that Casi0219860.1 and Casi0011940.1 are most likely to be involved in the biosynthesis of triterpenoid saponins that are glycosylated at the C-3 position in C. asiatica.
Despite the multiple pharmacological properties associated with triterpenes and triterpenoid saponins found in C. asiatica leaves, relatively few systematic studies have been undertaken to elucidate the biosynthesis of these compounds during their leaf developmental stages. In our investigation, we used the transcriptome and metabolome data to perform a comprehensive analysis to reveal the dynamic patterns of triterpenoid saponin accumulation and the identity of the key candidate genes associated with their biosynthesis in C. asiatica leaves. We found that the decrease in levels of the majority of triterpenes occurred only at the late stages of leaf development. However, the majority of triterpenoid saponins rapidly accumulated at early stages of leaf development, and then decreased but remained constant during the later periods. The levels of triterpenes and triterpenoid saponins showed different dynamic patterns at different leaf growth stages, indicating that the whole triterpenoid saponin biosynthetic pathway is relatively complex. Furthermore, 48 genes involved in the synthesis of the terpenoid backbone as well as 17 CYP450s and 26 UGTs genes were found to be associated with the backbone modifications of these compounds. By integrating metabolomics and transcriptomics analyses, we identified five candidate genes as having crucial roles in the biosynthesis of triterpenoid saponins in C. asiatica. The relative expression levels of these key genes were further verified by qRT-PCR. Overall, these results will provide a valuable reference for further research on the molecular mechanisms associated with triterpenoid saponin biosynthesis in C. asiatica as well as other medicinal plants.
The datasets presented in this study can be found in online repositories. The names of the repositories and accession number can be found at the Genome Sequence Archive (GSA), accession number: CRA012378 by using following link: https://ngdc.cncb.ac.cn/search/?dbId=gsa&q=CRA012378.
LW: Conceptualization, Data curation, Funding acquisition, Investigation, Writing – original draft. QH: Writing – original draft. CL: Investigation, Writing – review & editing, Funding acquisition. HY: Investigation, Writing – review & editing. GT: Data curation, Funding acquisition, Investigation, Writing – review & editing. SW: Data curation, Writing – review & editing. AE-S: Writing – review & editing. SS: Writing – review & editing. KZ: Investigation, Writing – review & editing. LP: Investigation, Writing – review & editing. ZZ: Data curation, Funding acquisition, Writing – review & editing. ML: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Natural Science Foundation of Guangxi Province (2023GXNSFAA026330 and 2020GXNSFAA159151), Guangxi Science and Technology Base and Special Talents (Guike AD22035026), Scientific Research Funding Project of Guangxi Botanical Garden of Medicinal Plants (GYJ202008), Innovative Team for Traditional Chinese Medicinal Materials Quality of Guangxi (GZKJ2305) and the Key Laboratory Construction Program of Guangxi Health commission (ZJC2020003).
We are very grateful to the kind administration of Guangxi Botanical Garden of Medicinal Plants, Nanning, China for providing us such a prestigious and well-equipped platform for research and development.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1295186/full#supplementary-material
Supplementary Figure 1 | The C. asiatica leaves at four different growth stages.
Supplementary Figure 2 | A correlation heatmap of all the samples (A); The metabolomics sample replicates resulting from PCA analysis (B).
Supplementary Figure 3 | The KEGG pathway annotation of the metabolites detected (A); The KEGG pathway annotations of the differentially expressed genes (B).
Supplementary Figure 4 | The hierarchical clustering for the dynamic change pattern of triterpenes (A) and triterpenoid saponins (B) in the C. asiatica at four leaf growth stages.
Supplementary Figure 5 | Upper: temporal expression patterns of the differentially accumulated triterpenes, Lower: a heatmap of triterpenes accumulation levels in different clusters (A); Upper: temporal expression patterns of the differentially accumulated triterpenoid saponins, Lower: a heatmap of the triterpenoid saponins accumulation levels in different clusters (B).
Supplementary Figure 6 | The transcriptomic sample replicates resulting from PCA analysis.
Supplementary Figure 7 | The GO classification of the differentially expressed genes.
Supplementary Figure 8 | The phylogenetic tree of A-type (A) and non-A-type (B) CYP450s from C. asiatica and Arabidopsis. The representative CYP450 family members from C. asiatica and Arabidopsis are marked with red circles and blue triangles, respectively.
Supplementary Figure 9 | The phylogenetic tree of UGTs from C. asiatica and Arabidopsis. The representative UGT family members from C. asiatica and Arabidopsis are marked with red circles and blue triangles, respectively.
Adam, P., Hecht, S., Eisenreich, W., Kaiser, J., Gräwert, T., Arigoni, D., et al. (2002). Biosynthesis of terpenes: studies on 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase. Proc. Natl. Acad. Sci. 99, 12108–12113. doi: 10.1073/pnas.182412599
Aharoni, A., Jongsma, M. A., Kim, T. Y., Ri, M. B., Giri, A. P., Verstappen, F. W. A., et al. (2006). Metabolic engineering of terpenoid biosynthesis in plants. Phytochem. Rev. 5, 49–58. doi: 10.1007/s11101-005-3747-3
Augustin, J. M., Kuzina, V., Andersen, S. B., Bak, S. (2011). Molecular activities, biosynthesis and evolution of triterpenoid saponins. Phytochem. 72, 435–457. doi: 10.1016/j.phytochem.2011.01.015
Basyuni, M., Oku, H., Tsujimoto, E., Kinjo, K., Baba, S., Takara, K. (2007). Triterpene synthases from the Okinawan mangrove tribe, Rhizophoraceae. FEBS J. 274, 5028–5042. doi: 10.1111/j.1742-4658.2007.06025.x
Bhavna, D., Jyoti, K. (2011). Centella asiatica: the elixir of life. Int. J. Res. Ayurveda. Pharm. 2, 431–438.
Botella-Pavía, P., Besumbes, Ó., Phillips, M. A., Carretero-Paulet, L., Boronat, A., Rodríguez-Concepción, M. (2010). Regulation of carotenoid biosynthesis in plants: evidence for a key role of hydroxymethylbutenyl diphosphate reductase in controlling the supply of plastidial isoprenoid precursors. Plant J. 40, 188–199. doi: 10.1111/j.1365-313X.2004.02198.x
Brinkhaus, B., Lindner, M., Schuppan, D., Hahn, E. G. (2000). Chemical, pharmacological and clinical profile of the East Asian medical plant Centella aslatica. Phytomedicine 7, 427–448. doi: 10.1016/s0944-7113(00)80065-3
Cárdenas, P. D., Almeida, A., Bak, S. (2019). Evolution of structural diversity of triterpenoids. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.01523
Chang, A., Pei, W., Li, S., Wang, T., Song, H., Kang, T., et al. (2022). Integrated metabolomic and transcriptomic analysis reveals variation in the metabolites and genes of Platycodon grandiflorus roots from different regions. Phytochem. Anal. 33, 982–994. doi: 10.1002/pca.3153
Chen, R., Harada, Y., Bamba, T., Nakazawa, Y., Gyokusen, K. (2012). Overexpression of an isopentenyl diphosphate isomerase gene to enhance trans-polyisoprene production in Eucommia ulmoides Oliver. BMC Biotechnol. 12, 78. doi: 10.1186/1472-6750-12-78
De Costa, F., Barber, C. J., Kim, Y., Reed, D. W., Zhang, H., Fett-Neto, A. G., et al. (2017). Molecular cloning of an ester-forming triterpenoid: UDP-glucose 28-O-glucosyltransferase involved in saponin biosynthesis from the medicinal plant Centella asiatica. Plant Sci. 262, 9–17. doi: 10.1016/j.plantsci.2017.05.009
Fukushima, E. O., Seki, H., Ohyama, K., Ono, E., Umemoto, N., Mizutani, M., et al. (2011). CYP716A subfamily members are multifunctional oxidases in triterpenoid biosynthesis. Plant Cell Physiol. 52 (12), 2050–2061. doi: 10.1093/pcp/pcr146
Han, J. Y., Chun, J.-H., Oh, S. A., Park, S.-B., Hwang, H.-S., Lee, H., et al. (2018). Transcriptomic analysis of Kalopanax septemlobus and characterization of KsBAS, CYP716A94 and CYP72A397 genes involved in hederagenin saponin biosynthesis. Plant Cell Physiol. 59, 319–330. doi: 10.1093/pcp/pcx188
Han, X. Y., Zhao, J. Y., Chang, X. C., Li, Q. Y., Deng, Z. X., Yu, Y. (2022). Revisiting the transcriptome data of Centella asiatica identified an ester-forming triterpenoid: UDP-glucose 28-O-glucosyltransferase. Tetrahedron 129, 133136. doi: 10.1016/j.tet.2022.133136
Haralampidis, K., Trojanowska, M., Osbourn, A. E. (2002). Biosynthesis of triterpenoid saponins in plants. Adv. Biochem. Eng. Biotechnol. 75, 31–49. doi: 10.1007/3-540-44604-4_2
Huang, Z. J., Liu, D. Q., Ge, F., Chen, C. Y. (2014). Advances in studies on key post-modification enzymes in triterpenoid saponins biosynthesis. Acta Botanica Boreali-Occidentalia Sin. 34, 2137–2144. doi: 10.7606/j.issn.1000.2014.10.2137
Iturbe-Ormaetxe, I., Haralampidis, K., Papadopoulou, K., Osbourn, A. E. (2003). Molecular cloning and characterization of triterpene synthases from Medicago truncatula and Lotus japonicus. Plant Mol. Biol. 51, 731–743. doi: 10.1023/A:1022519709298
James, J. T., Dubery, I. A. (2009). Pentacyclic triterpenoids from the medicinal herb, Centella asiatica (L.) Urban. Molecules 14, 3922–3941. doi: 10.3390/molecules14103922
Kajiwara, S., Fraser, P. D., Kondo, K., Misawa, N. (1997). Expression of an exogenous isopentenyl diphosphate isomerase gene enhances isoprenoid biosynthesis in Escherichia coli. Biochem. J. 324 (Pt 2), 421–426. doi: 10.1042/bj3240421
Kalita, R., Modi, M. K., Sen, P. (2018). RNAi mediated silencing of 3-hydroxy-3-methylglutaryl-CoA reductases (HMGR) in Centella asiatica. Gene Rep. 11, 52–57. doi: 10.1016/j.genrep.2018.02.004
Kalita, R., Patar, L., Shasany, A. K., Modi, M. K., Sen, P. (2015). Molecular cloning, characterization and expression analysis of 3-hydroxy-3-methylglutaryl coenzyme A reductase gene from Centella asiatica L. Mol. Biol. Rep. 42, 1431–1439. doi: 10.1007/s11033-015-3922-6
Kim, O. T., Ahn, J. C., Hwang, S. J., Hwang, B. (2005a). Cloning and expression of a farnesyl diphosphate synthase in Centella asiatica (L.) Urban. Mol. Cell 19, 294–299.
Kim, O. T., Jin, M. L., Lee, D. Y., Jetter, R. (2017). Characterization of the asiatic acid glucosyltransferase, UGT73AH1, involved in asiaticoside biosynthesis in Centella asiatica (L.) Urban. Int. J. Mol. Sci. 18, 2630. doi: 10.3390/ijms18122630
Kim, O. T., Kim, M. Y., Huh, S. M., Bai, D. G., Ahn, J. C., Hwang, B. (2005c). Cloning of a cDNA probably encoding oxidosqualene cyclase associated with asiaticoside biosynthesis from Centella asiatica (L.) Urban. Plant Cell Rep. 24, 304–311. doi: 10.1007/s00299-005-0927-y
Kim, O. T., Seong, N. S., Kim, M. Y., Hwang, B. (2005b). Isolation and characterization of squalene synthase cDNA from Centella asiatica (L.) Urban. J. Plant Biol. 48, 263–269. doi: 10.1007/BF03030521
Kim, O. T., Um, Y., Jin, M. L., Kim, J. U., Hegebarth, D., Busta, L., et al. (2018). A novel multifunctional C-23 oxidase, CYP714E19, is involved in asiaticoside biosynthesis. Plant Cell Physiol. 59 (6), 1200–1213. doi: 10.1093/pcp/pcy055
Krokida, A., Delis, C., Geisler, K., Garagounis, C., Tsikou, D., Peña-Rodríguez, L. M., et al. (2013). A metabolic gene cluster inLotus japonicusdiscloses novel enzyme functions and products in triterpene biosynthesis. New Phytol. 200, 675–690. doi: 10.1111/nph.12414
Kuwahara, Y., Nakajima, D., Shinpo, S., Nakamura, M., Kawano, N., Kawahara, N., et al. (2019). Identification of potential genes involved in triterpenoid saponins biosynthesis in Gleditsia sinensis by transcriptome and metabolome analyses. J. Nat. Med. 73, 369–380. doi: 10.1007/s11418-018-1270-2
Li, Y., Zhao, J., Chen, H., Mao, Y., Yang, Y., Feng, L., et al. (2022). Transcriptome level reveals the triterpenoid saponin biosynthesis pathway of Bupleurum falcatum L. Genes 13, 2237. doi: 10.3390/genes13122237
Liu, M., Dai, Y., Li, Y., Luo, Y., Huang, F., Gong, Z., et al. (2008). Madecassoside isolated from Centella asiatica herbs facilitates burn wound healing in mice. Planta Med. 74, 809–815. doi: 10.1055/s-2008-1074533
Liu, Q., Khakimov, B., Cárdenas, P. D., Cozzi, F., Olsen, C. E., Jensen, K. R., et al. (2019). The cytochrome P450 CYP72A552 is key to production of hederagenin-based saponins that mediate plant defense against herbivores. New Phytol. 222, 1599–1609. doi: 10.1111/nph.15689
Luo, Z. L., Zhang, K. L., Ma, X. J., Guo, Y. H. (2016). Research progress in synthetic biology of triterpen saponins. Chin. Tradit. Herbal Drugs 47, 1806–1814. doi: 10.7501/j.issn.0253-2670.2016.10.029
Mao, Y., Chen, H., Zhao, J., Li, Y., Feng, L., Yang, Y., et al. (2023). Molecular cloning, functional characterization and expression of the β-amyrin synthase gene involved in saikosaponin biosynthesis in Bupleurum chinense DC. J. Plant Biochem. Biotechnol. 32, 284–295. doi: 10.1007/s13562-022-00804-2
Matsuda, H., Morikawa, T., Ueda, H., Yoshikawa, M. (2001). Medicinal foodstuffs. XXVII. Saponin constituents of gotu kola (2): structures of new ursane-and oleanane-type triterpene oligoglycosides, centellasaponins B, C, and D, from Centella asiatica cultivated in Sri Lanka. Chem. Pharm. Bull. 49, 1368–1371. doi: 10.1248/cpb.49.1368
Miettinen, K., Pollier, J., Buyst, D., Arendt, P., Csuk, R., Sommerwerk, S., et al. (2017). The ancient CYP716 family is a major contributor to the diversification of eudicot triterpenoid biosynthesis. Nat. Commun. 8, 14153. doi: 10.1038/ncomms14153
Morita, M., Shibuya, M., Kushiro, T., Masuda, K., Ebizuka, Y. (2000). Molecular cloning and functional expression of triterpene synthases from pea (Pisum sativum) New α-amyrin-producing enzyme is a multifunctional triterpene synthase. Eur. J. Biochem. 267, 3453–3460. doi: 10.1046/j.1432-1327.2000.01357.x
Nguyen, K. V., Pongkitwitoon, B., Pathomwichaiwat, T., Viboonjun, U., Prathanturarug, S. (2019). Effects of methyl jasmonate on the growth and triterpenoid production of diploid and tetraploid Centella asiatica (L.) Urb. hairy root cultures. Sci. Rep. 9, 18665. doi: 10.1038/s41598-019-54460-z
Patel, K., Mishra, R., Patel, D. K. (2016). A review on phytopharmaceutical importance of asiaticoside. J. Coast. Life Med. 4, 1000–1007. doi: 10.12980/jclm.4.2016J6-161
Phillips, D. R., Rasbery, J. M., Bartel, B., Matsuda, S. P. (2006). Biosynthetic diversity in plant triterpene cyclization. Curr. Opin. Plant Biol. 9, 305–314. doi: 10.1016/j.pbi.2006.03.004
Pootakham, W., Naktang, C., Kongkachana, W., Sonthirod, C., Yoocha, T., Sangsrakru, D., et al. (2021). De novo chromosome-level assembly of the Centella asiatica genome. Genomics 113 (4), 2221–2228. doi: 10.1016/j.ygeno.2021.05.019
Prakash, V., Jaiswal, N., Srivastava, M. (2017). A review on medicinal properties of Centella asiatica. Asian J. Pharm. Clin. Res. 10, 69–74. doi: 10.22159/ajpcr.2017.v10i10.20760
Prasad, A., Mathur, A. K., Mathur, A. (2019). Advances and emerging research trends for modulation of centelloside biosynthesis in Centella asiatica (L.) Urban-A review. Ind. Crops Prod. 141, 111768. doi: 10.1016/j.indcrop.2019.111768
Puttarak, P., Dilokthornsakul, P., Saokaew, S., Dhippayom, T., Chaiyakunapruk, N. (2017). Effects of Centella asiatica (L.) Urb. on cognitive function and mood related outcomes: A Systematic Review and Meta-analysis. Sci. Rep. 7, 10646. doi: 10.1038/s41598-017-09823-9
Ramos-Valdivia, A. C., van der Heijden, R., Verpoorte, R. (1997). Isopentenyl diphosphate isomerase: a core enzyme in isoprenoid biosynthesis. A review of its biochemistry and function. Nat. Prod. Rep. 14, 591–603. doi: 10.1039/NP9971400591
Saito, K., Matsuda, F. (2010). Metabolomics for functional genomics, systems biology, and biotechnology. Annu. Rev. Plant Biol. 61, 463–489. doi: 10.1146/annurev.arplant.043008.092035
Sangwan, R. S., Tripathi, S., Singh, J., Narnoliya, L. K., Sangwan, N. S. (2013). De novo sequencing and assembly of Centella asiatica leaf transcriptome for mapping of structural, functional and regulatory genes with special reference to secondary metabolism. Gene 525, 58–76. doi: 10.1016/j.gene.2013.04.057
Sawai, S., Saito, K. (2011). Triterpenoid biosynthesis and engineering in plants. Front. Plant Sci. 2. doi: 10.3389/fpls.2011.00025
Seki, H., Tamura, K., Muranaka, T. (2015). P450s and UGTs: key players in the structural diversity of triterpenoid saponins. Plant Cell Physiol. 56, 1463–1471. doi: 10.1093/pcp/pcv062
Shang, Y., Huang, S. (2019). Multi-omics data-driven investigations of metabolic diversity of plant triterpenoids. Plant J. 97, 101–111. doi: 10.1111/tpj.14132
Sun, Z., Cunningham, F. X., Jr., Gantt, C. E. (1998). Differential expression of two isopentenyl pyrophosphate isomerases and enhanced carotenoid accumulation in a unicellular chlorophyte. Proc. Natl. Acad. Sci. U.S.A 95, 14585–14585. doi: 10.1073/pnas.95.19.11482
Sun, J., Zhang, Y. Y., Liu, H., Zou, Z., Zhang, C.-J., Zhang, X. H., et al. (2010). A novel cytoplasmic isopentenyl diphosphate isomerase gene from tomato (Solanum lycopersicum): Cloning, expression, and color complementation. Plant Mol. Biol. Rep. 28, 473–480. doi: 10.1007/s11105-009-0174-4
Thimmappa, R., Geisler, K., Louveau, T., O’Maille, P., Osbourn, A. (2014). Triterpene biosynthesis in plants. Annu. Rev. Plant Biol. 65, 225–257. doi: 10.1146/annurev-arplant-050312-120229
Wang, R., Ren, C., Dong, S., Chen, C., Xian, B., Wu, Q., et al. (2021). Integrated metabolomics and transcriptome analysis of flavonoid biosynthesis in safflower (Carthamus tinctorius L.) with different colors. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.712038
Wei, G., Dong, L., Yang, J., Zhang, L., Xu, J., Yang, F., et al. (2018). Integrated metabolomic and transcriptomic analyses revealed the distribution of saponins in Panax notoginseng. Acta Pharm. Sin. B 8, 458–465. doi: 10.1016/j.apsb.2017.12.010
Wen, L., Yun, X., Zheng, X., Xu, H., Zhan, R., Chen, W., et al. (2017). Transcriptomic comparison reveals candidate genes for triterpenoid biosynthesis in two closely related Ilex species. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00634
Weng, X. X., Zhang, J., Gao, W., Cheng, L., Shao, Y., Kong, D. Y. (2012). Two new pentacyclic triterpenoids from Centella asiatica. Helv. Chim. Acta 95, 255–260. doi: 10.1002/hlca.201100287
Xu, G., Cai, W., Gao, W., Liu, C. (2016). A novel glucuronosyltransferase has an unprecedented ability to catalyse continuous two-step glucuronosylation of glycyrrhetinic acid to yield glycyrrhizin. New Phytol. 212, 123–135. doi: 10.1111/nph.14039
Xu, R., Fazio, G. C., Matsuda, S. P. (2004). On the origins of triterpenoid skeletal diversity. Phytochem. 65, 261–291. doi: 10.1016/j.phytochem.2003.11.014
Xu, Y., Zhao, G., Ji, X., Liu, J., Zhao, T., Gao, Y., et al. (2022). Metabolome and transcriptome analysis reveals the transcriptional regulatory mechanism of triterpenoid saponin biosynthesis in soapberry (Sapindus mukorossi Gaertn.). J. Agric. Food Chem. 70, 7095–7109. doi: 10.1021/acs.jafc.2c01672
Yang, J. L., Hu, Z. F., Zhang, T. T., Gu, A. D., Gong, T., Zhu, P. (2018). Progress on the studies of the key enzymes of ginsenoside biosynthesis. Molecules 23, 589. doi: 10.3390/molecules23030589
Yuan, Y., Zuo, J., Zhang, H., Li, R., Yu, M., Liu, S. (2022). Integration of transcriptome and metabolome provides new insights to flavonoids biosynthesis in Dendrobium huoshanense. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.850090
Zhao, Y., Ma, Y., Li, J., Liu, B., Liu, X., Zhang, J., et al. (2022). Transcriptomics–metabolomics joint analysis: New highlight into the triterpenoid saponin biosynthesis in quinoa (Chenopodium quinoa Willd.). Front. Plant Sci. 13. doi: 10.3389/fpls.2022.964558
Zhao, C., Xu, T., Liang, Y., Zhao, S., Ren, L., Wang, Q., et al. (2015). Functional analysis of β-amyrin synthase gene in ginsenoside biosynthesis by RNA interference. Plant Cell Rep. 34, 1307–1315. doi: 10.1007/s00299-015-1788-7
Keywords: Centella asiatica, transcriptome, metabolome, triterpenoid saponin, candidate gene
Citation: Wan L, Huang Q, Li C, Yu H, Tan G, Wei S, El-Sappah AH, Sooranna S, Zhang K, Pan L, Zhang Z and Lei M (2024) Integrated metabolome and transcriptome analysis identifies candidate genes involved in triterpenoid saponin biosynthesis in leaves of Centella asiatica (L.) Urban. Front. Plant Sci. 14:1295186. doi: 10.3389/fpls.2023.1295186
Received: 15 September 2023; Accepted: 22 December 2023;
Published: 12 January 2024.
Edited by:
Gao Jihai, Chengdu University of Traditional Chinese Medicine, ChinaReviewed by:
Kewang Xu, Nanjing Forestry University, ChinaCopyright © 2024 Wan, Huang, Li, Yu, Tan, Wei, El-Sappah, Sooranna, Zhang, Pan, Zhang and Lei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Lei, bGVpbWluZ0BneHl5end5LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.